
Understanding the structural landscape of Mn-based MOFs formed with hinged pyrazole carboxylate linkers†
Josephine F.
Smernik
a,
Pol
Gimeno-Fonquernie
 a,
Jorge
Albalad
a,
Tyla S.
Jones
a,
Rosemary J.
Young
ab,
Neil R.
Champness
bc,
Christian J.
Doonan
a,
Jack D.
Evans
a and
Christopher J.
Sumby
*a
a,
Jorge
Albalad
a,
Tyla S.
Jones
a,
Rosemary J.
Young
ab,
Neil R.
Champness
bc,
Christian J.
Doonan
a,
Jack D.
Evans
a and
Christopher J.
Sumby
*a
aDepartment of Chemistry and the Centre for Advanced Nanomaterials, School of Physics, Chemistry and Earth Sciences, The University of Adelaide, Adelaide, SA 5005, Australia. E-mail: christopher.sumby@adelaide.edu.au
bSchool of Chemistry, The University of Nottingham, Nottingham, UK
cSchool of Chemistry, The University of Birmingham, Birmingham, UK
First published on 28th September 2023
Abstract
Metal–organic frameworks (MOFs) capable of post-synthetic metalation (PSMet) have garnered significant interest as supports for catalytic metals. The Mn-based MOF, MnMOF-1 ([Mn3(L2Me)3] where L2Me = bis-(4-carboxyphenyl-3,5-dimethylpyrazolyl)methane), has been an exemplar for studying PSMet. Herein we investigate the synthesis of Mn-based MOFs from related flexible ditopic pyrazole carboxylate links, along with the formation of MOFs with similar tetratopic hinged linkers. We show for the first time that MnMOF-1 is likely a kinetic or metastable phase and a newly identified 2D layered material (MnMOF-2D) is the thermodynamically favoured product for this metal–linker combination. Formation of a MnMOF-1 structure with shorter linkers is thwarted by steric clashes that preclude the formation of the Mn3 cluster. This observation prompted the use of density functional theory (DFT) simulations that showed the target material to be very dense, highly strained and thereby energetically unfavourable, but potentially, a hypothetical MnMOF-1 structure with a longer phenylethynyl spacer would be energetically feasible. Finally, the predominance of 2D MOFs formed with shorter flexible links encouraged us to use tetratopic hinged linkers to form 3D frameworks, which was vindicated by the successful synthesis of two new porous 3D Mn-based MOFs, MnMOF-L4 and MnMOF-L5. These results highlight that reticular synthesis of MOFs formed with flexible, non-linear linkers is challenging.
Introduction
Large internal surface areas and pore volumes, and chemically mutable structures have earmarked metal–organic frameworks (MOFs) as materials of interest for applications in gas storage,1,2 separations1,3 and catalysis.4,5 MOFs can be prepared from combinations of metal ions (nodes) and organic linkers, which connect the nodes into an extended structure.6 A salient feature of MOFs is their reticular design principles whereby isoreticular materials can be synthesised by changing the structure metrics of the linker.2,7 Exemplars of this approach abound in materials such as the IRMOF series,8 and the zirconium-based Universitetet i Oslo (UiO) frameworks.9 While families of isoreticular MOFs are not exclusively associated with linkers possessing rigid structures,10,11 there are limited examples constructed from flexible organic building blocks, such as pyrazole carboxylate linkers (Fig. 1a), and often the flexibility is introduced through post-synthetic modification.12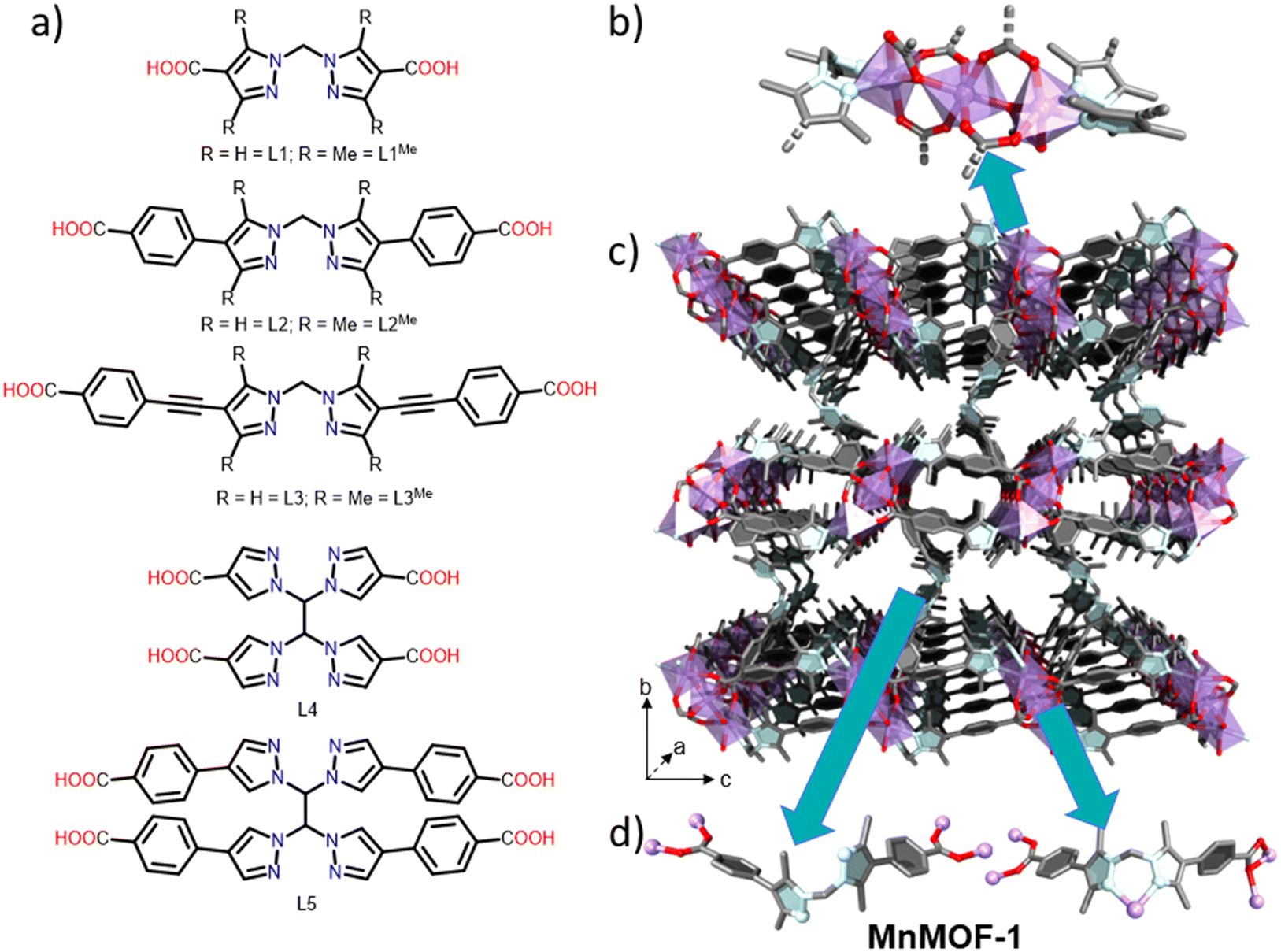 | ||
| Fig. 1 a) Summary of the ligands used, with all featuring bispyrazole and carboxylate coordinating groups. b–d) Structure of MnMOF-1: b) MnMOF-1 trinuclear manganese node capped by two bispyrazole groups. c) MnMOF-1 structure where 2D layers containing clusters and bispyrazole-coordinated ligands are connected to each other through ligands with non-coordinated bispyrazole groups. d) The two ligand types present in the structure of MnMOF-1. The first is a bridging ligand with non-coordinated bispyrazole groups and the second is the ligand present within the layers whose bispyrazole groups cap the Mn clusters. | ||
An example of a MOF made from a flexible linker is MnMOF-1 ([Mn3(L2Me)3], Fig. 1).13 MnMOF-1 is a 3D MOF (Fig. 1c) comprising flexible layers, formed from Mn trinuclear nodes (Fig. 1b) and coordinatively saturated flexible linkers (L2Me, Fig. 1a); finally, the Mn trinuclear nodes in the layers are bridged by a third molecule of L2Me possessing a vacant bis-pyrazole coordinating site (Fig. 1d). The material shows a significant degree of structural flexibility, converting from open and closed forms in response to solvent removal or exposure;14 and undergoes quantitative post-synthetic metalation (PSMet) with transition metals that modulate its flexibility.13,15 Coupled with the ability to reliably undertake single crystal X-ray diffraction (SCXRD) on the MOF crystals after PSMet, this MOF has enabled the study of novel adsorption switching processes,15 insights into reactivity of catalytic complexes,16–18 and photochemical reactions.19
The node in MnMOF-1 has been observed previously,20–22 and comprises a Mn trinuclear node that is bridged by six carboxylates and capped by four nitrogen donor atoms (Fig. 1b). Due to the relevance of Mn clusters in biology and magnetism,20,23 Mn3(O2CR)6(L)4 (R = acetate, benzoate; L = neutral O or N donor) and a variety of clusters formed from [Mn3O-(O2CR)6(L)3]2+ have been studied. Numerous Mn-based MOFs are also known including Mn2(dhtp) (Mn-MOF-74, Mn(dobdc), CPO-27(Mn)),24 Mn azolate materials (e.g. MAF-X25, [(Mn)2Cl2(BBTA)] where BBTA = benzo[1,2-d:4,5-d′]bis([1,2,3]triazole)-1,5-diide),25 and [Mn3(atpa)2(Hatpa)2] (H2atpa = 2-aminoterephthalic acid), the latter of which has a trinuclear node similar to that in MnMOF-1 but with all carboxylate donors.26
With this background, we set out to explore the possibility of reticulating the MnMOF-1 structure and to develop a further understanding of the principles associated with the formation of permanently porous MOFs from flexible linkers. Prior to this, we re-evaluated our synthetic protocols for MnMOF-1 and identified this as a likely kinetic or metastable phase which, depending on conditions, is in competition with formation of a 2D layered material, MnMOF-2D [Mn2(L2Me)2(H2O)2]. We report the synthetic conditions needed to exclusively form those two materials in a reproducible manner from L2Me. Turning to attempts to reticulate the MnMOF-1 structure, we encountered a range of competing phases with shorter links (L1) and significant solubility challenges with the longer variants (L3). These synthetic challenges prompted us to consider the accessibility of these structures by computational means which indicated that the MnMOF-1 structure is only strain-free and sterically possible for the intermediate length linkers (L2 and L2Me) and potentially the extended ligands L3 and L3Me. Finally, 2D frameworks formed with shorter link L1 suggested that a covalent linking strategy – that connects the hinge carbons of the ditopic linkers – might be a suitable approach to form additional 3D MOFs with Mn(II)-based nodes. Herein, we additionally report the synthesis, structures, and properties of MnMOF-L4 and MnMOF-L5, illustrating that permanently porous 3D frameworks from flexible linkers are achievable through this strategy.
Experimental
General experimental and characterisation
Unless otherwise stated, all chemicals were obtained from commercial sources and used as received. Solvents were dried using literature procedures and degassed with Ar prior to use. The ligands bis-(4-carboxypyrazolyl)methane (L1),27 bis-[4-(4-carboxyphenyl)pyrazolyl]methane (L2),28 bis-[4-(4-carboxyphenyl)-3,5-dimethylpyrazolyl]methane (L2Me),29 bis-[4-(4-carboxyphenylethynyl)pyrazolyl]methane (L3),30 and bis-[4-(4-carboxyphenylethynyl)-3,5-dimethylpyrazolyl]methane (L3Me)30 were synthesised as previously reported. The ligands 1,1-methylenebis(3,5-dimethyl-1H-pyrazole-4-carboxylic acid) (L1Me)31 and 1,1,2,2-tetrakis[4-(4-carboxyphenyl)pyrazolyl]ethane (L5)32 have been previously reported and were synthesised with some modifications. Full synthetic procedures for these compounds are included in the ESI† (Section S1).The synthesis of MnMOF-1 has been widely reported13 but is repeated here for completeness and to highlight the importance of the type of vials used for its synthesis.
Single crystals were mounted in Paratone-N oil on a MiTeGen micromount. Single-crystal X-ray data were collected under a variety of conditions and on several instruments or beamlines. Data for MnMOF-L1-1 was collected at 100 K on the MX133 beamline of the Australian Synchrotron using the Blu-ice software interface (λ = 0.71073 Å).34 Data for MnMOF-2D and MnMOF-L1-3 at room temperature (293 and 300 K respectively), and for MnMOF-L1Me at 150 K on an Oxford Diffraction X-Calibur diffractometer (λ = 0.71073 Å). Finally, MnMOF-L2, MnMOF-L4 and MnMOF-L5 was collected at 100 K on a Rigaku-Oxford Diffraction Synergy diffractometer (λ = 1.54056 Å). Details of this are further described in the crystallographic data tables in the ESI.† Absorption corrections were applied using multiscan methods and the structures solved using SHELXS35 or SHELXT,36 and refined by full-matrix least squares on F2 by SHELXL,37 interfaced through the programs X-Seed38 or OLEX2.39 In general, all atoms were refined anisotropically and hydrogen atoms were included as invariants at geometrically estimated positions, unless specified in the ESI.† Figures were produced using Diamond software.40 X-ray experimental data is given in Tables S1.1 and S1.2.† CIF data have been deposited with the Cambridge Crystallographic Data Centre, CCDC reference numbers CCDC 2221561–2221568 (see Tables S1.1 and S1.2† for the specific codes for each structure).
Powder X-ray diffraction (PXRD) data were collected on a Bruker Advanced D8 diffractometer (capillary stage) using Cu Kα radiation (λ = 1.54056 Å, 40 kW/40 mA, 2θ = 2–52.94°, phi rotation = 20 rotations per min at 1 s exposure per step, with 5001 steps using 0.5 mm glass capillaries for dried and post-adsorption samples and in DMF in 0.8 mm glass capillaries otherwise). PXRD data for MnMOF-L2 was collected on a Rigaku Synergy Diffractometer using Cu Kα radiation (λ = 1.54056 Å), 50 kW/1 mA, 2θ = 0–65.44°, 273 K, scan width: 300, exposure time: 300.0 s for each of 2 runs, at a detector distance of 60 mm.
FTIR spectra were collected on a Shimadzu IR spirit spectrometer using an ATR attachment (spectral range: 7800–350 cm−1).
Nuclear magnetic resonance (NMR) spectra were collected at 25 °C in deuterated solvents on an Agilent DD2 500 MHz NMR with a 5 mm OneNMR probe, using tetramethylsilane (TMS) signals as the internal reference standard. Solid MOF samples were digested in DCl/DMSO-d6 at 25 °C.
Gas adsorption measurements were performed on a Micromeritics 3-Flex surface area and pore size analyser. Prior to gas adsorption, the MOF materials underwent solvent exchange to EtOH at 25 °C by washing with EtOH (3×) followed by dry EtOH (1×) over a 2 day period. The materials were activated from EtOH by heating at 120 °C under vacuum for 3 hours. N2 and CO2 experiments were performed consecutively on the same sample with a 2 hour vacuum step in between to reactivate the materials.
Thermogravimetric analysis data was collected on an STA 449 F3 Jupiter analyser from 45–700 °C at 5 °C min−1 under 21% O2, 79% N2.
Linker and MOF synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 ethanol/water, and dried in air to give the ester-protected compound. Deprotection was achieved by boiling the white solid in 1 M NaOH overnight. The resulting solution was acidified with 4 M HCl to pH 2 to precipitate H4L4 as a white solid, which was subsequently collected, washed with water and acetone and dried under vacuum overnight. Yield: 1.8 g (40%). IR (cm−1): 1705, 1678, 1559, 1429, 1236, 1172, 1128; 1H NMR (500 MHz, DMSO-d6): 12.57 (br s, 4H, COOH), 8.68 (s, 4H, pyrH), 8.50 (s, 2H, N-CHR-N), 7.83 (2, 4H, pyrH).
:7 ethanol/water, and dried in air to give the ester-protected compound. Deprotection was achieved by boiling the white solid in 1 M NaOH overnight. The resulting solution was acidified with 4 M HCl to pH 2 to precipitate H4L4 as a white solid, which was subsequently collected, washed with water and acetone and dried under vacuum overnight. Yield: 1.8 g (40%). IR (cm−1): 1705, 1678, 1559, 1429, 1236, 1172, 1128; 1H NMR (500 MHz, DMSO-d6): 12.57 (br s, 4H, COOH), 8.68 (s, 4H, pyrH), 8.50 (s, 2H, N-CHR-N), 7.83 (2, 4H, pyrH).
Note: As in the synthesis of MnMOF-1, different phases have been observed for the same combination of reactants depending on the amount of water, loss of water during the reaction and reaction duration.
Computational methods
The CP2K software package was used to perform density functional theory (DFT) calculations.41 The exchange–correlation energy was evaluated in the Perdew–Burke–Ernzerhof (PBE) approximation,42 and the dispersion interactions were treated using the DFT-D3 approach.43 Four different grids and a plane-wave cut-off for the electronic density of 700 Ry was used for high accuracy. Valence electrons were treated by double-zeta valence polarized basis sets and norm-conserving Goedecker–Teter–Hutter pseudopotentials, all adapted for PBE (DZVP-GTH-PBE) for H, C, O and N or optimized for solids (DZVP-MOLOPT-SR-GTH) for Mn.44 Cell and atom positions were optimised from their respective crystal structures with default convergence criteria. Simulations of formation energy used the reported structure of MnO structure in the cubic phase.45Results and discussion
Further studies of MnMOF-1
Since the initial report of MnMOF-1 in 2014 (Fig. 1b–d), our group has subjected this material to a myriad of reactions and synthesis conditions. During these experiments, we periodically spotted the trace formation of a second crystalline phase by comparing the initial and post-reaction PXRD patterns (ESI† Fig. S5). Herein we report that a second phase (here referred as MnMOF-2D) can also be obtained as a pure phase product by increasing the reaction time during MnMOF-1 synthesis to over 72 h, heating to a higher temperature, undertaking the reaction in a pressurised vessel, as well as by leaving pristine sample in DMF for over a month (specific conditions in the Experimental section). These results hint at MnMOF-2D being the thermodynamically-stable phase of the Mn(II)-L2Me system, and towards the kinetic or metastable nature of MnMOF-1.MnMOF-2D (formula: [Mn2(L2Me)2(H2O)2]) crystalises in the monoclinic space group C2/c as large colourless block-shaped crystals, revealing a closely-packed layered motif (Fig. 2d). A linear disposition of square-pyramidal Mn(II) ions (Fig. 2a) is connected through four L2Me linkers via M–COO bonds, extending alternately above and below the planes of the 2D structure. The square-planar geometry of the carboxylates is completed by a capping axial water molecule. Importantly, the pyrazole rings in L2Me are non-coordinated and in an anti-configuration (Fig. 2c), which closely-packs adjacent layers in an alternating fashion. The manganese chains of one layer reside beneath the centre of the linker in the next layer (Fig. 2d), precluding any efforts of post-synthetically metalating MnMOF-2D crystals.
 | ||
| Fig. 2 Structure of MnMOF-2D, the 2D phase formed from the same linker as MnMOF-1. a) A Mn(II) ion chain where each Mn(II) ion is coordinated by four carboxylates in a square planar arrangement, with a water molecule as the capping ligand. b) A single layer of MnMOF-2D showing the Mn chains in the c axis linked in the a direction by L2Me ligands (C, grey; N, light blue; O, red; Mn, purple). Hydrogens have been omitted for clarity. c) An L2Me ligand with saturated carboxylate coordination sites and a non-coordinated bispyrazole unit in the anti position. d) Representation of the packing of layers in MnMOF-2D. Blue and yellow indicate separate layers. | ||
We also investigated the importance of the methyl groups in forming the MnMOF-1 structure. While the methyl groups provide steric hindrance around the pyrazole coordinating sites, as electron donors they improve the donor ability of the pyrazole nitrogen. Thus, we subjected the non-methyl linker L2 to synthesis conditions that are established for forming MnMOF-1. We found that the non-methyl linker L2 has significantly less solubility under the same water/DMF conditions than L2Me and forms a cloudy precipitate when water is added to the reaction medium. After heating the suspension at 100 °C for 24 hours, a few crystals of MnMOF-L2 were found in combination with amorphous material. Longer reaction times did not appear to provide a phase pure sample. MnMOF-L2 has a similar 2D close packed structure to MnMOF-2D (ESI† Fig. S1). Further structural details are provided in the ESI† Section S2.1. We posit, given the reduced solubility of L2, that it is not possible to obtain the equivalent material to MnMOF-1 due to it being a kinetic phase.
Attempted isoreticulation of MnMOF-1
After more thoroughly understanding the synthesis of MnMOF-1, we decided to attempt the preparation of analogues of MnMOF-1 with both shorter (L1) and longer (L3) linkers. The isoreticular synthesis of MOFs is a well-known technique to prepare MOFs with identical topologies but distinct properties (e.g., pore size, stability). Recently, we reported the synthesis of Zr-based metal–organic layers (MOLs) using the shorter non-methyl L1 linker.27 Given the availability of these links and demonstration that one of these materials can undergo facile PSMet, we initially focused our attempts to isoreticulate MnMOF-1 on the shorter links L1 and L1Me.We subjected L1Me to the synthetic conditions used to prepare MnMOF-1, namely reaction of 1.7 equivalents of MnCl2·4H2O with the ligand in a 2:1 DMF:water solvent mixture at 100 °C for 24 hours. The reaction yielded a new product as large colourless diamond-shaped crystals, tantalisingly like the crystals of MnMOF-1. SCXRD analysis revealed the resulting material to be a 3D material (MnMOF-L1Me, [Mn2(L1Me)2(H2O)4]) with free bis-pyrazole groups (Fig. 3 and S2†) but we were disappointed to note that it had linear cluster chains (Fig. 3) like MnMOF-2D and not the Mn3 nodes that are characteristic of MnMOF-1. MnMOF-L1Me, is further described in the associated ESI† (Section S2.2).
 | ||
| Fig. 3 Synthesis conditions and key structural features of materials made from the shorter linkers, L1Me and L1. Subtle variations in the DMF:water ratio and rate of water loss dictate the formation of three different phases for L1. From left to right, crystallographic representations of the Mn(II) clusters and ligand connectivities of MnMOF-L1Me, MnMOF-L1-1, MnMOF-L1-2 and MnMOF-L1-3 (C, grey; N, light blue; O, red; Mn, purple). Hydrogen atoms have been omitted for clarity. | ||
Given that the shorter length of L1Me and the steric bulk of four methyl groups might have been hindering the formation of the MnMOF-1 structure, we then investigated the non-methylated linker L1. Reactions of L1 with Mn salts under conditions that favour MnMOF-1 gave three distinct phases that are dependent on the ability to maintain water in the reaction under solvothermal conditions. Depending on the amount of water present in the reaction mixture, three different Mn clusters can be formed that led to three distinct phases (MnMOF-L1-1, MnMOF-L1-2 and MnMOF-L1-3). The different phases are formed under conditions where the initially added water is retained, a standard equivalent of water is used initially but some lost, and less water is used in the reaction initially (Fig. 3, S3 and S4†). The primary phase, MnMOF-L1-1, is a 3D structure which forms under longer reaction times and where more water is used (i.e. 1:2 H2O:DMF ratios or higher). MnMOF-L1-1 has a Mn2 cluster where each octahedral Mn(II) centre is coordinated to two pyrazoles, two mono coordinated carboxylates and two water molecules in the axial positions, Each non-coordinating oxygen of the carboxylate groups forms hydrogen bonding with a water molecule of the adjacent Mn(II) centre, this being the only interaction between both Mn(II) centres (Fig. 3). Reaction of MnCl2 with L1 under the same conditions but with non-pressurized vials leads to the formation of a new 2D phase (MnMOF-L1-2). The reduced amount of water leads to the formation of a different Mn2 cluster where both Mn(II) centres are now bridged by two carboxylates and a water molecule (Fig. 3). The mono-coordinated carboxylates present in one of the Mn(II) centres form hydrogen bonding with a capping water molecule coordinated to the same Mn(II) centre. Finally, when less water is used initially (1:4 H2O:DMF ratio) a very similar 2D phase is formed (MnMOF-L1-3). The Mn2 cluster is very similar to the one in the second phase but here it possesses a capping DMF ligand in place of a water bound to the Mn cluster. In this last structure, the mono-coordinated carboxylates are now forming hydrogen bonds to the bridging water. This small change in the orientation of the carboxylates leads to distinct overall structures despite both (MnMOF-L1-2 and MnMOF-L1-3) having the same linker connectivity (Fig. 3). Full structure descriptions are provided in the ESI† (Sections S2.3 and S2.4).
In summary, [Mn2(L1Me)2(H2O)4] is the only material able to be crystallised with L1Me. PXRD data confirms this material to be phase pure and the dried solid is stable due to its close-packed structure (ESI† Fig. S7). For L1 three distinct phases are encountered and PXRD analysis demonstrates that all three materials can be formed phase pure, although there are challenges in reliably forming MnMOF-L1-2 without contamination by MnMOF-L1-1. These three close-packed structures also result in materials that are stable to drying (ESI† Fig. S8–S10) but they were not further investigated.
Despite L1 and L1Me forming MOFs that have structural features (coordination environments, ligand binding modes) seen in MnMOF-1, neither ligand allowed us to isolate the targeted MnMOF-1 topology. This may be partly attributable to hindered rotation in the shorter linkers (lacking the phenyl spacer between the pyrazole and carboxylate donors); the importance of this was recently observed by us in Zr-based MOFs formed with L1 and L1Me where the chelating pyrazole site was shown not to be able to switch between syn and anti conformations.27 Additionally, and particularly for L1Me, the lack of the phenyl spacer brings the pyrazole groups close to the MOF nodes. Given these challenges we turned our attention to a computational approach to improve our understanding of the structure metrics that might allow the target MOF to form and to obtain insight into the possible formation of a MnMOF-1 type structure for shorter and longer links.
DFT simulations of isoreticular MnMOF-1 networks
The formation of isoreticular networks based on MnMOF-1 was investigated using dispersion-corrected density functional theory (DFT) simulations. Hypothetical frameworks featuring the ligands L1, L1Me, L2, L2Me (MnMOF-1 linker), L3 and L3Me, based on 1-for-1 ligand replacement with the MnMOF-1 structure, were generated. The atom positions and cell parameters of these structures were optimised using DFT methods, with no symmetry constraints applied. The resulting frameworks displayed no obvious signs of high energy strain, such as very close contacts or distorted molecular structures. This suggests that the formation of MnMOF networks could be possible with both shorter and longer ligands. The simulated structure for MnMOF-1 with this approach closely matches the crystal structure which demonstrates the accuracy of the DFT method.To quantify the ability for MnMOF networks to form, a hypothetical formation energy was computed, based on the reaction:
| ligand(g) + MnO(s) → MOF(s) + H2O(g) |
 | ||
| Fig. 4 A plot showing the formation energies of the simulated frameworks formed from L1, L1Me, L2, L2Me (MnMOF-1 linker), L3 and L3Me, based on 1-for-1 ligand replacement into the MnMOF-structure type. The orange squares show the formation energies for MOFs constructed from the methylated linkers and the blue dots are for the corresponding non-methyl linkers. From left to right (low to high density) are L3 and L3Me, L2 and L2Me, and the very high energy and dense potential structures formed with L1 and L1Me. MnMOF-1 is only reported for L2Me. | ||
Buoyed by these insights, we attempted the synthesis MnMOF-1 type structures with links L3 and L3Me.30 These linkers were previously used to form 2D layered MOFs with structures, related to CuMOF-1.28 Despite this previous synthetic success, the introduction of the ethynyl spacer significantly diminishes the solubility of the ligands in the reaction conditions used to form Mn-based MOFs, and all attempts under our hands failed to yield crystalline materials.
Towards porous Mn-based MOFs through covalent linking
Given that the shorter L1 and L2 links gave rise to several 2D layered structures, we hypothesised that linking these ligands together via the methylene hinge would facilitate formation of 3D materials possessing the desired structural features, namely flexibility and ability to undergo PSMet. This strategy was supported by our earlier efforts in the area with tetratopic linkers used to form 3D MOFs with Cu, Zn, and Cd.32 Thus, ligands L4 and L5 were prepared, the latter via a procedure similar to that previously reported,32 and the former via an analogous one step procedure starting with 4-carboxypyrazole ethyl ester in place of 4-iodopyrazole.Synthetic conditions similar to those required for the synthesis of MnMOF-1 were then used to form two new MOFs, [Mn2(L4)(H2O)3(DMF)]·(DMF)(H2O) (MnMOF-L4) and [Mn2(L5)]·4DMF (MnMOF-L5). MnMOF-L4 crystallised as colourless cube-shaped crystals in the monoclinic space group Pnma, forming a very similar Mn2 cluster to Mn-MOF-L1-3 and with the same linker connectivity. Six of the carboxylate oxygens are coordinated to a metal centre, the other two form hydrogen bonding with the bridging water molecule (Fig. 5a) and 50% of the bis-pyrazolyl groups coordinate a metal centre (Fig. 5b). Despite having a similar coordination environment to Mn-MOF-L1-3, the tetratopic connectivity of L4 leads to the formation of a 3D framework with 1D DMF-filled channels (5.7 × 6.2 Å) directed down the b axis (Fig. 5c).
 | ||
| Fig. 5 Structures of the MOFs made from the tetracarboxylate linkers L4 and L5. a) The Mn2 cluster of MnMOF-L4, which is very similar to that of MnMOF-L1-3 from the dicarboxylate linker L1. b) Ligand connectivity of MnMOF-L4. Half of the ligand has a non-coordinated bispyrazole and mono-coordinated carboxylates whereas the other half is fully saturated. c) Representation of MnMOF-L4 down the b axis, revealing the 1D pores (DMF has been removed for clarity). d) Mn cluster for MnMOF-L5, where two Mn atoms are capped by bispyrazoles and bridged by carboxylates. e) Ligand connectivity of MnMOF-L5 showing the fully occupied ligand coordination sites. f) Representation of the structure of MnMOF-L5 viewed down the b axis to show the wine rack type structure and 1D pores (DMF has been removed for clarity). In all structures C, grey; N, light blue; O, red; Mn, purple and hydrogen atoms have been omitted for clarity. g) PXRD data for MnMOF-L4 and MnMOF-L5 (bottom to top: MnMOF-L4 simulated – black; MnMOF-L4 as-synthesised – cyan; MnMOF-L4 activated – purple; MnMOF-L5 simulated – pink; MnMOF-L5 as-synthesised – red; MnMOF-L5 activated – orange). h) 77 K N2 and 195 K CO2 isotherms for MnMOF-L4 (black/squares and red/circles respectively) and MnMOF-L5 (cyan/triangles and purple/diamonds respectively). Filled shapes show adsorption and open shapes desorption. | ||
MnMOF-L5 is a 3D MOF, isoreticular with previously reported Zn and Cd-based MOFs.32 MnMOF-L5 crystallised in the monoclinic space group C2/c, forming clumps of colourless plate-shaped single crystals (Fig. S22†). MnMOF-L5 contains a Mn2 cluster where each Mn(II) centre is coordinated to two pyrazoles, a doubly connected carboxylate and two bridging carboxylates (Fig. 5d). The ligand lies in the ac plane, with both the carboxylate and bispyrazole groups coordinating Mn (Fig. 5e). This leads to columns of binuclear manganese nodes and the C–C bond of the linker alternating along the b-axis. In each ligand, one of the bispyrazole units coordinates the cluster above it and the other below it. The greater length of the ligand arms provides an open porous structure, with 1D channels (8.4 × 13.0 Å) aligned along the b-axis (Fig. 5f).
Both MnMOF-L4 and MnMOF-L5 are phase pure, with no loss in crystallinity but subtle changes in their PXRD patterns occurring upon solvent exchange, which is consistent with their flexible wine rack-like structures. To examine the permanent porosity of MnMOF-L4 and MnMOF-L5, we obtained N2 and CO2 adsorption isotherms for both materials. Activation of MnMOF-L4 and MnMOF-L5 from ethanol at 120 °C for 3 hours showed some minor structural changes and partial loss of crystallinity as evidenced by PXRD (Fig. 5g). Full activation of the frameworks, including the removal of coordinated DMF in the MnMOF-L4, is evidenced by TGA (no mass loss until the decomposition temperature: 310 °C for MnMOF-L4 and 420 °C for MnMOF-L5, Fig. S20 and S21†) and NMR analysis of the digested MOF samples post the adsorption experiments, which reveals no residual DMF. N2 adsorption at 77 K revealed that both frameworks are permanently porous with Brunauer–Emmett–Teller (BET) surface areas of 435.2 m2 g−1 for MnMOF-L4 and 950.9 m2 g−1 for MnMOF-L5 (Fig. 5h). Pore size calculations on the experimental isotherms reveal 6.4 Å pores for MnMOF-L4 and 11.8 Å pores for MnMOF-L5 (Fig. S19†). For MnMOF-L5, this represents a similar pore size and BET surface area to the isoreticular Zn structure.32 The 195 K CO2 isotherm (Fig. S21†) shows a gate opening behaviour that is typical of flexible MOFs.
Conclusions
Herein, we have shown that isoreticulating the structures of MOFs made of flexible linkers is nontrivial due to the significant degrees of freedom in the linkers. The variation in types of accessible Mn nodes, coupled with the ability of the bis-pyrazole site of the linkers to coordinate to the node, further complicates this pursuit. Our investigation of this family of materials has identified important synthetic parameters that can be used to form phase pure materials and further identified MnMOF-1 as being a kinetic or metastable phase that is in competition with the formation of a dense 2D phase, MnMOF-2D. Denser, or close packed phases are readily accessible for shorter ditopic linkers, with steric crowding precluding the formation of MnMOF-1 type structures, while solubility challenges prevented comprehensive investigation of longer linkers of this type. These observations around accessibility of the MnMOF-1 structure were supported by DFT simulations of the hypothetical isostructural materials.Consideration of the structures formed with the ditopic pyrazole carboxylate linkers suggested a need to use tetratopic variants as a strategy to form 3D frameworks. This approach was confirmed by the successful synthesis of two new porous 3D Mn-based MOFs, MnMOF-L4 and MnMOF-L5, which, while not being isoreticular, show an ability to systematically expand the pore volumes and surface areas through linker extension. Both materials are permanently porous, with the latter showing a reasonable degree of structural flexibility. MnMOF-L4 also retains a non-coordinated pyrazole group, as targeted, but due to an adjacent Mn node in the structure this is not accessible for metalation. These two materials demonstrate the rich coordination chemistry of pyrazole carboxylate links and we are actively using these ligands to form MOFs with a range of other metals.
Author contributions
J. F. Smernik, investigation, formal analysis, visualization, writing – original draft, writing – review & editing; P. Gimeno-Fonquernie, investigation, formal analysis, visualization, writing – review & editing; J. Albalad, investigation, writing – original draft; T. S. Jones, investigation; R. J. Young, investigation; N. R. Champness, supervision, writing – review & editing; C. J. Doonan, supervision, writing – review & editing; J. D. Evans, formal analysis, visualization, supervision, writing – review & editing; and C. J. Sumby, conceptualisation, formal analysis, supervision, visualization, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. J. S. and C. J. D. gratefully acknowledge the Australian Research Council for funding (DP190101402). J. D. E. was supported by a Ramsay Fellowship from the University of Adelaide and acknowledges an ARC DECRA fellowship (DE220100163). ZIH Dresden and Phoenix HPC service at the University of Adelaide are thanked for providing high-performance computing resources. This research was undertaken in part using the MX1 beamline at the Australian Synchrotron, part of ANSTO. J. F. S. and P. G.-F. gratefully acknowledge a Research Training Program Scholarship and University of Adelaide International Scholarship, respectively. P. G.-F. also acknowledges an Australian Institute of Nuclear Science and Technology Post-graduate Research Award. N. R. C. gratefully acknowledges support from the UK Engineering and Physical Sciences Research Council (EP/S002995/2).Notes and references
- H. Li, L. B. Li, R. B. Lin, W. Zhou, Z. J. Zhang, S. C. Xiang and B. L. Chen, EnergyChem, 2019, 1, 100006 CrossRef
.
- W. D. Fan, X. R. Zhang, Z. X. Kang, X. P. Liu and D. F. Sun, Coord. Chem. Rev., 2021, 443, 213968 CrossRef CAS
- X. Zhao, Y. Wang, D. S. Li, X. Bu and P. Feng, Adv. Mater., 2018, 30, e1705189 CrossRef PubMed
- D. Yang and B. C. Gates, ACS Catal., 2019, 9, 1779–1798 CrossRef CAS
- A. Dhakshinamoorthy, Z. Li and H. Garcia, Chem. Soc. Rev., 2018, 47, 8134–8172 RSC
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS PubMed
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed
- F. Niekiel, M. Ackermann, P. Guerrier, A. Rothkirch and N. Stock, Inorg. Chem., 2013, 52, 8699–8705 CrossRef CAS PubMed
- Z. J. Lin, J. Lu, M. Hong and R. Cao, Chem. Soc. Rev., 2014, 43, 5867–5895 RSC
- R. J. Marshall, T. Richards, C. L. Hobday, C. F. Murphie, C. Wilson, S. A. Moggach, T. D. Bennett and R. S. Forgan, Dalton Trans., 2016, 45, 4132–4135 RSC
- W. M. Bloch, A. Burgun, C. J. Coghlan, R. Lee, M. L. Coote, C. J. Doonan and C. J. Sumby, Nat. Chem., 2014, 6, 906–912 CrossRef CAS PubMed
- R. A. Peralta, M. T. Huxley, R. J. Young, O. M. Linder-Patton, J. D. Evans, C. J. Doonan and C. J. Sumby, Faraday Discuss., 2021, 225, 84–99 RSC
- J. Albalad, R. A. Peralta, M. T. Huxley, S. Tsoukatos, Z. Shi, Y. B. Zhang, J. D. Evans, C. J. Sumby and C. J. Doonan, Chem. Sci., 2021, 12, 14893–14900 RSC
- R. A. Peralta, M. T. Huxley, J. D. Evans, T. Fallon, H. J. Cao, M. X. He, X. S. Zhao, S. Agnoli, C. J. Sumby and C. J. Doonan, J. Am. Chem. Soc., 2020, 142, 13533–13543 CrossRef CAS PubMed
- R. A. Perlata, M. T. Huxley, Z. Shi, Y. B. Zhang, C. J. Sumby and C. J. Doonan, Chem. Commun., 2020, 56, 15313–15316 RSC
- A. Burgun, C. J. Coghlan, D. M. Huang, W. Chen, S. Horike, S. Kitagawa, J. F. Alvino, G. F. Metha, C. J. Sumby and C. J. Doonan, Angew. Chem., Int. Ed., 2017, 56, 8412–8416 CrossRef CAS PubMed
- R. J. Young, M. T. Huxley, L. Wu, J. Hart, J. O'Shea, C. J. Doonan, N. R. Champness and C. J. Sumby, Chem. Sci., 2023, 14, 9409–9417 RSC
- G. Christou, Acc. Chem. Res., 1989, 22, 328–335 CrossRef CAS
- G. Fernández, M. Corbella, J. Mahía and M. A. Maestro, Eur. J. Inorg. Chem., 2002, 2502–2510 CrossRef
- S. Menage, S. Vitols, P. Bergerat, E. Codjovi, O. Kahn, J. Girerd, M. Guillot, X. Solans and T. Calvet, Inorg. Chem., 1991, 30, 2666–2671 CrossRef CAS
- D. Maniaki, E. Pilichos and S. P. Perlepes, Front. Chem., 2018, 6, 461 CrossRef CAS PubMed
- W. Zhou, H. Wu and T. Yildirim, J. Am. Chem. Soc., 2008, 130, 15268–15269 CrossRef CAS PubMed
- P. Q. Liao, X. Y. Li, J. Bai, C. T. He, D. D. Zhou, W. X. Zhang, J. P. Zhang and X. M. Chen, Chem. – Eur. J., 2014, 20, 11303–11307 CrossRef CAS PubMed
- T. Ladrak, S. Smulders, O. Roubeau, S. J. Teat, P. Gamez and J. Reedijk, Eur. J. Inorg. Chem., 2010, 24, 3804–3812 CrossRef
- P. Gimeno-Fonquernie, W. Liang, J. Albalad, A. Kuznicki, J. R. Price, E. D. Bloch, C. J. Doonan and C. J. Sumby, Chem. Commun., 2022, 58, 957–960 RSC
- W. M. Bloch, R. Babarao, M. R. Hill, C. J. Doonan and C. J. Sumby, J. Am. Chem. Soc., 2013, 135, 10441–10448 CrossRef CAS PubMed
- W. M. Bloch, C. J. Doonan and C. J. Sumby, CrystEngComm, 2013, 15, 9663–9671 RSC
- A. Burgun, W. M. Bloch, C. J. Doonan and C. J. Sumby, Aust. J. Chem., 2017, 70, 566–575 CrossRef CAS
- N. P. Burlutskiy and A. S. Potapov, Molecules, 2021, 26, 413 CrossRef CAS PubMed
- C. J. Coghlan, C. J. Sumby and C. J. Doonan, CrystEngComm, 2014, 16, 6364–6371 RSC
- N. P. Cowieson, D. Aragao, M. Clift, D. J. Ericsson, C. Gee, S. J. Harrop, N. Mudie, S. Panjikar, J. R. Price, A. Riboldi-Tunnicliffe, R. Williamson and T. Caradoc-Davies, J. Synchrotron Radiat., 2015, 22, 187–190 CrossRef CAS PubMed
- T. M. McPhillips, S. E. McPhillips, H. J. Chiu, A. E. Cohen, A. M. Deacon, P. J. Ellis, E. Garman, A. Gonzalez, N. K. Sauter, R. P. Phizackerley, S. M. Soltis and P. Kuhn, J. Synchrotron Radiat., 2002, 9, 401–406 CrossRef CAS PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, 71, 3–8 CrossRef PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed
- L. J. Barbour, J. Supramol. Chem., 2001, 1, 189–191 CrossRef CAS
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS
-
Diamond, Crystal and Molecular Structure Visualization, Crystal Impact, Dr. H. Putz & Dr. K. Brandenburg GbR, Kreuzherrenstr. 102, 53227 Bonn, Germany, https://www.crystalimpact.de/diamond Search PubMed
- T. D. Kühne, M. Iannuzzi, M. Del Ben, V. V. Rybkin, P. Seewald, F. Stein, T. Laino, R. Z. Khaliullin, O. Schütt and F. Schiffmann, J. Chem. Phys., 2020, 152, 194103 CrossRef PubMed
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed
- S. Goedecker, M. Teter and J. Hutter, Phys. Rev. B: Condens. Matter, 1996, 54, 1703–1710 CrossRef CAS PubMed
- The Materials Project, 2020, DOI:10.17188/1193794.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional synthetic details, structure descriptions for additional crystal structures, PXRD data, FTIR spectra, gas adsorption, TGA, and SCXRD data, and NMR spectra of digested MOFs. CCDC 2221561–2221568. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ce00881a |
| ‡ Vials that controllably lose pressure are needed for the reproducible synthesis of MnMOF-1. This was observed because of syntheses of the MOF being conducted in different labs and by new lab members. While a number of different types of vials would be suitable, this is the link to the specific product we use: https://www.dwk.com/wheaton-liquid-scintillation-vials-pp-caps-attached-to-vials-glass-metal-foil-pulp-22-400-20-ml-986541. |
| § These vials were suitable for forming phase pure MnMOF-2D under identical conditions to those used for MnMOF-1 synthesis. https://www.dwk.com/wheaton-lab-file-sample-vials-12ml-standard-vials-with-caps-attached-ptfe-14b-rubber-w224585. |
| This journal is © The Royal Society of Chemistry 2023 |