
Cysteine-based fluorescence “turn-on” sensors for Cu2+ and Ag+†
V. Haridas*a,
P. P. Praveen Kumara and
Cherumuttathu H. Sureshb
aDepartment of Chemistry, Indian Institute of Technology Delhi, Hauz Khas-110016, New Delhi, India. E-mail: h-haridas@hotmail.com; haridasv@iitd.ac.in; Tel: +91-11-26591380
bInorganic and Theoretical Chemistry Section, CSTD, CSIR-National Institute for Interdisciplinary Science and Technology, Thiruvananthapuram – 695019, India
First published on 24th October 2014
Abstract
We designed and synthesized two metal ion binding molecules 3a and 3b based on cysteine. In 3a, pyrene is used as a fluorescent probe, while 3b contains tryptophan, which acts as a fluorescent probe as well as facilitates metal ion binding. Detailed spectroscopic, calorimetric, microscopic and computational studies revealed the binding mode and the plausible structures of the complexes.
Selective detection of metal ions using fluorescence spectroscopy is a method of choice particularly owing to its high sensitivity and simplicity of measurement.1 Detection by turn-on/off fluorescence will depend on the mode of interaction of the sensing molecule with the guest molecule.2 Therefore, the design and synthesis of fluorescent organic molecules with appropriate coordination pocket for the desired specificity for the metal ion are challenging. Proteins are highly superior in terms of their specificity and metal ion binding ability.3 The metal ion binding capacity of proteins is due to the specific amino acid side chains, for example, the well studied zinc finger proteins bind the zinc ion using the imidazole and the thiolate side chains of histidine and cysteine respectively.4 In a similar way, the indole moiety of tryptophan (Trp) is involved in metal binding in CusF protiens.5 However the ability of these amino acid side-chains are not completely exploited for the design of synthetic metal ion sensors.6
Here in this study, we use cysteine to design fluorescent sensors for metal ion binding. The C-terminus of the cysteine was conjugated to fluorophores such as pyrene or tryptophan to obtain compounds 3a or 3b respectively. Pyrene is an excellent fluorophore with a distinct monomer and excimer emission bands,7 whereas tryptophan is a naturally occurring amino acid which shows fluorescence as well as the ability to bind metal ion through its indole side chain. Trp can provide the most potent cation–π binding sites.8
In order to synthesize 3a and 3b, the thiol group of L-cysteine was protected as S-t-butylcysteine, which was then coupled with pyrene methylamine/L-tryptophan methyl ester (TrpOMe) under dicyclohexycarbodiimide (DCC) and N-hydroxysuccinimide (NHS) coupling condition to afford 3a and 3b respectively (Scheme 1).
 | ||
| Scheme 1 Synthesis of 3a and 3b. | ||
Compounds 3a and 3b were then investigated for their metal uptake potential. UV-visible spectroscopic studies revealed considerable changes in the spectra of 3a and 3b upon the addition of Cu2+ and Ag+ respectively (see ESI, Fig. S1†). Among the metal ions tested, addition of Cu2+ to 3a, resulted in formation of a new peak at 298 and 403 nm with two isobestic points, indicating the complex formation between 3a and Cu2+ (see ESI, Fig. S1c†). Studies on the complexing abilities of various metal cations (Na+, K+, Cs+, Ag+, Mg2+, Ba2+, Cd2+, Zn2+, Pb2+, Mn2+, Cu2+, Hg2+, Sn2+ and Fe3+) towards 3a demonstrated that it has higher selectivity for Cu2+ than other metal ions tested (see ESI, Fig. S1a†).
