Measuring the metabolome: current analytical technologies
Warwick B. Dunn*a, Nigel J. C. Baileyb and Helen E. Johnsonc
aBioanalytical Sciences Group, School of Chemistry, University of Manchester, Faraday Building, Sackville Street, P. O. Box 88, Manchester, UK M60 1QD. E-mail: Warwick.Dunn@manchester.ac.uk; Fax: 0161 2004556; Tel: 0161 2004414
bAnalytical and Discovery Technologies, SCYNEXIS Europe Ltd., Fyfield Business and Research Park, Fyfield Road, Ongar, Essex, UK CM5 0GS
cInstitute of Biological Sciences, University of Wales, Aberystwyth, Ceredigion, UK SY23 3DD
First published on 4th March 2005
Abstract
The post-genomics era has brought with it ever increasing demands to observe and characterise variation within biological systems. This variation has been studied at the genomic (gene function), proteomic (protein regulation) and the metabolomic (small molecular weight metabolite) levels. Whilst genomics and proteomics are generally studied using microarrays (genomics) and 2D-gels or mass spectrometry (proteomics), the technique of choice is less obvious in the area of metabolomics. Much work has been published employing mass spectrometry, NMR spectroscopy and vibrational spectroscopic techniques, amongst others, for the study of variations within the metabolome in many animal, plant and microbial systems. This review discusses the advantages and disadvantages of each technique, putting the current status of the field of metabolomics in context, and providing examples of applications for each technique employed.
 Warwick B. Dunn | Warwick Dunn currently works as a Research Associate in the Bioanalytical Sciences Group at the University of Manchester, employing mass spectrometry technologies to study microbial, plant and mammalian metabolomes. Previously he obtained BSc and PhD Analytical Chemistry based degrees at the University of Hull and has subsequently employed organic and isotope ratio mass spectrometry techniques in solving chemical and biological problems, in both industrial and academic environments. |
 Nigel J. C. Bailey | Nigel Bailey currently works as a senior research spectroscopist for SCYNEXIS Europe Ltd, Ongar, Essex. Previously he worked as a post-doctoral researcher in Jeremy Nicholson's group, Imperial College, London; developing NMR-based metabonomic approaches to the study of both mammalian and plant systems. He obtained his PhD in the same group, developing hyphenated NMR systems for the analysis of samples of agrochemical interest. Nigel obtained his BSc Analytical Chemistry degree from the University of Hull. |
 Helen E. Johnson | Helen Johnson obtained BSc and PhD degrees at the University of Wales, Aberystwyth and subsequently works as a Research Associate in the Institute of Biological Sciences at the same university. Current and previous research interests include studying plant metabolomes and silage production with high-throughput metabolomics approaches including FT-IR and DIMS coupled with multivariate mathematical modelling and machine learning. |
Introduction
In the post-genomics age, the analysis of the ‘omes’ (transcriptome, proteome and metabolome) has become widespread with the objective, among others, of large scale quantitative determination of RNA, proteins and metabolites to both establish gene function and ascertain the connectivity within the ‘omic’ hierarchy.1,2The metabolome is the final downstream product of the genome and is defined as the total quantitative collection of small molecular weight compounds (metabolites) present in a cell or organism which participate in metabolic reactions required for growth, maintenance and normal function.3–5 The estimated size of the metabolome is large (S. cerevisiae approximately 600 metabolites,6 plant kingdom up to 200 000 metabolites,7 though significantly lower numbers for different species, and analysis of the human metabolome reveals even greater complexity and number of metabolites detected). In comparison to the proteome or transcriptome, the metabolome is more diverse in chemical and physical properties because of the larger variations in atomic arrangements. Studies of the metabolome include the analysis of a wide range of chemical species, from low molecular weight polar volatiles such as ethanol, to high molecular weight polar glucosides, non-polar lipids and inorganic species.8 The range of metabolite concentrations can vary over nine orders of magnitude (pM–mM). These large variations in the nature and concentration of analytes to be studied provide challenges to all the analytical technologies employed in metabolomics strategies.
The terminologies used in the field do overlap and are at times incorrectly applied. The differences and similarities between ‘metabolomics’ and ‘metabonomics’ are topical and requires revision as to the clear definition of both. Metabolomics is generally defined as the analysis of intra and extra-cellular metabolites in simple biological systems (including microbial, plant and mammalian systems) generally using mass spectrometry and vibrational spectroscopy whereas metabonomics is defined as ‘the quantitative measurement of the dynamic multiparametric metabolic response of living systems to pathophysiological stimuli or genetic modification’9 and historically focuses on metabolic responses in environmental, marine and mammalian systems often using NMR spectroscopic detection.
During the last five years the scientific field of metabolomics has grown rapidly and become widely employed, though earlier applications were reported.10,11 The current knowledge concerning gene functionality is small3 and a large number of phenotypic12,13 and genotypic analyses14,15 are being undertaken. Other applications include biomarker determination indicative of disease, drug intervention or environmental stress,16–20 nutrigenomics and personal health assessments,21,22 clinical diagnostics,23,24 mode of action studies,25,26 metabolic engineering27 and the most widely anticipated goal of systems biology where the complete operation of the cell or organism is modelled by the integration of all ‘omics’ data.28 As more and more scientists become aware of the applicability and advantages of metabolomics, the application field is expected to expand further.
The metabolomics experiment (or metabolomics pipeline29) is composed of different stages (experimental design, sampling, sample preparation, sample analysis, data pre-processing and data processing) and several reviews are available.4,5,30–34 Data processing is of specific interest with general reviews5,13,29,33,35,36 and specific applications employing unsupervised9,37–39 and supervised multivariate analyses as well as evolutionary computing14,40,41 are available. All experimental stages should be carefully designed and executed to provide valid datasets and subsequently valid experimental conclusions and hypothesises. A number of different analytical strategies are employed, as shown in Table 1, with the ultimate goal of analysing a large fraction or all of the metabolites present. Realistically, a range of analytical technologies and not one will be employed to analyse all metabolites present.5,32,33 As a result of no single technology being ‘all encompassing’, there are currently no ‘set’ protocols to study metabolomics and there is a payoff between technologies and objectives (Fig. 1). It is the goal of researchers in the field to develop technologies that enable large scale, high-throughput screening, with the whole process being unbiased, robust, reproducible, sensitive and accurate.
 | ||
| Fig. 1 The ‘payoff’ between analytical technologies and the objectives of metabolomics. | ||
| Metabolomics |
| Non-biased identification and quantification of all metabolites in a biological system. The analytical technique(s) must be highly selective and sensitive. No one analytical technique, or combination of techniques, can currently determine all metabolites present in microbial, plant or mammalian metabolomes. |
| Metabonomics |
| The quantitative measurement of the dynamic multiparametric metabolic response of living systems to pathophysiological stimuli or genetic modification |
| Metabolite target analysis |
| Quantitative determination of one or a few metabolites related to a specific metabolic pathway after extensive sample preparation and separation from the sample matrix and employing chromatographic separation and sensitive detection. |
| Metabolite profiling/metabolic profiling |
| Analysis to identify and quantify metabolites related through similar chemistries or metabolic pathways. Normally employ chromatographic separation before detection with minimal metabolite isolation after sampling. |
| Metabolic fingerprinting |
| Global, rapid and high-throughput analysis of crude samples or sample extracts for sample classification or screening of samples. Identification and quantification is not performed. Minimal sample preparation. |
| Metabolic footprinting |
| Global measurement of metabolites secreted from the intra-cellular volume in to the extra-cellular spent growth medium. High-throughput method not requiring rapid quenching and time consuming extraction of intra-cellular metabolites for microbial metabolomics. |
The aim of this review is to describe and examine the stages of sampling, sample preparation, analysis and data processing in the metabolomics experiment. Particular emphasis will be placed on the most widely employed analytical technologies in metabolomics; mass spectrometry, NMR spectroscopy and vibrational spectroscopies.
Experimental design, sampling and sample preparation
Sample selection and preparation is a vital component of any metabolomics study as the biological systems studied are in constant metabolic flux (metabolic reaction half lives are generally less than 1 s) and there is the potential for metabolic changes to be observed that are not related to the scientific question being studied. The design of experiments is essential to ensure valid statistical conclusions.Sample size is dependent on the biological organism, level of environmental or biological control, laboratory or greenhouse size and finances available. Rigorous protocols must always be applied to ensure sampling is reproducible from day-to-day and month-to-month (sampling time, developmental or growth stage of cell or tissue, sample size, sample replicates). In microbial metabolomics the sample is small and the environment during growth is controlled and reproducible. In plant metabolomics management of the environment is more difficult even in controlled environment greenhouses as small variations (shade and light,42 diurnal changes in metabolism caused by photosynthesis,43 differences in the stage of growth of a plant,44 geographical variation, seasonal variation and harvesting procedures45) can all cause variation in biochemical status. The biological variation in human metabolomes is the largest of those discussed as it is the most difficult to observe in a controlled environment and differences in diet,22 diurnal changes, sex and estrus cycle,46 disease,20 lifestyle,47 time of sampling and drug dosing vehicle48 can all influence the metabolome. Groups can be fasted before sampling and matched controls are advisable, if possible, to reduce biological related variations separate to the variations derived from differences between healthy and diseased individuals. Nevertheless, research concludes that suitable controls regarding diet, lifestyle and time of sample collection should be put in place in order to allow metabolomic data to be used.
As in all biological experiments replication is essential to incorporate and assess inherent biological or analytical variation and also to provide a sample set representative of the whole population. The numbers of replicates used should be considered at the biological, extraction and machine or analytical level. High-throughput, short analysis time strategies (NMR, FT-IR, DIMS) are more suitable for larger replicate numbers than approaches resulting in longer analysis times, such as chromatographic techniques. Generally though, biological variation (variation observed from the analysis of different samples of the same biological origin) can be expected to be significantly greater than analytical variation (multiple analyses of one sample) for the techniques discussed in this review, including NMR spectroscopy49 and GC-MS.50
Different strategies for sampling of metabolites can be performed. Extracellular metabolites present in human or animal biofluids are sampled either non-invasively (urine) or invasively (serum, plasma, cerebrospinal fluid). The process of sampling can change the metabolome's composition, an increase in concentration of catecholamines concentrations in blood can be detected after invasive sampling, and is most likely caused by the fear of needles or anxiety of blood collection by the subject.51 Microbial extracellular metabolites excreted from cells (endometabolome) into growth medium (exometabolome) can be sampled and analysed employing metabolic footprinting40 as a high throughput, non-invasive microbial metabolomics strategy. Metabolites in the exometabolome are less readily available for biological reactions because of the large concentration dilution in growth medium and absence of enzymes. Alternatively, sampling of intra and extracellular microbial metabolites can be performed when aliquots of medium and cells are squirted into methanol held at −40 °C to quench metabolism followed by separation of the cell from the supernatant to allow analysis of both the endo and exometabolome.52 The study of urine and microbial metabolic footprints depict a picture of the metabolome during a period of metabolic and biological activity prior to sampling. For example, urinary metabolites are those produced over a period of time to be excreted from the body. Although time consuming and more technically demanding, the objective of sampling intracellular metabolites in microbes, plants or mammals is to provide a snapshot of the metabolome at the time of sampling rather than over a period of metabolic activity. Therefore to ensure rapid quenching of metabolic activity rapid freezing (in liquid nitrogen,53 freeze clamping54) or acidic treatments55 are undertaken. Extraction from cells can be an integral part of this quenching procedure or be performed at a later date.
Intracellular metabolite extraction procedures adopted will dictate the nature and levels of metabolites extracted. For non-targeted approaches the objectives are to extract the maximum number of metabolites from many chemical classes in a quantitative and non-biased manner with minimal losses of metabolites. For metabolic profiling, extraction is generally performed by the disruption of cell walls (lysation) by grinding, in for example a mortar and pestle with addition of liquid nitrogen to minimise metabolic activity, and subsequent distribution of metabolites into polar (methanol, water) and non-polar (chloroform, hexane, ethyl acetate) solvents followed by removal of the cellular residue.50 Other chemical extractions have been studied and compared including those for Escherichia coli metabolites where a cold (−40 °C) methanolic extraction procedure offered the greatest potential,56Arabidopsis thaliana57 and nonaqueous fractionation methods for selective extraction of glycolytic intermediates in potato tubers.58 Sample extraction should be designed and validated for the metabolites of interest before being applied as each provides problems. Rapid freezing can cause reduction in metabolite concentrations59 and wound metabolites to be produced. Freeze drying can cause metabolites to irreversibly bind to cell walls and acid treatments are not applicable to metabolites that are not stable at low pH. Sample preparation can be designed to include concentration steps to improve sensitivity. This is normally performed by drying and reconstitution in smaller solvent volumes. Stability in storage is an important factor. Either liquid or solid samples can be stored at −80 °C or drying can be performed which stops biological reactions as no medium is present to allow enzyme activation.
The preparation of samples for analysis is again dependent on the metabolomics strategy employed. For metabolic profiling and fingerprinting analyses, samples are either analysed directly without further separation of metabolites into subclasses, analysed after dilution or analysed after protein removal through precipitation.60 Targeted analyses ensure separation of the metabolome into chemical classes or separation of targeted metabolite(s) from the excess sample matrix (including other metabolites). There are solvent restrictions for all techniques discussed. For NMR spectroscopic analyses, solvents are deuterated (>10% v/v) and solvents that will provide multiple resonances are not applicable (hexane, ethanol, iso-propyl alcohol). In electrospray mass spectrometry solvents composed of non-volatile buffers can cause source contamination problems. In FT-IR water can present huge absorptions and hence the application of drying samples to remove water or attenuated total reflection (ATR) is employed. Further technique specific sampling considerations are outlined in the instrumental sections below.
