
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile one-pot synthesis of high-purity sodium antimony chalcogenides in polar solvents†
Saeed Ahmadi
Vaselabadi
,
Brynn
Benham
and
Colin A.
Wolden
 *
*
Chemical and Biological Engineering, Colorado School of Mines, Golden, Colorado 80401, USA. E-mail: cwolden@mines.edu
First published on 4th December 2024
Abstract
Alkali metal chalcogenides have emerged as a new class of inorganic materials with diverse applications in energy conversion and storage owing to their structural versatility and wide range of properties. Strategies are needed for simple and cost-efficient synthetic approaches that enable the composition and functional properties of these materials to be systematically tuned. Herein, we present a novel wet-chemistry approach to produce ternary Na-based metal chalcogenides with varying compositions. Phase-pure Na3SbCh4 (Ch = S, Se) solid-state electrolytes are synthesized in a single-step fashion by reacting an ethanolic solution of Na chalcogenides with appropriately selected metal halides at room temperature. This process simplifies the reaction protocols, improves yield, and decreases the raw material loss incurred in multistep systems by eliminating the need for phase-pure binary metal chalcogenides. The reaction mechanisms and impurity profile of various sodium metal chalcogenides introduced in this work were methodically investigated through characterization techniques such as X-ray diffraction (XRD) and Raman spectroscopy. Among the chalcogenides, synthesis of the sulfide compounds (∼99 wt% purity) was straightforward, achieving a yield of 92–95% whereas the selenides required more control to generate the appropriate mix of precursors, which resulted in a lower yield of 74–79% but with a high purity of 97.5–99.6 wt%. Electrochemical impedance spectroscopy of as-synthesized Na3SbCh4 (Ch = S, Se) showed a high ionic conductivity of 0.17–0.38 mS cm−1 and low activation energy of 0.19–0.21 eV comparable with other reports of solution-based synthesis. The one-pot scheme was successfully extended to the NaSbCh2 (Ch = S, Se) system, producing phase pure ternary sodium metal chalcogenides with tunable band gaps (1.6–1.8 eV) appropriate for solar energy conversion applications. The “one-pot” approach offers a simple yet economical route for scalable production of bulk sodium ternary chalcogenides at ambient conditions.
Introduction
Chalcogenides are essential classes of materials that include at least one chalcogen atom (Ch = S, Se, Te) in a reduced state. Unlike oxides, chalcogenides can form Ch–Ch bonds. Synthesis of a variety of chalcogenides is possible due to the reactivity of chalcogen anions with organic and inorganic cations such as alkali metals.1–4 Alkali metal chalcogenides have recently been the focus of intense research as a new class of inorganic materials for the energy conversion and storage sector. They have been explored extensively in various applications such as photovoltaics, thermoelectric, optoelectronics, and energy storage.1,5–8In this class of materials, slight differences in the elemental constitution, composition, and structure lead to different properties. For instance, sodium chalcogenides with Na3PnCh4 (Pn = P, Sb, As; Ch = S, Se) formula and their doped derivatives are being pursued as solid-state electrolytes for sodium-ion batteries due to their intrinsic high ionic conductivity and mechanical ductility. Originally based on sodium thiophosphate, Na3PS4, this class of solid-state electrolytes consists of polyanion polyhedra, i.e. SbCh4,3 with mobile Na+ distributed in interstices and form a crystalline structure in either cubic phase (I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3m space group symmetry) or tetragonal phase with P21c symmetry.9–15 As an example of the diversity of this class of materials, ternaries with the stoichiometric variation APnCh2 (A = Na, Li; Pn = Sb, Bi; Ch = S, Se) have received attention for their promising optical16–21 and thermoelectric5,22–24 properties. Specifically, they offer direct bandgaps suitable for solar light absorption, providing an abundant and environmentally friendly alternative.6,20
3m space group symmetry) or tetragonal phase with P21c symmetry.9–15 As an example of the diversity of this class of materials, ternaries with the stoichiometric variation APnCh2 (A = Na, Li; Pn = Sb, Bi; Ch = S, Se) have received attention for their promising optical16–21 and thermoelectric5,22–24 properties. Specifically, they offer direct bandgaps suitable for solar light absorption, providing an abundant and environmentally friendly alternative.6,20
A variety of multinary chalcogenides have been synthesized through classic solid-state synthesis with the direct reaction of pure elements or binary precursors at high temperatures. This method is generally time-intensive due to the slow diffusion of solid reactants and requires extremely high temperatures in air-free media like evacuated quartz ampoules. In addition, the products are limited to the most stable phases neglecting a diverse range of metastable phases only accessible at low temperatures.1,6 Alternatively, alkali metal chalcogenide or polychalcogenide fluxes have been employed at milder temperatures. However, the underlying mechanism in these systems is quite underexplored. Moreover, from the scale-up standpoint, this method requires high operational complexity, equipment maintenance, and safety considerations addressing the toxic gas releases with molten salt handling.25
Solution-based approaches, such as colloidal hot injection and solvothermal hydrothermal methods, have been widely utilized in synthesizing ternary alkali chalcogenides requiring a low-to-medium temperature range at near- and supercritical conditions.26 The colloidal method requires high boiling point solvents at elevated temperatures, and the products are usually contaminated with organic residues. Nanocrystalline alkali metal ternary I–V–VI2 materials are typically synthesized using colloidal hot-injection that facilitates control over nucleation and growth of nanomaterials however suffer from costly and toxic precursors as well as non-volatile solvents that are hard to fully remove.5,20,23
On the other hand, solvo/hydrothermal methods involve the production of chalcogenides at high pressure and temperature in an autoclave. This method has been used to synthesize alkali metal chalcogenides such as NaBiS2,27–29 AInS2 (A = Na, K),30 NaFeS2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 31 at temperatures ranging from 180–200 °C for extended periods (24–72 h), which utilized various chalcogen sources such as thiourea, L-cysteine, Na2S·9H2O, and NaOH.
31 at temperatures ranging from 180–200 °C for extended periods (24–72 h), which utilized various chalcogen sources such as thiourea, L-cysteine, Na2S·9H2O, and NaOH.
Overall, the liquid-phase synthetic approaches for multinary chalcogenides like Na3SbCh4 and NaSbCh2 are often costly and limited in scalability due to equipment and raw material (precursor and solvent) requirements. Inspired by the simple solvothermal reaction of Na chalcogenides such as Na2S with InCl3·4H2O in ethanol,30,32 we present a simple wet-chemical synthesis method for sodium ternary chalcogenides at room temperature. This approach is built on the hypothesis that the synthesis of the binary and ternary metal chalcogenides could be simplified to a single step, where precursors derived from the binary reaction are directly utilized to fabricate ternary Na-based chalcogenides. This approach, specifically, simplifies the current reaction protocols for Na3SbCh4 and NaSbCh2 by decreasing the raw material loss and improving the yield. The appropriate selection of precursors and solvents, as well as the impact of reaction parameters such as concentration and basicity, are investigated to optimize product purity.
First, we start by demonstrating the applicability of Na chalcogenide binary reagents to drive the single-step reaction with appropriate antimony salts in generating 314 chalcogenides with Na3SbCh4 (Ch = S, Se) stoichiometry in polar solvents. We further confirm their potential as solid-state electrolytes for sodium all-solid-state batteries. We then extend the same methodology to the 112 ternary compositions, i.e. NaSbCh2 (Ch = S, Se), confirming their semiconductor character with photoabsorption studies. The underlying chemistry and impurity profile of various compositions were investigated systematically through a complementary set of characterization techniques including scanning electron microscopy (SEM), X-ray diffraction (XRD), and Raman spectroscopy.
Experimental methods
Materials
Sodium sulfide hydrate (Na2S·xH2O, 60 wt%, Sigma-Aldrich), antimony(III) chloride (SbCl3, ACS, 99% min, Alfa Aesar), antimony(III) bromide (SbBr3, 99.5% metal basis, Thermo Scientific Chemicals), bismuth(III) bromide (BiBr3, >98%, Sigma-Aldrich), sodium borohydride (NaBH4, >98%, Sigma-Aldrich), sulfur (S, 99.999% trace metal basis, Thermo Scientific Chemicals), sodium (Na, Sigma-Aldrich), sodium hydroxide (NaOH, 98%, Thermo fisher scientific), selenium (Se, 99.99%, UMC), ethanol (EtOH, anhydrous, ≥99.5%, Sigma-Aldrich), dimethyl sulfoxide (DMSO, ACS, 99.9% min, Alfa Aesar), sodium selenate (Na2SeO4, anhydrous, 99.8+% metals basis, Thermo Scientific Chemicals), barium sulfate (BaSO4, 99%, precipitated, Alfa Aesar), and UHP grade argon (Ar, 99.999%, General Air) were used as received without purification. Se pellets were further ground using a mortar and pestle to reduce particle size. All procedures were carried out in an Ar-filled glovebox (<5 ppm of H2O) unless otherwise stated.Synthesis
| Sample name | Se solution | Molar ratio [Se]:[Sb]:[NaOH] |
Se conc. (M) | NaBH4 conc. (M) | Sb conc. (M) | NaOH (M) | Product | Size (nm) |
|---|---|---|---|---|---|---|---|---|
| A4-1-2 | A | 4:1:2 |
0.4 | 0.48 | 0.1 | 0.2 | NaSbSe2 (72%), Na3SbSe4 (28%) | — |
| A4-1-4 | A | 4:1:4 |
0.4 | 0.48 | 0.1 | 0.4 | Na3SbSe4 (90.8%), NaSbSe2 (9.2%) | 26.4 |
| A4-1-6 | A | 4:1:6 |
0.4 | 0.48 | 0.1 | 0.6 | Na3SbSe4 (99.3%), NaSbSe2 (0.7%) | 26.2 |
| A8-1-12 | A | 8:1:12 |
0.4 | 0.48 | 0.05 | 0.6 | Na3SbSe4 (99.6%), NaSbSe2 (0.4%) | 17.9 |
| B4-1-2 | B | 4:1:2 |
0.4 | 0.56 | 0.1 | 0.2 | NaSbSe2 (56.6%), Na3SbSe4 (9.2%), NaBr (34.2%) | — |
| B4-1-4 | B | 4:1:4 |
0.4 | 0.56 | 0.1 | 0.4 | NaSbSe2 (<100%), unknown impurity | 21.1 |
| B4-1-6 | B | 4:1:6 |
0.4 | 0.56 | 0.1 | 0.6 | Na3SbSe3 (∼82%), Na3SbSe4 (∼18%) | — |
| B8-1-12 | B | 8:1:12 |
0.4 | 0.56 | 0.05 | 0.6 | Na3SbSe4 (97.5%), NaSbSe2 (2.5%) | 37.3 |
The Na3SbSe4 reaction using Na2Se2 was attempted as follows: first, the Na2Se2 precursor solution was prepared as described previously.34 In brief, 110 mg (4.78 mmol) of Na was dissolved in 10 mL EtOH for approx. 1 h to form sodium ethoxide (EtONa) solution. Next, we added 29.4 mg (0.78 mmol) of NaBH4 to EtONa solution. In a three-neck flask, 430.6 mg (5.45 mmol) of Se was dispersed in 5 mL EtOH, and EtONa/NaBH4 solution was dripped slowly in Se dispersion for approx. 1 h and let it react overnight to generate ethanolic Na2Se2 solution. The reaction with similar stoichiometry to the A/B4-1-6 experiment was conducted. First, 333.3 mg (8.33 mmol) of NaOH was added to the Na2Se2 solution. After 30 min of stirring, 494.6 mg (1.37 mmol) of SbBr3 was added to the solution and the stirring continued overnight. A similar procedure was used to recover the sample powder as mentioned earlier (yield = ∼70%.)
:[Sb]:[NaOH] = 2:1:2 molar ratios. Similar recovery and purification as other selenides were implemented to obtain NaSbSe2 powder (yield = ∼80%).
Material characterization
Simultaneous thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were carried out on a TA Instruments SDT-Q600 model. For a typical run, 10 mg of sample was loaded into a pre-cleaned alumina pan and heated and cooled for one cycle under flowing Ar at 10 °C min−1 rate. X-ray diffraction (XRD) was performed with a Philips X'Pert X-ray diffractometer with Cu Kα radiation (λ = 0.15405 nm). Samples were prepared on a glass slide with protective tape covering the material to minimize air exposure. Rietveld refinement of the XRD patterns was conducted using the generalized structure analysis system (GSAS-II) software.35 A WiTec alpha 300 M Confocal Microscope/Raman Spectrometer employing a 100 mW 532 nm laser was used to obtain Raman spectra. Samples were mounted on a glass slide and sealed under a 0.1 mm quartz cover slip. The laser was focused through the coverslip onto the sample using a 20× objective, and spectra were collected using a CCD detector (Andor Technologies) at −60 °C. Field emission scanning electron microscopy (FESEM) images were collected on a JEOL JSM-7000F FESEM instrument equipped with energy-dispersive X-ray spectroscopy (EDX) for compositional analysis. To prepare the samples for SEM and EDX measurements, powder samples were placed onto an aluminum stub using double-sided carbon tape. An accelerating voltage of 5 kV was used for taking the SEM image while a higher voltage of 15–20 V was employed for EDX spectra collection. 77Se Nuclear Magnetic Resonance (NMR) spectroscopy was conducted on a JEOL ECA-500 500 MHz liquids-only spectrometer. A 1.5 M solution of sodium selenate (Na2SeO4) in D2O was used as standard. Fourier transform infrared (FTIR) spectroscopy was carried out on a Nicolet Summit FT-IR spectrometer using an attenuated total reflection (ATR) accessory equipped with a diamond crystal (16 scans, absorption mode, 4 cm−1 resolution). All sample preparation was done in an Ar glovebox.Electrochemical characterization
Pellets of Na3SbCh4 (Ch = S, Se) electrolytes were prepared for electrical conductivity measurement via conventional uniaxial pressing. For this purpose, 150–250 mg of the electrolyte was loaded into a 12 mm PEEK split cell with stainless steel plungers under a uniaxial fabrication pressure of 270 MPa and held for 5 min. Pellets were typically 0.6–0.8 mm thick. The pellets were contacted using stainless steel plungers as electrodes for electrochemical characterization. A Gamry Interface 1000E potentiostat was used to perform electrochemical impedance spectroscopy (EIS) measurements across a frequency range of 1 Hz to 1 MHz with a 10 mV perturbation with a stacking pressure of 75 MPa. Temperature-dependent EIS testing was performed by heating the split cell apparatus with an electrical heating element and allowing it to stabilize at the target temperature for 1.5 h. DC polarization measurements were conducted by applying multi-step potentials to the sample (04, 0.6, 0.8, and 1 V) and recording the transient current. The steady-state current was recorded after 2 h at each step potential, and the electrical conductivity was calculated using Ohm's law.Optical absorption characterization
A Cary 5G UV-Vis and NIR spectrophotometer, with a 200–1500 nm range was used to obtain diffuse reflectance data. Barium sulfate (BaSO4) was used as a reference with 100% reflectance. To prepare the samples, a 25:75% mixture of ternary powder and BaSO4 was ground in a pestle and mortar and pelletized into 10 mm pellets. The pellets were loaded on an integrating sphere attachment on the UV-Vis instrument. The reflectance versus wavelength data were collected to obtain the direct and indirect bandgaps of NaSbCh2 (Ch = S, Se) samples using the Kubelka–Munk transformation (F(R)) and Tauc plots (eqn (1) and (2)).36,37 | (1) |
| (F(R)hν)n ∝ (hν − Eg) | (2) |
Results and discussion
Na3SbCh4 (Ch = S, Se) synthesis and structural characterization
The main reagents commonly used to produce Na3SbCh4 are pure binary chalcogenides such as Na2Ch and Sb2Ch3. As we have shown in our previous reports, Sb2Ch3 can be prepared through simple metathesis reactions.11,12 However, the individual synthesis and purification of binary chalcogenides followed by their incorporation in ternary synthesis results in extensive raw materials loss and reductions in the overall reaction yield. Therefore, we submit a simplified protocol for the production and purification of Na3SbCh4 electrolytes by combining the binary and ternary reaction steps into a “one-pot” approach as illustrated in Fig. 1. | ||
| Fig. 1 Schematics comparing two-step and single-step (“one-pot”) reaction for Na3SbSCh4 (Ch = S, Se) synthesis in ethanol (created with BioRender.com). | ||
The choice of solvent is crucial in this process; first, the solvent must be able to dissolve all the precursors without any side reactions since this approach utilizes the products of the metathesis reaction as intermediates. Second, the solvent should facilitate the subsequent precipitation of the desired product with simple recovery steps based on the solubility criteria. Direct precipitation through filtration and centrifugation is typically more desirable compared to other energy-intensive solvent removal routines. Third, the reactants, products, and intermediates should be stable against solvents. Lastly, purification from the solvato complex should be attainable at low to moderate temperatures.
Sulfides
The standard solution-phase reaction of Na3SbCh4 is based on the nucleophilic attack of the chalcogenide ions on the binary compounds. In this approach, binary chalcogenides Sb2Ch3 are reacted with alkali metal polychalcogenides to form the salts of polyhedral anions of [SbCh4]3−. It is widely known that group (V) sulfides such as Sb2S3 dissolve in alkaline aqueous solutions to produce tetrahedral thiometallates in the form of [SbS3]3−. Furthermore, the addition of suitable cations can drive the formation of various polyanions in the presence of [SbS3]3− species.3 Previously, we utilized this concept by dissolving Sb2S3 in an alkaline solution of Na2S in EtOH. Dissolution of alkali metal sulfides such as Li2S and Na2S in highly polar alcohols results in the formation of hydrosulfide and alkoxide ions through alcoholysis creating an alkaline solution.32,38,39 The formed HS− anions act as the driving force for the formation of intermediate Na3SbS3 in ethanolic solution, and its subsequent oxidation to Na3SbS4 (eqn (3) and (S1)–(S4)†). The redox reaction is based on coupling Sb oxidation and sulfur reduction in ethanol. Sulfur solubility in ethanol further facilitates this reduction. | (3) |
The first approach to bypass the need for two binary precursors was to combine metathesis with ternary formation using methanol as the solvent and SbCl3, the most inexpensive and widely available antimony halide (eqn (4)):
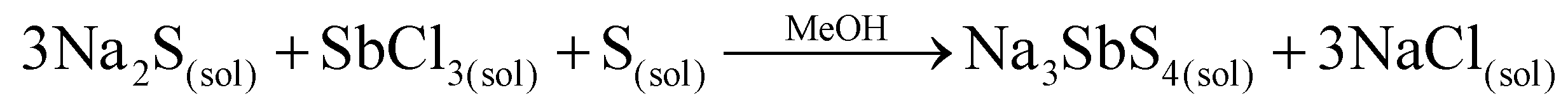 | (4) |
The reaction was successful, but since both products are highly soluble extensive washing with DMSO was required to recover phase pure Na3SbS4, making the approach expensive and inefficient. More details of the process and characterization are provided in the ESI (Fig. S1†).
This prompted us to search for other reagents and solvents that could resolve the NaCl separation issue. Interestingly, other halide precursors such as antimony bromide/iodide (SbX3, X = Br, I) are very soluble in alcohols, and their reaction with Na chalcogenides reagents results in the formation of highly soluble Na halides (NaX). For instance, NaBr solubility in EtOH is 2.496 g per 100 g EtOH.40 We previously showed that EtOH is a suitable solvent for ternary reaction since it improves overall ternary yield and retains comparable ionic conductivity to other synthetic approaches.12 Hence, we hypothesized that the following reaction (eqn (5)) with SbBr3 as the antimony source is thermodynamically favorable and could proceed in EtOH:
 | (5) |
Since Na3SbS4 is sparingly soluble in EtOH, this scheme provides direct precipitation of the sulfide at RT based on solubility criteria without any extra processing. The obtained powder was further dried at 150 °C to remove any remaining solvents. Fig. 2a shows the XRD patterns of powder collected at RT and after the 150 °C drying step. The crystal structure matches well with the tetragonal phase of Na3SbS4 (P21c space group, PDF 04-023-8842). The Rietveld refinement of the RT sample (Fig. S2†) shows a 98.8 wt% purity for Na3SbS4 with a 1.2% secondary Na3SbS3 phase.
 | ||
| Fig. 2 (a) XRD patterns and (b) corresponding Raman spectra Na3SbS4 recovered from one-pot reaction bromide halide at RT and 150°. (c) SEM micrograph, and (d) EDAX elemental mapping of Na3SbS4 at RT. | ||
The lattice parameters for Na3SbS4 (a = b = 7.1736 Å, c = 7.3041 Å, V = 375.877 Å3) are in good agreement with the values reported from Na3SbS4 synthesized through other methods.15,41 The average crystallite size of ∼31.2 nm for the primary particle is calculated from the Scherrer equation. As expected, the supernatant contains pure NaBr further confirming the feasibility of a one-pot reaction with bromide salt (Fig. S3†).
Fig. 2b exhibits the Raman spectra of corresponding Na3SbS4 samples and confirms the presence of tetrathioantimonate anions (SbS44−).42 The SEM images of the RT sample (Fig. 2c) feature large crystals (∼1–5 μm) that are formed through the agglomeration of primary nanoparticles. EDAX mapping confirms the homogeneity of the elemental constituents throughout the sample (Fig. 2d and S4†). FTIR spectroscopy (Fig. S5†) further demonstrated that mild heat treatment at 150 °C is required to remove residual solvents. In short, we showed that EtOH is an ideal solvent for this one-pot scheme that facilitates RT reactive precipitation of highly pure Na3SbS4 with high yield in a cost-effective and scalable approach.
Selenide
In our previous work, we presented a novel solution-based method to synthesize Sb2Se3 and Na3SbSe4 (314 phase) in polar solvents such as H2O and EtOH utilizing sodium hydroselenide (NaHSe) solution as the main resource for activated Se.11 In this process, elemental Se is reduced using NaBH4 in EtOH to generate NaHSe reagent at RT (eqn (S5)†). Then, crystalline Sb2Se3 is recovered from the metathesis reaction between NaHSe and SbCl3 followed by heat treatment at 300 °C (eqn (S6)†). Subsequently, Sb2Se3 is used as the precursor for the ternary reaction in conjunction with NaHSe(sol) and Se(s) reagents (eqn (6)).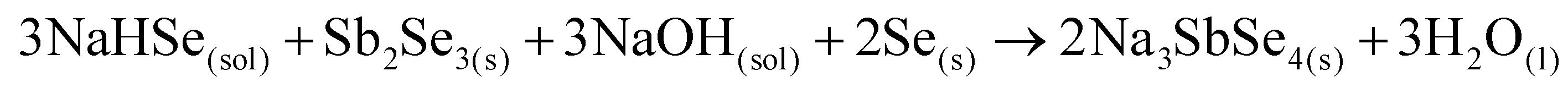 | (6) |
Similar to the sulfide synthesis detailed earlier, we attempted a one-pot reaction using antimony bromide as the precursor assuming that the nucleophilic attack of the activated Se on the halide can generate the ternary selenides directly as suggested in eqn (7). The formation of sodium bromide, analogous to the sulfide reaction, facilitates the direct precipitation of the t at RT through centrifugation and supernatant removal.
 | (7) |
In this reaction, EtOH serves multiple roles; first, the Se reduction with NaBH4 has been successfully reported in H2O and EtOH. However, the aqueous Se reduction is often accompanied by the formation of borax as a side product, which contaminates the process. Additionally, 314 phase is soluble in H2O further complicating its recovery. In this context, ethanol is the ideal solvent which also facilitates the reactive precipitation of the desired 314 phase during the final stage of reaction. Second, NaBH4 is moderately soluble in EtOH which leads to a slow decomposition and little loss of NaBH4, thereby preserving its effectiveness as a reducing agent over time.43 Third, Se reduction in ethanol produces a mildly alkaline environment contributing to the basicity needed to enhance the solubility of [SbChx] intermediates (eqn (8)).3,44
 | (8) |
Furthermore, it was found that the state of the NaHSe reagent impacted the reaction pathways. Initially, the expected “clear” solution44 was formed upon dropwise addition of NaBH4 solution (20% excess) to the Se suspension indicating the full reduction of Se after 1 h; however, the solution started to slowly turn light wine-red with color darkening as the stirring continued. Previous studies utilizing aqueous NaHSe have reported that the initial “clear” solution turns “wine-red” possibly due to its reactivity with water. The red color is attributed to the presence of amorphous selenium and polyselenide species in the solution.45,46 We observed similar behavior in ethanolic NaHSe solution even though the reaction was carried out in a glove box with minimal H2O (<5 ppm). Alternatively, injecting additional NaBH4 reverts the solution's state to “clear”. De Oliviera et al.47 also reported the occurrence of wine-red color and its reversibility upon adding excess NaBH4. They identified various Se species through organoselenide reactions involving reduced Se in ethanol and benzyl chloride. They concluded that by tuning the ratio of NaBH4 to Se, alternating concentrations of HSe− and Se22− species trapped through an alkylation reaction with benzyl chloride could be generated and identified via NMR studies. NMR is very suitable for characterizing the various Se species in solutions owing to the Se NMR active spin isotope.47–50
Similarly, we conducted 77Se NMR of the ethanolic Se solution to further understand the nature of the color change in the Se solution (Fig. 3). We prepared two Se solutions in ethanol for this experiment as detailed in the Experimental section (Fig. 3a). In the first solution (A), the Se was reduced with a 20% excess solution of NaBH4 in EtOH, generating a wine-red solution after 4 h. Small aliquots were separated for NMR analysis. The second solution (B) was prepared by titrating NaBH4 in solution A until the solution became “clear”. The Se concentration was maintained at 0.4 M for A and B solutions while NaBH4 concentrations were adjusted to 0.48 M and 0.56 M for A and B, respectively.
 | ||
| Fig. 3 (a) Photograph of activated Se solution used as Se precursors in Na3SbSe4 reactions, (b) 77Se NMR spectra of Se solutions (A: wine-red, and B: clear solution). | ||
The observed doublet peak at −495 ppm in the NMR profile for solutions A and B is in very good agreement with previous reports assigning it to HSe− monoselenide anion (Fig. 3b).47,50
The small shift to a lower chemical shift for solution B is possibly due to a slight change in the solvated environment ([B]NaBH4 = 0.56 M vs. [A]NaBH4 0.4 M), and the higher intensity of NMR signals for “clear” solution indicates a slightly higher concentration of HSe−1. Nevertheless, neither solution shows signs of other selenium species such as Se2− and Se22− anions. Cusick and Dance49 reported that they could not find any evidence of Se22− in the NMR spectrum of Na2Se2 in ethanol, since most probably it dissociates to HSe− and (Sex)2− or Se(s) species. Also, the diselenide Se22− is reported to be highly unstable while exposed to ambient oxygen and decomposes to monoselenide (Se2−) and Se(s).51,52 From these findings and our observation of the color shift in ethanolic Se, we can assume that the initial wine-red color is due to the formation of amorphous Se from Na2Se2 decomposition while most of the activated Se species are in form of HSe−1. The addition of extra NaBH4 further reduces the Se(s) and increases HSe−1 concentration reclaiming the clear solution. It is not feasible to conclusively pinpoint the water moisture (eqn (S8) and (S9)†) or oxygen (eqn (S10)†) as the source of decomposition and color shift in the Se solution. Despite the color shift, the concentration of activated Se in either state is constant; however, we can conclude that the monoselenide (HSe−1) is the highly dominant species in both solutions while appreciable amounts of unstable Se22− exist in the “wine-red” solution (A).
To systematically study the impact of color shift and reactant concentration, we designed a series of experiments with solutions A and B as the source of Se species and varying concentrations of Se and NaOH solutions. Fig. 4 compares XRD patterns of the one-pot reaction precipitate using (a) the wine-red and (b) clear NaHSe solutions. All powders were recovered at RT under vacuum without any post-synthesis heat treatment. The first set of reactions varied the NaOH concentration while fixing the [Se]:[Sb] ratio at 4:1. Our initial experiment was based on the stoichiometry shown in eqn (7) in which the concentration ratio of the precursors was [Se]:[Sb]:[NaOH] = 4:1:2. At this condition both solutions primarily generated NaSbSe2 (112 phase). Considering the Sb2Se3 as the intermediate formed in this scheme (eqn (S6)†), we can assume that hydrobromic acid (HBr) is also generated. Therefore, excess NaOH is required to neutralize the intermediate acid and maintain the basic condition required to drive the 314 phase reaction. In the case of the wine-red reagent increasing the NaOH ratio to 4 produced primarily Na3SbSe4, and further increasing to 6 eliminated the 112 phase (A4-1-6).
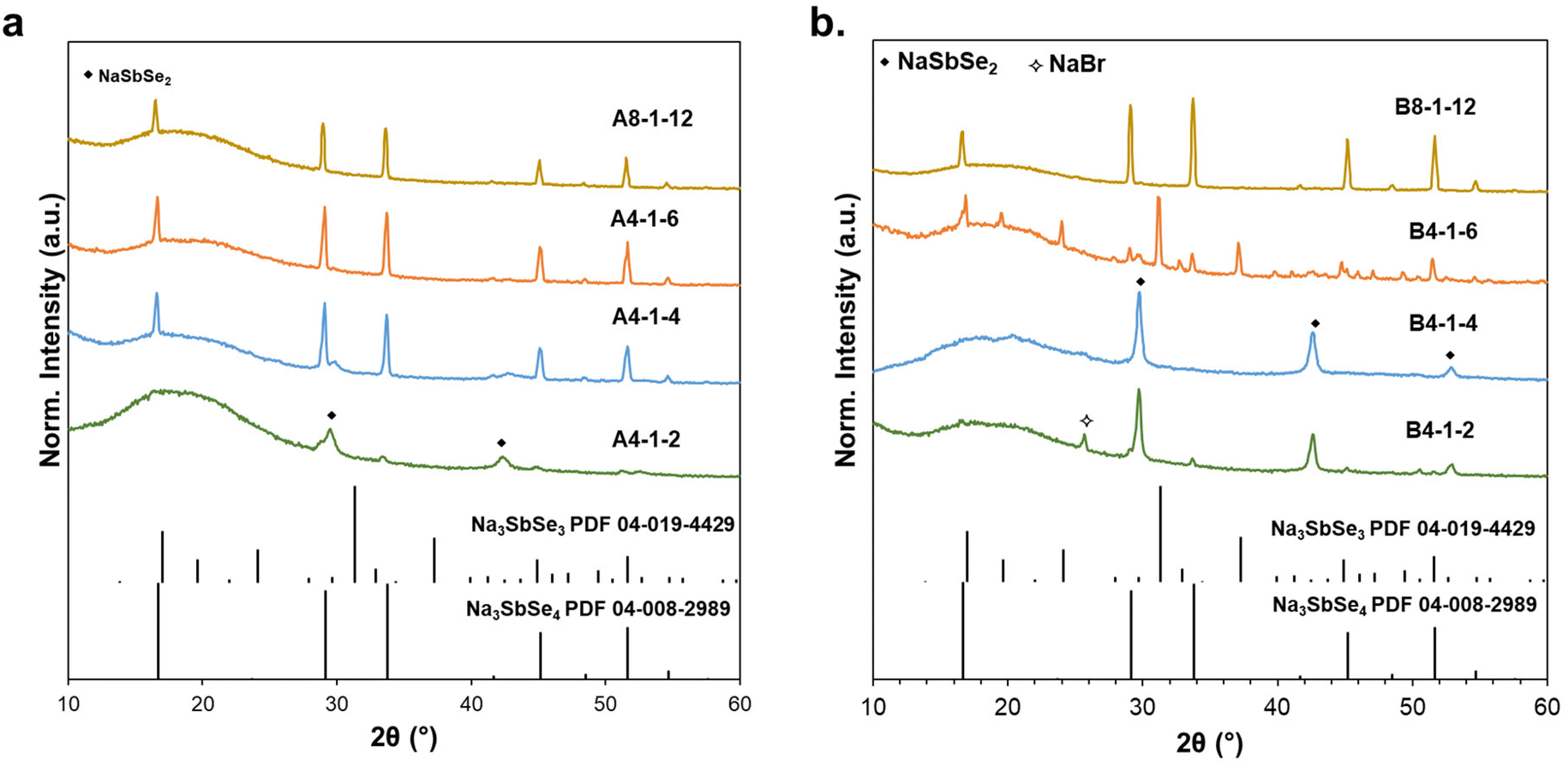 | ||
| Fig. 4 XRD patterns of Na3SbSe4 one-pot reaction precipitate with (a) “wine-red” (A) solution, and (b) “clear” (B) solution with varying [Se]:[Sb]:[NaOH] compositions. | ||
In contrast, with solution B the 112 phase persisted as NaOH was increased to 4, and at 6 the reaction produced a mixture of the 313 and 314 ternaries (B4-1-6). Optimization showed that the desired 314 phase could also be achieved through the use of excess Se. In this case the [Se]:[Sb] ratio was doubled to 8:1, and the [NaOH]:[Se] ratio was fixed to maintain solution pH and for consistency. Both solutions produced predominantly 314 with trace levels of the 112 phase, however slightly better purity was obtained with wine-red solution. Table 1 also summarizes all the reaction parameters, product composition as well as crystalline size determined from the Scherrer equation.
Although the purity of Na3SbSe4 for both A8-1-12 and B8-1-12 reactions was very similar, solution A proved superior in terms of reactant concentration and yield; in the case of A4-1-6, solution A produced comparable purity with a higher yield and lower Se concentration compared to reactions with solution B (B8-1-12 and B4-1-6). The prevalence of solution A was attributed to the presence of diselenide salts, and characterization of the supernatant showed that the Na2Se2 signal is more significant from the wine-red Se solution (A) further confirming our hypothesis (Fig. S6†). These results highlighted that reactions with solution (A), that included a mixture of activated Se species in the form of monoselenide and diselenide, proved to be favorable toward the production of the Na3SbSe4 phase with minimal quantities of precursors required. Another important observation was the absence of any gas evolution, which suggested that the proposed synthesis reaction (eqn (7)) is not correct. Based on our findings we propose that the presence of both NaHSe and selenide precursors promotes Na3SbSe4 formation:
 | (9) |
It is proposed that insufficient selenide leads to the formation of the 313 phase through eqn (10):
 | (10) |
 | (11) |
In studies on similar ternary chalcogenides, such as Cu3SbS4, it has been shown that a basic environment and the presence of oxidizing agents, like elemental chalcogens, are crucial for the oxidation of Sb(III) to Sb(V). This emphasizes the importance of additional sulfur in the Na3SbS4 reaction and the necessity of excess Se and NaOH in the selenide analog, Na3SbSe4.32,53 Schematics of possible Se species formed in ethanolic Se precursor solution and their subsequent products in the proposed one-pot reaction are depicted in Fig. 5a. Additional data supporting these conclusions is included in the ESI (Fig. (S7)–(S9)†).
 | ||
| Fig. 5 (a) Schematics of ethanolic Se species and ternary selenide products from “one-pot” approach. (b) Rietveld refinement of Na3SbSe4 sample recovered from A8-1-12 experiment. | ||
Fig. 5b shows the Rietveld refinement of the Na3SbSe4 sample recovered from the A8-1-12 experiment. The dominant crystalline phase (99.6 wt%) is cubic Na3SbSe4 with IQ3m space group as the typical high-temperature polymorph in Na3SbCh4 structure type. In this structure, the SbSe4 polyanion forms a body-centered sublattice with Na ions positioned in the octahedral voids (Fig. S10†).54 The sulfide counterpart forms the low-temperature tetragonal phase with the P21c space group with slight changes in polyhedra and Na positions (Fig. S10†). A small trace of NaSbSe2 (0.4 wt%) is also identified in the compound. SEM image and EDAX mapping of this sample is shown in Fig. S11.† Similar to the sulfide case, a mild heat treatment is carried out to remove any residual complexated solvents from Na3SbSe4 as demonstrated by the IR spectra in Fig. S12.†
Transport properties
The ionic transport of the ternary chalcogenides was characterized using electrochemical impedance spectroscopy (EIS). Fig. 6a and b shows representative room temperature Nyquist plots of Na3SbCh4 recorded from the one-pot reaction products. The impedance spectrum is fitted with an R-CPE element for the bulk resistance added to the CPE element contributing to the electronic resistance of the blocking electrodes (Table S1†). The ionic conductivity is calculated from the obtained resistance in the Nyquist plots. The ionic conductivity of Na3SbS4 (0.38 mS cm−1) and Na3SbSe4 (0.17 mS cm−1) from the one-pot reaction recovered at RT are in good agreement with our previous work utilizing the standard “two-pot” solution approach (Table S3†).11,12 | ||
| Fig. 6 Nyquist plots of (a) Na3SbS4, (b) Na3SbSe4, and (c) Arrhenius plot of ternary chalcogenides obtained from temperature-dependent impedance spectroscopy. The samples are recovered from the one-pot reaction in EtOH at RT. | ||
The kinetics of Na+ transport was studied via temperature-dependent EIS. The activation energy, derived from Arrhenius plots (Fig. 6c), shows almost identical values for both sulfides and selenides comparable with other reports in the literature.54–57 Even though a more facile ionic transport in selenide composition is predicted due to lattice expansion and softening,54 we see a lower ionic conductivity for selenide which is attributed to the presence of off-stoichiometry and impurity phases. The as-synthesized Na3SbS4 and Na3SbSe4 contain trace quantities of low-conductive Na3SbS3 and NaSbSe2 compounds, respectively, along with residual organic impurities. Furthermore, they possess lower crystallinity than highly crystalline chalcogenides developed from classical solid-state reactions, which may contribute to their inferior ionic conductivity (Table S3†).
DC polarization measurements were carried out to determine electronic conductivity. Fig. S13† shows the transient current time for sulfide and selenide at 0.6 V indicating the lower electronic conductivity obtained for ternary sulfide most likely due to different NaSbCh2 impurity concentrations. The obtained values from Ohm's law are 1.22 × 10−6 mS cm−1 and 2.11 × 10−5 mS cm−1 for Na3SbS4 and Na3SbSe4, respectively. The recorded ionic conductivity of ternary chalcogenides is 5–6 orders of magnitude higher than the electronic conductivity demonstrating the ideal characteristics of superionic conductors.
NaPnCh2 (Pn = Sb, Bi, Ch = S, Se) synthesis and structural characterization
Stoichiometric variation provides different functionalities in ternary alkali metal chalcogenides. For instance, NaSbCh2 materials, another variant composition of Na3SbCh4, are considered mixed ionic-electronic conductors with low band gaps, making them promising candidates for semiconductor applications. Conversely, they lack the superionic tendency of the latter composition largely due to an absence of Na-ion diffusion pathways in the lattice structure and a difference in their density of states. NaPnCh2 typically crystallizes with a disordered rock-salt cubic structure in which Na+/Sb3+ cations are positioned on the octahedral sites in a face-centered arrangement of chalcogenide ions, but other stable crystallographic structures like monoclinic and triclinic have also been reported.5,6,19–21Inspired by the side product formed in Na3SbCh4 synthesis, we turned to directly synthesizing NaSbCh2. Eqn (12) exhibits the simple scheme for the alkali metal sulfides:
| 2Na2S + PnBr3 → NaSbS2 + 3NaBr (Pn = Bi, Sb). | (12) |
This scheme was first tested using SbBr3 as the pnictogen precursor in ethanol. The XRD patterns of the precipitate recovered at RT (Fig. 7a) show a cubic phase of NaSbS2 (Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group) with broad peaks indicating the nanocrystalline nature of the primary particles (Fig. 7a). Heat treatment was carried out to remove any remaining solvated complexes and crystallize any possible amorphous phases. Interestingly, the initial cubic phase transitioned to a monoclinic (C2/c) phase after annealing at 300 °C. The thermal analysis of the sample obtained at RT through DSC also confirms the occurrence of a structural change at 245 °C (Fig. 7b). Raman spectra further verify this phase transition (Fig. S14†). Xia et al. reported a similar phase transition induced by heat treatment.58
m space group) with broad peaks indicating the nanocrystalline nature of the primary particles (Fig. 7a). Heat treatment was carried out to remove any remaining solvated complexes and crystallize any possible amorphous phases. Interestingly, the initial cubic phase transitioned to a monoclinic (C2/c) phase after annealing at 300 °C. The thermal analysis of the sample obtained at RT through DSC also confirms the occurrence of a structural change at 245 °C (Fig. 7b). Raman spectra further verify this phase transition (Fig. S14†). Xia et al. reported a similar phase transition induced by heat treatment.58
 | ||
| Fig. 7 (a) XRD patterns of NaSbS2 before and after annealing at 300 °C. (b) TGA-DSC scans of cubic NaSbS2 recovered at RT. (c) XRD patterns of NaSbSe2 recovered at RT and annealed at 300 °C. | ||
The polymorphism in NaSbS2 is highly dependent on the synthetic approach; even though the monoclinic phase18,59 is mainly observed through solid-state synthesis, metastable phases like cubic are mainly achieved through low-temperature solution-based methods.23,58,60,61 The annealed NaSbS2 powder was mostly agglomerated in large clusters with elemental constituents homogenously distributed within the compound (Fig. S15†).
Alternatively, the generalized form of one-pot reaction scheme for selenides analogs is suggested in eqn (13):
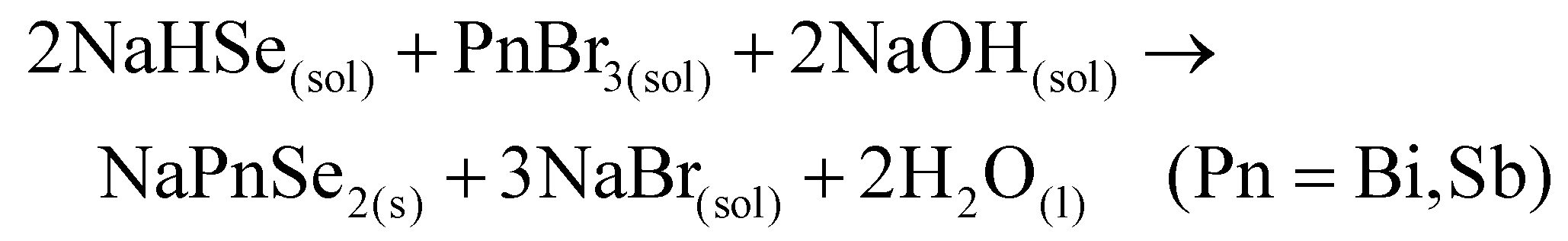 | (13) |
Previously, we showed that NaSbSe2 is the main impurity in Na3SbSe4 synthesis and is formed even without any NaOH precursor (Fig. S9†) with [Se]:[Sb] = 4:1 stoichiometry. Here, we shifted toward phase-pure NaSbSe2 synthesis based on eqn (13) which requires lower Se content. Our preliminary experiments showed that the choice of Se precursor solutions (A or B) does not have any meaningful impact on NaSbSe2 purity. Hence, all the following experiments were carried out with A (clear) solution. Fig. 7c displays the XRD patterns of NaSbSe2 products recovered from ethanolic solution at RT and further annealed at 300 °C. The highly pure NaSbSe2 sample adopts an F![[m with combining macron]](https://www.rsc.org/images/entities/char_006d_0304.gif) 3 m cubic structure with a = c = 5.9914 Å matching previous reports.5,62 Similar to other antimony-based chalcogenides, NaSbSe2 is highly crystalline at RT with extra annealing promotes its crystallinity. EDAX mapping further confirmed the purity of the 112 phase (Fig. S16†). In ESI,† we have presented our attempts to extend a similar methodology to synthesize phase-pure NaBiCh2. Table S2† summarizes all the synthetic conditions and parameters used for NaPnCh2 compounds. To investigate the potential of these ternary chalcogenides as candidates for solar energy conversion applications, diffuse reflectance spectroscopy was conducted to measure their optical bandgaps. As shown in Fig. 8, the onset of absorption for NaSbCh2 (Ch = S, Se) compounds begins around 800 nm. In the corresponding Tauc plots, the region with a linear increase in light absorption represents the characteristic of a semiconductor, and the x-axis intercept is used for band gap estimation.63 Both direct and indirect band gaps have been predicted and experimentally measured for NaSbCh223,61 compounds. The direct band gaps calculated for NaSbS2 (Fmm) and NaSbSe2 are 1.8 and 1.71 eV, while the indirect transition resulted in 1.58, and 1.64 eV, respectively. These values are in close agreement with the previous reports for nanocrystalline NaSbS2 (Fmm) and NaSbSe2 materials.19,20,23,28
3 m cubic structure with a = c = 5.9914 Å matching previous reports.5,62 Similar to other antimony-based chalcogenides, NaSbSe2 is highly crystalline at RT with extra annealing promotes its crystallinity. EDAX mapping further confirmed the purity of the 112 phase (Fig. S16†). In ESI,† we have presented our attempts to extend a similar methodology to synthesize phase-pure NaBiCh2. Table S2† summarizes all the synthetic conditions and parameters used for NaPnCh2 compounds. To investigate the potential of these ternary chalcogenides as candidates for solar energy conversion applications, diffuse reflectance spectroscopy was conducted to measure their optical bandgaps. As shown in Fig. 8, the onset of absorption for NaSbCh2 (Ch = S, Se) compounds begins around 800 nm. In the corresponding Tauc plots, the region with a linear increase in light absorption represents the characteristic of a semiconductor, and the x-axis intercept is used for band gap estimation.63 Both direct and indirect band gaps have been predicted and experimentally measured for NaSbCh223,61 compounds. The direct band gaps calculated for NaSbS2 (Fmm) and NaSbSe2 are 1.8 and 1.71 eV, while the indirect transition resulted in 1.58, and 1.64 eV, respectively. These values are in close agreement with the previous reports for nanocrystalline NaSbS2 (Fmm) and NaSbSe2 materials.19,20,23,28
 | ||
| Fig. 8 Solid-state optical diffuse reflectance spectra of (a) NaSbS2, and (c) NaSbSe2, and calculated direct and indirect band gaps from Tauc plots of (b) NaSbS2, and (d) NaSbSe2 compounds. | ||
Conclusions
In this work, we developed simple, solution-based protocols to produce sodium metal chalcogenides used in energy conversion and storage applications. Utilizing ethanolic solutions of activated chalcogens, we synthesized phase-pure Na3SbCh4 (Ch = S, Se) and NaPnCh2 (Pn = Sb, Bi, Ch = S, Se) compounds through reactive precipitation at RT with careful precursors selection and reaction optimization. Throughout this work, we observed that the underlying chemistry of chalcogen precursor and solvent interactions directly impact the phase purity of various ternary chalcogenides. Complementary analysis of the XRD and Raman spectroscopy was employed to shed light on the formation of the transient phases and the impact of precursor stoichiometry. Specifically, we identified the pivotal role of NaOH in enabling the redox mechanism of Sb(III) and Sb(V) in the Na3SbSe4 formation. Synthesis of the sulfide compound achieved a high yield of 92–95% with ∼99 wt% purity whereas the selenide reactions resulted in a lower yield of 74–79% (97.5–99.6 wt% purity). Electrochemical characterization of Na3SbCh4 compounds via impedance spectroscopy and chronoamperometry demonstrated high ionic conductivity (0.17–0.3 mS cm−1) as well as infinitesimal electronic conductivity further confirming their potential as solid-state electrolytes. Moreover, the one-pot protocol was extended to NaPnCh2 composition and resulted in highly pure ternaries with promising optical properties confirmed through diffuse reflectance UV-Vis spectroscopy. Although the one-pot solution presented in this study does not allow for precise control over the morphology or size of the chalcogenides, it is more scalable compared to other solution-based methods like solvothermal and colloidal hot injection, making it potentially more suitable for large-scale applications.Author contributions
The manuscript was written through the contributions of all authors. All authors have approved the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to sincerely thank Dr Yuan Yang for her assistance with NMR studies. This work was supported by the National Science Foundation through award 2219184. Some of the work was performed in the following core facility, which is a part of Colorado School of Mines’ Shared Instrumentation Facility (X-ray Diffraction & Computed Tomography: RRID: SCR_022053, Scanning Probe and Optical Microscopy: RRID: SCR_022048).References
- M. G. Kanatzidis, Discovery-Synthesis, Design, and Prediction of Chalcogenide Phases, Inorg. Chem., 2017, 56, 3158–3173 CrossRef CAS PubMed.
- M. G. Kanatzidis and S. P. Huang, Coordination chemistry of heavy polychalcogenide ligands, Coord. Chem. Rev., 1994, 130, 509–621 CrossRef CAS.
- W. S. Sheldrick and M. Wachhold, Chalcogenidometalates of the heavier Group 14 and 15 elements, Coord. Chem. Rev., 1998, 176, 211–322 CrossRef CAS.
- G. W. Drake and J. W. Kolis, The chemistry of mixed 15 16 main group clusters, Coord. Chem. Rev., 1994, 137, 131–178 CrossRef CAS.
- N. Kapuria, B. Nan, T. E. Adegoke, U. Bangert, A. Cabot, S. Singh and K. M. Ryan, Colloidal Synthesis of Multinary Alkali-Metal Chalcogenides Containing Bi and Sb: An Emerging Class of I–V–VI 2 Nanocrystals with Tunable Composition and Interesting Properties, Chem. Mater., 2023, 35, 4810–4820 CrossRef CAS PubMed.
- H. McKeever, N. N. Patil, M. Palabathuni and S. Singh, Functional Alkali Metal-Based Ternary Chalcogenides: Design, Properties, and Opportunities, Chem. Mater., 2023, 35, 9833–9846 CrossRef CAS PubMed.
- F. Strauss, J. H. Teo, J. Janek and T. Brezesinski, Investigations into the superionic glass phase of Li4PS4I for improving the stability of high-loading all-solid-state batteries, Inorg. Chem. Front., 2020, 7, 3953–3960 RSC.
- N. Zhao, D. Lu, J. Xu, K. Wu, H. Yu and H. Zhang, Greatly enhanced optical anisotropy in thiophosphates inspired by rational coupling of tetrahedra and ethane-like [P2S6]4− groups, Inorg. Chem. Front., 2024, 11, 4603–4610 RSC.
- H. Jia, L. Peng, C. Yu, L. Dong, S. Cheng and J. Xie, Chalcogenide-based inorganic sodium solid electrolytes, J. Mater. Chem. A, 2021, 5134–5148 RSC.
- Y. Tian, T. Shi, W. D. Richards, J. Li, J. C. Kim, S.-H. Bo and G. Ceder, Compatibility issues between electrodes and electrolytes in solid-state batteries, Energy Environ. Sci., 2017, 10, 1150–1166 RSC.
- S. A. Vaselabadi, K. Palmer, W. H. Smith and C. A. Wolden, Scalable Synthesis of Selenide Solid-State Electrolytes for Sodium-Ion Batteries, Inorg. Chem., 2023, 62, 17102–17114 CrossRef CAS PubMed.
- S. A. Vaselabadi, W. H. Smith and C. A. Wolden, Solution Synthesis of Sb2S3 and Na3SbS4 Solid-State Electrolyte, J. Electrochem. Soc., 2021, 168, 110533 CrossRef CAS.
- Y. Yang, S. Yang, X. Xue, X. Zhang, Q. Li, Y. Yao, X. Rui, H. Pan and Y. Yu, Inorganic All–Solid–State Sodium Batteries: Electrolyte Designing and Interface Engineering, Adv. Mater., 2023, 2308332, 1–24 Search PubMed.
- J. Huang, K. Wu, G. Xu, M. Wu, S. Dou and C. Wu, Recent progress and strategic perspectives of inorganic solid electrolytes: fundamentals, modifications, and applications in sodium metal batteries, Chem. Soc. Rev., 2023, 4933–4995 RSC.
- O. Maus, M. T. Agne, T. Fuchs, P. S. Till, B. Wankmiller, J. M. Gerdes, R. Sharma, M. Heere, N. Jalarvo, O. Yaffe, M. R. Hansen and W. G. Zeier, On the Discrepancy between Local and Average Structure in the Fast Na + Ionic Conductor Na2.9Sb0.9W0.1S4, J. Am. Chem. Soc., 2023, 145, 7147–7158 CrossRef CAS PubMed.
- R. McClain, C. D. Malliakas, J. Shen, J. He, C. Wolverton, G. B. González and M. G. Kanatzidis, Mechanistic insight of KBiQ2(Q = S, Se) using panoramic synthesis towards synthesis-by-design, Chem. Sci., 2021, 12, 1378–1391 RSC.
- B. A. Aragaw, J. Sun, D. J. Singh and M. W. Lee, Ion exchange-prepared NaSbSe2 nanocrystals: Electronic structure and photovoltaic properties of a new solar absorber material, RSC Adv., 2017, 7, 45470–45477 RSC.
- W. W. W. Leung, C. N. Savory, R. G. Palgrave and D. O. Scanlon, An experimental and theoretical study into NaSbS2 as an emerging solar absorber, J. Mater. Chem. C, 2019, 7, 2059–2067 RSC.
- A. Baqais, N. Tymińska, T. Le Bahers and K. Takanabe, Optoelectronic Structure and Photocatalytic Applications of Na(Bi,La)S 2 Solid Solutions with Tunable Band Gaps, Chem. Mater., 2019, 31, 3211–3220 CrossRef CAS.
- B. A. Rosales, M. A. White and J. Vela, Solution-Grown Sodium Bismuth Dichalcogenides: Toward Earth-Abundant, Biocompatible Semiconductors, J. Am. Chem. Soc., 2018, 140, 3736–3742 CrossRef CAS PubMed.
- A. M. Medina-Gonzalez, B. A. Rosales, U. H. Hamdeh, M. G. Panthani and J. Vela, Surface Chemistry of Ternary Nanocrystals: Engineering the Deposition of Conductive NaBiS2 Films, Chem. Mater., 2020, 32, 6085–6096 CrossRef CAS.
- T. J. Slade, J. A. Grovogui, J. J. Kuo, S. Anand, T. P. Bailey, M. Wood, C. Uher, G. J. Snyder, V. P. Dravid and M. G. Kanatzidis, Understanding the thermally activated charge transport in NaPbMSbQm +2(Q = S, Se, Te) thermoelectrics: Weak dielectric screening leads to grain boundary dominated charge carrier scattering, Energy Environ. Sci., 2020, 13, 1509–1518 RSC.
- A. M. Medina-Gonzalez, P. Yox, Y. Chen, M. A. S. Adamson, B. A. Rosales, M. Svay, E. A. Smith, R. D. Schaller, K. Wu, A. J. Rossini, K. Kovnir and J. Vela, Solution-Grown Ternary Semiconductors: Nanostructuring and Stereoelectronic Lone Pair Distortions in I–V–VI 2 Materials, Chem. Mater., 2022, 34, 7357–7368 CrossRef CAS.
- I. S. Khare, N. J. Szymanski, D. Gall and R. E. Irving, Electronic, optical, and thermoelectric properties of sodium pnictogen chalcogenides: A first principles study, Comput. Mater. Sci., 2020, 183, 109818 CrossRef CAS.
- X. Liu, N. Fechler and M. Antonietti, Salt melt synthesis of ceramics, semiconductors and carbon nanostructures, Chem. Soc. Rev., 2013, 42, 8237–8265 RSC.
- W. S. Sheldrick and M. Wachhold, Solventothermal Synthesis of Solid–State Chalcogenidometalates, Angew. Chem., Int. Ed. Engl., 1997, 36, 206–224 CrossRef CAS.
- H. Fei, Z. Feng and X. Liu, Novel sodium bismuth sulfide nanostructures: a promising anode materials for sodium-ion batteries with high capacity, Ionics, 2015, 21, 1967–1972 CrossRef CAS.
- P. Rong, S. Gao, H. Lu, S. Ren, M. Zhang, S. Jiao, Y. Zhang and J. Wang, Hierarchical Nanosheet-Based NaBiS2 Flowers for High-Performance and Self-Powered Broadband Photodetectors, ACS Appl. Nano Mater., 2022, 5, 11003–11010 CrossRef CAS.
- S. Kang, Y. Hong and Y. Jeon, A facile synthesis and characterization of sodium bismuth sulfide (NaBiS2) under hydrothermal condition, Bull. Korean Chem. Soc., 2014, 35, 1887–1890 CrossRef CAS.
- R. B. Zheng, J. H. Zeng, M. S. Mo and Y. T. Qian, Solvothermal synthesis of the ternary semiconductor AInS2 (A = Na, K) nanocrystal at low temperature, Mater. Chem. Phys., 2003, 82, 116–119 CrossRef CAS.
- V. G. Dileepkumar, P. S. Surya, C. Pratapkumar, R. Viswanatha, C. R. Ravikumar, M. R. Anil Kumar, H. B. Muralidhara, I. M. Al-Akraa, A. M. Mohammad, Z. Chen, X.-T. Bui and M. S. Santosh, NaFeS2 as a new photocatalytic material for the degradation of industrial dyes, J. Environ. Chem. Eng., 2020, 8, 104005 CrossRef CAS.
- J. F. W. Mosselmans, G. R. Helz, R. A. D. Pattrick, J. M. Charnock and D. J. Vaughan, A study of speciation of Sb in bisulfide solutions by X-ray absorption spectroscopy, Appl. Geochem., 2000, 15, 879–889 CrossRef CAS.
- W. H. Smith, J. Birnbaum and C. A. Wolden, Production and purification of anhydrous sodium sulfide, J. Sulfur Chem., 2021, 42, 426–442 CrossRef CAS.
- X. Yang, Q. Wang, Y. Tao and H. Xu, A Modified Method to Prepare Diselenides by the Reaction of Selenium with Sodium Borohydride, J. Chem. Res., 2002, 2002, 160–161 CrossRef.
- B. H. Toby and R. B. Von Dreele, GSAS-II : the genesis of a modern open-source all purpose crystallography software package, J. Appl. Crystallogr., 2013, 46, 544–549 CrossRef CAS.
- D. Xu, S. Shen, Y. Zhang, H. Gu and Q. Wang, Selective Synthesis of Ternary Copper–Antimony Sulfide Nanocrystals, Inorg. Chem., 2013, 52, 12958–12962 CrossRef CAS PubMed.
- P. Kubelka and F. Munk, An article on optics of paint layers, Tech. Phys., 1931, 1–16 Search PubMed.
- A. V. Kurzin, A. N. Evdokimov, V. S. Golikova and O. S. Pavlova, Solubility of sodium sulfide in alcohols, J. Chem. Eng. Data, 2010, 55, 4080–4081 CrossRef CAS.
- W. H. Smith, S. A. Vaselabadi and C. A. Wolden, Synthesis of high-purity Li2S nanocrystals via metathesis for solid-state electrolyte applications, J. Mater. Chem. A, 2023, 11, 7652–7661 RSC.
- S. P. Pinho and E. A. Macedo, Solubility of NaCl, NaBr, and KCl in water, methanol, ethanol, and their mixed solvents, J. Chem. Eng. Data, 2005, 50, 29–32 CrossRef CAS.
- L. Zhang, D. Zhang, K. Yang, X. Yan, L. Wang, J. Mi, B. Xu and Y. Li, Vacancy–Contained Tetragonal Na3SbS4 Superionic Conductor, Adv. Sci., 2016, 3, 1600089 CrossRef PubMed.
- W. Mikenda and A. Preisinger, Vibrational spectra of Na3SbS4, Na3SbS4·9H2O (Schlippe's salt) and Na3SbS4·9D2O, Spectrochim. Acta, Part A, 1980, 36, 365–370 CrossRef.
- H. C. Brown, E. J. Mead and B. C. Subba Rao, A Study of Solvents for Sodium Borohydride and the Effect of Solvent and the Metal Ion on Borohydride Reductions 1, J. Am. Chem. Soc., 1955, 77, 6209–6213 CrossRef CAS.
- D. L. Klayman and T. S. Griffin, Reaction of Selenium with Sodium Borohydride in Protic Solvents. A Facile Method for the Introduction of Selenium into Organic Molecules, J. Am. Chem. Soc., 1973, 95, 197–199 CrossRef CAS.
- S. Ganguli, S. Ghosh, G. Tudu, H. V. S. R. M. Koppisetti and V. Mahalingam, Design Principle of Monoclinic NiCo2Se4 and Co3Se4 Nanoparticles with Opposing Intrinsic and Geometric Electrocatalytic Activity toward the OER, Inorg. Chem., 2021, 60, 9542–9551 CrossRef CAS PubMed.
- U. K. Gautam, M. Nath and C. N. R. Rao, New strategies for the synthesis of t-selenium nanorods and nanowires, J. Mater. Chem., 2003, 13, 2845–2847 RSC.
- A. R. M. De Oliveira, L. Piovan, F. Simonelli, A. Barison, M. D. F. C. Santos and M. B. M. De Mello, A77Se NMR study of elemental selenium reduction using NaBH4, J. Organomet. Chem., 2016, 806, 54–59 CrossRef.
- J. D. Odom, W. H. Dawson and P. D. Ellis, Selenium-77 Relaxation Time Studies on Compounds of Biological Importance: Dialkyl Selenides, Dialkyl Diselenides, Selenols, Selenonium Compounds, and Seleno Oxyacids, J. Am. Chem. Soc., 1979, 101, 5815–5822 CrossRef CAS.
- J. Cusick and I. Dance, The characterization of [HSe]- and [Sex]2- ions by 77Se NMR, Polyhedron, 1991, 10, 2629–2640 CrossRef CAS.
- M. Bjoergvinsson and G. J. Schrobilgen, Homo- and heteropolychalcogenide anions Ch2-, HCh-, Ch22-, Ch32-, and Ch42- (Ch = selenium and/or tellurium): solution proton, selenium-77, tellurium-123 and -125 NMR study, Inorg. Chem., 1991, 30, 2540–2547 CrossRef CAS.
- Y. Abusa, P. Yox, S. D. Cady, G. Viswanathan, J. Opare-addo, E. A. Smith, Y. Mudryk, O. I. Lebedev, A. Perras, K. Kovnir, F. A. Perras and K. Kovnir, Make Selenium Reactive Again: Activating Elemental Selenium for Synthesis of Metal Selenides Ranging from Nanocrystals to Large Single Crystals, J. Am. Chem. Soc., 2023, 145, 22762–22775 CrossRef CAS PubMed.
- C. T. Irvine, N. Hoefer, M. J. Moser, R. A. Nelson, D. W. McComb and J. E. Goldberger, Diselenide Dianion's Dual Powers: PdSe2 Polymorph Control and Pd3Se10 Superatomic Crystal Creation, Chem. Mater., 2023, 35, 4404–4411 CrossRef CAS.
- F. Baum, T. Pretto, A. G. Brolo and M. J. L. Santos, Uncovering the Mechanism for the Formation of Copper Thioantimonate (SbV) Nanoparticles and Its Transition to Thioantimonide (SbIII), Cryst. Growth Des., 2018, 18, 6521–6527 CrossRef CAS.
- P. Till, M. T. Agne, M. A. Kraft, M. Courty, T. Famprikis, M. Ghidiu, T. Krauskopf, C. Masquelier and W. G. Zeier, Two-Dimensional Substitution Series Na3P1−xSbxS4−ySey : Beyond Static Description of Structural Bottlenecks for Na + Transport, Chem. Mater., 2022, 34, 2410–2421 CrossRef CAS.
- S. Xiong, Z. Liu, H. Rong, H. Wang, M. McDaniel and H. Chen, Na3SbSe4-xSx as Sodium Superionic Conductors, Sci. Rep., 2018, 8, 2–8 CrossRef PubMed.
- A. Banerjee, K. H. Park, J. W. Heo, Y. J. Nam, C. K. Moon, S. M. Oh, S.-T. T. Hong and Y. S. Jung, Na3SbS4: A Solution Processable Sodium Superionic Conductor for All-Solid-State Sodium-Ion Batteries, Angew. Chem., Int. Ed., 2016, 55, 9634–9638 CrossRef CAS PubMed.
- H. Wang, Y. Chen, Z. D. Hood, G. Sahu, A. S. Pandian, J. K. Keum, K. An and C. Liang, An Air-Stable Na3SbS4 Superionic Conductor Prepared by a Rapid and Economic Synthetic Procedure, Angew. Chem., Int. Ed., 2016, 55, 8551–8555 CrossRef CAS PubMed.
- Z. Xia, F. X. Yu, S. C. Lu, D. J. Xue, Y. S. He, B. Yang, C. Wang, R. Q. Ding, J. Zhong and J. Tang, Synthesis and characterization of NaSbS2 thin film for potential photodetector and photovoltaic application, Chin. Chem. Lett., 2017, 28, 881–887 CrossRef CAS.
- V. A. Bazakutsa, N. I. Gnidash, A. K. Kul'chitskaya and A. V. Salov, Photoelectric and optical properties of thin films of ternary chalcogenides of the form MeISbX2VI, Sov. Phys. J., 1975, 18, 472–475 CrossRef.
- M. Zubair, S. A. Ahad, I. S. Amiinu, V. A. Lebedev, M. Mishra, H. Geaney, S. Singh and K. M. Ryan, Colloidal synthesis of the mixed ionic-electronic conducting NaSbS2 nanocrystals, Nanoscale Horiz., 2023, 8, 1262–1272 RSC.
- P. C. Harikesh, A. Surendran, B. Ghosh, R. A. John, A. Moorthy, N. Yantara, T. Salim, K. Thirumal, W. L. Leong, S. Mhaisalkar and N. Mathews, Cubic NaSbS2 as an Ionic–Electronic Coupled Semiconductor for Switchable Photovoltaic and Neuromorphic Device Applications, Adv. Mater., 2020, 32, 1–9 CrossRef PubMed.
- B. Eisenmann and H. Schäfer, Über Seleno– und Telluroarsenite, –antimonite und –bismutite, Z. Anorg. Allg. Chem., 1979, 456, 87–94 CrossRef CAS.
- P. Makuła, M. Pacia and W. Macyk, How To Correctly Determine the Band Gap Energy of Modified Semiconductor Photocatalysts Based on UV–Vis Spectra, J. Phys. Chem. Lett., 2018, 9, 6814–6817 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qi02941k |
| This journal is © the Partner Organisations 2025 |