
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
J-dimers of phthalocyanine analogues: structural characterization and their use for determination of association constants between ligands and the central cation†
Jiri
Demuth
 a,
Stefan
Bednarik
a,
Radek
Machan
a,
Ivan
Mocak
a,
Tibor
Malinsky
a,
Mona
Abo El Dahabova
a,
Jakub
Holcak
a,
Miroslav
Miletin
a,
Jan
Labuta
bc,
Veronika
Novakova
a and
Petr
Zimcik
*a
a,
Stefan
Bednarik
a,
Radek
Machan
a,
Ivan
Mocak
a,
Tibor
Malinsky
a,
Mona
Abo El Dahabova
a,
Jakub
Holcak
a,
Miroslav
Miletin
a,
Jan
Labuta
bc,
Veronika
Novakova
a and
Petr
Zimcik
*a
aDepartment of Pharmaceutical Chemistry and Pharmaceutical Analysis, Faculty of Pharmacy in Hradec Kralove, Charles University, Heyrovskeho 1203, Hradec Kralove, 50005, Czech Republic. E-mail: zimcik@faf.cuni.cz
bInstitute of Organic Chemistry and Biochemistry (IOCB), Czech Academy of Sciences (CAS), Flemingovo nám. 2, 160 00 Prague, Czech Republic
cResearch Center for Materials Nanoarchitectonics (MANA), National Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan
First published on 31st December 2024
Abstract
The characterization of the stability of self-assembled supramolecular systems is critical for numerous applications that rely on non-covalent interactions between the components. However, in phthalocyanines (Pcs), the coordination of ligands to the central metal is typically not accompanied by significant spectral changes, complicating the determination of association constants (K1). In this study, we developed a reliable and widely applicable method based on fluorescence and absorption spectroscopy that allows straightforward determination of K1 for a broad range of ligands, from heterocyclic to purely aliphatic, with weak (K1 ∼ 102 M−1) to strong (K1 ∼ 107 M−1) binding affinities. The method benefits from the full characterization of unique J-dimers of Pcs, which are formed via coordination between the peripheral substituent of one molecule and the central metal of another Pc in a series of metal octa(dialkylamino)azaphthalocyanines. These J-dimers exhibit significantly red-shifted absorption bands (up to ∼710 nm) and retain red fluorescence with significant Stokes shifts (∼35–40 nm), making them ideal for spectroscopic analysis. The developed method allowed for the direct determination of dimerization constants (KD) by monitoring temperature-induced J-dimer disassembly. The determined KD values ranged from 108 to 1015 M−1, with the bulkiness of the coordinating substituents being the primary factor affecting the dimerization strength. The insights gained could be instrumental in the rational design of self-coordinating supramolecular systems that are important in, for example, energy and electron transfer processes.
Introduction
Phthalocyanines (Pcs), with a history spanning nearly a century, have been widely utilized in various applications, including pigments,1 nonlinear optics,2 dye-sensitized solar cells,3 and photodynamic therapy.4 In pursuit of expanding the functional scope of Pcs, their aza analogues, azaphthalocyanines (AzaPcs), have been synthesized.5 Among AzaPcs, tetrapyrazinoporphyrazines are the most extensively studied, finding use as sensors for metal cations6 or pH,7 photosensitizers,8 and quenchers of fluorescence in oligodeoxynucleotide probes.9 It is well established that the unique photophysical properties of these dyes, critical for such applications, are predominantly associated with their monomolecular form. However, the planar macrocyclic core of Pcs and AzaPcs exhibits a strong tendency to aggregate, leading to the formation of two distinct types of aggregates. The first and most commonly observed type is the H-aggregate, where molecules align in a parallel, sheet-like stacking arrangement. This type of aggregation results in a hypsochromic shift of the Q-band in the absorption spectrum, accompanied by a loss of fluorescence and other key photophysical properties (Fig. 1a). In contrast, the J-aggregate involves a slipped arrangement of molecules, characterized by a bathochromic shift of the Q-band, with the retention of most photophysical properties (Fig. 1b). Despite being prevalent in other structural types of dyes,10 the latter aggregates are exceedingly rare in Pcs where hydrophobic and π–π interactions strongly favor H-aggregate formation. In Pcs and AzaPcs, the J-aggregates are formed by the coordination of (non)peripheral substituents with the central cation of the neighboring molecule, thus enforcing the slipped formation. Despite the intriguing nature of these assemblies, reports of J-aggregates in Pcs and AzaPcs are exceedingly scarce and often limited to basic observations or specific applications,11–18 lacking comprehensive system characterization and analysis of underlying relationships. | ||
| Fig. 1 Basic differences between (a) H-aggregates and (b) J-aggregates in spatial arrangement, absorption, and emission spectra of Pcs and AzaPcs. Monomeric species are in red, and aggregated are in blue. Data adopted from ref. 17 (J-aggerates) and ref. 33 (H-aggregates). | ||
The central metal, playing a crucial role in J-dimers, is also widely utilized in other supramolecular assemblies of Pcs. Complexes of metal Pcs with various ligands coordinated to their central cations are well-documented in the literature. Pyridine is frequently employed as a coordinating ligand to reduce the formation of H-aggregates,13,19 while other nitrogen-donor ligands, such as imidazole, are utilized to modify compounds of interest, including fullerene,20 BODIPYs,21 and subphthalocyanines.22 These modifications, after coordination to the central cation of Pcs, often lead to the formation of functional non-covalently bonded dyads. Understanding the association constants between these ligands and the central cation is essential for the characterization and design of such supramolecular systems, which are applied in light harvesting,23,24 the development of organic solar cells,25 investigation of charge-transfer processes26,27 and other related fields. However, unlike in porphyrins, where ligand coordination induces significant spectral changes in the absorption spectra,28,29 the determination of association constants in Pcs is more challenging. The differences between the absorption spectra of free and ligand-coordinated Pcs are often minimal and, in some cases, nearly undistinguishable (also see below).30–32 In such instances, association constants are typically derived from secondary effects following ligand binding, such as electron or energy transfer to or from the ligand-attached molecule. However, these methods impose significant limitations on the range of studied ligands, as they must be functionalized with appropriate photon or electron donors/acceptors.
In this study, we developed a straightforward UV/Vis and fluorescence-based method to determine the association constants between a wide range of ligands and the central cation in Pcs and AzaPcs, leveraging the dissociation of J-dimers of these self-assembled molecules. This was allowed by the preparation of reliable mathematical models describing the assembly, followed by an in-depth investigation into the formation of J-aggregates in Pc analogues, focusing on their disassembly in relation to the substituents involved in the coordination and elucidating fundamental structural relationships.
Results and discussion
Design and synthesis
In this work, a series of zinc(II) AzaPcs of tetrapyrazinoporphyrazine type was designed (Fig. 2) to explore the impact of peripheral substitution bulkiness and flexibility on J-aggregate formation. The series included both symmetrical (1Zn–7Zn) and unsymmetrical (8Zn–10Zn) derivatives. The nitrogen in N,N-dialkylamino substituents acted as an N-donor, coordinating to the central zinc(II) of an adjacent molecule, thereby facilitating J-aggregate formation in non-coordinating solvents. The selection of AzaPc over Pc was primarily motivated by the synthetic availability of both the dicarbonitrile precursors and the final macrocycle, allowing the production of a wide range of molecules. In contrast, the synthesis of octa(dialkylamino)Pcs is typically very complex and yields are low,34 whereas the aza-analogues are readily available.17 | ||
| Fig. 2 Synthesized symmetrical (1–7) and unsymmetrical (8–10) AzaPcs. | ||
The synthesis of new compounds followed well-established procedures. Most precursors (Scheme 1) were synthesized from commercially available 5,6-dichloropyrazine-2,3-dicarbonitrile through nucleophilic substitution with secondary amines. This reaction was facilitated by strongly electron-deficient carbons at positions 5 and 6. The yields for these precursors were generally high, exceeding 70%, except compound 16, which had a lower yield of 48%. The reduced yield was likely due to its low solubility, leading to losses during purification. Of note, N,N,N′-trimethylethylenediamine was used instead of a secondary amine during the synthesis of compound 17, resulting in the formation of a stable six-membered ring followed by dealkylation.35 Compound 19 was synthesized by nucleophilic substitution of 5-chloropyrazine-2,3-dicarbonitrile (18), which was prepared according to a published procedure,36 with dimethylamine in ethanolic solution.
 | ||
| Scheme 1 Synthetic pathways: (i) dimethylamine (33% in EtOH), THF, r.t., 1 h; (ii) dipropylamine, THF, reflux, 2 h; (iii) dibutylamine, THF, reflux, 2 h; (iv) piperidine, THF, r.t., 1 h; (v) morpholine, THF, r.t., 1 h; (vi) N,N,N′-trimethylethylenediamine, K2CO3, THF, reflux, 2.5 h; (vii) dimethylamine (33% in EtOH), THF, r.t., 1 h; (viii) Li, BuOH, reflux, 30 min; and (ix) Zn(OAc)2, pyridine, reflux, 3 h. | ||
The preparation of metal-free AzaPcs was carried out using the Linstead method, initiated by lithium butoxide. The purification of symmetric compounds 1H2–7H2 was straightforward, with yields ranging from 11% to 58%. Lower yields were observed for 1H2, 6H2, and 7H2, primarily due to their poor solubility in most organic solvents. The isolation of low-symmetry compounds 8H2–10H2, produced by the statistical cyclotetramerization of two different precursors, was more challenging. It required the isolation of a single congener of AAAB type from a mixture of six possible congeners, which was achieved by column chromatography on silica, yielding 11% to 22%. The final step in the synthesis of all cyclotetramers involved the incorporation of a zinc(II) cation into the center of the metal-free macrocycle. This was accomplished by reacting the metal-free compounds with zinc(II) acetate in pyridine, yielding between 14% and 97%. Lower yields for 1Zn, 6Zn, and 7Zn were again attributed to their low solubility. To further extend our study to other central metals, magnesium(II) complex 4Mg was prepared by cyclotetramerization of the corresponding precursor using magnesium butoxide. All synthesized compounds were characterized by 1H and 13C NMR spectroscopy, and HRMS, with some exceptions due to the low solubility of some AzaPcs.
