
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Screen printed 3D microfluidic paper-based and modifier-free electroanalytical device for clozapine sensing†
Mohammad Hossein
Ghanbari
 ab,
Markus
Biesalski
b,
Oliver
Friedrich
c and
Bastian J. M.
Etzold
*ab
ab,
Markus
Biesalski
b,
Oliver
Friedrich
c and
Bastian J. M.
Etzold
*ab
aFriedrich-Alexander-Universität Erlangen-Nürnberg, Power-To-X Technologies, 90762 Fürth, Germany. E-mail: bastian.etzold@fau.de
bTechnische Universität Darmstadt, Ernst-Berl-Institute for Technical Chemistry and Macromolecular Science, Peter-Grünberg-Straße 8, 64287, Darmstadt, Germany
cInstitute of Medical Biotechnology, Department of Chemical and Biological Engineering, Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU), Paul-Gordan-Str. 3, 91052, Erlangen, Germany
First published on 4th October 2024
Abstract
The increasing demand in healthcare for accessible and cost-effective analytical tools is driving the development of reliable platforms to the customization of therapy according to individual patient drug serum levels, e.g. of anti-psychotics in schizophrenia. A modifier-free microfluidic paper-based electroanalytical device (μPED) holds promise as a portable, sensitive, and affordable solution. While many studies focus on the working electrode catalysts, improvements by engineering aspects e.g. of the electrode arrangement are less reported. In our study, we demonstrate the enhanced capabilities of the 3D electrode layout of μPED compared to 2D μPED arrangements. We especially show that screen printing can be employed to prepare 3D μPEDs. We conducted a comparison of different 2D and 3D electrode arrangements utilizing cyclic voltammetry in [Fe(CN)6]3−/4−, along with square-wave voltammetry for clozapine (CLZ) sensing. Our findings reveal that the utilization of the 3D μPED leads to an increase in both the electrochemically active surface area and the electron transfer rate. Consequently, this enhancement contributes to improve sensitivity in the CLZ sensing. The 3D μPED clearly outperforms the 2D μPED arrangement in terms of signal strength. With the 3D μPED under the optimized conditions, a linear dose–response for a concentration range from 7.0 to 100 μM was achieved. The limit of detection and sensitivity was determined to be 1.47 μM and 1.69 μA μM−1 cm−2, respectively. This evaluation is conducted in the context of detection and determination of CLZ in a human blood serum sample. These findings underscore the potential of the 3D μPED for future applications in pharmacokinetic analyses and clinical tests to personalize the management of schizophrenia.
1. Introduction
The healthcare landscape is evolving towards a new era where decentralized and rapid analysis plays a pivotal role. This shift is witnessing a decline in the widespread use of conventional clinical laboratory analyses in favor of cost-effective laboratory testing and decentralized point-of-care testing. Embracing novel sensing techniques is becoming increasingly appealing. Miniaturized, accessible, user-friendly, and equipment-free detection tools are significantly reducing decision-making time for subsequent testing or treatment. Moreover, the elimination of delays in test results due to specimen transport and preparation ensures rapid availability at the point of care.1–3 This facilitates direct diagnosis in medical facilities or primary care settings, enabling prompt responses and swift treatment decisions. Nevertheless, it remains imperative for these tests to maintain high accuracy and sensitivity, ensuring a reliable and quality diagnosis process. Reliable and less expensive sensors stand as promising solutions to fulfill these criteria, offering minimal sample volume requirements, along with miniaturized, portable devices, and user-friendly procedures. Furthermore, a range of detection techniques, including colorimetry, fluorimetry, and electrochemistry, are being employed to develop highly sensitive and accurate sensing devices.3–6Among various options, paper-based electrochemical detection tools present notable attributes including miniaturization, mobility, eco-friendliness, and cost-effectiveness, along with the capability to function in turbid and colorful environments.7–9 In line with this, the principles of Green Analytical Chemistry which have garnered significant attention over the past decade,4,10–13 strive to phase out plastic and testing cartridges currently employed in analytical methods due to their inability to be recycled or disposed of properly.
Moreover, cellulose's porous nature facilitates the creation of capillary-driven microfluidics and reagent-free devices.14–18 Paper has been instrumental in crafting innovative electrochemical sensors applicable across diverse fields, including environmental,19 agri-food,20 biomedical,21 and defense sectors.22 It is noteworthy to underscore that paper's utility extends beyond the simple replacement of other types of electrochemical sensors; it fundamentally enhances the functionality of electrochemical platforms.23 Paper-based electrochemical devices demonstrate the capability to detect target analytes in untreated samples.24 This approach has attracted significant interest from the scientific community across various electrochemical applications, such as sensing and biosensing applications.1,25
So far, screen-printed carbon-based electrodes (SPCEs) with 2D arrangements have found widespread utility across various scientific disciplines and engineering fields. Conversely, microfluidic electrochemical sensors (μCS) with possible 3D electrode arrangement and continuous analyte flow serve as an alternative to SPCEs, showcasing notable performance albeit receiving comparatively less attention in research and practical applications. The 2D electrode arrangement of SPCE causes the electrochemically active surface area (ECSA) to be transferred to the edge of the working electrode, as a result ECSA becomes smaller. On the other hand, μCS with a 3D electrode arrangement using graphite foil as electrodes, as well as paper strip as a spacer and microfluidic channel between electrodes and analyte carrier effectively enabled a high electrode utilization and additionally continuous mass transfer of analyte to detection sites. In fact, the 3D electrode arrangement allows the μCS to provide a larger electrochemically active surface area (ECSA) as the counter electrode is placed exactly on the surface of the working electrode where a paper strip is sandwiched between them. For instance, in a recent study, we compared the performance of SPCE and μCS setups for heavy metal sensing and depicted that those intrinsic properties of the μCS configuration lead to superior performance over SPCE in stability, selectivity, sensitivity using real sample tests.26 Although the 3D electrode arrangement of μCS is important for the superior results, up to now, graphite foil was pressed onto the paper strip to achieve this.8,26 This opens the question whether the electrodes could be printed onto/into the paper, as known from 2D SPCE, which would be a more scalable technique for mass production. Furthermore, we are interested whether the resulting sensors could also be employed for applications in pharmacokinetic analyses and clinical tests to personalize the management of, e.g., schizophrenia by sensing of clozapine (CLZ).