Excitation of 3a at 340 nm resulted in two emission bands at 378 and 399 nm corresponding to the monomer emission bands of pyrene.7 The addition of Cu2+ to 3a resulted in the formation of new emission band at 470 nm corresponding to the excimer of pyrene with a concomitant decrease in the monomer emission bands. The excimer band intensity increased upon the addition of Cu2+ indicating that binding of Cu2+ brings two pyrene moieties in close proximity. The observed fluorescence “turn-on” upon metal ion binding is a remarkable property of 3a, since heavy metals generally causes a quenching effect on the fluorescence (Fig. 1a and see ESI, Fig. S2a†).9 The complex formation between 3a and Cu2+ was further confirmed by mass spectrometric analysis, which showed a peak at 1066.3750 corresponding to [2·3a + Cu2+ + Na+] (see ESI, Fig. S3†). Job's plot further proved the formation of the 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex between 3a and Cu2+ (see ESI, Fig. S4†). The excitation spectra of 3a + Cu2+ showed a red shift of 7 nm indicating a static type excimer (see ESI, Fig. S5†).7 The fluorescence decay monitored at 390 nm and 470 nm indicated no change in the decay time of the free and complexed species, characteristic of static excimer formation (see ESI, Fig. S6 and Table S1†).10 Competitive binding studies of 3a with various metal ions (see ESI, Fig. S7†) showed that 3a can detect Cu2+ in the presence of other metal ions with a detection limit of 4 μM and a binding constant of 1.24 × 105 M−2.11
:1 complex between 3a and Cu2+ (see ESI, Fig. S4†). The excitation spectra of 3a + Cu2+ showed a red shift of 7 nm indicating a static type excimer (see ESI, Fig. S5†).7 The fluorescence decay monitored at 390 nm and 470 nm indicated no change in the decay time of the free and complexed species, characteristic of static excimer formation (see ESI, Fig. S6 and Table S1†).10 Competitive binding studies of 3a with various metal ions (see ESI, Fig. S7†) showed that 3a can detect Cu2+ in the presence of other metal ions with a detection limit of 4 μM and a binding constant of 1.24 × 105 M−2.11
 | ||
| Fig. 1 (a) Fluorescence spectra of 3a (1.2 × 10−5 M) with and without addition of Cu(ClO4)2 (1.2 × 10−4 M) in acetonitrile, λex = 340 nm. Addition of metal ion results in the enhancement of excimer band at 470 nm. (b) Fluorescence spectra of 3b (1.8 × 10−5 M) alone and upon the addition of AgClO4 (2 × 10−4 M) in acetonitrile, λex = 290 nm. The fluorescence intensity increases upon addition of AgClO4, indicating the “turn-on” behaviour. | ||
Trp-based compound 3b was designed with the notion that the indole moiety of tryptophan could bind metal ion by cation–π interactions. Among the various metal ions tested (Na+, K+, Cs+, Ag+, Mg2+, Ba2+, Cd2+, Zn2+, Pb2+, Mn2+, Cu2+, Hg2+, Sn2+, and Fe3+) for binding studies with 3b, a high selectivity was observed for Ag+ (see ESI, Fig. S1†). The absorbance at 285 nm of 3b decreased upon the addition of Ag+ with the concomitant formation of a new band at 314 nm with two isobestic points at 253 and 293 nm. The two isobestic points signified the presence of two species in equilibrium.12 The appearance of new absorption band at 314 nm is a result of the interaction of indole ring of Trp with Ag+ resulting in a strong complex (see ESI, Fig. S1†).13
The fluorescence intensity of 3b increased upon the addition of Ag+ (Fig. 1b). The binding studies revealed that 3b is selective towards Ag+ and Job's plot showed a 1:2 stoichiometry between 3b and Ag+ (see ESI, Fig. S2 and S4†). The mass spectrum displayed a peak at m/z 691.0371 (see ESI, Fig. S3†) corresponding to 3b + 2Ag+ further support the 1:2 complex formation. The binding constant of 1.2 × 108 M−2 was obtained by using Benesi–Hildebrand method (see ESI, Fig. S8†) with a detection limit of 8 μM.11 Competitive binding experiments revealed that 3b can detect Ag+ even in the presence of other cations (see ESI, Fig. S7†). The time resolved fluorescence studies showed that decay time for 3b (τ = 1.01) is the same as that of 3b + Ag+, indicating the formation of a complex between 3b and Ag+ (see ESI, Fig. S6 and Table S2†).10
Thermodynamics of binding was studied (Fig. 2) by isothermal calorimetry (ITC).14 3a was titrated with Cu2+ and the ITC data suggest the formation of 2:1 host–guest complex (2·3a:Cu2+). Similarly, ITC titration of 3b with Ag+ indicated a 1:2 host–guest (3b:2Ag+) complex. The affinity of the molecules 3a/3b towards Cu2+/Ag+ was enthalpy favored (see ESI, Table S3†). The negative free energy term ΔG = −30.18 kJ mol−1 for the 3a–Cu2+ indicates a thermodynamically favored process. The binding of 3b with Ag+ is also thermodynamically favored as indicated by free energy terms, ΔG1 = −29.10 kJ mol−1 and ΔG2 = −47.32 kJ mol−1. The binding constants obtained for 3a–Cu2+ and 3b–Ag+ by UV-visible, fluorescence and ITC methods are comparable.
