
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electroanalytical overview: the sensing of the mycophenolate mofetil and mycophenolic acid
Robert D.
Crapnell
 and
Craig E.
Banks
*
and
Craig E.
Banks
*
Faculty of Science and Engineering, Manchester Metropolitan University, Dalton Building, Chester Street, Manchester, M1 5GD, UK. E-mail: c.banks@mmu.ac.uk; Tel: +441612471196
First published on 17th October 2024
Abstract
In this review, we explore the electroanalytical determination of mycophenolate mofetil and mycophenolic acid. Mycophenolate mofetil is a prodrug of mycophenolic acid, which is an immunosuppressive agent used to lower the body's natural immunity in patients who receive organ transplants as well as to treat autoimmune conditions. Laboratory based analytical instrumentation provide a routine methodology to measure mycophenolate mofetil and its metabolites, but there is scope to develop in-the-field analytical measurements that are comparable to those from laboratory equipment. Electroanalysis provides an opportunity to provide highly selective and sensitive outputs but are cost-efficient and can support on-site analysis. In this review, we provide an electroanalytical overview of the current research directed toward the measurement of mycophenolate mofetil and mycophenolic acid, offering insights to future research.
1. Introduction to mycophenolate mofetil
Mycophenolate mofetil is a prodrug of mycophenolic acid, and classified as a reversible inhibitor of inosine monophosphate dehydrogenase (IMPDH);1,2Fig. 1 shows the chemical structures of both compounds. This drug is an immunosuppressant combined with other drugs such as corticosteroids to prevent organ rejection after hepatic, renal, and cardiac transplants and to treat autoimmune conditions such as lupus, lung fibrosis and Crohn's disease.1,3–6 Interestingly, mycophenolic acid was first isolated as a fermentation product of Penicillium brevicompactum cultures by Bartolomeo Gosio (see Fig. 1B)† in 1896.7,8 Mycophenolic acid received attention due to its antifungal, antitumor, antibacterial, antiviral and immunosuppressive properties based on data from early studies.8 Furthermore, the immunosuppressive effect of mycophenolate mofetil is, in fact, achieved due to its metabolite, mycophenolic acid. The oral bioavailability of mycophenolic acid is low, therefore it is administered as a prodrug, mycophenolate mofetil. Both mycophenolate mofetil and mycophenolic acid needs to be monitored by clinicians due to its potent immunosuppressive effects and potential side effects, which may include increased risk of infections and certain cancers.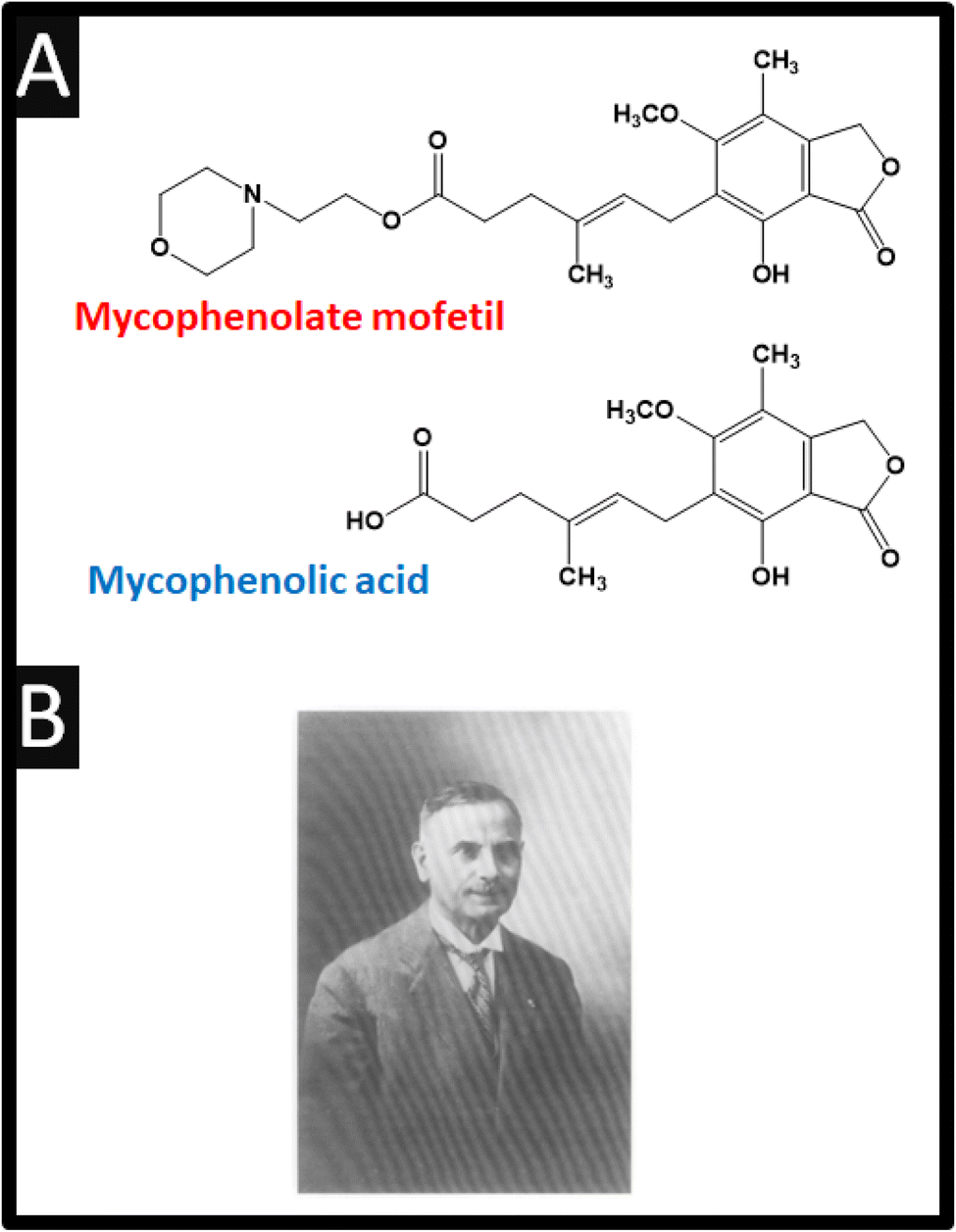 | ||
| Fig. 1 (A) Chemical structure of mycophenolate mofetil and mycophenolic acid. (B) Image of Bartolomeo Gosio. Image reproduced from ref. 7. Copyright 2001 Elsevier. | ||
The pharmacokinetics of mycophenolate mofetil report that when administrated orally, it is absorbed in the small intestine. After which, it is hydrolysed to mycophenolic acid by plasma esterases, reaching the peak plasmatic concentration within 60 to 90 minutes, noting that mycophenolate mofetil is undetectable in plasma.9,10 Mycophenolic acid is bound to albumin and its principal inactive metabolite is mycophenolic acid glucuronide. It is reported that 87% of mycophenolic acid is excreted in urine and 6% in faeces, with less than 1% of the administered dose of mycophenolate mofetil excreted as the active drug metabolite, mycophenolic acid.8 Mycophenolic acid has a half-life average of 17 hours where whole blood results indicate more than 99% of drug remains within plasma, supporting the rationale for the measurement of mycophenolic acid within serum or plasma.10 Therapeutic monitoring typically measures mycophenolic acid levels and associated metabolites as an aid in the management of mycophenolic acid therapy. Samples are usually taken at specific times post-dose e.g., 1–2 hours after dosing for therapeutic range for mycophenolic acid 1.0–3.5 μg mL−1 (∼3.1–11 μM) or mycophenolic acid glucuronide 35–100 μg mL−1 (∼70–201 μM) for a 2 g day−1 dose; greater than 25 μg mL−1 (∼50 μM) are classed as toxic.11 A 3 g day−1 dose may have plasma concentrations up to 5.0 μg mL−1 (∼10 μM).11 Mycophenolic acid has been shown that this is metabolized to mycophenolic acid β-D-glucuronide, mycophenolic acid acyl glucuronide and mycophenolic acid phenolic glucoside.12
Useful approaches have been reported for the quantification of mycophenolic acid and its metabolites in human plasma and urine to ensure to ensure therapeutic efficacy and minimise toxicity. This is particularly important in patients undergoing organ transplantation. For example, capillary electrophoretic13 and high performance liquid chromatography with UV (HPLC-UV)14 have been reported, as well as ultra-high performance chromatography – tandem mass spectrometry has been used to measure mycophenolic acid and its metabolites in human plasma and urine.15 Note that a protein precipitation solution comprising 30% of aqueous 0.2 M ZnSO4/70% methanol is needed where 200 μL of the human plasma/urine is mixed with 600 μL of the precipitation solution which are then centrifuged.15 Other approaches use a CEDIA® mycophenolic acid immunoassay, but it is reported that high performance chromatography – tandem mass spectrometry which have a superior specificity over immunoassays.15 Such approaches provide analytical techniques with high sensitivity and selectivity, but drawbacks include the requirement for highly skilled operators, high operational costs with and extensive analysis time involving pre-concentration step(s), calibration, preparation and sampling.16
An alternative approach that can rival the laboratory methods, as described above, is the use of electrochemistry. This is an influential tool for quantitative chemical analysis and finds applications in a wide range of fields allowing for real-time monitoring and precise measurements, a field that is termed as electroanalysis. In comparison to traditional laboratory instrumentation that require bulky, complex to perform, time-consuming and expensive instrumentation, electroanalysis provides an affordable and easy to use solution which are portable, rapid analysis times, yet provide sensitivity and selective approaches towards the analyte being measured. There are many available potentiostats that are hand-held, battery operated, and can be controlled by mobile devices via bluetooth allowing in situ measurement to be realised.17 In this review, we consider the use of electroanalytical approaches for the measurement of mycophenolate mofetil and mycophenolic acid.
