
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Transition metal-free, chemoselective arylation of thioamides yielding aryl thioimidates or N-aryl thioamides†
Piret
Villo
ab,
Gabriella
Kervefors
a and
Berit
Olofsson
 *a
*a
aDepartment of Organic Chemistry, Arrhenius Laboratory, Stockholm University, SE-106 91, Sweden. E-mail: berit.olofsson@su.se
bInstitute of Technology, University of Tartu, Tartu 50 411, Estonia
First published on 5th July 2018
Abstract
Reactions of secondary thioamides with diaryliodonium salts under basic, transition metal-free conditions resulted in chemoselective S-arylation to provide aryl thioimidates in good to excellent yields. Equimolar amounts of thioamide, base and diaryliodonium salt were sufficient to obtain a diverse selection of products within short reaction times. Reactions with thiolactams delivered N-arylated thioamides in good yield at room temperature.
Thioamides and their aryl thioimidate derivatives are important units in bioactive molecules, and in intermediates towards such compounds.1 The thioimidate moiety can also be found in heterocycles such as thiazoles and benzothiazoles,2 and in early stage solar absorbers.3 The aryl thioimidate scaffold has also been shown to regioselectively react with alkynes to give aryl thio azadienes.4 While alkylation of thioamides to access alkyl thioimidates is straightforward, the corresponding methodology to reach aryl thioimidates is much less developed.5 Conventional routes to access aryl thioimidates include reacting imidoyl chlorides, ketenimines or nitriles with thiophenols that already have an established S-aryl bond (Pinner synthesis).5c,6 A one-pot reaction between aryl thiol, hexamethyldisilazane and nitromethane gives related methaneimidothioate scaffolds.7 Formation of an aryl-sulfur bond in the synthesis of benzothiazoles can be achieved either via palladium-catalyzed S-arylation of thioacetamide with o-iodoanilines, followed by a cyclization;8 metal-free condensation of thioamides with 2-aminothiophenols,9 or an intramolecular cyclization of o-iodothiobenzanilide.10 Radical arylation of thioamides gave high yields of S-arylated products but could not be applied to thiolactams.11 To the best of our knowledge, the latter is the only report obtaining acyclic aryl thioimidates by direct S-arylation of thioamides. Diaryliodonium salts (diaryl-λ3-iodanes) are easily prepared hypervalent iodine reagents with low toxicity.12 They have frequently been employed to arylate heteroatom- or carbon nucleophiles,12,13 including the S-arylation of thioureas or closely related scaffolds under both metal-free,14 and copper-catalyzed conditions.15 While thioamides have never been arylated with diaryliodonium salts, iodine(III) reagents have been used to transfer alkenyl, alkynyl, trifluoromethyl, and nitrile functionalities to thioamides or similar compounds.16 Additionally, thioamides can be converted into benzothiazoles via oxidative arylation with bis(trifluoroacetoxyiodo)benzene.17 In our research line on hypervalent iodine chemistry, we have reported efficient, metal-free arylations of a range of heteroatom and carbon nucleophiles.18 Mechanistic aspects have been explored to expand the utility of these reagents in organic synthesis, also with focus on chemoselectivity using unsymmetric diaryliodonium salts.19 Reactions of nucleophiles with two possible arylation sites, e.g. enolates, amides, nitrite, oximes and quinolones, have proven highly selective towards C- and N-arylation, respectively.18b,c,19a,20 Continuing on this track, we explored the arylation of thioamides, and herein describe the highly selective S-arylation of secondary thioamides with diaryliodonium salts under basic, transition metal-free conditions (Scheme 1).
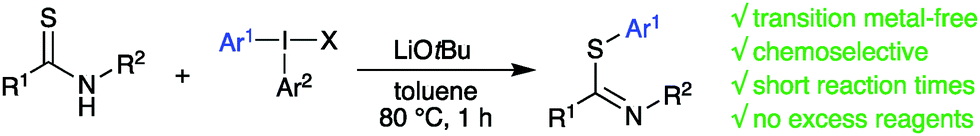 | ||
| Scheme 1 Arylation of thioamides with diaryliodonium salts. | ||
Initial screenings revealed that the reaction of thioamide 1a with diphenyliodonium triflate (2a) delivered phenyl thioimidate 3a in 30–40% yield with a selection of organic and inorganic bases in toluene at room temperature.21 Recovered 1a and side-products from either hydrolysis22 of 3a into the corresponding amide and thiol, or desulfurization,17,23 constituted the remaining mass. Remarkably, no N-arylated thioamide product was observed. Product 3a was isolated as an inseparable Z![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :E isomeric mixture in 93:7 ratio, based on NMR analysis and literature data on similar compounds.7,24 This is in accordance with alkylations of thioamides, which have been reported to proceed with S-functionalization.25 Based on these initial promising results, an extensive optimization was performed.21 A solvent screening with LiOtBu as base revealed that EtOAc, iPrOAc and CH2Cl2 gave comparable yields to reactions in toluene. The reaction temperature had a great impact on the reaction outcome, and reactions at 80 °C allowed the reaction time to be decreased from overnight to one hour (Table 1, entries 1–4). While running the reaction under argon atmosphere only influenced the outcome marginally (entry 5), degassing the solvent enhanced the yield considerably (entry 6). A counterion effect for the iodonium salt was also observed, where diphenyliodonium salts with OTf, OTs and Br outperformed iodonium salts with BF4, TFA and PF6.21
:E isomeric mixture in 93:7 ratio, based on NMR analysis and literature data on similar compounds.7,24 This is in accordance with alkylations of thioamides, which have been reported to proceed with S-functionalization.25 Based on these initial promising results, an extensive optimization was performed.21 A solvent screening with LiOtBu as base revealed that EtOAc, iPrOAc and CH2Cl2 gave comparable yields to reactions in toluene. The reaction temperature had a great impact on the reaction outcome, and reactions at 80 °C allowed the reaction time to be decreased from overnight to one hour (Table 1, entries 1–4). While running the reaction under argon atmosphere only influenced the outcome marginally (entry 5), degassing the solvent enhanced the yield considerably (entry 6). A counterion effect for the iodonium salt was also observed, where diphenyliodonium salts with OTf, OTs and Br outperformed iodonium salts with BF4, TFA and PF6.21
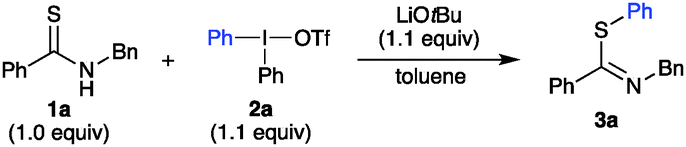
With the optimized conditions in hand, we looked into the scope of diaryliodonium salts with thioamide 1a (Scheme 2A). Symmetric and unsymmetric diaryliodonium salts were synthesized using efficient one-pot methodology,26 with phenyl, anisyl and trimethoxyphenyl (TMP) as “dummy” groups. The use of unsymmetric iodonium salts is often more atom efficient and cost effective, e.g. when highly functionalized or precious aryl groups are transferred.21 High chemoselectivity is crucial for this approach, i.e. selective transfer of only one of the aryl groups,19b,d and the arylation results with unsymmetric diaryliodonium salts proved superior to the corresponding symmetric salts in several S-arylations shown below.21 Electron deficient diaryliodonium salts generally behave well in heteroatom arylations, and the synthesis of p-CN product 3b indeed proceeded in excellent yield. Likewise, the p-NO2 product 3c was easily obtained, and surprisingly performed better in a non-degassed solvent.27
 | ||
| Scheme 2 Arylation scope with major isomer (Z) shown. Z:E ratios 91:9 to 96:4, except 3d (89:11), 3f (88:12), 3h (86:14), 3i (90:10), 3o (86:14), and 3q (87:13). Only one isomer obtained of 3w-4b. aIn non-degassed toluene. bNMR yield. cCombined yield. dAt rt for 16 h, 1.5 equiv. base. | ||
Further so, aryls with p-OCF3 (3d) and p-CF3 (3e) were delivered in good yields as well. Electron-withdrawing substituents in the meta- and ortho-positions were also tolerated, providing m-CF3 substituted 3f, with slightly lower Z:E ratio than previously observed; and o-F and o-COOMe decorated 3g–3h. Importantly, the introduction of aryl groups with a straightforward handle for further derivatization proved feasible, as exemplified by a p-N3 aryl moiety in 3i and halide-substituted products 3j, 3k, for which unsymmetric iodonium salts proved more efficient than the corresponding symmetric salts.21 Arylations with electron donating diaryliodonium salts can be demanding, as competing reaction pathways tend to give product mixtures.19c Pleasingly, this S-arylation proved compatible with such iodonium salts, and alkyl-substituted products 3l–3o were obtained in good yields, including the sterically congested 3n–3o. Furthermore, an anisyl group could easily be transferred (3p), and the synthesis of pyridyl product 3q was viable. Complete chemoselectivity was generally observed in the arylations, with minor amounts of dummy group transfer only in the synthesis of 3j.21 The reaction was demonstrated to have a considerable ortho-effect,28 with high yields and complete chemoselectivity in the synthesis of 3k and 3o.21 The scope of thioamide scaffolds was explored next (Scheme 2B). Both electron donating and withdrawing groups on the benzothioamide were well tolerated, delivering 3r and 3s in good yields. Contrary to the aryl thioamides, the arylation of alkylated thioamides resulted in S-arylated products 3t, 3u in moderate yield, along with a minor amount of N-arylation. The N-benzyl group could be replaced by a hexyl group to deliver 3w. More importantly, an easily removable Boc group was also tolerated, delivering S-arylated products 3w and 3x in high yields. Interestingly, reactions with this substrate in the presence of the weaker base potassium carbonate only gave recovered starting material, despite the more acidic NH proton.21 Furthermore, the heteroaromatic substrate pyridine-2-thiol proved suitable for this transformation, providing 3y in 78% yield. Cyclic thioamides, on the other hand, acted differently under the optimized conditions. Pyrrolidine-2-thione gave a mixture of N- and S-arylated products (4a, N:S ratio 1.5:1).21 Interestingly, the synthesis of 4b proceeded in excellent yield and complete N-selectivity at room temperature. This product class could give access to N-arylated amides after a one-step desulfurization.23b,29 Competition experiments were performed to evaluate the S-arylation in the presence of O- and N-nucleophiles that have been reported to undergo arylation with diaryliodonium salts under metal-free conditions.18c,30 Hence, reactions of thioamide 1a and Ph2IOTf were performed in the presence of amide 5 or 6, benzoic acid (7), or alcohol 8, and 2 equiv of base, to allow deprotonation of both nucleophiles (Scheme 3). The reactions proved to be completely chemoselective, with arylation of only thioamide 1a in all of the cases. S-Arylated thioimidate 3a was isolated in yields of 40–80%, with complete recovery of the competing nucleophile in all reactions.
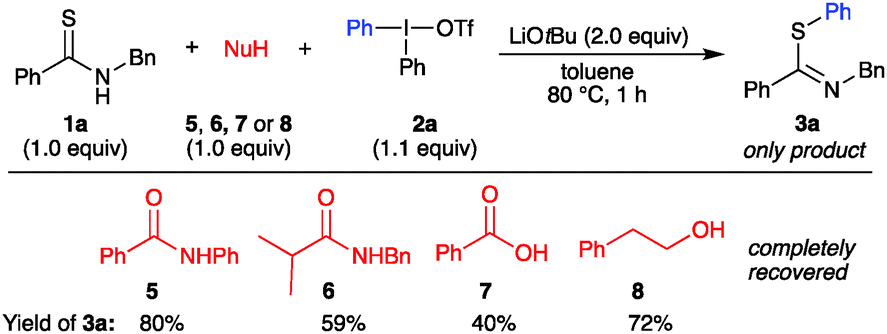 | ||
| Scheme 3 Competition experiments. | ||
Preliminary mechanistic investigations revealed that addition of aryne or radical scavengers (piperidine and 1,1-diphenylethylene, respectively) had negligible effect on the reaction outcome.21 Hence both an aryne pathway and a radical mechanism can be excluded. Based on previous experimental and mechanistic studies of enolates, amides and nitrite,18b,c,19a we propose that the reaction proceeds by deprotonation and ligand exchange to provide T-shaped intermediate A and/or B (Scheme 4). Ligand coupling in I–N intermediate A to form the S-arylated thioimidate 3 would proceed via a [2,3] rearrangement, whereas I–S intermediate B would undergo a [1,2] rearrangement to yield 3. Alternatively, intermediates A and B could yield N-aryl thioamide 4 through [1,2] and [2,3] rearrangement, respectively.
 | ||
| Scheme 4 Proposed mechanism for S- and N-arylation. | ||
The S-arylation selectivity observed for all acyclic thioamides, as well as the aromatic thioamide, can be rationalized by efficient conjugation of the nitrogen lone pair in the thioamide moiety, making the sulfur lone pairs most nucleophilic. The observed N-selectivity in arylation of thiolactams could be caused by less efficient conjugation due to cyclic constraints, making nitrogen a better nucleophile. While early literature proposed a rearrangement from S-arylated to N-arylated products,31 we observed constant N/S-arylation ratios throughout the reactions, making this mechanism unlikely.21
To conclude, we have developed a transition metal-free arylation of thioamides with diaryliodonium salts under basic conditions. Complete selectivity in favor of S-arylated products was observed with acyclic and aromatic thioamides, whereas thiolactams preferably were N-arylated. A wide scope of functional groups are tolerated in both thioamides and diaryliodonium salts. Furthermore, sterically congested aryl groups were transferred in high yield, and a considerable ortho-effect was observed. Efficient use of unsymmetric diaryliodonium salts have been demonstrated, with excellent chemoselectivity and improved yields compared to the corresponding symmetric reagents.
The Estonian Research Council (MOBTP59, PUTJD114) and the Swedish Research Council (2015-04404) are kindly acknowledged for financial support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- Recent applications of thioamide and thioimidate scaffolds:
(a) S. Julie, R. Patrick and T. Arnaud, Angew. Chem., Int. Ed., 2010, 49, 577–580 CrossRef PubMed
; (b) W. Zhang, J. Li, L.-W. Liu, K.-R. Wang, J.-J. Song, J.-X. Yan, Z.-Y. Li, B.-Z. Zhang and R. Wang, Peptides, 2010, 31, 1832–1838 CrossRef PubMed
- Reviews on synthesis of heterocycles:
(a) M. Witalewska, A. Wrona-Piotrowicz, A. Makal and J. Zakrzewski, J. Org. Chem., 2018, 83, 1933–1939 CrossRef PubMed
- A. L. Catherall, S. Harris, M. S. Hill, A. L. Johnson and M. F. Mahon, Cryst. Growth Des., 2017, 17, 5544–5551 CrossRef
- Y. Minami, H. Kuniyasu, A. Sanagawa and N. Kambe, Org. Lett., 2010, 12, 3744–3747 CrossRef PubMed
- Reviews on thioimidate synthesis:
(a) R. Roger and D. G. Neilson, Chem. Rev., 1961, 61, 179–211 CrossRef
- A. N. Kolontsova, M. N. Ivantsova, M. I. Tokareva and M. A. Mironov, Mol. Diversity, 2010, 14, 543–550 CrossRef PubMed
- K. Krzysztof and H. Grzegorz, Eur. J. Org. Chem., 2016, 4577–4585 Search PubMed
- K. Takagi, T. Iwachido and N. Hayama, Chem. Lett., 1987, 839–840 CrossRef
- K. R. Nivalkar and S. H. Mashraqui, Synth. Commun., 1996, 26, 3535–3542 CrossRef
- W. R. Bowman, H. Heaney and P. H. G. Smith, Tetrahedron Lett., 1982, 23, 5093–5096 CrossRef
-
(a) I. I. Kandror and I. O. Bragina, Izv. Akad. Nauk SSSR, Ser. Khim., 1982, 2121–2125 Search PubMed
- E. A. Merritt and B. Olofsson, Angew. Chem., Int. Ed., 2009, 48, 9052–9070 CrossRef PubMed
-
(a) K. Aradi, B. L. Tóth, G. L. Tolnai and Z. Novák, Synlett, 2016, 1456–1485 Search PubMed
-
(a) Z. Bianxiang, K. Yongqiang, Y. Lihua and C. Xia, ChemistrySelect, 2016, 1, 1529–1532 CrossRef
- H. Zhu, X. Liu, Y. Cheng, H.-Y. Peng, Y.-S. Li and Z.-B. Dong, Synthesis, 2018, 2247–2254 Search PubMed
- Selected references on functionalization of thioamides with iodine(III) reagents:
(a) M. Ochiai, S. Yamamoto, T. Suefuji and D.-W. Chen, Org. Lett., 2001, 3, 2753–2756 CrossRef PubMed
- N. K. Downer-Riley and Y. A. Jackson, Tetrahedron, 2008, 64, 7741–7744 CrossRef
-
(a) G. L. Tolnai, U. J. Nilsson and B. Olofsson, Angew. Chem., Int. Ed., 2016, 55, 11226–11230 CrossRef PubMed
-
(a) P.-O. Norrby, T. B. Petersen, M. Bielawski and B. Olofsson, Chem. – Eur. J., 2010, 16, 8251–8254 CrossRef PubMed
-
(a) X.-P. Ma, W.-M. Shi, X.-L. Mo, X.-H. Li, L.-G. Li, C.-X. Pan, B. Chen, G.-F. Su and D.-L. Mo, J. Org. Chem., 2015, 80, 10098–10107 CrossRef PubMed
- See the ESI† for details.
-
(a) W. Walter and J. Krohn, Chem. Ber., 1969, 102, 3786–3794 CrossRef
-
(a) S. Nobutaka, S. Kaniti, W. Satoshi, T. Noriyuki and I. Yasuji, Bull. Chem. Soc. Jpn., 1982, 55, 3351–3352 CrossRef
- Z. Guan, M. Nieger and A. Schmidt, Eur. J. Org. Chem., 2015, 4710–4719 CrossRef
- Y. Karibe, H. Kusama and N. Iwasawa, Angew. Chem., Int. Ed., 2012, 51, 6214–6218 CrossRef PubMed
-
(a) E. Lindstedt, M. Reitti and B. Olofsson, J. Org. Chem., 2017, 82, 11909–11914 CrossRef PubMed
- A preliminary substrate scope was performed without degassing the solvent, as this effect was discovered late in the project. See the ESI† for details.
- Y. Yamada and M. Okawara, Bull. Chem. Soc. Jpn., 1972, 45, 1860–1863 CrossRef
- I. Mohammadpoor-Baltork, M. M. Sadeghi and K. Esmayilpour, Phosphorus, Sulfur Silicon Relat. Elem., 2003, 178, 61–65 CrossRef
-
(a) T. B. Petersen, R. Khan and B. Olofsson, Org. Lett., 2011, 13, 3462–3465 CrossRef PubMed
- A. Varvoglis, Synthesis, 1984, 709–726 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, analytical data and NMR spectra of novel compounds. See DOI: 10.1039/c8cc04795b |
| This journal is © The Royal Society of Chemistry 2018 |