
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rare-earth metal complexes with redox-active formazanate ligands†
Da
Jin
,
Xiaofei
Sun
 ,
Alexander
Hinz
and
Peter W.
Roesky
*
,
Alexander
Hinz
and
Peter W.
Roesky
*
Institute of Inorganic Chemistry, Karlsruhe Institute of Technology (KIT), Engesserstr. 15, Geb. 30.45, 76131 Karlsruhe, Germany. E-mail: roesky@kit.edu
First published on 11th March 2022
Abstract
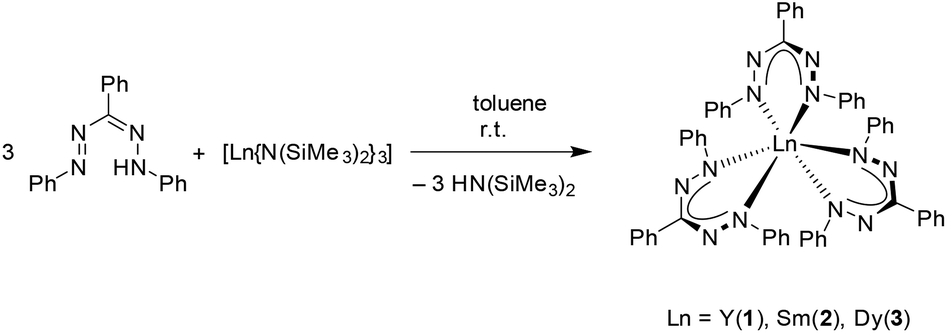
The synthesis and characterisation of rare-earth metal complexes with redox-active formazanate ligands are described. Deprotonation of the neutral formazan ligand L1H (L1 = PhNNC(Ph)NNPh) with [Ln{N(SiMe3)2}3] (Ln = Y, Sm, Dy) resulted in homoleptic tris(formazanate) complexes with the general formula [(L1)3Ln] (Ln = Y (1), Sm (2), Dy (3)), in which the central metal atom is coordinated by six N atoms, revealing a propeller-type structure. To generate heteroleptic complexes, a novel formazan ligand L2H (L2 = {PhNNC(4-tBuPh)NNPh}) was employed. Salt metathesis by using the trivalent precursors [SmCp*2(μ-Cl)2K(thf)] (Cp* = η5-C5Me5) or [LnCp2Cl]2 (Cp = η5-C5H5, Ln = Dy, Yb) and [L2K(thf)] formed mono(formazanate) complexes, [L2SmCp*2] (4) and [L2LnCp2] (Ln = Dy (5), Yb (6)), respectively. Unexpectedly, a redox reaction occurred between [L2K(thf)] and the divalent ytterbium precursor, [YbI2(thf)2], generating the trivalent ytterbium complex [(L2)3Yb] (7). When the neutral formazan ligand (L2H) reacted with [SmCp*2(thf)2], the oxidised samarium complex 4 was formed. These novel compounds were fully characterised and their electrochemical properties were explored by cyclic voltammetry.
Introduction
Formazans are a class of nitrogen-rich compounds featuring a Ar1–NH–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CR3–NN–Ar5 backbone. They were first reported more than one hundred years ago1 and have been studied extensively since the 1940s.2,3 Because of their intense colour, they have found application as dyes especially in chemical biology.4–6 Deprotonation of formazan results in the monoanionic chelating N-donor formazanate with a conjugated 5-membered π-system, which shows structural similarities to the well-known β-diketiminate ligands (Fig. 1).7 However, owing to the presence of four nitrogen atoms in the ligand framework, the LUMO of formazanates is low in energy, which may allow compounds coordinated by formazanates undergo ligand-based reductive activations under mild conditions.8,9 Although formazanates are similar in structure to other chelating N-donor ligands, surprisingly, their coordination chemistry has not been studied to the same extent. Only a selected few examples were reported in recent years.10,11 In 2007, Hicks and co-workers reported that formazanate boron diacetate compounds could be converted to radical anions, which was the first work demonstrating formazanates as redox-active ligands and set the stage for further exploration.12 With continuous efforts of chemists, a great number of formazanate complexes with different properties have been synthesised,13–15 and the reduction chemistry with formazanate ligands has shown that ligand-based storage of up to two electrons in a single formazanate ligand is feasible.16,17
CR3–NN–Ar5 backbone. They were first reported more than one hundred years ago1 and have been studied extensively since the 1940s.2,3 Because of their intense colour, they have found application as dyes especially in chemical biology.4–6 Deprotonation of formazan results in the monoanionic chelating N-donor formazanate with a conjugated 5-membered π-system, which shows structural similarities to the well-known β-diketiminate ligands (Fig. 1).7 However, owing to the presence of four nitrogen atoms in the ligand framework, the LUMO of formazanates is low in energy, which may allow compounds coordinated by formazanates undergo ligand-based reductive activations under mild conditions.8,9 Although formazanates are similar in structure to other chelating N-donor ligands, surprisingly, their coordination chemistry has not been studied to the same extent. Only a selected few examples were reported in recent years.10,11 In 2007, Hicks and co-workers reported that formazanate boron diacetate compounds could be converted to radical anions, which was the first work demonstrating formazanates as redox-active ligands and set the stage for further exploration.12 With continuous efforts of chemists, a great number of formazanate complexes with different properties have been synthesised,13–15 and the reduction chemistry with formazanate ligands has shown that ligand-based storage of up to two electrons in a single formazanate ligand is feasible.16,17
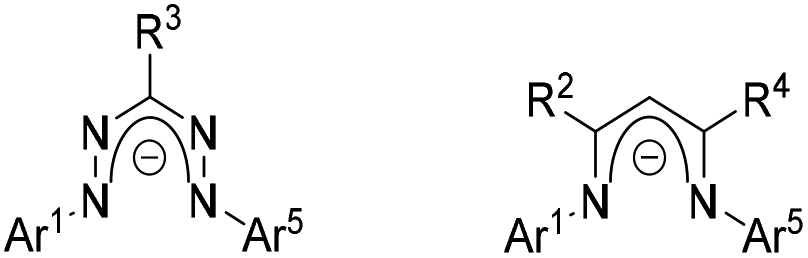 | ||
| Fig. 1 Formazanate ligand (left) and β-diketiminate ligand (right). | ||
Formazanates have been employed as ligands in both main group and transition metal chemistry over the past few decades.18 In contrast, their coordination chemistry toward the highly electropositive rare-earth elements has not yet been reported. There is still a gap in our knowledge about the basic coordination chemistry of formazanates. In earlier work, we have studied the coordination chemistry and application of rare-earth metal complexes with different N-donor ligands, such as amidinates and pyridines.19–23 In an effort to further develop rare-earth metal formazanate chemistry, we report the synthesis, diverse structures and redox properties of a series of rare-earth complexes bearing different formazanate ligands.
Results and discussion
The tris(formazanate) rare-earth metal compounds [(L1)3Ln] (L1 = PhNNC(Ph)NNPh, Ln = Y (1), Sm (2), Dy (3)) were synthesised by straightforward deprotonation of the neutral formazan ligand L1H with corresponding [Ln{N(SiMe3)2}3] (Ln = Y, Sm, Dy)24 in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio in toluene (Scheme 1). Suitable crystals for X-ray diffraction analysis were obtained by slow evaporation of toluene from the reaction mixture at room temperature in good yields (≥80%). All three complexes 1–3 are isostructural and crystallise solvent-free in the triclinic space group P
:1 ratio in toluene (Scheme 1). Suitable crystals for X-ray diffraction analysis were obtained by slow evaporation of toluene from the reaction mixture at room temperature in good yields (≥80%). All three complexes 1–3 are isostructural and crystallise solvent-free in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (Fig. 2 and 3). The metal atom in each complex is hexacoordinated by six nitrogen atoms of three formazanate ligands, resulting in a distorted octahedral geometry. These complexes adopt propeller-type structures and each is constituted by three non-planar six-membered heterocycles (NNCNNLn). The central metal is completely shielded by three ligands. Here, only the structure parameters of the samarium complex 2 are discussed in detail.
(Fig. 2 and 3). The metal atom in each complex is hexacoordinated by six nitrogen atoms of three formazanate ligands, resulting in a distorted octahedral geometry. These complexes adopt propeller-type structures and each is constituted by three non-planar six-membered heterocycles (NNCNNLn). The central metal is completely shielded by three ligands. Here, only the structure parameters of the samarium complex 2 are discussed in detail.
 | ||
| Scheme 1 Synthesis of tris(formazanate) rare-earth metal complexes 1–3. | ||
 | ||
| Fig. 2 Molecular structure of 2 in the solid state (left: front view; right: side view). All hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and bond angles [°]: Sm–N1 2.497(2), Sm–N4 2.373(2), Sm–N5 2.443(2), Sm–N8 2.434(2), Sm–N9 2.407(2), Sm–N12 2.435(2), N1–N2 1.302(3), N2–C1 1.356(3), N3–N4 1.317(3), N3–C1 1.338(3), N5–N6 1.310(3), N6–C2 1.347(3), N7–N8 1.318(3), N7–C2 1.352(3), N9–N10 1.313(3), N10–C3 1.363(3), N11–N12 1.300(3), N11–C3 1.358(3); N1–Sm–N4 68.02(6), N5–Sm–N8 65.12(6), N9–Sm–N12 69.59(6). | ||
 | ||
| Fig. 3 Molecular structures of 1 (left) and 3 (right) in the solid state. All hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and bond angles [°]: 1: Y–N1 2.315(2), Y–N4 2.429(2), Y–N5 2.377(2), Y–N8 2.392(2), Y–N9 2.351(2), Y–N12 2.382(2), N1–N2 1.315(2), N3–N4 1.303(2), N5–N6 1.320(2), N7–N8 1.316(2), N9–N10 1.322(2), N11–N12 1.306(2), N2–C1 1.336(2), N3–C1 1.362(2), N6–C2 1.345(2), N7–C2 1.351(2), N10–C3 1.359(2), N11–C3 1.358(2); N1–Y–N4 70.74(5), N5–Y–N8 66.71(5), N9–Y–N12 70.85(5). 3: Dy–N1 2.443(4), Dy–N4 2.327(5), Dy–N5 2.382(4), Dy–N8 2.385(4), Dy–N9 2.368(4), Dy–N12 2.379(4), N1–N2 1.305(6), N2–C1 1.355(6), N3–N4 1.313(6), N3–C1 1.335(7), N5–N6 1.307(6), N6–C2 1.356(7), N7–N8 1.309(6), N7–C2 1.351(6), N9–N10 1.315(6), N10–C3 1.365(6), N11–N12 1.316(6), N11–C3 1.355(6); N1–Dy–N4 70.46(14), N5–Dy–N8 66.29(14), N9–Dy–N12 70.44(14). | ||
Three formazanate ligands in complex 2 adopt the boat-like distortion where both the metal atom and the C atom are bent out of the plane defined by the four N atoms (Fig. 2). The dihedral angles between the defined plane of NSmN and NNNN in the ligand framework are 23°, 56° and 69°, respectively. The values of the bite angle of N–Sm–N in complex 2 (65.12(6)° to 69.59(6)°) are more obtuse than these in other homoleptic tris(guanidinates) samarium complexes (54.6(2)° to 55.62(7)°).25,26 The Sm–N bond lengths (2.373(2) Å to 2.497(2) Å) lie between representative covalent and donor–acceptor bonds.27,28 They are also comparable to those in other homoleptic Sm complexes with related bidentate N-ligands.29–31 Moreover, the C–N and N–N distances within the ligand frameworks are intermediate of the corresponding single and double bonds, indicating significant delocalisation of the π-electrons of the formazanate backbone.
NMR spectra were recorded for 1 and 2 but not for the highly paramagnetic Dy complex 3. In the 1H and 13C{1H} NMR spectra of 1 and 2, only one set of signals is observed for the formazanate ligands, respectively, suggesting the symmetrical coordination of the ligand to the metal atoms in solution. This is consistent with their molecular structures found in the solid state. Upon deprotonation and coordination toward metal atoms, the characteristic 1H NMR signals of the ligand L1H are shifted upfield (see ESI Fig. S1, S5 and S7†).
Cyclic voltammetry was employed to investigate the electrochemical properties. The voltammograms of the complexes 1–3 in THF solution (Fig. 4) show three (quasi-)reversible 1-electron redox-events in the range of −0.76 to −0.95, −1.51 to −1.71 and −2.22 to −2.54 V vs. Fc0/+ (also see ESI Table S2†). Surprisingly, the observed redox waves do not show the splitting of the waves expected for the presence of more than one redox active ligand, as for instance observed by Otten et al. for bis(formazanate)zinc complexes.32 The first reduction occurs at more positive potential than these of reported formazanate aluminium and zinc complexes,16,33 but at similar potential compared to the formazanate boron diacetate complex.12 The second redox wave occurs between the two reduction values (−1.55 V and −2.55 V) of the reported bis(formazanate)Zn complex and the third value is similar to the second value of bis(formazanate)Zn compound (−2.55 V).33 The third redox wave shows the least reversibility and may indicate slow decomposition or rearrangement of the reduced compound. Due to the great similarity of the redox potentials and CV graphs in three complexes and the absence of an additional wave in the Sm complex, the redox processes are likely to dominantly involve the formazanate ligand. The present shifts in redox potentials in the sequence Sm < Dy < Y are correlated with the ionic radius of the metals (Sm3+ > Dy3+ > Y3+) and hence, the average bite angle of the formazanate ligands (Sm 67.6° < Dy 69.1° < Y 69.4°). Provided a larger bite angle of the formazanates contributes to the stabilisation of a formal dianionic radical ligand, this trend in ligand bite angles provides a plausible explanation for the decreasing redox potentials the smaller the cations are.
 | ||
| Fig. 4 Cyclic voltammogram of 1–3 (THF, 0.1 M [Bu4N][PF6]) recorded at 250 mV s−1. | ||
Our attempts to isolate the reduction products of 2 by using common reducing agents such as cobaltocene or sodium at ambient temperature were unsuccessful, as they always led to complex mixtures. As the appearance of the reaction mixtures kept changing throughout the reduction process, there is a possibility that the reduced products are not stable under these conditions. To limit possible side-reactions and over-reduction, we decided to study heteroleptic rare-earth formazanate complexes bearing only one formazanate moiety. These were targeted in a second series of experiments by changing the stoichiometric ratio between the neutral formazan L1H and [Ln{N(SiMe3)2}3] in the reaction mixtures from 3:1 to 2:1 or 1:1. However, in all cases, according to NMR analysis of the crude product, homoleptic complexes were always formed as major constituent along with small amount of other minor products. The separation of those side products was not successful.
To circumvent this problem in the transamination reactions, we focused on the synthesis of mono(formazanate) rare-earth metal complexes by salt metathesis. Several combinations of lanthanide, formazanate and co-ligands were investigated, but on many occasions, problems with the crystallinity of the product compounds arose. To improve the crystallisation behaviour of the formazanate complexes, various substituent modifications were carried out on the formazan ligand framework. The best results were obtained with the novel formazanate ligand L2 (L2 = PhNNC(4-tBuPh)NNPh), bearing phenyl groups on the nitrogen atoms and a 4-tert-butylphenyl group on the central carbon atom. The corresponding formazan ligand L2H and its potassium salt [L2K(thf)] were prepared by modification of a published method.34,35 The salt metathesis reaction of [L2K(thf)] with [SmCp*2(μ-Cl)2K(thf)]36 or [LnCp2Cl]2 (Ln = Dy, Yb)37 in THF formed the corresponding mono(formazanate) complexes [L2SmCp*2] (4) and [L2LnCp2] (Ln = Dy (5), Yb (6)) (Scheme 2). Single crystals of compounds 4–6 were obtained from concentrated n-pentane solutions at room temperature.
 | ||
| Scheme 2 Synthesis of mono(formazanate) lanthanide complexes 4–6. | ||
Three mono(formazanate) lanthanide complexes 4–6 crystallise in the monoclinic system with space group P21/c (4) (Fig. 5) and P21 (5 and 6) (Fig. 6), respectively. The central lanthanide atom in each complex is in the centre of a distorted tetrahedron, coordinated by two nitrogen atoms of the formazanate ligand and two Cp* or Cp groups.
 | ||
| Fig. 5 Molecular structure of 4 in the solid state (left: front view; right: side view). All hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and bond angles [°]: Sm–N1 2.489(4), Sm–N4 2.475(5), N1–N2 1.306(6), N2–C1 1.335(7), N3–N4 1.290(6), N3–C1 1.349(7); N1–Sm–N4 72.15(15), N2–N1–Sm 133.3(4), N1–N2–C1 122.7(5), N4–N3–C1 122.3(5), N3–N4–Sm 134.3(4), N2–C1–N3 134.6(5). | ||
 | ||
| Fig. 6 Molecular structures of 5 (left) and 6 (right) in the solid state. All hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and bond angles [°]: 5: Dy–N1 2.370(8), Dy–N4 2.366(7), N3–N4 1.309(10), N3–C1 1.329(10), N1–N2 1.286(11), N2–C1 1.346(10); N1–Dy–N4 74.0(3), N2–N1–Dy 131.1(6), N1–N2–C1 123.0(8), N4–N3–C1 122.8(7), N3–N4–Dy 130.5(5), N2–C1–N3 131.0(8). 6: Yb–N1 2.37(2), Yb–N4 2.335(15), N1–N2 1.26(3), C1–N2 1.39(3), N3–C1 1.31(3), N3–N4 1.29(2); N1–Yb–N4 73.8(7), N2–N1–Yb 130.4(15), N1–N2–C1 122(2), N4–N3–C1 124(2), N3–N4–Yb 129.5(13), N3–C1–N2 130(2). | ||
As a representative example, the molecular structure of complex 4 is discussed in detail (Fig. 5). As shown by the equivalent N–N and C–N bond lengths, respectively, the bonding in the six-membered core is delocalised. The structure shows the expected η5-coordination of the cyclopentadienyl ligands, with the planar η5-rings exhibiting no significant distortion within the carbon framework. The Sm–Cring distances are in the range between 2.717(6) and 2.769(6) Å with the distance between the Sm and the ring-centroid being 2.454(9) and 2.471(4) Å. These data are in good agreement with other bis(cyclopentadienyl) complexes of trivalent samarium.38–40 The bond distances of Sm–N are similar to that of complex 2 and the angle of N–Sm–N (72.15(15)°) is a bit more obtuse than that of complex 2. In contrast to complex 2, the sum of inner angels of the six-membered heterocycle (NNCNNSm) in complex 4 is 719.35°, which is close to the ideal planar of 720°. But the corresponding sum of the internal angles in compounds 5 and 6 are 712.55° (Dy) and 709.74° (Yb), respectively, which indicate slight deviation from the planarity of [NNNN].
All three complexes 4–6 contain paramagnetic metal centres. NMR spectra with sharp and well-defined signals could only be obtained for the samarium complex 4. The 1H NMR spectrum of complexes 5 and 6 showed very broad signals owing to the paramagnetism of Dy(III) and Yb(III). In the 1H NMR spectrum of 4, two sharp singlets were observed at the high field δ 1.24 and 1.67 ppm, which could be assigned to the protons of the Cp* ring and tert-butyl groups, respectively.
The cyclic voltammograms of compounds 4–6 were measured in THF solution (Fig. 7). In contrast to the boron mono-formazanate complex [{PhNNC(p-tolyl)NNCPh}BPh2] where two redox processes were observed (−1.35 V and −2.26 V),17 in 4–6 there are three (−0.76 to −0.82, −1.51 to −1.60, −2.25 to −2.42 V, see ESI Table S3†). The two redox waves at similar potentials are found at more negative values for the lanthanide complexes. For this, both the bite angle of the ligand and the covalency of the interaction can play a role, as well as the difference in substitution of the phenyl rings, as p-tert-butyl groups cause a more electron-rich ligand scaffold. All three factors make L2 more difficult to further reduce and therefore lead to more negative redox potentials. Within the series of lanthanide complexes, the redox potentials follow the sequence Sm < Dy < Yb, which follows the same correlation with the ionic radius (Sm3+ > Dy3+ > Yb3+) as discussed for the L1 complexes. It is interesting to note that even the protic ligand L2H shows three redox processes with very similar characteristics to complexes 4–6 (ESI Fig. S19 and Table S3†) at −0.81, −1.55 and −2.29 V which further corroborates that the redox events in the lanthanide complexes almost exclusively occur on the formazanate ligands.
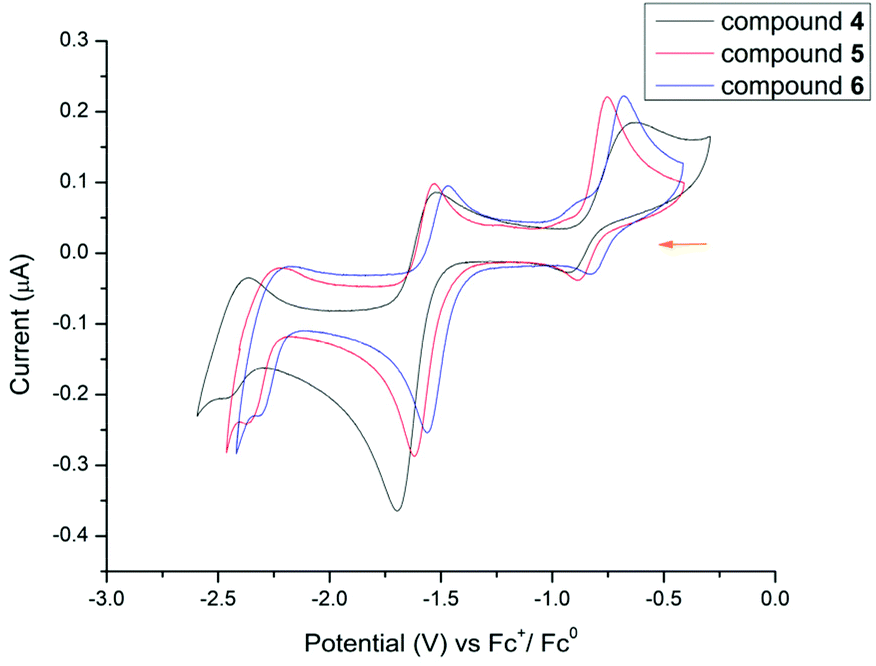 | ||
| Fig. 7 Cyclic voltammogram of 4–6 (THF, 0.1 M [Bu4N][PF6]) recorded at 250 mV s−1. | ||
Unfortunately, from attempted reactions of the chemical reduction of complex 4 by using cobaltocene only an oily product and no crystalline materials could be obtained. When sodium was used as a reducing agent, the lanthanide complex decomposed and a Na formazanate complex was formed. The Na complex could be isolated as crystalline material, but structural data were of low quality and only serve to confirm the connectivity in the complex (see ESI, Fig. S27†). Since its structure is related to the reported potassium formazanate,34 we did not pursue it any further. Attempted reductions with other reducing agents did not give the desired results.
Since the aforementioned reduction of formazanate lanthanide compounds did not progress smoothly and taking into account that divalent lanthanides are often used as reducing agents,41,42 we explored the reaction between divalent lanthanides complexes with formazanates. The potassium salt of the ligand [L2K(thf)] was treated with the divalent Yb halide complex [YbI2(thf)2] in THF.43 Unexpectedly, the homoleptic tris(formazanate) Yb compound [(L2)3Yb] (7) was formed (Scheme 3).
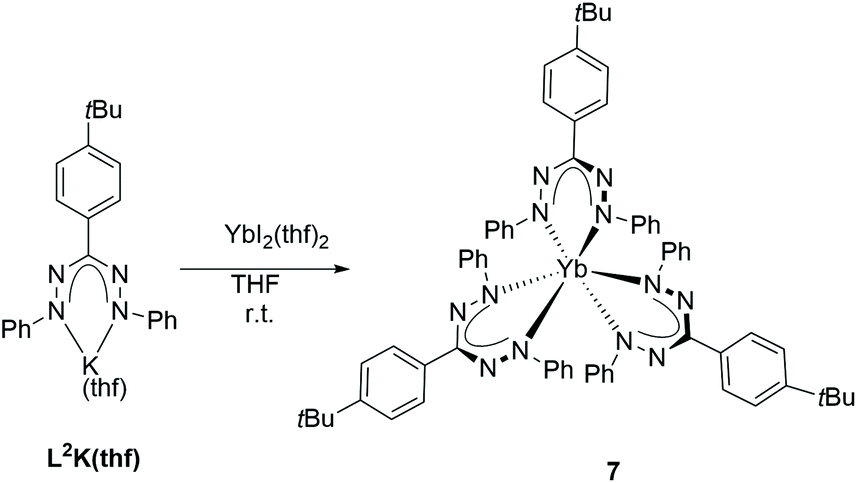 | ||
| Scheme 3 Synthesis of complex 7. | ||
The molecular structure was unambiguously determined by X-ray diffraction analysis (Fig. 8). It crystallises in the triclinic space group P. Except the small difference in the substituents on the ligand backbone, the structural parameters of 7 are closely related to those of the homoleptic complexes 1–3, which indicates that there are no electronic changes of the formazanate ligand moiety. The exact mechanism of the formation of 7 is unclear. Apparently, the presence of the formazanate ligand L2 in the coordination sphere of a divalent Yb(II) is not sufficient to stabilise the highly reactive metal centre and the formation of the trivalent complex is favoured. Such reactivity has been presented in the literature, as a similar redox reaction was observed when potassium amide [{KN(C6H3iPr2-2,6)(2-C5H3N-6-Me(OEt2)}2] was reacted with [SmI2(thf)2] and afforded a homoleptic Sm(III) trisamide.44 To further test the above mentioned hypothesis, L2H was treated directly with the divalent Sm compound [Cp*2Sm(thf)2],45 a similar behavior to the Yb complex was observed, as the trivalent Sm complex 4 formed, which could be identified according to the 1H NMR spectrum (Scheme 4).
 | ||
| Fig. 8 Molecular structure of 7 in the solid state. All hydrogen atoms and solvent molecules are omitted for clarity. Selected bond lengths (Å) and bond angles [°]: Yb–N1 2.324(2), Yb–N4 2.344(2), Yb–N5 2.372(2), Yb–N8 2.287(2), Yb–N9 2.311(2), Yb–N12 2.345(2), N1–N2 1.322(3), C1–N2 1.347(4), N3–C1 1.346(4), N3–N4 1.307(3), N5–N6 1.321(3), N6–C2 1.345(4), N7–C2 1.350(4), N7–N8 1.312(3), N9–N10 1.317(3), C3–N10 1.351(3), C3–N11 1.347(4); N1–Yb–N4 69.38(8), N5–Yb–N8 71.70(8), N9–Yb–N12 68.39(8). | ||
 | ||
| Scheme 4 Synthesis of complex 4 by redox reaction from [Cp*2Sm(thf)2]. | ||
The electrochemical properties of the homoleptic complex [(L2)3Yb] (7, Fig. 9) bear very close resemblance to heteroleptic [L2YbCp2] (6), both with respect to the potentials (−0.76, −1.53 and −2.32 V) and to the shape of the redox waves. In comparison with L1 complexes 1–3, the CV data show similarities with the smallest central atoms in accord with the correlation of larger ionic radius with more negative redox potentials.
 | ||
| Fig. 9 Cyclic voltammogram of 7 (THF, 0.1 M [Bu4N][PF6]) recorded at 250 mV s−1. | ||
UV-Vis spectra of compounds 1–7 were recorded in THF (Fig. 10). All compounds have high extinction coefficients (ε > 104 L mol−1 cm−1, ESI Table S3†), which is in agreement with previous reports.32,46 The tris-formazanate compounds 1–3 and 7 show intense absorption bands in the visible range of the spectrum between 400 and 600 nm as a result of π–π* transitions in the formazanate framework with an absorption maximum between 489 and 494 nm and a shoulder at 540 nm.
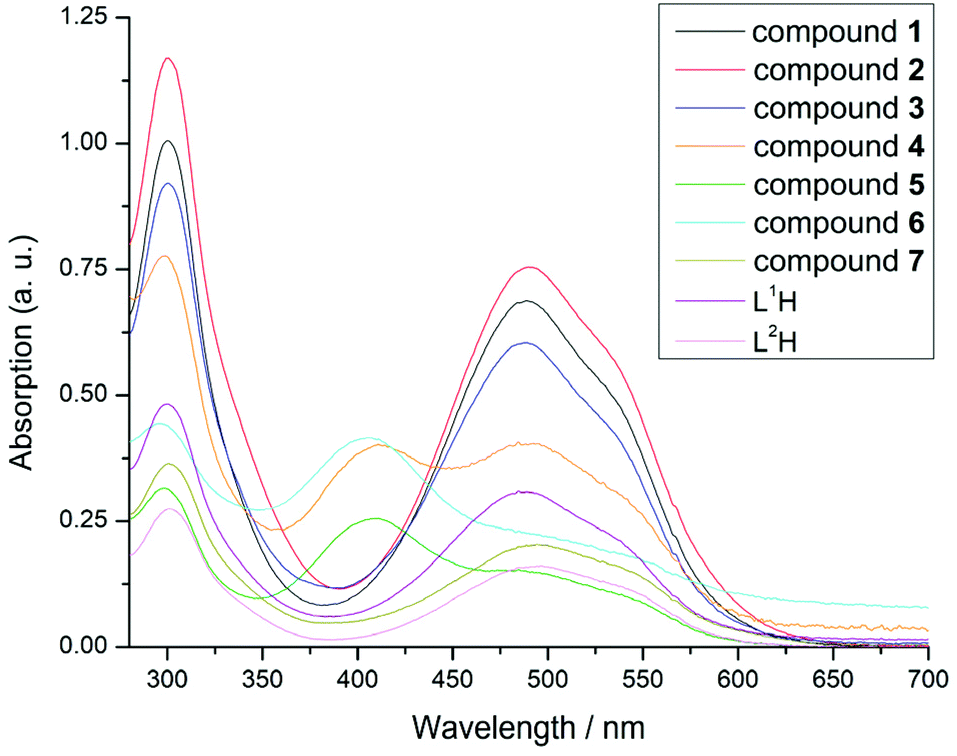 | ||
| Fig. 10 UV-Vis spectra of compounds 1–7 with ligands L(1/2)H in THF. | ||
The spectral formazanate framework with an absorption maximum between signature in each one is similar, in particular, closely resembling that of the protic ligand, suggesting these transitions are predominantly ligand-based in character, which is consistent with CV results. The spectra of mono-formazanate compounds 4–6 show additional maxima at 410 nm. However, the features observed in compounds 1–3 and 7 at 490 and 540 nm are still present in these spectra, albeit with lower extinction coefficients.
Conclusions
A series of tris(formazanate) and mono(formazanate) rare-earth metal complexes were synthesised and fully characterised. These complexes represent the first structurally characterised rare-earth complexes comprising formazanate ligands. Moreover, complexes 1, 2, 3 and 7 are the first examples of homoleptic tris(formazanate) complexes. The cyclic voltammograms of all complexes indicate three separate redox events, which are mainly attributed to the ligand framework. The shifts of redox potentials between these analogues are correlated with the ionic radius of the rare-earth metals. Although we could not isolate the corresponding reduction products, the obtained cyclic voltammograms on the rare-earth metal complexes could be used as a reference for the further electrochemical studies on this area.Experimental
General procedures
All manipulations of water- and air-sensitive compounds were performed with exclusion of moisture and oxygen in flame-dried Schlenk-type glassware either on a dual manifold Schlenk line, interfaced to a high vacuum (10−3 mbar) line or in an argon-filled MBraun glove box. All solvents were dried by using an MBraun solvent purification system (SPS 800), degassed and stored in vacuo over LiAlH4. Tetrahydrofuran was additionally distilled under nitrogen from potassium benzophenone ketyl before storage in vacuo over LiAlH4. C6D6 was vacuum transferred from sodium/potassium alloy into thoroughly dried glassware with the probe substance and flame sealed afterwards. NMR spectra were recorded on Bruker spectrometers (Avance III 300 MHz, Avance 400 MHz or Avance III 400 MHz) at 298k. Chemical shifts are referenced internally using signals of the residual protio solvent (1H) or the solvent (13C{1H}) and are reported relative to tetramethylsilane (1H, 13C{1H}). The multiplicity of the signals is indicated as s = singlet, d = doublet, dd = doublet of doublets, t = triplet, q = quartet, m = multiplet and br = broad. Assignments were determined on the basis of unambiguous chemical shifts, coupling patterns and 13C DEPT experiments or 2D correlations (1H–1H COSY, 1H–13C HMQC and 1H–13C HMBC). Elemental analyses were carried out with an Elementar Vario Micro cube from Elementar Analysensysteme GmbH. IR spectra were obtained on a Bruker Tensor 37 spectrometer equipped with a room temperature DLaTGS detector, a diamond ATR (attenuated total reflection) unit and a nitrogen-flushed chamber. In terms of their intensity, the signals were classified into different categories (vs = very strong, s = strong, m = medium, w = weak, and sh = shoulder). Cyclic voltammetry measurements were performed with a suitable potentiostat and electrochemical cell within a glovebox. We used a freshly polished Pt disk working electrode, a Pt wire as counter electrode, an Ag wire as (pseudo) reference electrode in a 0.1 M solution of [nBu4N][PF6] as electrolyte. Potentials were calibrated against the Fc/Fc+ couple as internal standard.1,3,5-Triphenylformazan (L1H) was obtained commercially and used as received. [L2K(thf)],34 [Ln{N(SiMe3)2}3],24 [SmCp*2(μ-Cl)2K(thf)],36 [LnCp2Cl]2,37 [YbI2(thf)2],43 and [Cp*2Sm(thf)2]45 were prepared following literature procedures. All other chemicals were obtained from commercial sources and used without further purification.
Synthesis of PhNNC(4-tBuPh)NNHPh (L2H)
PhNNC(4-tBuPh)NNHPh (L2H) was prepared by modification of a published method.35 Phenylhydrazine (1.622 g, 10 mmol) was combined with 4-tert-butyl benzaldehyde (1.081 g, 10 mmol) and ethanol (15 mL). 4-tert-Butylphenyl phenyl hydrazone formed as yellow precipitate upon stirring the mixture at ambient temperature overnight. The precipitate was collected by filtration after washing with cold ethanol several times. In a round bottom flask, the aniline (0.466 g, 5 mmol) was dissolved in hydrochloric acid (1.25 mL) and cooled to 0 °C in an ice bath. Sodium nitrite (0.345 g, 5 mmol) was dissolved in H2O (2 mL) and then added to the acidic aniline solution by a dropping funnel over 15 minutes. After the addition was complete, the diazonium salt solution was allowed to stir at 0 °C for further 30 minutes. In a separate flask, 4-tert butylphenyl phenyl hydrazone (1.260 g, 5 mmol) was dissolved in methanol (15 mL) and cooled in a salt-ice bath to −5 °C. The diazonium salt solution was then added to the hydrazone solution over 30 minutes. Additional ice was added during the addition to keep the solution at −5 °C. Once the addition of the diazonium salt was complete, the mixture was allowed to warm to room temperature and stirred overnight. Then, dark red precipitate was filtered off and purified by recrystallisation from ethanol.Yield: 0.997 g (0.28 mmol), 56%. 1H NMR (400 MHz, C6D6): δ (ppm) = 15.27 (s, 1H, NH), 8.29 (d, 2H, J = 8.3 Hz, tBuPh, m-H), 7.52 (d, 4H, J = 8.1 Hz, NPh, o-H), 7.46 (d, 2H, J = 8.3 Hz, tBuPh, o-H), 7.13 (d, 4H, J = 7.5 Hz, NPh, m-H), 6.97 (t, 2H, J = 7.3 Hz, NPh p-H), 1.29 (s, 9H, C(CH3)3). 13C{1H} NMR (101 MHz, C6D6): δ (ppm) = 150.7 (tBuPh ipso-C), 148.4 (CPh ipso-C), 142.1 (NCN), 135.4 (NPh ipso-C), 129.5 (NPh (meta)), 127.4 (NPh (para)), 126.5 (tBuPh (meta)), 125.6 (tBuPh (ortho)), 119.1 (NPh (ortho)), 34.6 (C(CH3)3), 31.5 (C(CH3)3). IR (ATR): ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) (cm−1) = 3053(w), 2954(s), 2901(w), 2864(w), 1596(m), 1506(vs), 1452(s), 1406(w), 1388(w), 1352(s), 1314(w), 1294(w), 1229(vs), 1206(s), 1181(m), 1162(m), 1110(w), 1071(w), 1033(s), 1007(m), 984(w), 915(w), 890(w), 836(s) 807(w), 767(m), 744(m), 719(m), 682(m), 655(m), 630(m), 549(w), 511(w), 481(w). Anal. Calc. (%) for C23H24N4 (356.47 g mol−1): C 77.50, H 6.79, N 15.72; found (%): C 77.29, H 6.32, N 15.72.
(cm−1) = 3053(w), 2954(s), 2901(w), 2864(w), 1596(m), 1506(vs), 1452(s), 1406(w), 1388(w), 1352(s), 1314(w), 1294(w), 1229(vs), 1206(s), 1181(m), 1162(m), 1110(w), 1071(w), 1033(s), 1007(m), 984(w), 915(w), 890(w), 836(s) 807(w), 767(m), 744(m), 719(m), 682(m), 655(m), 630(m), 549(w), 511(w), 481(w). Anal. Calc. (%) for C23H24N4 (356.47 g mol−1): C 77.50, H 6.79, N 15.72; found (%): C 77.29, H 6.32, N 15.72.
Synthesis of [{PhNNC(Ph)NNPh}3Ln] (Ln = Y (1), Sm (2), Dy (3))
To a mixture of L1H (0.360 g, 1.20 mmol) and [Ln{N(SiMe3)2}3] (Ln = Y, Sm, Dy) (0.40 mmol) toluene (50 mL) was added and the resulting mixture was stirred at room temperature for 18 h. All volatiles were removed under reduced pressure. The resulting solid was washed with cold n-pentane (5 mL) and dried in vacuo for 2 h affording the product as a dark red solid. Crystals suitable for X-ray diffraction analysis were obtained from slowly evaporation of toluene from the reaction mixture. (cm−1) = 3084(w), 3053(m), 3028(m), 1945(w), 1596(s), 1509(vs), 1491(vs), 1454(s), 1442(m), 1350(s), 1313(s), 1226(w), 1181(vs), 1161(s), 1098(s), 1071(s), 1042(s), 1017(s), 982(m), 914(m), 889(w) 843(w), 804(w), 750(vs), 686(s), 650(m), 632(m), 587(w), 542(w), 516(w), 498(m), 460(w). Anal. Calc. (%) for C57H45N12Y (986.98 g mol−1): C 69.37, H 4.60, N 17.03; found (%): C 69.22, H 4.61, N 16.60.
(cm−1) = 3084(w), 3052(m), 3028(m), 1944(w), 1653(s), 1636(vs), 1597(w), 1509(vs), 1491(vs), 1454(s), 1442(s), 13450(s), 1313(m), 1291(w), 1228(vs), 1181(s), 1162(s), 1149(s), 1098(w), 1071(m), 1042(s), 1017(s), 982(m), 914(m), 889(w), 844(w), 817(w), 804(w), 750(s), 686(s), 650(m), 632(m), 587(m), 542(w), 499(m), 459(w). Anal. Calc. (%) for C57H45N12Sm (1048.43 g mol−1): C 65.30, H 4.33, N 16.03; found (%): C 65.08, H 4.42, N 15.62.
(cm−1) = 3083(w), 3053(m), 3028(m), 1945(w), 1596(s), 1509(vs), 1491(vs), 1440(s), 1350(s), 1313(w), 1228(vs), 1180(s), 1158(s), 1097(w), 1071(m), 1042(m), 1017(s), 982(m), 914(m), 889(m), 805(w), 750(vs), 686(s), 650(m), 631(m), 586(m), 542(w), 498(m), 459(w). Anal. Calc. (%) for C57H45N12Dy (1060.57 g mol−1): C 64.55, H 4.28, N 15.85; found (%): C 64.71, H 4.27, N 15.53.
Synthesis of [{PhNNC(4-tBuPh)NNPh}SmCp*2] (4)
Method A: THF (10 mL) was added to a mixture of the potassium formazanate salt L2K(thf) (0.070 g, 0.15 mmol) and [SmCp*2(μ-Cl)2K(thf)] (0.090 g, 0.15 mmol) at room temperature. The resulting mixture was stirred overnight. All volatiles were removed under reduced pressure. The resulting solid was extracted with 15 mL of pentane and filtered. Concentration of the filtrate afforded dark red plate crystals suitable for X-ray diffraction analysis. Method B: L2H (0.053 g, 0.15 mmol) and [Cp*2Sm(thf)2] (0.085 g, 0.15 mmol) were combined in a Schlenk flask and dissolved in 15 mL of THF. The reaction mixture was stirred 6 hours. Then, the solvent was removed in vacuo and extracted by pentane. Crystals were obtained by concentrating the filtrate. (cm−1) = 3087(w), 3055(w), 2953(m), 2900(w), 2864(w), 1597(m), 1560(w), 1507(s), 1453(m), 1407(w), 1388(w), 1352(m), 1310(m), 1230(vs), 1204(s), 1180(s), 1144(s), 1111(s), 1071(m), 1033(m), 1006(m), 982(s), 915(w), 890(w), 836(m), 806(w), 767(m), 754(w), 742(m), 717(w), 680(m), 655(w), 630(m), 594(w), 574(w), 550(w), 482(w). Anal. Calc. (%) for C43H53N4Sm (776.29 g mol−1): C 66.53, H 6.88, N 7.22; found (%): C 66.18, H 6.58, N 7.23.
Synthesis of [{PhNNC(4-tBuPh)NNPh}LnCp2] (Ln = Dy (5), Yb (6))
Compounds 5 and 6 were synthesised using the same procedure as for 4 method A. THF (10 mL) was added to a mixture of the potassium formazanate salt [L2K(thf)] (0.070 g, 0.15 mmol) and [Cp2LnCl]2 (Ln = Dy, Yb) (0.5 eq.) at room temperature. The resulting mixture was stirred overnight. All volatiles were removed under reduced pressure. The resulting solid was extracted with 15 mL pentane and filtered. Concentration of the filtrate afforded dark red plate crystals suitable for X-ray diffraction analysis. (cm−1) = 3076(w), 3058(w), 3008(w), 2957(s), 2902(m), 2865(m), 1652(w), 1597(m), 1559(w), 1540(w), 1507(vs), 1481(br), 1453(s), 1406(w), 1391(w), 1353(m), 1313(w), 1295(w), 1269(w), 1226(vs), 1180(s), 1163(s), 1112(m), 1072(w), 1034(w), 1008(m), 985(s), 916(m), 890(w), 864(w), 837(m), 782(m), 766(m), 755(m), 744(m), 723(w), 682(w), 657(w), 632(w), 589(w), 573(w), 550(w), 510(w). Anal. Calc. (%) for C33H33N4Dy (648.16 g mol−1): C 61.15, H 5.13, N 8.64; found (%): C 60.24, H 5.00, N 8.66.
(cm−1) = 3080(w), 3003(w), 2960(m), 2902(w), 2865(w), 1671(w), 1653(w), 1583(w), 1560(w), 1540(w), 1508(w), 1479(m), 1455(w), 1394(w), 1362(w), 1331(w), 1297(m), 1273(s), 1225(vs), 1180(vs), 1159(s), 1115(s), 1079(m), 1046(w), 1009(w), 982(w), 888(w), 865(w), 842(m), 789(s), 757(s), 725(w), 688(m), 661(m), 617(w), 591(w), 557(w), 513(w). Anal. Calc. (%) for C33H33N4Yb (658.71 g mol−1): C 60.17, H 5.05, N 8.51; found (%): C 59.75, H 4.70, N 9.20.
Synthesis of [{PhNNC(4-tBuPh)NNPh}3Yb] (7)
THF (10 mL) was added to a mixture of [L2K(thf)] (0.198 g, 0.42 mmol) and [YbI2(thf)2] (0.121 g, 0.21 mmol) at room temperature. The resulting mixture was stirred overnight. All volatiles were removed under reduced pressure. The resulting solid was extracted with pentane and filtered. Crystals were obtained by concentrating the filtrate. (cm−1) = 3062(w), 3035(w), 2958(vs), 2902(m), 2866(m), 1699(w), 1651(w), 1634(w), 1597(m), 1560(w), 1540(w), 1508(vs), 1481(s), 1454(s), 1407(w), 1393(w), 1358(m), 1312(w), 1270(m), 1219(vs), 1185(vs), 1162(s), 1112(m), 1073(w), 1035(m), 1011(m), 982(w), 914(w), 890(w), 866(w), 837(m), 805(w), 758(m), 722(w), 687(m), 664(w), 633(w), 560(w), 486(w). Anal. Calc. (%) for C69H69N12Yb (1239.45 g mol−1): C 66.87, H 5.61, N 13.56; found (%): C 67.38, H 5.11, N 13.33.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. J. thanks the China Scholarship Council (no. 201906030178) for generous support. Financial support by the DFG-funded transregional collaborative research center SFB/TRR 88 “Cooperative Effects in Homo and Heterometallic Complexes (3MET)” project B3 is gratefully acknowledged.Notes and references
- A. Pinner, Ber. Dtsch. Chem. Ges., 1884, 17, 182–184 CrossRef.
- R. M. Rush and J. H. Yoe, Anal. Chem., 1954, 26, 1345–1347 CrossRef CAS.
- A. Nineham, Chem. Rev., 1955, 55, 355–483 CrossRef CAS.
- K. Grychtol and W. Mennicke, Ullmann's Encyclopedia of Industrial Chemistry, 2000, vol. 22, pp. 595–628 Search PubMed.
- J. C. Stockert, R. W. Horobin, L. L. Colombo and A. Blázquez-Castro, Acta Histochem., 2018, 120, 159–167 CrossRef CAS PubMed.
- E. Grela, J. Kozłowska and A. Grabowiecka, Acta Histochem., 2018, 120, 303–311 CrossRef CAS PubMed.
- L. Bourget-Merle, M. F. Lappert and J. R. Severn, Chem. Rev., 2002, 102, 3031–3066 CrossRef CAS PubMed.
- F. Milocco, S. Demeshko, F. Meyer and E. Otten, Dalton Trans., 2018, 47, 8817–8823 RSC.
- D. L. Broere, B. Q. Mercado, J. T. Lukens, A. C. Vilbert, G. Banerjee, H. M. Lant, S. H. Lee, E. Bill, S. Sproules and K. M. Lancaster, Chem. – Eur. J., 2018, 24, 9417–9425 CrossRef CAS PubMed.
- G. I. Sigeikin, G. N. Lipunova and I. G. e. Pervova, Russ. Chem. Rev., 2006, 75, 885–900 CrossRef CAS.
- G. Lipunova, T. Fedorchenko and O. Chupakhin, Russ. J. Gen. Chem., 2019, 89, 1225–1245 CrossRef CAS.
- J. B. Gilroy, M. J. Ferguson, R. McDonald, B. O. Patrick and R. G. Hicks, Chem. Commun., 2007, 2, 126–128 RSC.
- A. Mandal, B. Schwederski, J. Fiedler, W. Kaim and G. K. Lahiri, Inorg. Chem., 2015, 54, 8126–8135 CrossRef CAS PubMed.
- R. R. Maar, R. Zhang, D. G. Stephens, Z. Ding and J. B. Gilroy, Angew. Chem., Int. Ed., 2019, 58, 1052–1056 CrossRef CAS PubMed.
- F. Milocco, F. de Vries, I. M. Bartels, R. W. Havenith, J. Cirera, S. Demeshko, F. Meyer and E. Otten, J. Am. Chem. Soc., 2020, 142, 20170–20181 CrossRef CAS PubMed.
- R. Mondol and E. Otten, Inorg. Chem., 2019, 58, 6344–6355 CrossRef CAS PubMed.
- R. Mondol, D. A. Snoeken, M. C. Chang and E. Otten, Chem. Commun., 2017, 53, 513–516 RSC.
- J. B. Gilroy and E. Otten, Chem. Soc. Rev., 2020, 49, 85–113 RSC.
- A. Bremer, C. M. Ruff, D. Girnt, U. Müllich, J. Rothe, P. W. Roesky, P. J. Panak, A. Karpov, T. J. Müller and M. A. Denecke, Inorg. Chem., 2012, 51, 5199–5207 CrossRef CAS PubMed.
- T. S. Brunner, P. Benndorf, M. T. Gamer, N. Knöfel, K. Gugau and P. W. Roesky, Organometallics, 2016, 35, 3474–3487 CrossRef CAS.
- M. He, Z. Chen, E. M. Pineda, X. Liu, E. Bouwman, M. Ruben and P. W. Roesky, Eur. J. Inorg. Chem., 2016, 5512–5518 CrossRef CAS.
- J. Kratsch, B. B. Beele, C. Koke, M. A. Denecke, A. Geist, P. J. Panak and P. W. Roesky, Inorg. Chem., 2014, 53, 8949–8958 CrossRef CAS PubMed.
- M. Rastatter, A. Zulys and P. W. Roesky, Chem. Commun., 2006, 8, 874–876 RSC.
- D. Bradley, J. Ghotra and F. Hart, J. Chem. Soc., Chem. Commun., 1972, 6, 349–350 RSC.
- L. Zhou, Y. Yao, Y. Zhang, M. Xue, J. Chen and Q. Shen, Eur. J. Inorg. Chem., 2004, 2167–2172 CrossRef CAS.
- P. Tutacz, N. Harmgarth, F. Zörner, P. Liebing, L. Hilfert, F. Engelhardt, S. Busse and F. T. Edelmann, Z. Anorg. Allg. Chem., 2018, 644, 1653–1659 CrossRef CAS.
- E. D. Brady, D. L. Clark, J. C. Gordon, P. J. Hay, D. W. Keogh, R. Poli, B. L. Scott and J. G. Watkin, Inorg. Chem., 2003, 42, 6682–6690 CrossRef CAS PubMed.
- X. Xu, X. Xu, Y. Chen and J. Sun, Organometallics, 2008, 27, 758–763 CrossRef CAS.
- P. Benndorf, J. Kratsch, L. Hartenstein, C. M. Preuss and P. W. Roesky, Chem. – Eur. J., 2012, 18, 14454–14463 CrossRef CAS PubMed.
- M. Xue, R. Jiao, Y. Zhang, Y. Yao and Q. Shen, Eur. J. Inorg. Chem., 2009, 4110–4118 CrossRef CAS.
- D. Drees and J. Magull, Z. Anorg. Allg. Chem., 1994, 620, 814–818 CrossRef CAS.
- M.-C. Chang, P. Roewen, R. Travieso-Puente, M. Lutz and E. Otten, Inorg. Chem., 2015, 54, 379–388 CrossRef CAS PubMed.
- M. C. Chang, T. Dann, D. P. Day, M. Lutz, G. G. Wildgoose and E. Otten, Angew. Chem., Int. Ed., 2014, 53, 4118–4122 CrossRef CAS PubMed.
- R. Travieso-Puente, M.-C. Chang and E. Otten, Dalton Trans., 2014, 43, 18035–18041 RSC.
- H. Tezcan, Ş. Can and R. Tezcan, Dyes Pigm., 2002, 52, 121–127 CrossRef CAS.
- M. T. Gamer, G. Canseco-Melchor and P. W. Roesky, Z. Anorg. Allg. Chem., 2003, 629, 2113–2116 CrossRef CAS.
- R.-E. Maginn, S. Manastyrskyj and M. Dubeck, J. Am. Chem. Soc., 1963, 85, 672–676 CrossRef CAS.
- K. Vasudevan and A. H. Cowley, Chem. Commun., 2007, 33, 3464–3466 RSC.
- J. M. Veauthier, E. J. Schelter, C. N. Carlson, B. L. Scott, R. E. D. Re, J. Thompson, J. L. Kiplinger, D. E. Morris and K. D. John, Inorg. Chem., 2008, 47, 5841–5849 CrossRef CAS PubMed.
- W. J. Evans, E. Montalvo, D. J. Dixon, J. W. Ziller, A. G. DiPasquale and A. L. Rheingold, Inorg. Chem., 2008, 47, 11376–11381 CrossRef CAS PubMed.
- W. J. Evans, Coord. Chem. Rev., 2000, 206, 263–283 CrossRef.
- W. J. Evans, J. Organomet. Chem., 2002, 647, 2–11 CrossRef CAS.
- P. Girard, J. Namy and H. Kagan, J. Am. Chem. Soc., 1980, 102, 2693–2698 CrossRef CAS.
- K. W. Ku, C. W. Au, H.-S. Chan and H. K. Lee, Dalton Trans., 2013, 42, 2841–2852 RSC.
- W. J. Evans, T. A. Ulibarri, H. Schumann and S. Nickel, Inorg. Synth., 1990, 28, 297–300 CAS.
- J. B. Gilroy, P. O. Otieno, M. J. Ferguson, R. McDonald and R. G. Hicks, Inorg. Chem., 2008, 47, 1279–1286 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2120929–2120935. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2dt00456a |
| This journal is © The Royal Society of Chemistry 2022 |