
A Schiff-base-type vanadyl complex grafted on mesoporous carbon nitride: a new efficient catalyst for hydroxylation of benzene to phenol†
Jie Xu*,
Quan Jiang,
Jie-Kun Shang,
Yue Wang and
Yong-Xin Li*
Jiangsu Key Laboratory of Advanced Catalytic Materials and Technology, School of Petrochemical Engineering, Changzhou University, Gehu Road 1, Changzhou, Jiangsu 213164, PR China. E-mail: shine6832@163.com; liyxluck@163.com; Fax: +86-519-86330135; Tel: +86-519-86330135
First published on 21st October 2015
Abstract
Mesoporous graphitic carbon nitride (g-CN) was utilized as a new support to immobilize vanadyl(IV) acetylacetonate ([VO(acac)2]). The immobilized vanadyl complex materials (VOac-CND) have been thoroughly characterized by various techniques including N2 adsorption–desorption, XRD, SAXS, TEM, Raman, FT-IR, XPS and benzene-TPD. The characterization results showed that [VO(acac)2] had been successfully grafted on the surface of g-CN via the reaction between the carbonyl group of the acetylacetonate ligand and the amino groups of g-CN and thus transformed into a Schiff-base-type complex. Moreover, after the immobilization of [VO(acac)2], the ordered natures of the mesoporous structures and graphitic structures of the g-CN support have been well retained. The immobilization temperature has been found to be sensitive to the immobilization effect. As heterogeneous catalysts, the immobilized catalysts exhibited high performances in the direct hydroxylation reaction of benzene to phenol, affording a maximum phenol yield of ca. 20.0%. The catalytically active site for hydroxylation was proposed as the grafted vanadyl complex while the mesoporous g-CN played a crucial role in activating benzene.
1. Introduction
Phenol is one of the most important commodity chemicals, which is widely used as a basic intermediate in the production of resins, fibers, dyes, and medicines. Commercially, phenol is produced mainly based on the well-known three-step cumene process introduced by Hock and Lang in 1944,1,2 which however suffers some disadvantages including high pollution, low one-pass yield of phenol (<90%), low atomic efficiency, and a large amount of acetone as co-product.3 To address this issue, various alternative strategies have been proposed and up to now, one-step hydroxylation of benzene to phenol, featuring high atomic economy and high selectivity towards phenol, has been regarded as the ideal approach to replace the traditional cumene process.4In the cases of oxidants applied for the direct hydroxylation of benzene, air and oxygen are widely available. However, under such catalytic systems, a relatively high reaction temperature is necessarily demanded, which may lead to the deep oxidation of benzene. As for N2O, despite of its high selectivity (nearly 100%), the use of N2O is much more complex in industrial operation.5 Instead, using H2O2 as an oxidant is green and simple and more importantly, water is the only by-product.6,7 Notwithstanding, there remains a challenge to reach a high conversion (>10%) of benzene in the presence of H2O2. That is, phenol is liable to transforms into undesired by-products (e.g. hydroquinone, benzoquinone) due to the uncatalyzed deep oxidation of phenol.5 In this sense, how to design and develop a new and efficient catalyst, which can afford high activity and selectivity to phenol, is a hot topic for the catalytic hydroxylation of benzene.
In the past decade, a wide range of catalyst systems, including titanium silicalite sieves (TS-1), polyoxometalate (POM), graphene, carbon nanotubes etc., have been successfully exploited for the direct hydroxylation of benzene to phenol.7,8 Vanadyl(IV) acetylacetonate ([VO(acac)2]) is a typical efficient catalyst extensively employed for various oxidative transformations.2,9 However, the thorny issue associated with [VO(acac)2] lies in its intrinsically homogeneous property, especially the difficulty in catalyst separation and product purification.9,10 Alternatively, immobilization of [VO(acac)2] on other solids (including mesoporous silicas,2,11 amine-functionalized clay10,12 or carbon materials9), via the reactions between the carbonyl group of the acetylacetonate ligand and the amino groups of the solids surfaces, provides an effective strategy to utilize [VO(acac)2]. In spite of such success in the catalyst preparation, the final activities received in the corresponding oxidative reactions were not satisfactory. For instance, to achieve a conversion up to 25%, 8 h of reactions time was at least required.2 Furthermore, the leaching of the active component, i.e. VO(acac)2, still restrains the wide application of such grafted complex catalysts. In this context, it is of interest to explore a new material as an effective solid to immobilize [VO(acac)2].
As a fascinating material to complement traditional carbon materials, graphitic carbon nitride (g-CN) and its based materials have recently attracted tremendous attention in wide research communities.13,14 Owing to its combination of several unique physicochemical natures, g-CN material has been applied in numerous fields including photocatalysis,15 fuel cells,16 gas storage,17,18 and heterogeneous catalysis.19–21 As an analogue of graphite, g-CN possesses two-dimensional structures with π-conjugated planar layers that are constituted of tri-s-triazine moieties bridged by nitrogen atoms,22 and the feature enables g-CN with an inherent ability to activate aromatic molecules.23,24 As reported by Antonietti, g-CN materials showed outstanding catalysis in various benzene-involved reactions, such as Friedel–Crafts acylation,24 alkylation25 and oxidation of benzene by CO2.26 Very recently, Wang27 and Han4 reported that g-CN-based materials (V-g-C3N4, g-C3N4–H5PMo10V2O40) could also serve promising catalysts for oxidation of benzene to phenol. However, the catalytic activity obtained on the bare g-CN was limited.