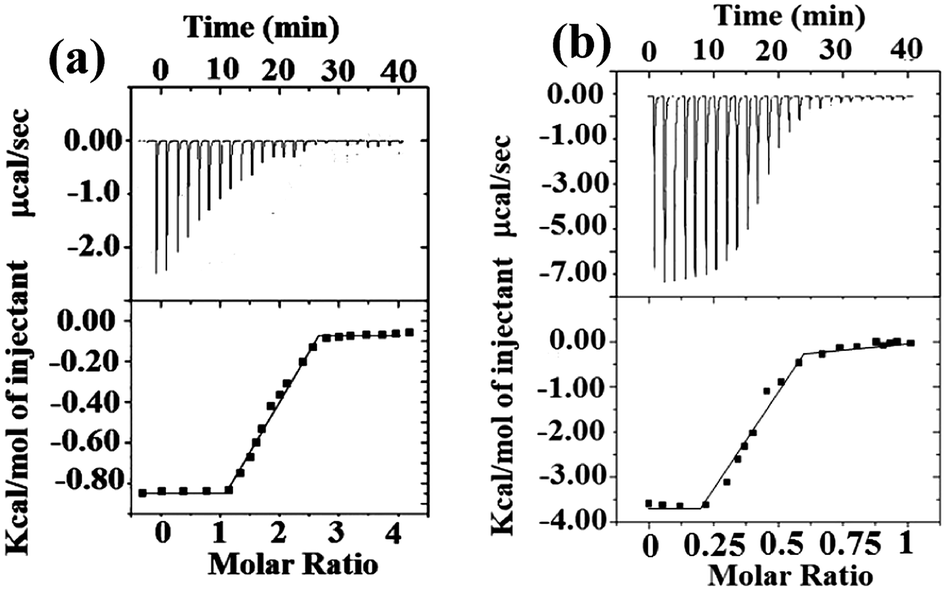 | ||
| Fig. 2 ITC titration data of (a) 3a [200 μM] with Cu2+ (n = 2.09, Ka = 1.95 × 105 M−1) (b) 3b [150 μM] with Ag+ (n = 0.48, Ka1 = 1.26 × 105 M−1 and Ka2 = 1.97 × 108 M−1). The titrations were performed at 25 °C in acetonitrile. Upper graphs represent experimental data and the bottom panels show the binding isotherms created by plotting the integrated heats from each injection, against the molar ratio of the compounds. Each titration experiment was composed of 19–21 successive injections of 5 μL of metal ions. | ||
Binding interactions of 3a and 3b with Cu2+ and Ag+ were also confirmed by electron microscopy (Fig. 3). The scanning electron microscopic (SEM) analysis of 3a showed fibrillar morphology, which changes to thick stick-like bars with a length of 824 nm (average of 50 bars) and width of 250 nm (average of 50 bars) as a result of binding with Cu2+. On the other hand, SEM of 3b showed spherical vesicles, comprised predominantly of two sizes; small vesicles having an average diameter of 458 nm (average of 20 vesicles) and bigger vesicles with an average diameter of 1.41 μm (average of 20 vesicles).
 | ||
| Fig. 3 SEM images of (a) 3a (b) 3a + Cu2+ (c) 3b (d) 3b + Ag+ in CH3CN. | ||
The remarkable ability of 3b to form vesicular self-assembly is attributed to the presence of indole ring.15 The presence of indole moiety can acts as a potent H-bond donor and facilitates π–π interactions leading to self-assembled structure. The metal ion binding perturbs the assembly and hence spherical vesicles changes to flower petal like morphology upon binding with Ag+ ion (Fig. 3). The completely altered morphologies of 3a and 3b with the addition of metal ions further support the interactions of the metal ions with the molecules. Atomic force microscopy (AFM) images also support similar observation (see ESI, Fig. S9†). Interestingly, the self-assembly to bar-shape and petal-like shapes in 3a–Cu2+ and 3b–Ag+ complexes indicate the presence of specific supramolecular interaction in the complexes.