Schizophrenia presents as a debilitating mental condition marked by disruptions in reality perception, often manifesting in hallucinations and behaviors impairing daily functioning.27,28 Among the medications for schizophrenia treatment, CLZ emerges as particularly promising.29–31 Unlike others, its efficacy can be gauged through blood tests, with therapeutic levels typically ranging from 1 to 3 μM in human blood tests32,33 which is a narrow therapeutic margin. Despite its widespread usage, CLZ administration is linked with significant adverse effects, underscoring the critical necessity for precise dosage management approaches. Various analytical techniques, including spectrophotometry and chromatography, are commonly employed to monitor CLZ levels in diverse formulations and real samples.32–34 Yet, despite their recognized accuracy and reliability, these methods are not without drawbacks. High-performance liquid chromatographic (HPLC) methods for instance, tend to be time-consuming and relatively costly due to the utilization of expensive reagents, equipment, and eluents while spectroscopic techniques may exhibit reduced sensitivity.27,28
In 2023, CLZ determination was conducted by Ghanbari et al. utilizing nickel-doped graphene nanotubes modified by a glassy carbon electrode (Ni@GRNT/GCE).35 The electrode's analytical performance was evaluated within a concentration range of 0.3 nM to 60 μM, achieving an LOD of 0.1 nM. Similarly, in another study, CLZ sensing was performed by employing a miniaturized unmodified toray paper-based electrochemical sensor.28 The electrode's analytical performance was assessed over a concentration range of 0.5 to 5.0 μM, yielding an LOD of 0.01 μM. Despite the extensive studies for CLZ sensing in static voltammetry using glassy carbon electrodes or SPCEs, their application to microfluidic paper-based electroanalytical devices (μPED) has been limited,30–35 and to the best of our knowledge, no study reports the fabrication of a modifier-free 3D μPED for CLZ sensing.
In this work, we introduce a low cost, sensitive and portable 3D μPED based on direct electrode printing on/in both sides of a flexible filter paper substrate to decrease the distance between the electrodes, which facilitates its use while benefiting from the microfluidic behavior of paper for CLZ sensing. Using electrode printing with varying 2D and 3D electrode arrangements and electrode dimensions are conducted to study the influence and compare the performance. As the 3D μPED arrangement showed superior performance, also the analysis of human blood serum samples was carried out and confirmed the excellent sensitivity and selectivity of such devices.
2. Experimental
2.1. Chemicals and apparatus
The chemicals and solvents utilized were of analytical-grade purity and were employed without further purification steps. These substances included clozapine (Thermo Fisher Scientific, UK), screen printable carbon paste (DYCOTEC, UK), conductive silver printing ink, resistivity 5–6 μΩ cm (Sigma-Aldrich, Australia), K3Fe(CN)6, 99.98% (Sigma-Aldrich, Taufkirchen, Germany), K4Fe(CN)6, 98.5% (Sigma-Aldrich, Taufkirchen, Germany), methanol (Sigma-Aldrich, France), filter paper grade 591 (Whatman™, China, standard, material: cellulose, basis weight: 161 g m−2, thickness: 350 μm, particle retention in liquid max.: 12 μm, flow rate: approx.: 45 s−1), buffer solution (Carl Roth, Germany), A4 glossy self-adhesive paper (Evergreen Goods Ltd, China), and other reagents were obtained from Sigma-Aldrich, Carl Roth or VWR. All solutions were prepared using double-distilled water. CLZ was dissolved in methanol to achieve a 1.0 mM CLZ stock solution.Scanning electron microscope (SEM) images were captured using a FEI-Helios NanoLab 600i FIB and were acquired by an Everhardt-Thornley detector (ETD) for secondary electrons (SE). Electrochemical investigations were performed employing a Multichannel Potentiostat (Ivium, Octostat 5000 and Vertex, Eindhoven, Netherlands) driven with IviumSoft software.
To evaluate the impact of different fabricated sensors, cyclic voltammograms (CVs) were carried out using 5.0 mM [Fe(CN)6]3−/4− and 0.1 M KCl as a redox probe solution. To assess the performance of various platforms in the enhancement of the surface area, ECSA, A (cm2), representing the surface roughness value. This was achieved by recording CV curves in the redox probe solution at different scan rates (v) ranging from 60 mV s−1 to 200 mV s−1 (Fig. S1†) and employing the Randles–Sevcik equation.36,37 Ultimately, the performance of 3D μPEDs was compared against 2D μPED.
2.2. Human blood serum sample preparation
Human blood serum samples were obtained from the Blood Bank (University Hospital Erlangen Transfusion Medicine and Hemostasiology Department) Erlangen, Germany. The samples were diluted with 0.1 M PBS at pH 8.0 at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5. To minimize interference from other substances at higher concentrations, precise concentrations of the CLZ were added into each diluted sample (1 mL of serum diluted with 4 mL of PBS). Subsequently, the SWV signals for each prepared solution were measured independently by the sensor utilizing standard addition procedure.
:5. To minimize interference from other substances at higher concentrations, precise concentrations of the CLZ were added into each diluted sample (1 mL of serum diluted with 4 mL of PBS). Subsequently, the SWV signals for each prepared solution were measured independently by the sensor utilizing standard addition procedure.