Characterization of J-dimers
The formation of supramolecular interactions was initially confirmed through NMR spectroscopy of the synthesized AzaPcs in various solvents. The NMR spectra of 3H2 and 3Zn are shown in Fig. 3 as representative examples. For the metal-free 3H2, 1H NMR spectra were recorded in both toluene-d8 and pyridine-d5 (Fig. 3a). In both solvents, the spectra displayed sharp and distinct triplets or quartets corresponding to the propyl group, indicative of the monomeric form of 3H2. This form was expected, as the absence of a central cation precludes any significant J-aggregation. Similar monomeric behavior was observed for 3Zn in pyridine-d5, attributed to the coordination of pyridine-d5 to the central zinc(II) ion (Fig. 3b). However, when 3Zn was analyzed in the non-coordinating toluene-d8, the 1H NMR spectrum became more complex, suggesting the formation of a supramolecular assembly (as schematically drawn in the inset to Fig. 3c). To further elucidate the structure of this assembly, the HSQC NMR technique was employed (Fig. 3c), allowing for the assignment of specific signals to their corresponding hydrogen atoms. The analysis revealed two strongly shielded sets of –N(CH2)– signals at δ = 3.04 and 2.89 ppm, which were attributed to the methylene signals of those –N(CH2)– involved in the coordination to the zinc(II) ion and the neighboring vicinal –N(CH2)–. The ring current effect of the macrocycle, which influenced the substituents positioned above the core, was responsible for this shielding. This effect was also evident for other hydrogens of the same substituent, even in the methyl groups observed at δ = 0.64 ppm. The ratio of the shielded –N(CH2)– signals (at δ = 3.04 and 2.89 ppm) to the unaffected signals (δ = 3.8–4.3 ppm) is approximately 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :24 as determined from the integral of HSQC spectra (see ESI, Fig. S1 and S2†). This ratio confirmed that only one quarter of the macrocycle is involved in the coordination, supporting the presence of the J-dimer structure connected by two coordination bonds as drawn in Fig. 3c. The data ruled out potential supramolecular stair-like chain, which would have exhibited a 16:16 ratio. DOSY NMR experiments further supported the presence of the supramolecular dimeric structure of 3Zn (Fig. S3a†), which can be disassembled upon the addition of NMI (Fig. S3b†).
:24 as determined from the integral of HSQC spectra (see ESI, Fig. S1 and S2†). This ratio confirmed that only one quarter of the macrocycle is involved in the coordination, supporting the presence of the J-dimer structure connected by two coordination bonds as drawn in Fig. 3c. The data ruled out potential supramolecular stair-like chain, which would have exhibited a 16:16 ratio. DOSY NMR experiments further supported the presence of the supramolecular dimeric structure of 3Zn (Fig. S3a†), which can be disassembled upon the addition of NMI (Fig. S3b†).
 | ||
| Fig. 3 NMR spectra of 3H2 (a) and 3Zn (b and c) in pyridine-d5 (black, upper spectra) and toluene-d8 (brown, lower spectra). Asterisks indicate residual solvent signal and signal of the non-deuterated solvent. (a) 1H NMR of 3H2 in pyridine-d5 and toluene-d8 (monomeric form in both solvents). (b) 1H NMR spectra of 3Zn in pyridine-d5 (monomeric form) and in toluene-d8 (dimeric form). Color arrows indicate which signals belong to which hydrogen. The resonance at 3.85 ppm in toluene-d8 is overlapped by a signal of water. (c) HSQC experiment of 3Zn in toluene-d8 and schematic illustration of the J-dimer formation. Green boxes (and green N,N-dipropylamino groups) indicate the hydrogens located above/below the macrocyclic core. | ||
The absorption and emission spectra provided further evidence for the formation of J-dimers of 3Zn. In pyridine, the absorption Q-band of 3Zn appeared as a single, sharp peak at λmax = 660 nm, characteristic of Pcs and their analogues in the monomeric state (Fig. 4b). This was accompanied by a corresponding mirrored emission peak at λem = 666 nm (Fig. 4c). In contrast, when measured in toluene, the absorption spectrum of 3Zn broadened and exhibited a bathochromic shift to λmax = 702 nm (Fig. 4b). The emission spectrum also showed a red-shifted peak at λem = 739 nm, with a significant Stokes shift of Δλ = 37 nm (Fig. 4b) that can be explained by increased flexibility of the dimeric structure of the chromophore not connected by covalent but coordination bonds. The excitation spectra measured in toluene perfectly overlapped the corresponding absorption spectra (Fig. S4†) confirming the origin of the fluorescence signal. These spectral changes are consistent with the formation of J-dimers in toluene, as supported by the NMR data. For comparison, the absorption spectra of metal-free 3H2 were consistent across both solvents (Fig. 4a), indicating that 3H2 remained in the monomolecular form. The observed splitting of the Q-band is a typical feature of the metal-free Pcs and their analogues, resulting from the reduced D2h symmetry of the macrocycle.
 | ||
| Fig. 4 (a) Absorption spectra of 3H2 (5 μM) in toluene (blue) and pyridine (red). Absorption (b) and emission spectra (λex = 588 nm) (c) of 3Zn (5 μM) in toluene (blue) and in pyridine (red). | ||
Photophysical properties
Following the confirmation of J-dimer formation, we proceeded with the photophysical characterization of all studied derivatives in both monomeric and J-dimer forms. The spectral behavior observed was consistent with that of 3Zn: the compounds exhibited monomeric characteristics in coordinating solvents, while in toluene, they formed J-dimers with nearly identical Q-band positions (Table 1). It is noteworthy that the presence of eight dialkylamino substituents significantly quenched the excited states through intramolecular charge transfer (ICT). ICT, a predominant relaxation pathway for (di)alkylamino AzaPcs, is highly efficient (particularly in polar solvents) and surpasses other relaxation mechanisms such as intersystem crossing or fluorescence.37 To evaluate the impact of different solvents on the properties of AzaPcs, we employed three solvents with distinct characteristics: DMF (a polar solvent where AzaPcs remain monomeric), toluene (a nonpolar solvent where AzaPcs form J-dimers), and toluene with 0.12 M pyridine (nonpolar, but where AzaPcs are in the monomeric form due to coordination to pyridine). Due to the low solubility of compound 7Zn, accurate determination of all photophysical parameters was not feasible.| DMF | Toluene | Toluene with 0.12 M pyridine | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| λ max, nm | λ em, nm | Φ Δ | Φ F | λ max, nm | λ em, nm | Φ Δ | Φ F | λ max, nm | λ em, nm | Φ Δ | Φ F | |
| a Concentration of pyridine was 0.62 M instead 0.12 M. b Cannot be determined due to insufficient solubility. c Taken from previous publication.17 | ||||||||||||
| 1Zn | 654 | 675 | 0.0033 | 0.0005 | 630/702 | 752 | 0.063 | 0.0089a | 655a | 663a | 0.034a | 0.0002a |
| 2Zn | 660 | 673 | <0.005c | <0.0001c |
635/699 | 738 | 0.28c | 0.027c | 659 | 664 | 0.056c | 0.0004c |
| 3Zn | 661 | 673 | 0.0049 | 0.000062 | 635/702 | 739 | 0.14 | 0.039 | 660 | 666 | 0.063 | 0.0056 |
| 4Zn | 661 | 673 | 0.0073 | <0.0001 | 635/708 | 756 | 0.26 | 0.029 | 660 | 675 | 0.081 | 0.0067 |
| 5Zn | 660 | 673 | <0.005 | 0.000048 | 640/706 | 752 | 0.10 | 0.0057 | 661 | 674 | 0.031 | 0.00092 |
| 6Zn | 656 | 673 | 0.0041 | 0.000052 | 632/702 | 737 | 0.36 | 0.033c | 657a | 661a | 0.044a | 0.0017 |
| 7Zn | 651 | 672 | —b | —b | —b | —b | —b | —b | 651a | 656a | —b | —b |
| 8Zn | 660 | 673 | 0.0080 | 0.00007 | 634/707 | 744 | 0.28 | 0.019 | 659 | 669 | 0.037 | 0.0029 |
| 9Zn | 665 | 673 | 0.0079 | 0.000050 | 636/722 | 766 | 0.35 | 0.022 | 665 | 674 | 0.061 | 0.0029 |
| 10Zn | 660 | 673 | 0.0064 | 0.00001 | 637/706 | 749 | 0.22 | 0.010 | 659 | 668 | 0.032 | 0.0017 |
First, all photophysical parameters were measured in DMF to assess the properties of the derivatives in their monomeric state. In DMF, the spectra of all studied compounds were consistent with those observed in pyridine (Table 1). The quantum yields for these compounds were close to zero as a result of highly efficient ICT facilitated by the highly polar solvent. In a nonpolar toluene/pyridine mixture, the ICT effect was diminished, leading to a notable increase in quantum yields, though they remained relatively low, with fluorescence quantum yields (ΦF) reaching up to 0.007 and singlet oxygen quantum yield (ΦΔ) ranging from 0.03–0.08. The formation of J-dimers in pure toluene further enhanced both quantum yields, with ΦF increasing up to 0.04 and ΦΔ rising significantly to between 0.06 and 0.36. This increase is likely due to the suppression of ICT within the supramolecular self-assembly of the J-dimers most likely due to changed properties of the donor (AzaPc) as coordination of one out of eight peripheral nitrogens cannot result in such a change.
