
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular design, synthesis, properties, and applications of organic triplet emitters exhibiting blue, green, red and white room-temperature phosphorescence
Mariia
Stanitska
 a,
Dmytro
Volyniuk
a,
Boris
Minaev
bc,
Hans
Agren
c and
Juozas V.
Grazulevicius
*a
a,
Dmytro
Volyniuk
a,
Boris
Minaev
bc,
Hans
Agren
c and
Juozas V.
Grazulevicius
*a
aDepartment of Polymer Chemistry and Technology, Kaunas University of Technology, Barsausko 59, LT-51423, Kaunas, Lithuania. E-mail: juozas.grazulevicius@ktu.lt
bBohdan Khmelnytsky National University of Cherkasy, Ukraine
cDepartment of Physics and Astronomy, Uppsala University, SE-751 20 Uppsala, Sweden
First published on 12th January 2024
Abstract
At room temperature, many organic compounds can emit light not only from singlet excited states but also from triplet excited states. This phenomenon is known as room-temperature phosphorescence (RTP). RTP has numerous potential applications in various areas, including industry, medicine, and daily life. RTP materials can be used in white light-emitting, memorising, sensing, bioimaging, semiconducting, 3D printing, microfluidic visualisation, quantum computing, X-ray detection, etc. They have advantages over fluorescent and metal-based phosphorescent materials. They are characterized by different emission spectra, longer lifetimes, and higher environmental sensitivity. However, challenges related to the tuning of emission colours, realization of dual emission, and meeting application-specific requirements still need to be overcome. Various molecular design strategies and synthetic approaches have been developed to overcome these challenges and to develop efficient RTP materials. This review article discusses recent advances in the synthesis and properties of RTP materials, focusing on emission colours and applications.
1. Introduction
With exponentially accelerating technological progress, the demand from humanity for efficient electroactive materials is rising since most modern electronic technologies that operate on integrated circuits require the utilization of semiconductors. A significant portion of semiconductors produced is employed in light-emitting devices, such as light-emitting diodes (LEDs) that have been proven to be the paramount light-generators in comparison to the alternative lighting sources. However, the most effective semiconductive materials up-to-date are inorganic, mainly consisting of silicon, germanium and binary compounds such as gallium arsenide.1 The fabrication of semiconductors involves the usage of hazardous substances, e.g., phosphorus, antimony, and arsenic.2 This poses a bunch of challenges for researchers such as achieving high device performance by replacing the inorganic constituents of LEDs with organic ones, while minimizing environmental risks.Researchers around the world try to make organic electroactive compounds compatible with inorganic ones in terms of efficiency. The main problem with organic light-emitting materials arises from spin-statistics: in simple fluorescent materials, emission can occur from only 25% of the formed excited states. The appealing solution to the problem was introduced by Forrest et al. in 19983 when they employed a platinum-centred phosphorescent organic complex in an electroluminescent device, thus taking a giant leap forward in lighting technology. Phosphorescent complexes usually consist of organic ligands that are coordinated by central heavy metal atoms, such as iridium, copper, platinum, etc. The presence of a transition metal makes phosphorescent emitters advantageous, as it enables the harvesting of 100% of the formed excited states. Strong spin–orbit coupling that is induced by a heavy metal atom facilitates intersystem crossing and promotes the harvesting of triplet excitons that are usually non-emissive in pure organic compounds. However, the presence of transition metal atoms in the molecules of emitters is quintessential and, at the same time, the most unwanted. Heavy metals such as iridium are expensive. Most of them, including copper and chromium, are environmentally unfriendly to the utmost degree. Once a sufficient amount of a compound containing such a metal is released into the environment, it disrupts the natural synthetic pathways of many bioactive compounds due to the strong complexing ability of the metal.3 Thus, the development of organic compounds with the ability to “activate” dark triplet excitons has appeared to be the major focus of research.