2.3. Fabrication of the 2D and 3D μPED
The configuration of the component for the custom-produced μPED are outlined in Schemes 1, S1 & S2.† The glossy paper stencil is used to create the mask for the electrode geometry. The design of stencil patterns for 3D μPEDs, and 2D μPED devices was generated using Autodesk Inventor 2023, the schematic geometry of the dimensions (mm) of 2D μPED and 3D μPEDs is outlined in Scheme S3.† Next, the computer-controlled laser cutting tool was employed to transfer the sensor pattern into the glossy paper stencil. This process finally prepares the masks for the sensors. For the 3D μPED, a filter paper was sandwiched between two stencils to minimize ohmic resistance and increasing the surface of the working electrode (WE) facing the counter electrode (CE), using carbon paste as the WE and the CE and conductive silver printing ink as the pseudo-reference electrode (RE) the electrodes were manually screen-printed on both side of paper (WE & RE on one side of the paper, CE on the backside of paper) then the stencils were removed with the help of a hair dryer. At the following step, a fluidic channel was generated by employing a clear nail polish onto/into the filter paper (by creating a hydrophobic zone). Next, the paper was placed into an oven at 70 °C for 30 min to cure the inks.9–14 At the following phase, the sensors were cut to the appropriate dimensions. To maintain cleanliness and prevent contamination, the poly(methyl methacrylate) (PMMA) substrate and sampling sponge were cleaned before μPED fabrication. Cleaning procedure involved sonication in ethanol and deionized water mixture (1:1) for 15 minutes at 300 W, followed by air-drying at room temperature before use. Next, the sensor was placed onto a PMMA substrate for user convenience. An analyte reservoir, composed of a piece of sponge at one end of the paper channel, held 2.0 mL of analyte solution, continuously supplying the analyte solution over a 15 minute period.8,26 To prevent concentration changes of the analyte due to water evaporation, a glass vial was used to envelope the sampling sponge. An absorbent pad was located at the opposite end of the paper channel to facilitate fluid wicking through the paper channel, which maintained a consistent analyte flow within the paper channel and allowed to collect the analyte passing through the detection sites (Scheme 2).8,26
 | ||
| Scheme 1 Scheme illustration the 3D μPED (d) assembling process and subsequent use in CLZ sensing. | ||
 | ||
| Scheme 2 (A) The schematic geometry of the dimensions (mm) for the 3D μPED, and (B) images of the fabricated sensor. | ||
2.4. Electrochemical measurements
Electrochemical investigations were carried out using a Potentiostat (Ivium, Octostat 5000 and Vertex, Eindhoven, Netherlands), and all experiments were conducted at room temperature. Cyclic voltammetry (CV) was utilized throughout the experiment via [Fe(CN)6]3−/4 as a redox probe solution to compare various platforms under a scan rate of 100 mV s−1 from −1.0 V to +1.0 V.CLZ detection was carried out via square wave voltammetry (SWV), with the following parameter settings: pulse height at 25 mV; step height at 10 mV; and frequency set to 25 Hz from −0.5 V to +1.0 V. To verify that the analyte solution adequately reached the electrode surfaces, electrochemical measurements were initiated following a 30 s holding time prior starting the 30 s accumulation time at open circuit potential for detecting CLZ. A Savitzky Golay algorithm was then applied to smooth the data using IviumSoft (11 points).
The limit of quantification (LOQ) and detection (LOD) were determined using the expression k·s/m.38 Here, “k” is assigned a value of 10 and 3 for LOQ and LOD, respectively. “s” and “m” represent the standard deviation of the blank's peak current (n = 4) and the slope of the calibration plot for CLZ, respectively.
Recovery quantifies the sensor's ability to accurately detect and measure a predetermined added amount of analyte within the complex matrix of the human blood serum sample. Typically expressed as a percentage, it is calculated using the equation:

3. Results and discussion
3.1. Physicochemical characterization of the sensor
The resulting morphology of different parts of the μPED were characterized by SEM. The images are given in Fig. 1. Fig. 1A depicts an SEM image for the paper surface before electrode printing where the shape and distribution of fibers and resulting porous structure can be clearly seen. Fig. 1B and C, show the electrodes’ surface (WE, CE and RE). No voids or agglomeration are visible, confirming the homogeneity of the coating resulting from the carbon paste and silver ink, respectively. It can be also observed from Fig. 1C that silver ink well distributed spherical silver particles, agglomerated in clusters. Fig. 1E and F, clearly show the edges created between paper and electrode from carbon paste, as well as electrode from silver ink, respectively. Fig. 1G shows a cross-sectional image of printing WE and CE on/in both sides of paper leading to the successful assembly of the printing electrodes. It can be seen from Fig. 1D that the porous structure of the paper substrate is completely filled with nail polish and the edges created between paper and hydrophobic barrier, which shall, as a hydrophobic barrier, limit the flow within the microfluidic application. Fig. 1H shows a cross-sectional image of the hydrophobic barrier which results in a good construction of the μPED device. In fact, it is fully impregnating the volume, but also showing an edge between the hydrophobic and hydrophilic area. Here, due to capillary forces, the edge is less straight compared to the electrodes and can vary by approx. 30 μm, which is sufficient for defining the microfluidic channels. | ||
