 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organosolv processing of Sitka spruce sawdust: large scale preparation of native-like lignin and ligninOX for valorisation†
Daniel J. Davidson‡
 a,
Geraud N. Sansom‡a,
Daniel M. Miles-Barretta,
David B. Cordesa,
Alexandra M. Z. Slawina,
James R. D. Montgomerya,
Tomas Lebla,
Ann Connorb,
Andrew M. Danbyc,
Mark J. Gronnowc,
Neil J. Parry*b,
Sarah L. Hosking*b,
David S. Grainger*b and
Nicholas J. Westwood*a
a,
Geraud N. Sansom‡a,
Daniel M. Miles-Barretta,
David B. Cordesa,
Alexandra M. Z. Slawina,
James R. D. Montgomerya,
Tomas Lebla,
Ann Connorb,
Andrew M. Danbyc,
Mark J. Gronnowc,
Neil J. Parry*b,
Sarah L. Hosking*b,
David S. Grainger*b and
Nicholas J. Westwood*a
aSchool of Chemistry and Biomedical Sciences Research Complex, University of St. Andrews and EaStCHEM, St Andrews, Fife, Scotland KY16 9ST, UK. E-mail: njw3@st-andrews.ac.uk
bUnilever Research and Development, Port Sunlight Laboratory, Quarry Road East, Bebington, Wirral, UK
cBiorenewables Development Centre, University of York, Unit 1 Hassacarr Close, Chessingham Park, Dunnington, York, YO19 5SN, UK
First published on 13th June 2025
Abstract
Sitka Spruce (SiS) dominates wood production in Scotland and represents an important source of wood in the UK. A systematic analysis of the lignin obtained from SiS sawdust using methano-, ethano-, butano- and isobutano-solv pretreatments was carried out. Detailed analysis of the resulting lignin using a range of methods (GPC, 31P after phosphitylation and HSQC NMR) and assessment of solvent costs enabled a comparison of the 4 pretreatment methods. The high quality of the lignin obtained reflects its stabilisation through alcohol incorporation at the α-position of the β-O-4 units. Scale up of the butanosolv pretreatment led to the controlled synthesis of a selectively oxidised form of the lignin (SiS ligninOX) on a relatively large scale. Additional insights into the detailed structure of ligninOX are presented. It is argued that this interesting, modified biopolymer may have significant potential for enhanced lignin valorisation.
Introduction
The lignin challenge
As one of the most abundant materials worldwide,1 lignin has long been in the spotlight as a potential source of feedstock chemicals and other useful products.2–10 The view that lignin should be of economic importance has inspired creative and exciting valorisation strategies. However, lignin’s heterogeneity, tendency to be modified during extraction, overall structural complexity and our lack of knowledge in terms of its physical–chemical properties all raise the level of the challenge. These factors (and others) provide a significant barrier to lignin use. An alternative view is that lignin’s intransigence makes the overall challenge of figuring out what to do with it more worthwhile, scientifically challenging and interesting.There remains a significant component of the lignin community that is interested in isolating a high quality, usable lignin from plant biomass (we estimated that there are approaching 100 publications in which the characterisation or use of isolated lignin is reported in the first 4 months of 2025 alone; for some examples see ref. 11–20). One argument in support of this approach is that it increases the flexibility inherent in future lignin use. For example, one portion of a batch of isolated lignin could be used in materials applications (through modification of the intact lignin polymer) whilst another portion of the same batch could be depolymerised to give relatively simple feedstock chemicals, such as 4-propylguaiacol. These biomass-derived compounds could then be used to prepare biologically active chemicals (including natural products), surfactants etc.21–25 Elegant approaches (typically referred to as lignin-first approaches) focus on an initial lignin depolymerisation step when dealing with plant biomass and are in the ascendancy in terms of the current thinking on lignin use.26–35
Sitka spruce-derived lignin
Here, we initially focus on isolating a high-quality lignin using an organosolv pretreatment. One key aspect of the report is that it focuses on the use of sawdust from Sitka spruce (Picea sitchensis), a highly abundant and commercially important softwood in the UK and worldwide. It was estimated that there are approximately 1 million tonnes of forestry residues available in the UK per annum, comprising roughly softwood and hardwood in a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio.36 These quantities exclude primary timber which is extracted for construction and paper and board manufacture. Approximately 25% of the 1 million tonnes of forestry residues corresponds to the brash and stumps. These components are retained in the forest as they deliver ecosystem services value and are difficult to extract. The remaining 750 kilotonnes is available but underexploited.36,37
:1 ratio.36 These quantities exclude primary timber which is extracted for construction and paper and board manufacture. Approximately 25% of the 1 million tonnes of forestry residues corresponds to the brash and stumps. These components are retained in the forest as they deliver ecosystem services value and are difficult to extract. The remaining 750 kilotonnes is available but underexploited.36,37
A large proportion of the softwood grown in the UK is Sitka spruce (SiS).38 To date, there are surprisingly few studies on the isolation, analysis and potential use of Sitka spruce-derived lignin. In an impressive report, Jarvis et al.39 described the use of an ethanosolv pretreatment to extract the lignin from SiS but the main focus of their work was not on the lignin but on the saccharide components of the biomass. Sitka spruce-derived lignins sit at the centre of our report and aspects of their isolation, stabilisation and modification (potentially en route to value-added products) are discussed in detail.
Softwood pretreatments
There are a wide range of options available to those interested in isolating lignin from plant biomass. The success (or not) of these methods clearly depends on the solubility of the lignin in the solvent used and on the impact that any lignin modification has on its overall solubility.40 There is also the concern that any lignin modification that occurs during extraction is counter-productive, in other words, it is equivalent to lignin degradation. One class of pretreatment technology involves the use of organic solvents and these methods are therefore known as organosolv pretreatments.Organosolv pretreatments are well studied for the extraction and isolation of lignin from softwoods as they afford lignin that has a near-native structure. Many solvents have been reported for organosolv preatments,41–51 including (but certainly not limited to) acetic acid,52–54 acetone,55–57 tetrahydrofuran,58–60 2-methyltetrahydrofuran (MeTHF),61,62 1,4-dioxane,63–65 and gamma-valerolactone (GVL).66–68 These pretreatments can give lignins in relatively high yields with significant retention of the important β-O-4 linkage (to enable subsequent processing). One of the major drawbacks of these methods that limits scaling up and use in industry is the inherent acid sensitivity of the native-like lignin structure. In addition, several of these solvents have toxicity issues or have a high boiling point that makes lignin purification more challenging. Alkosolv pretreatments, using alcohols as solvents, are a sub-section of organosolv pretreatments that resolve many of these issues.64,69–71 As a result, the use of most of the low molecular weight alcohols in lignin extraction has been reported (methanol,31,72–75 ethanol,76–81 butanol,62,82–90 and sec- and tert-butanols and propanols91–93). In almost all cases a very small portion of the solvent used is incorporated at the α-position of β-O-4 linkages within the lignin chain. This is an interesting chemical reaction, the thermodynamics and kinetics of which are understudied. The introduction of alkoxyl groups at the α-position, however, improves the acid stability and solubility of the resulting lignins. In fact, ethanosolv pretreatment of lignin has been used on a commercial scale previously.94 The use of alkosolv pretreatments for the delignification of softwoods has been well documented, in particular for Douglas fir biomass. As discussed above, very few reports on the use of Sitka spruce (SiS) biomass in alkosolv pretreatments exist with ethanosolv being the exception to date.39 Here, a systematic study compares a number of different alkosolv pretreatments of Sitka spruce sawdust and discusses which of the alcoholic solvents may be optimal (Fig. 1). These data are summarised through the use of a relatively accessible comparison method.
 | ||
| Fig. 1 An overview of the research presented and its relevance to the various sections of the Frontiers in physical chemistry for lignin valorisation Faraday Discussion. | ||
Downstream processing of alkosolv lignin
Having argued that the high level of alcohol incorporation at the α-position of β-O-4 linkages helps stabilise lignin during an alkosolv pretreatment, there is also a drawback. In cases where subsequent lignin depolymerisation is planned via oxidative methods, the α-position of the lignin is unfortunately protected from oxidation. Here we explore a solution to this problem on a significantly increased scale compared to previous reports (that used lignins from other biomass sources). Through careful conversion of the α-alkoxylated β-O-4 linkage in butanosolv lignin back to something like its native structure, selective oxidation of the now α-hydroxyl group in the β-O-4 linkage to the corresponding ketone is achieved on a >50 g scale. The reversion catalytic oxidation protocol that is reported, leads to over 100 g of the interesting lignin-derived oligomer referred to as ligninOX (Fig. 1). With significant amounts of this oxidised lignin available, a detailed analytical study using 2D NMR spectroscopic methods solves a structural conundrum present in the literature – the fate of the β-β linkage when lignin is oxidised. This type of advanced characterisation of biopolymers has a clear role to play in future valorisation pathways.Results
Systematic organosolv pretreatment of Sitka spruce sawdust
First, the lignin content of SiS sawdust was determined to enable the calculation of the lignin extraction efficiency using the different organosolv processes. An oven-dried sample of SiS sawdust was processed according to the CASA method,95 leading to a calculated theoretical maximum lignin yield of 21.3 weight percent (wt%, Fig. S1†). Methanol, ethanol, butanol and isobutanol were used as solvents in the organosolv pretreatments due to (i) the ability of the solvent to be incorporated into the lignin chain (this enables stabilisation of the lignin during the pretreatment96–99 and is desirable for some lignin applications100,101); (ii) a range of current solvent costs with the trend at the start of the work being methanol ≪ ethanol < isobutanol ≈ butanol, (iii) on-going academic interests (use of biobutanol50,102–104) and (iv) novelty as the isobutanosolv pretreatment has not been previously reported to the best of our knowledge. Sec- and tert-butanol and propanols were not investigated as these have been shown to be less efficient at lignin extraction, whilst having comparable costs to butanol.91 Alcohols with chain lengths >4 carbons were not selected due to the practical difficulty of removing them under reduced pressure.A standard pretreatment protocol was used for consistency with the SiS sawdust initially being suspended in the alcohol under investigation at a biomass loading of 9.5 mL g−1 and then heated to reflux. A 4 M aqueous solution of HCl (0.5 mL g−1 of biomass) was added and the reaction heated for a further 6 hours. After cooling, the suspension was filtered and the pulp washed with acetone. Neutralisation of the combined filtrates, concentration in vacuo and purification of the resulting solid by one of three different protocols was then carried out. The purification methods varied from (i) precipitation of an acetone/methanol (9:1) solution of the solid into water (bronze standard lignin) or (ii) a two step precipitation protocol where the isolated bronze lignin was redissolved in acetone/methanol (9:1) solution and precipitated into hexane/diethyl ether (1:1) (silver standard lignin) or (iii) the silver standard lignin was first treated with 0.1 M NaOH solution and then purified by column chromatography on silica gel (gold standard lignin). A slightly modified protocol was required for the pretreatments carried out in a pressure vessel with the key difference being the temperature used (20 °C above the atmospheric pressure boiling point of the alcohol). The pretreatment time (6 hours) and the work-up were unchanged. The results of this study are summarised in Table 1 above.