On the other hand, in terms of the tectonic units of g-CN, there exist abundant uncondensed amines, in the forms of –NH2 and –NH– groups, at its graphitic edges.28 Indeed, the characteristic compositions of amine species were usually utilized to enable g-CN as a solid base catalyst.19,28–30 Since the immobilization of [VO(acac)2] relies on the aliphatic amines located on the support while g-C3N4 has both innate amines and ability to activate benzene, we envision that g-CN could also be used as a solid to graft [VO(acac)2]. Inspired by this, in the present work, we have reported for the first time new Schiff-base-type materials by immobilizing [VO(acac)2] on mesoporous g-CN materials. In the direct hydroxylation of benzene to phenol, the grafted vanadyl complex catalysts demonstrated high and stable activity.
2. Experimental
2.1. Catalyst preparation
Mesoporous g-CN was prepared via a previously reported nanocasting approach using SBA-15 as a template and dicyandiamide (DCDA) as a precursor.30 In brief, 2.5 g of DCDA was dissolved in 10 mL of anhydrous ethylenediamine (EDA), and the solution was added dropwisely onto 1 g of dried SBA-15 (see the ESI† for detailed preparation routes). After the digestion for 6 h, the mixture was heated at 90 °C overnight. Next, the resultant solid was ground in a mortar, and heated at 2.5 °C min−1 up to 550 °C and then treated for further 4 h under N2 atmosphere. Afterwards, the as-synthesized dark powder was immersed into 200 mL of NH4HF2 solution (4 mol L−1) to etch SBA-15. Then, the dispersion was centrifuged and the precipitate was washed with water and ethanol for several times, and dried at 50 °C under vacuum. The resulting powder was designated as CND.175 mg of VO(acac)2 was dissolved into 40 mL of toluene, followed by addition of 0.6 g of dried CND. The mixture was transferred into a stainless autoclave and pressured with Ar up to 0.7 MPa, and then stirred under a desired temperature (80–120 °C) for 10 h. After that, the dispersion was filtrated and the precipitate was washed with toluene for several times. To eliminate the possibly residual VO(acac)2, the solid was further refluxed with chloroform for 3 h and washed for two times. After the subsequent drying under vacuum at 60 °C for 2 h, CND grafted with VO(acac)2 was obtained and labeled as VOac-CND-T, where T represented the reaction temperature between CND and VO(acac)2.
2.2. Sample characterization
Nitrogen adsorption–desorption isotherms were measured at −196 °C using a Micromeritics ASAP 2020 analyzer. The specific surface area was calculated according to the Brunauer–Emmet–Teller (BET) method, and pore size distribution was determined by the Barret–Joyner–Halenda method.X-ray diffraction patterns were recorded with a Rigaku D/max 2500 PC X-ray diffractometer equipped with a graphite monochromator (40 kV, 40 mA) using Ni-filtered Cu-Kα radiation (λ = 1.5418 Å).
Small angle X-ray scattering (SAXS) measurements were conducted on a Bruker NanoSTAR U SAXS system equipped with a high-resolution pinhole chamber using Cu-Kα radiation (40 kV, 35 mA).
Fourier transform infrared (FT-IR) spectra of the samples were collected in transmission mode at room temperature on a Bruker Tensor 27 spectrometer with a resolution of 4 cm−1, using 32 scans per spectrum in the region of 400–4000 cm−1. The mass ratio of every sample to KBr was constant at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :200.
:200.
X-ray photoelectron spectroscopy (XPS) measurements were performed using a Thermo ESCALAB 250XI spectrometer working in the constant analyzer energy mode with Mg Kα radiation as the excitation source. The atomic content of vanadium for each V-containing sample was calculated based on the peak areas (survey spectrum) of C, N, O, and V elements, and their corresponding atomic sensitivity factors.
Raman spectra were recorded on a Raman spectrometer (Jobin Yvon Lab Ram HR evolution) using 532 nm line as an excitation source.
Transmission electron microscopy (TEM) experiments were conducted on a JEOL 2010 electron microscope operating at 200 kV. Before being transferred into the TEM chamber, the samples dispersed in ethanol were deposited onto holey carbon films supported on Cu grids.
Benzene temperature-programmed desorption (benzene-TPD) experiments were performed on a Quantachrome ChemBET-3000 analyzer. 200 mg of the sample was pretreated at 350 °C for 1 h in dry He flow (30 mL min−1), cooled to 50 °C, and then exposed to He-containing benzene for 0.5 h. After purging the sample with He for 0.5 h, the TPD data based on the thermal conductivity detector (TCD) signals was recorded from 50 to 350 °C with a ramping rate of 10 °C min−1.