| Fig. 1 SEM images showing the morphology of the paper surface (A), WE and CE (B), RE (C) edges created with hydrophobic barrier (D), carbon paste (E) and silver ink (F). A cross-sectional view of the printing WE and CE on/in both sides of the paper (G) and resulting channels defined by hydrophobic barriers (H). | ||
3.2. Electrochemical characterization of electrodes
The possibility to easily vary the electrode size and arrangement with the screen-printing approach was employed to obtain different 2D and 3D μPEDs (Fig. 2A). To gain a deeper understanding of the position and ratio of the working and counter electrodes, four distinct sensors were fabricated. As illustrated in Scheme S3,† the working electrode geometry remains constant across all four sensors, while the size and position of the counter electrode vary. The ratio of electrodes is the same in design (a) and (b), while for design (b), the counter electrode is printed on the back of the paper to create a 3D structure. Designs (c) and (d) are also 3D arrangements. In design (c), the counter electrode area is roughly doubled compared to (b). The ratio of electrodes in design (c) and (d) are almost the same; however, in design (d), the counter electrode is printed exactly opposite of the working electrode on the back side of the paper. For assessing the quality of the electrodes and comparing the different electrode arrangements, CVs of platforms were recorded in the redox probe solution containing 5.0 mM of [Fe(CN)6]3−/4−. The inter-peak distance provides crucial information about the electrochemical reaction, with small distances indicating fast, reversible reactions, and larger distances suggesting slower or quasi-reversible processes. As can be seen in Fig. 2B, two well-defined redox peaks with a peak-to-peak separation (ΔEp) of around 35, 50, 55 and 38 mV are observed for the (a), (b), (c) and (d) designs, respectively. As illustrated in design (d), the redox couple exhibits a shift towards higher voltages. A potential explanation for this phenomenon could be the influence of altered local concentrations and, consequently, thermodynamic potentials, differences in kinetic processes, as well as mass transfer effects, but also differences at the quasi-reference electrode. Given that only a quasi-reference electrode is employed, it is possible that the geometrical changes may have an impact. With regard to design (d), it resulted that the CV is shifted in potential, rather than the redox peak moving closer together or further apart. This is an indication that reasons are less likely to be attributed to kinetic or mass transport factors, and more likely to be a consequence of the quasi-reference electrode. By employing the 3D μPED arrangements, peak currents increase markedly, the peak currents are around 20, 49, 52 and 58 μA for the (a), (b), (c) and (d) designs, respectively. This behavior is indicating that the 3D electrode layout reduces the distance between the electrodes which enhances the reversible nature of the electrochemical reaction. The resolution for the inter-peaks is relatively low for this setup, as the objective is not to conduct a comprehensive CV characterization. This is why SWV is employed e.g. for the analysis and not CV or LVS.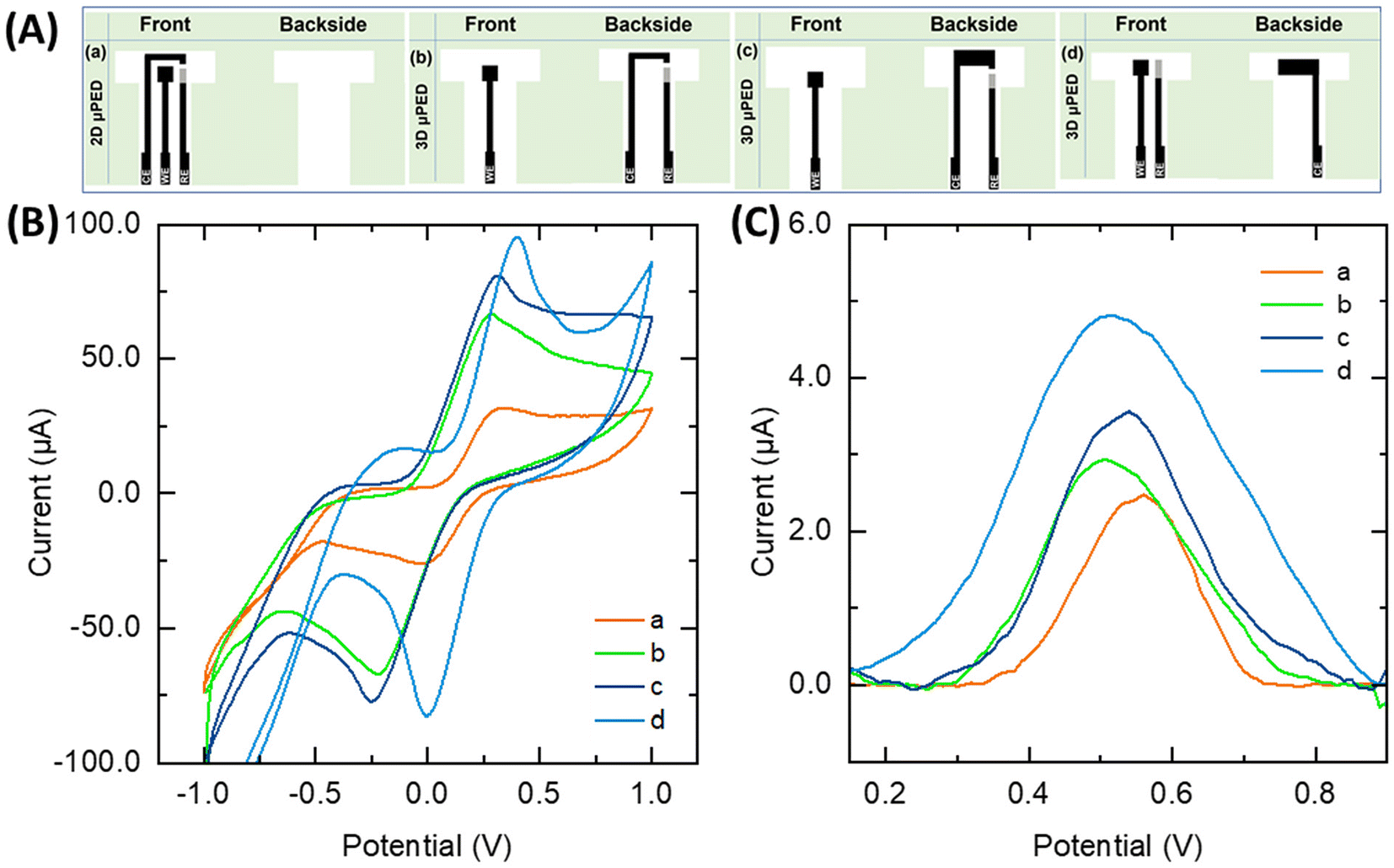 | ||
| Fig. 2 (A) The schematic of the prepared sensors. (B) CV of different platforms which were recorded in a redox probe solution consisting of 5.0 mM of [Fe(CN)6]3−/4− and 0.1 M KCl under a scan rate of 100 mV s−1. (C) The recorded SWVs for 50 μM CLZ under the optimum conditions. | ||
The ECSA determined with the redox probe of the different μPEDS (a), (b), (c), and (d) designs is 0.0184, 0.0185, 0.0297 and 0.0521 cm2, respectively. Despite the same geometrical working electrode size, the ECSA differs strongly, which is probably because of the different electric fields building up for the different arrangements and thus, the different utilization of the available working electrode area. The 2D electrode arrangement causes the ECSA to be transferred to the edge of the working electrode, as a result ECSA becomes smaller while the 3D electrode arrangement provides a larger ECSA as the counter electrode is placed exactly opposite to the working electrode on the back side of the paper, which improves the overall efficiency of the electrochemical reactions. Additionally, as illustrated in Fig. S2A,† the peak current has a correlation with the ECSA, which is in agreement with the Randles–Sevcik equation.36,37 Consequently, employing the 3D μPED, increases the ECSA and amplification the current signal of the platform which the highest ECSA was observed for the 3D μPED (d).