2. Electroanalytical sensing of mycophenolate mofetil
We have summarised all electroanalytical reports reported for the sensing of mycophenolate mofetil within Table 1, which reports useful linear ranges and low limits of detection (LoD), with the various modification of electrochemical substrates shown. The direct electrochemical oxidation of mycophenolate mofetil is limited as this results in high overpotential, slow kinetics, poor sensitivity and low selectivity; as one can observed within Table 1, these intrinsic problems are overcome through the use of surface modifications using various materials. For example, Solgi et al.18 report on the use of adsorptive anodic stripping differential pulse voltammetry using a magnetic Fe3O4 nanoparticles and functionalized carboxylated multi-walled carbon nanotubes modified glassy carbon electrode (GCE). This gave rise to a linear range of 0.05–200 μM with a LoD of 9 nM towards mycophenolate mofetil. The same group extends their direction for the sensing of mycophenolate mofetil opting for a molecularly imprinted polymer which produced a linear range of 9.9 nM–87 μM with a LoD of 7 nM and provides a simple, free of interference sensing approach.19| Electrode | Modification | Linear range | Limit of detection | Sample medium | Comments | Ref. |
|---|---|---|---|---|---|---|
| a Key: CPE, carbon paste electrode; Cu-1N-allyl-2-(2,5-dimethoxyphenyl)-4,5-diphenyl-1H-imidazole metal organic framework; Cu-porphyrin nanosheets, Cu(II) tetrakis(4-carboxyphenyl)porphyrin; ERGO, electroreduced graphene oxide; f-MWCNTs, functionalized (carboxylated) multi-walled carbon nanotubes; GCE, glassy carbon electrode; MOF/MWCNTs, multi-walled carbon nanotubes; SPE, screen-printed electrodes; SWCNTs, single wall carbon nanotubes; Zn–Co MOF, zinc–cobalt metal organic framework; GO, graphene oxide. | ||||||
| GCE | — | 0.5–750 μM | 0.148 μM | Pharmaceutical sample, human urine and serum | Mycophenolate mofetil | 20 |
| Pencil | MOF/MWCNTs | 8.5 nM–1.5 μM and 11 nM–1.7 μM | 2.8 nM and 3.6 nM | Human urine and plasma | Simultaneous determination of mycophenolate mofetil and tacrolimus | 21 |
| GCE | Fe3O4/f-MWCNTs | 0.05–200 μM | 9 nM | Human urine and serum | Mycophenolate mofetil | 18 |
| CPE | Molecularly imprinted polymer/MWCNTs | 9.9 nM–87 μM | 7 nM | Human urine and serum | Mycophenolate mofetil | 19 |
| CPE | Ionic liquid/SWCNTs/MgO | 0.1–450 μM | 0.07 μM | Pharmaceutical sample and human serum | Mycophenolate mofetil and tryptophan | 22 |
| GCE | ERGO | 40–15 μM | 11.3 nM | Pharmaceutical sample | Mycophenolate mofetil | 23 |
| Carbon cloth | 3-Aminopropyltriethoxysilane functionalized nickel cobaltite (NiCo2O4) | 10–100 nM and 1–100 μM | 1.23 nM | Artificial samples of blood serum and cerebrospinal fluid | Mycophenolate mofetil | 24 |
| Electrochemically assisted surface-enhanced Raman spectroscopy | Gold nanopillars | 1–50 μM | 1.7 μM | — | Mycophenolic acid | 25 |
| CPE | β-Cyclodextrin/multi-walled carbon nanotubes/cobalt oxide nanoparticles | 0.5–200 μM | 0.03 μM | Human urine and serum | Simultaneous determination of warfarin and mycophenolic acid | |
| SPE | Cu-prophyrin nanosheets | 1–200 μM | 10 nM | — | Mycophenolic acid | 26 |
| GCE | Zn–Co MOF/Ti3C2 MXene/Fe3O4-GO | 32 nM–8.9 μM | 21 nM | Grass silage | Mycophenolic acid | 27 |
| SPE | Violet phosphorene/porous carbon microsphere | 2.49–71.1 μM | 18.7 nM | Corn and wheat silage | Mycophenolic acid | 28 |
| GCE | MWCNTs | 5–160 μM; 2.5–60 μM | 0.9 μM; 0.4 μM | Human plasma and urine | Mycophenolate mofetil and mycophenolic acid | 29 |
| GCE | Chitosan – MWCNTs/Au nanoparticles | 0.001–0.1 μM | 0.05 μM | Rat plasma | Mycophenolic acid | 30 |
| Carbon cloth | (3-Aminopropyl)triethoxysilane (APTES)-functionalized Nb2CTx MXene nanosheets | 10–100 μM | 1 μM | Human serum | Mycophenolate mofetil | 31 |
| CPE | Poly(yellow PX4R) | 10–70 μM | 2 μM | Human urine | Simultaneous determination of mycophenolate mofetil and dopamine | 32 |
Other useful approaches report the use of an ionic liquid (n-hexyl-3-methylimidazolium hexafluoro phosphate) decorated carboxylated single wall carbon nanotube modified with magnesium oxide.22 The authors used commercially available single wall carbon nanotubes which are modified with magnesium oxide using a chemical hydrothermal methodology resulting in single crystalline nanoparticles. These are next mixed with graphite powder using diethyl ether as a solvent. After evaporation of solvent, these are combined with the ionic liquid and paraffin oil, which are then placed into a glass tube with a copper wire to connect this to the potentiostat.22 This electrode was explored towards the sensing of mycophenolate mofetil in the presence of tryptophan which gave a LoD of 0.07 μM and a linear range of 0.1–450 μM. This was further explored for the sensing of mycophenolate mofetil at a concentration of 5 μM where the potential interferent are added, namely: glucose, glycine, vitamin B9, isoleucine, vitamin B6, ascorbic acid, lithium, potassium, sodium, and chloride ions, all of which did not interfere. The authors demonstrated their sensor is useful toward real applications by measuring mycophenolate mofetil within a pharmaceutical sample. The pharmaceutical sample was prepared by grinding 10 tablets of mycophenolate mofetil by mortar and pestle which are dissolved in 100 mL 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 water/ethanol via ultrasonication. After ultrasonication, the solution was filtered using filter paper and it is ready for real sample analysis. Notably, they compared their sensor with HPLC which showed good agreement to both methodologies; this approach offers a rapid yet sensitive approach for the determination of mycophenolate mofetil within pharmaceutical samples that can be used routinely saving the problems of using HPLC (see introduction). This rapid yet sensitive approach has merits used in the routine sensing within pharmaceutical samples.
:1 water/ethanol via ultrasonication. After ultrasonication, the solution was filtered using filter paper and it is ready for real sample analysis. Notably, they compared their sensor with HPLC which showed good agreement to both methodologies; this approach offers a rapid yet sensitive approach for the determination of mycophenolate mofetil within pharmaceutical samples that can be used routinely saving the problems of using HPLC (see introduction). This rapid yet sensitive approach has merits used in the routine sensing within pharmaceutical samples.