| Entry | Sitka spruce lignin | Yield – wt% of total biomass (wt% of total lignin content) | Linkage content | GPC analysis | |||||
|---|---|---|---|---|---|---|---|---|---|
| Aliphatic hydroxyld (mmol g−1) | Total β-O-4e,f | β-5e | β-βe | Mn/Dag | Mw/Dag | PDIg | |||
| a The solubility of butanosolv lignin in organic solvents makes it uniquely applicable to a range of purification methods. This is not true of all organosolv lignins as solubility decreases with alcohol chain length.b Based on starting SiS bronze standard butanosolv lignin.c Isolated yield of gold standard lignin obtained by flash column chromatographic purification of silver standard lignin.d Data derived from quantitative 31P NMR analysis after lignin phosphitylation in mmol g−1 ± 1.7% (see ESI Fig. S2–S4, Tables S1 and S2† for more details).e Semi-quantitative determination using HSQC NMR analysis (see ESI Fig. S5 and S6†).f Value in parentheses indicates estimated percentage of β-O-4 linkages that are present in their α-alkoxylated form.g Determined from GPC analysis using a refractive index (RI) detector relative to polystyrene standards of known molecular weight ranging from 266 Da to 25000 Da. See ESI Fig. S7† for relevant spectra used in analysis. |
|||||||||
| 1 | Bronze methanosolv | 3.1 (14.6) | 3.36 ± 0.06 | 44 (91) | 10 | 6 | 1560 | 2690 | 1.72 |
| 2 | Bronze ethanosolv | 3.7 (17.4) | 3.02 ± 0.05 | 46 (91) | 9 | 5 | 1560 | 2260 | 1.44 |
| 3 | Bronze isobutanosolv | 7.8 (36.6) | 2.77 ± 0.05 | 49 (92) | 7 | 9 | 1710 | 2840 | 1.67 |
| 4 | Bronze butanosolv | 9.5 (44.6) | 2.54 ± 0.04 | 44 (93) | 6 | 9 | 2410 | 4360 | 1.81 |
| 5 | Silvera butanosolv | 7.8 (36.6) [82b] | 2.75 ± 0.05 | 51 (90) | 7 | 7 | 2050 | 3860 | 1.89 |
| 6 | Golda butanosolv | 5.7 (26.8) [60c] | 2.16 ± 0.04 | 51 (92) | 6 | 6 | 2740 | 6760 | 2.47 |
| 7 | Bronze pressurised methanosolv | 4.4 (20.6) | 3.44 ± 0.06 | 48 (94) | 9 | 5 | 2070 | 3220 | 1.55 |
| 8 | Bronze pressurised ethanosolv | 7.9 (37.1) | 3.03 ± 0.05 | 46 (91) | 7 | 9 | 1800 | 3040 | 1.69 |
Each lignin was analysed to determine: (i) the lignin yield (as wt% of both the total biomass and the CASA determined lignin content); (ii) lignin structure using the accurate and reliable method of quantitative 31P NMR analysis after phosphitylation105–107 (iii) relative linkage content (semi-quantitative) of the lignin using HSQC NMR methods; (iv) lignin molecular weight and polydispersity index (PDI) by GPC analysis. In brief, the calculated lignin yields were relatively low across all solvents under standard conditions with a maximum of approximately half of the lignin being removed using a single butanol pretreatment (Table 1, entries 1–4). As expected, the isolated yields of butanosolv lignin dropped as more extensive purification was carried out (c.f. entries 4, 5 and 6). The use of pressurised conditions did increase the lignin yields with the largest effect being on the ethanosolv pretreatment (c.f. entries 2 and 8).
The aliphatic hydroxyl content of all the bronze standard lignins was high (2.54–3.36 mmol g−1, c.f. analysis of an all β-O-4 model oligomer which gave a value of 6.40 ± 0.11 mmol g−1) as judged by 31P NMR analysis after lignin phosphitylation (Fig. S2–S4, Tables S1 and S2† and Scheme S1† for synthetic details for model oligomer synthesis in the ESI†).105,107 If it is assumed that the measured aliphatic hydroxyl group content is dominated by the β-O-4 linkages in the lignin, then it is clear that all of the pretreatments lead to lignins with high β-O-4 content (39–52% of the lignin linkages were β-O-4s). Due to the quantitative nature of this analytical method, it was decided to use this measurement in calculating a Lignin Value Factor (LVF) as discussed below. It appears that as the alcohol increases in size (from methanol to butanol) there is a significant decrease in aliphatic hydroxyl content (based on an estimated error of 1.7% for aliphatic hydroxyl content determination, see Table S2† legend). However, detailed interpretation of this data is more challenging. In general, HSQC NMR analysis agreed that the linkage content was consistently high and similar across the lignins (Fig. S5†), with a higher degree of alcohol incorporation into the β-O-4 linkages than previously reported (e.g. for ethanosolv of Douglas Fir sawdust82). This suggests that SiS sawdust may be particularly amenable to modification under the organosolv pretreatment conditions used here. The detailed analysis of an isobutanosolv lignin is reported for the first time (see Scheme S2 in ESI† for details of the model compound synthesis required for assignment and Fig. S6†). The changes in aliphatic hydroxyl content and total β-O-4 content observed in silver and gold standard butanosolv lignins compared to bronze standard butanosolv lignins were indicative of lignin purification. Removal of low molecular weight lignin and non-lignin components affords higher quality lignin but the additional steps required may not be necessary for all valorisation methodologies and so the end application should be considered prior to purification.
The molecular weights (Mn and Mw) of the SiS alkosolv lignins were similar for bronze methano-, ethano- and isobutanosolv lignins (Table 1, entries 1–3, Fig. S7†) with bronze butanosolv lignin having higher Mn and Mw values. This may be due to the increased temperature of the butanosolv pretreatment affording access to more recalcitrant lignin within the biomass. This likely contributes to the higher yield obtained of bronze butanosolv lignin. An increase in the molecular weight of the average lignin chain was also observed for the higher temperature bronze pressurised methanosolv and ethanosolv lignins (c.f. Mn, Mw, for entries 1 with 7 and for entries 2 with 8). As aliphatic hydroxyl group content was comparable across the four lignins, this suggested that the major change in the pressurised, higher temperature pretreatments was access to larger lignin chains. Relatively little variation was observed in the polydispersity (PDI, Table 1) across the lignins, indicating a comparable range of lignin could be accessed during each pretreatment. Whilst GPC-determined molecular weight is useful in predicting appropriate applications of these various lignins, these data were not used in the LVF calculations.
Having completed the pretreatment and analysis sections of the study, use of these data to allow direct comparison of the pretreatments was envisaged. It was decided to generate a single calculated value that accounted for the following factors: (i) isolated lignin yield (ideally high); (ii) volume of solvent used (ideally low); (iii) solvent cost (ideally low); (iv) aliphatic hydroxyl content of lignin (ideally high). The industrially relevant cost for each solvent was not trivial to determine. In the end, it was decided to use ECHEMI108 which tracks the cost of commodity chemicals. The only directly comparable figure for each solvent was the average domestic price in mainland China in Yuan per tonne without shipping. An assumption was therefore made that a theoretical large scale biorefinery carrying out organosolv pretreatment of biomass would be situated within mainland China with direct access to a solvent production plant requiring minimal (ideally zero) cost for solvent delivery. The cost of solvent according to ECHEMI is expressed in Yuan per tonne but would be more convenient as cost per volume and so was converted using the solvent density (Table S3†). The cost of solvents (Table S4†) and the exchange rate of Yuan to United States Dollar on 28th March 2024 and 28th March 2025 (1 Yuan = 0.1377 and 1 Yuan = 0.1384 USD, respectively) were used. Combining these factors into a single calculation provided a Lignin Value Factor (LVF) for each organosolv lignin. Eqn (1), as shown below, was used:
 | (1) |
Whilst containing a number of assumptions and simplifications, the LVFs for the pretreatments were calculated and the results are presented in Table 2 (Table S5† for additional details).
 |
The organosolv pretreatments could be divided into three LVF categories: low (<15 mmol per USD), moderate (15–20 mmol per USD) and high LVF (>20 mmol per USD). An initial assessment was carried out using the solvent costs and exchange rates on 28th March 2024 (Table 2). The bronze methanosolv, bronze pressurised methanosolv, and bronze pressurised ethanosolv pretreatments had high LVFs, with pressurised methanosolv having the highest LVF due to the low cost of methanol, the high aliphatic hydroxyl content of the lignin produced by this pretreatment, and the high yield of lignin per volume of solvent used. Bronze ethanosolv had a low LVF due to the low lignin yield and high solvent cost. Bronze isobutanosolv and bronze butanosolv had moderate LVFs. While the lignins yields are high in both these cases, rendering these the two most efficient pretreatments, the high solvent costs at that time lowered the LVFs. It was predicted that a decrease in cost of solvent of approximately 22% for isobutanol and 14% for butanol would move both isobutanosolv and butanosolv into the high LVF category. A decrease of solvent cost on this scale was thought to be possible as biobutanol production is becoming increasingly commercially viable. For example, Celtic Renewables109 based in Grangemouth, Scotland have recently demonstrated biobutanol production at a pilot scale. Further advances in the fermentation technology could reduce biobutanol costs, improving the LVF of butanosolv lignin. This was investigated by reevaluating the LVFs one year later based on solvent costs and exchange rates on 28th March 2025 (Tables 2 and S4†). Year to year, the spot price of methanol had increased 3%, ethanol had decreased 15%, isobutanol has decreased 3% and butanol had decreased 15%.108 As a result, no significant changes were identified in the LVFs for methanosolv, ethanosolv and isobutanosolv pretreatments. The butanosolv pretreatment had now moved into the high LVF category as the predicted price threshold had been passed. This now made the butanosolv pretreatment competitive with the atmospheric methanosolv pretreatment. This significant change in solvent costs in one year exemplifies the variation inherent in the LVF calculation and provides a useful rough measure of the response to changing economic and industrial factors. The stand-out LVF of pressurised methanosolv highlights a possible limitation of this methodology, as there are other factors associated with this pretreatment that argue against its use. For example, limitations on batch size and additional costs that are associated with running the reaction at higher pressures, as well as other process considerations, have not been taken into account.