3.3. Comparison of electrode arrangements, effect of scan rate and assessing pH influence for CLZ sensing
In evaluating the performance of the different electrode arrangements for CLZ sensing (Fig. 2D), the sensing and electrocatalytic properties of the platforms were examined in the presence of 50.0 μM CLZ at pH = 8.0 (0.1 M PBS) and accumulation time of 30 s and followed by sensing via SWV. All platforms distinctly exhibited an oxidation peak for CLZ. Notably, a clear difference between all sensor arrangements can be observed. A comparison of all 2D and 3D μPED, results show that (b), (c), and (d) designs increase the peak current by a factor of 1.3, 1.5, and 1.9, respectively, when compared to the (a) design. The previously noted increased ECSA with the 3D μPED (d) thus, leading to improve capabilities for detecting CLZ. The peak currents in CLZ sensing via SWV have a correlation with the ECSA which is graphically shown in Fig. S2B.†Therefore, the 3D μPED (d) arrangement was selected for further study and optimization. The following parameters were studied: (i) the effect of potential scan rate; (ii) the SWV parameter (data given in ESI†); and (iii) the pH of the supporting electrolyte.
For the best performing system, the additional characterization with CVs at different scan rates was carried out. To examine the kinetics parameters and electrochemical behavior of the sensor, the corresponding CVs were explored over scan rates from 60 to 250 mV s−1 in the electrolyte solution at pH 8.0, in the presence of 100.0 μM CLZ (Fig. 3A). A linear correlation was evident between the anodic peak current (Ipa) and the scan rate (ν) within the 60 to 250 mV s−1 range, denoted by an R2 value of 0.986, which shows the adsorptive nature of the electrochemical reaction for the oxidation of CLZ onto the WE surface of the sensor (Fig. 3B). Furthermore, with an increase in scan rate, a shift towards more positive potentials was observed, confirming the kinetics limitations inherent to the electrochemical reaction. The relationship between Epa and logν within the 60 to 250 mV s−1 range is linear, as illustrated in Fig. 3B. The corresponding linear regression equation is Epa = 0.108logν + 0.331, with a correlation coefficient of 0.990. The Tafel slope (b) can be determined from the Epavs. logν slope using the following equation:35,38

 | ||
| Fig. 3 (A) CVs of the proposed sensor in a solution containing 100 μM CLZ with 0.1 M PBS at pH = 8.0 for different scan rates; from inner to outer scan rates of 60, 80, 100, 150, 200 and 250 mV s−1, respectively. (B) Variation of Ipavs. ν and Epavs. logν; (C) SWVs for 30.0 μM CLZ at various pH values ranging from pH 6.0 to 10.0 (D) Effect of pH of CLZ solutions on Ipa and Epa. | ||
The determined Tafel slope recorded a value of 0.21 V. Employing the number of electrons engaged in the oxidation of CLZ in PBS at the sensor and the Tafel slope, the transfer coefficient (α) was calculated to be 0.9 using the following equation:36,38

The pH of the electrolyte solution is another critical factor in the catalytic determination of the target analyte, and the influence of electrolyte pH on the peak current of CLZ was investigated by adjusting the pH values from 6.0 to 10.0 using SWV (Fig. 3C). It was shown that the shape and peak potential of CLZ oxidation depend on electrolyte pH. Therefore, it is suggested that protons participate in the CLZ electro-oxidation reaction. As one can see from Fig. 3C, peak current showed a rapid rise as the pH values increased from 6.0 to 8.0. However, further increasing the pH to 10.0 led to a decrease in peak currents observed for CLZ measurement. The optimal pH identified for this study was 8.0. As one can see from Fig. 3D, by plotting oxidation peak potential against pH, a linear relationship between the oxidation peak potential and pH resulted with an R2 value of 0.981. The calculated slope closely aligns with the theoretical value of 59 mV, offering an equivalent contribution of protons and electrons (2e−–2H+) in the oxidation of CLZ at the sensor within the pH ranging from 6.0 to 10.0. Therefore, as shown in Scheme S4,† it supports the proposed mechanism of the electro-oxidation reaction of CLZ.31,36,38
3.4. Analytical performance
 | ||
| Fig. 4 (A) SWVs of proposed sensor for different concentrations of CLZ in 0.1 M PBS at pH = 8.0, (B) the calibration plot of peak current vs. CLZ concentration and current density vs. the different CLZ concentrations (error bars represent the standard deviation for the three different technical replicates (n = 3)). | ||
The outcomes demonstrate that the approach is rapid, cost-effective, and exhibits noteworthy sensitivity. The analytical characteristics of the sensor are compared with various electrochemical methods and devices in CLZ monitoring as presented in Table 1. The results indicate the proposed strategy is comparable in terms of LOD and/or response linear concentration range.