One approach has used electrochemically reduced graphene oxide and explored the sensing of mycophenolate mofetil. Graphene oxide was made via the usual Hummers' method, which is then drop-coated onto the surface of a GCE. It is then electrochemically reduced within pH 6 by potential cycling between +0.6 V to −1.6 V 25 times, producing 6 layers of individual graphene oxide sheets, after which this electrode is ready to use.23 The authors report that this produced two irreversible oxidation peaks at +0.84 V and +1.1 V (see later), which gave a linear response to mycophenolate mofetil of 40 nM–15 μM with a LoD of 11.3 nM. This approach was shown to be useful in the measurement of mycophenolate mofetil within pharmaceutical samples and exhibited a recovery of 99.34–98.00%.23
An interesting material has been used for the sensing of mycophenolate mofetil, as shown within Fig. 2, 3-aminopropyltriethoxysilane functionalized nickel cobaltite (NiCo2O4) has been fabricated using a solvothermal methodology which produces sea urchin type nanostructured material which has a size of 10.32 nm.24 This material is drop-casted onto a carbon cloth which gave a linear response of 10–100 nM and 1–100 μM with a LoD of 1.23 nM. This sensor was explored to interferents, namely: ascorbic acid, dopamine, glutamic acid, KCl, NaCl, glucose, glycine and alanine, in addition to immunosuppressant drugs, tacrolimus and cyclosporine. Using a concentration of 10 times more than mycophenolate mofetil, negligible response to the interferents was observed, highlighting the selectivity towards mycophenolate mofetil. The reproducibility and long-term storage stability of the electrodes were studied, which showed that over 21 days that the repeatability of the sensor is useful with only a standard deviation of 1.54%.24 Last, this sensor was explored within artificial blood serum samples and cerebrospinal fluid, where the authors made additions of mycophenolate mofetil which gave a linear range of 10–100 nM and a LoD of 6.8 nM. Further work needs to be conducted to assess its use within biological and clinical diagnostics.
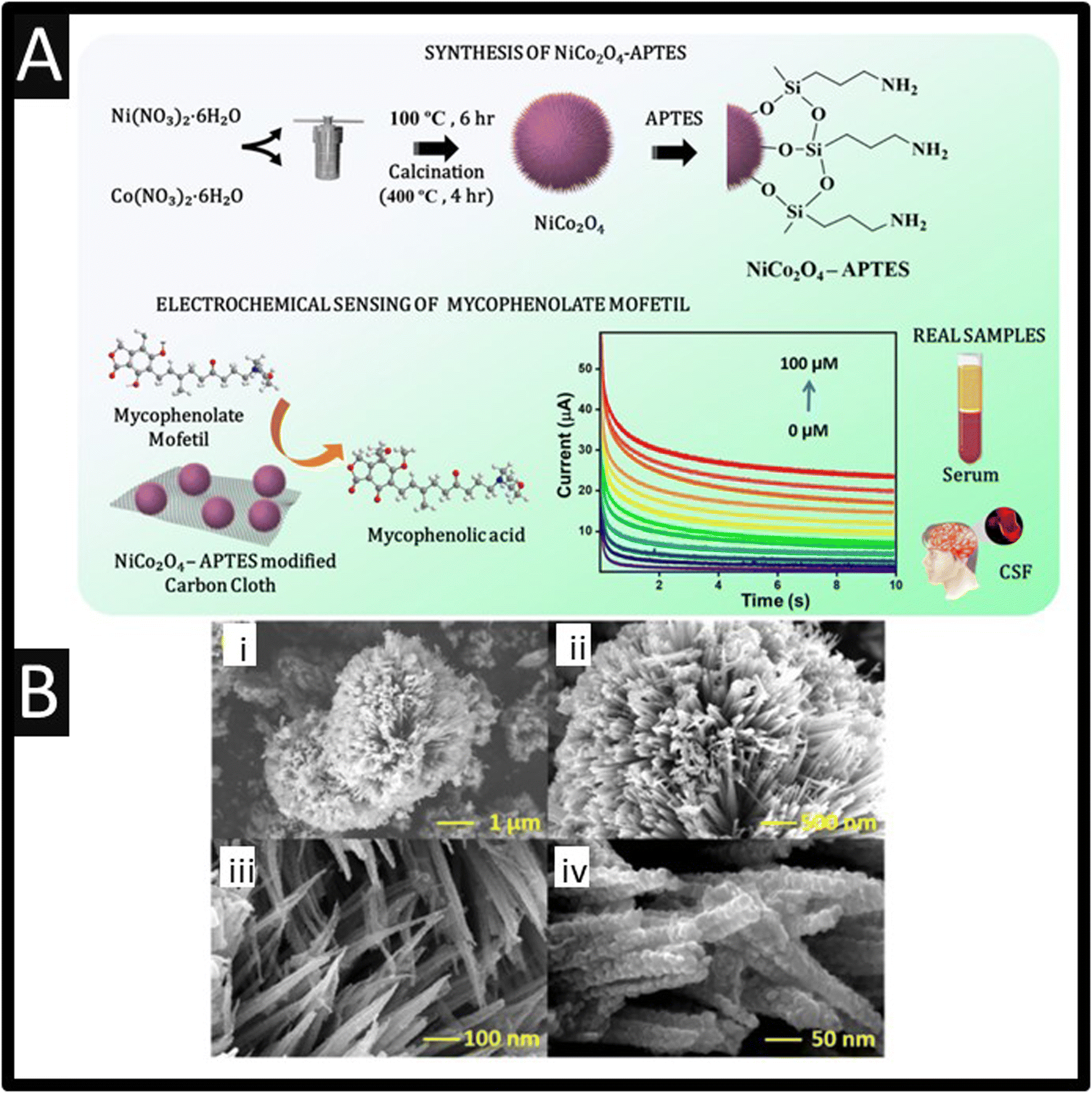 | ||
| Fig. 2 (A) Synthesis of 3-aminopropyltriethoxysilane modified NiCo2O4 and its application in the electrochemical sensing of mycophenolate mofetil. (B) SEM images of (i) sea urchin nanostructured NiCo2O4. (ii) Magnified image of the urchin model of NiCo2O4. (iii) Needles of the sea urchin model of NiCo2O4. (iv) Magnified image of nanoneedles showing the nanospheres. Figures reproduced from ref. 24 Copyright 2024 Royal Society of Chemistry. Creative Commons license. | ||
The electrochemical oxidation of mycophenolate mofetil has been studied using a GCE, where it can be observed, as shown within Fig. 3A, there are two oxidation peaks at +0.63 V and +0.92 V (vs. SCE). These show that on successive cyclic voltammetric sweeps, the signal is diminished where it is reported that this is due to the adsorption of the oxidized product on the electrode surface.20 Ideally, the authors need to refresh the electrode surface via mechanical polishing to ensure that the largest electroanalytical signal can be realised.