Having carried out this systematic assessment of different organosolv pretreatments, it was decided to study the structure and reactivity of the Sitka spruce butanosolv lignin.
A native-like Sitka spruce lignin
One advantage of organosolv lignins prepared using alcohols such as butanol (alkosolv lignins) is that incorporation of the alcohol at the α-carbon in the β-O-4 linkage stabilises the lignin during the pretreatment (Scheme 1). The use of the alcohol solvent decreases lignin degradation and condensation. This effect is comparable to the use of, for example, formaldehyde (or other aldehydes) in alternative organosolv pretreatments.96–99 In addition, the alcohol modification of the lignin also facilitates selective reaction at the γ-carbon in the β-O-4 linkage (Scheme 1). This is increasingly used in the synthesis of designer lignins for a range of interesting materials applications. A further advantage of alcohol incorporation is that it increases the lignin’s lipophilicity. This effect is significant for butanol (less so for methanol and ethanol) enabling (i) subsequent reactions in organic solvents; (ii) easier analytical characterisation of the lignin and even (iii) lignin purification by flash column chromatography (as described above when preparing gold standard butanosolv lignin).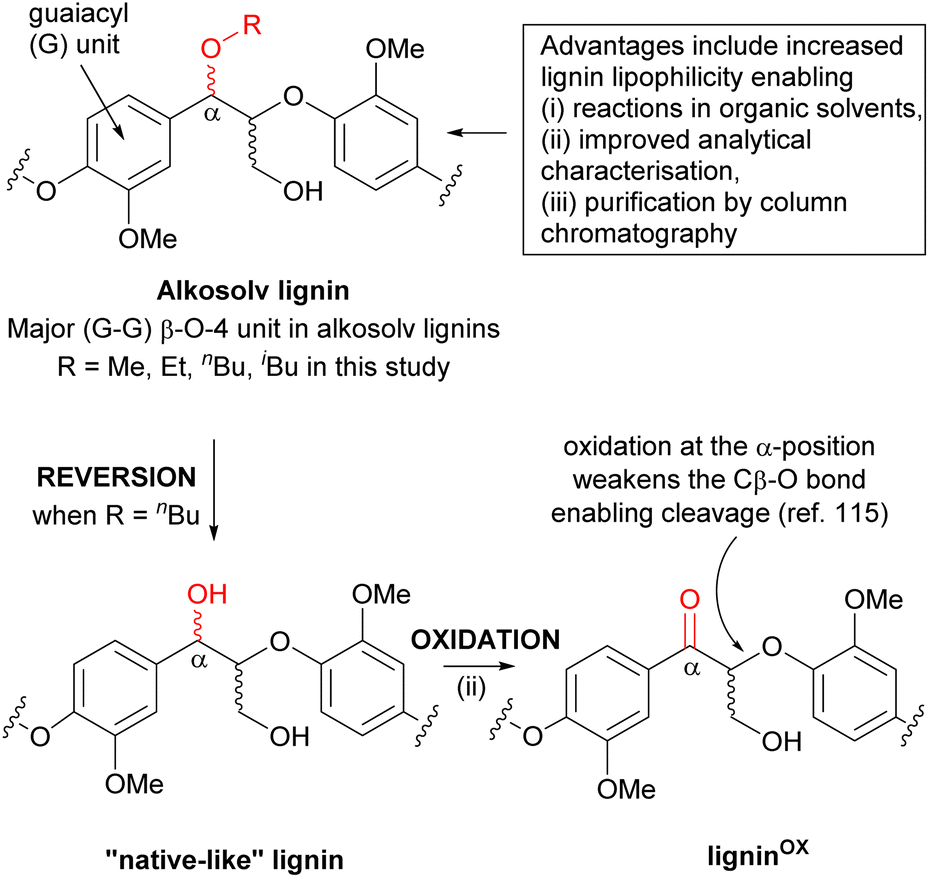 | ||
| Scheme 1 The presence of the α-modified β-O-4 linkage in alkosolv lignins provides several advantages but a key disadvantage arises from the inability to oxidise the α-position to the corresponding ketone. This oxidation is known to facilitate depolymerisation of the lignin. One solution is to convert the alkosolv lignin back to a “native-like” lignin by reinstalling the α-OH into the β-O-4 linkage (a process referred to as reversion). | ||
Whilst alkosolv lignins have been used directly in lignin depolymerisation,100,110–114 the α-modification in the β-O-4 linkage shuts down depolymerisation methods that require installation of a ketone at the α-position. This is important as it has been shown that α-oxidation leads to a significant weakening of the β-carbon to OAr bond in β-O-4 linkages, facilitating depolymerisation (Scheme 1).115 In fact, α-oxidation in β-O-4 linkages is essential in natural approaches to lignin degradation.116–121 One approach to resolving this limitation of alkosolv lignins is the controlled conversion of the modified β-O-4 linkages back to their native structure (with an α-OH group). This process has typically been referred to as “reversion” or “de-etherification” and provides an alternative (two step) approach to “native-like” lignins that are comparable to 1,4-dioxasolv lignins. Limited examples of reversion have been reported to date, and all of these are on small scales (1 g or less) and not on a Sitka spruce lignin.82,91 In the next phase of this study, large scale reversion was achieved leading to a “native-like” Sitka spruce lignin that was the substrate for a selective α-oxidation reaction. The overall goal was to prepare a well-defined Sitka spruce oxidised lignin (ligninα-OX) that could be a starting point for a range of potential valorisation methods.
Previous studies on 200 mg scale using Douglas Fir butanosolv lignin led to the desired reversion in 55–60 wt% yield.82 The reaction involved heating the lignin in a sealed tube at 100 °C in 1,4-dioxane and water (2:1) for 6 hours with hydrochloric acid as the catalyst. Repeating these conditions with Sitka spruce bronze butanosolv lignin also proved successful, although full exchange of the butanol for water at the α-position proved elusive (Table S6,† entry 2, Fig. S8B†). Carrying out the reaction at room temperature over 72 hours failed but shortening the reaction time from 6 to 2.5 hours (whilst maintaining the reaction temperature at 100 °C in a sealed tube, Table S6,† entry 4) improved the practicality of the reversion. Importantly, when the reversion reaction was repeated on a 5–8 gram scale, successful preparation of the “native-like” Sitka spruce lignin was achieved (Table S6,† entries 5–8). Translating these results to larger scale at the Biorenewables Development Centre (BDC, York) proved successful. In brief, in a typical reaction using a 2 L Hastelloy high pressure reactor, 60 g of lignin were added to the reactor followed by water, 1 M HCl(aq) (volume ratio 2:1, total of 333 mL) and 1,4-dioxane (667 mL). The reactor was sealed and then heated, with stirring, to 105 °C for 2.5 hours (autogenous pressure of ca. 0.2 barg was observed). After work up (see ESI† general procedure G) a 62 ± 2 wt% yield (average over 4 runs) of the desired “native-like” Sitka spruce lignin was obtained. Reversions on this scale proved reproducible (Fig. S9†).
Large scale preparation of Sitka spruce ligninOX
Lignin depolymerisation remains of interest in plans for the valorisation of this important component of biomass. Whilst reductive methods dominate this area, there is interest in (selective catalytic) oxidative processing of lignin as well as the subsequent depolymerisation methods. In fact, the preparation of a material often referred to as ligninOX may enable novel ways of using lignin in the future. We63,122 and others123–133 have studied the selective oxidation of hardwood lignins in the context of lignin depolymerisation. Here, it was hoped that some of the complexities inherent in hardwood lignins might be decreased by the fact that Sitka spruce is a softwood and therefore its lignin is dominated by guaiacyl (G)-aromatic units. However, there were initial concerns that a Sitka spruce softwood lignin would be more reactive under the oxidation conditions compared to the previously difficult to oxidise hardwoods.122 This increase in reactivity could lead to side reactions, a higher than desired degree of oxidation, or degradation of the highly oxidised lignin chain.Methods for the analysis of selectively oxidised lignins focus on the use of HSQC NMR.134–139 This analytical method separates signals in two dimensions enabling diagnostic crosspeaks to be identified. In the case of ligninOX, the observation of crosspeaks at δ1H/δ13C 5.65/81.7 and 5.92/81.6 ppm is used to read out on the success of an attempted oxidation of the α-OH to the corresponding ketone in G–G β-O-4 linkages (Scheme 1, for a very recent example see140). In brief, the α-oxidation of a G–G β-O-4 linkage that is adjacent to a non-oxidisable/not yet oxidised linkage (Oxidation Stage A in Fig. 2a) results in the crosspeak at δ1H/δ13C 5.65/81.7 ppm corresponding to the β-hydrogen (shown in red in Fig. 2a). If the adjacent linkage has been oxidised (including as shown in Fig. 2a for an adjacent oxidised β-O-4 linkage) then the observed crosspeak for the β-hydrogen is instead at δ1H/δ13C 5.92/81.6 ppm (Oxidation Stage B). Across our oxidation studies, a range of different outcomes were observed. These were classified into three categories termed underOX, goodOX and overOX (Fig. 2b, d and c respectively). Diagnostic crosspeaks for an underOX lignin were the presence of a crosspeak at 5.65/81.7 ppm but not at 5.92/81.6 ppm as well as crosspeaks at 4.76/71.6, 4.47/80.5 and 4.30/84.2 ppm (corresponding to the α- and β-protons of unreacted diastereomeric β-O-4 linkages). If oxidation was more successful and a goodOX lignin was formed then only two of the previously identified 5 crosspeaks were seen (at 5.65/81.7 and 5.92/81.6 ppm). Finally, if oxidation went too far and overOX lignin was formed then diagnostic (but as yet unassigned) additional crosspeaks at 7.56/129.2 and 7.63/132.1 ppm were observed (shown in insert in Fig. 2b and not present in other spectra) in addition to only one of the two crosspeaks (at 5.92/81.6 ppm) seen for goodOX lignin.