Stability of J-dimers and their disassembly
The stability of J-dimers is characterized using three constants in an approach similar to that published by García-Iglesias et al.12 The constant KL (Fig. 5a, eqn (1)) represents the apparent constant for the dimer disassembly by the coordinating ligand (L). This apparent constant is a composite of two underlying processes, which are the formation of the AzaPc J-dimer characterized by the dimerization constant (KD) (Fig. 5b, eqn (2)) and the association between the monomeric AzaPc's central cation and the coordinating ligand determined by the K1 constant (Fig. 5c, eqn (3)). Therefore, the apparent constant KL can be expressed in terms of KD and K1 stepwise constants as KL = K12/KD. For details regarding the derivation of the binding model, please refer to the binding models section in the ESI.† | ||
| Fig. 5 Schematics of the equilibrium processes and definition of corresponding equilibrium binding constants exemplified on 3Zn. (a) Dimer (AzaPc)2 disassembly by coordinating ligand L characterized by the KL constant. (b) AzaPc J-dimerization process characterized by the KD constant. (c) Monomeric AzaPc association with coordinating ligand L characterized by the K1 constant. | ||
To characterize the stability of the J-dimers in this series of AzaPcs, the dimerization constant (KD) was determined for each compound (Table 2). The concentration-dependent UV-Vis or fluorescence experiments (the dilution approach) could not be used due to the expected high dimerization constant KD where the monomeric and dimeric forms would not be visible in reasonable concentration ranges. Instead, KD was obtained by progressively disassembling the J-dimers through increasing temperature and monitoring the resulting changes in fluorescence spectra (Fig. 6b and Fig. S5, S6†), which allowed direct KD determination as well as enthalpy and entropy of the dimerization process (Table 2). So far, the KD values for Pc J-dimers were reported only in two cases, moreover approximated indirectly from titration with the ligand and the known value of K1 for structurally similar compounds.11,12 The details regarding our binding model and determination of KD at 23 °C are given in the Binding models section of the ESI.† The measurements were performed at a concentration in a range of 0.05–5.0 μM, tailored to cover most AzaPcs dimers’ stability range. Notably, the most stable J-dimers, 1Zn and 6Zn, did not disassemble even at the lowest measurable concentration (0.05 μM), which was close to the instrument's detection limit. For these compounds, KD was estimated using the later-determined K1 (see below) and values obtained from titration with a ligand and a combination of eqn (1) and (2), i.e., KD = K12/KL. Due to 7Zn's extremely low solubility, it was impossible to determine any constants for this compound, and it was excluded from further experiments. Temperature-induced changes could also be tracked in absorption spectra (Fig. 6a and Fig. S5†), however, due to the generally lower sensitivity of UV-Vis spectrophotometry in comparison to fluorescence methods, this was feasible only for 4Zn and 5Zn where higher concentrations (10 μM) were employed. The KD values obtained by both methods were consistent. The values of KD are summarized in Table 2 and graphically represented in Fig. 6e. From these results, it is evident that the primary factor influencing J-dimer stability is the bulkiness of the substituents on the N-donor moiety. There was an almost linear relationship between KD and the size of these substituents (Fig. 6e, black points), especially when considering only homologous series (1Zn–4Zn). Introducing an oxygen atom to the alkyl chain (5Zn) slightly increases the dimerization. To further explore the impact of bulky peripheral substituents that were not involved in coordination (such as dibutylamino groups that make up three-quarters of the molecule), unsymmetrical compounds 8Zn–10Zn with one quarter featuring smaller substituents were analyzed (due to steric reasons, it is expected that the smaller moiety will be responsible for coordination). Interestingly, their KD values (∼1011–1012 M−1) were significantly higher than that of the dibutyl-substituted 4Zn (KD = 1.51 × 108 M−1) but at the same time significantly smaller than that of dimethyl-substituted 1Zn (KD = 1.22 × 1015 M−1). This suggests that J-dimer stability was heavily driven by the smaller (coordinating) quarter of the molecule, however, it still had important contributions from the remaining substituents. This can be utilized in the design of J-dimers with finely tuned dimerization. Overall, the KD values of all derivatives observed were remarkably high, even for AzaPcs 4Zn and 5Zn with bulky substituents, aligning with the extreme stability of the few Pc J-dimers previously reported in the literature (KD ∼ 1011–1012 M−1).11,12 In contrast, H-dimers of Pcs and naphthalocyanines which were stabilized by simple π–π stacking, typically exhibit KD values in the range of 104–106 M−1 in various solvents.38,39
 | ||
| Fig. 6 Changes in the absorption (a) and emission (λex = 609 nm, isosbestic point) (b) spectra of 4Zn in toluene (c = 10 μM for UV/vis and 5 μM for emission) with increasing temperature. (c and d) Changes in the absorption (c) and emission (d, λex = 588 nm, isosbestic point) spectra of 4Zn (c = 10 μM for UV/vis and 5 μM for emission) upon J-dimer disassembly by the addition of pyridine. (e) Dependence of KD values on the number of atoms on nitrogen in the peripheral substituent (for 8Zn–10Zn, the number of atoms is considered four as it represents ¾ of all substituents). (f) Disassembly of J-dimers of all AzaPcs (c = 10 μM) upon the addition of pyridine, monitored as absorbance at the monomer band maximum. In all figures, dots represent experimental values, and lines represent the fit of the binding model. | ||
| K D [M−1] | K 1 (UV) [M−1] | K 1 (F) [M−1] | K L (UV) [M−1] | K L (F) [M−1] | ΔHc [J mol−1] |
ΔSd [J mol−1 K−1] |
|
|---|---|---|---|---|---|---|---|
| Ligand = pyridine.a Determined from the titration of J-dimer by pyridine and average K1 = 2.46 ± 0.90 × 105 M−1 (UV) and K1 = 2.89 ± 1.03 × 105 M−1 (F). n.d. = could not be determined.b Following KD values were obtained using absorption data: KD(4Zn) = 2.47 × 107 M−1, KD(5Zn) = 1.84 × 108 M−1.c Experimental error is under 10% except for 10Zn (18%).d Experimental error is under 10% for 4Zn, 5Zn, 8Zn and 9Zn, under 20% for 2Zn and 3Zn, and under 30% for 10Zn. | |||||||
| 1Zn | 1.24 × 1015a |
n.d. | n.d. | 4.26 × 10−5a |
6.86 × 10−5a |
n.d. | n.d. |
| 2Zn | 3.13 × 1011 | 1.54 × 105 | 3.17 × 105 | 7.61 × 10−2 | 3.22 × 10−1 | −1.23 × 105 | −1.97 × 102 |
| 3Zn | 5.32 × 109 | 2.90 × 105 | 2.62 × 105 | 1.58 × 101 | 1.29 × 101 | −1.23 × 105 | −2.30 × 102 |
| 4Zn | 1.51 × 108b |
4.24 × 105 | 4.58 × 105 | 1.19 × 103 | 1.40 × 103 | −1.21 × 105 | −2.53 × 102 |
| 5Zn | 1.71 × 109b |
2.67 × 105 | 2.45 × 105 | 4.14 × 101 | 3.51 × 101 | −1.22 × 105 | −2.35 × 102 |
| 6Zn | 3.73 × 1015a |
n.d. | n.d. | 1.20 × 10−5a |
2.24 × 10−5a |
n.d. | n.d. |
| 7Zn | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| 8Zn | 1.25 × 1011 | 1.60 × 105 | 1.62 × 105 | 2.05 × 10−1 | 2.10 × 10−1 | −1.38 × 105 | −2.54 × 102 |
| 9Zn | 3.09 × 1011 | 1.66 × 105 | 1.74 × 105 | 8.94 × 10−2 | 9.81 × 10−2 | −1.49 × 105 | −2.86 × 102 |
| 10Zn | 2.36 × 1012 | 2.63 × 105 | 4.05 × 105 | 2.94 × 10−2 | 6.95 × 10−2 | −1.74 × 105 | −3.55 × 102 |
To further investigate the stability of J-dimers in the presence of competing ligands, toluene solutions of J-dimers (at a concentration of 10 μM) were titrated with pyridine. Pyridine displaces the AzaPc N-donor from its coordination with the central zinc(II) ion, leading to the disassembly of the J-dimer. This process was tracked by observing changes in the absorption and emission spectra (Fig. 6c, d and Fig. S7, S8†). Specifically, the characteristic J-dimer band at around 702 nm (with corresponding emission at 750 nm) decreased, while the Q-band of the monomer at around 660 nm (with emission at 668 nm) increased, similarly to the temperature-induced disassembly described earlier. The spectra exhibited clear isosbestic points, indicating a direct transition between two states (monomer and dimer), which further confirmed the presence of a simple dimeric structure rather than more complex multimers. The data were then plotted as AzaPc's monomer absorbance versus pyridine concentration (Fig. 6f), indicating different J-dimer stability. Using the previously determined KD values for 2Zn–5Zn and 8Zn–10Zn, the titration data were fitted using a binding model containing both dimerization and association with ligand (see the Binding models section in the ESI†), which allowed for determining KL and K1. The titrations were analyzed using both absorbance and fluorescence changes, and the resulting KL values were in good agreement (Table 2). As expected, the order of KL values correlated closely with the KD values, demonstrating consistency across the series. Values of the determined K1 values (Table 2) were almost the same, yielding the average value of K1 = (2.46 ± 0.90) × 105 M−1 (from UV/vis) and K1 = (2.89 ± 1.03) × 105 M−1 (from fluorescence). The similarity of the ligand binding strength in the whole series of AzaPcs is the expected result since the electronic effects in all studied AzaPcs are the same and should not affect the strength of association between the ligand and AzaPc's central cation. The low variability of K1 values across such an extensive series of compounds confirms the validity of the measurements and the overall methodology. The average K1 values and data from pyridine-induced disassembly were then used to calculate KD and KL for 1Zn and 6Zn, where the high stability of the J-dimer prevented direct determination of KD through temperature changes. These experiments allowed for a comprehensive understanding of the stability and behavior of the J-dimers across the series, providing valuable insights into the factors influencing their formation and stability.
Determination of association constants with various ligands
As briefly mentioned in the Introduction section, determining K1 values for Pcs and their analogues is particularly challenging due to the limited spectral changes observed in both absorption and fluorescence spectra upon ligand binding. This difficulty can be demonstrated by the addition of several ligands to a toluene solution of zinc(II) octakis(tert-butylsulfanyl)AzaPc, which does not form J-dimers in toluene. In this case, almost no spectral changes were observed (refer to ESI, Fig. S15 and S16†). However, the situation is markedly different when J-dimers are present, as shown in our data above.Given the robustness of the newly developed method for determining association constants K1, we extended its application to a variety of N-, O-, and S-ligands (Table 3). The spectral changes in both absorption and fluorescence of 2Zn upon the addition of these ligands (Fig. S9–S11†) were similar to those observed with pyridine, as exemplified by the addition of diethylamine (Fig. 7a and b). The data were analyzed with fixed KD(2Zn) = 3.13 × 1011 M−1, allowing for the calculation of K1 for each ligand (Table 3). For ligands with lower affinity (i.e., lower K1) that failed to disassemble the J-dimers of 2Zn (e.g., THF, N,N-dimethylaniline, triethylamine Fig. 7c), J-dimers with lower KD (3Zn and 5Zn, KD ∼ 109 M−1) were utilized. As shown above, in the titration with pyridine, switching to AzaPc with different KD values did not affect the determined K1 value, which shows the expected consistency of our model (Table 3). This finding highlights the versatility of the method, enabling the determination of association constants for ligands across a wide range of K1 values by simply adjusting the strength of the J-dimer used in the experiments. The data revealed that the most potent ligand binding was observed with N-methylimidazole (NMI, K1(F) = 6.40 × 106 M−1), followed by other N-ligands. A comparison with the literature data available for pyridine and NMI, actually the only two ligands available in the literature to the best of our knowledge (see Table S1†), shows slightly higher values than those determined by other methods, however, they are still within a similar range. From the other ligands, it is notable that piperidine exhibited one of the strongest binding affinities, nearly matching that of NMI with K1(F) = 3.01 × 106 M−1. This suggests that piperidine could be a valuable alternative strong ligand for constructing non-covalently assembled supramolecular systems—a potential that has been relatively underexplored until now. The rigidity introduced by the ring closure in piperidine seems to play a crucial role, as a similar but more flexible secondary amine, diethylamine, showed a binding affinity two orders of magnitude lower K1(F) = 3.02 × 104 M−1. In contrast, the O-ligands demonstrated significantly lower affinity for the central zinc(II), with K1 ∼ 102–104 M−1. Surprisingly, ethanol, with a K1(F) = 1.06 × 104 M−1 showed two orders of magnitude higher binding ability than tetrahydrofuran (THF), which is commonly used to reduce aggregation in Pcs and their analogues. Lastly, the selected S-ligands (thiolan, butan-1-thiol) were unable to disassemble the J-dimers, indicating very low affinity for the central zinc(II). These findings underscore the utility of this novel method in elucidating the binding affinities of various ligands expanding the range of ligands that can be effectively studied.