2.3. Catalytic test
The hydroxylation reaction of benzene to phenol was performed in a two-neck round bottomed flask (25 mL). 50 mg of catalyst was added into 1 mL of benzene (11.2 mmol) and then mixed well. After the reactant was heated to 60 °C, 3 mL of aqueous H2O2 solution (30 wt%) was added into the mixture in 30 min via a peristaltic pump. The reaction was conducted for 4 h. After that, the mixture was centrifuged and analyzed by GC equipped with a SE-54 capillary column. The filtered catalyst was washed by acetonitrile (30 mL) for two times, dried at 50 °C for 2 h, and then investigated for its next running.The molar amount (n) for each component in the catalytic reaction was calculated by n = A × f, where A and f were the GC peak area and response factor, respectively. The conversion of benzene (Conv.) and selectivity to phenol (Sel.) were calculated as follows:

3. Results and discussions
3.1. Structure characterization
Fig. 1A shows the XRD patterns of CND and VOac-CND materials. Each sample displayed an apparent diffraction peak centered at 2θ = 25.9° (d = 0.343 nm), which was indexed as the interplanar stacking of graphitic structures, namely (002) plane.24 In addition, a shoulder peak with low intensity was also detected at ca. 2θ = 42°, further suggesting that CND and its grafted vanadyl materials possessed a high degree of intralayer condensation of graphitic phrase.31 In terms of the diffraction intensities of all the samples, the graphitic structures of the grafted VOac-CND materials have well remained, and the grafting of VO(acac)2 has not damaged the overall crystallinity of g-CN. Furthermore, referring to the XRD pattern of the pure VO(acac)2 sample (Fig. S1†), no clear diffraction line pertaining to VO(acac)2 has been observed in the patterns of all the VOac-CND materials. Besides XRD, Raman spectroscopy was also used to analyze the graphitic structures (Fig. S2†). The spectrum of CND revealed two bands centered at 1350 and 1551 cm−1, associated with disordered (D) and graphitic (G) modes of carbon, respectively. The ratio (ID/IG) of relative intensities for the two peaks was ca. 1.12, further verifying that the mesoporous g-CN contained comparatively high graphitic structures. After the introduction of VO(acac)2, the overall intensities of spectra declined whereas the ratios of ID/IG were still close to the above value.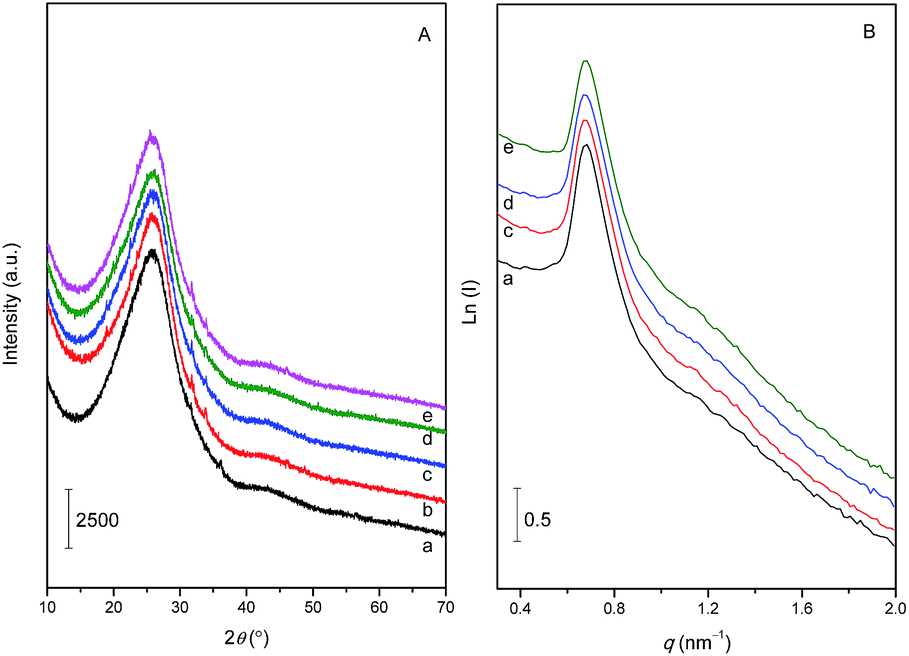 | ||
| Fig. 1 XRD (A), and SAXS patterns (B) of CND (a) and VOac-CND-T (b–e: 60, 80, 100, and 120) materials. | ||
SAXS technique was then employed to analyze the orderness of porosity of the materials. The SAXS pattern of SBA-15 template (Fig. S3†) showed four well-resolved scattering peaks in the range of q = 0.6–1.7 nm−1, corresponding to the (100), (110), (200), (210) planes of two-dimensional hexagonal structure (P6mm).32 Wherein, the lattice parameter (a0) calculated based on the (100) plane was ca. 10.2 nm. In the case of the negative replica, CND, the pattern demonstrated a peak and shoulder at q = 0.683 and 1.15 nm−1, respectively, along with lower intensities than those of SBA-15 (Fig. 1B). Meanwhile, the a0 of CND decreased to 9.2 nm, slighter smaller than the value gained in SBA-15. This means that after the hard-templating process, the orderness of SBA-15 template declined, probably due to the partial collapsed or shrinking of mesoporous walls, which was also found in other g-CN materials templated by mesoporous silicas.33,34 Nevertheless, the whole periodicity the mesoporous structures were successfully replicated. Interestingly, the introduction of vanadyl complex has not affected the original ordered mesostructures of CND, since both the scattering vectors of (100) planes and their intensities of VOac-CND samples showed no obvious change.
N2 adsorption–desorption isotherms of CND and VOac-CND materials are depicted in Fig. 2A. The pure CND exhibited typical IV curves with a steep H1 hysteresis loop at ca. 0.5–0.75 p/p0, inferring that the support had classical mesopores together with a narrow pore size distribution (Fig. 2B). After the incorporation of vanadyl complex, the quantity of adsorption of CND decreased obviously. Specifically, as the grafting temperatures were elevated from 60 to 120 °C, the pore volumes as well as surface areas of the obtained VOac-CND materials decreased monotonously from 380 to 328 m2 g−1 (Table 1). This implies that higher grafting temperatures would induce much more vanadyl species to be immobilized into the mesoporous pores of CND, thereby leading to the progressive decease of the pore sizes. However, no significant blockage of the mesopores after the grafting was found, as the corresponding pore size distributions still revealed concentrated peaks in the range of 4.5–5.0 nm (Fig. 2B).
 | ||
| Fig. 2 N2 adsorption–desorption isotherms (A) and pore size distributions (B) of CND and VOac-CND materials. | ||
The mesoporous morphology of SBA-15 and VOac-CND samples were characterized by TEM. The images of the parent SBA-15 material revealed highly ordered tubular arrays (Fig. 3A) and honeycomb-like hexagonal structures (Fig. 3B), corresponding to the (110) and (100) planes. On the other hand, the images of CND material presented strip-like (Fig. S4A†) and ordered slice-like (Fig. S4B†) arrangements. Wherein, the black region was g-CN walls templated by the mesopores of SBA-15, while the white one was derived from the original silica walls of SBA-15. Compared with SBA-15, a small amount of mesopores of CND underwent partial blockage. One reason accounting for this was the shrinkage of mesopores of CND during its rinsing or drying procedures. Additionally, under the exposure of high-speed electronic beam, the mesostructure walls of g-CN material was prone to be damaged, which was also found in our previous work.30,34 As for the grafted VOac-CND material, the TEM images (Fig. 3C and D) also exhibited similar patterns like those of CND. Overall, the TEM characterization manifested that the CND has replicated the ordered mesostructures of siliceous SBA-15 and moreover, after grafting vanadyl compound, the ordered structures have been reserved, in a good agreement with the above N2 adsorption–desorption and SAXS characterization.
 | ||
| Fig. 3 TEM images of SBA-15 (A & B) and VOac-CND-120 (C & D) materials. The left and right images were recorded along with the [110] and [100] directions, respectively. | ||
The chemical functions of CND and its grafted samples were analyzed using FT-IR spectroscopy. As described in Fig. 4, the FT-IR spectrum of the bare CND represented three distinct zones. The major and strong bands located in 1612 and 1284 cm−1 were attributed to aromatic ring modes and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching bonds,33,35 respectively. Whilst the low-intensity and broad bands in the range of 3382 to 3193 cm−1 were assigned to the physically adsorbed water molecules (νO–H) and N–H stretching of NH2 groups attached to the sp2 C atoms.35 Furthermore, the tiny yet apparent band at 2212 cm−1 was indexed as conjugated NC–N units in g-CN planar network.36 These FT-IR signals agreed well with mesoporous g-CN materials reported elsewhere.30,37 As far as VOac-CND samples, their FT-IR spectra exhibited similar characteristic bands to that of CND. However, the differences in the spectra of the pristine and grafted materials could be also identified. In detail, a weak yet unique band at 970 cm−1 emerged for each VOac-CND sample; moreover, the intensity of the band seems to be gradually enhanced with the increase of the grafting temperatures. According to the FT-IR spectra of vanadia-supported catalysts reported by Fehrmann et al.,38 the band at 970 cm−1 was actually derived from the stretching modes of VO bonds. Considering this fact, it can be inferred that VO(acac)2 compound has been successfully loaded on CND.
N stretching bonds,33,35 respectively. Whilst the low-intensity and broad bands in the range of 3382 to 3193 cm−1 were assigned to the physically adsorbed water molecules (νO–H) and N–H stretching of NH2 groups attached to the sp2 C atoms.35 Furthermore, the tiny yet apparent band at 2212 cm−1 was indexed as conjugated NC–N units in g-CN planar network.36 These FT-IR signals agreed well with mesoporous g-CN materials reported elsewhere.30,37 As far as VOac-CND samples, their FT-IR spectra exhibited similar characteristic bands to that of CND. However, the differences in the spectra of the pristine and grafted materials could be also identified. In detail, a weak yet unique band at 970 cm−1 emerged for each VOac-CND sample; moreover, the intensity of the band seems to be gradually enhanced with the increase of the grafting temperatures. According to the FT-IR spectra of vanadia-supported catalysts reported by Fehrmann et al.,38 the band at 970 cm−1 was actually derived from the stretching modes of VO bonds. Considering this fact, it can be inferred that VO(acac)2 compound has been successfully loaded on CND.
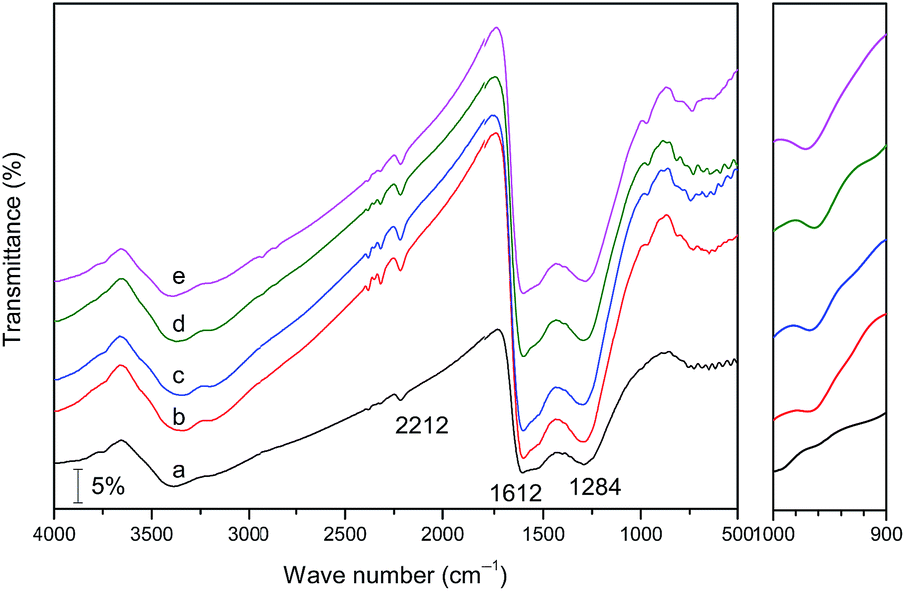 | ||
| Fig. 4 FT-IR spectra of CND (a) and VOac-CND-T (b–e: 60, 80, 100, and 120) materials. The right is the magnified region of 1000–900 cm−1. | ||
