
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
XAS investigation of silica aerogel supported cobalt rhenium catalysts for ammonia decomposition†
Karsten G.
Kirste
 a,
Said
Laassiri
b,
Zhigang
Hu
c,
Dragos
Stoian
d,
Laura
Torrente-Murciano
*c,
Justin S. J.
Hargreaves
*e and
Karina
Mathisen
*a
a,
Said
Laassiri
b,
Zhigang
Hu
c,
Dragos
Stoian
d,
Laura
Torrente-Murciano
*c,
Justin S. J.
Hargreaves
*e and
Karina
Mathisen
*a
aDepartment of Chemistry, Norwegian University of Science and Technology, Høgskoleringen 5, N-7491 Trondheim, Norway. E-mail: karina.mathisen@ntnu.no
bChemical & Biochemical Sciences, Green Process Engineering (CBS), Mohamed VI Polytechnic University, UM6P, 43150, Ben Guerir, Morocco
cDepartment of Chemical Engineering and Biotechnology, University of Cambridge, CB3 0AS, Cambridge, UK. E-mail: lt416@cam.ac.uk
dSwiss-Norwegian Beamline, European Synchrotron Radiation Facility, F-38043, Grenoble Cedex, France
eSchool of Chemistry, University of Glasgow, Joseph Black Building, Glasgow G12 8QQ, UK. E-mail: Justin.Hargreaves@glasgow.ac.uk
First published on 12th June 2020
Abstract
The implementation of ammonia as a hydrogen vector relies on the development of active catalysts to release hydrogen on-demand at low temperatures. As an alternative to ruthenium-based catalysts, herein we report the high activity of silica aerogel supported cobalt rhenium catalysts. XANES/EXAFS studies undertaken at reaction conditions in the presence of the ammonia feed reveal that the cobalt and rhenium components of the catalyst which had been pre-reduced are initially re-oxidised prior to their subsequent reduction to metallic and bimetallic species before catalytic activity is observed. A synergistic effect is apparent in which this re-reduction step occurs at considerably lower temperatures than for the corresponding monometallic counterpart materials. The rate of hydrogen production via ammonia decomposition was determined to be 0.007 molH2 gcat−1 h−1 at 450 °C. The current study indicates that reduced Co species are crucial for the development of catalytic activity.
1. Introduction
The effects of climate change are becoming more and more evident each day, with more extreme weather, more natural disasters and the increasing risk of the collapse of ecosystems.1 It has been argued that this change is connected to the use of fossil fuels and the need for new energy sources is increasingly becoming a major focus of attention.2 The so-called hydrogen economy is an alternative to the fossil fuel economy which would eliminate fossil fuel based emissions from distributed sources (e.g. vehicles). However, a major limitation in accomplishing the hydrogen economy is the current lack of existing infrastructure for the application of hydrogen directly as a fuel, as well as the safety risks associated with its transportation and storage.3 In recent years, the scientific community has investigated methods for hydrogen storage in porous materials. However, the capacity of such materials is still too low for large scale applications. An attractive alternative approach is the chemical storage of hydrogen in hydrides or organic molecules.3,4Within this context, ammonia as a hydrogen vector is promising due to its high hydrogen gravimetric content (17.6 wt%) and vapour pressure (10.6 bar at 300 K)5 compared to the alternative of pressurised hydrogen.6–8 Ammonia also has a low flammability range and there is existing infrastructure for ammonia distribution.7 Deployment of ammonia as a hydrogen vector requires the release of hydrogen at high rates and at low temperatures (such as 150–180 °C in relation to the working conditions of fuel cells) and currently the best catalyst for the process is ruthenium supported on graphitised carbon nanotubes (CNT).9–11 To achieve low temperature activity, promotion of the ruthenium active species with caesium is required.12,13 In this context, it is notable that ruthenium and caesium are scarce elements and the production costs of the elements are most likely too high for the use as an ammonia decomposition catalyst in vehicles.14,15 Accordingly, scientific interest is now focused on alternative catalysts based on non-noble metals. In low temperature ammonia decomposition the rate determining step has been demonstrated to be the desorption of nitrogen16 and in connection with this, metallic cobalt is presented as an attractive alternative due to the fact that its nitrogen binding energy is similar to ruthenium metal.17 The activity of cobalt catalysts has been shown to depend on particle size and dispersion,18,19 also cobalt–support interactions have been shown to be important in relation to activity20 with cobalt supported on silica being active.21 The importance of metallic cobalt dispersion and the texture of the silica matrix has been reported for cobalt containing mesosilicate systems by Varisli and co-workers.19,22
To further improve the properties of monometallic catalysts it has been suggested to form bimetallic systems, where one metal has too strong, nitrogen adsorption properties while the other has weaker nitrogen adsorption properties.23 In the context of bimetallic systems, notwithstanding the fact that rhenium is a scarce metal, it is interesting to note that cobalt together with rhenium has been reported to exhibit a synergistic effect with lower reduction temperatures for both metals being achieved.24–28 We have observed that unsupported Co–Re possesses ammonia decomposition activities comparable to Ru/CNT29 which, given that it possesses a very low surface area, suggests that successfully supporting the active phase might lead to further improvement in catalyst performance. Nanoparticles confined in micropores generally experience growth restriction due to pore confinement and enhanced bimetallic interaction.30 Silica aerogels are ultra-porous materials, with a structure of interconnected colloidal silica. The interconnected colloids form both mesopores (2–50 nm) and micropores (<2 nm) and exhibit properties such as high surface areas (600–1000 m2 g−1) and surface hydrophobicity.31 Methods for the introduction of rhenium into porous supports include covalently bonding rhenium complexes to support surfaces,32,33 atomic layer deposition from solution34 and the more widely preferred impregnation methods.27,35–38
In the present manuscript, we report the co-introduction of rhenium and cobalt onto an ambient pressure dried (APD) silica aerogel. In situ X-ray absorption spectroscopic (XAS) characterisation of the cobalt–rhenium phase during hydrogen production via ammonia decomposition is also reported to reveal the nature of the active species as well as the synergistic effect of cobalt–rhenium in the formation of such active species under reaction conditions.
2. Experimental
Introduction of CoRe to the APD silica aerogel
Cobalt rhenium supported on the silica aerogel was prepared by an approach based on the method reported by Kristiansen and co-workers.39 A sodium silicate solution (((NaO)x·SiO2), 8 wt% SiO2, Sigma Aldrich) was passed through a column of Amberlite 120H+ (Sigma Aldrich) with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio. Typically, cobalt(II) nitrate hexahydrate (Co(NO3)2·6H2O, Sigma Aldrich) and rhenium(VII) oxide (Re2O7, Sigma Aldrich) were dissolved in the ion exchanged sodium silicate solution before gelation was initiated by the addition of hexamethyldisilazane (HMDS, Sigma Aldrich) and hexamethyldisiloxane (HMDSO, Sigma Aldrich). Analogous silicate gels containing only cobalt and rhenium were synthesised as a comparison. All gels were immersed in n-heptane (VWR international) for 24 hours before decantation and drying at 65 °C for 1080 minutes, then at 85 °C for 180 min and finally further drying at 120 °C for 120 minutes employing a temperature ramp rate of 5 °C min−1. The thermal stability of the gels was tested by annealing a portion of them at 450 °C for 30 minutes or by calcination at 700 °C for 3 hours. The temperature ramp rate for both thermal treatments was 10 °C min−1 employing an atmosphere of static air. The theoretical molar composition of the synthesised cobalt–rhenium containing gel was 1 Si:0.8 HMDS:0.85 HMDSO:0.05 Co(II):0.05 Re(VII). The monometallic gels possessed comparable content of the metal component of interest.
:1 volume ratio. Typically, cobalt(II) nitrate hexahydrate (Co(NO3)2·6H2O, Sigma Aldrich) and rhenium(VII) oxide (Re2O7, Sigma Aldrich) were dissolved in the ion exchanged sodium silicate solution before gelation was initiated by the addition of hexamethyldisilazane (HMDS, Sigma Aldrich) and hexamethyldisiloxane (HMDSO, Sigma Aldrich). Analogous silicate gels containing only cobalt and rhenium were synthesised as a comparison. All gels were immersed in n-heptane (VWR international) for 24 hours before decantation and drying at 65 °C for 1080 minutes, then at 85 °C for 180 min and finally further drying at 120 °C for 120 minutes employing a temperature ramp rate of 5 °C min−1. The thermal stability of the gels was tested by annealing a portion of them at 450 °C for 30 minutes or by calcination at 700 °C for 3 hours. The temperature ramp rate for both thermal treatments was 10 °C min−1 employing an atmosphere of static air. The theoretical molar composition of the synthesised cobalt–rhenium containing gel was 1 Si:0.8 HMDS:0.85 HMDSO:0.05 Co(II):0.05 Re(VII). The monometallic gels possessed comparable content of the metal component of interest.
Physicochemical characterisation
Elemental analysis of the different materials was carried out with a high resolution inductively coupled plasma mass spectrometer (ICP-MS) Element 2 instrument supplied by Thermo Scientific. Sample dissolution was performed with concentrated nitric acid (HNO3, 1.5 mL) and hydrofluoric acid (HF, 40 wt%, 0.6 g). The samples were diluted with deionised water until a concentration of 0.1 M of the HNO3 was achieved.Powder X-ray diffraction (XRD) patterns were recorded using a Bruker D8 A25 DaVinci X-ray Diffractometer employing CuKα radiation with a LynxEye™ SuperSpeed Detector. The scans were collected in the angular range of 2θ = 5–60°, comprising 4142 steps, with a counting time of 40.32 s per step. The divergence slits were set to 0.1 mm.
Nitrogen adsorption/desorption isotherms and BET specific surface area were measured at liquid nitrogen temperature with a Micromeritics TriStar 3000 surface area and porosity analyser. The pore size distributions were calculated from the N2-adsorption branch, while average pore diameter was calculated from the BJH method based in the N2-desorption branch. The cumulative pore volume (Vc) was calculated from the BJH method at the relative pressure p/p0 = 0.98. The samples were degassed at 250 °C in vacuo for 12 hours prior to analysis. Filler rods were used to reduce measurement errors. The adsorption and desorption isotherms were acquired with 40 points each.
Temperature programmed reduction (TPR) experiments were carried out a Micromeritics Autochem II instrument equipped with a thermal conductivity detector (TCD). The samples were degassed under flowing argon for 20 min prior to TPR analyses up to 900 °C using a temperature ramp rate of 10 °C min−1 under a 50 mL min−1 flow of 5% H2/Ar.
X-ray absorption spectroscopy (XAS)
X-ray absorption spectroscopic data were acquired at the Swiss-Norwegian Beamlines (SNBL, BM31) at the European Synchrotron Radiation Facility (ESRF) for the Co K-edge (7709 eV) and the Re LIII-edge (10535 eV). The data were recorded in hybrid mode. SNBL is located on a bending magnet producing the white beam for the beamline with a Si(111) double crystal monochromator for EXAFS data collection. The cobalt K-edge and rhenium LIII-edge data were collected in transmission mode with the incident and transmitted intensities (I0, It and I2) being detected in ion chambers filled with, I0 (30 cm) 50% N2 + 50% He, and It (30 cm) with 85% N2 + 15% Ar at for the cobalt K-edge and for the rhenium LIII-edge I0 (30 cm) 100% N2, It (30 cm) 50% N2 + 50% Ar. Cobalt references (CoO, Co3O4, Co(OH)2, Co(NO3)2·6H2O and Co-foil) and rhenium references (Re-foil, ReO2, ReO3, KReO4 and NH4ReO4) were collected ex situ and were mixed with boron nitride for optimum absorption and placed in aluminium sample holders. The cobalt XAS data were measured in step scan from 7600 eV to 8300 eV with a step size of 0.5 eV and counting time 300 ms for the cobalt K-edge. The rhenium LIII-edge data were collected between 10350 eV and 11800 eV, with a step size of 0.5 eV and counting time 200 ms.
The in situ samples were placed in 0.9 mm in diameter quartz capillaries with quartz wool on either side. For all in situ measurements, capillary diameter, sample mass and gas flow were kept constant to ensure similar conditions at both edges. The capillaries were placed above a heater for temperature control of the reactor. A switching valve was used to ensure a quick change between pre-treatment gas and reaction gas. A Pfeiffer Omnistar mass spectrometer was used to continuously monitor the exhaust from the sample.
The in situ CoRe-aerogel samples were initially pre-treated in 75% H2 in Ar with a flow of 10 mL min−1 at 600 °C for one hour with a 10 °C min−1 ramp rate. After the pre-treatment, the samples were cooled to 200 °C and the feed gas through the capillary was switched to 5% NH3 in He with a flowrate of 10 mL min−1 before the samples were heated to 600 °C employing a 5 °C min−1 temperature ramp rate. EXAFS scans and XRD patterns were collected both at the beginning and at the end of the sample pre-treatment, as well as at 200 °C in a 75% H2 flow, in 5% NH3 at 200 °C and at 600 °C. XANES scans were collected during heating. The exit gas stream was continuously monitored by mass spectrometry.
X-ray absorption data treatment
Using the Athena Software from the IFFEFITT package40 the imported data were binned (Edge region −30 to 50 eV; Pre-edge grid 10 eV; XANES grid 0.5 eV, EXAFS grid 0.05 Å−1) and background extracted. χexpi(k) was extracted from the EXAFS part of the spectrum. The XANES spectra were normalised from 30–150 eV, multivariate curve resolution (MCR) data were normalised from 50 eV and to the end of the spectra and EXAFS spectra were normalised from 150 eV to the end of the spectra. The spectra were carefully deglitched and truncated when necessary. The threshold energy (E0) for cobalt was set to 0.5 of the normalised absorption edge step and it was chosen after any pre-edge or shoulder peaks. E0 for the rhenium samples was set to the first inflection point in the first derivative spectra. XANES spectra were energy corrected against the corresponding reference foil (Co = 7709 eV and Re = 10535 eV).