 | ||
| Fig. 3 (A) Cyclic voltammograms of 1.25 × 10−4 M mycophenolate mofetil in phosphate buffer (pH 6.0) at a scan rate 100 mV s−1. 1 to 5 are the successive voltammetric sweeps. (B) A summary of the electrochemical mechanism of mycophenolate mofetil. Figures are reproduced from ref. 20 Creative Commons license. | ||
The authors used bulk electrolysis for 6 h at a fixed potential of +1.2 V, which were isolated and subjected to mass spectral analysis. This showed three peaks at m/z values of 334, 320, and 302, respectively, corresponding to the fragmentation peaks of oxidation products of mycophenolate mofetil namely, (i), (ii), and (iii), respectively.20 The first oxidation (α1) peak, Fig. 3A, is attributed to electrochemical oxidation occurring at phenolic –OH followed by a nucleophilic attack at the para position to OH group by either water molecule or methanol.20 The second oxidation (α2) peak is attributed to oxidation of enolic –OH group – see Fig. 3B for an overview.20 The authors report a useful linear range of 0.5–750 μM with a LoD of 0.148 μM towards mycophenolate mofetil. The authors claim that their sensor is useful for the measurement of mycophenolate mofetil as evidence by analysing a pharmaceutical sample and also spiked urine and serum samples. Other approaches have reported the use of MXene nanosheets (Nb2CTx-APTES) towards mycophenolate mofetil which have been funcationsed with (3-aminopropyl) triethoxysilane which reports a linear range of 10–100 μM and a LoD of 1 μM.31 This sensor has promise as it has been shown to be successful in the measurement of mycophenolate mofetil within spiked human serum samples reported recoveries of 85.5–93.6%.31
3. Electroanalytical sensing of mycophenolic acid
Table 1 also summarises the sensing of mycophenolic acid, where it can be observed that various approaches have been reported. For example, using violet phosphorene which is a new 2-dimensional material and the most stable allotrope of phosphorus, or the use of MXene which is a class of two-dimensional inorganic compounds that consist of atomically thin layers of transition metal carbides, nitrides, or carbonitrides.33 Zhang and co-workers report the fabrication of a bulk modified screen-printed electrochemical platform which was modified with copper based porphyrin nanosheets of 6.6 nm thickness.26 Using this approach, a LoD of 10 nM is achievable but they are yet to explore their sensor in the measurement of mycophenolic acid in real samples. Notably, they used HPLC-MS/MS via chronoamperometry at an applied potential +0.67 V (vs. Ag/AgCl) within pH 7.4 which allowed them to confirm the electrochemical oxidation of mycophenolic acid involves a two-electron and two-hydrogen oxidation process, followed by a nucleophilic attack of water molecules at the para position of the hydroxyl group; see Fig. 4A.26 Furthermore, the electrochemical mechanism has been studied using electrochemistry coupled with liquid chromatography and tandem mass spectrometry (HPLC/ESI-MS/MS). This was used to study the oxidation products of mycophenolic acid using a boron-doped diamond electrode within pH 3.5 over the range of 0–3 V (vs. Pd/H2), which is summarised within Fig. 4B.34 Since the authors compared their electrochemical oxidation of mycophenolic acid against pH, we suggest that the mechanism is as suggested: see Fig. 4A.26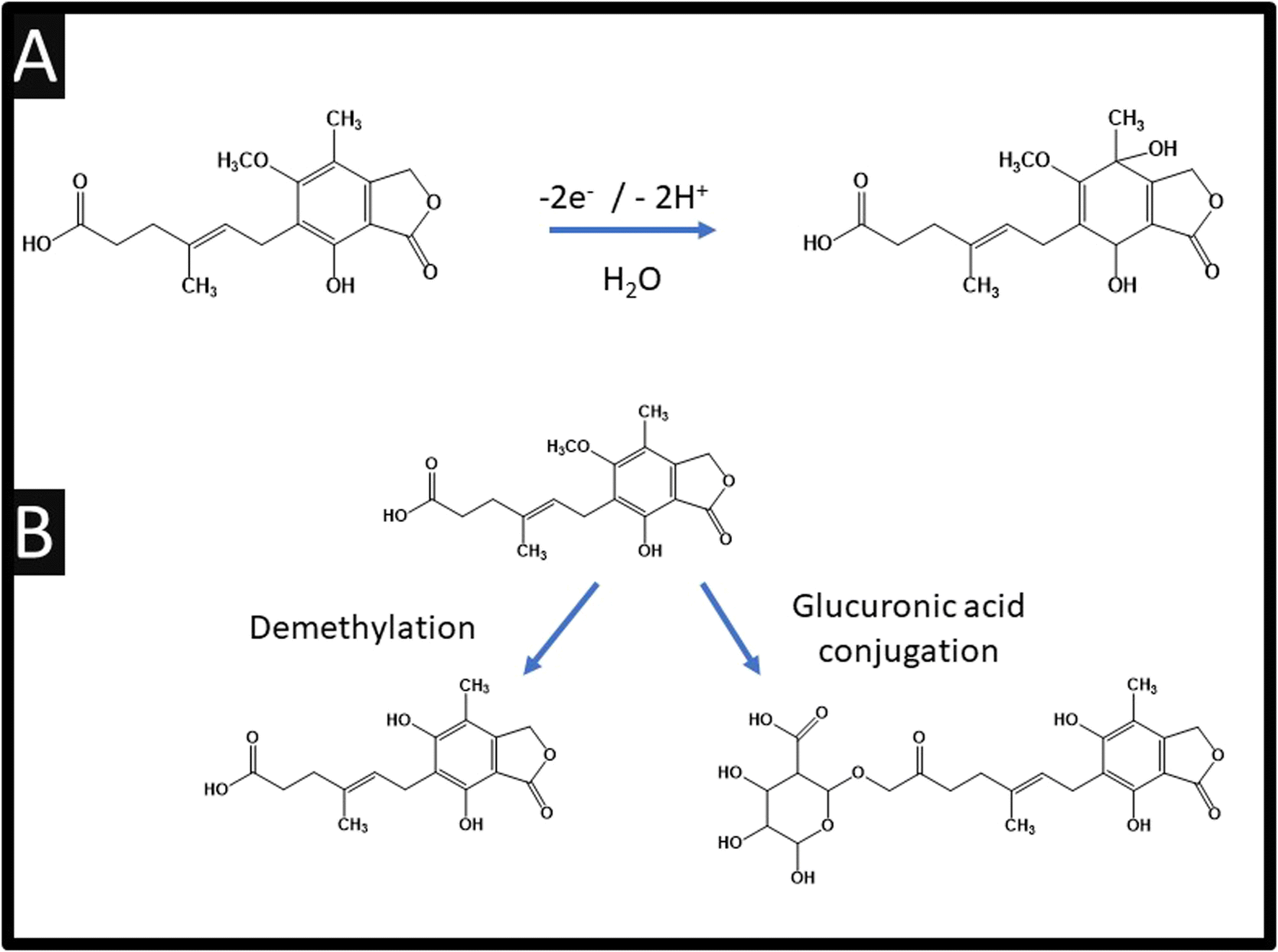 | ||
