
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Consequences of encapsulated vs. covalently attached Pd-NHC amphiphilic polymeric nanocatalysts in bioorthogonal catalysis
Lieke
de Jonge
,
Ibai
Hazenberg
,
Harshit
Singh
 ,
Tessa
Loman
,
A. P.
Prakasham
* and
Anja R. A.
Palmans
*
,
Tessa
Loman
,
A. P.
Prakasham
* and
Anja R. A.
Palmans
*
Department of Chemical Engineering & Chemistry and Institute for Complex Molecular Systems, Eindhoven University of Technology, P.O. Box 513, 5600 MB, Eindhoven, The Netherlands. E-mail: a.p.prakasham@tue.nl; a.palmans@tue.nl
First published on 26th January 2026
Abstract
A comparison between novel Pd-NHC (N-heterocyclic carbene) catalysts covalently and non-covalently attached to polymeric nanoparticles reveals that hydrophobic domains in the vicinity of the Pd catalysts aid catalytic activity, and covalent attachment reduces cytotoxicity, opening new avenues to tune bioorthogonal reactivity of TMC-based nanocarriers.
The field of bioorthogonal chemistry and catalysis has conveniently opened up numerous novel directions in biology, such as protein conjugation, cell engineering, drug delivery, antibody–drug conjugates, and in situ prodrug activation, to name a few.1–3 Typically, these non-natural reactions are performed in living systems without interfering with native biological processes, which uniquely distinguishes them as ‘bioorthogonal’ reactions. Transition metal catalysts (TMCs) hold tremendous potential in this field by the manifold of intriguing organic transformations they catalyse.4,5 However, their potential in complex biological environments has been hampered by toxicity, instability and deactivation, and poor solubility in aqueous media.6 As a result, nanocarriers have been engineered to incorporate TMCs and thereby enhance their stability and improve their biocompatibility in biological media.7,8 Of particular interest are amphiphilic heterograft polymers that collapse in water to form nanometer-sized nanoparticles (NPs) with hydrophobic interiors.9,10 These NPs carry the TMCs within their hydrophobic core, improving water compatibility and limiting exposure of TMCs to complex media.
Recently, several advanced nanocarriers have been developed for the Pd(0/II)-mediated bioorthogonal prodye and prodrug activation in complex environments (Table S12).11–14 We have previously developed amphiphilic heterograft polymers with PPh3 and bipyridine pendants that complex Pd(II) and found that the resulting NPs were capable of activating prodyes and prodrugs in water as well as in complex media.15 However, the polymers with pendant PPh3 were highly sensitive to oxidation to the corresponding oxide, requiring demanding formulation conditions to obtain the Pd(II)-based NPs. Both the nature of the ligand and the polymer's microstructure proved important for the catalyst's activity and stability, but activity was reduced in cell culture growth media. As part of our interest in using polymeric nanoparticles as carriers for bioorthogonal catalysis,16–18 we here focus on NHC ligands for the preparation of Pd(II) catalysts for bond cleavage activation of prodyes. Pd-NHC catalysts are highly effective in various C−X (X = C, N, O, S) bond forming reactions,19 and also function in water.20 Wegner et al. showed that incorporation of a Pd-NHC complex in the membrane of E. coli allowed the bioorthogonal cleavage of propargyl ethers.21 Their simple synthesis, stability, divergent structural motifs and high metal affinity make NHC a ligand of choice in organometallic chemistry. NHC-ligands were recently used by us in combination with Cu(I), showing excellent CuAAC activity in presence of 15 mM glutathione.22 Moreover, Cu(I)-NHC embedded in a polymer showed bioorthogonal activity in cellular media and mouse models.23 Here, we apply NHC ligands for complexing to Pd(II) and evaluate the activity in aqueous and complex media, and compatibility with mammalian cells to assess their suitability for bioorthogonal catalysis. Particularly, we unveil the intriguing differences between physically encapsulated and covalently attached Pd-NHC to polymeric NPs, highlighting the effect of the hydrophobic micro-environment on catalyst activity, toxicity and stability.
We designed our NHC ligand with the following features: (i) presence of a fused bicyclic motif for strong coordination and steric shielding for Pd (ii) azide moiety for covalent (‘click’) attachment to amphiphilic polymer and (iii) –CF3 label for easy NMR characterization within the polymer matrix. The –N3 and –CF3 group appended bicyclic 1,2,4-triazole based NHC ligand precursor (1) was synthesized from L-pyroglutamic acid through a sequence of manipulations (see SI for details, Scheme S1, Fig. S1-S4).24 Pd-NHC complex (2) was synthesized by reacting (1) with PdBr2 in presence of KBr, K2CO3 as base and pyridine as solvent for the reaction (Scheme 1). The pyridine in complex 2 is a labile, throw away ligand, which aids the incoming substrates during the catalysis process.25 The formation of complex (2) was confirmed by the disappearance of the characteristic NC![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) N resonance of the precarbene in the 1H NMR while the 19F{1H} signal was observed at −62.51 ppm for the –C
N resonance of the precarbene in the 1H NMR while the 19F{1H} signal was observed at −62.51 ppm for the –C![[F with combining low line]](https://www.rsc.org/images/entities/i_char_0046_0332.gif) 3 group (Fig. S5–S11).
3 group (Fig. S5–S11).
 | ||
| Scheme 1 Synthesis of Pd(II)-NHC complexes (2), (3) and covalently Pd-NHC functionalized amphiphilic (P2) polymers. | ||
In order to reinforce the Pd center further by the combined effect of both the phosphine and NHC ligands, complex (3) was made by replacing the Py ligand with a PPh3 ligand in complex (2) (Fig. S12–S19). The typical Pd coordinated cis-![[P with combining low line]](https://www.rsc.org/images/entities/i_char_0050_0332.gif) Ph3 signal was observed at 26.20 ppm in the 31P{1H} NMR (Fig. S17, Table S14). The presence of –N3 in Pd complexes (2) and (3) was confirmed using IR spectroscopy where the azido stretch appeared around 2113 cm−1 (Fig. S10 and S18).26
Ph3 signal was observed at 26.20 ppm in the 31P{1H} NMR (Fig. S17, Table S14). The presence of –N3 in Pd complexes (2) and (3) was confirmed using IR spectroscopy where the azido stretch appeared around 2113 cm−1 (Fig. S10 and S18).26
We incorporated the Pd complexes (2) and (3) into the amphiphilic nanoparticles by physical encapsulation as well as attached them to the heterograft polymers via a click reaction. For encapsulation, P1 polymer was used, which contained 20% n-dodecyl chains for hydrophobic collapse and 80% Jeffamine®M1000 for water solubility. For the covalent linking, we prepared polymer P2 with strained alkyne containing dibenzocyclooctyne (DBCO) grafts (12%) and dodecyl (22%) and Jeffamine (66%) as hydrophobic and hydrophilic grafts, respectively. Polymers P1,2 were made by post functionalization modification of the poly(pentafluoro-phenylacrylate) using previously optimized procedures and fully characterized (Fig. S20–S31).15 Using a strain promoted azide–alkyne cycloaddition (SPAAC) the azide functional Pd complexes were readily attached to the DBCO linked polymer backbone. Complete functionalization (12%) of P2 was done with Pd complexes (2) and (3) to obtain polymers P2@12% Pd-Py and P2@12% Pd-PPh3. In order to check the effect of catalyst concentration and polymer microstructure, half of the DBCO groups were functionalized, i.e., 6%, with (3) affording polymer P2@6% Pd-PPh3 (Scheme 1). Here, we also aimed to study the synergistic effect of NHC and PPh3 ligands on Pd, which is expected to enhance stability and activity in catalysis.27–29 After post functionalization, all polymers were purified using dialysis to remove unbound grafts/Pd/free pentafluorophenol and fully characterized (Fig. S32-S40).
The presence of polymer bound Pd was confirmed by NMR where, for instance, the –C3 group signal was observed at −62.48 ppm and the Pd bound Ph3 signal was observed at 26.69 ppm for P2@12% Pd-PPh3 in their respective 19F{1H} and 31P{1H} NMR (Fig. 1A). The Pd concentration in the polymer samples was quantified using inductively coupled plasma optical emission spectroscopy (ICP-OES). The measured Pd concentrations were in good agreement with the calculated Pd concentration. The higher concentration measured for P2@12% Pd-PPh3 suggests that the excess Pd complex used in the click reaction was not completely removed by dialysis (Fig. 1B). After formulating the obtained P1,2-based polymers into NPs in water, dynamic light scattering (DLS) showed hydrodynamic radii (RH) of the NPs around 6.5–11.6 nm, similar to the ones previously reported (Table S1 and Fig. S41).15
 | ||
| Fig. 1 (A) 19F{1H} and 31P{1H} NMR of P2@12% Pd-PPh3; (B) Pd quantification by ICP-OES measured in milliQ water (*) or 2% THF in milliQ water (#); (C) experimental SAXS curves (symbols) and worm-like chain form factor fits (lines) for Pd-based NPs. | ||
More detailed insights into the nature of the NPs were obtained from small angle X-ray scattering (SAXS) analysis in water (SI Section 4.4). The experimental SAXS curves (Fig. 1C) show global shapes indicative of conformationally stretched chains with limited flexibility for all NPs, similar to our previously studied copolymers lacking Pd complexes.30 The experimental data were fit to the previously applied semiflexible worm-like chain model.29 Comparison of the two encapsulated P1 NPs revealed that the form factor fits (details in SI, Table S4) were in good agreement with the experimental data. The Kuhn length lk, i.e. the distance over which the polymer is rigid, was around 9 nm for P1-based NPs containing encapsulated Pd catalysts, similar to the previously studied Jeffamine-based NPs with only hydrophobic grafts.30
The shape of the scattering curve obtained for P2@12% Pd-Py closely resembled that of P1-Pd-Py, indicating that nanoparticle shape and compactness are not significantly affected by physical encapsulation or covalent attachment. P2-based NPs showed some aggregation as evidenced by the upturn at low q values. This effect was more pronounced for the 12% functionalized particles and the PPh3 comprising polymers. To account for the aggregation, we adapted our fitting strategy by combining a power-law model with the flexible cylinder model to extract data from the scattering curves for P2-based polymers (Fig. 1C). This afforded good fits. Notably, the Kuhn lengths lk of P2-based polymers were comparable to the P1-based polymers, albeit that lk was slightly higher (≈16.0 nm) for P2@12% Pd-PPh3, suggesting a lower flexibility (Table S4).
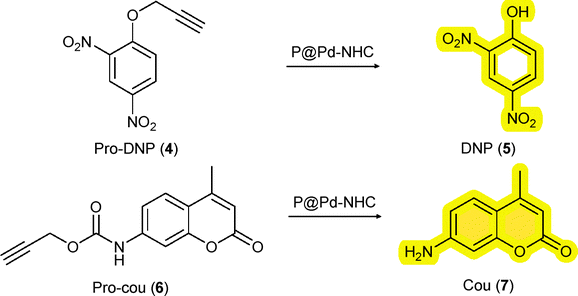
The hydrophobic interior of the NPs not only comprise the hydrophobic catalysts, but also sequester hydrophobic substrates in water.31 We applied two different prodyes, pro-2,4-dinitro phenol (4) and pro-coumarin (6) to test the TMC activation strategy as this allows real time monitoring of the catalytic process by absorption or fluorescence spectroscopy, respectively. To start with, we studied the depropargylation reaction of O-propargylated 2,4-dinitro phenol (4) using the new Pd-NHC NPs (Scheme 2). The reactions were performed in water (37 °C) using a substrate to catalyst S/C ratio of 10/3 (100 µM substrate, 30 µM Pd). The formation of the dye, 2,4-dinitro phenol (5) was monitored using UV-Vis spectroscopy at λ = 400 nm. The encapsulated P1 particles steadily cleaved the O-propargyl group of the pro-DNP in water over 12 h. P1-Pd-Py NPs showed slightly better activity than the P1-Pd-PPh3 NPs (Fig. 2A). Also the P2-based NPs showed good activity in water and both the covalently attached P2@12% Pd-Py and P2@12% Pd-PPh3 NPs showed similar behavior. Interestingly, increasing the polymer concentration while keeping the S/C ratio constant by comparing P2@12% Pd-PPh3 to P2@6% Pd-PPh3 (Fig. 2B) revealed notable differences, with the latter system showing a higher activity. This indicated that the presence of more hydrophobic compartments in P2@6% Pd-PPh3 benefits the rate of the deprotection reaction while keeping the overall Pd concentration constant. This is also obvious from the Pd to polymer (P) ratio, where Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :P is 24:1 for P2@12% Pd-PPh3 while it is 12:1 for P2@6% Pd-PPh3 with an overall [Pd] of 30 µM in both cases (Table S5). The enhanced activity observed for the P2@6% Pd-PPh3 NPs suggests that more hydrophobic domains are more beneficial than a higher local concentration of Pd. Additionally, the higher Kuhn length observed for P2@12% Pd-PPh3 suggested a less dynamic polymer chain adopting more compact conformations. Such rigidity can restrict segmental motion and diffusion pathways, partially shielding catalytic sites and reducing substrate accessibility, eventually leading to diminished catalytic activity. All the experiments were repeated at least four times to ensure reproducibility and consistency of the observed trend.
:P is 24:1 for P2@12% Pd-PPh3 while it is 12:1 for P2@6% Pd-PPh3 with an overall [Pd] of 30 µM in both cases (Table S5). The enhanced activity observed for the P2@6% Pd-PPh3 NPs suggests that more hydrophobic domains are more beneficial than a higher local concentration of Pd. Additionally, the higher Kuhn length observed for P2@12% Pd-PPh3 suggested a less dynamic polymer chain adopting more compact conformations. Such rigidity can restrict segmental motion and diffusion pathways, partially shielding catalytic sites and reducing substrate accessibility, eventually leading to diminished catalytic activity. All the experiments were repeated at least four times to ensure reproducibility and consistency of the observed trend.
 | ||
| Scheme 2 Model prodye activation using Pd nanoparticles. | ||
 | ||
| Fig. 2 (A) Kinetic profiles of encapsulated P1 and covalently attached P2 NPs in a pro-DNP (4) (100 µM) to DNP (5) conversion monitored by UV-Vis over time at λ = 400 nm in water at 37 °C using 30 µM of Pd; (B) T1/2 comparison of the P1 and P2 NPs in the formation of DNP (5); T1/2 (time taken for 50% formation of DNP (5) by UV-Vis, mean of multiple runs); (C) HPLC conversion of DNP (5) after 24 h. | ||
The average catalytic activity of different polymeric NPs was compared against T1/2, the time it takes to reach 50% formation of DNP (5), approximated by an absorbance of 0.3 at 400 nm in the UV-Vis spectrum (see SI section 5.1 for details). Fig. 2B showed that encapsulated P1-Pd-Py is the fastest and covalent P2@6% Pd-PPh3 is faster than P2@12% Pd-PPh3 when keeping the S/C ratio constant. Additionally, the formation of DNP (5) was quantified by HPLC after 24 h (SI section 5.2; Table S7). Analyzing all reactions after 24 h with a S/C ratio of 10/3 showed higher amounts of DNP formed for the covalent systems compared to the encapsulated systems (Fig. 2C). In fact, after 24 h, the best performing covalent system, P2@6% Pd-PPh3 showed full conversion, whereas the best performing encapsulated P1-Pd-Py had a conversion of 87% to DNP. In addition, P1-Pd-PPh3 showed a 58% formation of DNP. The results show that the covalent systems outperformed the encapsulated system. This aligns with our previous observations where a PdPPh3 complex encapsulated into NPs was slower than covalently crosslinked PdPPh3 complexes.15 However, the observed activity of these novel NHC-Pd-based NPs was lower (full conversion after 24 h) than our previous NPs (full conversion after 3.3 h, for same S/C 10/3), confirming the important role of the ligand and polymer microstructure for better catalytic activity.32 The differences between P1 and P2 extend beyond the mode of catalyst attachment and also include variations in Pd:P ratio, polymer graft composition and hydrophobic domain content, all of which may contribute to the observed differences in catalytic activity.
To assess the cytotoxicity of the novel catalytic systems, important for bioorthogonal reactivity, all Pd NPs were incubated with HeLa cells for 24 h. The cell viability was tested using the CCK-8 assay. The results showed that the encapsulated P1 NPs exhibited more pronounced toxicity than the covalently attached P2 NPs. P2@12% Pd-PPh3 NPs was the least toxic after 24 h of incubation (Fig. S61). Even at high Pd concentrations of 100 µM, the cell viability remained high. This finding emphasizes the beneficial effect of having covalently attached Pd catalysts in the polymer chain for better biocompatibility.
Having established that some of the novel catalytic NPs show good compatibility with mammalian cells, we finally tested the reactivity of the new catalysts in complex media. To this end, all reactions were repeated in PBS and the cell culture media, RPMI (preferred growth media for suspension cell lines) and DMEM (preferred for adherent cell lines). Cell culture media are a good predictor for catalyst activity in in vitro conditions.33 The catalytic activity was mostly retained in PBS with Pd-Py containing NPs showing enhanced activity. Unfortunately, activity was mostly lost in RPMI and DMEM (Fig. S47), also for the most active P2@6% Pd-PPh3. Changing the substrate from pro-DNP to pro-cou (6) (SI Section 5.3) containing a carbamate protecting group showed a similar trend in water, whereas in presence of cell culture media did not result in the formation of fluorescent dye (7). The loss of Pd activity in cell culture media prompted the investigation of deactivation pathways. Substrate activation studies in presence of 30 µM of various biomolecules revealed strong inhibition by L-histidine, L-cysteine and glutathione for both P1 and P2 NPs, consistent with Pd catalyst poisoning by amine and sulfur nucleophiles (Fig. S48). This is in line with the results of Wegner et al. where the catalytic reactions worked when performed in buffered conditions with E. coli, but failed for mammalian cells.21 Lastly, the reusability of the Pd NPs was evaluated by re-adding the substrate after the first run (15 h), and the Pd NPs remained active even after 30 h continuously cleaving the substrate in water (Fig. S49).
In summary, we developed novel Pd-NHC based polymeric nanoparticles and investigated their potential for bioorthogonal bond cleavage reactions. Covalently Pd-NHC attached NPs performed better than the encapsulated ones and the P2@6% Pd-PPh3 NPs with more hydrophobic micro-environment performed the best for prodye activation reactions in water. Additionally, the covalently linked P2 NPs were less toxic to HeLa cells than the physically encapsulated P1 NPs. Despite improved aqueous activity and reduced toxicity, Pd nanoparticles remained susceptible to deactivation by biological nucleophiles. Strategies such as the incorporation of chelate stabilizing ligands, polymer compartmentalization and cross-linking to enhance catalyst protection are underway in our laboratory.
Conflicts of interest
There are no conflicts to declare.Data availability
Supplementary information: experimental procedures, characterization and spectroscopic data; 1H, 13C{1H}, 19F{1H}, 31P{1H} NMR, MALDI-ToF-MS spectra of Pd-NHCs, GPC, DLS, ICP-OES, SAXS data of polymers, UV-Vis and fluorescence data, HPLC data of the kinetics runs are available in the electronic supplementary information (SI). See DOI: https://doi.org/10.1039/d5cc06993a. The SAXS data related to this article is available in DOI: https://doi.org/10.15151/ESRF-ES-2010314049.Acknowledgements
The project has received funding from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement no. 899987. We gratefully acknowledge support by The Dutch Research Council (NWO Gravitation; IPM program), the TU/e and Peter Lipman for ICP-OES analysis. We thank the staff of the ESRF and EMBL Grenoble for assistance and support in using beamline BM29 under proposal number MX-2677.References
- F. M. Zielke and F. P. J. T. Rutjes, Top. Curr. Chem., 2023, 381, 35 CrossRef CAS PubMed.
- W. Wang, X. Zhang, R. Huang, C.-M. Hirschbiegel, H. Wang, Y. Ding and V. M. Rotello, Adv. Drug Delivery Rev., 2021, 176, 113893 CrossRef CAS PubMed.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed.
- J. H. Docherty, T. M. Lister, G. Mcarthur, M. T. Findlay, P. Domingo-Legarda, J. Kenyon, S. Choudhary and I. Larrosa, Chem. Rev., 2023, 123, 7692–7760 Search PubMed.
- U. Bin Kim, D. J. Jung, H. J. Jeon, K. Rathwell and S. Lee, Chem. Rev., 2020, 120, 13382–13433 CrossRef PubMed.
- J. Miguel-Ávila, M. Tomás-Gamasa and J. L. Mascareñas, Trends Chem., 2023, 5, 474–485 CrossRef.
- A. A. H. Laporte and J. N. H. Reek, Chem. Rev., 2025, 125, 7223–7274 Search PubMed.
- F. Huang, J. Liu, M. Li and Y. Liu, J. Am. Chem. Soc., 2023, 145, 26983–26992 CrossRef CAS PubMed.
- A.-S. Glaive, C. Le Cœur, J.-M. Guigner, C. Amiel and G. Volet, Langmuir, 2024, 40, 2050–2063 Search PubMed.
- J. Odrobińska, M. Skonieczna and D. Neugebauer, Int. J. Mol. Sci., 2021, 22, 1202 CrossRef PubMed.
- R. Martínez, C. Carrillo-Carrión, P. Destito, A. Alvarez, M. Tomás-Gamasa, B. Pelaz, F. Lopez, J. L. Mascareñas and P. del Pino, Cell Rep. Phys. Sci, 2020, 1, 100076 CrossRef PubMed.
- E. Indrigo, J. Clavadetscher, S. V. Chankeshwara, A. Megia-Fernandez, A. Lilienkampf and M. Bradley, Chem. Commun., 2017, 53, 6712–6715 RSC.
- J. Li, J. Yu, J. Zhao, J. Wang, S. Zheng, S. Lin, L. Chen, M. Yang, S. Jia, X. Zhang and P. R. Chen, Nat. Chem., 2014, 6, 352–361 Search PubMed.
- B. J. Stenton, B. L. Oliveira, M. J. Matos, L. Sinatra and G. J. L. Bernardes, Chem. Sci., 2018, 9, 4185–4189 Search PubMed.
- A. Sathyan, S. Croke, A. M. Pérez-López, B. F. M. de Waal, A. Unciti-Broceta and A. R. A. Palmans, Mol. Syst. Des. Eng., 2022, 7, 1736–1748 Search PubMed.
- A. Sathyan, E. Archontakis, A. J. H. Spiering, L. Albertazzi and A. R. A. Palmans, Molecules, 2024, 29, 1850 CrossRef CAS PubMed.
- L. Deng, A. Sathyan, C. Adam, A. Unciti-Broceta, V. Sebastian and A. R. A. Palmans, Nano Lett., 2024, 24, 2242–2249 Search PubMed.
- A. Sathyan, T. Loman, L. Deng and A. R. A. Palmans, Nanoscale, 2023, 15, 12710–12717 Search PubMed.
- N. Muniyappan and S. Budagumpi, Inorg. Chim. Acta, 2026, 589, 122915 CrossRef CAS.
- E. Levin, E. Ivry, C. E. Diesendruck and N. G. Lemcoff, Chem. Rev., 2015, 115, 4607–4692 CrossRef CAS PubMed.
- T. Wegner, A. Dombovski, K. Gesing, A. Köhrer, M. Elinkmann, U. Karst, F. Glorius and J. Jose, Chem. Sci., 2023, 14, 11896–11906 RSC.
- A. P. Prakasham, H. Singh and A. R. A. Palmans, Chem. Commun., 2025, 61, 9697–9700 Search PubMed.
- D. Zheng, J. Tao, L. Jiang, X. Zhang, H. He, X. Shen, Y. Sang, Y. Liu, Z. Yang and Z. Nie, J. Am. Chem. Soc., 2025, 147, 998–1007 Search PubMed.
- J. Brand, J. Siles and J. Waser, Synlett, 2010, 881–884 CAS.
- M. Kumar Gangwar, S. Dey, A. P. Prakasham. and P. Ghosh, Polyhedron, 2021, 197, 115011 CrossRef CAS.
- J. Y. Park, H.-J. Kwon, S. Mondal, H. Han, K. Kwak and M. Cho, Phys. Chem. Chem. Phys., 2020, 22, 19223–19229 Search PubMed.
- E. Bortolamiol, G. Isetta, I. Caligiuri, N. Demitri, S. Paganelli, F. Rizzolio, T. Scattolin and F. Visentin, Eur. J. Inorg. Chem., 2023, e202300084 CrossRef CAS.
- A. Kumar, M. K. Gangwar, A. P. Prakasham, D. Mhatre, A. Ch Kalita and P. Ghosh, Inorg. Chem., 2016, 55, 2882–2893 Search PubMed.
- T. E. Schmid, D. C. Jones, O. Songis, O. Diebolt, M. R. L. Furst, A. M. Z. Slawin and C. S. J. Cazin, Dalton Trans., 2013, 42, 7345 Search PubMed.
- S. Wijker, D. Dellemme, L. Deng, B. Fehér, I. K. Voets, M. Surin and A. R. A. Palmans, ACS Macro Lett., 2025, 14, 428–433 CrossRef CAS PubMed.
- M. Artar, E. R. J. Souren, T. Terashima, E. W. Meijer and A. R. A. Palmans, ACS Macro Lett., 2015, 4, 1099–1103 CrossRef CAS PubMed.
- J. Willenbacher, O. Altintas, V. Trouillet, N. Knöfel, M. J. Monteiro, P. W. Roesky and C. Barner-Kowollik, Polym. Chem., 2015, 6, 4358–4365 RSC.
- Y. Liu, S. Pujals, P. J. M. Stals, T. Paulöhrl, S. I. Presolski, E. W. Meijer, L. Albertazzi and A. R. A. Palmans, J. Am. Chem. Soc., 2018, 140, 3423–3433 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |