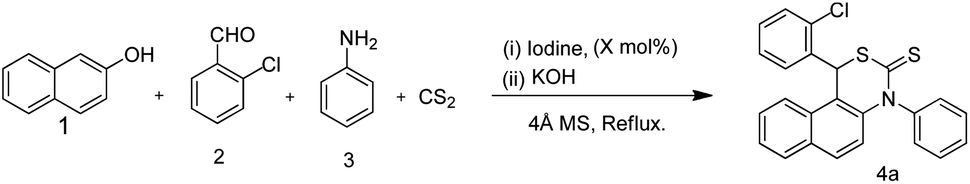
Domino condensation–heterocyclisation reactions: iodine catalyzed four component synthesis of 1,3-thiazine†
Munusamy Sathishkumar and
Kulathu Iyer Sathiyanarayanan*
Chemistry Division, School of Advanced Sciences, VIT University, Vellore-632014, Tamilnadu, India. E-mail: sathiya_kuna@hotmail.com; Fax: +91 4162243092
First published on 2nd December 2013
Abstract
An efficient iodine-catalyzed approach to synthesise 1,3-thiazine has been developed, and this synthetic methodology is economical and uses cheap and readily available starting materials. This is the first example of constructing 1,3-thiazine via a sequential thia-Michael addition and cyclodehydration.
The central theme of organic synthesis is the construction and cleavage of bonds in organic molecules. Modern synthesis involves the design of efficient synthetic protocol that minimizes the number of synthetic steps for the rapid generation of functionalized molecules with interesting properties.1 One approach to achieve this goal involves the development of eco-compatible, multicomponent procedures. Multicomponent reactions (MCRs) offer a wide range of possibilities for the construction of pre-defined highly complex molecules in a single step with high atom economy and straight forward experimental procedures.2,3 It has been reported that sulfur containing heterocycles exhibit activity against human immunodeficiency virus type (1), polio virus type (1), coxsackie virus type 3(Cox-3), vesicular stomatitis virus (VSV) and herpes simplex virus type 1(HSV-1).4 Cephalosporins are widely used β-lactams that contain 1,3-thiazine as the active core.5 Rhodamines such as 2-thioxo-1,3-thiazolidine-4-ones are sulfur/nitrogen heterocycles. These and typical 1,3-thiazines have antimalarial, antiviral, antitumor, anti-inflammatory, or herbicidal properties.6 Compounds bearing the dithiocarbamate group as part of the heterocyclic structure have been relatively less studied7 and the developments of a simple MCR protocol for these compounds are a highly desirable yet elusive goal. Several methods have been documented for the synthesis of 1,3-thiazine derivatives. One such strategy involves a Michael type addition of N-aryldithiocarbamic acid to an enone, generated in situ to afford the corresponding Michael adduct which on subsequent ring transformation yields the final product.8 In addition 1,3-thiazines have also been synthesized utilizing racemic α-chloro-β,ν-alkenote esters with in situ generated dithiocarbamates.9
Although the reported approaches are useful tools for the synthesis of 1,3-thiazine, most of them suffer from limitations such as expensive reagents, drastic reaction conditions and multi-step syntheses. Hence developing a more convenient, efficient, rapid and viable synthetic protocol for these renowned molecules is highly desirable. Owing to its numerous advantages eco-friendly iodine has been explored as a powerful catalyst for various organic transformations.10 In our continuation of research on the catalytic application of molecular iodine,11 we herein report an iodine-catalyzed novel method to assemble β-naphthol, aromatic aldehydes, aromatic anilines and carbondisulfide to provide 1,3-thiazine. Notably, this reaction does not require harsh conditions and proceeds with a variety of aldehydes with fewer by products (Scheme 1).
 | ||
| Scheme 1 Reaction for the formation of substituted [1,3]thiazines from S-nucleophiles (generated in situ from aniline and CS2), β-naphthol and benzaldehydes. | ||
Results and discussion
In our effort to design a simple MCR for 1,3-thiazine, we devised a simple strategy derived reterosynthetically and outlined in Scheme 2. On the grounds of reterosynthetic analysis, we concluded that coupling of 1, 2, 3 and CS2 would lead to the desired 1,3-thiazine. This optimized strategy involves formation of four new σ-bonds and one asymmetric carbon center through a thia-Michael12 addition and intermolecular cyclisation process.13 The starting materials are readily available for the synthesis of many 1,3-thiazine derivatives. | ||
| Scheme 2 Reterosynthetic analysis for 1,4-diphenyl-1H-naphthol[2,1-d][1,3]thiazine-3(4H)thione leading to easily available starting materials. | ||
Accordingly, β-naphthol, aromatic aldehydes, CS2 and aniline were taken as the substrate for the reaction. Our investigation started with the reaction between 1, 2, 3 and CS2 with a variety of catalysts in different solvents as well as under solvent free conditions. Among the various Lewis acid catalysts used, iodine afforded the targeted product 4a with a 92% yield. Some of the other Lewis acids could not trigger the reaction even after reflux (Table 1). When we used SnCl2 as a catalyst for this reaction we observed a lower yield of 1,3-thiazine. In addition to that we observed an insoluble solid being formed which we later assumed was a complex with the aid of IR spectra. We have not thoroughly investigated this as it is out of the scope of this manuscript. Having iodine as a good promoter in hand, next we tried to optimize its loading, and it was found that 20 mol% of iodine provided the best result.
|
||||
|---|---|---|---|---|
| Entrya | Catalyst (mol%) | Solvent | Time (h) | Yieldb (%) |
| a Reaction conditions: all the reactions were carried out on 4 mmol scale in 3 ml solvent.b Isolated yield. | ||||
| 1 | Iodine (20) | DCM | 12 | 40 |
| 2 | Iodine (20) | 1,2-DCE | 10 | 33 |
| 3 | Iodine (20) | Toluene | 16 | — |
| 4 | Iodine (20) | Benzene | 16 | — |
| 5 | Iodine (20) | Acetonitrile | 4 | 52 |
| 6 | Iodine (20) | Acetonitrile–ethanol | 4 | 92 |
| 7 | Iodine (15) | Acetonitrile–ethanol | 4 | 60 |
| 8 | Iodine (10) | Acetonitrile–ethanol | 4 | 45 |
| 9 | Iodine (5) | Acetonitrile–ethanol | 4 | 30 |
| 10 | SnCl2 | Acetonitrile–ethanol | 4 | 37 |
| 11 | FeCl2 | Acetonitrile–ethanol | 4 | 20 |
| 12 | CuSO4 | Acetonitrile–ethanol | 4 | — |
| 13 | CuO | Acetonitrile–ethanol | 4 | — |
| 14 | CuBr | Acetonitrile–ethanol | 4 | — |
| 15 | ZnCl2 | Acetonitrile–ethanol | 4 | — |
Reducing the iodine loading from 20 mol% to 5 mol% led to a significant decrease in the yield of 4a. To study the effect of the solvent on the reaction, we performed the model reaction in different solvents such as DCM, 1,2-DCE, toluene, benzene and acetonitrile. Out of these solvents acetonitrile gave the best yield of 4a (Table 1, entries 1–5). But by using acetonitrile alone we could not get the desired product in significant yield due to the low solubility of phenyldithiocarbamate in acetonitrile. Hence we added ethanol in a smaller proportion to the reaction mixture in order to dissolve the phenyldithiocarbamate and this increased the yield significantly (entry 6). With the intention to investigate a green approach for this synthesis, we carried out this reaction in solvent free conditions as well, but the results were not satisfactory.
The overall observation shows that the reaction proceeds well in acetonitrile with 20 mol% iodine catalyst on reflux. Having optimized the reaction conditions we first examined the reaction of a number of substituted benzaldehydes with phenyldithiocarbamate and β-naphthol. Generally the reaction proceeded well with this substrate to deliver substituted 1,3-thiazine (4a–4q) in good to excellent yields (Table 2, entries 1–17). The results demonstrated that both the electronic features and the orientation of the benzaldehydes have a limited influence on this reaction. We also extended the scope of the reaction to various anilines and the results are tabulated (Table 2, entries 8–17). Another notable characteristic of this reaction is that a wide range of functional groups such as fluoro, chloro, nitro and methoxy remain intact under these reaction conditions. To further extend the utility of this tandem reaction we tried the same reaction with aliphatic amines such as ethylamine, piperidine and morpholine but the reaction did not proceed at all.
|
|||
|---|---|---|---|
| Entrya | 4 | Yieldb (%) | Time (h) |
| a Reaction conditions: all the reactions were carried out on 4 mmol scale in acetonitrile (7 ml)–ethanol (3 ml).b Isolated yield. | |||
| 1 | 4a: R1 = 2-Cl, R2 = H. | 92 | 4 |
| 2 | 4b: R1 = 4-OCH3, R2 = H. | 71 | 4 |
| 3 | 4c: R1 = 3-NO2, R2 = H. | 86 | 3 |
| 4 | 4d: R1 = 4-Cl, R2 = H. | 79 | 3 |
| 5 | 4e: R1 = 4-CN, R2 = H. | 75 | 3 |
| 6 | 4f: R1 = 2-F, R2 = H. | 80 | 4 |
| 7 | 4g: R1 = 4-CH3, R2 = H. | 69 | 5 |
| 8 | 4h: R1 = 3-NO2, R2 = 2-Cl. | 78 | 4 |
| 9 | 4i: R1 = 2-Cl, R2 = 2-Cl. | 85 | 4 |
| 10 | 4j: R1 = 4-F, R2 = 2-Cl. | 81 | 3 |
| 11 | 4k: R1 = 4-Cl, R2 = 2-Cl. | 76 | 4 |
| 12 | 4l: R1 = 2-F, R2 = 2-Cl. | 71 | 3 |
| 13 | 4m: R1 = 4-Br, R2 = 2-Cl. | 73 | 4 |
| 14 | 4n: R1 = H, R2 = 4-F | 72 | 2.5 |
| 15 | 4o: R1 = H, R2 = 4-Br | 86 | 3 |
| 16 | 4p: R1 = H, R2 = 4-Cl | 77 | 3.5 |
| 17 | 4q: R1 = H, R2 = 3-NO2 | 80 | 4 |

Mechanistically, it can be ascertained that the reactions proceed via the formation of an ortho-quinonemethide12 from β-naphthol and an aldehyde. The subsequent thia-Michael addition of phenyldithiocarbamate13 followed by the cyclodehydration through the nucleophilic amination of the phenolic group14 would give the desired 1,3-thiazine product (Scheme 3).
 | ||
| Scheme 3 Proposed mechanism of the iodine-catalysed synthesis of 1,3-thiazine. | ||
Conclusion
In conclusion, we have reported an efficient and simple protocol for the synthesis of biologically important 1,4-diphenyl-1H-naphthol[2,1-d][1,3]thiazine-3(4H)thione via a thia-Michael addition13 and cyclodehydration14 with molecular iodine as the catalyst. This method offers many advantages such as short a reaction time, significant yield and easy availability of the catalyst at a low cost. It was gratifying to observe that this approach does not require any activated aldehydes or anilines in order to accomplish the reaction.Experimental section
General methods
All reactions were performed under reflux conditions and open to air. Melting points were determined in open capillary tubes and are uncorrected. 1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were recorded in CDCl3. Chemical shifts are given in δ-values referenced to TMS. All the yields mentioned in the experimental data are of isolated products unless otherwise mentioned.General procedure for the synthesis of 1,3-thiazine
A mixture of 2-chlorobenzadehyde (4 mmol) and β-naphthol (4 mmol) was mixed in a 50 ml two necked round bottom (RB) flask containing acetonitrile (7 ml) as the solvent. To this reaction mixture 20 mol% iodine was added and refluxed. After 10 minutes a mixture of CS2 (4 mmol) and aniline (4 mmol) in 3 ml ethanol and KOH (8 mmol) was added to the RB flask. The reaction mixture was allowed to reflux for 4 h. The completion of the reaction was monitored using thin layer chromatography (TLC). After the completion of the reaction, the reaction mixture was quenched with a 20% solution of sodium thiosulfate and extracted with ethyl acetate. The combined organic layer was dried over sodium sulfate, concentrated using evaporation and the settled white product was dried. Some of the derivatives which are obtained as gels were purified by using column chromatography on silica gel (60–120 mesh, ethyl acetate–n-hexane).Acknowledgements
Munusamy Sathishkumar thanks the Vellore Institute of Technology, Tamilnadu, India, for providing a Research Associateship. The DST-FIST NMR facility at VIT University is greatly acknowledged.Notes and references
-
(a) Multicomponent reactions, ed. J. P. Zhu and H. Bienayme, Wily-VCH, Weinheim, Germany, 2005, p. 2005 Search PubMed
; (b) S. L. Schreiber, Target-Oriented and Diversity-Oriented Organic Synthesis in Drug Discovery, Science, 2000, 287, 1964 CrossRef CAS
-
(a) D. Xing, C. Jing, X. Li, H. Qiu and W. Hu, Org. Lett., 2013, 15, 3578 CrossRef CAS PubMed
-
(a) P. Slobbe, E. Ruiiter and R. V. A. Orru, Med. Chem. Commun., 2013, 3, 1189 RSC
- H. G. Hahn, H. K. Rhee, C. K. Lee and K. J. Whang, Arch. Pharmacal Res., 2000, 23, 315 CrossRef CAS
-
(a) J. F. Fisher, S. O. Meroueh and S. Mobashery, Chem. Rev., 2005, 105, 395 CrossRef CAS PubMed
-
(a) T. Tomasic and L. P. Masic, Curr. Med. Chem., 2009, 16, 1596 CrossRef CAS
-
(a) J. E. Jansen and R. A. Mathes, J. Am. Chem. Soc., 1955, 77, 2866 CrossRef CAS
-
(a) L. S. Yadav and V. K. Rai, Tetrahedron, 2006, 62, 8029 CrossRef CAS PubMed
- A. M. Jacobine and G. H. Posner, J. Org. Chem., 2011, 76, 8121 CrossRef CAS PubMed
-
(a) H. Togo and S. Iida, Synlett, 2006, 2159 CrossRef CAS PubMed
- G. Ramachandran, N. S. Karthikeyan, P. Giridharan and K. I. Sathiyanarayanan, Org. Biomol. Chem., 2012, 10, 5343 CAS
-
(a) M. S. Singh, S. Samai and G. Chandra Nandi, Tetrahedron, 2012, 68, 1247 CrossRef PubMed
- K. Praveen Kumar, S. Satyanarayana, P. Lakshmi Reddy, G. Narasimhulu, N. Ravirala and B. V. Subba Reddy, Tetrahedron Lett., 2012, 53, 1738 CrossRef CAS PubMed
- R. S. Dowing, P. J. Kunkeler and H. van Bekkum, Catal. Today, 1997, 37, 121 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ra45454a |
| This journal is © The Royal Society of Chemistry 2014 |