| Fig. 4 (A) Electrochemical oxidation mechanism of mycophenolic acid; (B) schematic structures of possible oxidation products for mycophenolic acid. | ||
Warfarin and mycophenolic acid were determined simultaneously using a β-cyclodextrin/multi-walled carbon nanotubes/cobalt oxide nanoparticles modified carbon paste electrode.35 This electrode gave well-resolved electrochemical oxidation peaks and a linear range of 0.5–200 μM and a LoD of 0.03 μM. The author demonstrated that the presence of Co3O4 nanoparticles improved the electrocatalytic activity but also increased the surface area providing a suitable substrate for adsorbing drugs at the electrode surface.35 This sensor is explored further where they report the sensing of mycophenolic acid in human urine and serum. Human urine samples are collected from a 40 year-old man who had not used any mycophenolic acid (and warfarin) during his lifetime. The urine was filtered through a 0.45 μm filter and diluted 10 times with addition of buffer (pH = 5), then spiked with mycophenolic acid. This sensor exhibited recoveries across the range of 97.5–99.6%. In terms of the human blood serum samples, these were obtained from a local medical laboratory from volunteers who had not used any mycophenolic acid (and warfarin). The use of methanol is reported as sedimentation agent which was added to each serum sample. Next, the samples are centrifuged (10 min at 5000 rpm) for separation of the precipitated proteins. The clear supernatant layer was further filtrated through a 0.45 μm filter obtaining protein-free human serum sample where it is diluted in a ratio of 1:10 with buffer (pH = 5). This sensor exhibited recovery across the range of 98.7–101%. Further work needs to validate the sensors response against laboratory-based instrumentation.
The use of Raman fingerprint region and surface-enhanced Raman spectroscopy vibrational study of mycophenolic acid has been studied using gold nanopillars.25 As shown in Fig. 5A, the effect of electrochemical assisted surface-enhanced Raman spectroscopy when different charges are applied from +0.8 V to −0.8 V. It can be readily seen that the potential of +0.2 V corresponds to potential of zero charge and also the most intensity can be seen in acidic medium.25 Using optima electrochemical parameters which involved preconditioning with −0.1 V for 60 s then +0.8 V for 60 s, and surface-enhanced Raman spectroscopy detection during +0.2 V, this approach allows the sensing of mycophenolic acid over the range of 1–50 μM with a LoD of 1.7 μM (see Fig. 5B). Furthermore, the use of electrochemically assisted surface-enhanced Raman spectroscopy can be observed (Fig. 5B) where detection sensitivity can be reached by applying the optimum potentials to the surface-enhanced Raman spectroscopy chip, improving mycophenolic acid adsorption on the surface-enhanced Raman spectroscopy surface. This improvement, in sensitivity and repeatability, can be attributed to improved control of molecular attraction and orientation on the surface-enhanced Raman spectroscopy substrate through applied potentials.25 This is an interesting approach that should be considered further for the sensing of mycophenolic acid.