 | ||
| Fig. 2 Catalytic DDQ oxidation study using Sitka spruce lignin: (a) schematic representation of the oxidation of adjacent β-O-4 units within lignin including possible oxidation stages. The expected chemical shifts of the crosspeaks corresponding to the highlighted β-hydrogen (red) in the HSQC NMR spectra (d6-DMSO) of the possible products are shown. Initial oxidation of the α-hydroxyl to the corresponding ketone leads to a change in the chemical shift of the crosspeak corresponding to the β-hydrogen (to 5.65/81.7 ppm, oxidation stage A). Further oxidation of an adjacent β-O-4 unit leads to a further shift of this crosspeak to 5.92/81.6 ppm; (b–d) representative HSQC spectra highlighting the different lignin products that were observed, including the formation of an underOX lignin (reaction 1), a goodOX lignin (reaction 5) and an overOX lignin (reaction 4). Key observed crosspeaks are highlighted (hashed circles). Crosspeaks corresponding to the oxidation stages A and B highlighted in part (a) of the figure are emphasised by the inclusion of β′ and β’’ labels. (e) HSQC NMR analysis (d6-DMSO) of large scale oxidised SiS ligninOX highlighting (almost) complete oxidation of the α-hydroxyl in the β-O-4 linkages. This represents, to the best of our knowledge, the largest scale batch of ligninOX prepared to date. (f) Table of results from the oxidation optimisation study. All reactions were carried out at 85 °C in (1,4-dioxane/DME, 2:3). aEntry 1 used a lignin that was methylated on the phenolic oxygens before oxidation.114,142–144 The degree of oxidation was calculated using the HSQC NMR data. In brief, the integration of the crosspeak corresponding to the β-hydrogens in oxidised lignin was divided by the total integral for β-hydrogens in the β-O-4 linkages. A value of below 75% conversion to oxidised β-O-4 linkages (colour-coded purple) was judged as forming an underOX lignin whereas a value of above 75% (colour-coded green) was judged as forming goodOX lignin. The ratio of the integrations for the crosspeaks assigned to the β-hydrogens labelled β′′:β′ = oxidation stage B:oxidation stage A (units per 100 C9 units) was also measured and classified as forming underOX if the integral of the β′′ crosspeak was <1.0 (purple) or goodOX lignin if the integral was >1.0 (green). A measure of lignin over-oxidation (overOX) was provided by integrating crosspeaks at δ1H/δ13C 7.56/129.2 and 7.63/132.1 ppm in the HSQC NMR spectra. If the observed integration total was >10 units per 100 C9 units (red) then overOX lignin was formed. | ||
As no oxidation studies of this type have been reported on Sitka spruce lignin, to the best of our knowledge, initial studies used the model all G β-O-4 oligomer discussed above. Good levels of β-O-4 oxidation were obtained on a 200 mg scale using catalytic DDQ (0.3 equiv.) and tBuONO (0.3 equiv.) at 85 °C in dimethoxyethane (DME) and 2-methoxyethanol (3:2) with the oxygen (terminal oxidant) being supplied via a ballon (procedure based on a literature report using different lignins63). However, HSQC NMR analysis of the isolated oligomerOX showed that for some of the β-O-4 linkages one of the solvents (2-methoxyethanol) had been incorporated at the α-position (Fig. S10† for more details). A change of solvent mixture for the oxidation reaction to DME and 1,4-dioxane (3:2) was therefore used throughout the rest of the study. Even on a small scale (200 mg), oxygen mass transport problems were encountered. One attempt to overcome this involved actively squeezing balloons filled with oxygen into the reaction, forcing in additional oxygen as the reaction proceeded to provide sufficient dissolved oxygen in the system. Whilst this did aid conversion, a more viable alternative involved the use of compressed air (rather than oxygen) in combination with an autoclave. Compressed air was chosen due to its safer handling compared to the use of a pure oxygen atmosphere under these conditions.141 The results from this part of the optimisation study are presented in Fig. 2f (and Fig. S11–S13†). Initially, the experiment (Fig. 2f, entry 1) involved selectively methylating the phenolic oxygens in the reverted SiS lignin (using TBAF and MeI in DMSO114,142–144) before attempting the catalytic oxidation with DDQ and tBuONO (15 wt% of each) at 85 °C in DME/1,4-dioxane (3:2) under 5 atmospheres of compressed air. It was found, however, that this modification resulted in a lower than expected conversion to the desired ligninOX leading to the formation of underOX lignin (33% oxidation, ratio of β′′:β′ crosspeaks 0.39:1.00, Fig. 2b, reaction 1). Removing the pre-methylation step provided a way forward without having to use toxic reagents. An increased level of oxidation of the β-O-4 linkages was observed (71% oxidation, ratio of β′′:β′ crosspeaks 0.77:1.00) even with a lower amount of oxidising reagents (10% wt% DDQ and tBuONO, Fig. 2f, entry 2). Increasing the ratio of reagents (to 25 wt% DDQ and tBuONO) relative to the starting lignin resulted in the first acceptable batch of SiS ligninOX being formed (83% oxidation, ratio of β′′:β′ crosspeaks 1.24:1.00, goodOX, Fig. 2f, entry 3).
Increasing the concentration that the lignin oxidation reaction was run at from 20 g L−1 to 50 g L−1 provided additional insight. This change led to a higher level of oxidation (88%, ratio of β′′:β′ crosspeaks 1.65:1.00, entry 4). However, the crosspeak at 5.81/81.9 ppm was clearly present (Fig. 2c, reaction 4), as well as the two additional aromatic crosspeaks (7.56/129.3 and 7.62/132.1 ppm, insert in Fig. 2c). As increasing the concentration of this reaction meant decreasing the volume of solvent used, there was a corresponding increase in the volume of compressed air in the sealed system compared to other reactions. The formation of overOX lignin, in this case, was rationalised by the increased amount of available oxygen impacting on the catalytic cycle and therefore on successful DDQ regeneration. Further scale up to around 3 grams of lignin at the same concentration modified the amount of solvent required again (60 mL, entry 5). Under these conditions, the volume ratio of lignin in solution to compressed air in the sealed system was 60:40 and this was found to be almost optimal giving goodOX (Fig. 2d, reaction 5). By keeping this exact volume ratio, the oxidation was further improved by increasing the compressed air pressure to 10 atmospheres (entry 6). These conditions were found to be reasonably reproducible (entry 7). In collaboration with the BDC, the Sitka spruce lignin oxidation reaction was then run on a 60 grams scale with a good oxidation level being maintained. In the first of two runs (entry 8) the level of conversion (73%) was slightly lower than hoped for. However, an increase in reaction time from 16 to 20 hours (c.f. entries 8 and 9) delivered a high-quality sample of Sitka spruce ligninOX on a significant scale in 91 wt% yield (entry 9, and Fig. 2e). To the best of our knowledge, this is the first relatively large-scale preparation of ligninOX in a controlled manner. It is also the first time Sitka spruce ligninOX has been reported in the literature. In the next part of the study we took advantage of the availability of this material to study its structure in more detail.
The fate of the β-β linkage during softwood oxidation
The continued study of ligninOX enables a more detailed assessment of the conversion of other lignin linkages under the oxidative conditions used. This level of structural analysis is not only academically interesting but is likely to be important in the development of highly optimised lignin depolymerisation strategies in the future. We, and others, have reported previously on the oxidation of β-β and β-5 linkages, in addition to the abundant β-O-4 units.123,145–149 As expected, the large scale DDQ oxidation of SiS lignin reported here resulted in the disappearance of the NMR signals corresponding to both the β-β and β-5 linkages (for example in Fig. 2c the expected crosspeak corresponding to the β-5 linkage at δ1H/δ13C 5.60/88.4 ppm is absent in the final ligninOX product). Whilst the oxidation of the β-β linkage has been discussed previously, there remain uncertainties about the structure of the products formed.145 The next stage of the study was to revisit this issue.Due to the relatively low abundance of β-β linkages in SiS lignin, initial studies used compound 1 (Eudesmin), a model for the β-β linkage in a softwood (Scheme 2). Oxidation of 1 using 5 equivalents of DDQ in 1,4-dioxane at 80 °C for 3 hours resulted in the formation of two compounds that clearly had related structures. One of the compounds, compound 2, was proposed to contain an aldehyde functional group, whilst the other (compound 3) was tentatively assigned as the alcohol-containing precursor of 2. Treatment of the crude reaction mixture containing both 2 and 3 with Dess–Martin periodinane (DMP)150 led to the apparent conversion of 3 to 2, simplifying the reaction mixture. Whilst full characterisation of 2 was achieved (see ESI† for details), the structural assignment still proved challenging with several possible structures being proposed. Unfortunately, it was not possible to grow crystals of 2 that were of suitable quality for structure determination. However, in parallel studies using 4 (Yangambin), high quality crystals of the analogous aldehyde-containing compound 5 were obtained in 60% yield after DDQ and DMP treatment. X-ray crystallographic analysis of 5 showed that a furan substituted by both an aryl-ketone and an aldehyde functional group had formed (Scheme 2 and Fig. 3). Detailed comparison of the NMR analysis of the novel compound 5 with 2 was consistent with the structure of 2 being as shown (Scheme 2). Alcohols 3 and 6 were assigned the structures shown. In the light of these observations, it seems likely that structural reassignment of previously reported α-pyrones as products from related reactions should be revisited (for example see ref. 151).