Besides FT-IR, we have employed XPS measurement to further analyze the surface chemical information of the materials. The XPS survey (Fig. 5A) of CND showed the C, N, and O elements, which probably originated from the adsorbed water. In addition to the three peaks, the survey spectrum of VOac-CND material, for instance VOac-CND-120, also offered a low-intensity yet sharp signal near O 1s, which was attributed to the V 2p peak (ca. 516 eV). According to the peak areas under these elemental signals, the surface contents of vanadium per weight of VOac-CND-120 was 495 μmol gcatal.−1, close to the reported value of VO(acac)2 grafted on amine-functionalized active carbon.9 On the other hand, the nominal loading amount of VOac-CND-120 was ca. 1100 μmol gcatal.−1 (175 mg of VO(acac)2 mixed with 600 mg of CND), which meant that the amount of VO(acac)2 was enough to utilize the amino groups of CND. Fig. 5B gives the high-resolution spectra of V 2p. The similarity between the profiles of the free and grafted vanadyl complexes indicated that the complex had kept its integrity after immobilization on mesoporous g-CN. Nevertheless, as compared with the V 2p band of the pure VO(acac)2 (516.3 eV for 2p 3/2), a slight band shift to lower binding energy in VOac-CND-120 (516.0 eV for 2p 3/2) has been observed. It is apparent that the chemical environment of V atoms of vanadyl complex has undergone a change after immobilization, suggesting that the complex has reacted with g-CN. Nonetheless, one might think that vanadium in such VOac-CND materials existed as vanadia (V2O5) rather than vanadyl complex. Firstly, in the case of VO(acac)2, its decomposition temperature to transform into vanadia (V2O5) is at least 230 °C.39 In our experiment to prepare vanadyl complex, the grafting temperature we applied was less than 120 °C, far below the above transformation threshold. Meanwhile, after immobilization of VO(acac)2, no further calcination was demanded. Secondly, as mentioned above, the binding energy of V 2p 3/2 was 516.3 eV. By comparison, the corresponding location of V 2p 3/2 of vanadia (V2O5) was at least 516.7 eV, as reported in the work involving the supported vanadia materials.40,41 Given in these two points, it can be excluded that vanadium existed in the form of vanadia. Namely, like the vanadyl complexes anchored on amine-modified silicas,11,42 the CO group of acetylacetonate ligand of VO(acac)2 reacted with the terminal amines of g-CN, thus yielding a Schiff-base-type complex (Scheme 1). Wherein, as the electronegativity of N is slightly lower than that of O, the substitution of O by N in its ligand of vanadium led to the shift of V 2p band towards lower energy. Such variation in XPS profiles has also been found in other grafted vanadyl complexes.2
 | ||
| Fig. 5 XPS survey (A) and V 2p (B) spectra of CND (a), VOac-CND-120 (b), and VO(acac)2 (c) samples. | ||
 | ||
| Scheme 1 A possible Schiff-base-type vanadyl complex grafted on g-CN. | ||
3.2. Catalyst activity
Initially, a blank experiment was performed in the absence of catalyst (entry 1, Table 2); the result indicated that benzene could be rarely converted into the desired product, phenol. Alternatively, using the pure CND material (entry 2), the phenol productivity received was quite poor. In sharp contrast, VOac-CND complexes offered remarkable catalytic activity for the hydroxylation of benzene under the same reaction conditions. The target phenol contributed about 95% on the catalytic selectivities, along with a small amount of benzoquinone that probably originated from the deep oxidation of phenol. As summarized in Table 2, the catalytic conversions of benzene showed a direct dependence on the grafting temperatures. That is, higher temperatures applied in grafting VO(acac)2 on CND resulted in higher catalytic activities (entries 4–8). The maximum activity was achieved over VOac-CND-120, affording ca. 20% of phenol yield. As disclosed above, the grafting of VO(acac)2 on CND was essentially based on the reaction between terminal amine groups and CO groups of VO(acac)2. Therein, using higher grafting temperature would inevitably improve the loading amount of vanadia species, thus increasing the whole amounts of catalytically active species.
| Entry | Catalyst | Conv. (%) | Sel. (%) | Yield (%) |
|---|---|---|---|---|
| a Without any catalyst.b Reaction conditions: 1 mL of benzene (11.2 mmol), 3 mL of aqueous H2O2 solution (30 wt%), 6 mL of acetonitrile as solvent, Wcatal. = 50 mg, T = 60 °C, and t = 4 h.c 6.6 mg of the catalyst (equivalent to 25 μmol of vanadium).d Grafted with 150 mg of VO(acac)2.e Grafted with 100 mg of VO(acac)2.f Grafted with 50 mg of VO(acac)2. | ||||
| 1a | — | <1 | 100 | <1 |
| 2b | CND | 1.4 | 90.2 | 1.3 |
| 3c | VO(acac)2 | 12.0 | 96.8 | 11.6 |
| 4 | VOac-CND-60 | 6.5 | 97.4 | 6.3 |
| 5 | VOac-CND-80 | 8.7 | 94.3 | 8.2 |
| 6 | VOac-CND-90 | 9.8 | 94.4 | 9.3 |
| 7 | VOac-CND-100 | 10.5 | 95.4 | 10.0 |
| 8 | VOac-CND-120 | 20.7 | 96.8 | 20.0 |
| 9d | VOac-CND-120 | 18.6 | 97.0 | 18.0 |
| 10e | VOac-CND-120 | 10.9 | 98.6 | 10.7 |
| 11f | VOac-CND-120 | 5.2 | 99.3 | 5.1 |
Besides grafting temperature, the catalytic activity was found to be related to the grafting amount of VO(acac)2. With the increase of the amount from 50 to 150 mg (entries 9–11), the final benzene conversion increased remarkably but leveled off when the grafting amount was up to 175 mg. This can infer that, at that stage, the terminal amino groups of g-CN have been almost fully utilized by reaction with VO(acac)2. Accordingly, the FT-IR spectra (Fig. S5†) of the samples grafted with various amounts of VO(acac)2 clearly presented a progressive increase of the intensity of the band at 970 cm−1 as the feeding amount of VO(acac)2 was elevated.