The binding modes of 3a with Cu2+ and 3b with Ag+ was investigated by UB3LYP/6-31G(d) level of density functional theory (DFT) and B3LYP/Gen1 level of DFT, respectively where Gen1 corresponds to 6-31G(d) basis set for H, C, N, O and S atoms and LanL2DZ basis set with ‘f’ polarization function for Ag atom.16 The calculations were done using the Guassian 09 suite of programs.17 Several possible 2:1 complexation modes of Cu2+ and 1:2 complexation modes of Ag+ were studied (see ESI, Fig. S10–S12†). The most stable complex for 3a with Cu2+ (3a–Cu2+–3a) and 3b with Ag+ (Ag+–3b–Ag+) are shown in Fig. 4.
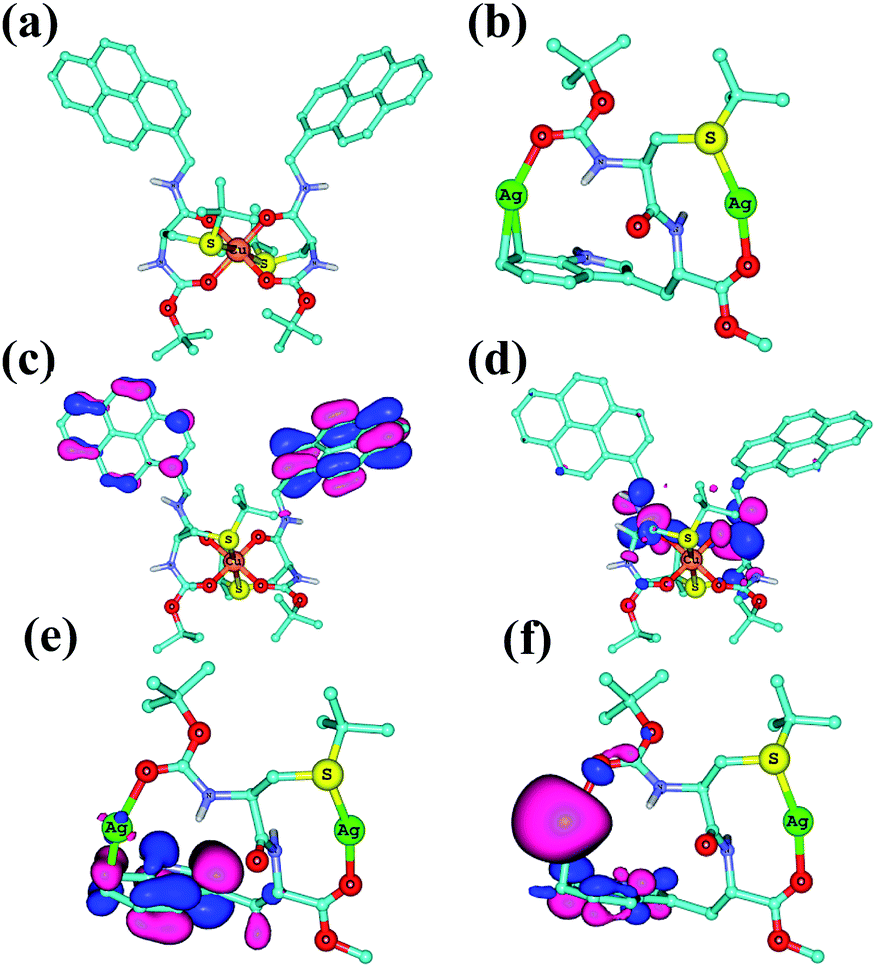 | ||
| Fig. 4 B3LYP/6-31G(d) optimized structure of (a) 3a–Cu2+–3a and (b) Ag+–3b–Ag+. (c) HOMO of 3a–Cu2+–3a, (d) LUMO of 3a–Cu2+–3a. (e) HOMO of Ag+–3b–Ag+ and (f) LUMO of Ag+–3b–Ag+. Many hydrogen atoms are omitted for clarity. | ||
The 3a–Cu2+–3a shows an octahedral geometry (Fig. 4a), wherein Cu2+ displays a coordination to four carbonyl oxygens of amides in the equatorial positions with an average –O–Cu2+– distance of 1.96 Å and also coordination to two sulfur atoms in the apical positions with average –S–Cu2+– distance of 2.86 Å. The pyrene rings are oriented towards the same side, suggesting the possibility of excimer formation. This is as expected from experimental results from fluorescence studies which indicated the formation of excimer upon Cu2+ binding (Fig. 1a). The HOMO of 3a–Cu2+–3a is located on the pyrene rings, while the LUMO was centered on the amide bonds (Fig. 4c and d).