 | ||
| Scheme 2 Oxidation of eudesmin 1 and yangambin 4 with excess DDQ in 1,4-dioxane at 80 °C leading to the formation of the novel furan products. | ||
 | ||
| Fig. 3 Views of the structure obtained from single crystal X-ray diffraction (thermal ellipsoid plot 50% probability) of furan-containing aldehyde product 5 resulting from reaction of yangambin 4 with excess DDQ in 1,4-dioxane at 80 °C. (a) View of 5 with selected crystallographic numbering; (b) view illustrating the inter-ring angles in 5. Selected bond lengths (Å), angles (°) and torsions (°): C1–Cα 1.483(4), Cβ–Cβ′ 1.445(3), Cγ–O2 1.359(3), Cα′–O2 1.372(3), C1′–Cα′ 1.456(3), Cα–O1 1.228(3), Cγ′–O3 1.203(3); C1–Cα–Cβ 119.9(2), Cβ–Cγ–O2 110.3(2), C1′–Cα′–Cβ′ 137.3(2), Cα′–Cβ′–Cγ′ 132.6(2), Cβ′–Cα′–O2 108.7(2); C1–Cα–Cβ–Cγ −46.3(4), C1′–Cα′–Cβ′–Cγ′ 1.8(5), Cα–Cβ–Cγ–O2 172.7(2), Cγ′–Cβ′–Cα′–O2 174.6(3), Cβ–Cβ′–Cγ′–O3 175.0(3). See CCDC 2445448 and ESI† for experimental details. | ||
In an attempt to confirm that the β-β linkage in lignin is converted to the corresponding furan structure on oxidation to ligninα-OX, inspection of the aldehyde region of the HSQC NMR analysis of SiS ligninα-OX proved challenging. This is likely due to the fact that there are relatively low amounts of the β-β linkage in this softwood lignin. However, confirmation that this transformation does occur in lignin was obtained by comparing the 2D HSQC and HMBC spectra of aldehyde 5 with beech ligninα-OX (Fig. 4). Beech is a hardwood that contains predominantly S units and an increased relative number of β-β linkages compared to SiS lignin. In brief, 1,4-dioxasolv beech ligninα-OX, prepared using 3 weight equivalents of DDQ as described previously,122 displayed a broad cross peak at 1H/13C δ 10.17/187 ppm in the 2D HSQC NMR analysis (Fig. 4b(i)). An overlay of this HSQC spectrum with that of aldehyde 5 (Fig. 4a(i) and c(i) for overlay) supported the presence of the furan aldehyde moiety in the ligninα-OX sample. HMBC NMR analysis further supported this with clear correlations between the aldehyde proton (Hγ′) and Cβ and Cα′ being observed in both the model compound 5 and beech ligninα-OX. A relatively weak 2J correlation between Hγ′ and Cβ′ was also seen in both samples although this signal is very weak in the beech ligninα-OX sample (Fig. 4 for chemical structures and legend for more detail). Overall, it seems likely that the disappearance of the β-β linkages on formation of SiS ligninα-OX results from formation of the analogous furan unit. This raises interesting questions for the future on whether it is possible to isolate this furan unit (or a structure derived from it) on depolymerisation of ligninα-OX.
 | ||
| Fig. 4 Identifying the furan-aldehyde unit in SiS ligninOX. Selected regions of the HSQC NMR (1H/13C/δ 10.05–10.25/185–189 ppm, 700 MHz, d6-DMSO) analysis of: (a) (i) aldehyde 5 and (b) (i) beech ligninOX and (c) (i) an overlay of the two spectra indicating the presence of an aldehyde crosspeak in both. Selected regions of the HMBC NMR analysis (1H/13C/δ 10.05–10.25/110–170 ppm, 700 MHz, d6-DMSO) of: (A) (ii) aldehyde 5; (B) (ii) beech ligninOX (generated using 3 wt. eq. of DDQ) and (c) (ii) an overlay of the two spectra indicating 3 key crosspeaks resulting from various strength couplings. For atom labelling see structures at top of figure. | ||
Discussion and conclusions
Dramatic progress in the chemistry and biology of lignin has been made in the last two decades, building on and extending the pioneering studies of Payen, Shulze, Klason, Freudenberg, Nimz, Adler, Hibbert, Kratzl, Tischenko, Nakano and many others. This resurgence has been driven by technological advances and a focus on the use of plant biomass rather than fossil fuels for chemical applications. In one approach to the isolation of lignin, a pretreatment of the biomass is carried out. In a perfect world, this would deliver native lignin (with a structure almost identical to that found in the plant) in quantitative yield. Delivering this continues to prove a considerable challenge but a wide range of creative approaches have been applied (including excellent ionosolv methods152–155). One of these involves the use of organic solvents in what is known as an organosolv pretreatment. In many cases, there is no stabilisation of the lignin during extraction and this often results in extensive degradation of the native lignin leading to the generation of a very intransigent material. The use of an alcohol (for example ethanol or butanol) in the pretreatment appears to simplify the challenge as by incorporation of the alcohol at the α-position of the β-O-4 linkages, the lignin is stabilised and alkosolv pretreatments often deliver reasonable yields of good quality lignin. Whilst considerable progress has been made, there remain details and biomass types that have been understudied in the context of alkosolv pretreatments. Here we focused on one such case, the isolation of alkosolv lignins from the softwood Sitka spruce (SiS).It is perhaps surprising that more work on the pretreatment of Sitka spruce residues is not found in the literature, given its strategic importance in the UK forestry industry. Here we show that alkosolv SiS lignin can be obtained relatively easily and suggest that the economics of which alcohol should be used is in a state of flux. For example, just a year ago it was clear that butanosolv processing of Sitka spruce sawdust at atmospheric pressure was not competitive with an analogous methanosolv pretreatment. More recently this differentiation has become less obvious. During our studies we used a relatively simple method of assessing the competitiveness of different pretreatments (calculation of a lignin value factor). Whilst the proposed LVF is far from the most detailed way of carrying out this comparison, we believe it is useful in engaging colleagues and peers in discussions on lignin isolation strategies. Following an excellent suggestion during the review of this manuscript, an even simpler method than the one selected here was considered that focused on cost per kilogram of lignin prepared (see Table S7 and eqn (S1)† for details and results of this assessment in the context of this work). It may also be preferable to carry out a second-generation calculation that converts a wider range of factors into a single key performance indicator (KPI).156 Additional factors could include the use of a space–time yield calculation as an improved method of maximising the use of yield information and accounting for the strengths and limitations of the pressurised vessels used in some of the pretreatments.157,158 Other factors moving forward could also include (i) an assessment of the relative ease of downstream purification, (ii) recycling of the different alcohol solvents, and (iii) a measure of the quality of the cellulose pulp obtained (for example,60,82 it is standard practice to use the total amount of glucose produced from the pulp using commercially available enzyme preparations as a measure of pulp quality). The question of whether to include an LVF calculation for each different biomass being considered for a particular process could also be addressed in future versions of this approach.
Through collaboration with the BDC, it also proved possible to convert the butanoslv SiS lignin back to a “native-like” lignin via reversion. Whilst still not fully optimised, the method described here was scalable and reproducibly delivered relatively large quantities of SiS lignin for study. The term “native-like” is used as the β-O-4 linkages in the reverted lignin now have the same structure as they (predominantly) do in the wood with a hydroxyl functional group at the α-position. A more detailed comparison of this lignin (using 2D NMR methods) with that of, for example, a milled-wood lignin would be a useful further extension of this work. Regardless, it was then possible to oxidise the β-O-4 linkages, converting the α-hydroxyl to a ketone. This delivers a material usually referred to as ligninOX and, to the best of our knowledge, is the first time that ligninOX has been prepared from Sitka spruce. The fact that this oxidation was relatively reproducible on a 60 gram scale opens up numerous options for further exploring this interesting modified biopolymer. There has been some doubt cast on whether it is possible to prepare ligninOX by the method used here, but these studies demonstrates that it is possible on a multigram scale for an industrially important plant biomass source.
One question that has arisen when thinking about the structure of ligninOX is whether its formation should be considered a selective reaction. In terms of the chemistry at the β-O-4 linkage, it is selective (α-instead of γ-hydroxyl oxidation), however, in terms of oxidation of different linkages, it is not. It has been noted previously that in addition to oxidation of the β-O-4 linkage both the β-β and β-5 linkages are oxidised. Here, we have clarified what happens to the β-β linkages reporting for the first time its conversion to an interesting keto-furan aldehyde/alcohol unit.
Is ligninOX relevant to future valorisation strategies? Whilst the answer to this is more likely negative than positive at this time, it remains instructive to think about what the advantages and disadvantages of ligninOX are. Whilst the three-step synthesis is a challenge, the route used here (via an alkosolv lignin that is reverted and oxidised) is reproducible and scalable (at least up to the 60 gram scale). The upside is the reactive functionality that is present in ligninOX. The material prepared here is soluble in a number of organic solvents and it should therefore be possible to react the ketone using a wide range of standard organic reactions. A range of potential materials applications are possible.159 In addition, studies are on-going aimed at optimising the depolymerisation of this material. This should deliver highly functionalised monomeric units for use in the synthesis of bioactive compounds and surfactants etc. The possible isolation of novel monomers isolated from other processed linkages (e.g. the β-β linkage, all be it in low yield) seems feasible. Overall, the possibilities and complications that lignin chemistry continues to deliver will ensure that a wide number of pathways forward will continue to be explored. We hope that this manuscript addresses some issues of interest and builds on previous excellent Faraday Discussions based on lignin.160,161
Data availability
The research data supporting this publication can be accessed at https://doi.org/10.17630/fc7eab7c-0839-4ab1-a36d-489b2f266206.Author contributions
Investigation: DJD, GNS, DMM-B, DBC, AMZS, JRDM, TL, AC, AMD; methodology: DJD, GNS, DMM-B, DBC, AMD, NJW; conceptualisation: NJW, DSG, SLH, NJP; supervision: NJW, DSG, SLH, NJP, MJG, TL, AMZS; funding acquisition: SLH, DSG, NJW, NJP; project administration: NJW, SLH, DSG, NJP; visualisation: DJD, NJW, GNS, DMM-B, DBC, writing-original draft: NJW, DJD, GNS; writing – review and editing: All authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Innovate UK (grant 10027463, Unilever PLC, BDC and NJW), the Industrial Biotechnology Innovation Centre (IBioIC, DMM-B PhD) and EaSI-CAT at the University of St Andrews (DJD PhD) for funding. We would also like to thank Mrs Caroline Horsburgh for mass spectrometry analysis and Woodworks, Mold, UK for Sitka spruce wood.References
- F. Souto and V. Calado, Green Chem., 2022, 24, 8172–8192 RSC.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110(6), 3552–3599 CrossRef CAS PubMed.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 Search PubMed.
- J. S. Luterbacher, D. Martin Alonso and J. A. Dumesic, Green Chem., 2014, 16, 4816–4838 Search PubMed.
- W. Schutyser, T. Renders, S. Van Den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 1246843 Search PubMed.
- S. Zheng, Z. Zhang, S. He, H. Yang, H. Atia, A. M. Abdel-Mageed, S. Wohlrab, E. Baráth, S. Tin, H. J. Heeres, P. J. Deuss and J. G. de Vries, Chem. Rev., 2024, 124, 10701–10876 CrossRef CAS PubMed.