 | ||
| Fig. 7 Disassembly of the J-dimer of 2Zn in toluene (10 μM) by the addition of diethylamine monitored by UV-vis (a) and fluorescence (b) spectra at 23 °C. (c) Disassembly of the J-dimer of 2Zn in toluene (10 μM) by the addition of various coordinating ligands monitored in UV-vis as maximum absorbance of the monomeric form. In all figures, dots represent experimental values, and lines represent the fit of the binding model. | ||
| Ligand | Zn(II) (UV) | Zn(II) (F) | Mg(II) (UV) | Mg(II) (F) |
|---|---|---|---|---|
| a Determined using 2Zn. b Determined using 4Mg. c Determined using 3Zn. d Determined using 5Zn. | ||||
| N-Methylimidazol | 5.82 × 106a |
6.40 × 106a |
1.93 × 106b |
2.28 × 106b |
| Piperidine | 3.00 × 106a |
3.01 × 106a |
2.04 × 105b |
2.03 × 105b |
| 4,4′-Bipyridine | 4.22 × 105a |
5.21 × 105a |
3.79 × 104b |
3.88 × 104b |
| Pyridine | 1.54 × 105a |
3.17 × 105a |
1.66 × 104b |
1.79 × 104b |
| Diethylamine | 2.44 × 104a |
3.02 × 104a |
3.08 × 104b |
3.20 × 104b |
| Pyrazine | 8.26 × 103a |
1.04 × 104a |
2.36 × 103b |
3.39 × 103b |
| Ethanol | 2.92 × 103a |
1.06 × 104a |
n.d. | n.d. |
| Triethylamine | 4.11 × 103c |
4.03 × 103c |
7.82 × 104b |
7.08 × 104b |
| Tetrahydrofuran | 5.77 × 102d |
6.15 × 102d |
2.59 × 103b |
3.35 × 103b |
| N,N-Dimethylaniline | 3.65 × 102d |
3.32 × 102d |
n.d. | n.d. |
| Thiolan | n.d. | n.d. | n.d. | n.d. |
| Butanthiol | n.d. | n.d. | n.d. | n.d. |
To further validate the determination of the K1 value by an alternative method, we explored changes in 1H NMR during titration with NMI (with the highest K1 among studied ligands), using AzaPc 3Zn in toluene-d8 (1 mM) as a representative example (Fig. 8). During the titration, the methyl signals of the J-dimer, initially observed at chemical shifts δ = 1.25, 1.14 and 0.64 ppm gradually decreased in intensity as the corresponding signal for the monomeric form started to appear at δ = 1.06 ppm. This behavior was consistent across other signals, clearly indicating that the disassembly of the J-dimer as NMI was introduced. K1 was determined based on the fitting of the binding model to the integrated intensity of resonances at δ = 0.64 and 1.14 ppm, which are attributed to the J-dimer methyl group, and with KD = 5.32 × 109 M−1. The determined K1 = 9.04 × 106 M−1 corresponds well to K1 for NMI determined by spectroscopic methods (see also Table 3): K1(F) = 5.82 × 106 M−1 and K1(UV) = 6.40 × 106 M−1. This consistency between NMR and spectroscopic methods reinforces the reliability of our results and highlights the versatility of the approaches used in characterizing the interaction between the ligands and the central zinc(II) cation in these supramolecular systems.
 | ||
| Fig. 8 The course of titration of 3Zn in toluene-d8 (1 mM) by NMI at 23 °C. Blue numbers and arrows belong to the dimeric form. Red numbers and arrows belong to monomers. Inset: disassembly of the J-dimer of 3Zn by NMI as monitored by integrated intensity of resonances due to the J-dimer at δ = 0.64 ppm (squares) and δ = 1.14 ppm (circles). Dots represent experimental values, and lines represent the fit of the binding model. | ||
However, the supramolecular systems are not always based only on zinc(II) Pcs. Other central metals can be utilized as well. The same method was therefore subsequently applied to the magnesium(II) complex of the AzaPc derivatives, specifically to compound 4Mg, which was chosen as the representative with potentially suitable KD value for determination of K1 with a larger number of ligands. Compound 4Mg efficiently formed J-dimers, similar to its zinc(II) counterpart (Fig. S12 and S13†). Using the binding models and methods described above, the dimerization constant was determined to be KD(4Mg) = 9.88 × 1010 M−1. This value is significantly higher than that observed for the corresponding zinc(II) complex (KD(4Zn) = 1.51 × 108 M−1), indicating a stronger dimerization tendency for the magnesium(II) complexes with smaller central cations. Following this, 4Mg was used in titrations with selected N-ligands to determine K1, which are listed in Table 3. When comparing these values with those of the zinc(II) analogues (also shown in Table 3), it was observed that the association constants for the magnesium(II) complexes were consistently lower. This trend suggests that while magnesium(II) complexes have a stronger tendency to form J-dimers (as indicated by higher KD values), their interaction with ligands, as measured by K1, is weaker compared to their zinc(II) counterparts. This difference might be attributed to the central metal ions’ different electronic properties and coordination environments, affecting their ability to interact with external ligands.
To further explore the effects of peripheral substituents on the association constant K1 and further extend the variability in the used compounds, 20Zn (Fig. 9 and Fig. S14†) was synthesized from precursor 19 and 5,6-bis(pentan-3-ylsulfanyl)pyrazine-2,3-dicarbonitrile. The presence of one N,N-dimethylamino substituent in 20Zn induced J-dimer formation in toluene, consistent with the behavior observed for previous AzaPcs. The bulky pentan-3-yl substituents were responsible for good solubility in organic solvents and tuning the dimerization constant as suggested above. The compound was also designed so that the electronic effects of alkylsulfanyl substituents differ significantly from those of dialkylamino groups, as indicated by Hammett substituent constants (data for the closest available substituent are σp = 0.07 and σp = −0.93 for –SCH(CH3)2 and –N(C3H7)2, respectively).40 The presence of just one (instead of eight) N,N-dialkylamino fragment (donor for ICT) also had a positive effect on the fluorescence intensity that reached values of ΦF = 0.044 (J-dimer in toluene) and ΦF = 0.23 (monomer in toluene with 0.12 M pyridine). By application of the above procedures, KD for 20Zn was determined to be 1.65 × 1010 M−1, relatively closely correlating with the KD = 3.09 × 1011 M−1 of compound 9Zn, which features the same N,N-dimethylamino binding moiety but with different rest of the molecule containing six N,N-dibutylamino fragments. The J-dimer of 20Zn in toluene was then disassembled by pyridine and NMI (Fig. 9), allowing the determination of K1 values from both UV-vis and fluorescence data. The binding constants obtained were K1(UV) = 6.05 × 104 M−1, K1(F) = 5.22 × 104 M−1 and K1(UV) = 9.73 × 106 M−1, K1(F) = 8.13 × 106 M−1 for pyridine and NMI, respectively. When compared with K1 values for octa(dialkylamino)ZnAzaPcs in Table 3, there was only a slight difference, despite significant variation in the electronic effects of the substituents. This finding suggests that peripheral substituents have a rather small influence on the ligand-binding strength of the central cation. Consequently, the K1 values obtained here can be considered broadly applicable, regardless of the specific peripheral substitution in the studied Pc/AzaPc molecules. However, for precise determination of K1 values for the target Pc/AzaPc molecules and ligands of interest in the future, the Pcs/AzaPcs analogues that form J-dimers should be designed structurally as close as possible to the original target molecule. Then, the temperature-induced J-dimer disassembly (KD) and titration of J-dimer solution with a ligand (KL) can be used as a proxy for the determination of direct ligand binding strength (K1) using UV-Vis or fluorescence spectroscopy methods.
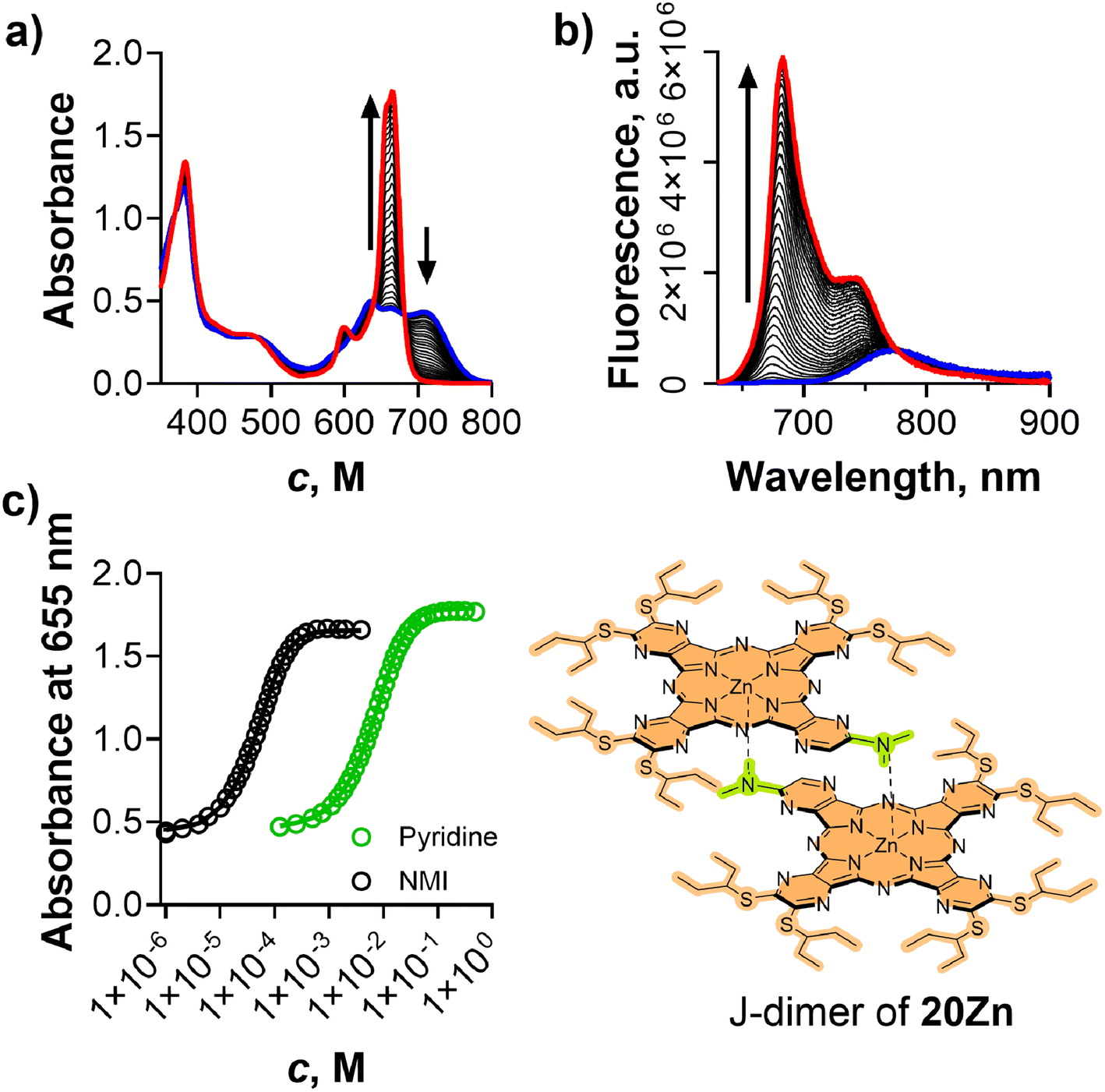 | ||
| Fig. 9 (a and b) Disassembly of the J-dimer of 20Zn in toluene (10 μM) by pyridine monitored by UV-vis (a) and fluorescence (b) spectra at 23 °C. (c) Disassembly of the J-dimer of 20Zn in toluene (10 μM) by pyridine (green) and NMI (black) monitored by absorbance of its monomeric form. | ||
Conclusion
In this study, we reported the formation of unique J-dimers within a series of alkylamino-substituted AzaPcs and quantified their dimerization constants (KD). Our findings confirmed that the spatial bulkiness of the AzaPc's peripheral substituent coordinating to the central cation plays a pivotal role in the stability of J-dimers, thereby influencing the corresponding KD values with further fine-tuning by the rest of the peripheral substituents. The J-dimers can be disassembled by various competing ligands that bind to the central cation. The association constants (K1) of these ligands were independent of the J-dimer stability, allowing the use of J-dimers as interchangeable probes for determining K1 values of ligands with varying binding strengths. Consequently, the disassembly of J-dimers offers a straightforward method for determining K1 for a wide range of ligands, ranging from heterocyclic to purely aliphatic ones, without the need for complex synthetic modifications, such as the addition of fluorophores or electron donors/acceptors. Among the ligands investigated in this work, piperidine emerges as a particularly effective candidate, exhibiting binding efficiency to zinc(II) AzaPc comparable to that of the widely used NMI. The significant spectral changes observed during the formation and disassembly of J-dimers, coupled with the consistently low variability in the K1 values determined above across the entire series, underscore the reliability and robustness of the method. It opens its potential for the reliable determination of association constants between the central cation of Pcs (and their analogues) and the various ligands. Understanding these association constants could facilitate the rational design of self-coordinating supramolecular systems currently employed in applications such as light harvesting, solar cell development, and electron transfer processes.Experimental section
General
All organic solvents were of analytical grade. Anhydrous butanol used for the cyclotetramerization was freshly distilled from magnesium before use. All other chemicals for the syntheses were purchased from certified suppliers (i.e., Sigma-Aldrich, TCI Europe, Acros, and Merck) and used as received. TLC was performed on Merck aluminum sheets coated with silica gel 60 F254. Merck Kieselgel 60 (0.040–0.063 mm) was used for column chromatography. The 1H and 13C NMR spectra were recorded on a Varian VNMR S500 (Agilent Technologies, Santa Clara, USA) spectrometer or a Jeol JNM-ECZ600R spectrometer (Jeol, Akishima, Japan). The chemical shifts are reported as δ values in ppm and are indirectly referenced to Si(CH3)4via the residual signal from the solvent. J values are given in Hz. IR spectra were measured on Nicolet 6700 (Thermo Scientific, USA). The UV-Vis spectra were recorded using a Shimadzu UV-2600 spectrophotometer (Shimadzu, Kyoto, Japan). Fluorescence spectra were recorded using an FS5 or FLS-1000 spectrofluorometer (Edinburgh Instruments, Edinburgh, UK). HRMS spectra were recorded on UHPLC system Acquity UPLC I-class (Waters, Milford, USA) coupled to the high-resolution mass spectrometer (HRMS) Synapt G2Si (Waters, Manchester, UK) based on Q-TOF. Chromatography involved an Acquity UPLC Protein BEH C4 (2.1 × 50 mm, 1.7 μm, 300 Å) column using gradient elution with ACN and 0.1% formic acid at a flow-rate of 0.4 ml min−1. Electrospray ionization was operated in the positive ion mode. The ESI spectra were recorded in the range of 50–5000 m/z using leucine-enkephalin as a lock mass reference and sodium iodide for external calibration or in the range of 50–1200 m/z using leucine-enkephalin as a lock mass reference and sodium formate for external calibration. The following compounds were prepared according to published procedures: AzaPcs 2H2,412Zn,415H2,175Zn,17 5-chloropyrazine-2,3-dicarbonitrile,36 and 5,6-bis(pentan-3-ylthio)pyrazine-2,3-dicarbonitrile.42Synthesis
:1, Rf = 0.61). After completion, the reaction was filtered and evaporated under reduced pressure. The product was purified by recrystallization from MeOH. Yield: 933 mg (86%), yellow crystals. 1H NMR (500 MHz, CDCl3) δ 3.02 (s, 12H). 13C NMR (126 MHz, CDCl3) δ 147.64, 120.45, 114.87, 38.82. HRMS (ESI): m/z calculated for C10H13N6 [M + H]+: 217.1196; found: 217.1206. IR (ATR) ν 2970 (CH), 2227 (CN), 1531, 1504, 1422, 1400, 1319, 1189, 1141 cm−1.
General procedure for synthesis of metal-free cyclotetramers
For symmetrical AzaPcs, the precursor (12–16, 1 equivalent) was dissolved in freshly distilled butanol (∼5 ml per 1 mmol of precursors), heated to reflux, and metal lithium (7 equivalents) was added to the boiling mixture. For unsymmetrical AzaPcs, precursor A (12, 17, 19, 1 equivalent) and compound 14 (3 equivalents) were dissolved in freshly distilled butanol (∼5 ml per 1 mmol of all precursors), heated to reflux, and metal lithium (28 equivalents) was added to the boiling mixture. The reaction was refluxed for 30 minutes in both cases, and then butanol was evaporated under reduced pressure. The solid residue was dissolved in a mixture of dichloromethane (∼50 mL per 1 mmol of precursor), water was added (∼25 mL per 1 mmol of precursor) and acidified with HCl (HCl was added in 1.2 equivalents per equivalent of lithium) before being transferred to a separation funnel. The mixture was extracted with dichloromethane (3 × 75 mL). The organic layers were collected, dried over anhydrous sodium sulfate, and evaporated. Purification was performed by column chromatography on silica (the mobile phases for each compound are mentioned below). Finally, the product was dissolved in a minimal amount of dichloromethane, added dropwise into methanol (100 mL), and precipitated in a freezer for 24 hours. The solid was collected and dried under vacuum.:1). Yield:140 mg (16%), a purple solid. 1H NMR (500 MHz, CDCl3/pyridine-d5 – 3:1) δ 3.40 (s, 48H), −1.46 (s, 2H). 13C NMR (126 MHz, CDCl3/pyridine-d5 – 3:1) δ 151.50, 39.51 (some aromatic signals were not detected); HRMS (ESI): m/z calculated for C40H51N24 [M + H]+: 867.4723; found: 867.4711.
:1, Rf = 0.67). Yield: 232 mg (58%), a purple solid. 1H NMR (500 MHz, CDCl3) δ 3.99 (t, J = 7.3 Hz, 32H), 1.79 (h, J = 7.3 Hz, 32H), 1.00 (t, J = 7.3 Hz, 48H), −1.35 (s, 2H). 13C NMR (126 MHz, CDCl3) δ 149.93, 147.17, 140.02, 50.70, 21.25, 11.90. HRMS (ESI): m/z calculated for C72H115N24 [M + H]+: 1315.9731; found: 1315.9723.
:1). Yield: 736 mg (42%), a purple solid. 1H NMR (500 MHz, pyridine-d5) δ 4.10 (t, J = 7.4 Hz, 32H), 1.84 (p, J = 7.5 Hz, 32H), 1.53 (h, J = 7.4 Hz, 32H), 1.02 (t, J = 7.4 Hz, 48H), −1.41 (s, 2H). 13C NMR (126 MHz, pyridine-d5) δ 150.59, 141.02, 48.93, 30.46, 21.04, 14.25 (one aromatic signal was not detected); HRMS (ESI): m/z calculated for C88H147N24 [M + H]+: 1540.2235; found: 1540.2224.
:1, Rf = 0.23). Yield: 131 mg (33%), a purple solid. 1H NMR (500 MHz, pyridine-d5) δ 4.11–3.54 (m, 32H), 1.89–1.69 (m, 32H), 1.69–1.51 (m, 16H). Due to solubility reasons, the signals in 13C NMR were not detected. HRMS could not be performed due to solubility reasons.
:1, Rf = 0.41). Yield: 76 mg (11%), a green solid. 1H NMR (500 MHz, CDCl3/pyridine-d5 – 2:1) δ 4.57–3.06 (m, 64H), −1.49 (s, 2H). Due to solubility reasons, the signals in 13C NMR were not detected. HRMS (ESI): m/z calculated for C56H67N24O8 [M + H]+: 1203.5568; found: 1203.5547.
:1). The second most lipophilic spot was isolated. Yield: 381 mg (22%), a purple solid. 1H NMR (500 MHz, pyridine-d5) δ 4.09 (q, J = 7.4 Hz, 16H), 3.97 (t, J = 7.4 Hz, 8H).3.28 (s, 12H), 1.88–1.79 (m, 16H), 1.78–1.68 (m, 8H), 1.60–1.48 (m, 16H), 1.48–1.38 (m, 8H), 1.06–0.93 (m, 36H), −1.40 (s, 2H). 13C NMR (126 MHz, pyridine-d5) δ 151.5, 150.7, 150.6, 141.3, 141.2, 141.0, 49.0, 48.9, 48.7, 39.5, 30.5, 30.3, 21.1, 21.1, 21.0, 14.3, 14.2 (some aromatic signals were not detected). HRMS (ESI): m/z calculated for C76H123N24 [M + H]+: 1372.0357; found: 1372.0344.
:1). Second most lipophilic spot was isolated. Yield: 220 mg (11%), a purple solid. 1H NMR (600 MHz, pyridine-d5) δ 8.98 (s, 1H), 4.15–4.08 (m, 16H), 4.00 (dt, J = 23.3, 7.4 Hz, 8H), 3.45 (s, 6H), 1.90–1.81 (m, 16H), 1.77–1.69 (m, 8H), 1.59–1.50 (m, 16H), 1.47–1.37 (m, 8H), 1.04 (q, J = 7.4 Hz, 24H), 0.96 (dt, J = 28.3, 7.4 Hz, 12H), −1.31 (s, 2H). 13C NMR (150 MHz, pyridine-d5) δ 156.15, 150.93, 150.88, 150.74, 142.88, 142.66, 139.53, 139.37, 138.98, 132.91, 48.86, 48.77, 48.45, 38.05, 30.32, 30.24, 30.12, 20.91, 20.86, 20.76, 14.10, 14.07, 13.99. HRMS (ESI): m/z calculated for C74H118N23 [M + H]+: 1328.9935; found: 1328.9902.
:1). Second most lipophilic spot was isolated. Yield: 326 mg (20%), a purple solid. 1H NMR (500 MHz, pyridine-d5) δ 4.09 (dt, J = 9.9, 7.3 Hz, 16H), 3.95 (t, J = 7.4 Hz, 8H), 3.62 (s, 6H), 3.49 (s, 4H), 1.89–1.78 (m, 16H), 1.74–1.65 (m, 8H), 1.58–1.47 (m, 16H), 1.47–1.37 (m, 8H), 1.02 (q, J = 7.1 Hz, 24H), 0.96 (t, J = 7.3 Hz, 12H), −1.44 (s, 2H). 13C NMR (126 MHz, pyridine-d5) δ 150.9, 150.5, 150.3, 146.6, 142.7, 142.6, 139.3, 138.4, 49.0, 48.9, 48.4, 46.7, 37.0, 30.5, 30.3, 21.1, 21.0, 14.3, 14.3, 14.2. HRMS (ESI): m/z calculated for C76H121N24 [M + H]+: 1370.0201; found: 1370.0172.
General procedure for the synthesis of a zinc(II) cyclotetramer
Metal-free cyclotetramer (1 equivalent) was dissolved in pyridine (8 mL per 0.1 mmol of the metal-free compound), and zinc(II) acetate (10 equivalents) was added. The reaction mixture was refluxed for 3 hours, then pyridine was evaporated under reduced pressure. The crude residue was dissolved in dichloromethane (50 mL) and washed with distilled water (3 × 50 mL). The organic phase was separated, dried over anhydrous sodium sulfate, filtered, and evaporated under reduced pressure. Column chromatography on silica was performed to purify the products (mobile phases for each AzaPc are written in the following paragraphs). Finally, the product was dissolved in a minimal amount of dichloromethane, added dropwise into methanol (100 mL), and precipitated in a freezer for 24 hours. The solid was collected and dried under vacuum.:1 → 10:1). Yield: 28 mg (26%), a blue solid. 1H NMR (600 MHz, pyridine-d5) δ 3.21 (s, 48H). 13C NMR (150 MHz, pyridine-d5) δ 165.43, 151.23, 150.92, 142.76, 134.10, 129.88, 39.31 (one aromatic signal was not detected or overlapped with solvent signal). HRMS could not be performed due to solubility reasons.
:1). Yield: 98 mg (42%), a blue solid. 1H NMR (600 MHz, pyridine-d5) δ 3.89 (d, J = 7.8 Hz, 32H), 1.76–1.64 (m, 32H), 0.95 (t, J = 7.4 Hz, 48H). 13C NMR (150 MHz, pyridine-d5) δ 151.13, 142.45, 50.86, 21.30, 11.98 (one aromatic signal was not detected or overlapped with solvent signal). HRMS (ESI): m/z calculated for C72H113N24Zn [M + H]+: 1377.8866; found: 1377.8844.
:1 → 10:1). Yield: 566 mg (97%), a blue solid. 1H NMR (500 MHz, pyridine-d5) δ 4.07 (t, J = 7.5 Hz, 32H), 1.80 (q, J = 3.6 Hz, 32H), 1.51 (h, J = 7.4 Hz, 32H), 0.99 (t, J = 7.4 Hz, 48H). 13C NMR (126 MHz, pyridine-d5) δ 151.04, 142.43, 48.77, 30.24, 20.85, 14.05 (one aromatic signal was not detected or overlapped with signal of solvent); HRMS (ESI): m/z calculated for C88H145N24Zn [M + H]+: 1602.1370; found: 1602.1343.
:1). Yield: 21 mg (14%), a dark green solid. NMR spectra and HRMS could not be performed for reasons of low solubility.
:1). Yield: 40 mg (50%), a dark blue solid. NMR spectra and HRMS could not be performed for reasons of low solubility.
:1). Yield: 220 mg (74%), a dark blue solid. 1H NMR (500 MHz, pyridine-d5) δ 4.14–4.05 (m, 16H), 3.97 (t, J = 7.4 Hz, 8H), 3.27 (s, 12H), 1.88–1.76 (m, 16H), 1.75–1.65 (m, 8H), 1.58–1.48 (m, 16H), 1.48–1.39 (m, 8H), 1.05–0.94 (m, 36H), 13C NMR (126 MHz, pyridine-d5) δ 151.2, 151.2, 151.1, 150.9, 150.8, 142.6, 142.5, 142.5, 48.8, 48.8, 48.5, 39.2, 30.3, 30.3, 30.1, 20.9, 20.8, 20.8, 14.1, 14.1, 14.0 (some aromatic signals were not detected). HRMS (ESI): m/z calculated for C76H121N24Zn [M + H]+: 1433.9492; found: 1433.9454.
:1). Yield: 95 mg (45%), a dark blue solid. 1H NMR (600 MHz, pyridine-d5) δ 8.97 (s, 1H), 4.13–4.07 (m, 16H), 3.99 (dt, J = 20.8, 7.4 Hz, 8H), 3.43 (s, 6H), 1.87–1.80 (m, 16H), 1.74–1.67 (m, 8H), 1.58–1.49 (m, 16H), 1.46–1.36 (m, 8H), 1.04–0.98 (m, 24H), 0.96 (t, J = 7.4 Hz, 6H), 0.91 (t, J = 7.4 Hz, 6H). 13C NMR (150 MHz, pyridine-d5) δ 155.9, 152.1, 151.5, 150.8, 150.5, 142.7, 142.5, 142.4, 142.4, 139.5, 135.0, 132.7, 48.9, 48.8, 48.4, 48.4, 38.0, 30.3, 30.2, 30.1, 20.9, 20.8, 14.1, 14.0 (some aromatic signals were not detected). HRMS (ESI): m/z calculated for C74H116N23Zn [M + H]+: 1390.9070; found: 1390.9034.
:1). Yield: 280 mg (96%), a dark blue solid. 1H NMR (500 MHz, pyridine-d5) δ 4.09 (p, J = 6.5 Hz, 16H), 3.96 (t, J = 7.4 Hz, 8H), 3.60 (s, 6H), 3.45 (s, 4H), 1.87–1.77 (m, 16H), 1.68 (p, J = 6.6 Hz, 8H), 1.59–1.47 (m, 16H), 1.41 (h, J = 7.4 Hz, 8H), 1.00 (q, J = 7.3 Hz, 24H), 0.94 (t, J = 7.4 Hz, 12H). 13C NMR (126 MHz, pyridine-d5) δ 152.3, 151.3, 150.9, 150.8, 149.7, 145.8, 142.7, 142.6, 142.6, 141.2, 49.0, 49.0, 48.5, 46.9, 37.0, 30.5, 30.2, 21.1, 21.1, 21.0, 20.5, 20.3, 20.1, 14.3, 14.2. HRMS (ESI): m/z calculated for C76H119N24Zn [M + H]+: 1431.9336; found: 1431.9302.
:1:1, 30 ml) and stirred for 30 min at r.t. Followed by extraction to dichloromethane (3 × 30 ml). Organic phases were collected and washed with 5% sodium bicarbonate solution (50 ml). The organic phase was collected and dried over anhydrous sodium sulfate, filtered, and evaporated. The final product was purified by column chromatography on silica (eluent: dichloromethane/methanol (400:1)). Yield: 82 mg (40%), a dark blue solid. 1H NMR (600 MHz, pyridine-d5) δ 4.08 (t, J = 7.5 Hz, 32H), 1.81 (p, J = 7.4 Hz, 32H), 1.52 (h, J = 7.4 Hz, 32H), 1.00 (t, J = 7.4 Hz, 48H). 13C NMR (151 MHz, pyridine-d5) δ 150.7, 142.8, 48.8, 30.3, 20.9, 14.1 (one aromatic signal was not detected due to overlap with residual solvent signal). HRMS (ESI): m/z calculated for C88H145MgN24 [M + H]+: 1562.1929; found: 1562.1902.
:1, Rf = 0.40). Finally, the product was scratched from flask walls to methanol, filtered, dried and washed with hexane on filter paper. Yield: 131 mg (11%), a dark green solid. 1H NMR (600 MHz, pyridine-d5) δ 8.94 (s, 1H), 5.07–4.97 (m, 4H), 4.85–4.82 (m, 1H), 4.66 (p, J = 6.4 Hz, 1H), 3.42 (s, 6H), 2.27–2.21 (m, 16H), 2.17–2.12 (m, 2H), 2.09–1.99 (m, 6H), 1.40 (m, 24H), 1.32 (t, J = 7.4 Hz, 6H), 1.20 (t, J = 7.3 Hz, 6H), −1.70 (s, 2H). 13C NMR (151 MHz, pyridine-d5) δ 159.78, 159.61, 158.96, 158.82, 158.62, 158.52, 156.10, 145.28, 145.21, 144.73, 144.52, 142.58, 142.52, 142.36, 50.18, 50.07, 49.91, 49.82, 49.70, 37.97, 27.59, 27.54, 27.43, 27.32, 11.87, 11.75, 11.45. HRMS (ESI): m/z calculated for C56H76N17S6 [M + H]+: 1178.4788; found: 1178.4783.
:1, Rf = 0.26). Finally, the product was scratched from flask walls to methanol, filtered, dried and washed with hexane on filter paper. Yield: 15 mg (28%), a dark green solid. 1H NMR (600 MHz, CDCl3/pyridine-d5 – 3:1) δ 9.00 (s, 1H), 5.08 (p, J = 6.5 Hz, 1H), 5.05–4.97 (m, 4H), 4.89 (p, J = 6.3 Hz, 1H), 3.74 (s, 6H), 2.34–2.20 (m, 24H), 1.51–1.48 (m, 6H), 1.48–1.43 (m, 24H), 1.41–1.38 (m, 6H). 13C NMR (151 MHz, CDCl3/pyridine-d5 – 3:1) δ 158.16, 157.98, 157.83, 155.81, 153.09, 151.76, 150.97, 150.85, 150.52, 145.22, 145.11, 144.90, 144.73, 132.61, 49.76, 49.35, 49.15, 38.30, 27.16, 11.57, 11.28. HRMS (ESI): m/z calculated for C56H74N17S6Zn [M + H]+: 1240.3923; found: 1240.3903.
Determination of the quantum yield of singlet oxygen
Quantum yields of singlet oxygen production (ΦΔ) were determined by the comparative method based on the decomposition of a chemical trap for singlet oxygen (1,3-diphenylisobenzofuran) and using unsubstituted zinc(II) phthalocyanine as a reference compound (ΦΔ = 0.56 (DMF),43 0.58 (toluene)44). Details of the method are described elsewhere.45 All the determinations were performed in triplicate, and the data represent the mean of the measurements. The estimated experimental error was ±10%. In the case of the toluene/pyridine mixture, 1% (v/v) pyridine solution in toluene was used, which corresponds to 0.12 M pyridine in toluene (unsubstituted zinc(II) phthalocyanine in toluene was used as the reference), unless stated otherwise.Determination of the quantum yield of fluorescence
Fluorescence quantum yields (ΦF) were determined on an FLS 1000 spectrofluorometer (Edinburg Instruments) by the comparative method46 using unsubstituted zinc(II) phthalocyanine as a reference compound (ΦF = 0.32 (THF) (ref. 47)). The determination of ΦF values was performed in triplicate, and the data represent the mean of these measurements. The estimated experimental error was ±10%. Absorption of the samples at the excitation wavelength was kept below 0.05 and at a Q band maximum below 0.1 to avoid the inner filter effect. The results of ΦF were corrected for the refractive indices of the solvents. In the case of the toluene/pyridine mixture, 1% (v/v) pyridine solution in toluene was used, which corresponds to 0.12 M pyridine in toluene, unless stated otherwise.Determination of KDvia temperature
All synthesized compounds were subjected to a fluorescence temperature dependency experiment in toluene to determine KD. The experiments were performed at different concentrations (ranging between 5.0–0.05 μM) in dependence on the stability of the J-dimer. All experiments are presented in Fig. S5 and S6.† The excitation wavelength was always chosen in an isosbestic point close to 600 nm. The temperature was gradually changed from 10 °C to 100 °C in 5 °C steps with 5 min intervals between each measurement to allow equilibration. For compounds 4Zn and 5Zn, the spectral changes were also monitored in absorption spectra at 10 μM concertation.Determination of KL for pyridine in series with different peripheral substituents
Stock solutions of AzaPcs were prepared in toluene (100 μM for 2–5Zn, 8–10Zn, 4Mg, and 20Zn; 2 μM for 1Zn, and 25 μM for 6Zn). The stock solution was added to the cuvette with toluene to reach the desired concentration (see below), and then pyridine was added stepwise. During titration, changes in Q-band absorption (increasing band of the monomer) were observed, and titration was stopped when the plateau phase was reached. Absorption and emission (excitation at isosbestic point close to 600 nm) spectra were measured after each addition of pyridine. Concentration in the cuvette was 15 μM for 3Zn; 10 μM for AzaPcs 2Zn, 4Zn, 5Zn, 8–10Zn, 4Mg, and 20Zn; 2 μM for 1Zn and 6Zn. Changes in the Q-band absorption maxima of the monomer were used to determine KL. In emission spectra, the decrease in dimer emission and increase in monomer emission were used to determine KL. All experiments were performed at 23 °C.Determination of KL for a series of different ligands
Stock solutions of selected AzaPcs were prepared in toluene (100 μM). The stock solution was added to the cuvette with toluene to reach a concentration of 10 μM, and then the ligand was added stepwise. During titration, changes in Q-band absorption (increasing band of the monomer) were observed, and titration was stopped when the plateau phase was reached. Absorption and emission (excitation at the isosbestic point close to 600 nm) spectra were measured after each ligand addition. Changes in the Q-band absorption maxima of monomer were used to determine KL. In emission spectra, the decrease in dimer emission and increase in monomer emission were used to determine KL. All experiments were performed at 23 °C.NMR titration of 3Zn
A solution of 3Zn in toluene-d8 (1 mM, 600 μL) was taken in the NMR cuvette, and 1H spectrum was measured. Then 3 μL of 40 mM solution N-methylimidazole (in toluene-d8) was added into the cuvette, gently mixed, and the 1H spectrum was measured again. The previous step was repeated until no changes in the 1H spectrum were observed. Finally, 3 μL of 1 M N-methylimidazole was added to ensure that compound 3Zn was in the monomeric form. KL was determined by fitting the binding model to the integrated intensity of resonances at δ = 0.64 ppm and δ = 1.14 ppm, which belong to the J-dimer. The experiments were performed at 25 °C.Author contributions
Jiri Demuth: investigation, data curation, formal analysis, writing – original draft, methodology, and supervision. Stefan Bednarik: investigation. Radek Machan: investigation. Ivan Mocak: investigation. Tibor Malinsky: investigation. Mona Abo El Dahabova: investigation. Jakub Holcak: investigation. Miroslav Miletin: supervision. Jan Labuta: methodology and formal analysis. Veronika Novakova: supervision, visualization, writing – review and editing, and funding acquisition. Petr Zimcik: conceptualization, funding acquisition, methodology, project administration, and writing – review and editing,Data availability
Data for this article, including NMR data and absorption and fluorescence spectra are available at Zenodo at: https://doi.org/10.5281/zenodo.14047683.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the Czech Science Foundation (23-06177S) and the project New Technologies for Translational Research in Pharmaceutical Sciences /NETPHARM, project ID CZ.02.01.01/00/22_008/0004607, co-funded by the European Union. VN, MM and JD would also like to acknowledge financial support from the Ministry of Education, Youth and Sports (ERC CZ programme, LL2318). This work was also supported by the World Premier International Research Center Initiative (WPI Initiative), MEXT, Japan. Financial support from Charles University (SVV 260 666) is gratefully acknowledged as well.References
- R. Christie and A. Abel, Phthalocyanine pigments: general principles, Phys. Sci. Rev., 2021, 6, 671–677 Search PubMed.
- M. Yahya, Y. Nural and Z. Seferoglu, Recent advances in the nonlinear optical (NLO) properties of phthalocyanines: A review, Dyes Pigm., 2022, 198, 109960 CrossRef CAS.
- M. Urbani, M. E. Ragoussi, M. K. Nazeeruddin and T. Torres, Phthalocyanines for dye-sensitized solar cells, Coord. Chem. Rev., 2019, 381, 1–64 CrossRef CAS.
- M. Macháček, K. A. Carter, F. Kostelanský, D. Miranda, A. Seffouh, J. Ortega, T. Šimůnek, P. Zimčík and J. F. Lovell, Binding of an amphiphilic phthalocyanine to pre-formed liposomes confers light-triggered cargo release, J. Mater. Chem. B, 2018, 6, 7298–7305 RSC.
- M. P. Donzello, C. Ercolani, V. Novakova, P. Zimcik and P. A. Stuzhin, Tetrapyrazinoporphyrazines and their metal derivatives. Part I: Synthesis and basic structural information, Coord. Chem. Rev., 2016, 309, 107–179 CrossRef CAS.
- L. Lochman, M. Machacek, M. Miletin, S. Uhlirova, K. Lang, K. Kirakci, P. Zimcik and V. Novakova, Red-Emitting Fluorescence Sensors for Metal Cations: The Role of Counteranions and Sensing of SCN- in Biological Materials, ACS Sens., 2019, 4, 1552–1559 CrossRef CAS.
- M. Karlikova, V. Cermakova, J. Demuth, V. Valer, M. Miletin, V. Novakova and P. Zimcik, Magnesium tetrapyrazinoporphyrazines: tuning of the pKa of red-fluorescent pH indicators, Dalton Trans., 2019, 48, 6162–6173 RSC.
- B. Ghazal, M. Machacek, M. A. Shalaby, V. Novakova, P. Zimcik and S. Makhseed, Phthalocyanines and Tetrapyrazinoporphyrazines with Two Cationic Donuts: High Photodynamic Activity as a Result of Rigid Spatial Arrangement of Peripheral Substituents, J. Med. Chem., 2017, 60, 6060–6076 CrossRef CAS PubMed.
- J. Demuth, R. Kucera, K. Kopecky, Z. Havlinova, A. Libra, V. Novakova, M. Miletin and P. Zimcik, Efficient synthesis of a wide-range absorbing azaphthalocyanine dark quencher and its application to dual-labeled oligonucleotide probes for quantitative real-time polymerase chain reactions, Chem. – Eur. J., 2018, 24, 9658–9666 CrossRef CAS.
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, J-Aggregates: From Serendipitous Discovery to Supramolecular Engineering of Functional Dye Materials, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef PubMed.
- K. Kameyama, M. Morisue, A. Satake and Y. Kobuke, Highly fluorescent self-coordinated phthalocyanine dimers, Angew. Chem., Int. Ed., 2005, 44, 4763–4766 CrossRef CAS.
- M. Garcia-Iglesias, K. Peuntinger, A. Kahnt, J. Krausmann, P. Vazquez, D. Gonzalez-Rodriguez, D. M. Guldi and T. Torres, Supramolecular assembly of multicomponent photoactive systems via cooperatively coupled equilibria, J. Am. Chem. Soc., 2013, 135, 19311–19318 CrossRef CAS PubMed.
- F. Bächle, C. Maichle-Mössmer and T. Ziegler, Helical Self-Assembly of Optically Active Glycoconjugated Phthalocyanine J-Aggregates, ChemPlusChem, 2019, 84, 1081–1093 CrossRef.
- A. Y. Tolbin, V. E. Pushkarev, L. G. Tomilova and N. S. Zefirov, Monohydroxyphthalocyanines as potential precursors to create nanoscale optical materials, J. Porphyrins Phthalocyanines, 2017, 21, 128–134 CrossRef CAS.
- A. Tolbin, V. Pushkarev, M. Sedova, S. Maklakov and L. Tomilova, Aggregation of slipped-cofacial, phthalocyanine J-type dimers: Spectroscopic and AFM study, Spectrochim. Acta, Part A, 2018, 205, 335–340 CrossRef CAS PubMed.
- V. Novakova, P. Zimcik, K. Kopecky, M. Miletin, J. Kunes and K. Lang, Self-assembled azaphthalocyanine dimers with higher fluorescence and singlet oxygen quantum yields than the corresponding monomers, Eur. J. Org. Chem., 2008, 3260–3263 CrossRef CAS.
- J. Demuth, M. Miletin, R. Kucera, A. Ruzicka, Z. Havlinova, A. Libra, V. Novakova and P. Zimcik, Self-assembly of azaphthalocyanine-oligodeoxynucleotide conjugates into J-dimers: towards biomolecular logic gates, Org. Chem. Front., 2020, 7, 445–456 RSC.
- J. Demuth, M. Miletin, M. Machan, M. Kantor, P. Zimcik and V. Novakova, Synthesis and J-Dimer Formation of Tetrapyrazinoporphyrazines with Different Functional Groups for Potential Biomolecular Probe Applications, ChemPlusChem, 2020, 85, 527–537 CrossRef CAS.
- J. Sessler, J. Jayawickramarajah, A. Gouloumis, G. Pantos, T. Torres and D. Guldi, Guanosine and fullerene derived de-aggregation of a new phthalocyanine-linked cytidine derivative, Tetrahedron, 2006, 62, 2123–2131 CrossRef CAS.
- M. A. Lebedeva, T. W. Chamberlain and A. N. Khobystov, Harnessing the Synergistic and Complementary Properties of Fullerene and Transition-Metal Compounds for Nanomaterial Applications, Chem. Rev., 2015, 115, 11301–11351 CrossRef CAS PubMed.
- V. Bandi, M. E. El-Khouly, V. N. Nesterov, P. A. Karr, S. Fukuzumi and F. D'Souza, Self-Assembled via Metal–Ligand Coordination AzaBODIPY–Zinc Phthalocyanine and AzaBODIPY–Zinc Naphthalocyanine Conjugates: Synthesis, Structure, and Photoinduced Electron Transfer, J. Phys. Chem. C, 2013, 117, 5638–5649 CrossRef CAS.
- H. Xu and D. K. P. Ng, Construction of subphthalocyanine-porphyrin and subphthalocyanine-phthalocyanine heterodyads through axial coordination, Inorg. Chem., 2008, 47, 7921–7927 CrossRef CAS.
- M. El-Khouly and S. Fukuzumi, Light harvesting a gold porphyrin-zinc phthalocyanine supramolecular donor-acceptor dyad, Photochem. Photobiol. Sci., 2016, 15, 1340–1346 CrossRef CAS PubMed.
- B. Platzer, B. Berna, M. Bischetti, D. Cicero, R. Paolesse, S. Nardis, T. Torres and D. Guldi, Exploring the Association of Electron-Donating Corroles with Phthalocyanines as Electron Acceptors, Chem. – Eur. J., 2022, 28, e202103891 CrossRef CAS PubMed.
- N. Bichan, E. Ovchenkova, A. Tsaturyan and T. Lomova, Spectral properties of supramolecular systems based on cobalt(II)/manganese(III) phthalocyanine and fullero[60]pyrrolidines with PET, New J. Chem., 2020, 44, 11262–11270 RSC.
- B. Berna, B. Platzer, M. Wolf, G. Lavarda, S. Nardis, P. Galloni, T. Torres, D. Guldi and R. Paolesse, Panchromatic Light Harvesting and Stabilizing Charge-Separated States in Corrole-Phthalocyanine Conjugates through Coordinating a Subphthalocyanine, Chem. – Eur. J., 2020, 26, 13451–13461 CrossRef CAS PubMed.
- S. Seetharaman, J. Follana-Berná, L. Martín-Gomis, G. Charalambidis, A. Trapali, P. Karr, A. Coutsolelos, F. Fernández-Lázaro, A. Sastre-Santos and F. D'Souza, Sequential, Ultrafast Energy Transfer and Electron Transfer in a Fused Zinc Phthalocyanine-free-base Porphyrin-C60 Supramolecular Triad, ChemPhysChem, 2019, 20, 163–172 CrossRef CAS.
- X. W. Wang, S. S. Nurttila, W. I. Dzik, R. Becker, J. Rodgers and J. N. H. Reek, Tuning the Porphyrin Building Block in Self-Assembled Cages for Branched-Selective Hydroformylation of Propene, Chem. – Eur. J., 2017, 23, 14769–14777 CrossRef CAS PubMed.
- S. Shao, H. Gobeze, I. De Silva, J. Schaffner, G. Verbeck, P. Karr and F. D'Souza, Photoinduced Energy and Electron Transfer in a ‘Two-Point’ Bound Panchromatic, Near-Infrared-Absorbing Bis-styrylBODIPY(Zinc Porphyrin)2 - Fullerene Self-Assembled Supramolecular Conjugate, Chem. – Eur. J., 2024, 30, e202401892 CrossRef CAS.
- B. Przybyl and J. Janczak, 4 + 1 Complexes of zinc phthalocyanine with pyridine derivative ligands, Dyes Pigm., 2015, 118, 102–109 CrossRef CAS.
- B. Przybyl and J. Janczak, Complexes of zinc phthalocyanine with monoaxially coordinated irnidazole-derivative ligands, Dyes Pigm., 2016, 130, 54–62 CrossRef CAS.
- M. E. El-Khouly, L. M. Rogers, M. E. Zandler, G. Suresh, M. Fujitsuka, O. Ito and F. D'Souza, Studies on intra-supramolecular and intermolecular electron-transfer processes between zinc naphthalocyanine and imidazole-appended fullerene, ChemPhysChem, 2003, 4, 474–481 CrossRef CAS.
- J. Kollar, M. Machacek, M. Halaskova, J. Lenco, R. Kucera, J. Demuth, M. Rohlickova, K. Hasonova, M. Miletin, V. Novakova and P. Zimcik, Cationic Versus Anionic Phthalocyanines for Photodynamic Therapy: What a Difference the Charge Makes, J. Med. Chem., 2020, 63, 7616–7632 CrossRef CAS.
- Y. X. Chen, W. Cao, K. Wang and J. Z. Jiang, Unprecedented Phthalocyanines Bearing Eight Di-butylamino Peripheral Substituents: Synthesis, Spectroscopy, and Structure, Inorg. Chem., 2015, 54, 9962–9967 CrossRef CAS PubMed.
- K. Kopecky, V. Novakova, M. Miletin, L. Plistilova, P. Berka and P. Zimcik, Synthesis and properties of azaphthalocyanines with 1,2,3,4-tetrahydropyrazino[2,3-b]pyrazine moiety, Macroheterocycles, 2011, 4, 171–176 CrossRef.
- A. Nakamura, T. Ataka, H. Segawa, Y. Takeuchi and T. Takematsu, Studies on Herbicidal 2,3-Dicyanopyrazines .1. Structure-Activity Relationship of Herbicidal 2,3-Dicyano-5-Substituted Pyrazines, Agric. Biol. Chem., 1983, 47, 1555–1560 CAS.
- V. Novakova, P. Zimcik, M. Miletin, L. Vachova, K. Kopecky, K. Lang, P. Chábera and T. Polívka, Ultrafast Intramolecular charge transfer in tetrapyrazinoporphyrazines controls the quantum yields of fluorescence and singlet oxygen, Phys. Chem. Chem. Phys., 2010, 12, 2555–2563 RSC.
- M. T. M. Choi, P. P. S. Li and D. K. P. Ng, A direct comparison of the aggregation behavior of phthalocyanines and 2,3-naphthalocyanines, Tetrahedron, 2000, 56, 3881–3887 CrossRef CAS.
- M. Kostka, P. Zimcik, M. Miletin, P. Klemera, K. Kopecky and Z. Musil, Comparison of aggregation properties and photodynamic activity of phthalocyanines and azaphthalocyanines, J. Photochem. Photobiol., A, 2006, 178, 16–25 CrossRef CAS.
- C. Hansch, A. Leo and R. W. Taft, A survey of Hammett substituent constants and resonance and field parameters, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- P. Petrik, P. Zimcik, K. Kopecky, Z. Musil, M. Miletin and V. Loukotova, Protonation and deprotonation of nitrogens in tetrapyrazinoporphyrazine macrocycles, J. Porphyrins Phthalocyanines, 2007, 11, 487–495 CrossRef CAS.
- S. Bednarik, J. Demuth, J. Kernal, M. Miletin, P. Zimcik and V. Novakova, Tuning Electron-Accepting Properties of Phthalocyanines for Charge Transfer Processes, Inorg. Chem., 2024, 63, 8799–8806 CrossRef CAS.
- U. Michelsen, H. Kliesch, G. Schnurpfeil, A. K. Sobbi and D. Wohrle, Unsymmetrically substituted benzonaphthoporphyrazines: A new class of cationic photosensitizers for the photodynamic therapy of cancer, Photochem. Photobiol., 1996, 64, 694–701 CrossRef CAS.
- A. Ogunsipe, D. Maree and T. Nyokong, Solvent effects on the photochemical and fluorescence properties of zinc phthalocyanine derivatives, J. Mol. Struct., 2003, 650, 131–140 CrossRef CAS.
- V. Novakova, M. Miletin, T. Filandrová, J. Lenčo, A. Růžička and P. Zimcik, Role of Steric Hindrance in the Newman-Kwart Rearrangement and in the Synthesis and Photophysical Properties of Arylsulfanyl Tetrapyrazinoporphyrazines, J. Org. Chem., 2014, 79, 2082–2093 CrossRef CAS PubMed.
- V. Novakova, P. Zimcik, K. Kopecky, M. Miletin, J. Kuneš and K. Lang, Self-Assembled Azaphthalocyanine Dimers with Higher Fluorescence and Singlet Oxygen Quantum Yields than the Corresponding Monomers, Eur. J. Org. Chem., 2008, 3260–3263 CrossRef CAS.
- P. Zimcik, V. Novakova, K. Kopecky, M. Miletin, R. Z. U. Kobak, E. Svandrlikova, L. Váchová and K. Lang, Magnesium Azaphthalocyanines: An Emerging Family of Excellent Red-Emitting Fluorophores, Inorg. Chem., 2012, 51, 4215–4223 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, a binding model, absorption and emission spectra, and a table with literature data for association constants. See DOI: https://doi.org/10.1039/d4qi02834a |
| This journal is © the Partner Organisations 2025 |