The Ag+–3b–Ag+ structure shown in Fig. 4b can be considered as a bicyclic metallomacrocycle (see ESI, Fig. S12† for seven other binding modes and Table S4†). One of the Ag+ ions is coordinated by one –S– atom and one amide carbonyl, while the second Ag+ ion is coordinated by indole ring of tryptophan residue (Fig. 4b) and another amide carbonyl. The –S–Ag+– and –C–Ag+– distances are 2.48 Å and 2.50 Å respectively, while the average –CO–Ag+ distance is 2.21 Å. The HOMO of Ag+–3b–Ag+ is a π-orbital of indole moiety while the LUMO is centered on one of the Ag atoms (Fig. 4e and f).
The binding mode of 3b with Ag+ was supported by 1H NMR as well as by 13C NMR titrations (Fig. 5 and see ESI, Fig. S13†).18 Addition of 0.1 equiv. Ag+ resulted in an initial shift of –S–CH2– protons, further addition 0.5 equiv. Ag+ resulted in splitting of the –S–CH2– signal. The tryptophan aromatic protons (Hg + Hk + Hj) broadened and shifted down field (see ESI, Fig. S13†). Similarly, the 13C NMR spectra recorded in CD3CN showed down field shift for the aromatic carbon atoms as well as for the –S–CH2 carbon atom. The prominent deshielding effect was observed for the ester (j) as well as for the Boc-carbonyls (e) 0.58 and 0.55 ppm respectively (see ESI, Table S5†). The –S–CH2– carbon atom (c) deshielded by 0.17 ppm, suggesting the involvement of –S– atom in the binding. The aromatic carbon atoms ‘p’ and ‘q’ of the indole ring are deshielded by 0.47 and 0.21 ppm respectively (see ESI, Table S5† for chemical shifts of carbon atoms), indicating the involvement of these carbons in binding Ag+ as was supported by the DFT calculations. Theoretically computed NMR results using the GIAO method19 also matched very well with the experimental NMR values (see ESI, Table S5†). Thus both the NMR data and the DFT calculations indicate an η2-type coordination of Ag+ with aromatic ring of Trp.13
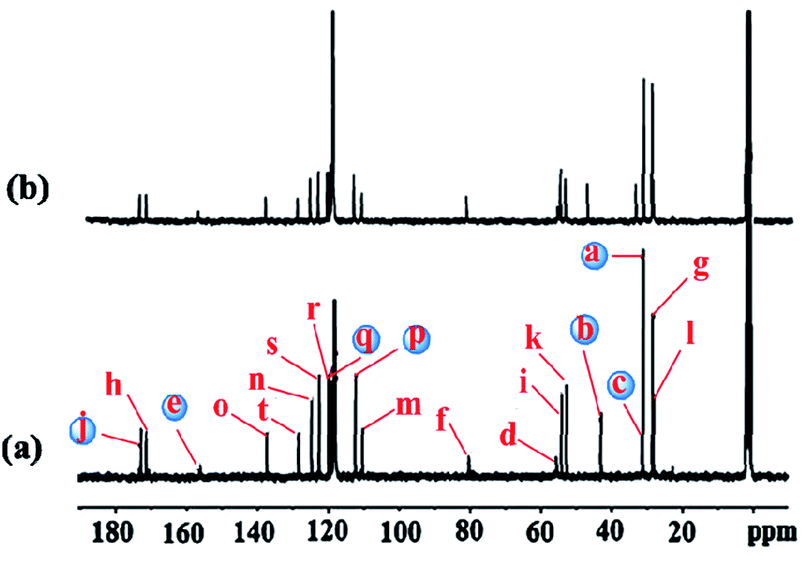 | ||
| Fig. 5 13C NMR spectra (75 MHz) of (a) 3b (b) 3b + Ag+ ion in CD3CN. The blue coloured carbon atoms are involved in Ag+ binding. | ||
In conclusion, we have designed and synthesized cysteine based fluorescent “turn-on” sensors 3a and 3b for Cu2+ and Ag+ respectively. The design and experimental studies illustrated here highlight that cysteine is a potential amino acid for the design of novel metal ion sensors. Transformation of cysteine to systems with specific metal ion harboring capability is noteworthy. The results presented here will be useful for the design of novel metal-binding peptide systems. Such peptide systems may find a variety of biomedical applications.