Thermally activated delayed fluorescence (TADF) allows gathering 75% of “dark” triplet excitons via non-radiative internal conversion of triplet excited states to singlet ones (reverse intersystem crossing4). However, this is not the only way to exploit triplet excitons in pure organic compounds. Room-temperature phosphorescence (further abbreviated as RTP) is another excellent mechanism of complete utilization of electrogenerated excited states.5
When discussing RTP and TADF, it is important to differentiate between the various interpretations of the phosphorescence and luminescence phenomena. Throughout history, the term “phosphorescence” has been used to describe several mechanisms with distinct physical origins. The word “phosphorescence” comprises a distinction between long-lasting afterglow and fast emission (fluorescence) in a general optical sense. In such a phenomenological context, the term “phosphorescence” is still often used to describe different mechanistic photo-processes in adjacent fields of physics and chemistry. The distinction based on the lifetime of the fast and slow emission in solid-state physics concerning the luminescence of inorganic and organic materials leads to the “phosphorescence” notion assignment to electronic mechanisms of a completely different physical nature.6 The long-lasting emission of inorganic solids is related to the trapping of excitons by lattice impurities, while organic RTP and TADF of organic compounds are determined by spin selection of spontaneous emissive and non-radiative processes. Organic RTP as well as a more general low-temperature phosphorescence phenomenon refer to the radiative transition from the first excited triplet (T1) state to the ground singlet (S0) state of a photo-chemically stable molecule. This spin-forbidden T1 → S0 triplet–singlet emission typically has a lower transition moment and a rate constant ca. a million times lower than that of the spin-allowed S1–S0 transition, i.e., fluorescence.
The growing research interest in organic compounds exhibiting RTP is depicted in Fig. 1a. Although TADF still remains the paramount strategy to utilize triplet excitons that has resulted in a number of scientific publications exceeding those related to RTP, the progress in the investigations of the RTP phenomenon is more rapid. For example, in 2019 RTP publications covered barely one-third of the number of papers about emission from triplet excitons whereas in 2020, this number increased up to one half. It is evident from Fig. 1b that in 2020 and in 2021, the number of publications on TADF increased by 1.09 and 1.16 times, respectively, while the number of articles on RTP increased by 1.5 times (here dividing the number of publications from the current year by the number from the previous year).
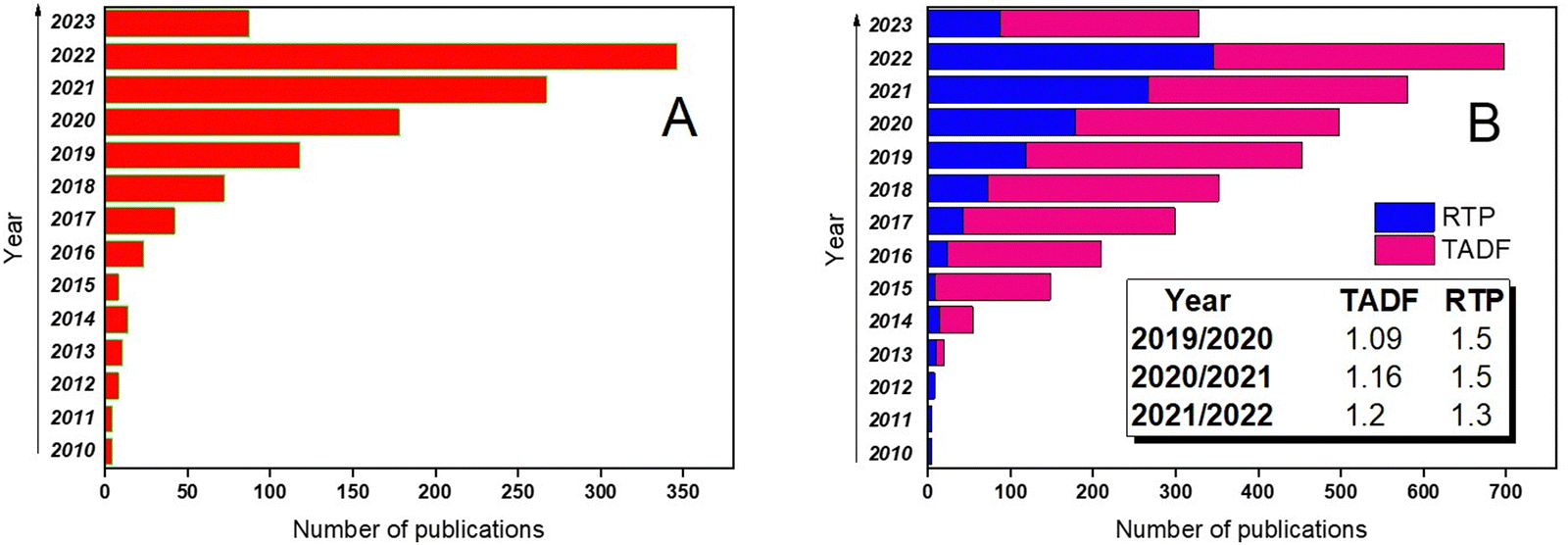 | ||
| Fig. 1 (a) Progress in RTP scientific papers in the 2010–2023 timeframe and (b) comparative progress in TADF and RTP scientific papers in the same timeframe. Adapted from the Scopus database. | ||