- T. Q. Yuan, F. Xu and R. C. Sun, J. Chem. Technol. Biotechnol., 2013, 88, 346–352 Search PubMed.
- W. Lan and J. S. Luterbacher, ACS Cent. Sci., 2019, 5, 1642–1644 CrossRef CAS PubMed.
- D. D. S. Argyropoulos, C. Crestini, C. Dahlstrand, E. Furusjö, C. Gioia, K. Jedvert, G. Henriksson, C. Hulteberg, M. Lawoko, C. Pierrou, J. S. M. Samec, E. Subbotina, H. Wallmo and M. Wimby, ChemSusChem, 2023, 16, e202300492 CrossRef CAS PubMed.
- R. Li, Z. Zhang, X. Wang, X. Zhao, M. Li, H. Xu and C. Huang, Green Chem., 2025, 27, 3044–3063 RSC.
- H. Liao, X. Meng, Y. Pu, A. Ragauskas and J. Zhang, Sep. Purif. Technol., 2025, 358, 130221 CrossRef CAS.
- D. F. de Waard, P. D. Kouris, M. D. Boot and E. J. M. Hensen, ACS Sustainable Chem. Eng., 2025, 13, 3269–3279 CrossRef CAS PubMed.
- Y. Mao, I. Tarhanli, G. Owen, C. S. Lee, E. Senses and E. Binner, Ind. Crops Prod., 2025, 226, 120614 CrossRef CAS.
- L. Schoofs, D. Weidener, W. Leitner, H. Klose and P. M. Grande, ChemSusChem, 2025, 18, e202401063 CrossRef CAS PubMed.
- W. Lan, L. P. Y. Lam, A. Lui and C. Lo, Curr. Opin. Plant Biol., 2025, 85, 102703 CrossRef CAS PubMed.
- Y. Zheng, X. Kang, Z. You, Y. Li, Y. Huang, T. He, T. Su, A. J. Ragauskas, Z. Li, Q. Wang and X. Song, Int. J. Biol. Macromol., 2025, 290, 138807 CrossRef CAS PubMed.
- Y. Pan, W. Chen, Q. Kang, L. Hao, J. Lu and J. Zhu, Bioprocess Biosyst. Eng., 2025, 48, 367–379 CrossRef CAS PubMed.
- Y. Li, H. Sun, T. Mu and M. Garcia-Vaquero, Biomass Bioenergy, 2025, 193, 107594 CrossRef CAS.
- H. D. Zamora Zamora, C. de Freitas, D. Pasquini, F. Masarin and M. Brienzo, Bioenergy Res., 2025, 18, 8 CrossRef CAS.
- E. Blondiaux, J. Bomon, M. Smoleń, N. Kaval, F. Lemière, S. Sergeyev, L. Diels, B. Sels and B. U. W. Maes, ACS Sustainable Chem. Eng., 2019, 7, 6906–6916 CrossRef CAS.
- S. D. Karlen, V. I. Timokhin, C. Sener, J. K. Mobley, T. Runge and J. Ralph, ChemSusChem, 2024, 17, e202400234 CrossRef CAS PubMed.
- D. J. Davidson, O. Lynard, D. M. Miles-Barrett, G. N. Sansom, F. Tedesco, F. Watson and N. J. Westwood, Arkivoc, 2024, 4, 202312087 Search PubMed.
- X. Liu, F. P. Bouxin, J. Fan, V. L. Budarin, C. Hu and J. H. Clark, ChemSusChem, 2020, 13, 4296–4317 CrossRef CAS PubMed.
- A. Afanasenko and K. Barta, iScience, 2021, 24, 102211 CrossRef CAS PubMed.
- E. M. Anderson, M. L. Stone, R. Katahira, M. Reed, G. T. Beckham and Y. Román-Leshkov, Joule, 2017, 1, 613–622 CrossRef CAS.
- A. De Santi, M. V. Galkin, C. W. Lahive, P. J. Deuss and K. Barta, ChemSusChem, 2020, 13, 4468–4477 Search PubMed.
- A. W. Bartling, M. L. Stone, R. J. Hanes, A. Bhatt, Y. Zhang, M. J. Biddy, R. Davis, J. S. Kruger, N. E. Thornburg, J. S. Luterbacher, R. Rinaldi, J. S. M. Samec, B. F. Sels, Y. Román-Leshkov and G. T. Beckham, Energy Environ. Sci., 2021, 14, 4147–4168 RSC.
- X. Liu, H. Li, L. P. Xiao, R. C. Sun and G. Song, Green Chem., 2019, 21, 1498–1504 RSC.
- O. E. Ebikade, N. Samulewicz, S. Xuan, J. D. Sheehan, C. Wu and D. G. Vlachos, Green Chem., 2020, 22, 7435–7447 RSC.
- R. N. Nishide, J. H. Truong and M. M. Abu-Omar, ACS Omega, 2021, 6, 8142–8150 CrossRef CAS PubMed.
- D. Lebedeva, S. Hijmans, A. P. Mathew, E. Subbotina and J. S. M. Samec, ACS Agric. Sci. Technol., 2022, 2, 349–358 CrossRef CAS.
- J. H. Jang, J. Callejón Álvarez, Q. S. Neuendorf, Y. Román-Leshkov and G. T. Beckham, ACS Sustainable Chem. Eng., 2024, 12, 12919–12926 CrossRef CAS PubMed.
- F. Bugli, A. Baldelli, S. Thomas, M. Sgarzi, M. Gigli, C. Crestini, F. Cavani and T. Tabanelli, ACS Sustainable Chem. Eng., 2024, 12, 16638–16651 CrossRef CAS PubMed.
- M. M. Abu-Omar, K. Barta, G. T. Beckham, J. S. Luterbacher, J. Ralph, R. Rinaldi, Y. Román-Leshkov, J. S. M. Samec, B. F. Sels and F. Wang, Energy Environ. Sci., 2021, 14, 262–292 RSC.
- S. Searle and C. Malins, Availability of cellulosic residues and wastes in the EU, Washington, DC 2025, USA, 2013, https://theicct.org/wp-content/uploads/2021/06/ICCT_EUcellulosic-waste-residues_20131022.pdf, accessed 27/04/2025.
- G. Kerr, D. Williams, J. Haufe and J. Walmsley, Q. J. For., 2021, 115(2), 98–106 Search PubMed.
- B. Ditchburn and A. Brewer, GB 25-year forecast of standing coniferous volume and increment, https://cdn.forestresearch.gov.uk/2022/02/nfi-statistical-analysis-report_gb-25-year-forecast-standing-coniferous-volume_1kbi6uo.pdf, accessed 27/04/2025.
- F. P. Bouxin, S. D. Jackson and M. C. Jarvis, Bioresour. Technol., 2014, 151, 441–444 CrossRef CAS PubMed.
- Q. Ma, C. Yu, Y. Zhou, D. Hu, J. Chen and X. Zhang, Int. J. Biol. Macromol., 2024, 256, 128506 Search PubMed.
- T. K. Bedru, B. T. Meshesha, S. A. Mohammed, A. G. Demesa and M. Jayakumar, Int. J. Chem. Eng., 2025, 3120449, DOI:10.1155/ijce/3120449.
- D. Di Francesco, K. R. Baddigam, S. Muangmeesri and J. S. M. Samec, Green Chem., 2021, 23, 9401–9405 Search PubMed.
- C. Nitsos, R. Stoklosa, A. Karnaouri, D. Vörös, H. Lange, D. Hodge, C. Crestini, U. Rova and P. Christakopoulos, ACS Sustainable Chem. Eng., 2016, 4, 5181–5193 CrossRef CAS.
- P. Paulsen Thoresen, H. Lange, C. Crestini, U. Rova, L. Matsakas and P. Christakopoulos, ACS Omega, 2021, 6, 4374–4385 CrossRef CAS PubMed.
- P. Giannì, H. Lange and C. Crestini, ACS Sustainable Chem. Eng., 2020, 8, 7628–7638 CrossRef PubMed.
- M. Karlsson, J. Romson, T. Elder, Å. Emmer and M. Lawoko, Biomacromolecules, 2023, 24, 2314–2326 CrossRef CAS PubMed.
- G. Tofani, E. Jasiukaitytė-Grojzdek, M. Grilc and B. Likozar, Green Chem., 2024, 26, 186–201 RSC.
- C. Sun, G. Song, Z. Pan, M. Tu, M. Kharaziha, X. Zhang, P. L. Show and F. Sun, Bioresour. Technol., 2023, 368, 128356 CrossRef CAS PubMed.
- S. C. Rabelo, P. Y. S. Nakasu, E. Scopel, M. F. Araújo, L. H. Cardoso and A. C. da Costa, Bioresour. Technol., 2023, 369, 128331 CrossRef CAS PubMed.
- X. Meng, Y. Wang, A. J. Conte, S. Zhang, J. Ryu, J. J. Wie, Y. Pu, B. H. Davison, C. G. Yoo and A. J. Ragauskas, Bioresour. Technol., 2023, 368, 128280 CrossRef CAS PubMed.
- J. A. Ferreira and M. J. Taherzadeh, Bioresour. Technol., 2020, 299, 122695 CrossRef CAS PubMed.
- F. Pereira Marques, A. K. Lima Soares, D. Lomonaco, L. M. Alexandre e Silva, S. Tédde Santaella, M. de Freitas Rosa and R. Carrhá Leitão, Int. J. Biol. Macromol., 2021, 175, 304–312 CrossRef CAS PubMed.
- S. Başakçılardan Kabakcı and M. H. Tanış, Biomass Convers. Biorefin., 2021, 11, 2869–2880 CrossRef.
- J. Snelders, E. Dornez, B. Benjelloun-Mlayah, W. J. J. Huijgen, P. J. de Wild, R. J. A. Gosselink, J. Gerritsma and C. M. Courtin, Bioresour. Technol., 2014, 156, 275–282 CrossRef CAS PubMed.
- A. T. Smit, A. van Zomeren, K. Dussan, L. A. Riddell, W. J. J. Huijgen, J. W. Dijkstra and P. C. A. Bruijnincx, ACS Sustainable Chem. Eng., 2022, 10, 6012–6022 CrossRef CAS PubMed.
- A. Smit and W. Huijgen, Green Chem., 2017, 19, 5505–5514 RSC.