Acknowledgements
We thank the Department of Science and Technology (DST and DST-FIST) for the financial support. PPPK thanks University Grants Commission (UGC), New Delhi for the fellowship.Notes and references
- (a) L. Fabbrizzi and A. Poggi, Chem. Soc. Rev., 1995, 24, 197–202 RSC; (b) B. Valeur and I. Leray, Coord. Chem. Rev., 2000, 205, 3–40 CrossRef CAS; (c) L. Prodi, F. Bolletta, M. Montalti and N. Zaccheroni, Coord. Chem. Rev., 2000, 205, 59–83 CrossRef CAS; (d) A. P. de Silva, D. B. Fox, A. J. M. Huxley and T. S. Moody, Coord. Chem. Rev., 2000, 205, 41–57 CrossRef CAS.
- B. Schazmann, N. Alhashimy and D. Diamond, J. Am. Chem. Soc., 2006, 128, 8607–8614 CrossRef CAS PubMed; J. Wang, X. Qian and J. Cui, J. Org. Chem., 2006, 71, 4308–4311 CrossRef PubMed; Z. Xu, Y. Xiao, X. Qian, J. Cui and D. Cui, Org. Lett., 2005, 7, 889–892 CrossRef PubMed; H. Yuasa, N. Miyagawa, T. Izumi, M. Nakatami, M. Izumi and H. Hashimoto, Org. Lett., 2004, 6, 1489–1492 CrossRef PubMed; Z. C. Wen, R. Yang, H. He and Y. B. Jiang, Chem. Commun., 2006, 106–108 RSC; S. K. Kim, S. H. Lee, J. Y. Lee, R. A. Bartsch and J. S. Kim, J. Am. Chem. Soc., 2004, 126, 16499–16506 CrossRef PubMed.
- T. M. Desilva, G. Veglia, F. Porcelli, A. M. Prantner and S. J. Opella, Biopolymers, 2002, 64, 189–197 CrossRef CAS PubMed; G. Veglia, F. Porcelli, T. Desilva, A. Prantner and S. J. Opella, J. Am. Chem. Soc., 2000, 122, 2389–2390 CrossRef.
- A. Klug, Annu. Rev. Biochem., 2010, 79, 213–231 CrossRef CAS PubMed; A. Klug, Q. Rev. Biophys., 2010, 43, 1–21 CrossRef PubMed; D. Jantz, B. T. Amann, G. J. Gatto Jr and J. M. Berg, Chem. Rev., 2004, 104, 789–799 CrossRef PubMed; J. H. Laity, B. M. Lee and P. E. Wright, Curr. Opin. Struct. Biol., 2001, 11, 39–46 CrossRef.
- Y. Xue, A. V. Davis, G. Balakrishnan, J. P. Stasser, B. M. Staehlin, P. Focia, T. G. Spiro, J. E. Penner-Hahn and T. V. O'Halloran, Nat. Chem. Biol., 2008, 4, 107–109 CrossRef CAS PubMed; K. J. Franz, Nat. Chem. Biol., 2008, 4, 85–86 CrossRef PubMed; I. R. Loftin, S. Franke, N. J. Blackburn and M. M. Mcevoy, Protein Sci., 2007, 16, 2287–2293 CrossRef PubMed.