| Method | Technique | Deposition time (s) | Potential (mV) | pH | Liner range (nM) | LOD (nM) | Real samples | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Differential pulse voltammetry. b Toray paper electrochemical device. c Chronoamperometry. d Titanium oxide nanoparticle. e Carbon pate electrode. f Lab-in-a-pencil graphite microfluidic sensing electrode. g Multi walled carbon nanotube. h Nafion-modified multi walled carbon nanotube. i Square-wave voltammetry. j Tungsten trioxide nanoparticles hydride by α – terpineol. | ||||||||
| Graphene modified Au | DPVa | — | 340 | 7.4 | — | 700 | Blood | 39 |
| TPEDb | CAc | — | 320 | 7.4 | 5.0 × 10+2–5.0 × 10+3 | 10 | Blood | 28 |
| TiO2NPd/CPEe | DPV | 420 | 370 | 9.0 | 5 × 10+2–45 × 10+3 | 61.0 | Tablet | 31 |
| μFSEf | Amperometry | — | 320 | 7.4 | 1.0 × 10+2–1.0 × 10+4 | 24.0 | Blood | 32 |
| Ion selective electrode | Potentiometry | — | — | 4.5–8.0 | 1.0 × 10+4–1.0 × 10+7 | 3700 | Tablet | 40 |
| MWCNTg/CPE | Potentiometry | — | — | 3.5–5.0 | 1.0 × 10+3–1.0 × 10+7 | 1000 | Tablet | 41 |
| Nf/MWCNT on CTSh | SWVi | 60 | 150 | 7.4 | 1.0 × 10+2–5.0 × 10+3 | 83 | Blood | 42 |
| MWWTj/GCE | SWV | — | 462 | 7.0 | 1.0 × 10+2–2.0 × 10+3 | 30.0 | Urine and blood | 38 |
| 3D μPED | SWV | 30 | 520 | 8.0 | 7.0 × 10 +3 –1.0 × 10 +5 | 1470 | Blood serum | This work |
To analyze biological samples employing the proposed sensor, an examination of the influence of common electrochemical species in human blood was conducted via SWV. Selectivity studies were performed to assess the sensor's capability for CLZ detection in the presence of potential interfering substances. In order to accomplish this, solutions containing 12.0 μM CLZ at 0.1 M PBS and pH 8.0 were tested alongside other substances including 24 μM glucose, 24 μM uric acid, 24 μM theophylline, 24 μM ascorbic acid, and a solution containing all at the same time. Results show with the mentioned interfering species did not significant interfere with 12.0 μM CLZ, and sensor was able to detect CLZ even in the presence of these interfering substances. The peak currents normalized to the peak current without further substances (I/I0) are indicated in Fig. 5. I and I0 represent SWV peak current responses of CLZ in the presence and absence of interferences, respectively. The sensor displayed a minimal current variation and exhibited significant selectivity in detecting CLZ in the presence of glucose, uric acid, theophylline, and ascorbic acid as common interfering substances.
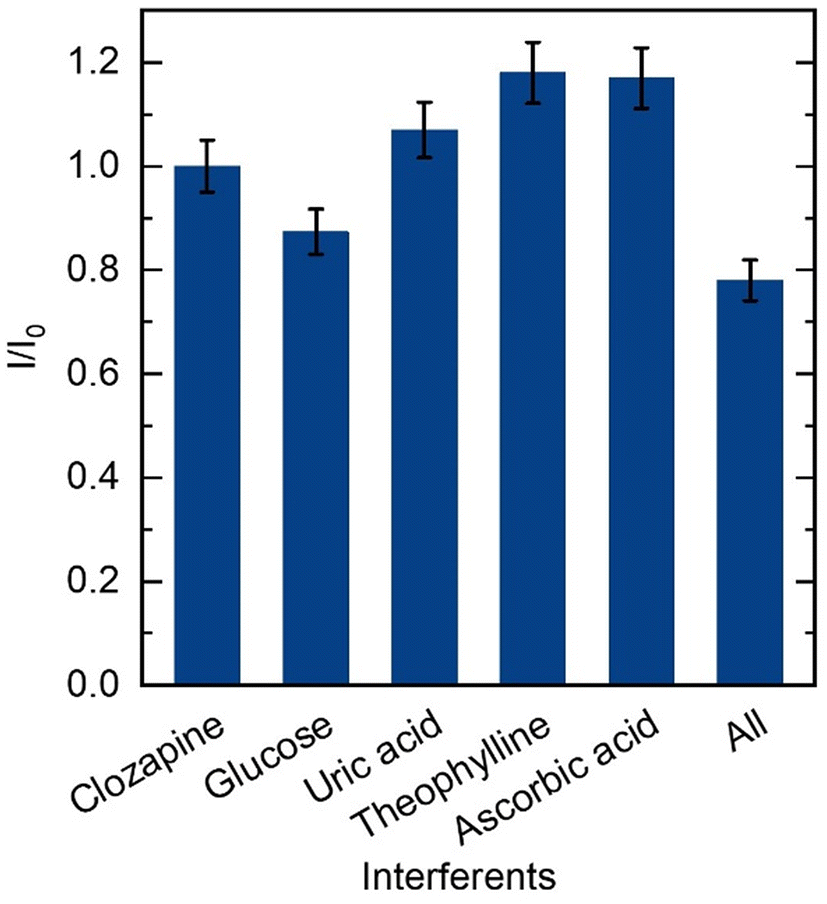 | ||
| Fig. 5 Peak current for 12.0 μM CLZ in 0.1 M PBS at pH 8.0 detection compared to expect peak current when various possible interfering biomolecules were present with 24.0 μM as single interfering molecule or a mixture of 24.0 μM of glucose, uric acid, theophylline, and ascorbic acid each (error bars represent the standard deviation for the three different technical replicates (n = 3)). | ||
3.5. Human blood serum sample analysis
To demonstrate the analysis of CLZ in real sample environment, we conducted voltametric sensing of CLZ in human blood serum sample under the optimal conditions using the SWV technique. Due to the presence of various biomolecules in serum that are absent in pure buffer solution, a calibration plot was generated for the determination of CLZ. This plot showed that the peak currents of CLZ increase linearly with concentration from 10.0 to 20.0 μM (Fig. 6). | ||
| Fig. 6 (A) SWVs of the proposed sensor in human blood serum sample for various concentrations of CLZ in 0.1 M PBS (pH = 8.0). (B) The calibration plot of peak current vs. CLZ concentration and current density vs. the different CLZ concentrations. | ||
After adding a specific CLZ concentration, the SWV signal was measured. The obtained concentration values were then compared with the spiked concentrations in the serum samples to calculate RSD values from three measurements. Importantly, the peak intensity increased proportionally with the CLZ concentration ranging from 10.0 to 15.0 μM. As depicted in Table 2 and Fig. S5,† the recovery was ranging from 104.3% to 99.8%. Therefore, there is clear evidence of good alignment between the proposed sensor and the standard method. Additionally, taking into account the various factors that influence CLZ levels in plasma for schizophrenia patients, including gender, age, and drug dosage, several studies in the literature have reported drug concentrations ranging from 48–1304 ng ml−1.31,35,38 Consequently, these fundings also demonstrated that the presence of various biomolecules in blood serum samples has a minimal impact on the sensing performance of the device.