MCR plus the alternating least-square (ALS) mathematical algorithm was used to provide the pure response profile of the chemical constituent (species) of the unresolved mixture. The range in energy for the MCR-analysis was 7700–7770 eV for the spectra at the Co K-edge and 10520–10570 eV for the spectra at the Re LIII-edge. To determine the number of principal components able to mimic the system (i.e. rank-analysis), a built-in method based on a singular value decomposition (SVD) approach was used along with a principal component analysis (PCA) of the time resolved data.41,42 Specifically, the time-resolved spectra with a high level of band overlap were translated into a set of chemically meaningful pure-component spectra with respective concentration profiles. Positive constraints for both concentration and spectra profiles and closure constraints for the concentration (i.e. no mass transfer; constant concentration of the absorber, Co/Re, throughout the time resolved experiment) were utilised. Nowadays, MCR is commonly used as a blind source separation method (no reference spectra) to process large datasets generated in labs and synchrotron facilities all over the world. For a detailed description of the methods employed, software and usage, the reader is guided to Jaumot et al.,43,44 Ruckbush et al.45 and Voronov et al.46
EXAFS least-squares refinements were carried out using DL-EXCURV.47 DL-EXCURV fits the theoretical curve (χth(k)) to the experimentally obtained curve (χexp(k)) using the curved wave theory. The fit parameter reported for each refinement procedure is given by the statistical R-factor, defined as:
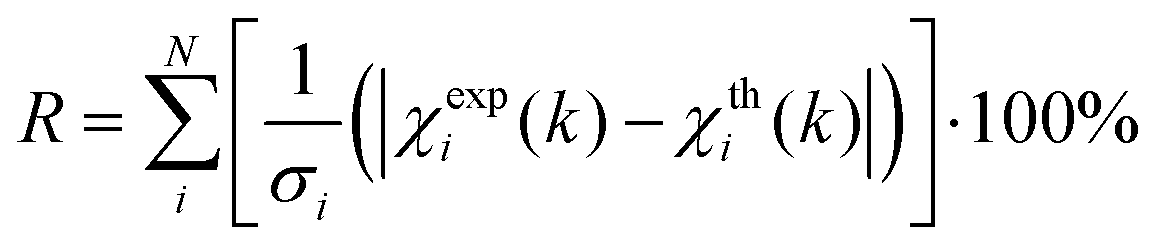
DL-EXCURV calculated ab initio phase shifts were used and verified by using reference compounds. The least-squares refinements were carried out in typical wave number k range 2–11 Å−1 for cobalt and k-range 3–10 Å−1 for rhenium using k3-weighted data. The EXAFS data were analysed with a shell-by-shell approach. A shell was added and then refined before a new shell was added and both shells were refined again (either together or shell wise) until the best fit was found. In cases where the k range was limited, because of the experimental conditions, shells with low coordination numbers that appeared during the refinements were fixed. For one sample at the Re LIII-edge it was necessary to decouple the Debye–Waller factor and coordination number by finding the best fit for both k2- and k3-weighting. This was also verified by comparing the Debye–Waller factor with a suitable reference.
The reduced fraction (Fred) from the EXAFS data was evaluated by calculating the oxidised fraction (Fox) between NCo–O(after PT)/NCo–O (annealed or as-synthesised) and subtracting this from 1. The NCo–O from both the as-synthesised and the annealed sample was used as reference points. The least squares linear combination feature in Athena was used to determine linear combinations (LC).40
Ammonia decomposition reaction
Ammonia decomposition reactions were carried out in a continuous differential packed bed reactor using 25 mg of catalyst diluted in a silica bed. The reactor system was equipped with mass flow and temperature controllers. All reactor tubing was heated to ∼50 °C to avoid any ammonia condensation and related corrosion issues. Prior to each reactor run, the catalysts were pre-reduced under a H2 flow at 330 °C for 1 hour (unless differently stated). After pre-reduction, the temperature was returned to ambient under a H2 flow. Then, the reaction temperature was ramped from room temperature up to 580 °C under 2.5 mL min−1 NH3 and 6 mL min−1 He (corresponding to a GHSV of 6000 mLNH3 gcat−1 h−1). The reactor exit gas was analysed using an on-line gas chromatograph fitted with a Hayesep Q column and employing a thermal conductivity detector. Mass balances were found to be within ±10%.3. Results
General characterisation of aerogel materials
The synthesised aerogels were characterised by XRD, N2 adsorption/desorption isotherms, ICP-MS and TPR. The thermal stability of the material, after annealing and calcination, was also investigated by employing the same techniques. Monometallic equivalents and a plain silica aerogel were studied for comparison. The elemental compositions (Table 1) of cobalt in the Co-gel and the CoRe-gel were determined to be ∼3.0 ± 0.1 wt% for both materials. However, differences in the rhenium content were observed in the monometallic (Re-gel) and the bimetallic (CoRe-gel). The CoRe-gel has an increased content of rhenium of 2.7 wt% when combined with cobalt in the synthesis compared to 1.0 wt% in the monometallic Re-gel. The specific surface areas of the silica aerogels in the present study are high (588–688 m2 g−1) and generally comparable to those reported by Kristiansen and co-workers.39The thermal stability of the CoRe-aerogel was tested by annealing the material to 450 °C in static air especially with attention to the surface area and porosity. The CoRe-aerogel was also tested at a higher temperature (700 °C) to be comparable to that employed for bulk CoRe.48,49 The XRD pattern of the CoRe-aerogel presented in Fig. 1 indicates the presence of the ammonium perrhenate (NH4ReO4) phase (PDF 000-010-0252, ICDD, 2020) which may have formed from the reaction between the dissolved Re2O7 precursor and ammonium ions derived from the reacted HMDS. The NH4ReO4 phase is lost as the sample is heat treated during annealing and calcination. The XRD pattern of the as-synthesised Co-gel (ESI,† S1) is amorphous in nature and continues to be amorphous after the annealing and calcination. However, the XRD pattern of the as-synthesised Re-gel (ESI,† S1) possesses reflections corresponding to ammonium perrhenate, but the overall intensity is lower than for the CoRe-gel which corresponds to the lower metal content of the rhenium gel. As observed in the case of its CoRe counterpart, the annealed and calcined Re-gels exhibit amorphous XRD patterns. Rhenium(VII) oxide and ammonium perrhenate are known to be volatile above 300 °C when supported on alumina.50 In the calcined CoRe-gel (3 hours, 700 °C in static air, 10 °C min−1 heating rate) volatilisation of rhenium is observed where the amount of rhenium is 0.5 wt%. However, the volatilisation and loss of rhenium is not observed in the annealed CoRe-gel (30 minutes, 450 °C in static air, 10 °C min−1 heating rate) as the rhenium content is unchanged compared to the as-synthesised CoRe-gel. The cobalt content in the CoRe-gel and the Co-gel (Fig. 1 and ESI,† Table S1) is stable with an apparent small increase of cobalt content after annealing and calcination compared to the as-synthesised sample. This is associated with the methylated surface being burnt off, the decomposition of the anions of the cobalt precursor (NO3−), and desorption/decomposition of residues of ammonia and the ammonium ions introduced from the surface modification during synthesis.
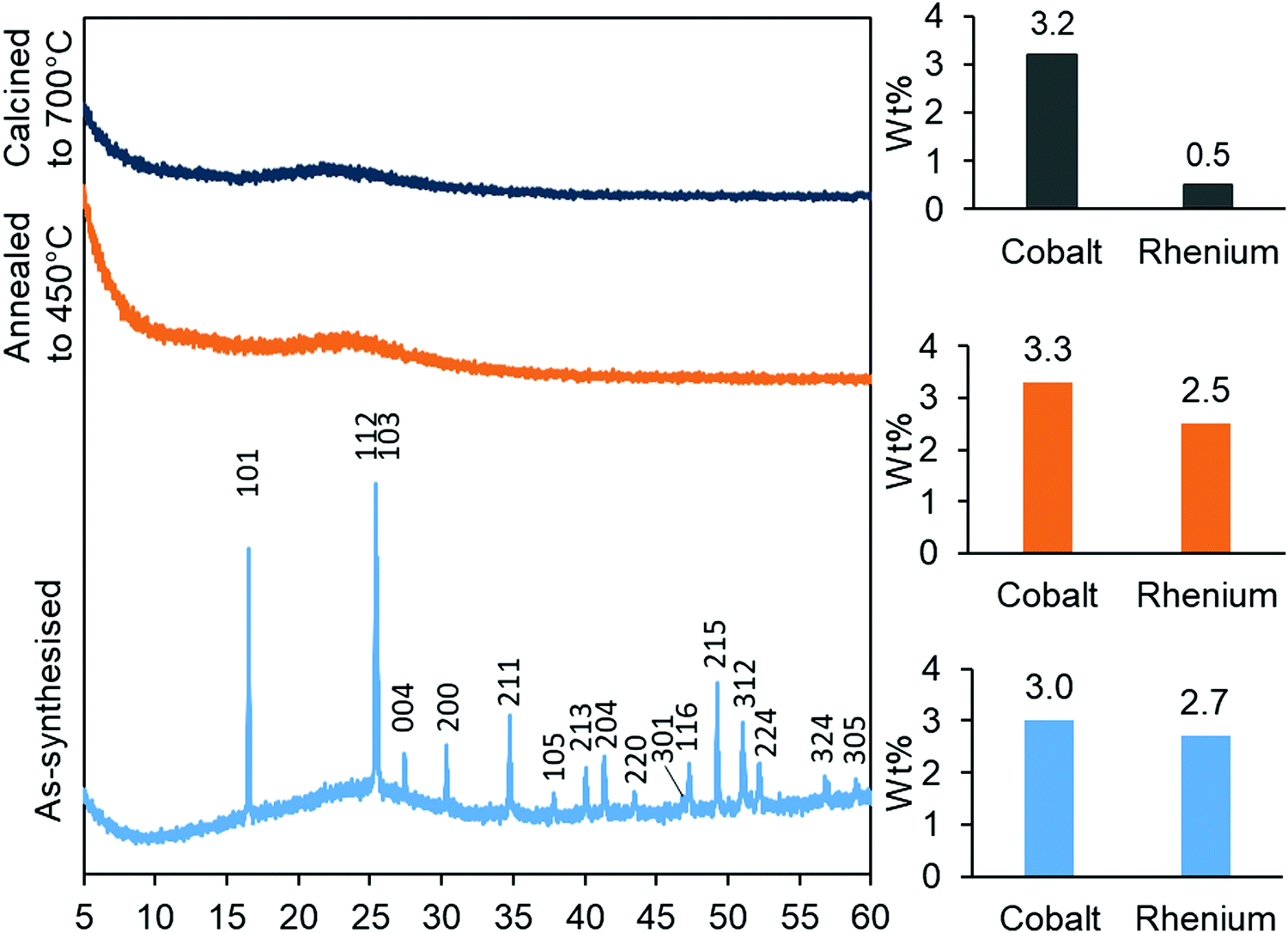 | ||
| Fig. 1 XRD patterns (left) of the as-synthesised, annealed and calcined CoRe-aerogels and their metal contents (right) after their respective thermal treatments. | ||
The nitrogen adsorption–desorption-isotherm of the as-synthesised CoRe-gel (Fig. 2) shows a combination of type I and type IV (IUPAC classification) isotherm characteristics as expected for silica aerogels being both micro- and mesoporous. Similarly, the isotherm of the Co-gel (ESI,† S2) is a type I and type IV composite isotherm. Both the Co-gel and the CoRe-gel have hystereses similar in shape which resemble a combination of type H2 and type H4 indicative of poorly defined pore sizes and slits.51 The pore size distributions (ESI,† S3) show the majority of the pore volume in the material originates from small mesopores and micropores.
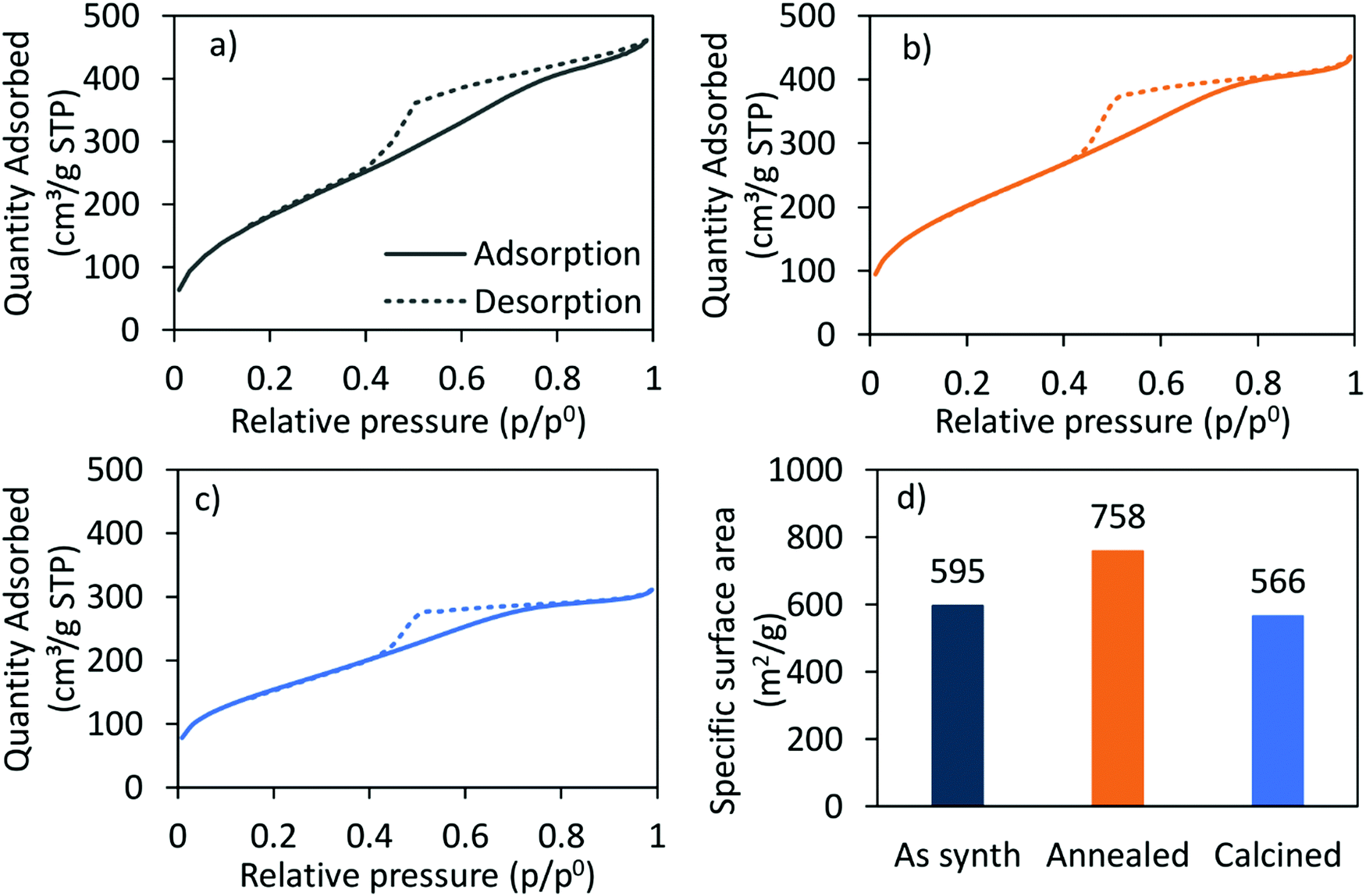 | ||
| Fig. 2 The N2-adsorption (solid)/-desorption (dashed) isotherms of the (a) as-synthesised, (b) annealed, (c) calcined CoRe-aerogel and (d) the corresponding BET surface areas. | ||
The hysteresis loops are also present in the annealed and the calcined CoRe-gel and Co-gel materials (Fig. 2 and ESI,† S2) confirming the thermal stability of their mesoporous nature up to 700 °C for 3 hours. However, the isotherms of the CoRe- and Co-gels become more similar to type I in nature with an H2 hysteresis, indicative of microporous materials, upon thermal treatment. A decrease in hysteresis loop size with higher temperature thermal treatment indicative of partial loss of mesopores is observed for the Co- and the CoRe-gels.
There is an apparent increase in the BET specific surface area (Fig. 2) after annealing the CoRe-aerogel and the same trend is also observed for the plain gel, Co-gel and the Re-gel (ESI,† S3), although caution must be applied to such analyses given the microporous nature of the materials which is a limitation for BET analysis. This apparent increase may be related to the oxidation of the hydrophobic surface and/or unblocking pores. Interestingly, the specific surface area decreases for the calcined gels which may arise from pore shrinkage due to dehydroxylation.
The temperature programmed reduction profiles of the CoRe-gel and the monometallic Re-gel and Co-gel counterparts are shown in Fig. 3. The Co-gel displays only one very small and almost negligible peak (Peak 1) in the TPR profile confirming only partial reduction. This reduction event at 387 °C correlates with the reduction of Co3O4 to CoO27 suggesting stabilisation of the Co2+ species in the silica gel framework. On the other hand, the Re-gel, presents two small reduction peaks at 295 °C and 290 °C, respectively. It is important to note that the H2 consumptions in the mono-metallic catalysts are very small compared to the Co–Re-gel system as shown in Table 2, not only being related to the differences of metal loading but more importantly to the enhanced reducibility of the CoRe-gel catalyst. Indeed, the Co-gel catalyst is only reduced 6.9% from Co3O4 to CoO while the Re-gel catalyst reduces ∼34%. The combination of CoRe enhances the combined reduction to 53.1% (Table 2). Such cooperative interaction of the reduction process is reflected in the TPR profile of the CoRe-gel. The reduction of both cobalt and rhenium takes place at very similar temperatures of 250–400 °C with the peak being fitted by two overlapping peaks (as shown by the red and blue profiles). Rhenium species reduce first, leading to the reduction of Co3O4 species at lower temperature than the counterpart Co-gel catalyst due to hydrogen spill over from rhenium.52 The negative peaks in the three TPR profiles at ∼700 °C was also observed by Guzci et al.53 for Re–Co/SiO2 and attributed to decomposition induced desorption of H2 and smaller organic compounds.
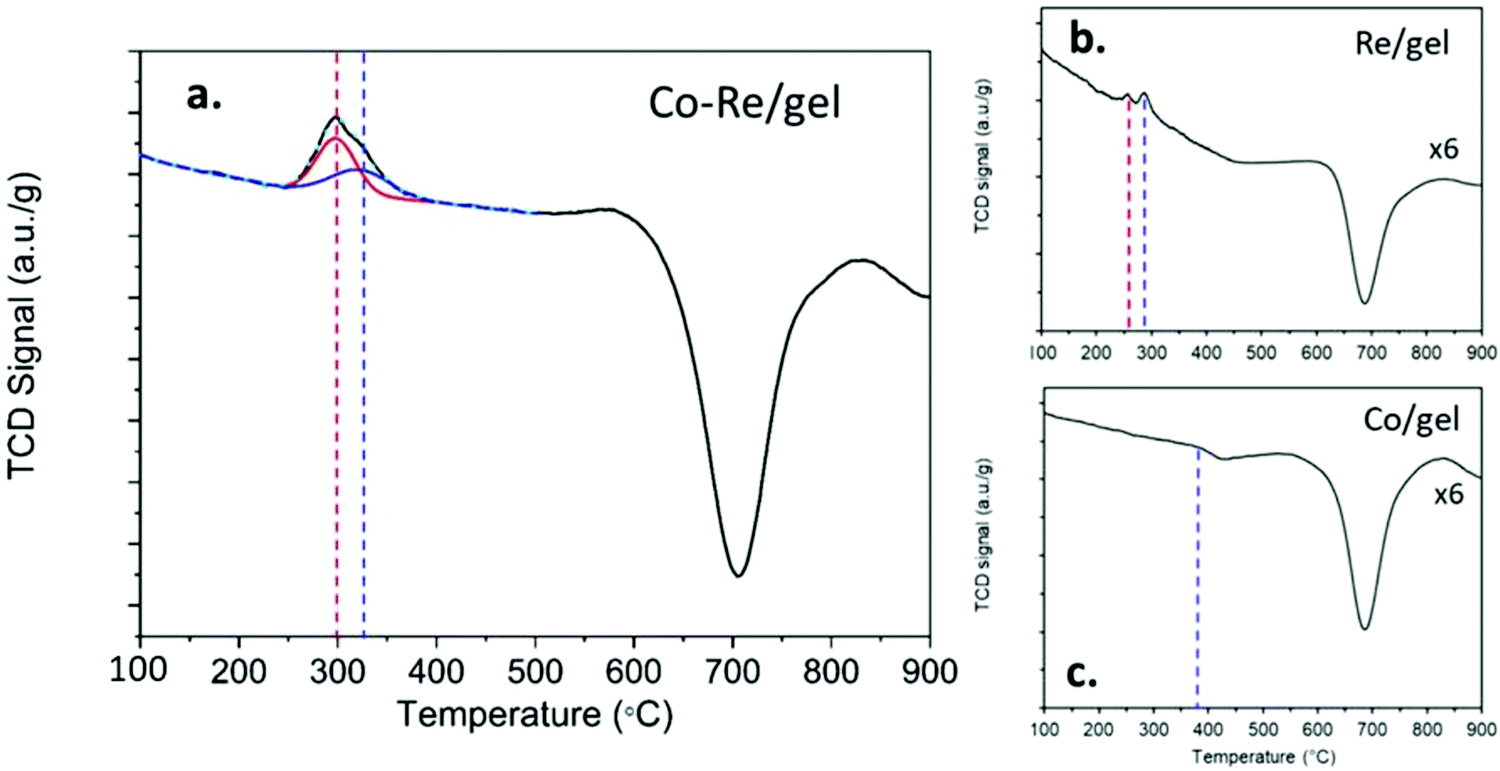 | ||
| Fig. 3 Temperature programmed reduction (TPR) of the as-synthesised catalysts (a) 2.7 wt% Co–3 wt% Re on silica aerogel (CoRe/gel), (b) 1.0 wt% Re on silica aerogel (Re/gel) and (c) 2.9 wt% Co on silica aerogel (Co/gel). The TCD signal of (b) and (c) have been multiplied 6 times to show any H2 consumption peaks. | ||
| Catalyst | H2 consumed (cm3 g−1) | Peak 1 | Peak 2 | Metal reduction (%) |
|---|---|---|---|---|
| Co-gel | 0.28 | 387 | 7 | |
| Re-gel | 0.45 | 255 | 290 | 34 |
| CoRe-gel | 4.08 | 298 (60%) | 322 (40%) | 53 |
X-ray absorption spectroscopy
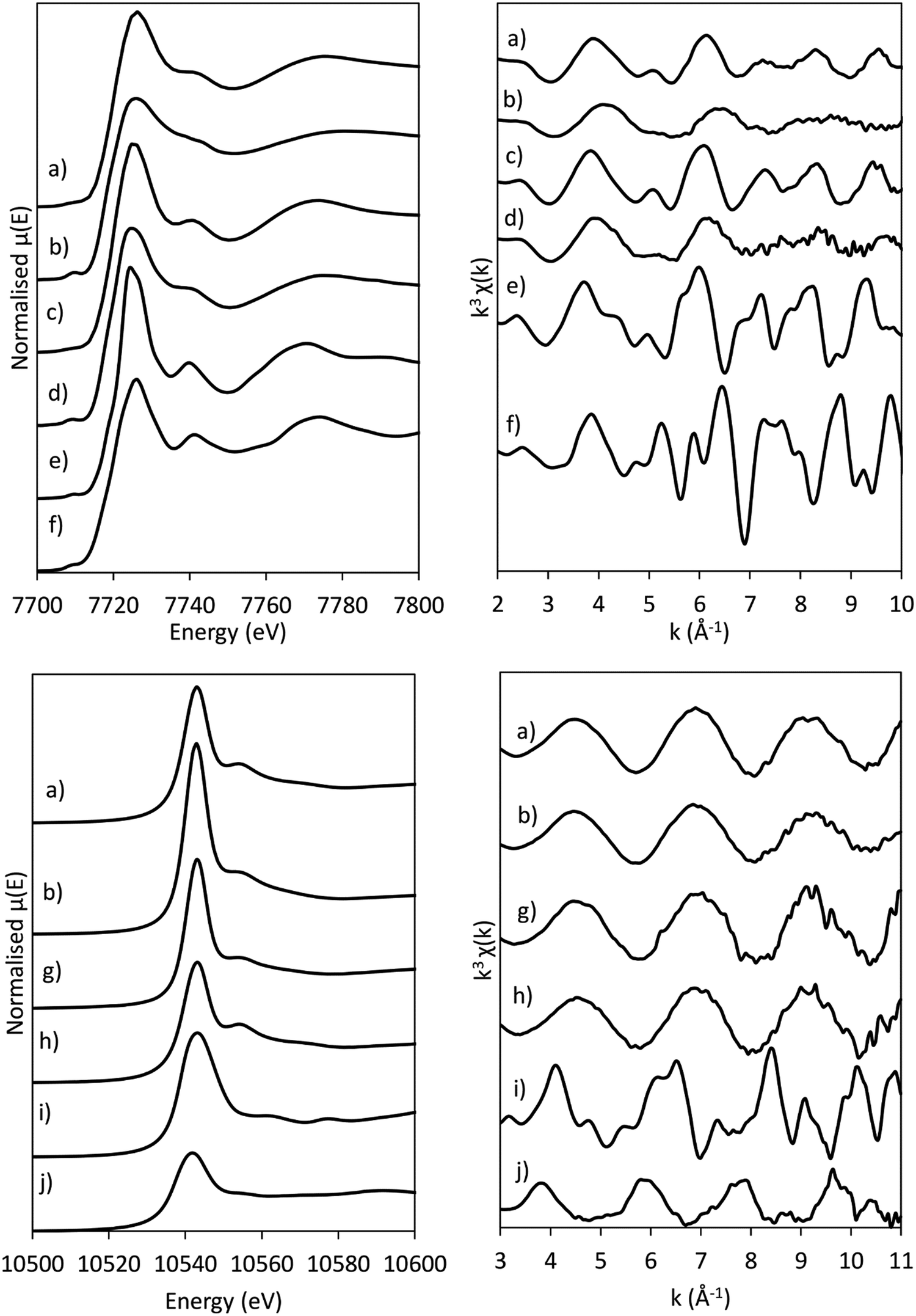 | ||
| Fig. 4 XANES spectra (left) and k3-weighted chi-curves (right) of the (a) as-synthesised CoRe-gel, (b) annealed CoRe-gel, (c) as-synthesised Co-gel, (d) annealed Co-gel, (e) Co(OH)2, (f) CoO, (g) annealed Re-gel, (h) NH4ReO4, (i) ReO3 and (j) ReO2 at the Co K-edge (top) and at the Re LIII-edge (bottom). | ||
The presence of Co(OH)2 in the as-synthesised CoRe-gel is further confirmed by the EXAFS analysis (Table 3 and Fig. 5) which yields an average multiplicity of 5.3 for the Co–O pair at 2.08 Å and 6 in average multiplicity for the Co⋯Co pair at 3.12 Å. The Re-environment in the CoRe-gel possesses an average multiplicity for the Re–O pair of 4.2 at 1.73 Å correlating well with the NH4ReO4 phase as found with XRD. There are no reflections in the XRD pattern which can be attributed to cobalt containing phases, however the presence of a Co⋯Co backscattering pair from EXAFS indicates that some of the cobalt is present as nanophase CoOx or Co(OH)2.
| Treatment | Shell | N | R (Å) | 2σ2 (Å2) | E F (eV) | R (%) | Δk | AFAC |
|---|---|---|---|---|---|---|---|---|
| a DW-value was fixed. b Shell was kept as a constant and not fitted, due to experimental conditions and constraints in k-range. | ||||||||
| As-synthesised | Co–O | 5.3(5) | 2.080(9) | 0.023(4) | −6.5(2) | 32 | 2–10 | 0.66 |
| Co…Co | 6(2) | 3.122(9) | 0.027(6) | |||||
| Re–O | 4.2(1) | 1.734(3) | 0.0005a | −13.6(9) | 19 | 3–11 | 0.64 | |
| Annealed at 450 °C | Co–O | 3.5(5) | 2.01(1) | 0.021(6) | −6.5(9) | 49 | 2–10 | 0.66 |
| Re–O | 4.1(2) | 1.718(3) | 0.002(1) | −10.0(7) | 15 | 3–10 | 0.64 | |
| H2/Ar at 600 °C | Co–Ob | 0.5 | 1.95 | 0.001 | −8.9 | 42 | 2–9.5 | 0.79 |
| Co–Co | 1.4(4) | 2.414(7) | 0.020(6) | |||||
| Co–Re | 2.8(6) | 2.70(2) | 0.04(1) | |||||
| Re–Ob | 0.5 | 1.90 | 0.01(7) | −11(1) | 38 | 3–11 | 0.8 | |
| Re–Co | 3.0(8) | 2.53(1) | 0.034(6) | |||||
| Re–Re | 3(1) | 2.74(1) | 0.030(8) | |||||
| NH3 at 600 °C | Co–O/N | 1.2(3) | 1.96(2) | 0.017(8) | −8.9(6) | 45 | 2–9.5 | 0.79 |
| Co–Re | 2.7(6) | 2.58(1) | 0.023(5) | |||||
| Re–O/N | 1.1(2) | 1.99(2) | 0.020(7) | −12.7(6) | 29 | 3–10 | 0.8 | |
| Re–Co | 6(1) | 2.608(5) | 0.041(4) | |||||
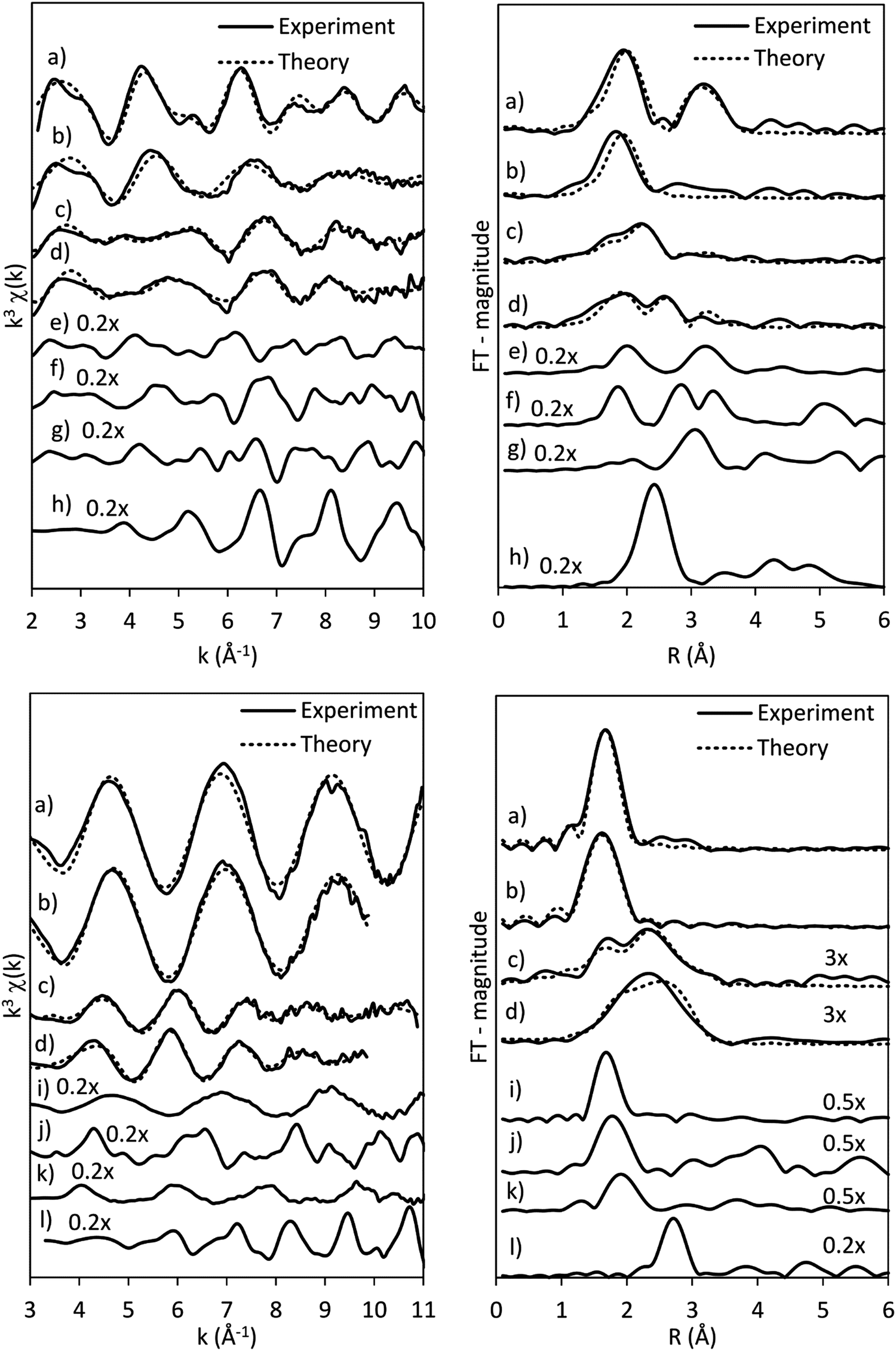 | ||
| Fig. 5 Results of EXAFS analysis of the CoRe-gel (a) as-synthesised (b) annealed at 450 °C (c) pre-treated at 600 °C in H2 and (d) in 5% NH3 at 600 °C with selected references (e) Co(OH)2, (f) Co3O4, (g)CoO, (h) Co-foil, (i) NH4ReO4, (j) ReO3, (k) ReO2 and (l) Re-foil at the Co K-edge (top) and Re LIII-edge (bottom) with the k3-weighted chi-curves (left) and FT-magnitude (right). Signals have been multiplied with factors given in the figure for better evaluation. | ||
The annealed CoRe-gel (Fig. 6) reveals a Co–O pair at 2.01 Å with an average coordination number of 3.5. The fitting of the second shell was unsuccessful, but the identity of the backscattered signal from this R-range was identified to be a Co⋯Si contribution by analysis of the Fourier filtered data (ESI,† Tables S2 and S4). The second Co–O–Co coordination shell is lost after annealing and this dramatic change suggests a dispersion of the CoOx/Co(OH)x species and subsequent interaction with the silica phase resulting in cobalt migrating into the gel network. The rhenium environment (Table 3 and Fig. 5) with Re–O pair at bond distance of ∼1.72 Å and an average coordination number of 4.1 is apparently not significantly changed by annealing, although there is a contraction of the Re–O bond distance observed indicating a stronger interaction between Re and O. From XRD the crystallinity is seen to be lost corresponding to decomposition of the NH4ReO4 phase.
 | ||
| Fig. 6 Schematic of the CoRe-gel after annealing with the major species consisting of rhenium (blue), cobalt (green) and oxygen (red). | ||
Speciation of cobalt and rhenium in silica aerogels during H2 reduction
MCR-analysis of the pre-treatment in 75% H2 flow was performed to investigate the reduction path of cobalt and rhenium. MCR-analysis provides the different components of a system and their concentrations based on the spectra themselves with no external references (see the ESI† for the choice of number of components pp. 15–19). The reduction of the CoRe-gel is seen in both elements at ∼240 °C as evident from the MCR-analysis (Fig. 7) and rhenium is largely reduced after 1 hour at 600 °C. Another rhenium component is found in the analysis which is formed from 240 °C and is at its maximum relative concentration from 350–425 °C and is not present at the end of the pre-treatment. This component (ESI,† Fig. S5) lacks the feature at 10554 eV, which is characteristic for Re(VII)-species and is believed to be Re(IV/VI) due to its similarity with these species. The reduction of cobalt is initiated at ∼220 °C and is slower than for rhenium and continues after the CoRe-gel reaches 600 °C. The starting component of cobalt is not fully reduced and stabilises only after ∼60 minutes at 600 °C. The Co(II*)-component in Fig. 7 is believed to be a different Co(II) species from the characteristic pre-edge peak from comparison with references (ESI,† Fig. S5 and S6). Co(II*) is present in the final state of the CoRe-gel. The components Co(II*) and Re(IV/VI) from the MCR analysis may also be related to species formed due to metal–silica aerogel interactions.
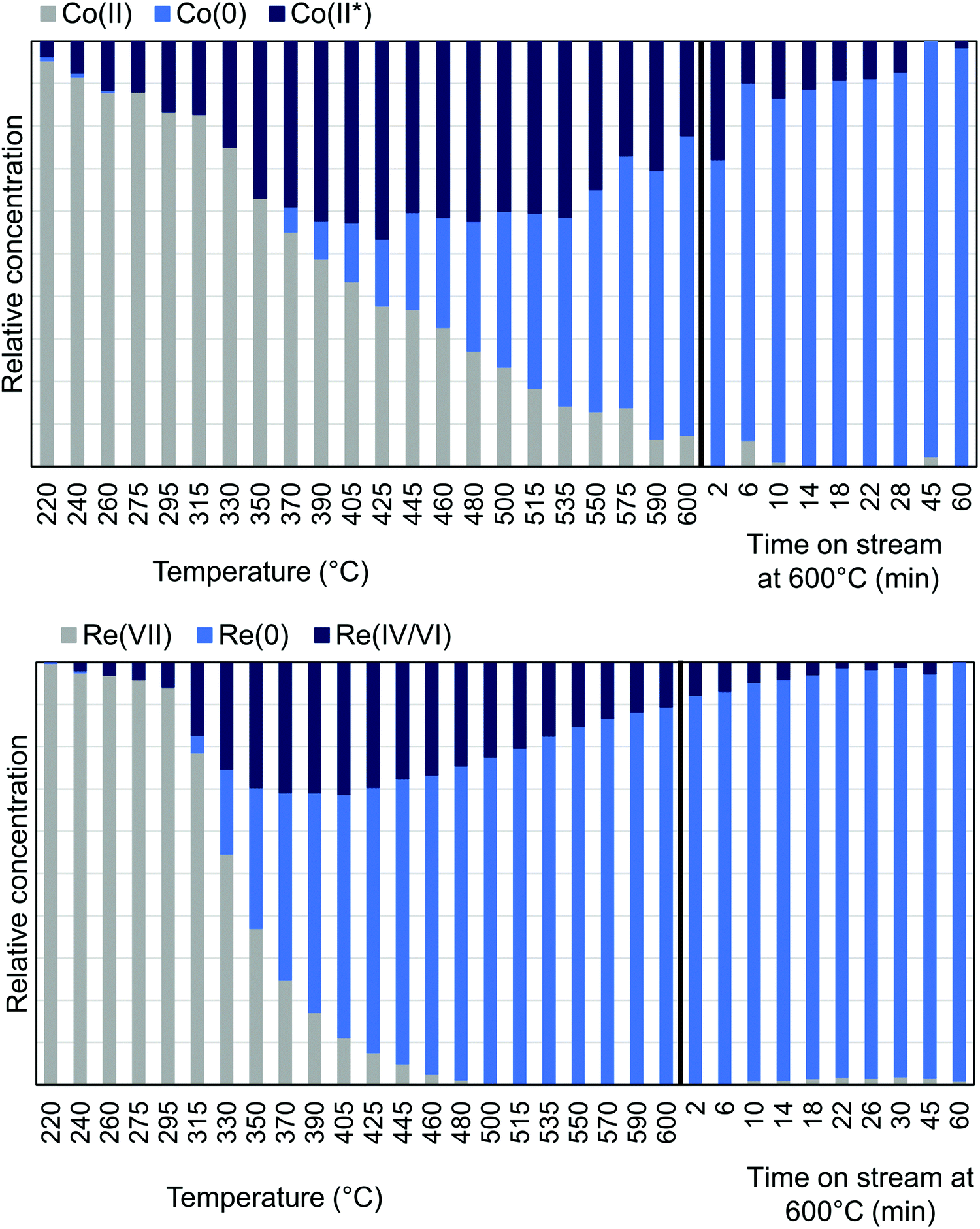 | ||
| Fig. 7 MCR-analysis of the pre-treatment of CoRe-gel from the Co K-edge (top) and Re LIII-edge (bottom) with the relative concentrations of the components. | ||
The normalised and energy shifted XANES of the pre-treated Co-gel and pre-treated CoRe-gel (ESI,† S7 and S8) at 600 °C in 75% H2 show the synergistic effect of rhenium and cobalt. While the Co-gel is not reduced after the pre-treatment, with a prominent white line feature (7726 eV) and a pre-edge peak (7710 eV) characteristic for Co2+ species being present, the CoRe-gel is reduced and resembles the Co-foil. However, the CoRe-gel has a small white line intensity at 7727 eV compared to bulk Co-metal. This difference was also observed in the unsupported CoRe material.25,29
The results of the EXAFS analysis (Table 3 and Fig. 5, 8) of pre-treated CoRe-gel supports the MCR-analysis confirming an 80–90% reduction. The fit was significantly improved by adding a short distance Co–O shell (NCo–O = 0.5) at 1.95 Å. This shell was fixed and not part of the final refinements due to limited k-range. EXAFS analysis of rhenium suggests 80–90% of the rhenium is reduced and this contrasts with the MCR which show that rhenium is fully reduced. This difference is due to an addition of a constant Re–O contribution (NRe–O = 0.5, at 1.9 Å) in the refinements. The presence of unreduced small cobalt/rhenium-containing oxidic clusters and/or a strong cobalt/rhenium interaction with the silica aerogel support can be inferred. There are monometallic contributions found with 1.4 Co–Co at ∼2.4 Å and 3 Re–Re contributions at 2.74 Å. In addition, there are both Co–Re and Re–Co contributions found at 2.7 Å (NCo–Re = 2.8) and at ∼2.53 Å (NRe–Co = 3) respectively. Several refinement strategies have carefully been tested such as fixed bond distances and mixed sites function in DL_EXCURV, but due to the experimentally constrained k-range neither was successful in providing more similar bonding distances.
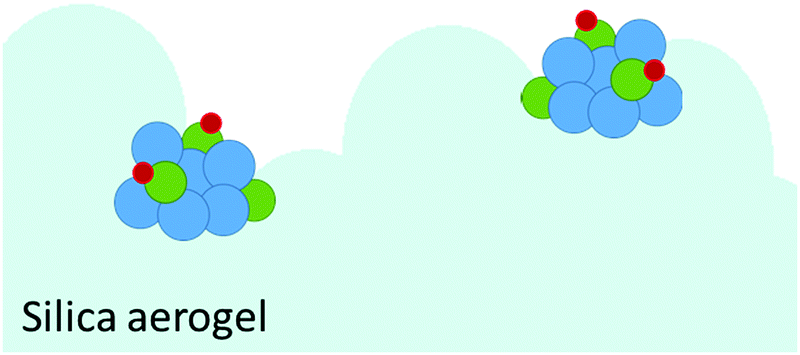 | ||
| Fig. 8 Schematic of the CoRe-gel in 75% H2 at 600 °C with the major species consisting of rhenium (blue), cobalt (green) and oxygen (red). | ||
Cobalt and rhenium speciation during ammonia treatment
After the pre-treatment in H2 at 600 °C, the CoRe-gel was cooled to 200 °C in the same atmosphere and there were no observable changes in the XAS spectra of either cobalt or rhenium. When switching to ammonia at 200 °C, a large increase in the white line intensity in the Co XANES for the CoRe-gel was observed, confirming oxidation to Co(II/III) species. There are only small observable changes in the Re XANES (Fig. 9) suggesting only minor changes in the local environment of Re when switching to ammonia at 200 °C. Partial oxidation of the Co species in the Co-gel is also observed (Fig. 9). The partial oxidation could be attributed to the interaction of N and/or O species with the metal phases. | ||
| Fig. 9 XANES-spectra before and after switching from H2 to NH3 from the Co K-edge (left) and Re LIII-edge (right). | ||
Catalytic activity for ammonia decomposition
The conversion for ammonia decomposition (Fig. 10) for the CoRe-gel was 50% at ∼550 °C and, by comparison, the monometallic counterparts only reach 15% at 580 °C. Hence it can be concluded that presence of both metals significantly improves the activity. By comparison, bulk CoRe1.6 has a lower temperature (435 °C) for 50% conversion under comparable reaction conditions.29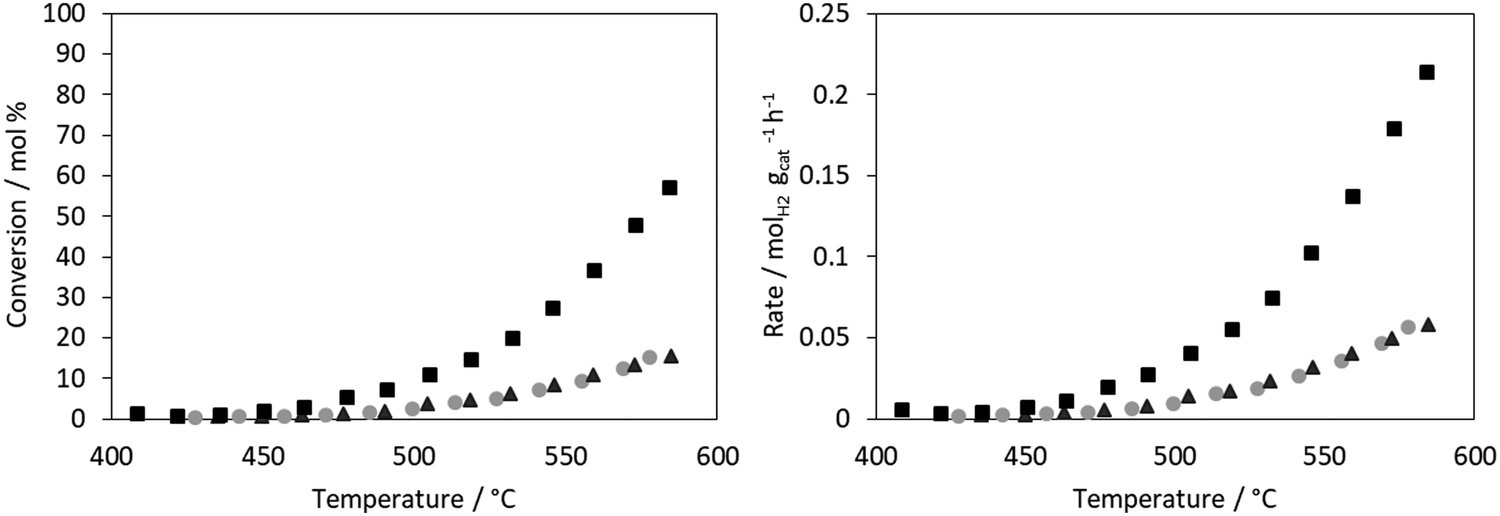 | ||
Fig. 10 Catalytic activity (left) and rate of reaction (right) of ▲ Co-aerogel (1.7 wt% Co),  Re-aerogel (1.5 wt% Re), ■ CoRe gel (3 wt% Co–2.7 wt% Re). Reaction conditions: 2.5 mL min−1 NH3, 6 mL min−1 He, 25 mg catalyst, GHSV: 6000 h−1. Catalysts are pre-reduced at 330 °C under H2 flow. Re-aerogel (1.5 wt% Re), ■ CoRe gel (3 wt% Co–2.7 wt% Re). Reaction conditions: 2.5 mL min−1 NH3, 6 mL min−1 He, 25 mg catalyst, GHSV: 6000 h−1. Catalysts are pre-reduced at 330 °C under H2 flow. | ||
Co-only and Re-only supported catalysts present similar ammonia decomposition catalytic activity. However, it is important to highlight the synergetic effect achieved by the CoRe gel in agreement with the characterisation results, although it should be cautioned that the pre-reduction temperature applied is lower than that employed in the XAS experiments.
Cobalt and rhenium speciation during NH3-decomposition
In situ XAS was collected during ammonia decomposition over the CoRe-gel up to 600 °C to investigate the active CoRe species (Fig. 12). The XANES (ESI,† S9) of the CoRe-gel in 5% ammonia shows an increase in the white line features at both the Co K-edge and Re LIII-edge increase in the temperature range between 200–400 °C, indicating an oxidation of both cobalt and rhenium. Interestingly, when increasing the temperature from 400–600 °C in reaction gas (Fig. 11) a reduction of both elements is observed by the decrease in the white line features. Linear combinations of the CoRe-gel in 5% ammonia at 600 °C (ESI,† S10) using the as-synthesised CoRe-gel and corresponding Co-/Re-foil show that the active material cannot be expressed as a combination of the two. The white line feature at 7726.5 eV at the Co K-edge was also reported for CoRe25,54 and CoPt in NaY zeolite,55 CoPT/C56 where bimetallic interaction is suggested. Therefore, the XANES strongly indicate bimetallic Co–Re interaction when the CoRe-gel is active for ammonia decomposition. The XANES of the Re-environment (ESI,† S11) in the active CoRe-gel display a white line feature at ∼10540 eV which matches the position of the white line feature in the Re-foil. The features seen in the references NH4ReO4 (10554 eV), ReO3 (10562 eV and 10577 eV) and ReO2 (10554 eV) are not found for the Re-environment in the CoRe-gel at 600 °C in 5% NH3. The oxidised fraction of the CoRe-gel in 5% NH3 during ammonia decomposition is 20–30% for both cobalt and rhenium based upon the average multiplicities from EXAFS (Table 3).
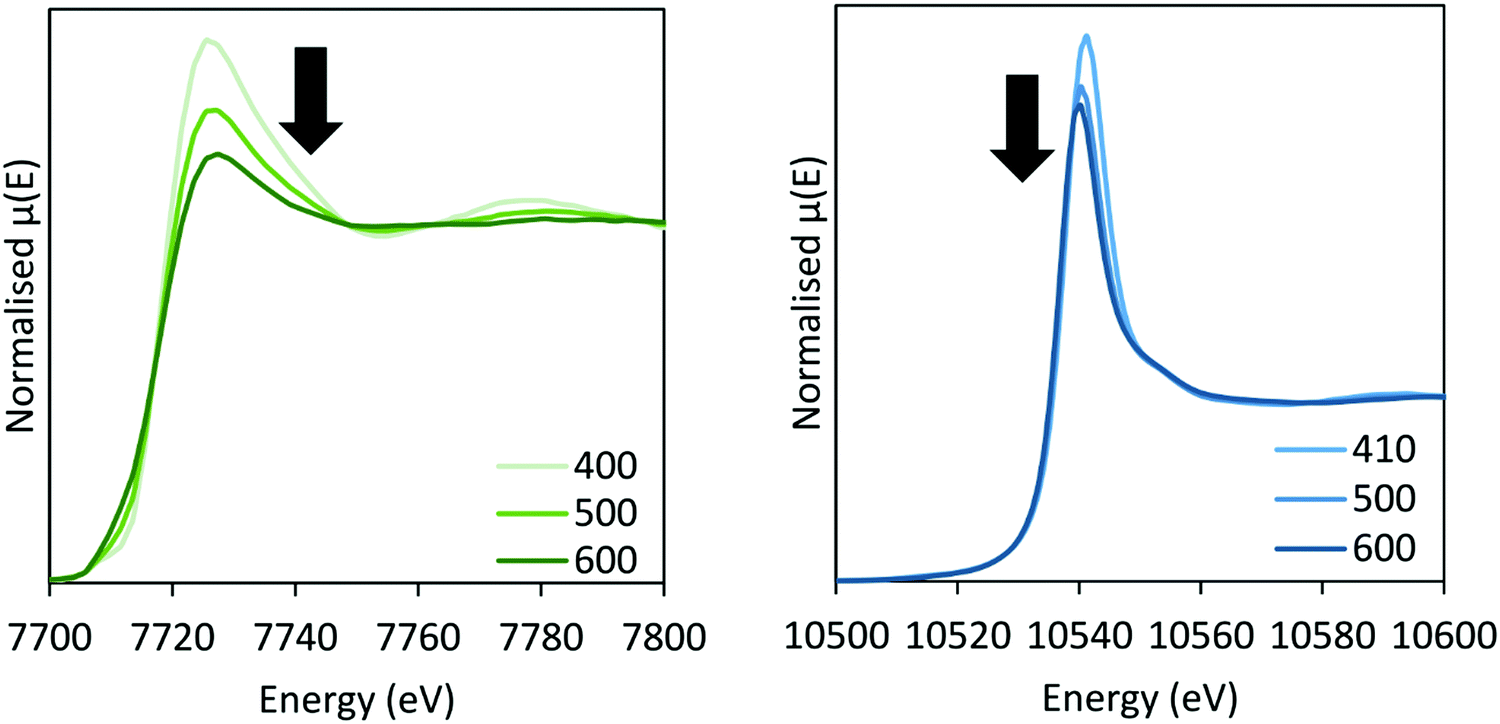 | ||
| Fig. 11 XANES of the CoRe-gel during ammonia decomposition at the Co K-edge (left) and Re LIII-edge (right) between 400–600 °C in 5% NH3. | ||
As presented, the EXAFS analysis of CoRe-gel at the end-point (Table 3 and Fig. 5) reveals no monometallic contributions, although a Co-Co contribution was detected in the EXAFS analysis but this was not included due to limited k-range due to experimental constraints. The average coordination number of the bimetallic Co–Re contribution is mostly unchanged at 2.7 with a shortening of the bonding distance from 2.70 Å to 2.58 Å. Interestingly, the Re–Co contribution increased in average coordination number to 6. The bonding distance of the Re–Co elongates from 2.53 Å to 2.61 Å during ammonia decomposition and this might be due to some monometallic Re–Re contribution in the fitted Re–Co shell. However, the Re–Re could not be fitted and we believe this is due to the experimental constraints making the Re–Co and Re–Re shell inseparable. The Re–Co and Re–Re shells were attempted to be fitted with the mixed sites function in DL_EXCURV, but this was not successful as there was no convergence for the distribution of the Re–Re/–Co backscattering pair.
4. Discussion
Cobalt promoted rhenium introduction in silica aerogels
As a consequence of the hydrophilic nature of [ReO4−], it can be difficult to introduce rhenium onto supports by methods other than by impregnation.32–34,57 In this study, the uptake of rhenium nearly triples when cobalt is co-introduced during the sol–gel stage. In the as-prepared CoRe-gel, cobalt forms nanoparticulate CoOx/Co(OH)x as evidenced by the Co–O–Co distances apparent in the EXAFS measurements, while rhenium is present as NH4ReO4 as seen in the XRD patterns. Ammonium perrhenate is also observed in the XRD pattern for the Re-gel, indicating that the speciation of rhenium is not affected by the presence of cobalt. EXAFS at the Co K-edge reveals similar local environments for the Co-gel and the CoRe-gel with a Co–O shell and a Co⋯Co shell with relatively high multiplicity (N ∼ 6 and for the Co-gel N ∼ 5) which is strongly indicative of the presence of a CoOx/Co(OH)2 phase. Differences in the XANES (Fig. 4) were further investigated by linear combination as it is this region which is more sensitive to structural changes. The best fit for the cobalt species in the Co-gel was obtained using a mixture of references CoO (0.55) and Co(OH)2 (0.45) (ESI,† Table S3 and Fig. S12). Interestingly, the best fit for the CoRe-gel was obtained with fractions of only 0.2 of Co(OH)2 and 0.25 of CoO while the remainder of the cobalt species (0.55) resembles the surroundings of the annealed CoRe-gel. Hence, the presence of rhenium precludes Co–O–Co bonds and strongly suggests single cobalt species possibly interacting with the framework. The annealed CoRe-gel was also introduced as a “non-particulate” Co2+ reference when fitting the Co-gel, but this was not successful.Synergistic effect of cobalt and rhenium during annealing
Typical behaviour of metal precursors on a 2D carrier, detrimental to catalytic activity, is oxide formation and particle growth during thermal treatment. A 3D carrier may inhibit particle growth due to its microporous nature, and for ideal catalyst speciation control is desired over the chosen temperature range. The gels applied in this study were therefore annealed to 450 °C while monitoring the metal speciation using XAS. A major observation in this work is the loss of the long-range order corresponding to ammonium perrhenate in the CoRe-gel after annealing. This cannot be explained by the volatilisation of rhenium as the rhenium content apparently remains unchanged. Concurrently, results from EXAFS-analysis at the Re LIII-edge reveal no change after annealing. Together these results suggest thermal dispersion and migration of perrhenate particles resulting in ReVII-species present as [ReO4]– single entities. Rønning et al.57 and Bazin et al.35 reported the RRe–Osupport to be ∼2.0 Å for CoRe/Al2O3 systems and, by contrast, the Re–O bond distance in the CoRe-gel is much shorter (1.72–1.74 Å) corresponding with that found in the first Re–O shell of ammonium perrhenate.
The synergistic relationship between cobalt and rhenium is consolidated by the dispersion of NH4ReO4 coinciding with the disappearance of the Co–O–Co backscattering shell in the CoRe-gel during annealing. Results from EXAFS refinements suggest that CoOx/Co(OH)2 species undergo thermal disruption while forming highly dispersed [⋯Co(–O)3⋯]-species. In contrast, the Co-species in the Co-gel remain mostly unchanged during annealing where a Co–O backscattering shell (N ∼ 4–5, R ∼ 2.1) and a Co–O–Co backscattering shell (N ∼ 7–5, R ∼ 3.09–3.14) were fitted (Fig. 13). In the Re-gel, a loss of crystallinity is observed confirming thermal dispersion as observed for the CoRe-gel. Compared to the CoRe-gel, the rhenium content of the Re-gel is halved upon annealing, confirming the volatilisation of [ReO4]– and the stabilisation of the Re-species in the silica aerogel in the presence of cobalt species. Hence, the presence of rhenium promotes the dispersion of particulate CoOx/Co(OH)2 to single site Co2+ while cobalt clearly assists in anchoring [ReO4]− entities during thermal treatment. The Co–O bond distance decreases from 2.11 Å to 2.00 Å after annealing which could be ascribed to Co2+ ions migrating into the framework. Similar results were reported for cobalt ion exchanged CoNaKPHI (zeolite) where a decrease in Co–O bond distance from 2.09 Å to 2.02 Å58 is explained by Co2+ present as the hexaaquacobalt(II)-ion complex in the cages of the zeolite upon dehydration migrating into more uncoordinated positions in the pores of the zeolite.
 | ||
| Fig. 12 Schematic of the CoRe-gel in 5% ammonia at 600 °C with the major species consisting of rhenium (blue), cobalt (green) and oxygen/nitrogen (red). | ||
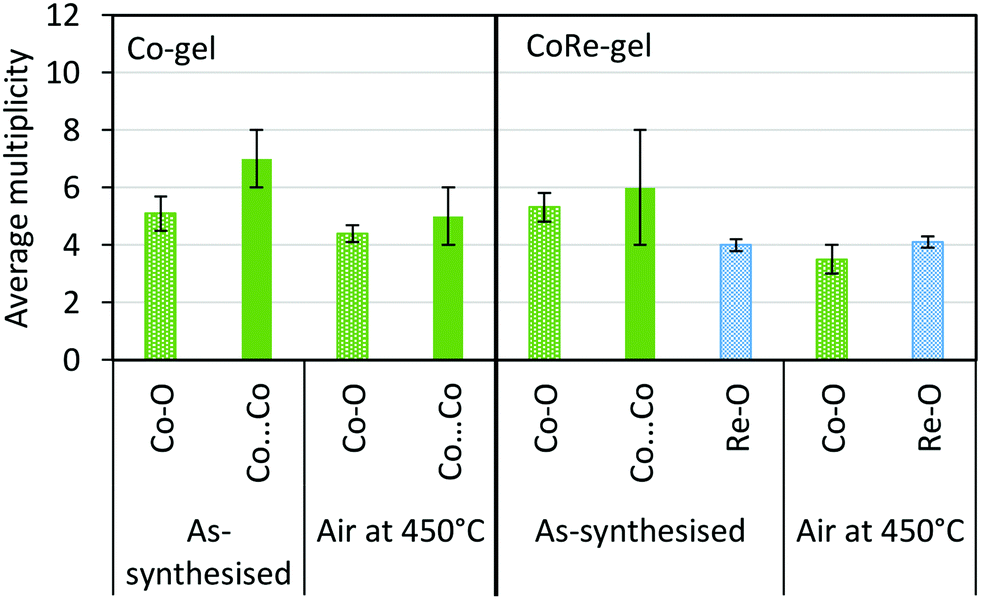 | ||
| Fig. 13 Average multiplicity of the Co-gel and CoRe-gel before and after annealing in air to 450 °C. | ||
The thermal stability of the porous network in silica aerogels is an important consideration for the catalysts. When the CoRe-gel, Co-gel and Re-gel are treated at 450 °C in static air, an increase in specific surface area is observed in all cases. This can be explained by the removal of some of the methyl groups as seen by a decrease in the intensity of the methyl vibrations (2963 cm−1 and 2906 cm−1) in DRIFTS (see ESI,† Fig. S13 and S14).
Formation of CoRe bimetallic nanoclusters in silica aerogels
We have recently demonstrated that hydrogen pre-treatment of unsupported CoRe1.6 resulted in formation of highly active CoRe-surface pairs for the ammonia decomposition reaction.29 Depositing CoRe on a high surface area support such as silica aerogel is desirable as it ensures high dispersion, potentially increasing the concentration of accessible active sites. In situ XAS during hydrogen pre-treatment shows that the rhenium(VII) species starts to reduce at ∼240 °C while the reduction of the cobalt(II) species commences at 220 °C. Both the reduction of rhenium and cobalt is incomplete even after 1 hour at 600 °C as seen by the constant shells of Co–O shell (N = 0.5) and by the Re–O (N = 0.5) from EXAFS that was improving the fit at both edges. Similarly, TPR experiments show that reduction occurs over two steps between 290–380 °C indicating that most of the species are reduced in this temperature range. Interestingly, the Co–O bond distance decreases from 2.01 Å to 1.95 Å after reduction which is shorter than in cobalt(II) oxide. Moen et al.59 incorporated Co2+ into the framework of SAPO-34 yielding Co–O bonding distances of 1.94 Å which they claimed to be consistent with framework substitution. We therefore believe that the remaining Co–O contribution at 1.95 Å in the CoRe-gel is tightly bound to the silica gel structure.Bimetallic contribution in the CoRe-gel is confirmed from EXAFS analysis with average coordination numbers of NCo–Re = 2.8 and NRe–Co = 3. In addition, monometallic backscattering pairs were also fitted (NCo–Co= 1.4 and NRe–Re = 3) yielding a total metallic contribution of Ntotalmetallic ∼ 4–6 for both metals. These results indicate a higher degree of Co–Re mixing for the aerogel deposited CoRe-phase compared to the bulk CoRe1.629 as the Co–Re shell could not be fitted in the latter. Estimates for the average particle size from XAS based on the work of de Graaf et al.60 was approximated to be <25 Å. However, the in situ XRD pattern (ESI,† S15) shows small broad reflections confirming that the CoRe-phase is present as nanoparticles and a larger size is expected.25,61 By comparison, particles (2 nm from TEM) were reported by Guczi et al.53 for CoRe deposited on monolithic SiO2. Guczi et al.53,62 also report the requirement of higher temperatures (>450 °C) for complete reduction of the CoRe-phase which is likely to be related to different particle sizes and metal–support interactions. Interestingly, the cobalt in the Co-gel (ESI,† S7) remains as Co2+ during the pre-treatment with a prominent white line feature and characteristic pre-edge feature. This is also confirmed by EXAFS (ESI,† Tables S4 and S16) where a Co–O backscattering shell and a Co–O–Co backscattering shell could be fitted after hydrogen pre-treatment. Matsuzaki et al.52 found that well dispersed Co(II)-species on a SiO2-surface form strong interactions with the support and decrease in reducibility in agreement with the observed lack of reduction of the Co-gel during treatment in 75% H2. Clearly, rhenium promotes reduction of cobalt at lower temperatures with subsequent synergistic formation of bimetallic CoRe-nanoparticles.
Re-oxidation of cobalt and rhenium in NH3 and the active phase
The CoRe-gel achieved 50% conversion for the ammonia decomposition (Fig. 10) at ∼550 °C, contrasting with the rather inactive monometallic counterparts (both achieve 10% at 550 °C). The unsupported CoRe1.6 phase demonstrates promising performance for ammonia decomposition, showing conversions comparable to those of the highly active ruthenium systems.29 The limitations for the bulk CoRe phase are related to its very low surface area (<0.5 m2 g−1) and hence its area normalised activity is very high. Therefore, dispersion of the CoRe phase on a high surface area support could potentially increase the concentration of accessible active sites, which is further warranted by the relative high cost of rhenium.Even if immediate re-oxidation occurs for both rhenium and cobalt when switching to ammonia, as seen by the increase of the white line intensity in the XANES (Fig. 9), both metals are again reduced to the metallic state between 415–600 °C in 5% ammonia and catalytic activity commences at 450 °C (Fig. 11). By comparison, the considerably less active Co-gel remains mainly as Co(II) during both pre-treatment and ammonia decomposition supporting the claim that maintaining cobalt in the reduced state is crucial for activity. The observed re-oxidation of the CoRe-gel is also observed for the unsupported CoRe1.629 when switching to 5% NH3 in He, however, in this case, the degree of re-oxidation is considerably lower. Thus, the smaller particle size observed in the CoRe-gel results in the improved reversibility of the Re(VII) ↔ Re(0) and Co(II) ↔ Co(0) valence states.
Despite the fact that there is a remaining oxidised fraction in the CoRe-gel, linear combination (ESI,† S10) of the as-synthesised gel and the Co-/Re-foil to CoRe-gel in 5% NH3 at 600 °C resulted in poorly fitted curves meaning bimetallic interaction. This is further supported by there being no similar oxidic rhenium references (ReIV, ReVI and ReVII (as perrhenate)). The position of the white line in the Re-foil and the CoRe-gel was similar indicating mostly a metallic environment.
While no changes are observed in the in situ XRD (ESI,† S15) studies during ammonia decomposition, the EXAFS shows that the Co–O contribution is 1.2 as the CoRe bimetallic nanoparticles are reduced; however, the CoRe-phase remains more oxidised than in H2. This is also emphasised by the fitted Re–O/N contribution of 1.1 in ammonia an increase compared to the Re–O/N backscattering pair present during hydrogen treatment, corresponding to 25–30% oxidised phase.
The bimetallic contribution in the EXAFS spectra is confirmed by Fourier filtering the respective ranges in real space (Fig. 14 and 15) at both the Co K-edge and the Re LIII-edge. Most noticeable, adding a bimetallic shell significantly reduces the R-factor and the shape of the backscattered signal cannot be expressed by a monometallic contribution. The apparent variation in the experimental curve in Fig. 14 is due to Ef being fitted out of range for the Co–Co-model (<−20 eV). The results of the fitted back transformed chi curves (ESI,† Table S5) are in accordance with the EXAFS-analysis (Table 3 and Fig. 5) with Co–Re/Re–Co contribution (NCo–Re = 1.3 Å and NRe–Co = 3.9 Å) at more similar bonding distance of 2.58 Å at the Co K-edge and 2.60 Å at the Re LIII-edge as the best fits for the isolated chi curves. While the quantitative information obtained from the Fourier filtered data should be viewed with great care, a qualitative assessment works as a fingerprint as Co and Re display very different backscattered waves.
 | ||
| Fig. 14 Fourier filtered and fitted backscattered signal originating from the metallic contribution (real space range: 2.1–3.15 Å) of the CoRe-gel at 600 °C in 5% NH3 at the Co K-edge. | ||
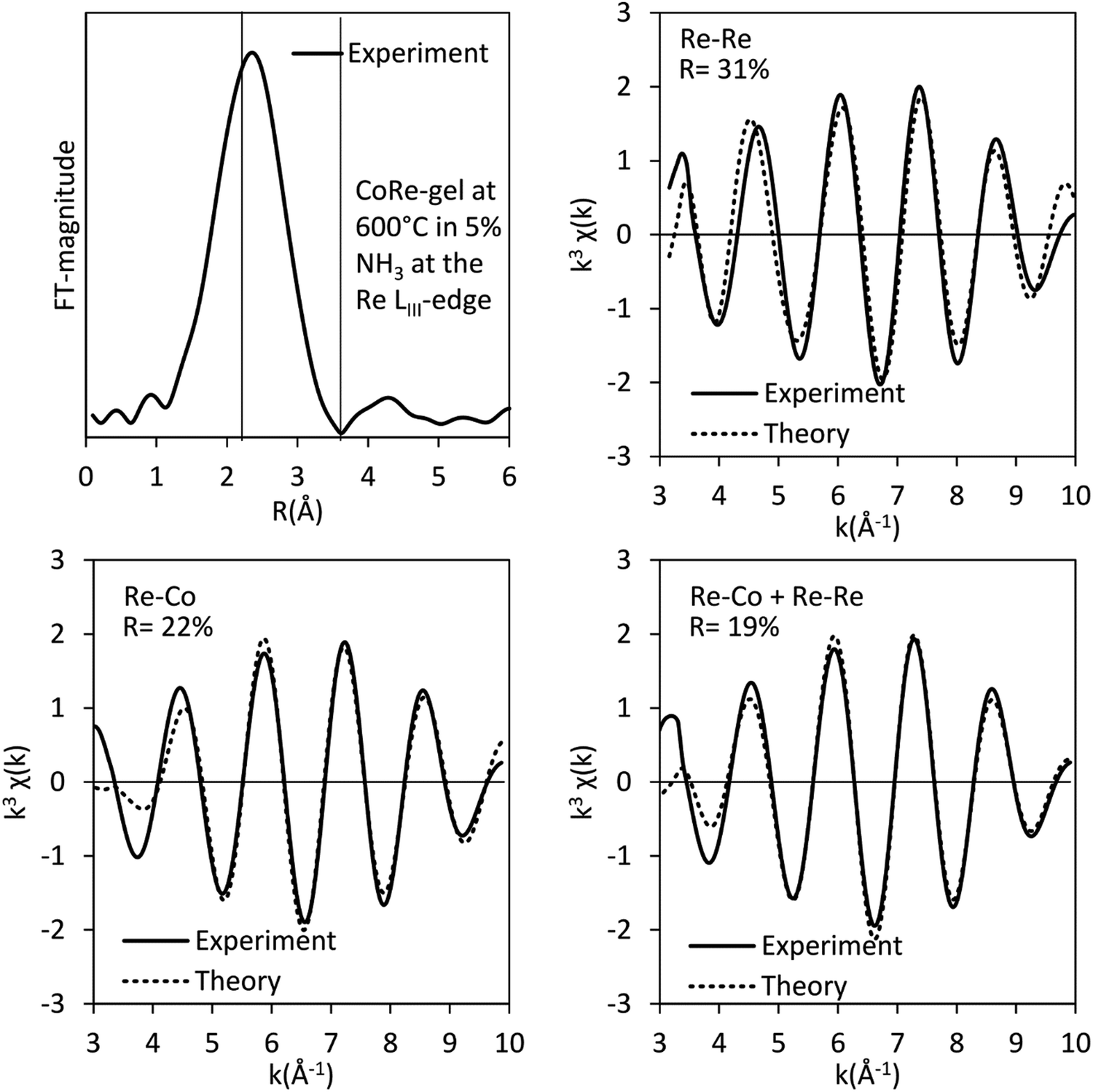 | ||
| Fig. 15 Fourier filtered and fitted backscattered signal originating from metallic contribution (real space range: 2–3.4 Å) for the CoRe-gel at 600 °C in 5% NH3 at the Re LIII-edge. | ||
The EXAFS results after pre-treatment in hydrogen at 600 °C suggest that two types of nanoparticles are present in the CoRe-gel, namely bimetallic CoRe particles and monometallic Re particles. This can be deduced from the Re–Re contribution (Fig. 16) prior to ammonia decomposition. The average particle size of the bimetallic CoRe species is estimated to be stable and are expected to be <25 Å, and accordingly the reflections of the in situ XRD (ESI,† S15) do not increase in intensity.
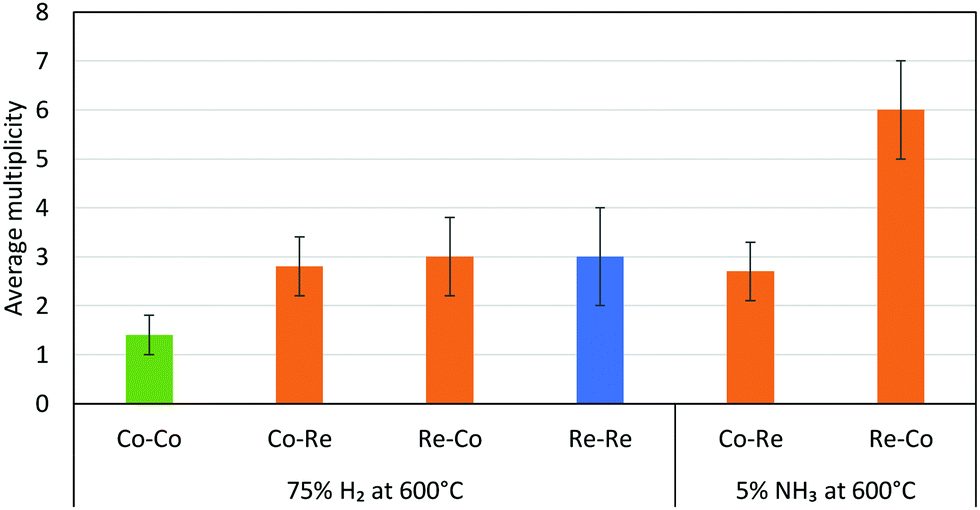 | ||
| Fig. 16 Average multiplicities of the CoRe-gel after pre-treatment in H2 and during ammonia decomposition. The Me–O/N shells were removed for clarification. | ||
The stark difference between the bulk and supported CoRe-phase is the superior speciation control in the latter where only bimetallic particles are present during ammonia decomposition. By contrast the bulk phase consists of 20% bimetallic particles together with monometallic Co and Re species of varying size. In terms of size control, none of the particles found in the CoRe-gel were larger than 2.5 nm. Hence, depositing CoRe in a silica aerogel leads to improved control over size and speciation. Future avenues for exploration would include direct observation of the particle size/size distribution and/or the application of chemisorption methods in this respect.
5. Conclusions
In this study we have presented a route for co-introduction for rhenium and cobalt species within the porous structure of a silica aerogel resulting in dispersed bimetallic Co–Re nanoclusters active for the release of hydrogen via ammonia decomposition. The CoRe-gel is thermally stable up to 450 °C with the presence of cobalt preventing volatilisation of rhenium. In return, the presence of rhenium promotes the dispersion of particulate CoOx/Co(OH)2 to single site Co2+ with concurrent formation of [ReO4]− entities during annealing. During pre-treatment in 75% H2, bimetallic CoRe and monometallic rhenium particles form whereas for the monometallic analogues the reducibility is limited. Re-oxidation occurs for both rhenium and cobalt in ammonia, seen by the appearance of Co/Re–O/N shells in the EXAFS. However, both metals are again reduced to the metallic state between 415–600 °C in 5% ammonia. The active phase for ammonia decomposition consists of bimetallic CoRe species estimated to <25 Å in average particle size.Conflicts of interest
There are no conflicts of interest to declare in relation to this work.Acknowledgements
The authors would like to acknowledge The Norwegian University of Science and Technology and the Norwegian Resource Council for grants supporting the Swiss-Norwegian Beamlines (SNBL), the Anders Jahre fund for promotion of science and A/S M. H. Lundgreens Enkes Fond for funding. The assistance of beamline scientist W. van Beek is very much appreciated. JSJH and SL wish to acknowledge financial support from the EPSRC through EP/L02537X/1 and LTM wishes to acknowledge financial support from the EPSRC through EP/L020423/2 and EP/N013778/1.References
- V. Masson-Delmotte, P. Zhai, H. O. Pörtner, D. Roberts, J. Skea, P. R. Shukla, A. Pirani, W. Moufouma-Okia, R. P. C. Péan, S. Connors, J. B. R. Matthews, Y. Chen, X. Zhou, M. I. Gomis, E. Lonnoy, T. Maycock and M. Tignor, IPCC, 2018: Summary for Policymakers, in Global warming of 1.5 °C. An IPCC Special Report on the impacts of global warming of 1.5 °C above pre-industrial levels, ed. T. Waterfield, World Meteorological Organization, 2018.
- Core writing team and R. K. Pachauri, IPCC, 2014: Climate Change 2014: Synthesis Report. in Contribution of Working Groups I, II and III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, ed. L. A. Meyer, IPCC, 2014.
- L. Schlapbach and A. Zuttel, Hydrogen-storage materials for mobile applications, Nature, 2001, 414, 353–358 CrossRef CAS PubMed.
- U. Eberle, M. Felderhoff and F. Schuth, Chemical and physical solutions for hydrogen storage, Angew. Chem., Int. Ed., 2009, 48, 6608–6630 CrossRef CAS PubMed.
- CRC handbook of chemistry and physics, 100th (Internet version 2019), CRC Press/Taylor & Francis, Boca Raton, Fla Search PubMed.
- S. Satyapal, J. Petrovic, C. Read, G. Thomas and G. Ordaz, The U.S. Department of Energy's National Hydrogen Storage Project: Progress towards meeting hydrogen-powered vehicle requirements, Catal. Today, 2007, 120, 246–256 CrossRef CAS.
- T. E. Bell and L. Torrente-Murciano, H2 Production via Ammonia Decomposition Using Non-Noble Metal Catalysts: A Review, Top. Catal., 2016, 59, 1438–1457 CrossRef CAS.
- K. T. Møller, T. R. Jensen, E. Akiba and H.-W. Li, Hydrogen – A sustainable energy carrier, Prog. Nat. Sci.: Mater. Int., 2017, 27, 34–40 CrossRef.
- A. K. Hill and L. Torrente-Murciano, In-situ H-2 production via low temperature decomposition of ammonia: Insights into the role of cesium as a promoter, Int. J. Hydrogen Energy, 2014, 39, 7646–7654 CrossRef CAS.
- T. E. Bell, G. W. Zhan, K. J. Wu, H. Zeng and L. Torrente-Murciano, Modification of Ammonia Decomposition Activity of Ruthenium Nanoparticles by N-Doping of CNT Supports, Top. Catal., 2017, 60, 1251–1259 CrossRef CAS PubMed.
- Z. G. Hu, J. Mahin, S. Datta, T. E. Bell and L. Torrente-Murciano, Ru-Based Catalysts for H-2 Production from Ammonia: Effect of 1D Support, Top. Catal., 2019, 62, 1169–1177 CrossRef CAS.
- A. K. Hill and L. Torrente-Murciano, Low temperature H2 production from ammonia using ruthenium-based catalysts: Synergetic effect of promoter and support, Appl. Catal., B, 2015, 172–173, 129–135 CrossRef CAS.
- Z. Hu, J. Mahin and L. Torrente-Murciano, A MOF-templated approach for designing ruthenium–cesium catalysts for hydrogen generation from ammonia, Int. J. Hydrogen Energy, 2019, 44, 30108–30118 CrossRef CAS.
- P. C. K. Vesborg and T. F. Jaramillo, Addressing the terawatt challenge: scalability in the supply of chemical elements for renewable energy, RSC Adv., 2012, 2, 7933–7947 RSC.
- T. E. Bell and L. Torrente-Murciano, H-2 Production via Ammonia Decomposition Using Non-Noble Metal Catalysts: A Review, Top. Catal., 2016, 59, 1438–1457 CrossRef CAS.
- L. Wang, Y. Zhao, C. Liu, W. Gong and H. Guo, Plasma driven ammonia decomposition on a Fe-catalyst: eliminating surface nitrogen poisoning, Chem. Commun., 2013, 49, 3787–3789 RSC.
- A. Boisen, S. Dahl, J. Norskov and C. Christensen, Why the optimal ammonia synthesis catalyst is not the optimal ammonia decomposition catalyst, J. Catal., 2005, 230, 309–312 CrossRef CAS.
- L. Torrente-Murciano, A. K. Hill and T. E. Bell, Ammonia decomposition over cobalt/carbon catalysts-Effect of carbon support and electron donating promoter on activity, Catal. Today, 2017, 286, 131–140 CrossRef CAS.
- D. Varisli and N. G. Kaykac, COx free hydrogen production over cobalt incorporated silicate structured mesoporous catalysts, Appl. Catal., B, 2012, 127, 389–398 CrossRef CAS.
- H. Zhang, Y. A. Alhamed, W. Chu, Z. B. Ye, A. AlZahrani and L. Petrov, Controlling Co-support interaction in Co/MWCNTs catalysts and catalytic performance for hydrogen production via NH3 decomposition, Appl. Catal., A, 2013, 464, 156–164 CrossRef.
- L. H. Yao, Y. X. Li, J. Zhao, W. J. Ji and C. T. Au, Core–shell structured nanoparticles (M@SiO2, Al2O3, MgO; M = Fe, Co, Ni, Ru) and their application in COx-free H2 production via NH3 decomposition, Catal. Today, 2010, 158, 401–408 CrossRef CAS.
- D. Varisli and N. G. Kaykac, Hydrogen from ammonia over cobalt incorporated silicate structured catalysts prepared using different cobalt salts, Int. J. Hydrogen Energy, 2016, 41, 5955–5968 CrossRef CAS.
- C. J. Jacobsen, S. Dahl, B. S. Clausen, S. Bahn, A. Logadottir and J. K. Norskov, Catalyst design by interpolation in the periodic table: bimetallic ammonia synthesis catalysts, J. Am. Chem. Soc., 2001, 123, 8404–8405 CrossRef CAS PubMed.
- D. Schanke, S. Vada, E. A. Blekkan, A. M. Hilmen, A. Hoff and A. Holmen, Study of Pt-Promoted Cobalt Co Hydrogenation Catalysts, J. Catal., 1995, 156, 85–95 CrossRef CAS.
- K. Mathisen, K. G. Kirste, J. S. J. Hargreaves, S. Laassiri, K. McAulay, A. R. McFarlane and N. A. Spencer, An In Situ XAS Study of the Cobalt Rhenium Catalyst for Ammonia Synthesis, Top. Catal., 2018, 61, 225–239 CrossRef CAS PubMed.
- T. Matsuzaki, K. Takeuchi, T. Hanaoka, H. Arawaka and Y. Sugi, Effect of Transition-Metals on Oxygenates Formation from Syngas over Co/Sio2, Appl. Catal., A, 1993, 105, 159–184 CrossRef CAS.
- N. Kumar, E. A. Payzant, K. Jothimurugesan and J. J. Spivey, Combined in situ XRD and in situ XANES studies on the reduction behavior of a rhenium promoted cobalt catalyst, Phys. Chem. Chem. Phys., 2011, 13, 14735–14741 RSC.
- N. Kumar, K. Jothimurugesan, G. G. Stanley, V. Schwartz and J. J. Spivey, In Situ FT-IR Study on the Effect of Cobalt Precursors on CO Adsorption Behavior, J. Phys. Chem. C, 2011, 115, 990–998 CrossRef CAS.
- K. Kirste, K. McAulay, T. E. Bell, D. Stoian, S. Laassiri, A. Daisley, J. S. J. Hargreaves, K. Mathisen and L. Torrente-Murciano, COx-free hydrogen production from ammonia – mimicking the activity of Ru catalysts with unsupported Co-Re alloys Under review, 2020.
- M. Tian, A. Buchard, S. A. Wells, Y. N. Fang, L. Torrente-Murciano, A. Nearchou, Z. L. Dong, T. J. White, A. Sartbaeva and V. P. Ting, Mechanism of CO2 capture in nanostructured sodium amide encapsulated in porous silica, Surf. Coat. Technol., 2018, 350, 227–233 CrossRef CAS.
- N. Hüsing and U. Schubert, Aerogels—Airy Materials: Chemistry, Structure, and Properties, Angew. Chem., Int. Ed., 1998, 37, 22–45 CrossRef.
- G. S. Mishra, E. C. B. A. Alegria, A. J. L. Pombeiro and L. M. D. R. S. Martins, Highly Active and Selective Supported Rhenium Catalysts for Aerobic Oxidation of n-Hexane and n-Heptane, Catalysts, 2018, 8, 114 CrossRef.
- R. Huang, Y. Peng, C. Wang, Z. Shi and W. Lin, A. Rhenium-Functionalized Metal-Organic Framework as a Single-Site Catalyst for Photochemical Reduction of Carbon Dioxide, Eur. J. Inorg. Chem., 2016, 4358–4362 CrossRef CAS.
- X. Y. She, J. H. Kwak, J. M. Sun, J. Z. Hu, M. Y. Hu, C. M. Wang, C. H. F. Peden and Y. Wang, Highly Dispersed and Active ReOx on Alumina-Modified SBA-15 Silica for 2-Butanol Dehydration, ACS Catal., 2012, 2, 1020–1026 CrossRef CAS.
- D. Bazin and L. John, ReCo/NaY and ReCo/Al2O3 bimetallic catalysts in situ Exafs study and catalytic activity, Catal. Lett., 2002, 84, 169 182 CrossRef.
- E. Rytter, A. U. R. Salman, N. E. Tsakoumis, R. Myrstad, J. Yang, S. Lögdberg, A. Holmen and M. Rønning, Hydrophobic catalyst support surfaces by silylation of γ-alumina for Co/Re Fischer-Tropsch synthesis, Catal. Today, 2018, 299, 20–27 CrossRef CAS.
- B. C. Vicente, R. C. Nelson, A. W. Moses, S. Chattopadhyay and S. L. Scott, Interactions Involving Lewis Acidic Aluminum Sites in Oxide-Supported Perrhenate Catalysts, J. Phys. Chem. C, 2011, 115, 9012–9024 CrossRef CAS.
- F. Hayashi and M. Iwamoto, Ammonia synthesis over rhenium supported on mesoporous silica MCM-41, Microporous Mesoporous Mater., 2011, 146, 184–189 CrossRef CAS.
- T. Kristiansen, K. Mathisen, M. A. Einarsrud, M. Bjorgen and D. G. Nicholson, Single-Site Copper by Incorporation in Ambient Pressure Dried Silica Aerogel and Xerogel Systems: An X-ray Absorption Spectroscopy Study, J. Phys. Chem. C, 2011, 115, 19260–19268 CrossRef CAS.
- B. Ravel and M. Newville, ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- A. Tsoukalou, P. M. Abdala, D. Stoian, X. Huang, M.-G. Willinger, A. Fedorov and C. R. Müller, Structural Evolution and Dynamics of an In2O3 Catalyst for CO2 Hydrogenation to Methanol: An Operando XAS-XRD and In Situ TEM Study, J. Am. Chem. Soc., 2019, 141, 13497–13505 CrossRef CAS PubMed.
- A. Martini, E. Borfecchia, K. A. Lomachenko, I. A. Pankin, C. Negri, G. Berlier, P. Beato, H. Falsig, S. Bordiga and C. Lamberti, Composition-driven Cu-speciation and reducibility in Cu-CHA zeolite catalysts: a multivariate XAS/FTIR approach to complexity, Chem. Sci., 2017, 8, 6836–6851 RSC.
- J. Jaumot, A. de Juan and R. Tauler, MCR-ALS GUI 2.0: New features and applications, Chemom. Intell. Lab. Syst., 2015, 140, 1–12 CrossRef CAS.
- J. Jaumot, R. Gargallo, A. de Juan and R. Tauler, A graphical user-friendly interface for MCR-ALS: a new tool for multivariate curve resolution in MATLAB, Chemom. Intell. Lab. Syst., 2005, 76, 101–110 CrossRef CAS.
- C. Ruckebusch and L. Blanchet, Multivariate curve resolution: a review of advanced and tailored applications and challenges, Anal. Chim. Acta, 2013, 765, 28–36 CrossRef CAS PubMed.
- A. Voronov, A. Urakawa, W. van Beek, N. E. Tsakoumis, H. Emerich and M. Ronning, Multivariate curve resolution applied to in situ X-ray absorption spectroscopy data: an efficient tool for data processing and analysis, Anal. Chim. Acta, 2014, 840, 20–27 CrossRef CAS PubMed.
- S. Tomic, B. G. Searle, A. Wander, N. M. Harrison, A. J. Dent, J. F. W. Mosselmans and J. E. Inglesfield, New Tools for the Analysis of EXAFS: The DL EXCURV Package, CCLRC Technical Report, Council for the Central Laboratory of the Research Councils, UK, 2005.
- R. Kojima and K. Aika, Rhenium containing binary catalysts for ammonia synthesis, Appl. Catal., A, 2001, 209, 317–325 CrossRef CAS.
- K. McAulay, J. S. J. Hargreaves, A. R. McFarlane, D. J. Price, N. A. Spencer, N. Bion, F. Can, M. Richard, H. F. Greer and W. Z. Zhou, The influence of pre-treatment gas mixture upon the ammonia synthesis activity of Co-Re catalysts, Catal. Commun., 2015, 68, 53–57 CrossRef CAS.
- F. D. Hardcastle, I. E. Wachs, J. A. Horsley and G. H. Via, The structure of surface rhenium oxide on alumina from laser Raman spectroscopy and X-ray absorption near-edge spectroscopy, J. Mol. Catal., 1988, 46, 15–36 CrossRef.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Reporting Physisorption Data for Gas Solid Systems with Special Reference to the Determination of Surface-Area and Porosity (Recommendations 1984), Pure Appl. Chem., 1985, 57, 603–619 CAS.
- T. Matsuzaki, K. Takeuchi, T.-A. Hanaoka, H. Arawaka and Y. Sugi, Effect of transition metals on oxygenates formation from syngas over Co/SiO2, Appl. Catal., A, 1993, 105, 159–184 CrossRef CAS.
- L. Guczi, G. Stefler, L. Borkó, Z. Koppány, F. Mizukami, M. Toba and S. Niwa, Re-Co bimetallic catalysts prepared by sol/gel technique: characterization and catalytic properties, Appl. Catal., A, 2003, 246, 79–86 CrossRef CAS.
- T. K. Das, G. Jacobs, P. M. Patterson, W. A. Conner, J. Li and B. H. Davis, Fischer–Tropsch synthesis: characterization and catalytic properties of rhenium promoted cobalt alumina catalysts, Fuel, 2003, 82, 805–815 CrossRef CAS.
- L. Guczi, D. Bazin, I. Kovács, L. Borkó, Z. Schay, J. Lynch, P. Parent, C. Lafon, G. Stefler, Z. Koppány and I. Sajó, Structure of Pt–Co/Al2O3 and Pt–Co/NaY Bimetallic Catalysts: Characterization by In Situ EXAFS, TPR, XPS and by Activity in Co (Carbon Monoxide) Hydrogenation, Top. Catal., 2002, 20, 129–139 CrossRef CAS.
- N. M. Bertero, A. F. Trasarti, B. Moraweck, A. Borgna and A. J. Marchi, Selective liquid-phase hydrogenation of citral over supported bimetallic Pt–Co catalysts, Appl. Catal., A, 2009, 358, 32–41 CrossRef CAS.
- M. Rønning, D. G. Nicholson and A. Holmen, In situ EXAFS study of the bimetallic interaction in a rhenium-promoted alumina-supported cobalt Fischer–Tropsch catalyst, Catal. Lett., 2001, 72, 141–146 CrossRef.
- A. M. Beale and G. Sankar, Understanding the Crystallization of Nanosized Cobalt Aluminate Spinel from Ion-Exchanged Zeolites Using Combined in Situ QEXAFS/XRD, Chem. Mater., 2006, 18, 263–272 CrossRef CAS.
- A. Moen, D. G. Nicholson, M. Ronning, G. M. Lamble, J. F. Lee and H. Emerich, X-Ray absorption spectroscopic study at the cobalt K-edge on the calcination and reduction of the microporous cobalt silicoaluminophosphate catalyst CoSAPO-34, J. Chem. Soc., Faraday Trans., 1997, 93, 4071–4077 RSC.
- J. de Graaf, A. J. van Dillen, K. P. de Jong and D. C. Koningsberger, Preparation of Highly Dispersed Pt Particles in Zeolite Y with a Narrow Particle Size Distribution: Characterization by Hydrogen Chemisorption, TEM, EXAFS Spectroscopy, and Particle Modeling, J. Catal., 2001, 203, 307–321 CrossRef CAS.
- A. M. Alexander, J. S. J. Hargreaves and C. Mitchell, The Denitridation of Nitrides of Iron, Cobalt and Rhenium Under Hydrogen, Top. Catal., 2013, 56, 1963–1969 CrossRef CAS.
- L. Guczi, G. Stefler, Z. Schay, I. Kiricsi, F. Mizukami, M. Toba and S. Niwa, Structure and catalytic properties Of cobalt-rhenium bimetallic catalysts prepared by sol/gel method and by ion exchange in NaY zeolite, in Studies in Surface Science and Catalysis, ed. A. Corma, F. V. Melo, S. Mendioroz and J. L. G. Fierro, Elsevier, 2000, pp. 1097–1102 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp00558d |
| This journal is © the Owner Societies 2020 |