- R. M. F. Batista, R. C. M. Ferreira, M. M. M. Raposo and S. P. G. Costa, Tetrahedron, 2012, 68, 7322–7330 CrossRef CAS PubMed; L. N. Neupane, J. Y. Park, J. H. Park and K. H. Lee, Org. Lett., 2012, 15, 254–257 CrossRef PubMed; M. H. Yang, P. Thirupathi and K. H. Lee, Org. Lett., 2011, 13, 5028–5031 CrossRef PubMed.
- (a) A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515–1566 CrossRef CAS PubMed; (b) F. M. Winnik, Chem. Rev., 1993, 93, 587–614 CrossRef CAS.
- D. A. Dougherty, Acc. Chem. Res., 2013, 46, 885–893 CrossRef CAS PubMed; D. K. Chakravorty, B. Wang, M. N. Ucisik and K. M. Merz Jr, J. Am. Chem. Soc., 2011, 133, 19330–19333 CrossRef PubMed; D. A. Dougherty, Science, 1996, 271, 163–168 Search PubMed.
- L. Fabbrizzi, M. Licchelli, P. Pallavicini, A. Perotti, A. Taglietti and D. Sacchi, Chem.–Eur. J., 1996, 2, 75–82 CrossRef CAS; D. J. S. Birch, K. Suhling, A. S. Holmes, T. Salthammer and R. E. Imhof, Pure Appl. Chem., 1993, 65, 1687–1692 CrossRef.
- G. Petroselli, M. L. Dantola, F. M. Cabrerizo, C. Lorente, A. M. Braun, E. Oliveros and A. H. Thomas, J. Phys. Chem. A, 2009, 113, 1794–1799 CrossRef CAS PubMed; C. B. Murphy, Y. Zhang, T. Troxler, V. Ferry, J. J. Martin and W. E. Jones Jr, J. Phys. Chem. B, 2004, 108, 1537–1543 CrossRef.
- L. Ma, H. Li and Y. Wu, Sens. Actuators, B, 2009, 143, 25–29 CrossRef CAS PubMed; H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2707 CrossRef.
- G. Scheibe, Angew. Chem., Int. Ed., 1937, 50, 212–219 CrossRef CAS; M. D. Cohen and E. Fischer, J. Chem. Soc., 1962, 3, 3044–3052 RSC.
- I. R. Loftin, N. J. Blackburn and M. M. McEvoy, J. Biol. Inorg. Chem., 2009, 14, 905–912 CrossRef CAS PubMed; O. Kuhl and W. Hinrichs, ChemBioChem, 2008, 9, 1697–1699 CrossRef PubMed.
- V. D. Jadhav and F. P. Schmidtchen, Org. Lett., 2005, 7, 3311–3314 CrossRef CAS PubMed; A. Cooper, Curr. Opin. Chem. Biol., 1999, 3, 557–563 CrossRef.
- E. Abel, S. L. De Wall, W. B. Edwards, S. Lalitha, D. F. Covey and G. W. Gokel, J. Org. Chem., 2000, 65, 5901–5909 CrossRef CAS PubMed; E. Abel, M. F. Fedders and G. W. Gokel, J. Am. Chem. Soc., 1995, 117, 1265–1270 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed; A. W. Ehlers, M. Böhme, S. Dapprich, A. Gobbi, A. Höllwarth, V. Jonas, K. F. Köhler, R. Stegmann, A. Veldkamp and G. Frenking, Chem. Phys. Lett., 1993, 208, 111–114 CrossRef.
- M. J. Frisch, et al., Gaussian 09, Revision C.01, 2010 Search PubMed.
- Y. Li and C. M. Yang, J. Am. Chem. Soc., 2005, 127, 3527–3530 CrossRef CAS PubMed; H. Sun and E. Oldfield, J. Am. Chem. Soc., 2004, 126, 4726–4734 CrossRef PubMed.
- K. Wolinski, J. F. Hilton and P. Pulay, J. Am. Chem. Soc., 1990, 112, 8251–8260 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental procedures, DFT studies, spectroscopic data, NMR (1H and 13C) and ITC titration data. See DOI: 10.1039/c4ra10936h |
| This journal is © The Royal Society of Chemistry 2014 |