 | ||
| Fig. 5 (A) Dependence of the 1327 cm−1 mycophenolic acid band intensity on the PBS buffer pH and on the potential applied to the surface-enhanced Raman spectroscopy substrate. Mycophenolic acid concentration was 100 μM. Potential-dependant spectral changes of mycophenolic acid. 100 μM in PBS pH 2.7, after wetting in a mycophenolic acid free PBS pH 2.7 solution (−0.8 V for 60 s followed by +0.8 V for 60 s) when the potential was changed from +0.8 V to −0.8 V with 0.2 V steps (vs. Ag/AgCl). The black arrows indicate the direction of potential changes during potential sweeps. The error bars in (a) represent the standard deviation of 3 measurements. The vertical dashed lines in (b) highlight the position of mycophenolic acid characteristic peaks. (B) Calibration curves obtained for mycophenolic acid with the optimised electrochemically assisted surface-enhanced Raman spectroscopy conditions (EC-SERS, black curve, preconditioning of −0.1 V for 60 s followed by +0.8 V for 60 s, and surface-enhanced Raman spectroscopy detection during 0.2 V) and without any potential applied (red curve). Comparison of the surface-enhanced Raman spectroscopy and electrochemically assisted surface-enhanced Raman spectroscopy spectra of mycophenolic acid at lower concentrations, highlighting the gain in sensitivity obtained with electrochemically assisted surface-enhanced Raman spectroscopy. The in-house built tabletop spectrophotometer used to acquire the spectra is displayed. Figure reproduced from ref. 25 Copyright 2024 Elsevier. | ||
The use of multi-walled carbon nanotubes (outer diameter between 70 and 90 nm and inner diameter between 5 and 9 nm) deposited upon a GCE have been reported for the simultaneous determination of mycophenolate mofetil and its active metabolite, mycophenolic acid using adsorptive differential pulse voltammetry.29 As shown in Fig. 6A, two electrochemical profiles can be observed at +0.87 V and +1.1 V (vs. Ag/AgCl), which are well resolved and are superior to that of a bare glassy carbon electrode. This sensor reports the simultaneous determination of mycophenolate mofetil and its active metabolite, mycophenolic acid reporting linear ranges of 5–160 μM and 2.5–60 μM, and LoDs of 0.9 μM and 0.4 μM respectively. The repeatability and stability of the sensor were investigated by cyclic voltammetric measurements of 10 μM mycophenolate mofetil and 30 μM mycophenolic acid, where it was found that relative standard deviation (RSD) for successive assays was 1.4% and 1.2% and retained 95% of the initial response after 2 weeks suggesting that the sensor has good stability and reproducibility. The sensor was explored towards interferents, where no interference is reported for the sensing of mycophenolate mofetil and mycophenolic acid in the presence of glucose, fructose, ascorbic acid, starch, calcium, magnesium, and carbonate. This sensor was evaluated in the sensing of mycophenolate mofetil and mycophenolic acid within spiked urine and serum samples which was directly compared with HPLC, showing excellent correlation. This sensor has could be applied to the determination of mycophenolate mofetil and mycophenolic acid in biological samples giving a selectivity and sensitivity approach.
 | ||
| Fig. 6 (A) Differential pulse and cyclic voltammograms of 4 μM mycophenolic acid and 20 μM mycophenolate mofetil. pH 5 with (i) MWCNTs/GCE and (ii) bare GCE. Figure reproduced from ref. 29. Copyright 2014 Elsevier. (B) An overview of chitosan – MWCNTs/gold nanoparticles/GCE sensor. Figure reproduced from ref. 30. Copyright 2023 Elsevier. | ||
A notable approach has reported a chitosan altered multi-walled carbon nanotube with gold nanoparticle modified electrode towards mycophenolic acid.30 A GCE was polished using alumina and placed into a gold salt solution where gold nanoparticles are formed via electrochemical precipitation through holding the potential negative for 10 seconds. Next, chitosan and multi-walled carbon nanotubes are drop-cast onto the gold nanoparticle glassy carbon surface and the sensor is ready to use. This sensor gave rise to a linear range of 0.001–0.1 μM towards mycophenolic acid with a LoD of 0.05 μM. This sensor is evaluated in spiked rat plasma. Blood samples are obtained from healthy rats, where they precipitated the proteins with acetonitrile and centrifuged at 12000 rpm for 10 min. Using the supernatant as a blank plasma sample, it is diluted five times diluted in a phosphate-buffered solution (pH 6). The spiked mycophenolic acid is evaluated at 3, 10 and 30 μM which are directly compared with HPLC showing good correlation. This sensor noted that the analysis took 15 min or less and it is much cheaper and quicker than the HPLC method (60–120 min) suggesting that the electrochemical approach has potential for practical applications.30
4. Conclusions
We have overviewed the use of electroanalytical sensors for the determination of mycophenolate mofetil and mycophenolic acid. Researchers have used a wide range of material modifications, mainly onto the surface of glassy carbon electrodes. To fully utilise the advantages that electrochemistry can provide, a movement toward low-cost electrodes that do not require treatment such as screen-printed or additively manufactured electrodes are recommended. In comparison to laboratory-based instrumentation, which measures all metabolites within human plasma/serum, in the case of electrochemistry, only two have been determined, e.g., mycophenolate mofetil and mycophenolic acid. The electrochemistry field needs to expand its use to measure all metabolites, within real samples and needs to compare their analysis against laboratory-based instruments; chromatography with electrochemical detection is suggested. The use of electrochemistry has the possibility to provide sensitive and selective approaches to the measurements of the target analytes while being economical, portable and is quicker than laboratory-based instrumentation; but yet there is more to do to ensure that electrochemistry can realise its potential and validate electrochemical results to ensure confidence in the approach.Data availability
No data was used for the research described in the article.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.References
- A. C. Allison, Lupus, 2005, 14(Suppl 1), s2–s8 CrossRef CAS.
- C. E. Staatz and S. E. Tett, Clin. Pharmacokinet., 2007, 46, 13–58 CrossRef CAS.
- P. Fickert, T. A. Hinterleitner, H. H. Wenzl, B. W. Aichbichler and W. Petritsch, Am. J. Gastroenterol., 1998, 93, 2529–2532 CrossRef CAS.
- M. Walsh, M. James, D. Jayne, M. Tonelli, B. J. Manns and B. R. Hemmelgarn, Clin. J. Am. Soc. Nephrol., 2007, 2, 968–975 CrossRef CAS.
- J. J. Lipsky, Lancet, 1996, 348, 1357–1359 CrossRef CAS.
- M. Behrend, Expert Opin. Invest. Drugs, 1998, 7, 1509–1519 CrossRef CAS.