There are review articles on organic materials exhibiting RTP either covering a wide range of structural peculiarities and utility areas, or discussing narrow topics such as the work of metal-free polymer-based materials,7 chiral organic materials8 and co-crystals exhibiting RTP,9 as well as boron-containing RTP compounds.10 Despite the fact that the emission colours of organic compounds exhibiting RTP can significantly impact their potential applications, there is a lack of literature analysing and classifying them based on the emission colours. In this review, we have categorized the latest and most effective organic emitters exhibiting RTP based on the colour of their phosphorescence. Only low-molar-mass purely organic RTP compounds are discussed, excluding polymers, metalorganic complexes and quantum dots. We cover both theoretical and experimental approaches used for the development of organic RTP compounds, exhibiting blue, green, red, and white emissions. We also briefly discuss their potential applications.
2. Theory of room-temperature phosphorescence
The theoretical analysis of phosphorescence requires adiabatic calculations of the S and T excited states’ structure and electronic wave functions with an obligatory account of internal magnetic interactions (spin–orbit coupling, SOC). Very often, it is sufficient to account for SOC through perturbation theory,6,12 at least for the T1 → S0 spontaneous emission lifetime estimation. For calculations involving non-radiative transition rates and quenching channels, the electronic–vibrational and spin–vibronic couplings should be taken into account as additional perturbations. The quadratic response (QR) formalism in terms of the Dalton code11 is often used now in quantum-chemical calculations of phosphorescence.6 The phosphorescent T1–S0 transition can acquire the dipole activity by “intensity borrowing” from the spin-allowed S0–Sn and T1–Tm transitions calculated under the general nonrelativistic Born–Oppenheimer approximation6via spin–orbit coupling perturbation.The RTP definition being applied until recent time resembles a “delayed luminescence that persists after the removal of the excitation source”.6 But according to the modern IUPAC rule, a significant number of processes of delayed luminescence of inorganic metal salt semiconductors should be excluded from our scope, even though they are considered originally as producing real genuine phosphorescence.11 Several luminescence delay processes can follow the accumulation of the photochemical reaction products, thus tracking the chemical kinetics of photo-transformations. This luminescence delay could be connected with exciton migration between traps in crystals and does not depend on spin prohibition for optical transition. The white phosphorus in the moist air can be slowly oxidized to make it luminescent; such long-persistent emission erroneously contributed to the origin of the term “phosphorescence”, although it has nothing in common with the modern RTP notion. Thus, the quite general RTP phenomenon of molecular spectroscopy was fallaciously connected with the particular inorganic reaction of slowly oxidized and glowing white phosphorus. In doped metal sulphides and some heated minerals, the electron–hole recombination in the p–n junctions of these semiconductors, which leads to long-lasting emission, has nothing in common with molecular phosphorescence.