- E. Araque, C. Parra, J. Freer, D. Contreras, J. Rodríguez, R. Mendonça and J. Baeza, Enzyme Microb. Technol., 2008, 43, 214–219 CrossRef CAS.
- L. Liang, Y. Y. Wang, S. Bhagia, V. Sethuraman, Z. Yang, X. Meng, N. Bryant, L. Petridis, J. C. Smith, S. V. Pingali, N. C. Gallego, Y. Pu, B. R. Evans, H. M. O’neill, B. H. Davison and A. J. Ragauskas, ACS Sustainable Chem. Eng., 2022, 10, 9041–9052 CrossRef CAS.
- L. G. Nair and P. Verma, Biomass Bioenergy, 2024, 191, 107454 CrossRef CAS.
- X. Xu, K. Wang, Y. Zhou, C. Lai, D. Zhang, C. Xia and A. Pugazhendhi, J. Fuels, 2023, 338, 127361 CrossRef CAS.
- T. vom Stein, P. M. Grande, H. Kayser, F. Sibilla, W. Leitner and P. Domínguez de María, Green Chem., 2011, 13, 1772–1777 RSC.
- E. Viola, F. Zimbardi, M. Morgana, N. Cerone, V. Valerio and A. Romanelli, Processes, 2021, 9, 2051 CrossRef CAS.
- C. S. Lancefield, O. S. Ojo, F. Tran and N. J. Westwood, Angew. Chem., Int. Ed., 2015, 54, 258–262 CrossRef CAS PubMed.
- P. J. Deuss, C. S. Lancefield, A. Narani, J. G. De Vries, N. J. Westwood and K. Barta, Green Chem., 2017, 19, 2774–2782 RSC.
- X. Li, Y. Xia, X. Hu, Q. Liu, W. Li, L. Yan and L. Ma, Ind. Crops Prod., 2023, 201, 116896 CrossRef CAS.
- J. S. Luterbacher, A. Azarpira, A. H. Motagamwala, F. Lu, J. Ralph and J. A. Dumesic, Energy Environ. Sci., 2015, 8, 2657–2663 RSC.
- F. Cheng, S. Liu, S. D. Karlen, H. Kim, F. Lu, J. Ralph, L. M. Vázquez Ramos, G. W. Huber and J. A. Dumesic, Green Chem., 2023, 25, 336–347 RSC.
- V. I. Timokhin, M. Regner, A. H. Motagamwala, C. Sener, S. D. Karlen, J. A. Dumesic and J. Ralph, ACS Sustainable Chem. Eng., 2020, 8, 17427–17438 CrossRef CAS.
- M. F. Li, S. Yang and R. C. Sun, Bioresour. Technol., 2016, 200, 971–980 CrossRef CAS PubMed.
- K. J. Yong and T. Y. Wu, Bioresour. Technol., 2023, 384, 129238 CrossRef CAS PubMed.
- R. Li, Y. Zheng, X. Zhao, Q. Yong, X. Meng, A. Ragauskas and C. Huang, Green Chem., 2023, 25, 2505–2523 RSC.
- L. Zhong, C. Wang, M. Xu, X. Ji, G. Yang, J. Chen, S. Janaswamy and G. Lyu, Energy Fuels, 2021, 35, 5039–5048 CrossRef CAS.
- D. Sarkar, I. J. Santiago and J. V. Vermaas, Chem. Eng. Sci., 2023, 272, 118587 CrossRef CAS.
- M. A. A. Farid, I. Ibrahim, J. Lease, T. Tsubota and Y. Andou, Bioresour. Technol. Rep., 2023, 24, 101622 CrossRef CAS.
- L. Chen, Z. Liang, X. Zhang, L. Zhang, S. Wang, C. Chen, L. Zeng and D. Min, Int. J. Biol. Macromol., 2022, 222, 1423–1432 CrossRef CAS PubMed.
- C. Zhang, C. Y. Ma, L. H. Xu, Y. Y. Wu and J. long Wen, Int. J. Biol. Macromol., 2021, 183, 1362–1370 CrossRef CAS PubMed.
- H. Amiri, K. Karimi and H. Zilouei, Bioresour. Technol., 2014, 152, 450–456 Search PubMed.
- M. Parchami, S. Agnihotri and M. J. Taherzadeh, Bioresour. Technol., 2022, 362, 127764 CrossRef CAS PubMed.
- F. Carvalheiro, L. C. Duarte, F. Pires, V. Van-Dúnem, L. Sanfins, L. B. Roseiro and F. Gírio, Energies, 2022, 15, 5654 CrossRef CAS.
- Z. Zhou, F. Lei, P. Li and J. Jiang, Biotechnol. Bioeng., 2018, 115, 2683–2702 CrossRef CAS PubMed.
- K. Tekin, N. Hao, S. Karagoz and A. J. Ragauskas, ChemSusChem, 2018, 11, 3559–3575 CrossRef CAS PubMed.
- C. S. Lancefield, I. Panovic, P. J. Deuss, K. Barta and N. J. Westwood, Green Chem., 2017, 19, 202–214 RSC.
- I. Panovic, C. S. Lancefield, D. Phillips, M. J. Gronnow and N. J. Westwood, ChemSusChem, 2019, 12, 542–548 CrossRef CAS PubMed.
- D. S. Zijlstra, J. de Korte, E. P. C. de Vries, L. Hameleers, E. Wilbers, E. Jurak and P. J. Deuss, Front. Chem., 2021, 9, 655983 CrossRef CAS PubMed.
- H. Liao, P. Wen, B. Feng, S. Wang and J. Zhang, ACS Sustainable Chem. Eng., 2024, 12, 12986–12996 CrossRef CAS.
- M. Morgana, E. Viola, F. Zimbardi, N. Cerone, A. Romanelli and V. Valerio, Processes, 2021, 9, 1093 CrossRef CAS.
- L. F. Del Rio, R. P. Chandra and J. N. Saddler, Appl. Biochem. Biotechnol., 2010, 161, 1–21 CrossRef CAS PubMed.
- H. Teramura, K. Sasaki, T. Oshima, F. Matsuda, M. Okamoto, T. Shirai, H. Kawaguchi, C. Ogino, K. Hirano, T. Sazuka, H. Kitano, J. Kikuchi and A. Kondo, Biotechnol. Biofuels, 2016, 9, 27 CrossRef PubMed.
- M. O. Razi and S. Sasmal, Biomass Convers. Biorefin., 2022, 12, 5221–5228 CrossRef CAS.
- Q. Schmetz, H. Teramura, K. Morita, T. Oshima, A. Richel, C. Ogino and A. Kondo, ACS Sustainable Chem. Eng., 2019, 7, 11069–11079 CrossRef CAS.
- D. S. Zijlstra, C. W. Lahive, C. A. Analbers, M. B. Figueirêdo, Z. Wang, C. S. Lancefield and P. J. Deuss, ACS Sustainable Chem. Eng., 2020, 8, 5119–5131 CrossRef CAS.
- Z. Zhang, W. O. S. Doherty and I. M. O’Hara, ACS Sustainable Chem. Eng., 2017, 5, 5284–5292 CrossRef CAS.
- B. Wang, X. J. Shen, J. L. Wen, L. Xiao and R. C. Sun, Int. J. Biol. Macromol., 2017, 97, 447–459 CrossRef CAS PubMed.
- E. Kendall Pye, Tappi J., 1991, 74(3), 113–117 Search PubMed.
- F. Lu, C. Wang, M. Chen, F. Yue and J. Ralph, Green Chem., 2021, 23, 5106–5112 RSC.
- Y. M. Questell-Santiago, M. V. Galkin, K. Barta and J. S. Luterbacher, Nat. Rev. Chem., 2020, 4, 311–330 CrossRef CAS PubMed.
- S. Zheng, S. Sun, L. P. Manker and J. S. Luterbacher, Acc. Chem. Res., 2025, 58(6), 877–892 CrossRef CAS PubMed.
- J. Behaghel De Bueren, F. Héroguel, C. Wegmann, G. R. Dick, R. Buser and J. S. Luterbacher, ACS Sustainable Chem. Eng., 2020, 8, 16737–16745 CrossRef CAS.
- L. Shuai, M. T. Amiri, Y. M. Questell-Santiago, F. Heroguel, Y. Li, H. Kim, R. Meilan, C. Chapple, J. Ralph and J. S. Luterbacher, Science, 2016, 354(6310), 329–333 CrossRef CAS PubMed.
- D. J. Davidson, F. Lu, L. Faas, D. M. Dawson, G. P. Warren, I. Panovic, J. R. D. Montgomery, X. Ma, B. G. Bosilkov, A. M. Z. Slawin, T. Lebl, A. Chatzifragkou, S. Robinson, S. E. Ashbrook, L. J. Shaw, S. Lambert, I. Van Damme, L. D. Gomez, D. Charalampopoulos and N. J. Westwood, ACS Sustainable Chem. Eng., 2023, 11, 14323–14333 CrossRef CAS PubMed.
- I. Panovic, D. M. Miles-Barrett, C. S. Lancefield and N. J. Westwood, ACS Sustainable Chem. Eng., 2019, 7, 12098–12104 Search PubMed.
- F. Foltanyi, J. E. Hawkins, I. Panovic, E. J. Bird, T. M. Gloster, C. S. Lancefield and N. J. Westwood, Biomass Bioenergy, 2020, 141, 105680 CrossRef CAS.
- Y. Guo, Y. Liu, M. Guan, H. Tang, Z. Wang, L. Lin and H. Pang, RSC Adv., 2022, 12, 18848–18863 RSC.
- S. B. Bankar, S. A. Survase, H. Ojamo and T. Granström, RSC Adv., 2013, 3, 24734–24757 RSC.
- A. Granata and D. S. Argyropoulos, J. Agric. Food Chem., 1995, 43, 1538–1544 CrossRef CAS.
- B. Saake, D. S. Argyropoulos, O. Beinhoff and O. Faix, Phytochemistry, 1996, 43(2), 499–507 CrossRef CAS.
- X. Meng, C. Crestini, H. Ben, N. Hao, Y. Pu, A. J. Ragauskas and D. S. Argyropoulos, Nat. Protoc., 2019, 14, 2627–2647 CrossRef CAS PubMed.
- ECHEMIhttp://www.echemi.com/, accessed on dates in Table S4†.
- For example Celtic Renewables, http://www.celtic-renewables.com, accessed 27/04/2025.