- R. Bentley, in Advances in Applied Microbiology, Academic Press, 2001, vol. 48, pp. 229–250 Search PubMed.
- H. Park, J. Clin. Aesth. Dermatol., 2011, 4, 18–27 Search PubMed.
- R. Bullingham, S. Monroe, A. Nicholls and M. Hale, J. Clin. Pharmacol., 1996, 36, 315–324 CrossRef CAS.
- D. W. Holt, Ann. Clin. Biochem., 2002, 39, 173–183 CrossRef CAS.
- https://ltd.aruplab.com/Tests/Pub/2010359 .
- M. Shipkova, C. P. Strassburg, F. Braun, F. Streit, H.-J. Gröne, V. W. Armstrong, R. H. Tukey, M. Oellerich and E. Wieland, Br. J. Pharmacol., 2001, 132, 1027–1034 CrossRef CAS.
- K. Ohyama, N. Kishikawa, H. Nakagawa, N. Kuroda, M. Nishikido, M. Teshima, H. To, T. Kitahara and H. Sasaki, J. Pharm. Biomed. Anal., 2008, 47, 201–206 CrossRef CAS.
- H. Danafar and M. Hamidi, Adv. Pharm. Bull., 2015, 5, 563–568 CrossRef CAS.
- J. Klepacki, J. Klawitter, J. Bendrick-Peart, B. Schniedewind, S. Heischmann, T. Shokati, U. Christians and J. Klawitter, J. Chromatogr. B, 2012, 883–884, 113–119 CrossRef CAS.
- C. Jin Mei and S. Ainliah Alang Ahmad, Arab. J. Chem., 2021, 14, 103303 CrossRef CAS.
- R. D. Crapnell, I. V. Arantes, J. R. Camargo, E. Bernalte, M. J. Whittingham, B. C. Janegitz, T. R. Paixão and C. E. Banks, Microchim. Acta, 2024, 191, 96 CrossRef CAS.
- M. B. Gholivand and M. Solgi, Anal. Biochem., 2017, 520, 1–8 CrossRef CAS.
- H. Momeneh and M. B. Gholivand, Anal. Biochem., 2018, 557, 97–103 CrossRef CAS.
- S. N. Prashanth, K. C. Ramesh and J. Seetharamappa, Int. J. Electrochem., 2011, 2011, 193041 Search PubMed.
- M. H. Mahnashi, A. M. Mahmoud, S. A. Alkahtani, R. Ali and M. M. El-Wekil, Anal. Bioanal. Chem., 2020, 412, 355–364 CrossRef CAS.
- M. Ashjari, H. Karimi-Maleh, F. Ahmadpour, M. Shabani-Nooshabadi, A. Sadrnia and M. A. Khalilzadeh, J. Taiwan Inst. Chem. Eng., 2017, 80, 989–996 CrossRef CAS.
- P. S. Narayan, N. L. Teradal, S. Jaldappagari and A. K. Satpati, J. Pharm. Anal., 2018, 8, 131–137 CrossRef.
- K. Niyas, B. Richard, M. Ankitha and P. A. Rasheed, Analyst, 2024, 149, 3615–3624 RSC.
- E. Dumont, J. J. Castillo, C. E. Rozo, G. Zappalá, R. Slipets, L. H. E. Thamdrup, T. Rindzevicius, K. Zor and A. Boisen, Sens. Actuators, B, 2024, 417, 136126 CrossRef CAS.
- M. Deng, W. Jin, W. Yang, L. Tian, X. Gao, X. Wang, L. Feng, M. Janyasupab, B. Zhou and Y. Zhang, ACS Appl. Nano Mater., 2023, 6, 22979–22988 CrossRef CAS.
- Y. Ge, M. B. Camarada, P. Liu, M. Qu, Y. Wen, L. Xu, H. Liang, E. Liu, X. Zhang, W. Hao and L. Wang, Sens. Actuators, B, 2022, 372, 132627 CrossRef CAS.
- Y. Ge, P. Liu, Q. Chen, M. Qu, L. Xu, H. Liang, X. Zhang, Z. Huang, Y. Wen and L. Wang, Biosens. Bioelectron., 2023, 237, 115454 CrossRef CAS.
- T. Madrakian, M. Soleimani and A. Afkhami, Mater. Sci. Eng., C, 2014, 42, 38–45 CrossRef CAS.
- Y. Xiong, S. Zhu, H. Zhao, J. Li, Y. Li, T. Gong, Y. Tao, J. Hu, H. Wang and X. Jiang, Anal. Biochem., 2023, 677, 115265 CrossRef CAS.
- P. V. Vaishag, M. Ankitha and P. A. Rasheed, ACS Appl. Nano Mater., 2023, 6, 23324–23331 CrossRef CAS.
- G. S. Sumanth, B. E. Kumara Swamy, K. Chetankumar and S. C. Sharma, Sens. Technol., 2024, 2, 2361612 CrossRef.
- R. D. Crapnell and C. E. Banks, Talanta Open, 2021, 4, 100065 CrossRef.
- M. Szultka-Mlynska and B. Buszewski, J. Pharm. Biomed. Anal., 2019, 176, 112799 CrossRef CAS.
- M.-B. Gholivand and M. Solgi, Anal. Chim. Acta, 2018, 1034, 46–55 CrossRef CAS.
Footnote |
| † Bartolomeo Gosio (1863–1944) is an Italian physician who made diverse but yet important contributions to microbiology. Gosio discovered “Gosio Gas” where he showed that fungal volatilization of arsenic into trimethylarsine and furthermore showed that penicillium brevicompactum produced mycophenolic acid and furthermore discovered an antibacterial compound against the anthrax bacterium. See an overview of Gosio: ref. 7. R. Bentley, in Advances in Applied Microbiology, Academic Press, 2001, vol. 48, pp. 229–250. |
| This journal is © The Royal Society of Chemistry 2024 |