According to the modified Jablonsky diagram (Fig. 2a), the lowest-lying triplet excited molecular state (T1) could be populated through the sequence of the following processes:
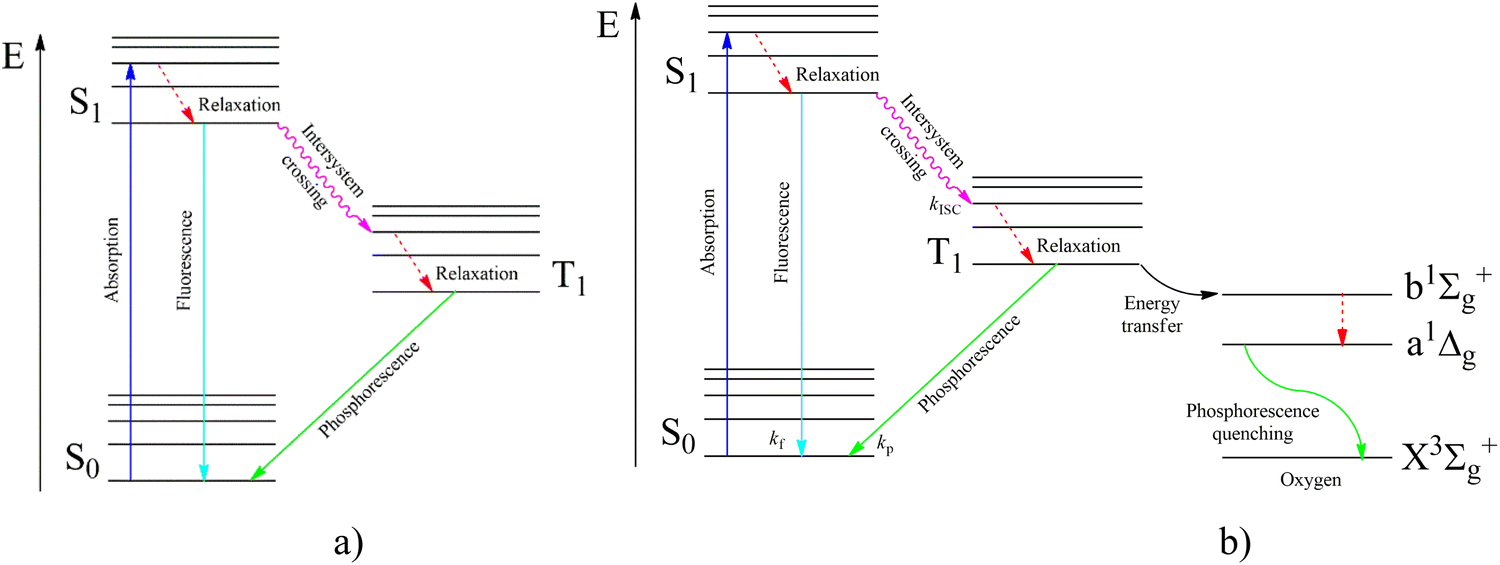 | ||
| Fig. 2 Jablonsky diagram of fluorophores in an inert atmosphere (a) and in the presence of oxygen (b). | ||
(1) the starting incident UV light absorption (S0 → Sk) leads to the resonance excitation of a highly excited Sk state followed by
(2) a fast internal conversion (IC) to the lowest singlet excited state (S1) with the rate constant (kIC) which is typically high (kIC > 1012 s−1) for numerous fluorophores; the kIC rate constant usually follows the so-called Kasha rule, which posits that fluorescence emission (S* → S0) more frequently originates from the S1 state;
(3) additionally, the nonradiative transition S1∼ → T1 called intersystem crossing (ISC) produces the T1 state population with the kISC rate constant;
(4) the ISC between the higher-lying singlet (Sk) and triplet (Tn) states followed by the subsequent internal conversion (Tn–T1) with a high kIC rate constant in some molecules could also lead to the T1 state population and to the subsequent phosphorescent emission.
(5) the fluorescence channel (S0 → Sk) with the rate constant (kf) is the main radiative process that competes with the S1∼ → T1 intersystem crossing kISC rate of relaxation leading to T1 state production.
(6) besides such T1 triplet state optical pumping, the lowest triplet can be produced by an electric current through an organic semiconductor solid as a result of electron–hole recombination in specially fabricated organic light-emitting diodes (OLEDs).
(7) after the optical or electrical T1 state population, the nonradiative T1∼ → S0 relaxation (knr) rate strongly competes with the phosphorescent T1 → S0 radiation (kp) being dependent on temperature, crystal packing and vibrational modes.11 The T1 → S0 emissive transition is spin forbidden according to the electric dipole selection rule that renders the phosphorescence process less probable. This makes phosphorescence less intense than the fast spin-allowed fluorescent emission in pure organic fluorophores. For the nonradiative ISC relaxation T1∼ → S0, the electron–vibrational coupling plays a much more crucial role than for spontaneous phosphorescent light emission. The late ISC process strongly depends on the temperature and aggregation form of the environment (much more temperature dependent than the S1∼ → T1 intersystem crossing having a smaller energy gap). Thus, for the ISC relaxation T1∼ → S0 process the spin prohibition is not as severe as for light emission being of a different physical origin. Spin prohibition is based on Pauli's exclusion principle and the ISC rate still follows the general quantum mechanical trend and obeys the spin conservation law. Apparently, an additional SOC perturbation is necessary in both processes to violate spin selection since SOC induces S–T mixing. Non-radiative processes are more dependent on the rigidity of a large molecular environment around the particular phosphorescent chromophore and the vibrational activity of many degree of freedom including many phonons. Radiative transitions are “more localized” depending on the molecular symmetry point group and local electronic wave functions. Spin–lattice relaxation and ISC processes provide vibrational energy flow and embrace many structural variables. Much more spin–vibronic perturbations are involved in ISC calculations than in radiative T–S transition calculations; thus, the selection rules for ISC relaxation are less severe. Both “activating” and “promoting” vibrational modes participate in the non-radiative energy flow during ISC processes. Taking them into account, one can see much greater possibility to add new SOC perturbations in order to reduce restrictions in the spin selection rule for the ISC transitions. SOC perturbations for radiative T–S transitions are much more limited by the smaller number of electronic and vibrational parameters.
In Fig. 2b, we present the influence of dissolved molecular oxygen on the photophysics of organic luminophores which shows blue or green phosphorescence; the T1 state energy is larger than 1.63 eV, which is the second singlet state energy level of oxygen O2(1Σ+g). The first excited singlet oxygen state O2(1Δg) level lies 0.98 eV above the ground triplet state O2(3Σ−g). Paramagnetic triplet oxygen in the ground O2(3Σ−g) state can effectively quench phosphorescence by colliding with the triplet-excited T1 organic molecule in a solvent; the process is limited by diffusion since it is not forbidden by the spin selection rule. The energy transfer to the singlet excited O2(1Σ+g) oxygen with simultaneous T1∼ → S0 quenching of the luminophore is a spin-allowed process
| T1 (↑↑) + O2(3Σ−g) (↓↓) → S0 (↑↓) + O2(1Σ+g) (↑↓) |
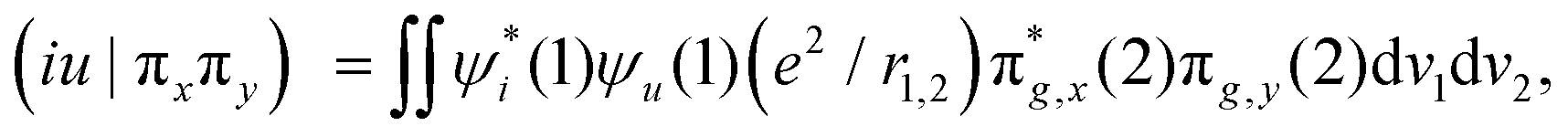 | (1) |
The singlet–singlet internal relaxation in oxygen O2(1Σ+g) → O2(1Δg) is very fast and UV excitation of the luminophore often leads to the singlet oxygen O2(1Δg) production which being very reactive can oxidize the singlet S0 ground-state of organic molecules. In contrast to atmospheric oxygen O2(3Σ−g), which shows sluggish chemical reactivity in the absence of fire (and radicals), the singlet oxygen O2(1Δg) molecule has no spin restrictions for interaction with diamagnetic organic substances.