- Q. Wang, L. P. Xiao, Y. H. Lv, W. Z. Yin, C. J. Hou and R. C. Sun, ACS Catal., 2022, 12, 11899–11909 CrossRef CAS.
- Z. Wang, Z. Zhang, H. Wang and P. J. Deuss, Chem Catal., 2022, 2, 1407–1427 CAS.
- G. Xiao, J. R. D. Montgomery, C. S. Lancefield, I. Panovic and N. J. Westwood, Chem.–Eur. J., 2020, 26, 12397–12402 CrossRef CAS PubMed.
- Z. Zhang, D. S. Zijlstra, C. W. Lahive and P. J. Deuss, Green Chem., 2020, 22, 3791–3801 RSC.
- Z. Zhang, C. W. Lahive, D. S. Zijlstra, Z. Wang and P. J. Deuss, ACS Sustainable Chem. Eng., 2019, 7, 12105–12116 CAS.
- S. Kim, S. C. Chmely, M. R. Nimlos, Y. J. Bomble, T. D. Foust, R. S. Paton and G. T. Beckham, J. Phys. Chem. Lett., 2011, 2, 2846–2852 CrossRef CAS.
- G. T. Beckham, C. W. Johnson, E. M. Karp, D. Salvachúa and D. R. Vardon, Curr. Opin. Biotechnol., 2016, 42, 40–53 CrossRef CAS PubMed.
- E. Masai, S. Kubota, Y. Katayama, S. Kawai, M. Yamasaki and N. Morohoshi, Biosci., Biotechnol., Biochem., 1993, 57(10), 1655–1659 CrossRef CAS PubMed.
- E. Masai, Y. Katayama and M. Fukuda, Biosci., Biotechnol., Biochem., 2007, 71(1), 1–15 CrossRef CAS PubMed.
- T. D. H. Bugg, Chem. Commun., 2024, 60, 804–814 RSC.
- D. L. Gall, J. Ralph, T. J. Donohue and D. R. Noguera, Curr. Opin. Biotechnol., 2017, 45, 120–126 CrossRef CAS PubMed.
- P. Picart, H. Liu, P. M. Grande, N. Anders, L. Zhu, J. Klankermayer, W. Leitner, P. Domínguez de María, U. Schwaneberg and A. Schallmey, Appl. Microbiol. Biotechnol., 2017, 101, 6277–6287 CrossRef CAS PubMed.
- H. Guo, D. M. Miles-Barrett, A. R. Neal, T. Zhang, C. Li and N. J. Westwood, Chem. Sci., 2018, 9, 702–711 RSC.
- A. Rahimi, A. Azarpira, H. Kim, J. Ralph and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 6415–6418 CrossRef CAS PubMed.
- A. Das, A. Rahimi, A. Ulbrich, M. Alherech, A. H. Motagamwala, A. Bhalla, L. Da Costa Sousa, V. Balan, J. A. Dumesic, E. L. Hegg, B. E. Dale, J. Ralph, J. J. Coon and S. S. Stahl, ACS Sustainable Chem. Eng., 2018, 6, 3367–3374 Search PubMed.
- H. Luo, E. P. Weeda, M. Alherech, C. W. Anson, S. D. Karlen, Y. Cui, C. E. Foster and S. S. Stahl, J. Am. Chem. Soc., 2021, 143, 15462–15470 Search PubMed.
- J. D. Nguyen, B. S. Matsuura and C. R. J. Stephenson, J. Am. Chem. Soc., 2014, 136, 1218–1221 CrossRef CAS PubMed.
- M. D. Kärkäs, I. Bosque, B. S. Matsuura and C. R. J. Stephenson, Org. Lett., 2016, 18, 5166–5169 CrossRef PubMed.
- G. Magallanes, M. D. Kärkäs, I. Bosque, S. Lee, S. Maldonado and C. R. J. Stephenson, ACS Catal., 2019, 9, 2252–2260 CrossRef CAS.
- C. Zhang, H. Li, J. Lu, X. Zhang, K. E. Macarthur, M. Heggen and F. Wang, ACS Catal., 2017, 7, 3419–3429 CrossRef CAS.
- W. Lan, J. B. de Bueren and J. S. Luterbacher, Angew. Chem., Int. Ed., 2019, 58, 2649–2654 CrossRef CAS PubMed.
- L. Hong, A. Spielmeyer, J. Pfeiffer and H. A. Wegner, Catal. Sci. Technol., 2020, 10, 3008–3014 RSC.
- C. Sun, L. Zheng, W. Xu, A. V. Dushkin and W. Su, Green Chem., 2020, 22, 3489–3494 RSC.
- D. N. Dülger, Z. Yuan, S. K. Singh, S. Omolabake, C. R. Czarnecki, S. Nikafshar, M. Li, V. E. Bécsy-Jakab, S. Park, S. Park, M. Nejad, S. S. Stahl, E. L. Hegg and D. B. Hodge, Ind. Eng. Chem. Res., 2024, 63, 6182–6193 CrossRef.
- J. Ralph and B. R. Adams, J. Wood Chem. Technol., 1983, 3, 183–194 CrossRef CAS.
- J. L. Wen, S. L. Sun, B. L. Xue and R. C. Sun, Materials, 2013, 6, 359–391 CrossRef PubMed.
- J. Ralph, R. F. Helm, S. Quideau and R. D. Hatfield, J. Chem. Soc., Perkin Trans. 1, 1992, 2961–2969 RSC.
- J. Ralph, R. D. Hatfield, S. Quideau, R. F. Helm, J. H. Grabber and H.-J. G. Jung, J. Am. Chem. Soc., 1994, 116, 9448–9456 CrossRef CAS.
- R. M. Ede and J. Ralph, Magn. Reson. Chem., 1996, 34, 261–268 CrossRef CAS.
- M. Sette, H. Lange and C. Crestini, Comput. Struct. Biotechnol. J., 2013, 6, e201303016 CrossRef PubMed.
- Y. Zhang, W. Zheng, B. Jiang, Z. Li, F. Jiang, J. Cheng, J. Liu, Y. Jin and C. Zhang, ACS Sustainable Chem. Eng., 2025, 13, 5764–5766 CrossRef CAS.
- P. M. Osterberg, J. K. Niemeier, C. J. Welch, J. M. Hawkins, J. R. Martinelli, T. E. Johnson, T. W. Root and S. S. Stahl, Org. Process Res. Dev., 2015, 19, 1537–1543 CrossRef CAS PubMed.
- S. Sen, S. Patil and D. S. Argyropoulos, Green Chem., 2015, 17, 1077–1087 RSC.
- D. M. Miles-Barrett, J. R. D. Montgomery, C. S. Lancefield, D. B. Cordes, A. M. Z. Slawin, T. Lebl, R. Carr and N. J. Westwood, ACS Sustainable Chem. Eng., 2017, 5, 1831–1839 CrossRef CAS.
- F. Lu and J. Ralph, Plant J., 2003, 35, 535–544 CrossRef CAS PubMed.
- F. Tran, C. S. Lancefield, P. C. J. Kamer, T. Lebl and N. J. Westwood, Green Chem., 2015, 17, 244–249 RSC.
- Z. Fang and M. S. Meier, Org. Biomol. Chem., 2018, 16, 2330–2341 RSC.
- C. W. Lahive, C. S. Lancefield, A. Codina, P. C. J. Kamer and N. J. Westwood, Org. Biomol. Chem., 2018, 16, 1976–1982 RSC.
- T. Elder, Energy Fuels, 2014, 28, 1175–1182 CrossRef CAS.
- M. Wang, L. H. Li, J. M. Lu, H. J. Li, X. C. Zhang, H. F. Liu, N. C. Luo and F. Wang, Green Chem., 2017, 19, 702–706 RSC.
- D. B. Dess and J. C. Martin, J. Am. Chem. Soc., 1991, 113, 7277–7287 CrossRef CAS.
- R. S. Ward, A. Pelter, I. R. Jack, P. Satyanarayana, B. V. Gopala Rao and P. Subrahmanyan, Tetrahedron Lett., 1981, 22(41), 4111–4114 CrossRef CAS.
- A. R. Abouelela, S. Y. Tan, G. H. Kelsall and J. P. Hallett, ACS Sustainable Chem. Eng., 2020, 8, 14441–14461 CrossRef CAS.
- L. Weigand, S. Mostame, A. Brandt-Talbot, T. Welton and J. P. Hallett, Faraday Discuss., 2017, 202, 331–349 RSC.
- G. F. De Gregorio, C. C. Weber, J. Gräsvik, T. Welton, A. Brandt and J. P. Hallett, Green Chem., 2016, 18, 5456–5465 RSC.
- A. Brandt, J. Gräsvik, J. P. Hallett and T. Welton, Green Chem., 2013, 15, 550–583 RSC.
- R. Ammara, L. Fradette and J. Paris, Chem. Eng. Res. Des., 2016, 115, 107–115 CrossRef CAS.
- M. E. Leblebici, G. D. Stefanidis and T. Van Gerven, Chem. Eng. Process., 2015, 97, 106–111 CrossRef CAS.
- A. Lazaridou, L. R. Smith, S. Pattisson, N. F. Dummer, J. J. Smit, P. Johnston and G. J. Hutchings, Nat. Rev. Chem., 2023, 7, 287–295 CrossRef CAS PubMed.
- I. Delidovich, P. J. C. Hausoul, L. Deng, R. Pfützenreuter, M. Rose and R. Palkovits, Chem. Rev., 2016, 116, 1540–1599 CrossRef CAS PubMed.
- A. Brandt-Talbot and L. Weigand, Faraday Discuss., 2017, 202, 11–30 RSC.
- D. Argyropoulos, H. Bitter, A. Brandt-Talbot, V. Budarin, C. Chesi, J. Clark, M. Coma, C. Crestini, B. Dale, I. Graca, J. Hallett, C. Hu, X. Huang, G. Huber, T. Hughes, A. Hunt, E. Kontturi, Y. Luo, M. Mascal, A. Matharu, V. Matveeva, A. Mount, X. Ouyang, R. Rinaldi, G. Rothenberg, J. Samec, S. Sarkanen, C. M. Seidel, C. Stevens, V. Thaore, K. Waldron, K. Wilson, F. Xie and D. S. Zijlstra, Faraday Discuss., 2017, 202, 371–389 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2445448. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5fd00074b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |