
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The influence of the ligand chelate effect on iron-amine-catalysed Kumada cross-coupling†
Robin B.
Bedford
*a,
Peter B.
Brenner
a,
David
Elorriaga
a,
Jeremy N.
Harvey
b and
Joshua
Nunn
a
aSchool of Chemistry, University of Bristol, Cantock's Close, Bristol, BS8 1TS, UK. E-mail: r.bedford@bristol.ac.uk
bQuantum Chemistry and Physical Chemistry Section, Department of Chemistry, University of Leuven (KU Leuven), Celestijnenlaan 200F – box 2404, Leuven, Belgium
First published on 16th June 2016
Abstract
The application of a variety of iron complexes with chelating amine ligands as pre-catalysts in the representative cross-coupling of 4-tolylmagnesium bromide with cyclohexyl bromide was investigated. The results from this study indicate the performance of the pre-catalyst is inversely proportional to the strength of the chelate or macrocyclic effect of the amine ligand, as determined by the propensity of the ligand to be displaced from the iron centre by reaction with excess benzyl magnesium chloride. The findings from this study are consistent with a catalytic cycle wherein the chelating amine ligand is not coordinated to the iron centre during turnover.
Introduction
While there is no doubt that palladium-catalysed cross-coupling is an exceptionally powerful synthetic methodology (Scheme 1(a)),1 the benefits of the use of palladium are offset by its high price, relative toxicity and the environmental impact of its extraction.2 Accordingly there has been much recent3 effort expended on developing iron-based catalysts for cross-couplings,4 due to the far lower cost, toxicity and supply issues associated with iron. By far the most widely studied iron-catalysed cross-coupling reaction is the Kumada reaction, that is the coupling of Grignard reagents with alkyl or aryl halides and related electrophilic substrates (Scheme 1(b)).4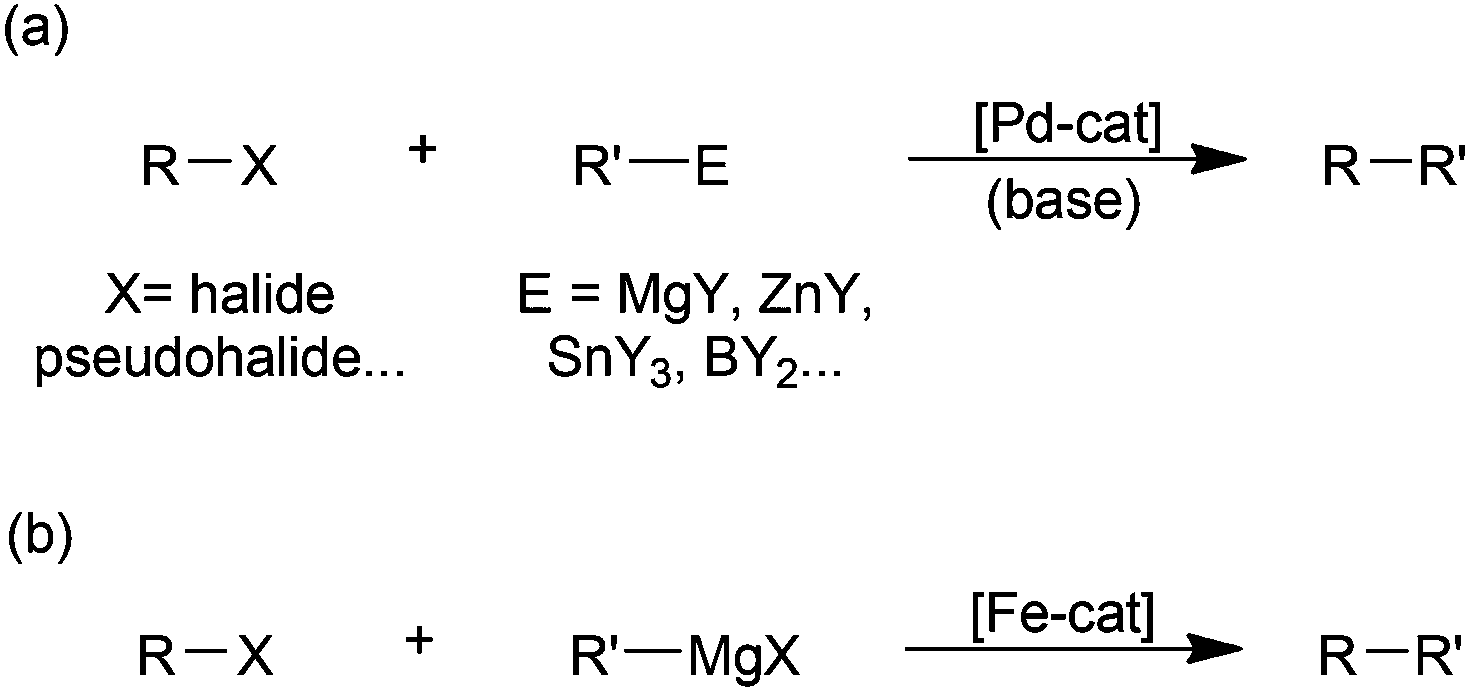 | ||
| Scheme 1 (a) Generalised palladium-catalysed cross-coupling; (b) the iron-catalysed Kumada reaction. | ||
While several classes of complexes and ligands have been investigated, amine complexes of iron have been found to be particularly useful pre-catalysts in a range of iron-catalysed Kumada reactions, often giving high yields and good selectivity for the cross-coupled product. For instance Nakamura and Nakamura showed that 1,2-bis(dimethylamino)ethane, TMEDA, could be employed to good effect, providing that it was used in stoichiometric or greater amounts with respect to the Grignard substrate and that the amine–Grignard mixture was added slowly to the reaction mixture, typically with a syringe pump.5 We,6 and subsequently Cahiez,7 demonstrated that TMEDA, as well as other amine ligands, could actually be used in catalytic quantities and that slow addition was not always required for good activity.
Clearly, given the successes enjoyed with TMEDA and other chelating amines, it is important to establish the role(s) played by the ligands in the catalytic cycle of the iron-catalysed Kumada reaction. In the first study to address this issue, Nagashima and co-workers investigated the reaction of 3 equivalents of mesitylMgBr with FeCl3 and excess TMEDA.8 They showed that this gave the bis-mesityl complex 1, which reacts with octyl bromide to give the cross-coupled product and the second mesityl complex 2. Reaction of 2 with the Grignard reagent regenerates 1. On the basis of these observations, the authors proposed the catalytic cycle outlined in Scheme 2.
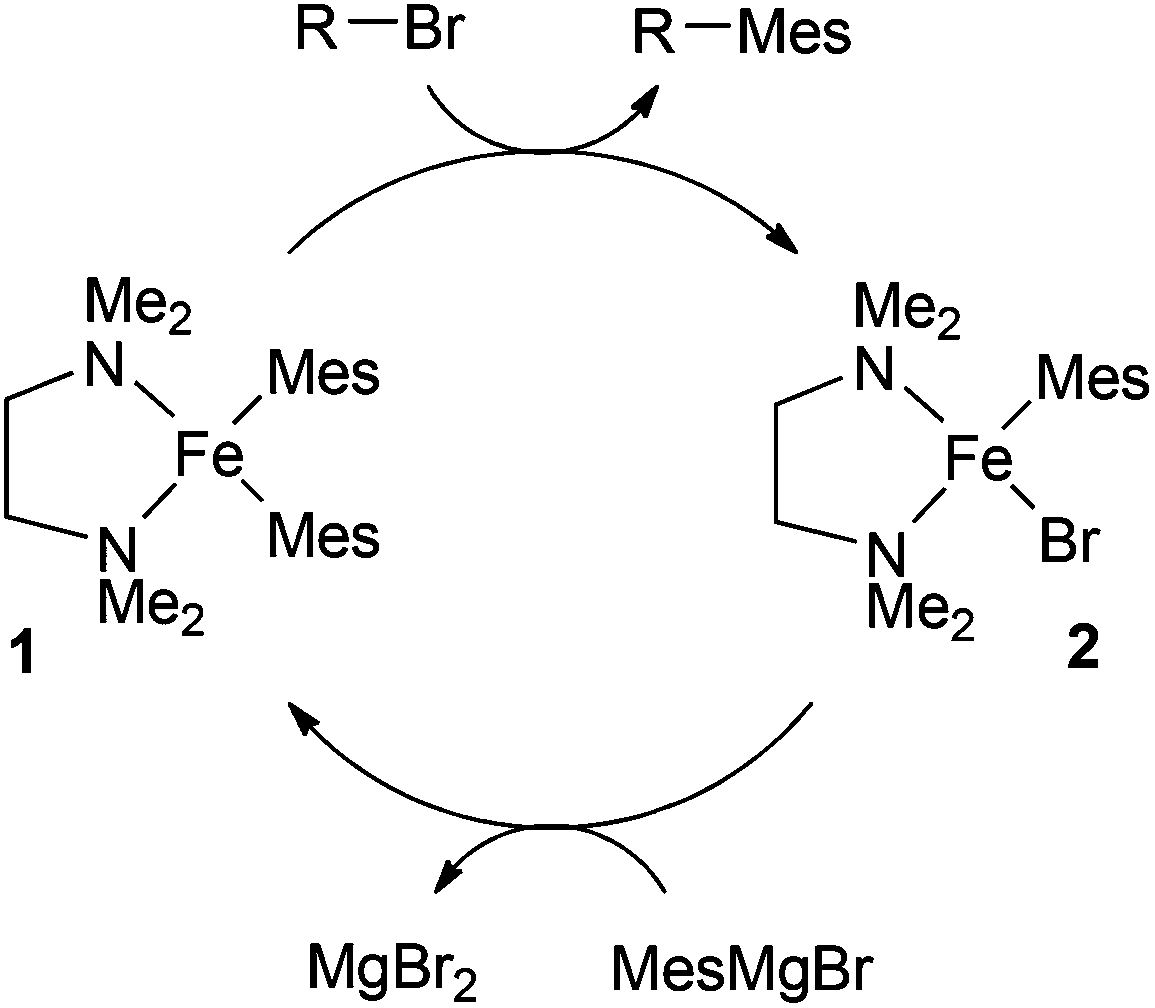 | ||
| Scheme 2 Nagashima's proposed catalytic cycle.8 | ||
By contrast, we demonstrated that in the presence of excess MesMgBr, such as would be seen under catalytic conditions, complex 1 reacts to generate the homoleptic ‘ate’ complex 3 (Scheme 3).9 Indeed 1H NMR studies revealed that during catalysis only complex 3 is observed and not complex 1. Furthermore, complex 3 reacts far faster than complex 1 with OctBr, effectively eliminating 1 as a feasible catalytic intermediate in the primary cycle. We instead proposed the cycle shown in Scheme 3,10 based on the observations above and the finding that the same amount of cross-coupled product is generated in the absence of TMEDA, but that slightly increased amounts of side-products are obtained, suggesting a competing, non-selective pathway, possibly involving iron nanoparticles.9 In this proposed pathway, the amine ligand is not involved in the primary catalytic cycle, but rather it intercepts the thermally fragile intermediates derived from the reaction of the intermediate 4 with the electrophilic coupling partner, generating 2 before further decomposition can occur. Subsequent reaction of 2 with excess MesMgBr regenerates 3via the intermediate 1.
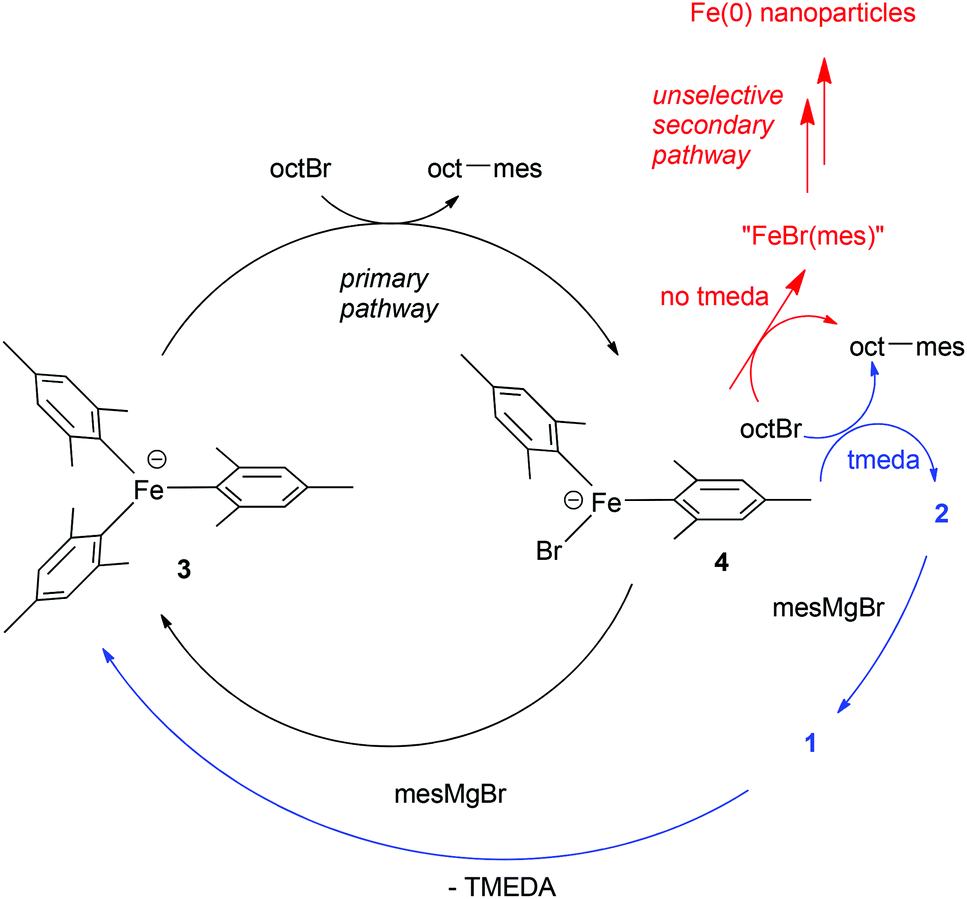 | ||
| Scheme 3 Alternative catalytic pathway.10 | ||
Ultimately, these model studies are limited because mesityl Grignard is a poor, non-representative substrate in iron-catalysed cross-couplings, with low yields of the cross-coupled product furnished after protracted reaction times.8,9 Accordingly, we decided to investigate the effect of varying the amine-donor ligands in the presence of a smaller, far more representative aryl nucleophile. Clearly, if the mechanism in Scheme 3 is still operative with less bulky Grignard reagents, and the amine is required to dissociate from the iron centre in order for catalysis to proceed, then there should be an inverse dependence of catalytic activity on chelate complex stability. The results from this study support this hypothesis and are presented below.
Results and discussion
Iron-amine chelate complexes
The chelating amine complexes selected for study are shown in Fig. 1. Leigh and co-workers showed that the TMEDA adducts 5 and 6 both form in the reaction of TMEDA with FeCl2, crystallising at different rates.11 Therefore while we isolated the complex 6, in the catalytic studies (see below) mixtures of TMEDA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :FeCl2 were used instead. The previously reported complex 7 contains the diamine ligand (−)-sparteine.12 The cage-like structure of sparteine gives much higher conformational rigidity compared with TMEDA, which in turn should lead to higher chelate complex stability. The tetradentate ligand in complex 8, HMTETA, should also give more stable iron complexes than those containing TMEDA, due to the higher denticity of the ligand. Complex 8 was prepared by reaction of HMTETA with FeCl2 in THF at reflux temperature for 8 hours, and the single crystal X-ray structure of the complex is shown in Fig. 2. As can be seen, the complex adopts a cis coordination geometry, in contrast to the TMEDA complex 5.
:FeCl2 were used instead. The previously reported complex 7 contains the diamine ligand (−)-sparteine.12 The cage-like structure of sparteine gives much higher conformational rigidity compared with TMEDA, which in turn should lead to higher chelate complex stability. The tetradentate ligand in complex 8, HMTETA, should also give more stable iron complexes than those containing TMEDA, due to the higher denticity of the ligand. Complex 8 was prepared by reaction of HMTETA with FeCl2 in THF at reflux temperature for 8 hours, and the single crystal X-ray structure of the complex is shown in Fig. 2. As can be seen, the complex adopts a cis coordination geometry, in contrast to the TMEDA complex 5.
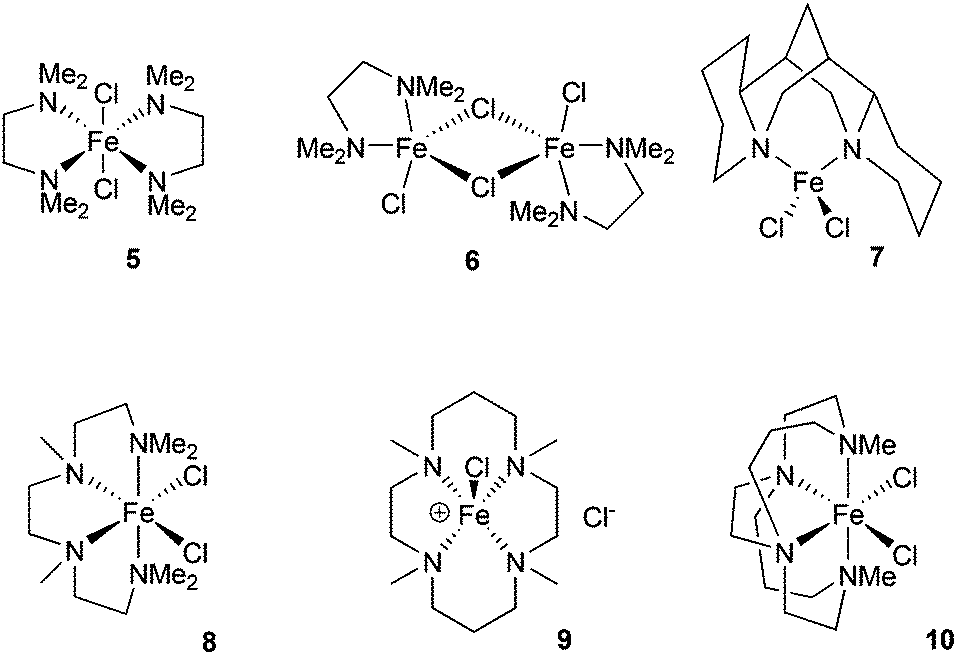 | ||
| Fig. 1 The iron complexes examined. | ||
In order to increase complex stability further, the tetra-methylcyclam (TMC)-containing complex 9 was prepared by warming the ligand with FeCl2 in acetonitrile at 40 °C for 90 minutes. The macrocyclic nature of the TMC ligand should confer higher stability of resultant complexes compared with the acyclic HMTETA. The crystal structure of 9 (Fig. 2) revealed a five-coordinate cationic complex, with chloride counter ion, in which the TMC ligand occupies both axial coordination sites and two of the equatorial ones, with the third equatorial position occupied by chloride.
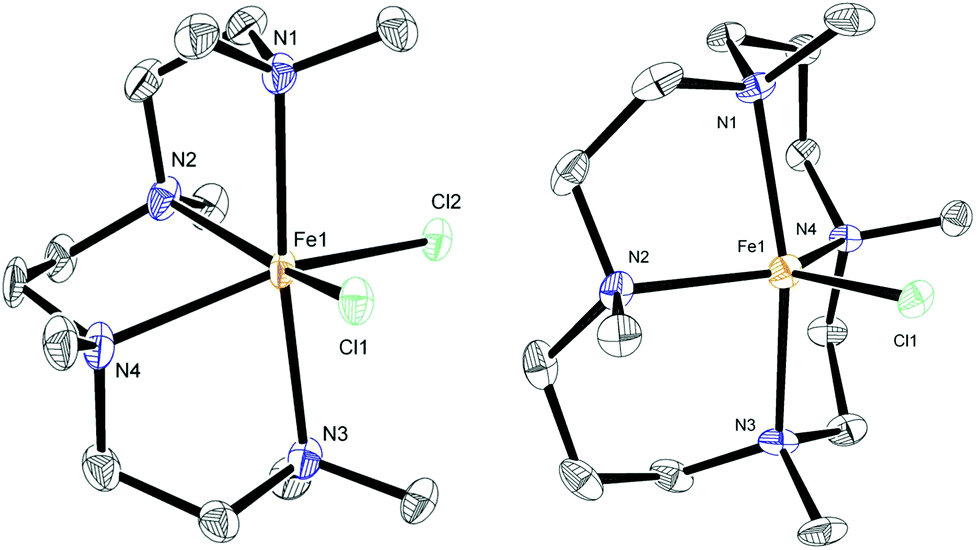 | ||
| Fig. 2 X-ray crystal structures of complexes 8 and 9. Thermal ellipsoids set at 50% probability; hydrogen atoms, chloride counter-ion of 9 and three molecules of MeCN solvate in 9 omitted for clarity. | ||
Finally, the previously reported complex 10 was examined,13 which contains an ethylene-bridged cyclam ligand (Me2EBC). In this case the bicyclic nature of the ligand would be expected to confer even greater macrocylic stabilisation, compared with the TMC ligand, giving by far the most stable complex of the series.
Catalytic studies
The complexes 7–10, as well as the species formed in situ from FeCl2 and TMEDA in THF were tested as pre-catalysts in the representative cross-coupling of 4-tolyl magnesium bromide with cyclohexylbromide, under two sets of conditions. In the first set of (A), the reaction was performed at 0 °C with slow addition of the Grignard reagent using a syringe pump, conditions similar to those reported by Nakamura.5 Meanwhile in the second set of conditions (B) the Grignard reagent was added rapidly and the reaction was warmed to 40 °C, to mirror the conditions that we previously exploited.6 The results from this study are summarised in Table 1.|
|
||||
|---|---|---|---|---|
| Entry | [Fe-cat] | Conditionsa | Conversion to 11b, % | Appearance at end of reaction |
| a Conditions, A: CyBr (1.0 mmol), 4-tolylMgBr (drop-wise, 1.2 mmol), [Fe-cat] (0.05 mmol), THF, 0 °C, 1 h; B: CyBr (1.0 mmol), 4-tolylMgBr (2.0 mmol), [Fe-cat] (0.05 mmol), Et2O, 40 °C, 1 h. b Conversion to 11 determined by 1H NMR spectroscopy (1,3,5-trimethoxybenzene internal standard). c 2.0 mmol. d 0.1 mmol. | ||||
| 1 | FeCl2 + excess TMEDAc | A | 93 | Heterogeneous |
| 2 | FeCl2 + 2TMEDAd | B | 77 | Heterogeneous |
| 3 | 7 | A | 51 | Heterogeneous |
| 4 | 7 | B | 54 | Heterogeneous |
| 5 | 8 | A | 19 | Heterogeneous |
| 6 | 8 | B | 53 | Heterogeneous |
| 7 | 9 | A | 15 | Homogeneous |
| 8 | 9 | B | 3 | Homogeneous |
| 9 | 10 | A | 1 | Homogeneous |
| 10 | 10 | B | 3 | Homogeneous |
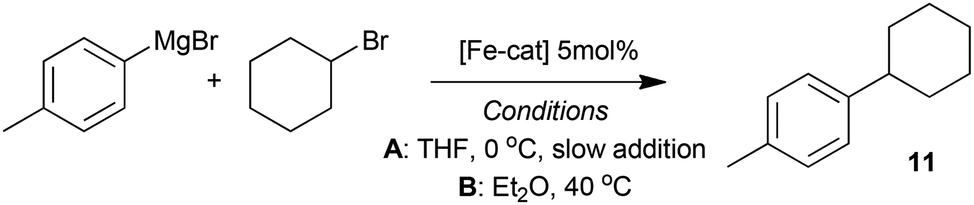
Compared with the excellent and good conversions observed after 1 hour with the TMEDA-containing system under slow and rapid addition of the Grignard reagent respectively (entries 1 and 2) it is clear that much lower activity is seen with the pre-catalyst 7, which contains the more conformationally rigid diamine ligand sparteine (entries 3 and 4). Even worse activity is observed with complex 8, which contains the tetradentate chelating ligand HMTETA, under slow addition conditions (entry 5). Interestingly some activity is recovered when the reaction was repeated with rapid addition of the Grignard reagent.
In all the examples above, the catalysis is accompanied by the formation of black, heterogeneous reaction mixtures, most likely due to the formation of iron nanoparticles, which have previously been demonstrated to be catalytically competent in Kumada14 and related cross-coupling reactions.15 By contrast, the reactions catalysed by complexes 9 or 10 remained homogeneous over the course of the reactions under both sets of conditions, and in all cases gave very little of the cross-coupled product 11. It is clear that an increase in the chelate or macrocyclic complex stability leads to a sharp decrease in catalyst performance. This is despite the fact that, in all the cases examined, the pre-catalysts should be capable of providing the cis-disposed reactive sites that might be envisaged as necessary for catalytic activity if an iron-amine-based cycle such as that shown in Scheme 2 were operative.
Based on both the relative productivities of the pre-catalysts and the appearance of the reaction mixtures, it is tempting to conclude that the amine ligands do indeed dissociate from the iron centre prior to its entry into the primary catalytic cycle, as hypothesised and outlined in Scheme 3. In this scenario, amine dissociation would be triggered by reaction with excess Grignard reagent, therefore we next focussed on establishing the ease and extent of ligand dissociation from the amine complexes in the presence of a representative Grignard reagent.
Reactions with benzyl Grignard reagent
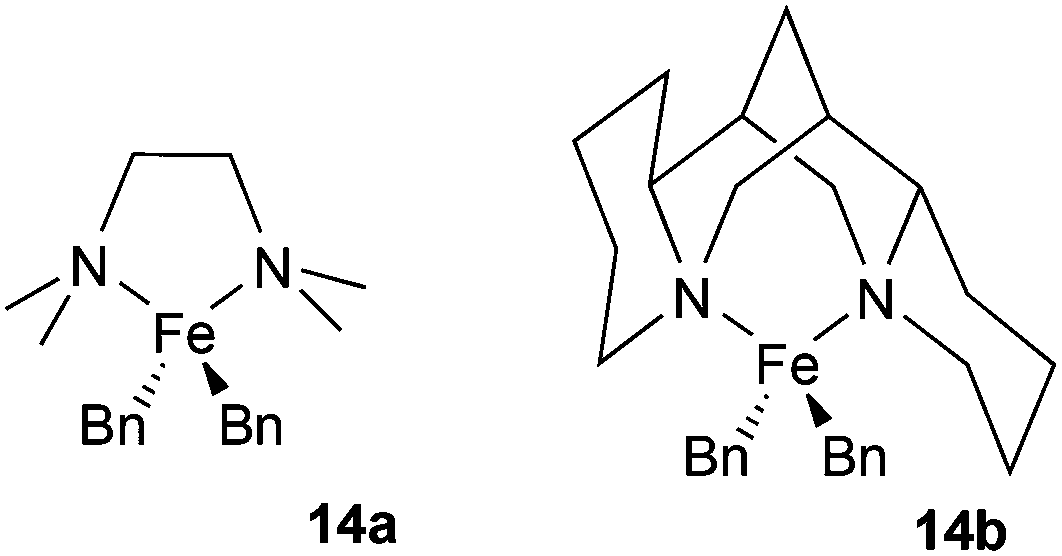
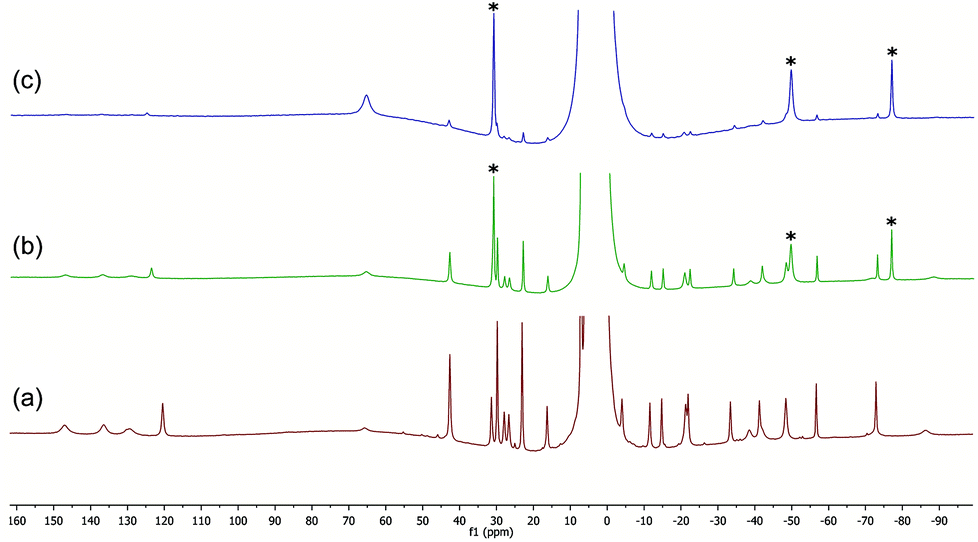
We previously found that the reaction of either FeCl2 or FeCl2/TMEDA with excess 4-tolyl Grignard at low temperature (−30 °C) gives species tentatively assigned as the homoleptic arylferrates [FeAr3]− and [FeAr4]2− respectively, as characterised by 1H NMR spectroscopy. In the latter case the TMEDA ligand is not coordinated to the paramagnetic iron centre, but instead most likely coordinates to the magnesium salt counter-ion. Whilst these species have so far proven too thermally fragile for us to isolate, we found that we were able to isolate and characterise the comparable Fe(II) benzyl-containing anionic ‘ate’ complex, [FeBn3]− (12), as well as the Fe(III) analogue [FeBn4]− (13), and probe its significance as a possible active intermediate in cross-coupling.9 In view of the improved stability of iron-benzyl adducts compared to iron-4-tolyl intermediates, we decided to explore the reactivity of the iron amine-complexes listed above with benzyl Grignard.Fig. 3 shows the 1H NMR spectra recorded on addition of varying amounts of BnMgCl to complex 6 in THF. Complex 6 is NMR silent, but the addition of one equivalent of the Grignard reagent gives a spectrum assigned as that of the complex [FeClBn(TMEDA)], 15, which could be isolated in modest yield when the reaction was repeated on a larger scale. Addition of a further 0.5 or 1 equivalent of the Grignard reagent to the NMR-scale reaction mixture led to increasing amounts of a species previously identified by Sen and co-workers as the bis-benzyl complex 14a.16 As described previously,9 the addition of excess BnMgCl rapidly led to the formation of the ‘ate’ complex 12.
 | ||
| Fig. 3 (a) 1H NMR spectrum of complex 6 in THF (NMR silent). (b) Part of spectrum of complex 15, formed in situ from 6 and 1 equiv. BnMgCl/Fe (peak at 1067 ppm omitted). (c) and (d) Addition of a further 0.5 and 1 equivalent of BnMgCl/Fe. Peaks marked ‘*’ correspond to [FeBn2(TMEDA)], (14a). | ||
 | ||
| Fig. 4 (a) 1H NMR spectrum of complex 14b in THF. (b) 14b + 20 BnMgCl, after 10 min. (c) As above, after 5 h; peaks marked with ‘*’ correspond to ‘ate’ complex 12. | ||
The 1H NMR spectrum of the HMTETA-containing complex 8 in THF at room temperature is shown in Fig. 5(a), whilst (b) shows the spectrum after addition of 2 equivalents of BnMgCl. The latter spectrum is more complicated and is likely to correspond to the formation of one or more iron-benzyl/HMTETA adducts. On addition of a further two equivalents of the Grignard reagent, the tetraamine ligand was lost from the iron centre and the ‘ate’ complex 12 was once again the only paramagnetic iron-containing complex observed (spectrum (c)). The data clearly show that, as with both the TMEDA- and sparteine-containing complexes, the presence of excess Grignard reagent favours the dissociation of HMTETA and the concomitant formation of the amine-ligand-free homoleptic ‘ate’ complex 12.
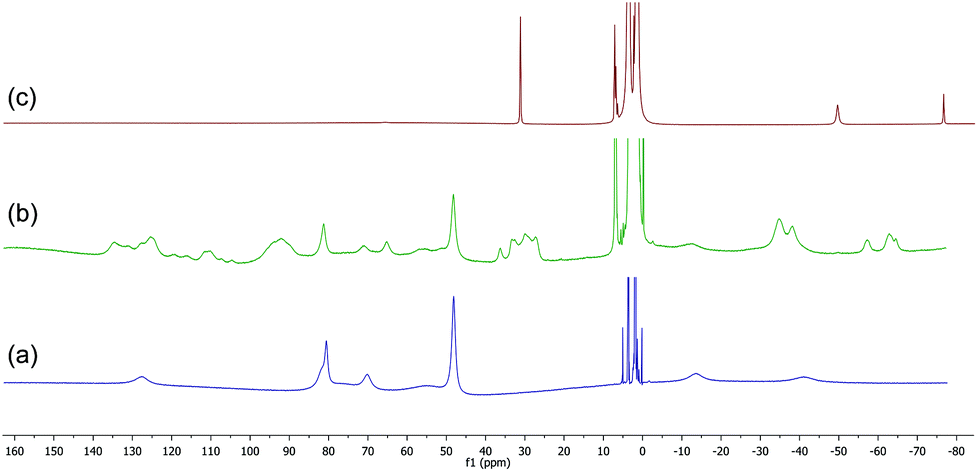 | ||
| Fig. 5 (a) 1H NMR spectrum of complex 8. (b) and (c) Spectra recorded after addition of 2 and 4 equiv. BnMgCl respectively. | ||
The 1H spectra of macrocyclic complexes 9 and 10 are shown in Fig. 6 (spectra (a) and (c) respectively), meanwhile the spectra of the red and orange reaction mixtures recorded 10 minutes after addition of excess BnMgCl (10 equivalents) to 9 and 10 are shown in plots (b) and (d) respectively. It is clear that new paramagnetic complexes were formed in both cases, again presumably containing benzyl ligands in addition to the macrocyclic amine ligands. However, in stark contrast to all of the reactions discussed above with iron complexes of TMEDA, sparteine or HMTETA, none of the homoleptic anionic complex 12 was observed suggesting that the macrocyclic ligands were not displaced by the Grignard reagent. Indeed, no 12 was observed when the spectra were run again after 24 hours.
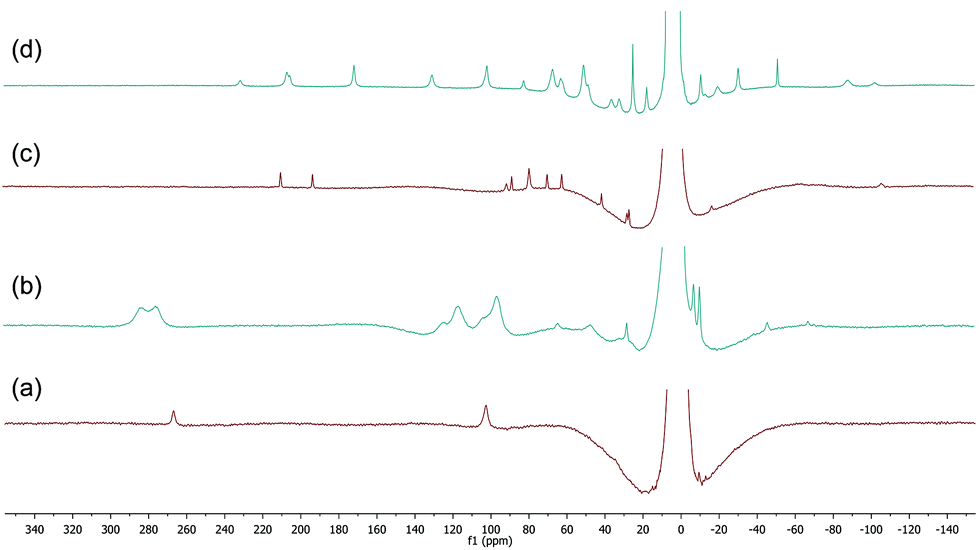 | ||
| Fig. 6 (a) and (c) 1H NMR spectra of complexes 9 and 10 respectively (THF, r.t.); (b) and (d) spectra recorded 10 minutes after addition of 10 equiv. BnMgCl to 9 and 10 respectively. | ||
Conclusions
The lability of the amine ligands of the complexes 5–10 in the presence of excess benzyl Grignard reflects decreasing catalytic activity in the coupling of cyclohexylbromide with 4-tolylmagnesium bromide, with the least labile systems showing the poorest catalytic activity. Of particular note, whilst the complexes 9 and 10 do react with benzyl Grignard, the amine ligands are not displaced by excess Grignard, unlike the amine ligands in the other complexes examined. These two complexes show only very poor activity as catalysts in the cross-coupling reactions and it is important to note that in these cases the reaction mixtures stay homogeneous throughout the catalysis. Conversely TMEDA, sparteine and HMTETA are all displaceable by excess benzyl Grignard, and their complexes are catalytically competent with an activity in the order HMTETA < sparteine < TMEDA reflecting the decreasing chelate effect of the ligands. In these cases the catalytic reactions all turned black, suggestive of the formation of iron nanoparticles.Taken together the data reported here provide further strong evidence that amine ligands are not coordinated to the iron centre in the primary catalytic cycle of Kumada reactions employing iron-amine pre-catalysts, lending support to variants of the catalytic cycle outlined in Scheme 3.
Experimental
General
All reactions were carried out under a dry atmosphere of nitrogen using standard Schlenk line and glove box techniques, unless otherwise specified. Anhydrous solvents were obtained from a Grubbs solvent drying system, degassed and stored over molecular sieves in a flame dried Strauss flask. Commercial reagents were used as received without further purification unless otherwise specified. The ligand Me2EBC was prepared according to a literature method.171H NMR spectra were recorded on a Jeol Lambda 300, Jeol ECP 400, Varian 400-MR, or Varian 500 spectrometer. Half-height peak widths for paramagnetic compounds are reported in Hz in parentheses after the chemical shift. Elemental analyses were obtained from the microanalytical service at the University of Bristol. Infrared spectra were recorded on a PerkinElmer Spectrum 100 FTIR Spectrometer, selected frequencies are reported.Cross-coupling of 4-tolylMgBr with cyclohexyl bromide Table 1, conditions A
Cyclohexyl bromide (123 μl, 1.0 mmol), the appropriate iron complex (0.05 mmol) and where appropriate, TMEDA (300 μl, 2.0 mmol) were stirred for 3 minutes in THF (2.0 ml) held at 0 °C. 4-TolylMgBr (1.2 mmol) was added dropwise with stirring over 30 min using a syringe pump and the reaction mixture was stirred for a further 30 min at 0 °C. The reaction was quenched by addition of H2O (5 ml) and the organic products extracted with CH2Cl2 (3 × 10 ml). 1,3,5-Trimethoxybenzene (168 mg, 1.0 mmol) was added as an internal standard and the conversion to cross-coupled product was determined by 1H NMR spectroscopy. Conditions B. Cyclohexyl bromide (123 μl, 1.0 mmol), the relevant iron complex (0.05 mmol) and where appropriate, TMEDA (15 μl, 0.1 mmol) were stirred for 3 minutes in Et2O (3.0 ml) at room temperature. 4-TolylMgBr (2.0 mmol) was added in one portion and the reaction mixture heated at reflux temperature for 1 h. Workup and determination of conversion to cross-coupled product as above.NMR studies of the reaction of iron-amine chelate complexes with BnMgCl (Fig. 3–6)
The appropriate iron-amine complex (0.5 mmol) was dissolved in THF (2 ml), BnMgCl (0.84 M in THF, specified amount) was added at room temperature, the reaction mixture stirred for 10 min, a sample was removed and the 1H NMR was recorded.Preparation of [FeClBn(TMEDA)], 15
Complex 6 (0.972 g, 2.0 mmol) was dissolved in THF (5.0 ml), BnMgCl added (2.0 ml, 2.0 M in THF, 4.0 mmol) and the reaction mixture stirred for 10 min to give an orange solution. Cooling the solution at −20 °C gave 15 as an orange solid (402 mg, 33.7%). 1H NMR (400 MHz, 25.0 °C, THF) δ 1066.8 (3823 Hz, 4H), 121.6 (2168), 102.7 (892), 99.4 (763), 93.3 (688), 88.1 (1897), 30.4 (73, 4H), −36.3 (337, 4H), −61.9 (76, 2H). Anal. Found: C, 52.58; H, 8.02; N, 8.95. Calcd for C26H46Cl2Fe2N4: C, 52.28; H, 7.76; N, 9.38.Preparation of [FeBn2((−)-sparteine)], 14b
A literature preparation was followed.12 To 7 (0.203 g, 0.562 mmol) was added diethyl ether (5 ml) and the mixture stirred. To this was added KCH2C6H5 (0.146 g, 1.12 mmol) in diethyl ether (10 ml) and the reaction mixture was stirred for 6 h to give a yellow solution. This was filtered through celite and the solvent removed in vacuo to yield 14b as a yellow solid (0.151 g, 57.1%). The 1H NMR spectroscopic data (benzene-d6) were in agreement with reported values,12 and were also recorded in THF. 1H NMR (400 MHz, 25.0 °C, THF) δ 932.8 (2433, 1H), 896.2 (2107, 1H), 827.8 (2401, 1H), 777.7 (1626, 1H), 350.9 (343, 1H), 341.2 (371, 1H), 218.9 (315, 1H), 147.3 (958, 1H), 136.8 (794, 1H), 129.6 (1203, 1H), 120.8 (215, 1H), 43.2 (135, 2H), 31.7 (134, 1H), 29.9 (99, 2H), 28.4 (168, 1H), 27.1 (187, 1H), 23.1 (103, 2H), 16.7 (150, 1H), 7.1 (93, 1H), 6.0 (105, 1H), −3.6 (177, 1H), −11.3 (134, 1H), −14.6 (122, 1H), −21.4 (311, 1H), −21.8 (161, 1H), −33.3 (137, 1H), −38.3 (575, 1H), −41.0 (165, 1H), −41.8 (649, 1H), −48.3 (277, 1H), −56.7 (87, 1H), −72.9 (104, 2H), −86.0 (1139, 1H), −148.1 (1208, 1H).Acknowledgements
We thank the EPSRC for a PhD studentship (JN) and postdoctoral support (PBB and DE, grant number EP/K012258/1).Notes and references
- (a) Metal-Catalyzed Cross-Coupling Reactions, ed. A. de Meijere and F. Diederic, Wiley-VCH, 2nd edn, 2008 Search PubMed; (b) Handbook of Organopalladium Chemistry for Organic Synthesis, ed. E. Negishi, Wiley, 2002 Search PubMed.
- M. Albrecht, R. Bedford and B. Plietker, Organometallics, 2014, 33, 5619 CrossRef CAS.
- It should be noted that iron-catalysed cross-couplings pre-date analogous palladium-catalysed processes. For seminal examples see: (a) G. Vavon and P. Mottez, C. R. Hebd. Seances Acad. Sci., 1944, 218, 557 CAS; (b) W. C. Percival, R. B. Wagner and N. C. Cook, J. Am. Chem. Soc., 1953, 75, 3731 CrossRef CAS; (c) M. Tamura and J. K. Kochi, J. Am. Chem. Soc., 1971, 93, 1487 CrossRef CAS; (d) M. Tamura and J. Kochi, Synthesis, 1971, 303 CrossRef CAS; (e) S. M. Neumann and J. K. Kochi, J. Org. Chem., 1975, 40, 599 CrossRef CAS; (f) R. S. Smith and J. K. Kochi, J. Org. Chem., 1976, 41, 502 CrossRef CAS.
- Recent reviews: (a) I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170 CrossRef CAS PubMed; (b) R. B. Bedford and P. B. Brenner, Top. Organomet. Chem., 2015, 50, 19 CrossRef CAS; (c) E. Nakamura, T. Hatakeyama, S. Ito, K. Ishizuka, L. Ilies and M. Nakamura, Org. React., 2014, 83, 1 CAS.
- M. Nakamura, K. Matsuo, S. Ito and E. Nakamura, J. Am. Chem. Soc., 2004, 126, 3686 CrossRef CAS PubMed.
- R. B. Bedford, D. W. Bruce, R. M. Frost and M. Hird, Chem. Commun., 2005, 4161 RSC.
- G. Cahiez, V. Habiak, C. Duplais and A. Moyeux, Angew. Chem., Int. Ed., 2007, 46, 4364 CrossRef CAS PubMed.
- D. Noda, Y. Sunada, T. Hatakeyama, M. Nakamura and H. Nagashima, J. Am. Chem. Soc., 2009, 131, 6078 CrossRef CAS PubMed.
- R. B. Bedford, P. B. Brenner, E. Carter, P. M. Cogswell, M. F. Haddow, J. N. Harvey, D. M. Murphy, J. Nunn and C. H. Woodall, Angew. Chem., Int. Ed., 2014, 53, 1804 CrossRef CAS PubMed.
- R. B. Bedford, Acc. Chem. Res., 2015, 48, 1485 CrossRef CAS PubMed.
- S. C. Davies, D. L. Hughes, G. J. Leigh, J. R. Sanders and J. S. de Souza, J. Chem. Soc., Dalton Trans., 1997, 1981 RSC.
- S. C. Bart, E. J. Hawrelak, A. K. Schmisseur, E. Lobkovsky and P. J. Chirik, Organometallics, 2004, 23, 237 CrossRef CAS.
- T. J. Hubin, J. M. McCormick, S. R. Collinson, M. Buchalova, C. M. Perkins, N. W. Alcock, P. K. Kahol, A. Raghunathan and D. H. Busch, J. Am. Chem. Soc., 2000, 122, 2512 CrossRef CAS.
- R. B. Bedford, M. Betham, D. W. Bruce, S. A. Davis, R. M. Frost and M. Hird, Chem. Commun., 2006, 1398 RSC.
- R. B. Bedford, P. B. Brenner, E. Carter, T. W. Carvell, P. M. Cogswell, T. Gallagher, J. N. Harvey, D. M. Murphy, E. C. Neeve, J. Nunn and D. R. Pye, Chem. – Eur. J., 2014, 20, 7935 CrossRef CAS PubMed.
- D. H. Hill and A. Sen, J. Am. Chem. Soc., 1988, 110, 1650 CrossRef CAS.
- E. H. Wong, G. R. Weisman, D. C. Hill, D. P. Reed, M. E. Rogers, J. S. Condon, M. A. Fagan, J. C. Calabrese, K. Lam, I. A. Guzei and A. L. Rheingold, J. Am. Chem. Soc., 2000, 122, 10561 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic experimental and data. CCDC 1478297 and 1478298. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt01823h |
| This journal is © The Royal Society of Chemistry 2016 |