| No. | Primary CLZ (μM) | Added CLZ (μM) | Found CLZ (μM) | Recovery (%) |
|---|---|---|---|---|
| 1 | — | 0.0 | 0.0 | 100 |
| 2 | — | 10.0 | 10.43 | 104.3 |
| 3 | — | 15.0 | 14.97 | 99.8 |
4. Conclusion
This study shows that screen printing of 3D electrode arrangements is suitable for the fabrication of low cost but high performing μPEDs. The electrode arrangement and design have a distinct influence on the sensor performance. For the specific CLZ example, the 3D μPED described in this article provides a simple, fast, and low-cost platform for the quantitive detection of analytes, both biological and inorganic compounds, in real samples. This type of device offers at least six distinct advantages: (i) it is light-weight, portable and disposable, (ii) it is flexible, (iii) it prevents physical contaminants, such as unsolved particulates in the sample, from fouling the electrode within the sample cell, (iv) it prevents dissolved gas in aqueous samples from forming bubbles and adsorbing on the electrode surface, (v) it ensures continuous flow of the aqueous solution along the paper and across the electrodes surface through capillary forces, thereby maintaining a fresh sample solution on the electrode surface without requiring manual stirring and (vi), the 3D electrode layout decreases distance between the electrodes which decrease the resistance resulting in enhanced sensitivity for sensing. The sensor is capable of detecting CLZ over a wide linear range, low LOD, a larger electrochemical effective surface area, and high sensitivity. We believe our findings effectively meet the demands of point-of-care detection for original samples in environmental monitoring, food analysis, pharmacokinetic analyses and clinical tests.Author contributions
MHG: Conceptualization, visualization, investigation, methodology, validation, writing original draft; MB: Funding acquisition, review & editing; OF: Review & editing; BE: Conceptualization, funding acquisition, supervision, review & editing.Data availability
All data presented in the main article and in the ESI† are available from an open access repository. Source data are provided at Zenodo. https://doi.org/10.5281/zenodo.13325148.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge funding for part of the work through the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Projektnummer 465690040 and Projektnummer 405469627. BE acknowledges the support by the Bavarian State Ministry for Science and Arts through the Distinguished Professorship Program. The authors also acknowledge Universitätsklinikum Erlangen, Transfusionsmedizinische Abteilung, Blutbank, Ms Ida-Sophie Löber, and Ms Maria Leidenberger for facilitating human blood serum sample analysis. The cartoon images presented in the graphical abstract were designed by the Freepik and the iStock.com/id-work.References
- V. Mazzaracchio, et al., A smart paper-based electrochemical sensor for reliable detection of iron ions in serum, Anal. Bioanal. Chem., 2023, 415(6), 1149–1157 CrossRef PubMed.
- T. Yan, et al., Flexible biosensors based on colorimetry, fluorescence, and electrochemistry for point-of-care testing, Front. Bioeng. Biotechnol., 2021, 9, 753692 CrossRef PubMed.
- J. Yang, et al., Detection platforms for point-of-care testing based on colorimetric, luminescent and magnetic assays: A review, Talanta, 2019, 202, 96–110 CrossRef PubMed.
- A. Gałuszka, Z. Migaszewski and J. Namieśnik, The 12 principles of green analytical chemistry and the SIGNIFICANCE mnemonic of green analytical practices, TrAC, Trends Anal. Chem., 2013, 50, 78–84 CrossRef.
- P. M. Nowak, R. Wietecha-Posłuszny and J. Pawliszyn, White Analytical Chemistry: An approach to reconcile the principles of Green Analytical Chemistry and functionality, TrAC, Trends Anal. Chem., 2021, 138, 116223 CrossRef.
- N. A. Meredith, et al., Based analytical devices for environmental analysis, Analyst, 2016, 141(6), 1874–1887 RSC.
- E. Noviana and C. S. Henry, Simultaneous electrochemical detection in paper-based analytical devices, Curr. Opin. Electrochem., 2020, 23, 1–6 CrossRef CAS.
- L.-L. Shen, et al., Modifier-free microfluidic electrochemical sensor for heavy-metal detection, ACS Omega, 2017, 2(8), 4593–4603 CrossRef CAS PubMed.
- A. E. F. Oliveira, A. C. Pereira and M. A. C. de Resende, Fabrication of Low–cost Screen–printed Electrode in Paper Using Conductive Inks of Graphite and Silver/Silver Chloride, Electroanalysis, 2023, 35(2), e202200093 CrossRef CAS.
- A. M. de Campos, et al., Design and fabrication of flexible copper sensor decorated with bismuth micro/nanodentrites to detect lead and cadmium in noninvasive samples of sweat, Chemosensors, 2022, 10(11), 446 CrossRef CAS.
- M. A. Ehsan, S. A. Khan and A. Rehman, Screen-printed graphene/carbon electrodes on paper substrates as impedance sensors for detection of coronavirus in nasopharyngeal fluid samples, Diagnostics, 2021, 11(6), 1030 CrossRef CAS PubMed.
- Z. Fu, et al., Rolling reliability of polyurethane and polyurethane-acrylic ICAs interconnections on printed stretchable electronics, Microelectron. Reliab., 2021, 119, 114067 CrossRef CAS.
- S. Malik , et al., A fringing field based screen-printed flexible capacitive moisture and water level sensor, 2020 IEEE International Conference on Flexible and Printable Sensors and Systems (FLEPS), IEEE, 2020.
- M. Medina-Sánchez, et al., Eco-friendly electrochemical lab-on-paper for heavy metal detection, Anal. Bioanal. Chem., 2015, 407, 8445–8449 CrossRef.
- D. M. Cate, et al., Recent developments in paper-based microfluidic devices, Anal. Chem., 2015, 87(1), 19–41 CrossRef CAS.