On the hand, the pure VO(acac)2 compound exhibited a moderate phenol yield (11.6%, entry 3). Note, the weight of VO(acac)2 was indeed based on the actual loading amount of vanadyl species on VOac-CND-120, as analyzed by XPS. In view of their catalyst weights, the catalytic activities obtained over VO(acac)2 was exactly superior than VOac-CND-120. Nonetheless, it is worth noting that the reaction system employing VO(acac)2 was solely homogeneous, suffering intrinsic difficulty in catalyst–product separation.
The influence of reaction conditions on the catalytic performances of VOac-CND in the hydroxylation reactions was further investigated. As described in Fig. 6A, the reaction temperatures were found to be sensitive to the catalytic activity. Upon increasing the temperature, the conversions of benzene were enhanced progressively, whereas the selectivity to phenol still remained above 95%. However, much higher temperatures above 60 °C led to a negative impact on the catalytic activity, mainly due to the rapid decomposition of H2O2. The catalytic activity was also dependant on reaction time (Fig. 6B). Under the conditions adopted in the present study, the highest activity was received at 4 h, and further prolonging the hydroxylation reaction showed no apparent improvement for its productivity. Furthermore, the effect of catalyst amount on the catalytic performances has also been carried out (Fig. 6C), and the corresponding evaluation exhibited that adding more catalysts (10–50 mg) could facilitate the activity, while the benzene conversions were improved smoothly under excessive use of the VOac-CND catalyst. In addition to the optimization of reaction conditions, the recyclability and reproductivity were also important parameters to evaluate a heterogeneous catalyst. Given this viewpoint, the VOac-CND-120 catalyst has been submitted with several consecutive runs, and the corresponding results are listed in Table 3. During the four runs, the selectivities to phenol were above 95%, and the benzene conversions were higher than 19%, showing almost no apparent loss of catalytic activity in such heterogeneous catalytic examination. According to N2 adsorption–desorption (Fig. S6, and Table S1†) and FT-IR characterization (Fig. S7†) for the spent VOac-CND-120 catalyst after four runs, its surface area, pore size, and main chemical bonding have been well reserved, confirming that the grafted vanadyl complex was stable in the catalytic reactions.
 | ||
| Fig. 6 Influence of reaction temperatures (A), time (B), and catalyst amounts on the catalytic activities of VOac-CND-120. Reaction condition: 1 mL of benzene (11.2 mmol), 3 mL of aqueous H2O2 solution (30 wt%), 6 mL of acetonitrile as solvent. | ||
| Run | Conv. (%) | Sel. (%) | Yield (%) |
|---|---|---|---|
| a Reaction conditions: 1 mL of benzene (11.2 mmol), 3 mL of aqueous H2O2 solution (30 wt%), 6 mL of acetonitrile as solvent, Wcatal. = 50 mg, T = 60 °C, and t = 4 h. | |||
| 1 | 20.7 | 96.8 | 20.0 |
| 2 | 19.8 | 95.4 | 18.8 |
| 3 | 19.6 | 95.0 | 18.6 |
| 4 | 19.0 | 94.7 | 18.0 |
Since the CND material possesses abundant aliphatic amines at the edges of its graphitic sheets, which could be utilized to graft VO(acac)2, it is of interest to synthesize g-CN materials grafted with wide transition-metal complexes. Envisioned by this, we thereafter grafted another acetylacetonate-containing transition-metal compounds, for instance copper(II) acetylacetonate ([Cu(acac)2]) on CND. The synthesized Cuac-CND sample showed similar textual properties and chemical functionalities with VOac-CND-120 (Table S1, Fig. S6 and S8†). As expected, Cuac-CND-120 demonstrated a moderate benzene conversion (12.1%, Table 4), together with a high selectivity (97.4%) to phenol. In view of this tentative test, it is anticipated that mesoporous g-CN could serve as a new support for wide transition-metal complexes. In this work, in addition to mesoporous g-CN, SBA-15 and carbon nanotubes have been also applied as supports to immobilize [VO(acac)2] (see the ESI† for the detailed preparation routes. FT-IR spectra of the bare SBA-15, SBA-15-NH2, and VOac-SBA-15-NH2 materials are given in Fig. S9†). As summarized in Table 4, under the present reaction conditions, VOac-SBA15-NH2 and VOac-CNTs-NH2 showed poor catalytic activities. Upon adding the catalyst amounts, a moderate benzene conversion (11.2%) was retrieved in the case of VOac-SBA15-NH2; however, the activity received over VOac-CNTs-NH2 was still low. The comparison strongly indicated that, in the hydroxylation of benzene catalyzed by immobilized vanadyl complexes, besides the reaction conditions, the support itself also played an important role for the catalytic activity.
| Entry | Catalyst | Conv. (%) | Sel. (%) | Yield (%) |
|---|---|---|---|---|
| a Reaction conditions: 1 mL of benzene (11.2 mmol), 3 mL of aqueous H2O2 solution (30 wt%), 6 mL of acetonitrile as solvent, Wcatal. = 50 mg, T = 60 °C, and t = 4 h.b Reaction conditions: 1 mL of benzene (11.2 mmol), 3 mL of aqueous H2O2 solution (30 wt%), 6 mL of acetonitrile as solvent, Wcatal. = 200 mg, T = 60 °C, and t = 4 h. | ||||
| 1a | VOac-CND-120 | 20.7 | 96.8 | 20.0 |
| 2a | Cuac-CND-120 | 12.1 | 97.4 | 11.7 |
| 3a | VOac-SBA15-NH2 | <1 | 100 | <1 |
| 4b | VOac-SBA15-NH2 | 11.2 | 81.4 | 9.0 |
| 5a | VOac-CNTs-NH2 | <1 | 100 | <1 |
| 6b | VOac-CNTs-NH2 | 4.7 | 100 | 4.7 |
In addition, we have also calculated the turnover frequency (TOF, grams of synthesized phenol per gram of the total catalyst per hour) value in the case of entry 1 of Table 4, and the resultant value was ca. 1.1 h−1. It is worth noting that in the cases of previously reported supported vanadia catalysts (e.g. V/clay,43 VO/TiO2,44 V-g-C3N4,4 and VO/MCM-41-NH2 (ref. 45)), their TOF values were in the range of 0.14–0.73 h−1, whereas the reaction conditions adopted such as temperatures were similar. Given this point, the catalytic activity received over the present grafted vanadyl complex catalyst was relatively high.
To probe a potential effect of the support on the hydroxylation reaction, benzene-TPD was conducted to investigate the adsorption capability of benzene on SBA-15 and CND samples. As depicted in Fig. 7, both SBA-15 and its grafted vanadyl complex represented minor desorption peaks at ca. 120 °C, which should be due to physically adsorbed benzene molecules. In sharp contrast, CND offered a pronounced broad yet high-intensity peak centered at 150 °C. Referring to the textual parameters of SBA-15 and CND (Tables S1† and 1), the surface area of CND (488 m2 g−1) was actually lower than that of its counterpart (564 m2 g−1). This means that the desorption peak acquired in CND was probably contributed from the chemical rather than physical adsorption of benzene. Based on the benzene-titration experiment, the adsorbed amount of benzene on CND was ca. 28 μmol gcatal.−1, far beyond the value gained on SBA-15 (ca. 1.9 μmol gcatal.−1). Obviously, the mesoporous g-CN possessed a strong interaction with benzene, definitely owing to the aforementioned conjugated tri-s-triazine units of g-CN, as also reported by Wang et al.46 Interestingly, the capability to adsorb benzene molecules remained even after the immobilization of [VO(acac)2]. Based on the reported work involving hydroxylation of benzene catalyzed by vanadyl complexes,2,45 a simple catalytic mechanism can be speculated. Initially, H2O2 oxidized V4+ into a peroxo vanadyl radical, i.e. V5+–O–O˙. On the other hand, benzene was absorbed by mesoporous g-CN support. Afterwards, the activated benzene underwent an insertion of oxygen atom in one of its C–H bonds by V5+–O–O˙, therein yielding phenol and V4+. Finally, the V4+ species was regenerated by another oxidation by H2O2.
 | ||
| Fig. 7 Benzene-TPD profiles of SBA-15 (a), VOac-SBA15-NH2 (b), CND (c), and VOac-CND-120 (d). | ||
4. Conclusion
In summary, we have synthesized an immobilized Schiff-base-type vanadyl complex using mesoporous graphitic carbon nitride as a new support. According to the characterization results of FT-IR and XPS, the immobilization of [VO(acac)2] was based on the reaction of acetylacetonate ligand and the amino groups of g-CN, and high temperatures could facilitate the immobilization efficiency. In the catalytic hydroxylation of benzene, the VOac-CND materials provided high and stable catalytic conversion of benzene (20.7%) along with high selectivity (96.8%) to phenol. Moreover, the catalytic performance was comparably high than that obtained over vanadyl complex grafted on amine-functionalized SBA-15. The mesoporous g-CN material acted not only as a support for immobilizing [VO(acac)2] but also a crucial component in activating benzene.Acknowledgements
This work was supported by National Natural Science Foundation of China (21203014), Postgraduate Innovation Project of Jiangsu Province (KYLX_1097), Jiangsu Key Laboratory of Advanced Catalytic Materials and Technology (BM2012110), and the Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.Notes and references
- P. Borah, A. Datta, K. T. Nguyen and Y. Zhao, Green Chem., 2015 10.1039/c1035gc01194a.
- P. Borah, X. Ma, K. T. Nguyen and Y. Zhao, Angew. Chem., Int. Ed., 2012, 51, 7756–7761 CrossRef CAS PubMed.
- B. Guo, L. Zhu, X. Hu, Q. Zhang, D. Tong, G. Li and C. Hu, Catal. Sci. Technol., 2011, 1, 1060–1067 CAS.
- G. Ding, W. Wang, T. Jiang, B. Han, H. Fan and G. Yang, ChemCatChem, 2013, 5, 192–200 CrossRef CAS.
- J.-H. Yang, G. Sun, Y. Gao, H. Zhao, P. Tang, J. Tan, A.-H. Lu and D. Ma, Energy Environ. Sci., 2013, 6, 793–798 CAS.
- T. Jiang, W. Wang and B. Han, New J. Chem., 2013, 37, 1654–1664 RSC.
- J. Xu, Q. Jiang, T. Chen, F. Wu and Y.-X. Li, Catal. Sci. Technol., 2015, 5, 1504–1513 CAS.
- X. Ye, Y. Cui and X. Wang, ChemSusChem, 2014, 7, 738–742 CrossRef CAS PubMed.
- B. Jarrais, A. R. Silva and C. Freire, Eur. J. Inorg. Chem., 2005, 4582–4589 CrossRef CAS.
- C. Pereira, K. Biernacki, S. L. H. Rebelo, A. L. Magalhãesa, A. P. Carvalho, J. Piresb and C. Freire, J. Mol. Catal. A: Chem., 2009, 312, 53–64 CrossRef CAS.
- C. Pereira, J. F. Silva, A. M. Pereira, J. P. Araújo, G. Blanco, J. M. Pintado and C. Freire, Catal. Sci. Technol., 2011, 1, 784–793 Search PubMed.
- C. Pereira, A. R. Silva, A. P. Carvalho, J. Pires and C. Freire, J. Mol. Catal. A: Chem., 2008, 283, 5–14 CrossRef CAS.
- J. Zhu, P. Xiao, H. Li and S. A. C. Carabineiro, ACS Appl. Mater. Interfaces, 2014, 6, 16449–16465 CAS.
- J. Xu, H.-T. Wu, X. Wang, B. Xue, Y.-X. Li and Y. Cao, Phys. Chem. Chem. Phys., 2013, 15, 4510–4517 RSC.
- F. Su, S. C. Mathew, G. Lipner, X. Fu, M. Antonietti, S. Blechert and X. Wang, J. Am. Chem. Soc., 2010, 132, 16299–16301 CrossRef CAS PubMed.
- Y. Zheng, J. Liu, J. Liang, M. Jaroniec and S. Z. Qiao, Energy Environ. Sci., 2012, 5, 6717–6731 CAS.
- S. S. Park, S.-W. Chu, C. Xue, D. Zhao and C.-S. Ha, J. Mater. Chem., 2011, 21, 10801–10807 RSC.
- K. S. Lakhi, W. S. Cha, S. Joseph, B. J. Wood, S. S. Aldeyab, G. Lawrence, J.-H. Choy and A. Vinu, Catal. Today, 2015, 243, 209–217 CrossRef CAS.
- F. Su, M. Antonietti and X. Wang, Catal. Sci. Technol., 2012, 2, 1005–1009 CAS.
- M. B. Ansari, B.-H. Min, Y.-H. Mo and S.-E. Park, Green Chem., 2011, 13, 1416–1421 RSC.
- Y. Gong, M. Li, H. Li and Y. Wang, Green Chem., 2015, 17, 715–736 RSC.
- Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–89 CrossRef CAS PubMed.
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J.-O. Müller, R. Schlögl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893–4908 RSC.
- F. Goettmann, A. Fischer, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2006, 45, 4467–4471 CrossRef CAS PubMed.
- F. Goettmann, A. Fischer, M. Antonietti and A. Thomas, Chem. Commun., 2006, 4530–4532 RSC.
- F. Goettmann, A. Thomas and M. Antonietti, Angew. Chem., Int. Ed., 2007, 46, 2717–2720 CrossRef CAS PubMed.
- Z. Long, Y. Zhou, G. Chen, W. Ge and J. Wang, Sci. Rep., 2014, 4, 3651 CAS.
- X. Jin, V. V. Balasubramanian, S. T. Selvan, D. P. Sawant, M. A. Chari, G. Q. Lu and A. Vinu, Angew. Chem., Int. Ed., 2009, 48, 7884–7887 CrossRef CAS PubMed.
- J. Xu, K. Shen, B. Xue and Y.-X. Li, J. Mol. Catal. A: Chem., 2013, 372, 105–113 CrossRef CAS.
- J. Xu, T. Chen, Q. Jiang and Y.-X. Li, Chem.–Asian J., 2014, 9, 3269–3277 CrossRef CAS PubMed.
- J. P. Paraknowitsch, A. Thomas and M. Antonietti, J. Mater. Chem., 2010, 20, 6746–6758 RSC.
- D. Zhao, J. Sun, Q. Li and G. D. Stucky, Chem. Mater., 2000, 12, 275–279 CrossRef CAS.
- Q. Li, J. Yang, D. Feng, Z. Wu, Q. Wu, S. S. Park, C.-S. Ha and D. Zhao, Nano Res., 2010, 3, 632–642 CrossRef CAS.
- J. Xu, T. Chen, X. Wang, B. Xue and Y.-X. Li, Catal. Sci. Technol., 2014, 4, 2126–2133 CAS.
- S. N. Talapaneni, S. Anandan, G. P. Mane, C. Anand, D. S. Dhawale, S. Varghese, A. Mano, T. Mori and A. Vinu, J. Mater. Chem., 2012, 22, 9831–9840 RSC.
- M. J. Bojdys, J.-O. Müller, M. Antonietti and A. Thomas, Chem.–Eur. J., 2008, 14, 8177–8182 CrossRef CAS PubMed.
- G. P. Mane, D. S. Dhawale, C. Anand, K. Ariga, Q. Ji, M. A. Wahab, T. Mori and A. Vinu, J. Mater. Chem. A, 2013, 1, 2913–2920 CAS.
- S. B. Kristensen, A. J. Kunov-Kruse, A. Riisager, S. B. Rasmussen and R. Fehrmann, J. Catal., 2011, 284, 60–67 CrossRef CAS.
- R. Nenashev, N. Mordvinova, V. Zlomanov and V. Kuznetsov, Inorg. Chem., 2015, 51, 891–896 CAS.
- K. N. Rao, B. M. Reddy, B. Abhishek, Y.-H. Seo, N. Jiang and S.-E. Park, Appl. Catal., B, 2009, 91, 649–656 CrossRef CAS.
- V. D. Dasireddy, H. B. Friedrich and S. Singh, Appl. Catal., A, 2013, 467, 142–153 CrossRef CAS.
- K. M. Parida, S. Singha and P. C. Sahoo, J. Mol. Catal. A: Chem., 2010, 325, 40–47 CrossRef CAS.
- X. Gao and J. Xu, Appl. Clay Sci., 2006, 33, 1–6 CrossRef CAS.
- D. Xu, L. Jia and X. Guo, Catal. Lett., 2012, 142, 1251–1261 CrossRef CAS.
- C.-H. Lee, T.-S. Lin and C.-Y. Mou, J. Phys. Chem. B, 2003, 107, 2543–2551 CrossRef CAS.
- Y. Gong, P. Zhang, X. Xu, Y. Li, H. Li and Y. Wang, J. Catal., 2013, 297, 272–280 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra21438f |
| This journal is © The Royal Society of Chemistry 2015 |