Mass spectrometry
As an analytical tool mass spectrometry is widely employed and standard texts detailing theory and applications are available.61 In metabolomic applications mass spectrometry provides both sensitive detection and metabolite identification via mass spectrum interpretation and comparison or molecular formulae determination via accurate mass measurements. Hyphenated chromatography-mass spectrometry instrumentation (gas chromatography, high performance liquid chromatography, capillary electrophoresis) enables better metabolite identification and quantification. However, unlike NMR spectroscopy and FT-IR spectroscopy it is also destructive. In the future the utilization of HPLC-NMR-MS will provide the ability to undertake more precise metabolite identification while still maintaining sensitivity via the mass spectrometer detector.62Direct injection mass spectrometry (DIMS)
DIMS provides rapid, high throughput analyses of crude samples or sample extracts with typical analysis times of 1–3 min and up to 1000 analyses per day.63 Infusion of solutions with syringe pumps can be performed64 or automation with HPLC systems used.40 The technique usually employs electrospray mass spectrometers operating at µl min−1 solvent flow rates to detect polar metabolites, though the higher flow rate atmospheric pressure chemical ionisation (APCI) has been employed to analyse less polar and non-polar metabolites, or to improve sensitivity.65,66Different analytical issues should be considered when employing DIMS, including matrix effects and metabolite identification. Matrix effects67 (otherwise referred to as ionisation suppression or enhancement) should be assessed when analysing complex biological samples without chromatographic separation. Here the efficiency of ionisation in the liquid phase and subsequent transfer of ions from liquid to gas phase (droplet formation and desolvation) can be affected by the presence of other chemical species. The sensitivity and accuracy of quantification between samples of differing matrix composition (including differences between matrices for sample and standards) can be compromised. For samples of similar composition it is generally accepted that this effect is minimal, though further studies are required. Isotopic analogues of the metabolites of interest can be used as internal standards to compensate for any matrix effects so to enable accurate quantification, as is observed in disease diagnosis. Alternatively in plant and microbial metabolomics relative peak areas may be compared, with the assumption that matrix composition variation is low and between-sample reproducibility is good. Metabolites are generally preferentially ionised in only positive or negative ion modes, but not both. Therefore, for a non-biased approach, both ion modes should be used, though for metabolic profiling of one chemical functionality (for example characterisation of olive oils64) one ion mode can be chosen. For plant and microbial metabolomics µM concentrations can be detected with benchtop mass spectrometers68 and in clinical diagnostics µM and nM detection limits are observed.23
A typical mass spectrum of a S. cerevisiae metabolic footprint is shown in Fig. 2(a). Instrumental conditions were optimised to ensure minimal in-source ion fragmentation was observed. These mass spectra provide the ability, normally through chemometric based analyses, to discriminate between samples of different genotype or phenotype. The mass spectra are highly complex in the numbers of peaks detected and ranges of metabolite concentrations. A wide range of ions can be formed ([M + H]+, [M + NH4]+, [M + Na]+, [M + K]+, [M − H]−, [M − Cl]−, [2M + H]+ and [2M − H]−) depending on the sample matrix and ionisation modifiers added. The addition of formic acid can increase protonation though the high salt content of biological samples (up to mM concentration) can greatly enhance the presence of salt adducts. The majority of ions are detected with m/z less than 400 Da, as would be expected from observing metabolites which are defined as low MW compounds.69 Electrospray and APCI techniques are soft ionisation methods and hence molecular ions with few fragment ions are observed. This means chromatographic separation is not required.70
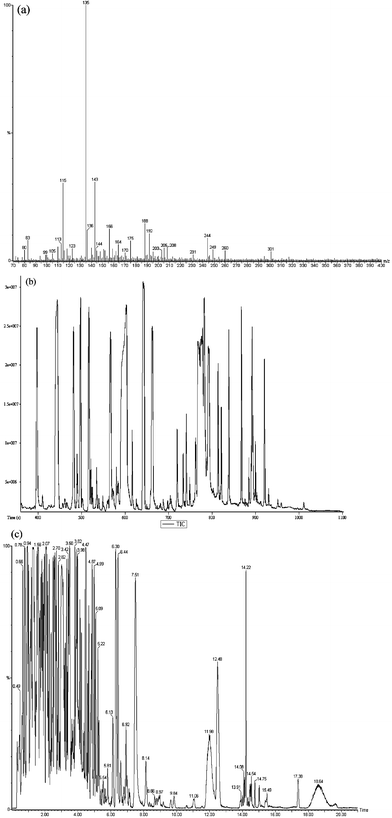 | ||
| Fig. 2 Typical mass spectrometer outputs as raw data plots. (a) Typical DIMS positive ion metabolic footprint of Saccharomyces cerevisiae. (b) Total ion chromatogram (TIC) of human plasma employing GC-TOF-MS. (c) Typical negative ion HPLC-MS base peak intensity (BPI) chromatogram for human urine collected on Waters Acquity UPLC™ system. | ||
Global analyses to discriminate between samples of differing biological status in microbial14 and plant71 systems are commonly observed. Early developments of microbial DIMS included chemotaxonomic discrimination of crude fungal extracts of Pencillium cultures via identification of strain specific secondary metabolites72 and the guidance of culture conditions through the assessment of secondary metabolites of actinomycetes.73 Discrimination of Escherichia coli wild-type and tryptophan mutant strains has been performed by DIMS analysis of the metabolic footprints40 and further work has used metabolic footprinting to discriminate between different physiological states of S. cerevisiae. This methodology has also provided discrimination between single gene knockout mutants of S. cerevisiae which have closely related areas of metabolism.14 Bacterial characterisation and identification using DIMS on cell free extracts has been studied74 and species specific peaks were identified by tandem mass spectrometry for strains of Escherichia coli, Bacillus spp and Brevibacillus laterosporus. Further research by these authors used intact microorganisms of Escherichia coli and Bacillus cereus to discriminate to below species level.75
In the more complex plant metabolome, DIMS has been employed in studying Pharbitis nil leaf sap where discrimination between different photoperiods was observed.71 Medicinal plant extracts have been characterized with the detection and identification of a wide range of secondary metabolites including anthocyanins, isoflavones, flavonolglycosides, terpenes, caffeoyl-quinic acids, ginsenosides, catechins, flavones and flavanones.68 Specific region dependent metabolites, mainly organic acid derivatives, were identified by tandem mass spectrometry in the characterization of European, American and African propolis resins.76 Fourier transform ion cyclotron resonance mass spectrometry (FTICR-MS) is a powerful tool.77 Accurate exact mass measurements (mass accuracy < 1 ppm) can be used to identify metabolites of interest where all metabolites are completely mass resolved (high specificity) and FTICR-MS is currently the only technique capable of the required mass resolutions (greater than 100 000 FWHM). However, structural isomers of the same molecular weight can not be resolved. Lower metabolite concentrations can also be detected when compared to other mass spectrometers. Aharoni et al., have employed FTICR-MS in sensitive, nontargeted metabolome analysis of strawberry fruits and tobacco flowers and to determine the identities of metabolites shown to differ between developmental stages of strawberry fruit.78
For data analysis, mass bins of constant size are generally employed. Data from all mass spectrometer instruments can be combined into unit mass bins and further analysed (i.e. all the responses for all peaks in mass range 99.501–100.500 are summed into mass bin 100).71 However, for data collected on high resolution mass spectrometers this can lead to loss of biological information where many metabolites contribute to the response for a single mass bin (for example, glutamine and lysine have the same nominal mass but different monoisotopic masses). A method has been developed which employs high resolution TOF mass spectra and a matching algorithm to provide automated comparison and classification of the mass spectra of different Penicillium species.79 In many other applications, raw data is normally expressed as mass lists (mass vs. intensity) and these can be easily exported into other software packages (such as Microsoft Excel™, MatLab™, Pirouette™) as text files for further data processing or analysis.
Clinical diagnostic applications provide screening of large sample numbers through targeted analyses of specific metabolites indicative of metabolism disorders. Samples are normally collected as blood63 or urine,80 spotted on filter paper and extracted into methanolic solutions containing isotopic analogues as internal standards, to enable isotope dilution techniques to be used for quantification.23 Some applications derivatise metabolites of interest to improve sensitivity or produce more readily ionisable derivatives, for example butylated derivatives of amino acids.23 These samples are then infused directly into an electrospray tandem mass spectrometer. Tandem mass spectrometry provides both specificity to the analysis of these crude samples, where each unit mass may be composed of more than one metabolite (isobaric interferences) and hence influence quantification and identification accuracy, and also enhancements in sensitivity by improvements in the signal to noise (S/N) ratio of measurements. A number of groups have applied the technique for newborn screening in Saudi Arabia,63 USA,81 Australia82 and Japan,83 all analysing between 23000 and 700000 samples in their studies. A wide range of metabolites are employed in disease diagnosis and are discussed elsewhere.23,84 Postnatal diagnosis is centred around the analysis of carnitine, acetylcarnitines and amino acids (normally methylated or butylated derivatives) to determine diseases including FAODs (fatty acid oxidation defectives) and organic acidemias.23,84–86 Postmortem diagnosis by analysis of bile fluids or urine have indicated metabolic disorders that resulted in sudden infant death syndrome.87 Determining the normal variation in any population is important and studies on reference ranges and cut-off values have been undertaken.63 In all of these applications introduction of internal standards and further automated data processing using, for example, computer-assisted metabolic profiling algorithm (CAMPA)63 provides data either as ratios of different metabolites which indicate the presence or absence of a disease or as full quantification by employing isotope dilution techniques.
Gas chromatography-mass spectrometry (GC-MS)
First employed over 20 years ago in disease diagnosis,88 GC-MS based metabolomic applications have grown rapidly for the analysis of volatile and thermally stable polar and non-polar metabolites. Analyses progress by high resolution chromatographic separations, at temperatures of up to 350 °C, on capillary gas chromatography columns. Subsequent sensitive detection of separated components is performed by electron impact35 or chemical ionisation89 mass spectrometry. Metabolite identification or confirmation is performed by retention time or index comparisons with pure compounds and mass spectral interpretation or comparison using retention index/mass spectral library databases.90 Metabolites can be classified into two classes; volatile metabolites not requiring chemical derivatisation and non-volatile metabolites requiring chemical derivatisation.Volatile samples are those that do not require chemical derivatisation to enable elution through gas chromatographs. Sampling protocols include direct collection and analysis of the sample headspace,91 collection of breath from mammals,92,93 absorption of metabolites from headspace or liquid samples onto solid absorbents94 or SPME fibres,95 and solvent extraction from liquid and solid samples.96 These samples are then directly analysed, generally without further sample preparation. Animal or human breath is normally collected as exhaled breath condensates (EBC) (which can also contain less volatile metabolites93), are stored frozen before analysis to minimise loss of volatiles, and are employed in health issues including workplace risk assessments97 and disease diagnostics.98 A separate non-invasive clinical diagnostic tool involves the diagnosis of infections including Helicobacter pylori. Here a 13C-urea containing bolus is consumed by a patient and the 13C/12C isotope ratio of CO2 in exhaled breath is determined pre- and post-consumption by gas chromatography-isotope ratio mass spectrometry (GC-IRMS). The difference in the isotope ratio pre-and post-consumption determines the presence or absence of infection.99 Stable isotopes, especially 13C, can also be used in studying flux and kinetics in metabolic pathways,100 though generally for metabolites requiring derivatisation. Increased production of terpenoids in tomato plants after spider mite infection has been observed after sampling and analysis of volatiles.101 Patel et al. have used GC-MS to study the influence of 20 different yeast strains on the volatile compounds produced from Symphony wines.102
In comparison, the analysis of non-volatile metabolites has largely been applied for metabolic profiling where multiple classes of metabolites (amino and organic acids, sugars, phosphorylated metabolites, amines, alcohols, lipids and others) are determined in one or a few analyses. Sample preparation is more extensive and includes sample drying, which can result in loss of volatile metabolites. Subsequent two-stage chemical derivatisation is used to induce volatility and thermal stability50 and can be used for several different classes of metabolites (for example OH, NH and SH functional groups present in carboxylic and amino acids, alcohols, amines, amides, thiols, sulfo-acids). Derivatisation generally involves oxime formation (with O-alkylhydroxylamines) followed by trimethylsilylation (N-acetyl-N-(trimethylsilyl)-trifluoroacetamide (MSTFA)) to replace active hydrogens on polar functional groups with less polar trimethylsilyl (TMS) groups and therefore increase volatility by reduction of dipole–dipole interactions. Other more specific derivatisations are available for targeted analysis.103 Derivatisation can produce a number of derivatised products for each metabolite which contain more than one active proton. Derivatisation is efficient, quantitative and reproducible. It should also be rapid and employ low temperatures so as not to be biased against non-derivatised metabolites that are not stable at higher temperatures. Oxime/silylation based derivatisations are time consuming (1–3 h) and the stability of derivatised samples is an issue. Silylation is a reversible reaction especially in the presence of water. All samples should be fully dry, excess silylating reagent can be added to react with any water present and samples should be analysed soon after derivatisation. Ideally an automated derivatisation procedure (in-vial derivatisation)104 could be performed so that there is minimal time between sample derivatisation and analysis to ensure no or minimal sample degradation. With this fact and the presence of blank related peaks90 the number of metabolites detected can be over estimated. A typical total ion current chromatogram of human serum is shown in Fig. 2(b). Closed-loop, multi-objective optimisation of GC-MS instrumentation for metabolomic analysis has been undertaken for the multi-parameter optimisation of GC-MS instruments.105 More than 900 peaks were detected after optimisation.
Samples are analysed with small liquid sample injection volumes (approximately 1 µl) on high resolution capillary columns (30 or 60 m columns with 5–50% phenyl stationary phases are generally used) and allow sensitive analyses (µM–nM limits of detection). Splitless injections (all of volatilised sample introduced on to column) are experimentally preferential for trace analysis to provide the highest sensitivity,106 though the wide range of metabolite concentrations and molecular weight discrimination in splitless injections means that split injections (fraction of volatilised sample introduced on to column) are also used.13 Electron-impact mass spectrometers are almost exclusively used as these provide molecular ion fragmentation to produce a mass spectrum indicative of the metabolite's structure. For this reason tandem mass spectrometry is rarely employed, though some applications are observed when chemical ionisation is used, which produces minimal ion fragmentation.107 The mass spectrometer employed can influence the sensitivity of detection. Quadrupole instruments can be used in single ion monitoring (SIM) modes to enhance their sensitivity (and generally have wider dynamic ranges) than TOF or ion trap instruments.108 However, SIM applications require previous information on the metabolites present in samples analysed and so introduces bias in metabolic profiling applications. TOF instruments provide full mass scan abilities and a complete mass spectrum per data point collected. As all metabolites are detected, good sensitivity without the need of de novo knowledge of metabolites present can be attained. Unique or unusual metabolites can therefore be identified post-analysis. In metabolic profiling methods, the sample analyses are normally performed followed by preparation of data processing methods using samples from typical sample classes to ensure all metabolites are reported.
The application of retention indices aids correct metabolite identification by alignment of chromatograms. Sample analyses over weeks and months will provide variations in retention times through instrumental parameter variations. Raw data can be processed in different ways. Deconvolution software (freely available AMDIS software package (http://chemdata.nist.gov/mass-spc/amdis/) or software provided with commercial instrumentation (for example, LECO ChromaTof™)) now enables shorter analysis times (reduced from over 60 min50 to less than 15 min13) and metabolites that are not fully chromatographically resolved to be determined.109,110 Deconvolution methodology uses the hypothesis that the mass spectrum of a pure component remains consistent across a chromatographic peak and uses single ion chromatograms as models for the peak shapes of components (which also allows ions unique to each co-eluting derivatised metabolite to be used for quantification). Therefore co-eluting peaks with different mass spectra, apexes separated by less than one second and trace components present in large excess of other components can be detected.13 Care must be taken with metabolites of similar mass spectra (structural isomers) to ensure chromatographic separation as mass spectra will be similar and deconvolution processes can be ineffective. Other software packages are available for deconvolution of GC-MS and LC-MS chromatograms including Waters MarkerLynx™, Spectralworks AnalyzerPro™ and metAlign. Alternatively analysis of raw data without deconvolution can be performed. The MSFACTS (Metabolomics Spectral Formatting, Alignment and Conversion Tools) software has been designed to provide alignment of chromatographic datasets and extract information from raw chromatographic ASCII data files without deconvolution procedures.111 Other methodologies not requiring deconvolution processes have also been described.112,113
The commercial availability (or more commonly non-availability) of metabolites greatly influences the efficiency of both metabolite identification and full quantification. Of approximately 600 metabolites thought to act in biological pathways in S. cerevisiae approximately 200 can be commercially purchased. The number of metabolites present in plant and human metabolomes are greater and lower percentages of commercially available metabolites are likely. Therefore full quantification is not possible for all metabolites of interest in a metabolic profiling application and normally full quantification is only undertaken in more targeted approaches of a few metabolites.114 More commonly semi-quantification is performed with the use of internal standard(s), including isotopic analogues, to provide response ratios of metabolites (peak area metabolite/peak area internal standard).35 The second issue is the availability of pure mass spectra to experimentally construct mass spectral libraries. Commercially available libraries (for example, NIST/EPA/NIH library), although extensive, do not contain a large number of metabolites perceived possible from studying metabolic pathway networks. Metabolite specific libraries are required and are being produced within the community but are limited to the metabolites commercially available or those that can be identified from mass spectral interpretation.
In the pharmaceutical field GC-MS was employed before the wide ranging applications of HPLC-MS for targeted drug and metabolite profiling.115 More recently applications include profiling of organic acids in urine and blood to ascertain inborn errors of metabolism, organic acidurias (of which there are 50 known disorders that can be identified), fatty acid oxidation and neurometabolic disorders have been reported.116,117 In the last 5 years metabolomics has grown extensively in the application of GC-MS through proof of concept papers35,50,118 and there are an expanding number of applications of which only a range are discussed here. GC-MS based metabolic profiling has compared four Arabidopsis genotypes and showed each genotype exhibited a different metabolite profile35 and hexose phosphorylation has been shown to diminish for transgenic tomato plants over expressing hexokinase.119 Finally, silent phenotypes of potatoes have been distinguished from their parental background by employing metabolic profiling.13 The same approach has recently been employed in microbial metabolomics to study the effect of different growth conditions on Corynebacterium glutamicum.120
High performance liquid chromatography-mass spectrometry (HPLC-MS)
Technological limitations of HPLC-MS hindered its initial development and wide spread applications. These limitations included the issues of introducing liquids at high pressure into low pressure vacuum regions and the analysis of thermally labile analytes at high temperatures. Although the use of other ionisation techniques such as thermospray were available,121 it was not until the late 1980's that these issues were addressed with the commercial advent of atmospheric pressure ion sources (electrospray and APCI). These ion sources perform ionisation at atmospheric pressure with subsequent transfer of ions from atmospheric to vacuum pressure, while avoiding overloading of the vacuum system. Also low operating temperatures enable the analysis of thermo-labile compounds.Today, HPLC-MS is the standard analytical tool in pharmaceutical qualitative and quantitative analysis of potential pharmaceuticals and their related metabolites for studies from initial discovery to large scale production. A large mass of developed and validated methods are available.122,123 However, metabolomics (and specifically metabonomics) is defined as the study of endogenous metabolites, not drug-related metabolites. Non-targeted analyses of endogeneous metabolites in clinical and pharmaceutical environments aim to detect, in a non-biased manner and after minimal sample preparation, large numbers of metabolites in a high-throughput approach (10–30 min analysis times). Objectives include the identification of biomarkers (medical diagnosis) related to healthy vs. diseased states (or less commonly biomarkers indicative of toxicity or different stages of a disease or indicative of successful drug intervention). The reader is reverted to a recent review.124 Strain, diurnal and gender differences and their influence on the metabolome of mouse urine has been assessed, with identification of interesting metabolites by exact mass measurements on TOF instrumentation.125 Toxicity and metabolism of candidate pharmaceuticals has been studied in an early proof of concept application126 and also in the study of cyclosporine A-induced changes in the metabolome of rat urine.127 Here both HPLC-TOF/MS and high field 1H NMR spectroscopy were used and HPLC-MS provided complementary information to that of the NMR spectroscopy, showing that different analytical platforms and their combined use in studies is advantageous. Metabolites highlighted as being specific to toxicity or metabolism differences can be identified by accurate mass measurements (as shown by the study time course of onset of nephrotoxicity127) or by tandem mass spectrometry. Currently there are a few limited mass spectral libraries available128 though none of the size of GC-MS libraries. The production of metabolite specific mass spectral libraries is being undertaken and research has shown that mass spectra collected on different instruments and instrument types are similar.129
Issues with HPLC-MS, especially for metabolic profiling rather than targeted analysis, include the chromatographic resolution, effect of matrix effects (ionisation suppression) on co-eluting metabolites and influence of column chemistries employed.
High resolution separations of complex metabolomic samples will provide more descriptive data and can be performed with capillary columns, where in theory, as the column internal diameter and packing particle size decrease the chromatographic resolution will increase. Due to a reduction in band broadening, there will also be a greater S/N ratio, and thus an increase in sensitivity. Increased chromatographic resolution has been used in plant metabolomics in the study of the model species Arabidiposis130 and include metabolite identification with Q-TOF instrumentation.131 Improved chromatographic resolution provides reduced co-elution of metabolites and probable reduction of ionisation suppression. As an alternative ultrahigh pressure liquid chromatography (UPLC™) systems which employ ultrahigh pressure LC systems with small diameter particle (<2 µm) columns132 can be used. A typical negative ion base peak ion (BPI) chromatogram of human urine analysed on a Waters UPLC™ system is shown in Fig. 2(c). Over 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 peaks are typically detected for human urine and serum samples, though not all will be metabolites.
000 peaks are typically detected for human urine and serum samples, though not all will be metabolites.
Column chemistry is also important as most published applications use C18 reversed phase assays with solvents that are compatible with electrospray ionisation instrumentation. However, polar metabolites can be eluted in the void volume without chromatographic retention and therefore separation is not achieved and ionisation suppression can be problematic. Different column chemistries are being assessed including hydrophilic interaction liquid chromatography (HILIC) in the study of Cucuribita maxima leaves, where oligosaccharides, glycosides, amino sugars, amino acids and sugar nucleotides were all detected.133 Other weak ion-exchange chemistries columns are expected to become commercially available (including the newly available Waters Atlantis™ metabonomics column). Derivatisation of metabolites to provide improved chromatographic resolution or enhanced detection sensitivity have been observed. For example, derivatisation of low MW amines and carboxylic acids using quaternary nitrogen compounds has been studied.134
Applications of HPLC-MS in plant and microbial metabolomics are small in number. Many examples can be found of targeted analyses and the objective of this review is not to discuss all of these applications. One example from the plant arena is the determination of apple polyphenols and glucosides, including the determination of the number of hydroxyl and sugar groups and the substitution pattern of molecules with a photodiode array detector and mass spectrometer in series.135 This application also highlights that other detectors are used, including UV and photodiode array detection for the quantification of isoprenoids in transgenic tomatoes and Arabidopsis thaliana.136 Also combinations of UV, mass spectrometry and NMR spectroscopic detection have been employed for on-line structural investigations of plant metabolites.137 In microbial metabolomics HPLC-tandem mass spectrometry has been used for the rapid profiling (<4 min per sample) of amino acids during Moniliella pollinis fermentations.138 In metabolic profiling strategies, applications are much less developed, mainly due to the problems of chromatographic resolution, column chemistries and possible ionisation suppression effects discussed earlier. Two strategies are available; the rapid analysis using reversed phase gradient elution with minimal chromatographic resolution of these complex samples and PCA based data analysis, as observed for metabonomic studies though not yet reported in the literature for other applications. Alternatively it may be possible to employ the use of capillary columns or UPLC systems discussed above.
Capillary electrophoresis-mass spectrometry (CE-MS)
CE-MS operates in a similar manner to HPLC-MS with chromatographic separation of ionic species in the liquid phase by application of high voltages, followed by detection employing electrospray mass spectrometry.139 The technique has the potential to be applied widely in the field of metabolomics due to its high chromatographic resolution, sensitivity and fast separation of charged metabolites. However, the technique is not, as of yet, well developed in metabolomics though issues including the use of non-volatile ionics as the mobile phase have been overcome with the introduction of coaxial liquid sheaths.140 The number of instruments in analytical laboratories is also lower when compared to GC-MS and HPLC-MS. The majority of applications are again targeted analyses, for example, the analysis of amino acids in human urine.141 For the less biased metabolic profiling applications few studies have been published. Quantitative metabolome analysis has been performed on Bacillus subtilis extracts with detection of 1692 metabolites of which approximately 150 were identified.142 The method provided evidence of changes in metabolites during sporulation. Urinary profiling to discriminate between acute and chronic cadmium toxicity in rats, with multivariate analysis used to discriminate between the classes has been described.143 Finally hyphenation of capillary LC and capillary electrophoresis for Bacillus subtilis metabolomics has also been studied.144Emerging mass spectrometry technologies
Applications of soft ionisation techniques involving lasers to provide sample volatilisation and ionisation are currently being studied and developed. Here samples mixed in a matrix and spotted onto a target plate (matrix assisted laser desorption/ionisation, MALDI), or alternatively spotted directly onto a plate without being diluted with a matrix (desorption/ionisation on silicon, DIOS), are vapourised and ionised by the application of laser energy to the matrix or sample plate.145 Matrices, normally used for protein analysis, are chromophoric and so absorb the laser energy and volatilise and ionise the sample.61 However, the matrices are normally low MW chromophores that will interfere in the analysis of low MW metabolites. The application of matrixless analyses is preferable and DIOS146,147 is the method of choice. Silicon absorbs UV radiation efficiently, acts in a similar manner to the matrix in MALDI, and therefore allows matrixless analyses on plates that are re-usable after cleaning. Although in early stages of development, the technique has the potential to provide rapid, high-throughput global analyses.Finally, the application of orthogonal multi-dimensional separations will have a large impact on the sensitivity and number of metabolites detected in the future, as 2D-gels has for proteomics, through improved chromatographic resolution and increased S/N ratios.148 GC×GC-TOF-MS is commercially available, though as of yet, not largely employed in metabolomics. TOF instruments are normally employed because of fast acquisition rates, though quadrupole mass spectrometers are also used.149,150 The application of HPLC-HPLC-MS and HPLC-CE-MS151 is in practice, though limited to the pharmaceutical industry.152 All these are employed to improve chromatographic resolution of highly complex biological samples though with the need for complex informatics technologies.
Nuclear magnetic resonance spectroscopy
Nuclear magnetic resonance (NMR) spectroscopy is one of the most widely used structural analysis techniques available in the analytical chemist's toolbox. Many good textbooks are available for general information, theory, application and typical chemical shift values.153 NMR spectroscopy is a technique that has been heavily utilised for metabolomics research in many areas for over 20 years34,154,155 and benefits from the fact that it is a specific and yet non-selective technique. This means that each separate resonance observed in an NMR spectrum is specific to a particular compound, and provides a wealth of structural information regarding the components of a sample. A combination of chemical shift (indicates the nature of the chemical environment in which a particular nucleus is located), spin–spin coupling (indicates the number and nature of nearby nuclei, and thus connectivity information), and relaxation or diffusion (gives an indication of the size of a molecule and also the large-scale environment in which a molecule is located) all allow rapid identification of any components regarded as interesting in the analysis. At the same time, the analysis is non-destructive, and does not require pre-selection of the analysis conditions, such as ion source conditions for mass spectrometry or chromatographic operating conditions (stationary phase, mobile phase, temperature).Sample preparation
NMR-based metabolomic experiments typically operate using solution state samples such as biofluids or solvent extracts following sample preparation or sample extraction. In general, preparing samples for NMR analysis is a straightforward procedure. It may be necessary to buffer the pH of the sample,156 and add a small amount of D2O, or other deuterated solvent (chloroform, acetonitrile or methanol), to allow for deuterium locking—a procedure whereby the effect of magnetic field drift is controlled—but otherwise many samples may be used ‘as is’. 3–5 mm diameter NMR tubes containing a few hundred microlitres of solvent are used. Within this basic process however, there are large differences in the actual protocol followed, depending on the nature of a particular study. Derivatisation of analytes is not necessary and using the technique of magic angle spinning (MAS), even intact tissues can be analysed.Experimental aspects of NMR data acquisition
By employing high-throughput NMR techniques, upward of a few hundred samples may be analysed in a day. This approach uses flow-injection NMR (FI-NMR) to sequentially load each sample into the NMR spectrometer, analyse the sample, and either dispose of or recover the sample, with rinsing of the probe prior to analysis of the next sample. When analysing samples, it is necessary to apply some kind of solvent suppression method in order to reduce the residual protonated solvent resonance normally present in great excess compared to the analytes. This results in some spectral loss, at the frequency and the immediate surrounding area of the water resonance, in turn resulting in loss of spectral information. In addition to reducing the solvent signal's intensity, however, there can be additional attenuation of peaks elsewhere in the spectrum. Whilst various methods for performing this suppression exist, the effects are generally less than the effect caused by a toxicological insult.157 It was shown however, that it is possible to obtain data clustering from rat urine based solely on the method of solvent suppression used.157 Several published studies also report on the effect of inter-spectrometer variation.157–159 In cases where spectrometer variation does have a larger influence than a more subtle toxicological effect, data processing procedures may be applied to reduce this influence.159Alternative NMR experiments are available to customise data acquisition depending on the nature of the samples. Plasma samples contain a combination of both macromolecular (result in a broad envelope of resonances over several ppm in spectral width) and low molecular weight species (produce discrete, well resolved and reasonably sharp resonances) and therefore signals from macromolecules typically obscure resonances in the spectrum from small molecules. Whilst both large molecules (for example lipoproteins) and smaller metabolites (amino acids, carbohydrates) are of interest in terms of metabolomics, it is useful to be able to eliminate the broader signals in order to observe the resonances otherwise obscured. By using a Carr–Purcell–Meiboom–Gill (CPMG) pulse train, the different relaxation properties of small versus large molecules are utilised to effectively remove resonances from macromolecules whilst retaining those from smaller molecules. Optimisation experiments performed using the CPMG sequence when applied to a typical metabonomic analysis have been undertaken and concluded that whilst in general the CPMG experiment can be varied to produce the optimum NMR spectrum with respect to low molecular weight analytes, care should be taken due to variability in protein levels in samples, which may impact on quantitation.160 Different relaxation-edited experiments have been applied to improve the detection of low molecular weight species in blood plasma161 through the use of relaxation-edited 1- and 2-D NMR spectra to remove broad resonances in biological samples and also allow higher receiver gains to be employed. This has the added advantage of greater sensitivity and thus detection and identification of low concentration and low molecular weight species.
In addition to relaxation-edited experiments, several other 1- and 2-D NMR experiments have been reported in relation to metabolomic analyses, principally for the resolution of closely resonating signals.37,162 One approach for reducing congestion and thus extracting more information is J-resolved spectroscopy which separates the chemical shift information and spin–spin couplings onto separate axes, resulting in a 2 dimensional plot. Reconstitution of the 2-D plot to form a 1-D ‘skyline plot’ results in a spectrum that effectively is proton-decoupled. Applying this approach to metabolomic data, results in tighter clustering of sample classes than may be achieved using a conventional 1-D spectrum.37 In addition to reducing congestion in the spectrum, J-resolved spectra also benefit from the exclusion of broad resonances from macromolecules, in a similar way to the CPMG approach. One disadvantage however, is the increased acquisition time (typically 20 min sample−1). An extended policy employed by the COMET project, a collaboration involving Imperial College (London, UK) and six global pharmaceutical companies, is to acquire four separate experiments for plasma or serum samples.162 The suite of experiments comprised a water suppressed spectrum showing the entire collection of both large and small molecular species, a CPMG experiment to reduce the macromolecular component, a J-resolved experiment as discussed above and a diffusion ordered spectroscopy (DOSY) experiment where the low molecular weight compounds are removed, leaving the macromolecules for analysis.
Whilst the 1H nucleus is the most commonly used for metabolomic studies involving NMR spectroscopy, the 13C nucleus has also been employed for such analyses. In 13C NMR spectroscopy, the chemical shift dispersion is twenty times greater, and spin–spin interactions are removed by decoupling. These properties offer the potential to greatly simplify the spectrum acquired in terms of resonance overlap. In addition, for aqueous samples, there is no requirement for solvent suppression, which as discussed above, can result in the loss of some spectral information in 1H NMR studies. Despite these advantages, the low sensitivity of 13C NMR (due to its lower natural abundance and gyromagnetic ratio, 13C is 3 orders of magnitude less sensitive than 1H) prevents its routine use with complex extracts. One approach to get around this limitation is to use 13C-enriched samples, and to follow metabolic flux as the 13C, for example from glucose, is chemically transferred to other endogenous metabolites.163,164 Recently, developments in the area of NMR spectrometer hardware have improved sensitivity dramatically, allowing the potential of 13C NMR spectroscopy to be used for metabolomic studies. One of the main sources of noise in NMR spectroscopic measurement is the electronics used for the detection of the NMR signal. By reducing the temperature of the electronics by bathing them in a cryogen such as liquid helium, it is possible to achieve up to a 16-fold gain in the signal to noise ratio per scan.165 It has been demonstrated that 13C NMR spectra acquired using a Cryoprobe™ (Bruker Biospin, Germany) had approximately twice the signal to noise of a conventional probe with a much longer acquisition time (17 h versus 30 min).165 The 13C NMR spectra obtained from rat urine after dosing with hydrazine are shown in Fig. 3. It can be seen that the spectra are markedly different, with several new resonances, in addition to resonances representing for example, citrate and taurine, varying in intensity between groups. 1H–13C correlation spectra (heteronuclear single quantum correlation, HSQC) were acquired to aid signal assignment, with good quality spectra acquired in ca. 4.5 h.
 | ||
| Fig. 3 Typical 500 MHz cryogenic probe 13C NMR spectra of rat urine samples taken at 48 h post dosing with hydrazine. (A) Control, (B) low dose, 30 mg kg−1, (C) high dose, 90 mg kg−1. Total acquisition time 30 min sample−1. Reproduced with permission from ref. 166. | ||
Sensitivity issues, especially relating to low volumes of available sample have also improved by using NMR probes with reduced detection volumes. Such probes allow the 1H NMR spectroscopic analysis of a few microlitres of sample, and so are particularly useful in experiments that are sample limited, such as rodent cerebrospinal fluid (CSF) studies. A 1 mm microlitre probe, for example, allowed the analysis of just 2 µl of CSF (diluted to a total volume of 5 µl).166 Similarly, a nanoprobe (Varian, CA, USA) was used to study 20 µl of CSF obtained by in vivo microdialysis to study brain neurochemistry.167
In addition to the solution state approaches discussed, NMR spectroscopy also has the advantage of being able to analyse intact tissues through a technique called magic angle spinning (MAS). In conventional NMR, the analysis of a solid or semi-solid sample, for example a section of brain or kidney tissue, would result in very broad lines and loss of spectral information due to the inhomogeneity of the sample. In the technique of MAS-NMR however, the sample is spun very fast (typically a few kHz) at an angle of 54.7° (the so called ‘magic angle’) to the direction of the magnetic field. This has the effect of eliminating the broad lines caused by such inhomogeneity, thus restoring the information content of the spectrum. MAS is an extremely powerful tool for metabolomics analysis. Where mass spectrometry techniques require destructive metabolite extraction and non-destructive FT-IR analysis is less sensitive, this approach means that intact tissues may be studied non-destructively and without the need for tissue extracts. Not only does this result in a more complete picture in terms of metabolite content being observed, but it is also possible to start observing compartmentation of metabolites.168 It can be seen in Fig. 4, for example, that the MAS NMR spectra of rat cardiac tissue and mitochondria (Fig. 4a and 4c) tend to be dominated by lipids (signals around 0.89 and 1.29 ppm), which are not observed in the spectra from tissue extracts (Fig. 4b and 4d). In addition, low molecular weight metabolites are readily observed in the extracts, but not in the intact tissues. This suggests that many of the metabolites are in highly restricted environments, such as the viscous conditions inside intact mitochondria, and also possibly as a result of enzyme complexation. This approach to metabonomics has so far been limited to mammalian applications, such as studying the effects of toxic insult on renal or hepatic tissues in the rat,169,170 or for comparative biochemical studies of small mammals.171 Non-mammalian applications of MAS-NMR spectroscopy do exist,172 and so it seems likely that MAS-NMR based metabolomic analyses applied to, for example plant samples, will be reported in the future.
 | ||
| Fig. 4 600 MHz 1H NMR solvent suppressed spectra. (a) MAS-NMR spectrum of intact rat cardiac tissue. (b) NMR spectrum of an extract of rat cardiac tissue. (c) MAS 1H NMR spectrum of rat heart mitochondria. (d) NMR spectrum of an extract of heart mitochondria. Reproduced with permission from ref. 168. | ||
Finally, it is worth noting that NMR spectroscopy is also a very powerful tool for structural elucidation in addition to the profiling roles outlined above. This means that following metabolomic analysis, it is possible to determine the identity of the resonances of interest. Although not as large as the efforts in genomics and proteomics to produce universally accessible databases of protein sequences, several tables of metabolite assignments found in the analysis of biological fluids have been published.173–176 In addition, 1- and 2-D NMR experiments offer the potential to identify previously unknown metabolites. A combination of previously reported assignments and newly reported identities was used in the assignment of spectra for the profiling of genetically modified tomato extracts.177
Data analysis
NMR-based metabolomic datasets are very large, in terms of the datapoints per sample (typically 32k or 64k), and also the number of samples and resulting spectra (from dozens to thousands, often including replicates) acquired in order to obtain meaningful results. Strategies must therefore be developed for the analysis and interpretation of such data once it has been acquired using the methods outlined above. The area of data analysis is a large one and will only briefly be discussed here. Interested readers are directed to the review by Lindon et al. for further coverage of some of the issues155 or to reviews highlighted in the Introduction.Data manipulation typically starts with some form of ‘bucketing’ whereby the spectrum is split into discrete regions (typically between 0.02 and 0.04 ppm in width), which are then integrated to return a list of integral values for each spectrum, essentially reducing the spectrum to a bar chart. Whilst this reduces the resolution of the data, it has the advantage of reducing the effects of pH variation between samples. Increasingly, work is being carried out to use all datapoints in the spectrum, but to use some kind of algorithm to align the peaks present and thus eliminate unwanted variation.178,179 It is also necessary to apply some kind of scaling to the data in order to ensure that the relative importance of each variable within the dataset is suitable. In NMR spectroscopic data, although the integral values across a spectrum are proportional to concentration and the number of resonances present, the largest resonances would, without scaling, have the largest effect in further methods of data analysis and variable stability scaling (VAST) offered advantages over previously employed scaling methods in terms of the resulting multivariate modelling and calibration achieved.180 The paper also compares VAST with orthogonal signal correction (OSC). OSC is a technique that has been successfully employed to filter confounding data such as physiological or experimental variation out of datasets to enable more meaningful models to be built.181
NMR applications
The previous sections demonstrate the potential that NMR spectroscopy has in the field of metabolomics. As far as NMR-based metabonomics is concerned, mammalian toxicology has been widely studied.9,154 A typical example is the extensive study of the toxicity of the model hepatotoxin, hydrazine, in the rat by analysis of urine and blood pre-dose to 7 days post-dose along with conventional clinical chemistry and liver histopathology.16 Dose dependent trajectory plots showed increasing metabolic insult and recovery with Krebs cycle intermediates being shown to be decreased in dosed animals. In addition, it was shown that there is a dose dependent increase in levels of 2-aminoadipic acid, a molecule previously shown to be a neurotoxin.A limited number of metabonomic studies have been applied to human metabolism,49 including the biochemical impact of dietary isoflavones.22 This study looked at the effects of soy isoflavones consumption on the blood plasma profiles of five healthy pre-menopausal women. The results indicated that even where human studies include a tightly controlled diet regime, inter-individual variation is still a large confounding factor (which has also been reported elsewhere49,181). This confounding variation was removed using OSC and resulted in clear differentiation based on dietary status due to increases in 3-hydroxybutyrate, N-acetyl glycoproteins and a consistent effect on the lipoprotein profile, in particular, a decrease in choline and an increase in glycerol and CH3 lipoprotein groups. Carbohydrate levels were also affected by the soy diet.
Environmental applications are also now starting to report the use of metabonomics, particularly for the monitoring of environmental toxins. In addition to monitoring ground water and soil samples for industrial pollutants and toxins, it is also possible to monitor indigenous species for signs of toxic insult. Earthworms for example, due to their consumption of organic matter, are widely used as ecotoxicological test organisms. Whilst gross toxic effects can be monitored through observation of earthworm mortality, low levels of toxins will affect the biochemical profile of earthworms, without affecting the overall earthworm population. The metabonomic effects of a series of fluoro-anilines (4-fluoroaniline, 4FA, 3,5-difluoroaniline, 3,5DFA and 2-fluoro-4-methylalanine, 2F4MA) on earthworms (Eisenia veneta) has been performed and PCA analyses indicated two modes of action one for 4FA and one for 2F4MA and 3,5DFA.182 A combination of 1H and 13C 1D and 2D NMR experiments, along with FTICR mass spectrometry allowed the identification of novel biomarkers for these toxins. This demonstrated the power of metabonomics to non-selectively determine the presence of biomarkers for a particular toxin, and secondly, the potential for the combination of NMR and MS together to solve complicated structural problems. The impact of environmental stressors have also been studied in the aquatic environment. The effect of the poorly understood fatal disease withering syndrome on the biochemical profiles of Abalone (Haliotis rufescens) has been studied for healthy, stunted and diseased organisms by analysis of different tissue extracts (muscle, digestive gland, hemolymph).17 The three classes (healthy, stunted and diseased) were separated by PCA analysis and analysis of the loadings indicated an increase in homarine (N-methylpicolinic acid) in diseased muscle and a decrease in adenylates and aromatic amino acids.
In phytochemical applications, work has been reported in two main areas; metabolism, toxicology and mode of action research and quality control and profiling type applications. The determination of mode of action of xenobiotics on corn (Zea mays) has been reported.183 Mode of action is of importance as it provides information regarding the metabolic pathway a compound is affecting, determines whether an analogue is acting in the same way as a parent, and allows the classification of novel lead compounds. The authors applied neural network analysis to interpret the data and when classifying mode of action, this approach correctly classified 64% of the spectra, returning an unknown class 30% of the time, and providing a wrong answer just 6% of the time. A subgroup of samples was consistently either classified as unknown, or classified wrongly as a result of only subtle differences in the NMR spectra. This was overcome with a second model and resulted in a 20% increase in recognition of the mode of action.
NMR spectroscopic analysis is gaining a lot of popularity in the area of quality control. Rather than pinpointing a particular biomarker, this approach uses the entire spectrum as a fingerprint for comparison between samples of different origin. The rapid analysis time and wide coverage of endogenous metabolites means it is ideal for discriminating variation, geographic variation, variation between manufacturers, genetic modifications and so on. The quality of different green teas (191 teas from 6 different countries though mainly China) have been assessed using this approach.184 Variation in chemical shifts caused by caffeine resonances required spectra alignment before data analysis and shows the advantage of alignment on the quality of data and further data analysis. Geographical location classification was generally unsuccessful though attempts to classify the Chinese teas according to the quality of the tea were successful, separating high quality tea (Longjing, 38 different teas) from all the other samples (77 teas). Analysis of the loadings indicated that the higher quality teas had higher levels of theanine, theogallin, epicatechin gallate, gallic acid, caffeine and theobromine. These teas had lower levels of fatty acids, quinic acid, sucrose and epigallocatechin when compared to the lower quality teas.
Finally, limited work has been used to study metabolic footprints in microbial systems. Functional ANalysis by Co-responses in Yeast (FANCY) has been performed to study silent phenotypes in yeast Saccharomyces cerevisiae where a single gene knockout has no effect on growth rate but does affect the metabolome and hence allow clustering of strains of similar gene function.12 Although an initial enzymatic analysis indicated differences in intracellular concentrations of glycolytic intermedicates, NMR-based metabolomics allowed the more complete coverage of the metabolic profile that allows the examination of silent phenotypes.
Vibrational spectroscopy
Techniques such as infrared (IR) and Raman spectroscopy are valuable analytical tools that are applied in a wealth of applications spanning a broad range of biological disciplines. A majority of publications are observed in the areas of microbial, plant and medical research, and in light of this literature the importance of IR and Raman spectroscopy as metabolic fingerprinting tools is now being realised. Although these vibrational spectroscopies are not as sensitive as mass spectrometric methods and do not enable the compilation of a list of metabolite identities in complex samples, they do provide a relevant metabolomics tool. Currently, Fourier transform-infrared (FT-IR) is the more widely used technique to obtain spectral fingerprints of biological samples representing a snap-shot of the biochemistry at a given time. Both techniques have the benefit of enabling the rapid (particularly with respect to FT-IR), reagentless, non-destructive analysis of complex biological samples, so facilitating high-throughput screening and providing unbiased measurements of the whole system.30 Analyses are also comparatively inexpensive. The term high-throughput is often misused with respect to analytical techniques. However, FT-IR is truly high-throughput in terms of both sample preparation and analysis time (less than 1 min sample−1).Optical spectroscopy predominantly measures the vibrations and rotations of molecular functional groups that result from the energy exchange when radiation interacts with a sample. This interaction results in an increase of molecular energy which can produce three different transitions: electronic excitation, vibrational change and rotational change. The type of event depends on the wavelength of the incident radiation. IR spectroscopy as its name suggests utilises the IR region of the spectrum (12,000 cm−1 to 10 cm−1 wavenumbers), whereas Raman spectroscopy utilises a monochromatic beam usually having a wavelength within the visible or UV regions of the spectrum (ranging from 1 µm to 10 nm). The IR region is divided into three sub-regions, near-IR (NIR), mid-IR (MIR) and far-IR. The boundaries between these are not clearly defined but MIR is generally considered to range from 4,000 to 400 cm−1, with NIR being at wavenumbers above 4,000 cm−1 and far-IR at wavenumbers below 400 cm−1 and into the microwave region. The two methods are complimentary, measuring different types of transitions resulting due to either changes in molecular configuration or changes of electron distribution within the molecule. There is a wealth of literature describing in great detail IR spectroscopy, Raman spectroscopy and the Fourier transform. Key publications detailing biological applications of IR spectroscopy185–188 and Raman spectroscopy189–192 are available.
Raman spectroscopy
Raman spectroscopy operates by the detection of scattered energy after the passage of monochromatic radiation through a transparent substance. A majority of the scattered energy has the same frequency as the incident radiation and is termed Rayleigh scattering, but a small proportion consists of discrete frequencies above and below that of the incident radiation and this is termed Raman scattering, which results from an inelastic collision in which there is the exchange of energy between the photon and the molecule.193 If the molecule gains energy the scattered photons emitted are at a lower frequency than the incident radiation and this is termed Stokes scattering, which is typically more intense. However, if the molecule loses energy then a higher frequency radiation is emitted, termed anti-Stokes scattering. Fig. 5 shows a Stokes Raman spectrum, wavelength shift against intensity, collected from a bacterial suspension of Escherichia coli. | ||
| Fig. 5 The Raman spectrum of a clinical isolate of the UTI pathogen Escherichia coli, collected using a 785 nm system (courtesy of R. Jarvis, University of Manchester, UK). | ||
In order for a molecule to be Raman active, such that it is susceptible to Raman scattering, there must be a change in the molecular polarizability caused by internal vibration. An incident electric field induces an electric dipole moment which is a separation of charge within the molecule and under these conditions the molecule is said to be polarised. Electrons within the molecule are more easily displaced along a specific axis, producing a polarizability ellipsoid. Raman scattering is a measure of the changes in the magnitude or direction of this ellipsoid. For IR the molecular vibration must produce a change in the electric dipole of the molecule.189
The incident beam of radiation used for Raman spectroscopy is typically within the visible or UV region of the electromagnetic spectrum, commonly using a He–Ne laser at 632.8 nm or an argon laser at 514.5 or 488.0 nm. It is the development of a range of inexpensive lasers that has increased the breadth of Raman spectroscopy. These enable a range of wavelengths to be utilised, so fluorescence of many samples can be avoided, whilst providing a pure monochromatic beam which, due to its narrow diameter, can be focused onto a small sample whilst still delivering sufficient power. Classical Raman spectroscopy of biological samples often results in a weak spectral intensity with a high degree of interference due to sample fluorescence at wavelengths within the visible region, and hence requires long collection times to try and maximise the signal.195 The development of enhancement techniques such as surface enhanced Raman scattering (SERS),196,197 and the use of lasers emitting radiation in the NIR region with 830 nm198,199 or 1064 nm excitation200 or in the UV region with 244 nm excitation194 aid in minimising fluorescence and enhancing signal intensity.
Until the 1980's Raman spectroscopy was overlooked in the field of biological sciences. However, over the past decade there have been an increasing number of publications demonstrating the potential of Raman spectroscopy for the identification and characterisation of microorganisms. Work by a number of authors applied a range of vibrational spectroscopies, including Raman, for the classification of a number of clinical isolates including Enterococcus, Staphylococcus and Escherichia.195,199,201 Resonance Raman and SERS have both been applied for the discrimination of urinary tract infection (UTI) bacteria194,196,202 and more unusually FT-Raman has been used for the analysis of cell walls in fungi.203 The application of vibrational spectroscopies for the identification of microorganisms of medical relevance have been reviewed195 and it was proposed that it was the advances in IR spectroscopy with the introduction of the interferometer and Fourier transform that rekindled interest in Raman spectroscopy.
The use of Raman spectroscopy for the study of complex biological systems outside the area of microbiology is still in its infancy but the potential of using 1064 nm excitation has been demonstrated in studies of the biochemical analysis of honey204 and for the analysis of plant pigments and essential oils.200 In contrast, IR spectroscopy has been applied for biomedical diagnostics, characterisation of both microorganisms and plants, adulteration and quality assurance, biomarker discovery and biochemical responses.
Infrared spectroscopy
In the 1950s IR spectroscopy was applied within the field of microbiology principally for the classification of microorganisms as demonstrated in a publication by Goulden et al. where IR absorption spectroscopy was used to classify 76 lactobacilli strains into different sub-types.205 In these studies, each wavelength within the IR region in question was measured sequentially by use of a conventional prism and grating monochromators. IR technology at this time had several drawbacks particularly with respect to analysis time, reproducibility and sensitivity, resulting in loss of interest in IR for biological and medical applications.186 By the 1980's interest was regained due to the development of the interferometer and the mathematical Fourier transform. These processes enable the simultaneous and almost instantaneous recording of the whole spectrum from the IR and into the microwave regions.189IR spectra are typically shown as absorbance plotted against wavenumber (Fig. 6). The use of absorbance is favoured over transmittance as the absorbance is proportional to concentration at a given wavelength (Beer's Law). An IR spectrum consists of many bands originating from the vibrational motion within the molecule due to the absorption of incident radiation. Bands due to rotational motion are absent from the spectra of biological samples as the samples tend to be in the condensed form, as solids, liquids or solutions, so only vibrational motion is observed. The features of the spectra, the number of bands, frequency, intensity and half-widths are characteristic so giving a fingerprint unique for the sample.186 Five major regions have been highlighted within the 4000 to 600 cm−1 MIR range (Fig. 6),206 these being broadly termed as the fatty acid region (3100 to 2800 cm−1), the amide region (1700 to 1500 cm−1) which can be divided into amide I and amide II bands, a region from 1200 to 1250 cm−1 exhibiting mixed vibrations from carboxylic groups of proteins and PO2− of phosphodiesters, the polysaccharide region (1200 to 900 cm−1) and a mixed region consisting of a variety of weak features. This later region (labelled E in Fig. 6) is termed the (bacterial) fingerprint region as although difficult to interpret it is often characteristic for bacterial classification.206
 | ||
| Fig. 6 An example FT-IR absorbance spectrum showing the major regions of interest and the types of molecular vibrations observed, where A is the fatty acid region, B is the amide region, C is a mixed region, D is the carbohydrate region and E is the bacterial fingerprint region. | ||
Typically, spectral fingerprints are collected spanning either this MIR region or the NIR region (wavenumbers above 4000 cm−1). Despite the close proximity of these two regions different attributes are measured. MIR measures fundamental molecular vibrations providing data containing chemical and structural information about the sample, which is amenable to direct interpretation.185 In contrast, NIR measures overtones and combination-band absorption characteristics of CH, NH and OH groups, producing spectra containing broad overlapping features which are not interpretable at a chemical level.207 For this reason NIR has been widely used to generate calibration models for the quantification or prediction of a single or series of attributes within the sample, for example in the study of forage quality and composition of animal feeds,208,209 for the quantification of fat content in chicken,210 for the non-destructive prediction of sugar content in fruit,211,212 non-invasive analysis of blood metabolites e.g. glucose213 and for detection of metabolites in faeces.214 Metabolomic applications of IR spectroscopy currently favour the use of the MIR region as this provides greater chemical and structural information about the sample.188,215,216 One disadvantage is that water is strongly absorbed within the MIR region producing broad bands in the spectra which mask the characteristic biochemical fingerprint. Several strategies can be employed to overcome this problem, the simplest and probably most commonly employed is drying the sample prior to analysis as described in many publications using aluminium plates216–221 or ZnSe (zinc selenide) plates.222 Another alternative is to use an optics accessory such as an attenuated total reflectance (ATR) cell,223 which enables the direct, non-destructive analysis of aqueous samples such as for the study of spoilage organisms on meat224 and for the classification of basil chemotypes.225
Many applications of FT-IR use it as a metabolic fingerprinting method, one of a number of strategies for metabolomics (Table 1). The aim of metabolic fingerprinting is to enable the high-throughput screening of crude metabolite mixtures and to use these biochemical fingerprints, coupled with multivariate mathematical modelling,215,226 to model relationships between samples and to classify according to their origin or biological provenance,7 or in response to a stimuli.216
The primary applications of FT-IR spectroscopy to study complex biological systems are in the field of microbiology, typically with respect to biomedical and industrial applications.186 The high-throughput reagentless nature of FT-IR makes it ideal for the rapid identification of clinically significant bacterial isolates for example, Eubacterium associated with oral infections,227 the differentiation between Candida isolates,218 the characterisation of Streptococcus and Enterococcus species219 and UTI isolates.228 In addition to focusing on bacterial classification, research has also used FT-IR to monitor bioprocesses (industrial fermentations) for the quantification of metabolite production.221,229
The potential of FT-IR as a medical diagnostics tool has been demonstrated by the application of FT-IR microspectroscopy to study cell proliferation in colonic biopsies230,231 and for the identification of possible biomarkers for malignancy.232,233 Disease diagnosis by FT-IR is sometimes referred to as ‘infrared pathology’234 and includes detection of arthritic disorders,235 diabetes236 and scrapie.237 FT-IR spectroscopy has also been used as a diagnostic tool for quality assurance within the food industry, applied to meat,224,238 jam,239 soft drinks,240 beer222 and cocoa.241 The use of this technique as a rapid fingerprinting method has now been adopted in the plant sciences. Applications range from the discrimination and classification of plants, including the study of Arabidopsis cell wall mutants,242,243 to studies of plant responses to abiotic stresses244 where chemical changes in leaves of ice plants and Arabidopsis in response to salt stress were studied. In later work FT-IR was used for the identification of potential salt-stress biomarkers in tomato.216 In an ecological study modelling of the biochemical changes occurring at the plant–plant interface was undertaken220 and modelling of the plant–microbe interface and the biochemical changes occurring during silage fermentations has also been demonstrated.215 Finally the technique has been employed in combination with DIMS in the metabolic footprinting of mutants of Escherichia coli, to study the discrimination of those with altered tryptophan metabolism.40
Although diverse in subject, the same basic objective applies in all these studies, to rapidly obtain a global biochemical fingerprint of the sample at a given time, which is characteristic and hence descriptive for that sample. Due to the complexity of these spectral fingerprints, a wide range of multivariate data analysis methods are often applied for the derivation of models and the identification of discriminatory features within the data.227,229,230,235 Spectroscopic metabolic fingerprinting coupled with multivariate data analysis methods, provides an inductive experimental approach in which hypotheses are derived as the output rather than constituting the input, so providing direction for future research.245 There is currently no single analytical technology which meets all the criteria required to answer the questions in metabolomics. Vibrational spectroscopy, with current focus particularly on FT-IR, provides a starting point in the hierarchy of analytical approaches required to unravel the complexities of the metabolome.
Combined applications and other technologies
Whilst as outlined above, several analytical technologies are being used to study metabolomics problems, it seems likely that ultimately it will be some combination of these that will prove to be most useful in the exploration of the metabolome. Noteborn et al. for example, employed off-line HPLC followed by NMR analysis to look at differences in transgenic tomatoes.45 Transgenic potatoes have also been studied recently employing 1H NMR spectroscopic and HPLC-UV data for a series of potatoes with genetic modifications affecting different metabolic pathways.246 In addition to mass spectrometric detection of liquid chromatography separations, electrochemical (EC) detection also has potential in the metabolomic analysis of samples. EC detectors analyse only redox active molecules within a sample. Whilst this initially appears to be restrictive, it does mean that those compounds that are directly relevant to biochemical transformations within the body as a result of drug metabolism or toxic insult are monitored, with less meaningful molecules essentially filtered out.247 One major advantage of EC detection is that of sensitivity. Limits of detection in the picogram to femtogram range are readily achievable, leading to a sensitive and targeted technique for the redox compounds present in biological samples.248 Whilst high throughput is also touted as an advantage of EC detection, with single sample analyses possible in around 5 min, it is found in practice that more complicated gradient methods and exotic mobile phases are implemented. This results in better chromatographic resolution, and enhanced data information, but with run times of up to perhaps 100 min.248 Whilst reported applications are currently limited, those that are published demonstrate the technique's ability to detect approximately 600 redox metabolites in complex matrices such as mitochondrial extracts. During the next decade other technologies will emerge to be employed to study the metabolome.Acknowledgements
WD and HJ wish to thank the BBSRC for financial support. WD wishes to thank Douglas Kell for in-depth discussions and also Hilary Major at Waters MS Technologies Centre and Nick Bukowski at Anatune for analyses and helpful discussions.References
- J. O. Castrillo and S. G. Oliver, J. Biochem. Mol. Biol., 2004, 37, 93–106 Search PubMed.
- G. Colebatch, B. Trevaskis and M. Udvardi, New Phytol., 2002, 153, 27–36 Search PubMed.
- S. G. Oliver, M. K. Winson, D. B. Kell and F. Baganz, Trends Biotechnol., 1998, 16, 373–378 CrossRef CAS.
- G. G. H. a. R. Goodacre, Metabolic Profiling: Its Role in Biomarker Discovery and Gene Function Analysis, Kluwer Academic Publishers, London, 2003 Search PubMed.
- R. Goodacre, S. Vaidyanathan, W. B. Dunn, G. G. Harrigan and D. B. Kell, Trends Biotechnol., 2004, 22, 245–252 CrossRef CAS.
- J. Forster, I. Famili, P. Fu, B. O. Palsson and J. Nielsen, Genome Res., 2003, 13, 244–253 CrossRef CAS.
- O. Fiehn, Comp. Funct. Genomics, 2001, 2, 155–168 Search PubMed.
- B. Lahner, J. M. Gong, M. Mahmoudian, E. L. Smith, K. B. Abid, E. E. Rogers, M. L. Guerinot, J. F. Harper, J. M. Ward, L. McIntyre, J. I. Schroeder and D. E. Salt, Nat. Biotechnol., 2003, 21, 1215–1221 CrossRef CAS.
- J. K. Nicholson, J. C. Lindon and E. Holmes, Xenobiotica, 1999, 29, 1181–1189 CrossRef CAS.
- J. K. Nicholson, J. A. Timbrell and P. J. Sadler, Mol. Pharmacol., 1985, 27, 644–651 CAS.
- S. I. Goodman, Am. J. Hum. Genet., 1980, 32, 781–792 CAS.
- L. M. Raamsdonk, B. Teusink, D. Broadhurst, N. S. Zhang, A. Hayes, M. C. Walsh, J. A. Berden, K. M. Brindle, D. B. Kell, J. J. Rowland, H. V. Westerhoff, K. van Dam and S. G. Oliver, Nat. Biotechnol., 2001, 19, 45–50 CrossRef CAS.
- W. Weckwerth, M. E. Loureiro, K. Wenzel and O. Fiehn, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 7809–7814 CrossRef CAS.
- J. Allen, H. M. Davey, D. Broadhurst, J. K. Heald, J. J. Rowland, S. G. Oliver and D. B. Kell, Nat. Biotechnol., 2003, 21, 692–696 CrossRef CAS.
- J. Taylor, R. D. King, T. Altmann and O. Fiehn, Bioinformatics, 2002, 18, S241–S248 Search PubMed.
- A. W. Nicholls, E. Holmes, J. C. Lindon, J. P. Shockcor, R. D. Farrant, J. N. Haselden, S. J. P. Damment, C. J. Waterfield and J. K. Nicholson, Chem. Res. Toxicol., 2001, 14, 975–987 CrossRef CAS.
- M. R. Viant, E. S. Rosenblum and R. S. Tjeerdema, Environ. Sci. Technol., 2003, 37, 4982–4989 CrossRef CAS.
- N. J. C. Bailey, M. Oven, E. Holmes, J. K. Nicholson and M. H. Zenk, Phytochemistry, 2003, 62, 851–858 CrossRef CAS.
- R. S. Plumb, C. L. Stumpf, J. H. Granger, J. Castro-Perez, J. N. Haselden and G. J. Dear, Rapid Commun. Mass Spectrom., 2003, 17, 2632–2638 CrossRef CAS.
- J. T. Brindle, H. Antti, E. Holmes, G. Tranter, J. K. Nicholson, H. W. L. Bethell, S. Clarke, P. M. Schofield, E. McKilligin, D. E. Mosedale and D. J. Grainger, Nat. Med., 2002, 8, 1439–1444 CrossRef CAS.
- J. B. German, M. A. Roberts and S. M. Watkins, J. Nutr., 2003, 133, 4260–4266 CAS.
- K. S. Solanky, N. J. C. Bailey, B. M. Beckwith-Hall, A. Davis, S. Bingham, E. Holmes, J. K. Nicholson and A. Cassidy, Anal. Biochem., 2003, 323, 197–204 CrossRef CAS.
- M. S. Rashed, J. Chromatogr. B, 2001, 758, 27–48 CrossRef CAS.
- I. Matsumoto and T. Kuhara, Mass Spectrom. Rev., 1996, 15, 43–57 CAS.
- N. Aranibar, B. K. Singh, G. W. Stockton and K. H. Ott, Biochem. Biophys. Res. Commun., 2001, 286, 150–155 CrossRef CAS.
- J. Allen, H. M. Davey, D. Broadhurst, J. J. Rowland, S. G. Oliver and D. B. Kell, Appl. Environ. Microbiol., 2004, 70, 6157–6165 CrossRef CAS.
- C. Bro and J. Nielsen, Metab. Eng., 2004, 6, 204–211 CrossRef CAS.
- W. Weckwerth, Annu. Rev. Plant Biol., 2003, 54, 669–689 Search PubMed.
- M. Brown, W. B. Dunn, D. I. Ellis, R. Goodacre, J. Handl, J. D. Knowles, S. O'Hagan, I. Spasic and D. B. Kell, Metabolomics, 2005, 1, 39–51 Search PubMed.
- D. B. Kell, Curr. Opin. Microbiol., 2004, 7, 296–307 CrossRef CAS.
- J. K. Nicholson and I. D. Wilson, Nat. Rev. Drug Discov., 2003, 2, 668–676 CrossRef CAS.
- O. Fiehn, Plant Mol. Biol., 2002, 48, 155–171 CrossRef CAS.
- L. W. Sumner, P. Mendes and R. A. Dixon, Phytochemistry, 2003, 62, 817–836 CrossRef CAS.
- J. C. Lindon, E. Holmes, M. E. Bollard, E. G. Stanley and J. K. Nicholson, Biomarkers, 2004, 9, 1–31 CrossRef CAS.
- O. Fiehn, J. Kopka, P. Dormann, T. Altmann, R. N. Trethewey and L. Willmitzer, Nat. Biotechnol., 2000, 18, 1157–1161 CrossRef CAS.
- O. Fiehn and W. Weckwerth, Eur. J. Biochem., 2003, 270, 579–588 CrossRef CAS.
- M. R. Viant, Biochem. Biophys. Res. Commun., 2003, 310, 943–948 CrossRef CAS.
- L. Eriksson, E. Johansson, N. Kettaneh-Wold and S. Wold, Multi- and megavariate data analysis: principles and applications, Umetrics AB, Umea, Sweden, 2001 Search PubMed.
- N. J. C. Bailey, J. Sampson, P. J. Hylands, J. K. Nicholson and E. Holmes, Planta Med., 2002, 68, 734–738 CrossRef CAS.
- N. N. Kaderbhai, D. I. Broadhurst, D. I. Ellis, R. Goodacre and D. B. Kell, Comp. Funct. Genomics, 2003, 4, 376–391 Search PubMed.
- E. Holmes, J. K. Nicholson and G. Tranter, Chem. Res. Toxicol., 2001, 14, 182–191 CrossRef CAS.
- F. W. Ma and L. L. Cheng, Plant Sci., 2003, 165, 819–827 CrossRef CAS.
- L. L. Cheng and F. W. Ma, J. Am. Soc. Hortic. Sci., 2004, 129, 313–320 Search PubMed.
- D. C. Boyes, A. M. Zayed, R. Ascenzi, A. J. McCaskill, N. E. Hoffman, K. R. Davis and J. Gorlach, Plant Cell, 2001, 13, 1499–1510 CrossRef CAS.
- H. Noteborn, A. Lommen, R. C. van der Jagt and J. M. Weseman, J. Biotechnol., 2000, 77, 103–114 CrossRef CAS.
- M. E. Bollard, E. Holmes, J. C. Lindon, S. C. Mitchell, D. Branstetter, W. Zhang and J. K. Nicholson, Anal. Biochem., 2001, 295, 194–202 CrossRef CAS.
- H. Jernstrom, T. L. Klug, D. W. Sepkovic, H. L. Bradlow and S. A. Narod, Carcinogenesis, 2003, 24, 991–1005 CrossRef CAS.
- B. M. Beckwith-Hall, E. Holmes, J. C. Lindon, J. Gounarides, A. Vickers, M. Shapiro and J. K. Nicholson, Chem. Res. Toxicol., 2002, 15, 1136–1141 CrossRef CAS.
- E. M. Lenz, J. Bright, I. D. Wilson, S. R. Morgan and A. F. P. Nash, J. Pharm. Biomed. Anal., 2003, 33, 1103–1115 CrossRef CAS.
- U. Roessner, C. Wagner, J. Kopka, R. N. Trethewey and L. Willmitzer, Plant J., 2000, 23, 131–142 CrossRef CAS.
- E. Grouzmann, C. Cavadas, D. Grand, M. Moratel, J. F. Aubert, H. R. Brunner and L. Mazzolai, Pfluegers Arch., 2003, 447, 254–258 CrossRef CAS.
- J. I. Castrillo, A. Hayes, S. Mohammed, S. J. Gaskell and S. G. Oliver, Phytochemistry, 2003, 62, 929–937 CrossRef CAS.
- J. L. Ward, C. Harris, J. Lewis and M. H. Beale, Phytochemistry, 2003, 62, 949–957 CrossRef CAS.
- J. R. Evans and T. C. Vogelmann, Plant Cell Environ., 2003, 26, 547–560 CrossRef CAS.
- R. E. Hausler, K. L. Fischer and U. I. Flugge, Anal. Biochem., 2000, 281, 1–8 CrossRef.
- R. P. Maharjan and T. Ferenci, Anal. Biochem., 2003, 313, 145–154 CrossRef.
- J. Gullberg, P. Jonsson, A. Nordstrom, M. Sjostrom and T. Moritz, Anal. Biochem., 2004, 331, 283–295 CrossRef CAS.
- E. M. Farre, A. Tiessen, U. Roessner, P. Geigenberger, R. N. Trethewey and L. Willmitzer, Plant Physiol., 2001, 127, 685–700 CrossRef CAS.
- C. Wittmann, J. O. Kromer, P. Kiefer, T. Binz and E. Heinzle, Anal. Biochem., 2004, 327, 135–139 CrossRef CAS.
- C. A. Daykin, P. J. D. Foxall, S. C. Connor, J. C. Lindon and J. K. Nicholson, Anal. Biochem., 2002, 304, 220–230 CrossRef CAS.
- J. H. Gross, Mass Spectrometry: A Textbook, Springer, Berlin, 2004 Search PubMed.
- J. C. Lindon, J. K. Nicholson and I. D. Wilson, J. Chromatogr. B, 2000, 748, 233–258 CrossRef CAS.
- M. S. Rashed, M. P. Bucknall, D. Little, A. Awad, M. Jacob, M. Alamoudi, M. Alwattar and P. T. Ozand, Clin. Chem., 1997, 43, 1129–1141 CAS.
- R. Goodacre, S. Vaidyanathan, G. Bianchi and D. B. Kell, Analyst, 2002, 127, 1457–1462 RSC.
- P. Frycak, R. Huskova, T. Adam and K. Lemr, J. Mass Spectrom., 2002, 37, 1242–1248 CrossRef CAS.
- O. V. Konstantinova, A. P. Antonchick, N. J. Oldham, V. N. Zhabinskii, V. A. Khripach and B. Schneider, Collect. Czech. Chem. Commun., 2001, 66, 1729–1734 CrossRef CAS.
- R. King, R. Bonfiglio, C. Fernandez-Metzler, C. Miller-Stein and T. Olah, J. Am. Soc. Mass Spectrom., 2000, 11, 942–950 CrossRef CAS.
- P. Mauri and P. Pietta, J. Pharm. Biomed. Anal., 2000, 23, 61–68 CrossRef CAS.
- D. B. Kell, Curr. Opin. Microbiol., 2004, 7, 296–307 CrossRef CAS.
- H. F. Schroder, Trends Anal. Chem., 1996, 15, 349–362 CAS.
- R. Goodacre, E. V. York, J. K. Heald and I. M. Scott, Phytochemistry, 2003, 62, 859–863 CrossRef CAS.
- J. Smedsgaard and J. C. Frisvad, J. Microbiol. Methods, 1996, 25, 5–17 CrossRef CAS.
- J. A. Zahn, R. E. Higgs and M. D. Hilton, Appl. Environ. Microbiol., 2001, 67, 377–386 CrossRef CAS.
- S. Vaidyanathan, D. B. Kell and R. Goodacre, J. Am. Soc. Mass Spectrom., 2002, 13, 118–128 CrossRef CAS.
- R. Goodacre, J. K. Heald and D. B. Kell, FEMS Microbiol. Lett., 1999, 176, 17–24 CrossRef CAS.
- A. Sawaya, D. M. Tomazela, I. B. S. Cunha, V. S. Bankova, M. C. Marcucci, A. R. Custodio and M. N. Eberlin, Analyst, 2004, 129, 739–744 RSC.
- S. C. Brown, G. Kruppa and J.-L. Dasseux, Mass Spectrom. Rev., 2005, 24, 223–231 CrossRef.
- A. Aharoni, C. H. Ric De Vos, H. A. Verhoeven, C. A. Maliepaard, G. Kruppa, R. Bino and D. B. Goodenowe, OMICS, 2002, 6, 217–234 Search PubMed.
- M. E. Hansen and J. Smedsgaard, J. Am. Soc. Mass Spectrom., 2004, 15, 1173–1180 CrossRef CAS.
- J. J. Pitt, M. Eggington and S. G. Kahler, Clin. Chem., 2002, 48, 1970–1980 CAS.
- E. W. Naylor and D. H. Chace, J. Child Neurol., 1999, 14, S4–S8 Search PubMed.
- V. Wiley, K. Carpenter and B. Wilcken, Acta Paediatr., 1999, 88, 48–51 CrossRef CAS.
- Y. Shigematsu, I. Hata, Y. Kikawa, M. Mayumi, Y. Tanaka, M. Sudo and N. Kado, J. Chromatogr. B, 1999, 731, 97–103 CrossRef CAS.
- D. H. Chace, Chem. Rev., 2001, 101, 445–477 CrossRef CAS.
- K. Gempel, M. Kottlors, M. Jaksch, K. D. Gerbitz and M. F. Bauer, J. Inherited Metab. Dis., 1999, 22, 941–942 Search PubMed.
- K. H. Carpenter and B. Wilcken, J. Inherited Metab. Dis., 1999, 22, 840–841 Search PubMed.
- M. S. Rashed, P. T. Ozand, M. J. Bennett, J. J. Barnard, D. R. Govindaraju and P. Rinaldo, Clin. Chem., 1995, 41, 1109–1114 CAS.
- K. Tanaka, A. Westdull, D. G. Hine, T. B. Lynn and T. Lowe, Clin. Chem., 1980, 26, 1847–1853 CAS.
- H. J. Leis, G. Fauler, G. N. Rechberger and W. Windischhofer, Curr. Med. Chem., 2004, 11, 1585–1594 CAS.
- C. Wagner, M. Sefkow and J. Kopka, Phytochemistry, 2003, 62, 887–900 CrossRef CAS.
- A. Vikram, B. Prithiviraj, H. Hamzehzarghani and A. Kushalappa, J. Sci. Food Agric., 2004, 84, 1333–1340 CrossRef CAS.
- C. H. Deng, X. M. Zhang and N. Li, J. Chromatogr. B, 2004, 808, 269–277 CrossRef CAS.
- P. Cap, J. Chladek, F. Pehal, M. Maly, V. Petru, P. J. Barnes and P. Montuschi, Thorax, 2004, 59, 465–470 CrossRef CAS.
- N. Yassaa, E. Brancaleoni, M. Frattoni and P. Ciccioli, J. Chromatogr. A, 2001, 915, 185–197 CrossRef CAS.
- A. Mallouchos, M. Komaitis, A. Koutinas and M. Kanellaki, J. Agric. Food Chem., 2002, 50, 3840–3848 CrossRef CAS.
- S. Patel and T. Shibamoto, J. Agric. Food Chem., 2002, 50, 5649–5653 CrossRef CAS.
- S. Ghittori, A. Alessio, S. Negri, L. Maestri, P. Zadra and M. Imbriani, Ind. Health, 2004, 42, 226–234 CrossRef CAS.
- M. Phillips, R. N. Cataneo, T. Cheema and J. Greenberg, Clin. Chim. Acta, 2004, 344, 189–194 CrossRef CAS.
- D. Y. Graham and P. D. Klein, Gastroenterol. Clin. North Am., 2000, 29, 885–892 Search PubMed.
- B. Christensen, A. K. Gombert and J. Nielsen, Eur. J. Biochem., 2002, 269, 2795–2800 CrossRef CAS.
- M. R. Kant, K. Ament, M. W. Sabelis, M. A. Haring and R. C. Schuurink, Plant Physiol., 2004, 135, 483–495 CrossRef CAS.
- S. Patel and T. Shibamoto, J. Food Compos. Anal., 2003, 16, 469–476 CrossRef CAS.
- J. Drozd, Chemical derivatization in gas chromatography, Elsevier, Amsterdam, 1981 Search PubMed.
- Y. C. Fiamegos, C. G. Nanos, J. Vervoort and C. D. Stalikas, J. Chromatogr. A, 2004, 1041, 11–18 CrossRef CAS.
- S. O'Hagan, W. B. Dunn, M. Brown, J. Knowles and D. B. Kell, Anal. Chem., 2005, 77, 290–303 CrossRef CAS.
- K. Robards, P. R. Haddad and P.E. Jackson, Principles and Practice of Modern Chromatographic Methods, Academic Press, London, 1997 Search PubMed.
- A. Muller, P. Duchting and E. W. Weiler, Planta, 2002, 216, 44–56 CrossRef CAS.
- B. K. Choi, D. M. Hercules, T. L. Zhang and A. I. Gusev, LC-GC North Am., 2001, 19, 514–524 CAS.
- S. E. Stein, J. Am. Soc. Mass Spectrom., 1999, 10, 770–781 CrossRef CAS.
- J. M. Halket, A. Przyborowska, S. E. Stein, W. G. Mallard, S. Down and R. A. Chalmers, Rapid Commun. Mass Spectrom., 1999, 13, 279–284 CrossRef CAS.
- A. L. Duran, J. Yang, L. J. Wang and L. W. Sumner, Bioinformatics, 2003, 19, 2283–2293 Search PubMed.
- P. Jonsson, J. Gullberg, A. Nordstrom, M. Kusano, M. Kowalczyk, M. Sjostrom and T. Moritz, Anal. Chem., 2004, 76, 1738–1745 CrossRef CAS.
- J. E. Katz, D. S. Dumlao, S. Clarke and J. Hau, J. Am. Soc. Mass Spectrom., 2004, 15, 580–584 CrossRef CAS.
- C. Birkemeyer, A. Kolasa and J. Kopka, J. Chromatogr. A, 2003, 993, 89–102 CrossRef CAS.
- R. F. Staack, D. S. Theobald and H. H. Maurer, Ther. Drug Monit., 2004, 26, 441–449 CrossRef CAS.
- T. Kuhara, J. Chromatogr. B, 2002, 781, 497–517 CrossRef CAS.
- M. D. Carlson, Curr. Opin. Neurol., 2004, 17, 133–138 CrossRef.
- U. Roessner, A. Luedemann, D. Brust, O. Fiehn, T. Linke, L. Willmitzer and A. R. Fernie, Plant Cell, 2001, 13, 11–29 CrossRef CAS.
- U. Roessner-Tunali, B. Hegemann, A. Lytovchenko, F. Carrari, C. Bruedigam, D. Granot and A. R. Fernie, Plant Physiol., 2003, 133, 84–99 CrossRef CAS.
- S. Strelkov, M. von Elstermann and D. Schomburg, Biol. Chem., 2004, 385, 853–861 CrossRef CAS.
- W. A. Korfmacher, J. P. Freeman, T. A. Getek, J. Bloom and C. L. Holder, Biomed. Environ. Mass Spectrom., 1990, 19, 191–201 CAS.
- T. V. Olah, D. A. McLoughlin and J. D. Gilbert, Rapid Commun. Mass Spectrom., 1997, 11, 17–23 CrossRef CAS.
- M. Jemal, S. Khan, D. S. Teitz, J. A. McCafferty and D. J. Hawthorne, Anal. Chem., 2001, 73, 5450–5456 CrossRef CAS.
- I. D. Wilson, R. Plumb, J. Granger, H. Major, R. Williams and E. M. Lenz, J. Chromatogr. B, 2005, 817, 67–76 CrossRef CAS.
- R. Plumb, J. Granger, C. Stumpf, I. D. Wilson, J. A. Evans and E. M. Lenz, Analyst, 2003, 128, 819–823 RSC.
- R. S. Plumb, C. L. Stumpf, M. V. Gorenstein, J. M. Castro-Perez, G. J. Dear, M. Anthony, B. C. Sweatman, S. C. Connor and J. N. Haselden, Rapid Commun. Mass Spectrom., 2002, 16, 1991–1996 CrossRef CAS.
- E. M. Lenz, J. Bright, R. Knight, I. D. Wilson and H. Major, J. Pharm. Biomed. Anal., 2004, 35, 599–608 CrossRef CAS.
- J. L. Josephs, Rapid Commun. Mass Spectrom., 1995, 9, 1270–1274 CAS.
- J. L. Josephs and M. Sanders, Rapid Commun. Mass Spectrom., 2004, 18, 743–759 CrossRef CAS.
- V. V. Tolstikov, A. Lommen, K. Nakanishi, N. Tanaka and O. Fiehn, Anal. Chem., 2003, 75, 6737–6740 CrossRef CAS.
- E. von Roepenack-Lahaye, T. Degenkolb, M. Zerjeski, M. Franz, U. Roth, L. Wessjohann, J. Schmidt, D. Scheel and S. Clemens, Plant Physiol., 2004, 134, 548–559 CrossRef CAS.
- R. Plumb, J. Castro-Perez, J. Granger, I. Beattie, K. Joncour and A. Wright, Rapid Commun. Mass Spectrom., 2004, 18, 2331–2337 CrossRef CAS.
- V. V. Tolstikov and O. Fiehn, Anal. Biochem., 2002, 301, 298–307 CrossRef CAS.
- S. J. Barry, R. M. Carr, S. J. Lane, W. J. Leavens, S. Monte and I. Waterhouse, Rapid Commun. Mass Spectrom., 2003, 17, 603–620 CrossRef CAS.
- R. M. Alonso-Salces, K. Ndjoko, E. F. Queiroz, J. R. Ioset, K. Hostettmann, L. A. Berrueta, B. Gallo and F. Vicente, J. Chromatogr. A, 2004, 1046, 89–100 CrossRef CAS.
- P. D. Fraser, M. E. S. Pinto, D. E. Holloway and P. M. Bramley, Plant J., 2000, 24, 551–558 CrossRef CAS.
- J. L. Wolfender, K. Ndjoko and K. Hostettmann, J. Chromatogr. A, 2003, 1000, 437–455 CrossRef CAS.
- J. J. Dalluge, S. Smith, F. Sanchez-Riera, C. McGuire and R. Hobson, J. Chromatogr. A, 2004, 1043, 3–7 CrossRef CAS.
- P. Schmitt-Kopplin and M. Frommberger, Electrophoresis, 2003, 24, 3837–3867 CrossRef CAS.
- R. D. Smith, H. R. Udseth, C. J. Barinaga and C. G. Edmonds, J. Chromatogr., 1991, 559, 197–208 CrossRef CAS.
- T. Soga, Y. Kakazu, M. Robert, M. Tomita and T. Nishioka, Electrophoresis, 2004, 25, 1964–1972 CrossRef CAS.
- T. Soga, Y. Ohashi, Y. Ueno, H. Naraoka, M. Tomita and T. Nishioka, J. Proteome Res., 2003, 2, 488–494 CrossRef CAS.
- S. Zomer, C. Guillo, R. G. Brereton and M. Hanna-Brown, Anal. Bioanal. Chem., 2004, 378, 2008–2020 CrossRef CAS.
- L. Jia, B. F. Liu, S. Terabe and T. Nishioka, Anal. Chem., 2004, 76, 1419–1428 CrossRef CAS.
- C. Wittmann and E. Heinzle, Eur. J. Biochem., 2001, 268, 2441–2455 CrossRef CAS.
- W. G. Lewis, Z. X. Shen, M. G. Finn and G. Siuzdak, Int. J. Mass Spectrom., 2003, 226, 107–116 CrossRef CAS.
- D. B. Wall, J. W. Finch and S. A. Cohen, Rapid Commun. Mass Spectrom., 2004, 18, 1403–1406 CrossRef CAS.
- A. Guttman, M. Varoglu and J. Khandurina, Drug Discovery Today, 2004, 9, 136–144 CrossRef CAS.
- R. A. Shellie and P. J. Marriott, Analyst, 2003, 128, 879–883 RSC.
- L. M. Blumberg, J. Chromatogr. A, 2003, 985, 29–38 CrossRef CAS.
- C. R. Evans and J. W. Jorgenson, Anal. Bioanal. Chem., 2004, 378, 1952–1961 CrossRef CAS.
- C. J. Venkatramani and Y. Zelechonok, Anal. Chem., 2003, 75, 3484–3494 CrossRef CAS.
- H. Gunther, NMR Spectroscopy, John Wiley, London, 1995 Search PubMed.
- J. C. Lindon, E. Holmes and J. K. Nicholson, Prog. Nucl. Magn. Reson. Spectrosc., 2004, 45, 109–143 CrossRef CAS.
- J. C. Lindon, E. Holmes and J. K. Nicholson, Prog. Nucl. Magn. Reson. Spectrosc., 2001, 39, 1–40 CrossRef CAS.
- M. Defernez and I. J. Colquhoun, Phytochemistry, 2003, 62, 1009–1017 CrossRef CAS.
- B. C. M. Potts, A. J. Deese, G. J. Stevens, M. D. Reily, D. G. Robertson and J. Theiss, J. Pharm. Biomed. Anal., 2001, 26, 463–476 CrossRef CAS.
- H. C. Keun, T. M. D. Ebbels, H. Antti, M. E. Bollard, O. Beckonert, G. Schlotterbeck, H. Senn, U. Niederhauser, E. Holmes, J. C. Lindon and J. K. Nicholson, Chem. Res. Toxicol., 2002, 15, 1380–1386 CrossRef CAS.
- N. J. C. Bailey, M. Oven, E. Holmes, M. H. Zenk and J. K. Nicholson, Spectrosc.: Int. J., 2004, 18, 279–287 Search PubMed.
- Q. N. Van, G. N. Chmurny and T. D. Veenstra, Biochem. Biophys. Res. Commun., 2003, 301, 952–959 CrossRef CAS.
- H. R. Tang, Y. L. Wang, J. K. Nicholson and J. C. Lindon, Anal. Biochem., 2004, 325, 260–272 CrossRef CAS.
- J. C. Lindon, J. K. Nicholson, E. Holmes, H. Antti, M. E. Bollard, H. Keun, O. Beckonert, T. M. Ebbels, M. D. Reilly, D. Robertson, G. J. Stevens, P. Luke, A. P. Breau, G. H. Cantor, R. H. Bible, U. Niederhauser, H. Senn, G. Schlotterbeck, U. G. Sidelmann, S. M. Laursen, A. Tymiak, B. D. Car, L. Lehman-McKeeman, J. M. Colet, A. Loukaci and C. Thomas, Toxicol. Appl. Pharmacol., 2003, 187, 137–146 CrossRef CAS.
- J. Kikuchi, K. Shinozaki and T. Hirayama, Plant Cell Physiol., 2004, 45, 1099–1104 CrossRef CAS.
- G. Baverel, A. Conjard, M. F. Chauvin, B. Vercoutere, A. Vittorelli, L. Dubourg, C. Gauthier, C. Michoudet, D. Durozard and G. Martin, Biochimie, 2003, 85, 863–871 CrossRef CAS.
- H. C. Keun, O. Beckonert, J. L. Griffin, C. Richter, D. Moskau, J. C. Lindon and J. K. Nicholson, Anal. Chem., 2002, 74, 4588–4593 CrossRef CAS.
- J. L. Griffin, A. W. Nicholls, H. C. Keun, R. J. Mortishire-Smith, J. K. Nicholson and T. Kuehn, Analyst, 2002, 127, 582–584 RSC.
- P. Khandelwal, C. E. Beyer, Q. Lin, P. McGonigle, L. E. Schechter and A. C. Bach, J. Neurosci. Methods, 2004, 133, 181–189 CrossRef CAS.
- M. E. Bollard, A. J. Murray, K. Clarke, J. K. Nicholson and J. L. Griffin, FEBS Lett., 2003, 553, 73–78 CrossRef CAS.
- S. D. Huhn, C. M. Szabo, J. H. Gass and A. E. Manzi, Anal. Bioanal. Chem., 2004, 378, 1511–1519 CrossRef CAS.
- S. Garrod, E. Humpher, S. C. Connor, J. C. Connelly, M. Spraul, J. K. Nicholson and E. Holmes, Magn. Reson. Med., 2001, 45, 781–790 CrossRef CAS.
- J. L. Griffin, L. A. Walker, S. Garrod, E. Holmes, R. F. Shore and J. K. Nicholson, Comp. Biochem. Physiol., B: Biochem. Mol. Biol., 2000, 127, 357–367 CrossRef CAS.
- H. R. Tang, P. S. Belton, A. Ng and P. Ryden, J. Agric. Food Chem., 1999, 47, 510–517 CrossRef CAS.
- T. W. Fan, Prog. Nucl. Magn. Reson. Spectrosc., 1996, 28, 161–219 CrossRef CAS.
- J. K. Nicholson, P. J. D. Foxall, M. Spraul, R. D. Farrant and J. C. Lindon, Anal. Chem., 1995, 67, 793–811 CrossRef CAS.
- J. C. Lindon, J. K. Nicholson and J. R. Everett, Annu. Rep. NMR Spectrosc., 1999, 38, 1–88 Search PubMed.
- A. P. Sobolev, A. Segre and R. Lamanna, Magn. Reson. Chem., 2003, 41, 237–245 CrossRef CAS.
- G. Le Gall, I. J. Colquhoun, A. L. Davis, G. J. Collins and M. E. Verhoeyen, J. Agric. Food Chem., 2003, 51, 2447–2456 CrossRef CAS.
- J. Forshed, I. Schuppe-Koistinen and S. P. Jacobsson, Anal. Chim. Acta, 2003, 487, 189–199 CrossRef CAS.
- G. C. Lee and D. L. Woodruff, Anal. Chim. Acta, 2004, 513, 413–416 CAS.
- H. C. Keun, T. M. D. Ebbels, H. Antti, M. E. Bollard, O. Beckonert, E. Holmes, J. C. Lindon and J. K. Nicholson, Anal. Chim. Acta, 2003, 490, 265–276 CrossRef CAS.
- B. M. Beckwith-Hall, J. T. Brindle, R. H. Barton, M. Coen, E. Holmes, J. K. Nicholson and H. Antti, Analyst, 2002, 127, 1283–1288 RSC.
- J. G. Bundy, E. M. Lenz, N. J. Bailey, C. L. Gavaghan, C. Svendsen, D. Spurgeon, P. K. Hankard, D. Osborn, J. A. Weeks and S. A. Trauger, Environ. Toxicol. Chem., 2002, 21, 1966–1972 CrossRef CAS.
- K. H. Ott, N. Aranibar, B. J. Singh and G. W. Stockton, Phytochemistry, 2003, 62, 971–985 CrossRef CAS.
- G. Le Gall, I. J. Colquhoun and M. Defernez, J. Agric. Food Chem., 2004, 52, 692–700 CrossRef CAS.
- P. R. Griffiths and J. A. de Haseth, Fourier transform infrared spectroscopy, John Wiley & Sons, New York, 1986 Search PubMed.
- D. Naumann, D. Helm, H. Labischinski and P. Giesbrecht, in Modern techniques for rapid microbiological analysis, ed. W. H. Nelson, VCH publishers, New York, 1991, pp. 43–96 Search PubMed.
- J. K. Gillie, J. Hochlowski and G. A. Arbuckle-Keil, Anal. Chem., 2000, 72, 71R–79R CAS.
- D. I. Ellis, G. G. Harrigan and R. Goodacre, in Metabolic profiling: Its role in biomarker discovery and gene function analysis, ed. G. G. Harrigan and R. Goodacre, Kluwer Academic Publishers, Boston, 2003, pp. 111–124 Search PubMed.
- C. N. Banwell, Fundamentals of Molecular Spectroscopy, McGraw-Hill Book Company, London, 1983 Search PubMed.
- P. R. Carey, Biochemical applications of Raman and resonance Raman spectroscopies, Academic Press, New York, 1982 Search PubMed.
- P. R. Carey, in Modern Physical Methods in Biochemistry, ed. A. Neuberger and L. L. M. Van Deenen, Elsevier Science Publishers, Amsterdam, 1988, pp. 27–64 Search PubMed.
- W. H. Nelson and J. F. Sperry, in Modern techniques for rapid microbiological analysis, ed. W. H. Nelson, VCH publishers, New York, 1991, pp. 97–144 Search PubMed.
- N. Everall, B. King and I. Clegg, Chem. Br., 2000, July, 40–43 Search PubMed.
- R. M. Jarvis and R. Goodacre, FEMS Microbiol. Lett., 2004, 232, 127–132 CrossRef CAS.
- K. Maquelin, C. Kirschner, L. P. Choo-Smith, N. van den Braak, H. P. Endtz, D. Naumann and G. J. Puppels, J. Microbiol. Methods, 2002, 51, 255–271 CrossRef CAS.
- R. M. Jarvis, A. Brooker and R. Goodacre, Anal. Chem., 2004, 76, 5198–5202 CrossRef CAS.
- M. Moskovits, Rev. Mod. Phys., 1985, 57, 783–826 CrossRef CAS.
- K. Maquelin, L. P. Choo-Smith, L. L. M. Van Deenen, H. P. Endtz, B. Smith, R. Bennett, H. A. Bruining and G. J. Puppels, Anal. Chem., 2000, 72, 12–19 CrossRef CAS.
- L. P. Choo-Smith, K. Maquelin, T. van Vreeswijk, H. A. Bruining, G. J. Puppels, N. A. Ngo Thi, C. Kirschner, D. Naumann, D. Ami, A. M. Villa, F. Orsini, S. M. Doglia, H. Lamfarraj, G. D. Sockalingum, M. Manfait, P. Allouch and H. P. Endtz, Appl. Environ. Microbiol., 2001, 67, 1461–1469 CrossRef CAS.
- B. Schrader, H. H. Klump, K. Schenzel and H. Schulz, J. Mol. Struct., 1999, 509, 201–212 CrossRef CAS.
- C. Kirschner, K. Maquelin, P. Pina, N. A. Ngo Thi, L. P. Choo-Smith, G. D. Sockalingum, C. Sandt, D. Ami, F. Orsini, S. M. Doglia, P. Allouch, M. Mainfait, G. J. Puppels and D. Naumann, J. Clin. Microbiol., 2001, 39, 1763–1770 CrossRef CAS.
- R. M. Jarvis and R. Goodacre, Anal. Chem., 2004, 76, 40–47 CrossRef CAS.
- H. G. M. Edwards, N. C. Russell, R. Weinstein and D. D. Wynn-Williams, J. Raman Spectrosc., 1995, 26, 911–916 CAS.
- L. F. C. de Oliveira, R. Colombara and H. G. M. Edwards, Appl. Spectrosc., 2002, 56, 306–311 CrossRef CAS.
- J. D. S. Goulden and M. E. Sharpe, J. Gen. Microbiol., 1958, 19, 76–86 CAS.
- D. Naumann, C. P. Shultz and D. Helm, in Infrared Spectroscopy of Biomolecules, ed. H. H. Mantsch and D. Chapman, Wiley-Liss. Inc., 1996, pp. 279–310 Search PubMed.
- P. S. Belton, A. M. Saffa and R. H. Wilson, Analyst, 1987, 112, 1117–1120 RSC.
- G. Xiccato, A. Trocino, J. L. De Boever, L. Maertens, R. Carabano, J. J. Pascual, J. M. Perez, T. Gidenne and L. Falcao-E-Cunha, Anim. Feed Sci. Technol., 2003, 104, 153–168 CrossRef CAS.
- G. C. Marten, J. S. Shenk and F. E. Barton, Near infrared reflectance spectroscopy (NIRS): Analysis of forage quality, National Technical Information Service, Springfield, Virginia, 1989 Search PubMed.
- F. Abeni and G. Bergoglio, Meat Sci., 2001, 57, 133–137 CrossRef.
- R. W. Cho, M. R. Sohn and Y. K. Kwon, J. Near Infrared Spectrosc., 1998, 6, 75–78 Search PubMed.
- J. Guthrie and K. Walsh, Aust. J. Exp. Agric., 1999, 39, 73–80 Search PubMed.
- H. M. Heise, A. Bittner and R. Marbach, Clin. Chem. Lab. Med., 2000, 38, 137–145 Search PubMed.
- T. Nakamura, T. Takeuchi, A. Terada, Y. Tando and T. Suda, Int. J. Pancreatol., 1998, 23, 137–143 Search PubMed.
- H. E. Johnson, D. Broadhurst, D. B. Kell, M. K. Theodorou, R. J. Merry and G. W. Griffith, Appl. Environ. Microbiol., 2004, 70, 1583–1592 CrossRef CAS.
- H. E. Johnson, D. Broadhurst, R. Goodacre and A. R. Smith, Phytochemistry, 2003, 62, 919–928 CrossRef CAS.
- A. C. McGovern, R. Ernill, B. V. Kara, D. B. Kell and R. Goodacre, J. Biotechnol., 1999, 72, 157–167 CrossRef CAS.
- E. M. Timmins, S. A. Howell, B. K. Alsberg, W. C. Noble and R. Goodacre, J. Clin. Microbiol., 1998, 36, 367–374 CAS.
- R. Goodacre, E. M. Timmins, P. J. Rooney, J. J. Rowland and D. B. Kell, FEMS Microbiol. Lett., 1996, 140, 233–239 CrossRef CAS.
- E. Gidman, R. Goodacre, B. Emmett, A. R. Smith and D. Gwynn-Jones, Phytochemistry, 2003, 63, 705–710 CrossRef CAS.
- K. C. Schuster, R. Goodacre, J. R. Grapes and M. Young, J. Ind. Microbiol. Biotechnol., 2001, 27, 314–321 CrossRef CAS.
- M. C. Curk, F. Peladan and J. C. Hubert, FEMS Microbiol. Lett., 1994, 123, 241–248 CrossRef CAS.
- J. Schmitt and H. Flemming, Int. Biodeterior. Biodegrad., 1998, 41, 1–11 CrossRef CAS.
- D. I. Ellis, D. Broadhurst, D. B. Kell, J. J. Rowland and R. Goodacre, Appl. Environ. Microbiol., 2002, 68, 2822–2828 CrossRef CAS.
- H. Schulz, B. Schrader, R. Quilitzsch, S. Pfeffer and H. Kruger, J. Agric. Food Chem., 2003, 51, 2475–2481 CrossRef CAS.
- A. D. Shaw, M. K. Winson, A. M. Woodward, A. C. McGovern, H. M. Davey, N. Kaderbhai, D. Broadhurst, R. J. Gilbert, J. Taylor, E. M. Timmins, R. Goodacre and D. B. Kell, in Advances in Biochemical Engineering/Biotechnology, ed. T. Scheper, Springer-Verlag, Berlin, Heidelberg, 1999, pp. 84–113 Search PubMed.
- B. K. Alsberg, W. G. Wade and R. Goodacre, Appl. Spectrosc., 1998, 52, 823–832 CrossRef CAS.
- R. Goodacre, E. M. Timmins, R. Burton, N. Kaderbhai, A. M. Woodward, D. B. Kell and P. J. Rooney, Microbiology (UK), 1998, 144, 1157–1170 Search PubMed.
- A. C. McGovern, D. Broadhurst, J. Taylor, N. Kaderbhai, M. K. Winson, D. A. Small, J. J. Rowland, D. B. Kell and R. Goodacre, Biotechnol. Bioeng., 2002, 78, 527–538 CAS.
- R. K. Sahu, S. Argov, E. Bernshtain, A. Salman, S. Walfisch, J. Goldstein and S. Mordechai, Scand. J. Gastroenterol., 2004, 39, 557–566 CrossRef CAS.
- A. Salman, R. K. Sahu, E. Bernshtain, U. Zelig, J. Goldstein, S. Walfisch, S. Argov and S. Mordechai, Vib. Spectrosc., 2004, 34, 301–308 CrossRef CAS.
- M. Huleihel, A. Salman, V. Erukhimovitch, J. Ramesh, Z. Hammody and S. Mordechai, J. Biochem. Biophys. Methods, 2002, 50, 111–121 CrossRef CAS.
- S. Mordechai, R. K. Sahu, Z. Hammody, S. Mark, K. Kantarovich, H. Guterman, A. Podshyvalov, J. Goldstein and S. Argov, J. Microsc., 2004, 215, 86–91 CrossRef CAS.
- M. Diem, S. Boydston-White and L. Chiriboga, Appl. Spectrosc., 1999, 53, 148A–161A CAS.
- H. H. Eysel, M. Jackson, A. Nikulin, R. L. Somorjai, G. T. D. Thomson and H. H. Mantsch, Biospectroscopy, 1997, 3, 161–167 CrossRef CAS.
- W. Petrich, B. Dolenko, J. Fruh, M. Ganz, H. Greger, S. Jacob, F. Keller, A. E. Nikulin, M. Otto, O. Quarder, R. L. Somorjai, A. Staib, G. Warner and H. Wielinger, Appl. Opt., 2000, 39, 3372–3379 Search PubMed.
- J. Schmitt, M. Beekes, A. Brauer, T. Udelhoven, P. Lasch and D. Naumann, Anal. Chem., 2002, 74, 3865–3868 CrossRef CAS.
- J. McElhinney, G. Downey and C. O'Donnell, J. Food Sci., 1999, 64, 587–591 CAS.
- M. Defernez and R. H. Wilson, J. Sci. Food Agric., 1995, 67, 461–467 CAS.
- F. J. Rambla, S. Garrigues, N. Ferrer and M. de la Guardia, Analyst, 1998, 123, 277–281 RSC.
- R. Goodacre and E. Anklam, J. Am. Oil Chem. Soc., 2001, 78, 993–1000 CAS.
- L. M. Chen, N. C. Carpita, W. D. Reiter, R. H. Wilson, C. Jeffries and M. C. McCann, Plant J., 1998, 16, 385–392 CrossRef CAS.
- G. Mouille, S. Robin, M. Lecomte, S. Pagant and H. Hotte, Plant J., 2003, 35, 393–404 CrossRef CAS.
- J. Yang and H. E. Yen, Plant Physiol., 2002, 130, 1032–1042 CrossRef CAS.
- D. B. Kell and S. G. Oliver, BioEssays, 2003, 26, 99–105.
- M. Defernez, Y. M. Gunning, A. J. Parr, L. V. T. Shepherd, H. V. Davies and I. J. IColquhoun, J. Agric. Food Chem., 2004, 52, 6075–6085 CrossRef CAS.
- R. Kaddurah-Daoul, C. Beecher, B. S. Kristal, W. R. Matson and D. J. Bogdanov, Pharmagenomics, 2004, 4 Search PubMed.
- B. S. Kristal, K. E. Vigneau-Callahan and W. R. Matson, Anal. Biochem., 1998, 263, 18–25 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2005 |