- A. W. Martinez, et al., Diagnostics for the developing world: microfluidic paper-based analytical devices, Anal. Chem., 2010, 3–10 CrossRef CAS.
- L.-L. Shen, et al., based microfluidic aluminum–air batteries: toward next-generation miniaturized power supply, Lab Chip, 2019, 19(20), 3438–3447 RSC.
- L.-L. Shen, et al., Towards best practices for improving paper-based microfluidic fuel cells, Electrochim. Acta, 2019, 298, 389–399 CrossRef CAS.
- C.-T. Kung, et al., Microfluidic paper-based analytical devices for environmental analysis of soil, air, ecology and river water, Sens. Actuators, B, 2019, 301, 126855 CrossRef CAS.
- F. Arduini, et al., Sustainable materials for the design of forefront printed (bio) sensors applied in agrifood sector, TrAC, Trends Anal. Chem., 2020, 128, 115909 CrossRef CAS.
- E. Noviana, et al., Emerging applications of paper-based analytical devices for drug analysis: A review, Anal. Chim. Acta, 2020, 1116, 70–90 CrossRef CAS PubMed.
- F. Arduini, Nanomaterials and cross-cutting technologies for fostering smart electrochemical biosensors in the detection of chemical warfare agents, Appl. Sci., 2021, 11(2), 720 CrossRef CAS.
- F. Arduini, Electrochemical paper-based devices: when the simple replacement of the support to print ecodesigned electrodes radically improves the features of the electrochemical devices, Curr. Opin. Electrochem., 2022, 101090 CrossRef CAS.
- S. Cinti, et al., Low-cost and reagent-free paper-based device to detect chloride ions in serum and sweat, Talanta, 2018, 179, 186–192 CrossRef CAS PubMed.
- L. O. Orzari, et al., Lab-made disposable screen-printed electrochemical sensors and immunosensors modified with Pd nanoparticles for Parkinson's disease diagnostics, Microchim. Acta, 2024, 191(1), 1–15 CrossRef.
- M. H. Ghanbari, et al., Superior performance of N-doped carbon Nanoonions/Nafion based microfluidic electrochemical Cd2+ sensor when compared to Screen-Printed Carbon-Based electrode devices, Microchem. J., 2024, 110506 CrossRef.
- M. H. Ghanbari, et al., Electrochemical determination of the antipsychotic medication clozapine by a carbon paste electrode modified with a nanostructure prepared from titania nanoparticles and copper oxide, Microchim. Acta, 2019, 186, 1–10 CrossRef PubMed.
- S. Kumar, et al., A miniaturized unmodified toray paper-based electrochemical sensing platform for antipsychotic drug analysis, Sens. Actuators, A, 2023, 360, 114520 CrossRef.
- D. L. Kelly, et al., Blood draw barriers for treatment with clozapine and development of a point-of-care monitoring device, Clin. Schizophr. Relat. Psychoses, 2018, 12(1), 23–30 CrossRef PubMed.
- H. Ben-Yoav, et al., An electrochemical micro-system for clozapine antipsychotic treatment monitoring, Electrochim. Acta, 2015, 163, 260–270 CrossRef.
- M. H. Mashhadizadeh and E. Afshar, Electrochemical investigation of clozapine at TiO2 nanoparticles modified carbon paste electrode and simultaneous adsorptive voltammetric determination of two antipsychotic drugs, Electrochim. Acta, 2013, 87, 816–823 CrossRef CAS.
- M. Senel and A. Alachkar, Lab-in-a-pencil graphite: A 3D-printed microfluidic sensing platform for real-time measurement of antipsychotic clozapine level, Lab Chip, 2021, 21(2), 405–411 RSC.
- C. Frahnert, M. L. Rao and K. Grasmäder, Analysis of eighteen antidepressants, four atypical antipsychotics and active metabolites in serum by liquid chromatography: a simple tool for therapeutic drug monitoring, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2003, 794(1), 35–47 CrossRef CAS PubMed.
- N. Y. Hasan, et al., Stability indicating methods for the determination of clozapine, J. Pharm. Biomed. Anal., 2002, 30(1), 35–47 CrossRef CAS PubMed.
- M. H. Ghanbari, Z. Norouzi and A. Amiri, Application of nickel-doped graphene nanotubes to modified GCE as a sensitive electrochemical sensor for the antipsychotic drug clozapine in spiked human blood serum samples, J. Iran. Chem. Soc., 2023, 1–11 Search PubMed.
- A. J. Bard, L. R. Faulkner and H. S. White, Electrochemical methods: fundamentals and applications, John Wiley & Sons, 2022 Search PubMed.
- M. P. Siswana, K. I. Ozoemena and T. Nyokong, Electrocatalysis of asulam on cobalt phthalocyanine modified multi-walled carbon nanotubes immobilized on a basal plane pyrolytic graphite electrode, Electrochim. Acta, 2006, 52(1), 114–122 CrossRef CAS.
- M. R. Fathi and D. Almasifar, Electrochemical sensor for square wave voltammetric determination of clozapine by glassy carbon electrode modified by WO3 nanoparticles, IEEE Sens. J., 2017, 17(18), 6069–6076 CAS.
- M. Kang, et al., Reliable clinical serum analysis with reusable electrochemical sensor: Toward point-of-care measurement of the antipsychotic medication clozapine, Biosens. Bioelectron., 2017, 95, 55–59 CrossRef CAS.
- A. S. Al Attas, Novel PVC membrane selective electrode for the determination of clozapine in pharmaceutical preparations, Int. J. Electrochem. Sci., 2009, 4(1), 9–19 CrossRef CAS.
- M. R. Ganjali, et al., Nano-composite clozapine potentiometric carbon paste sensor based on biomimetic molecular imprinted polymer, Int. J. Electrochem. Sci., 2012, 7(5), 4756–4765 CrossRef CAS.
- M. Senel, Electrochemistry Test Strip as Platform for In Situ Detection of Blood Levels of Antipsychotic Clozapine in Finger-Pricked Sample Volume, Biosensors, 2023, 13(3), 346 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4an01136h |
| This journal is © The Royal Society of Chemistry 2024 |