Spin quantization and theoretical backgrounds of the Jablonsky diagram
All-electronic wave functions as solutions of the nonrelativistic Schrödinger equation for a molecule under Born–Oppenheimer approximation should be the eigen-functions of the total spin operator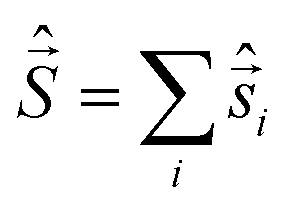 , where
, where 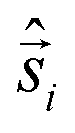 represents the single-electron Pauli matrices for the ith electron.5
represents the single-electron Pauli matrices for the ith electron.5
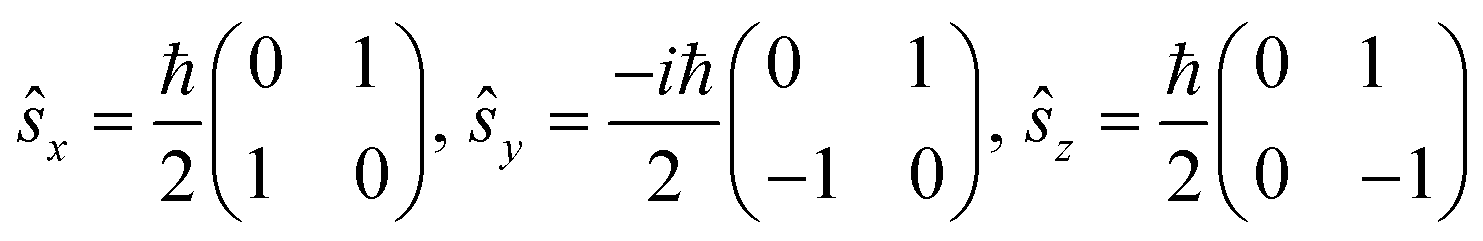 The single-electron Pauli functions
The single-electron Pauli functions 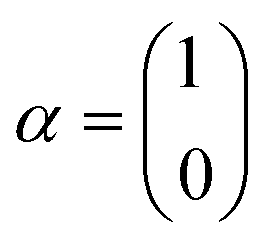 and
and 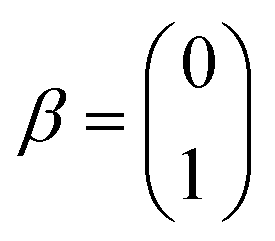 are eigen-functions of the ŝz and
are eigen-functions of the ŝz and 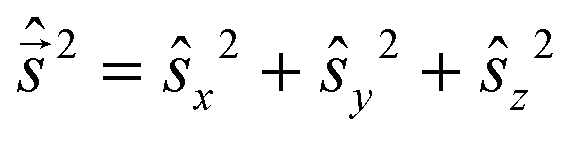 operators. In a many-electron molecule, the total wave function satisfies the following equations for the total spin
operators. In a many-electron molecule, the total wave function satisfies the following equations for the total spin 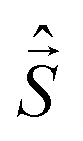 operator (of its square and z-projection operators)
operator (of its square and z-projection operators)
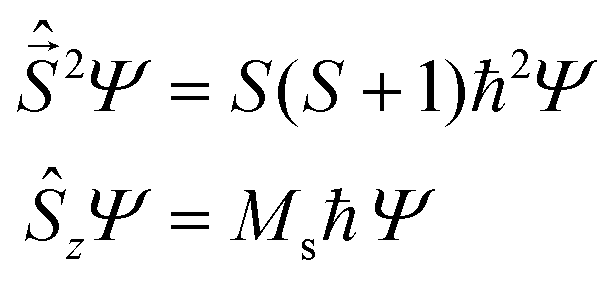 | (2) |
 | (3) |
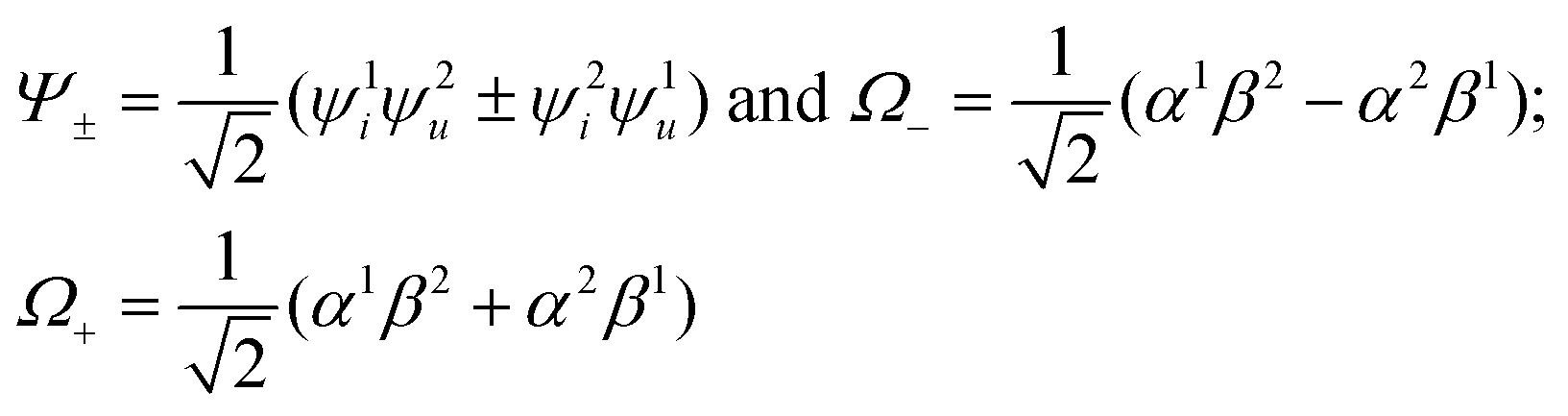 | (4) |
In the framework of the electric-dipole approximation, the S–T transitions are strictly forbidden since the transition dipole moment is equal to
| MS–T = 〈Ψ+Ω−|M|Ψ−Ω+〉 = 0. | (5) |
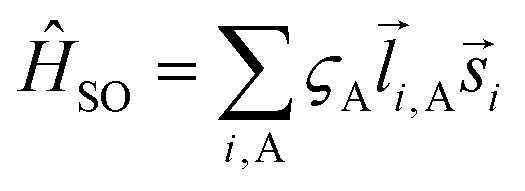 . The wave function for two electrons can be presented in the following form:
. The wave function for two electrons can be presented in the following form: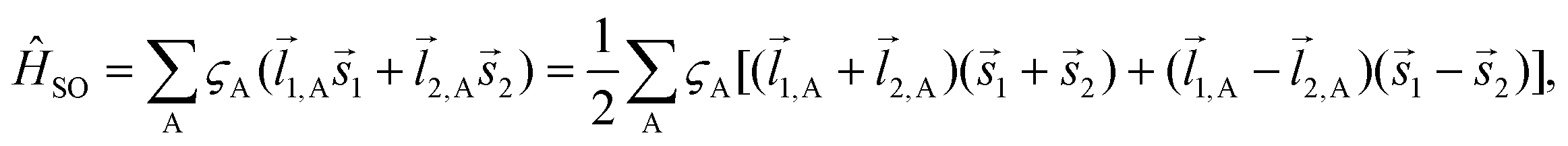 | (6) |
![[l with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_006c_20d1.gif) i,A,
i,A,![[s with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0073_20d1.gif) i represent the orbital and spin angular momenta for the i-th electron, respectively. Eqn (6) includes artificial separation of the SOC operator into two parts: one being symmetrical with respect to permutation of two electrons, which can mix triplet states, and the other – is antisymmetric. The latter can mix S0 and T(nπ*) states, each including different permutation symmetry for their orbital and spin functions. Thus, the SOC matrix element (ME) 〈1ΨS|ĤSO|3ΨT〉 is proportional to a single-electron orbital integral
i represent the orbital and spin angular momenta for the i-th electron, respectively. Eqn (6) includes artificial separation of the SOC operator into two parts: one being symmetrical with respect to permutation of two electrons, which can mix triplet states, and the other – is antisymmetric. The latter can mix S0 and T(nπ*) states, each including different permutation symmetry for their orbital and spin functions. Thus, the SOC matrix element (ME) 〈1ΨS|ĤSO|3ΨT〉 is proportional to a single-electron orbital integral 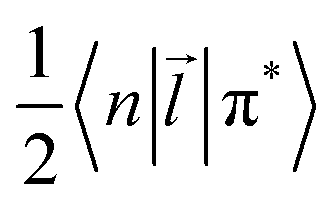 between two molecular orbitals. Since the operator produces rotation of the π* MO in the direction of the orthogonal n-orbital, the SOC ME is nonzero and includes large contribution from the heteroatom (the owner of the lone pare) which typically possesses larger internal SOC ςA constants than carbon atoms. That is why the dyes with a low-lying 3(nπ*) triplet state provide more intense phosphorescence than pure hydrocarbons. This is also a background for the well-known El-Sayed rule.5 The SOC ME determines a mixture of the lowest singlet and triplet states according to perturbation theory
between two molecular orbitals. Since the operator produces rotation of the π* MO in the direction of the orthogonal n-orbital, the SOC ME is nonzero and includes large contribution from the heteroatom (the owner of the lone pare) which typically possesses larger internal SOC ςA constants than carbon atoms. That is why the dyes with a low-lying 3(nπ*) triplet state provide more intense phosphorescence than pure hydrocarbons. This is also a background for the well-known El-Sayed rule.5 The SOC ME determines a mixture of the lowest singlet and triplet states according to perturbation theory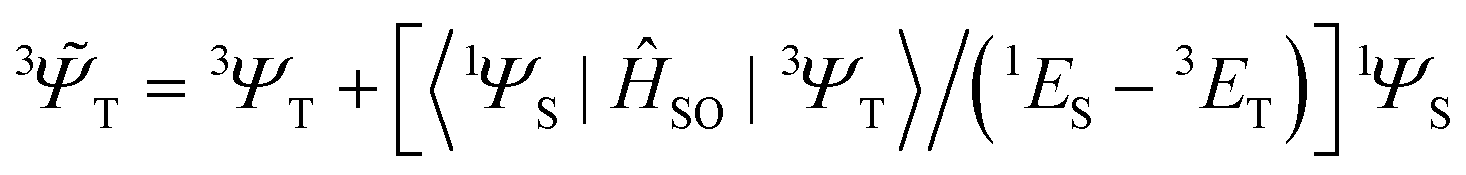 | (7) |
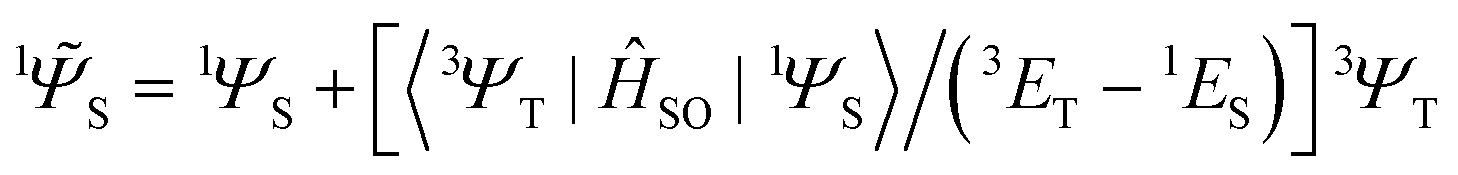 | (8) |
![[capital Psi, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e11c.gif) determines the perturbed wave functions accounting for SOC perturbation. Since 〈1ΨS|ĤSO|3ΨT〉* = 〈3ΨT|ĤSO|1ΨS〉 the electric-dipole transition moment (EDTM) between these states is equal
determines the perturbed wave functions accounting for SOC perturbation. Since 〈1ΨS|ĤSO|3ΨT〉* = 〈3ΨT|ĤSO|1ΨS〉 the electric-dipole transition moment (EDTM) between these states is equal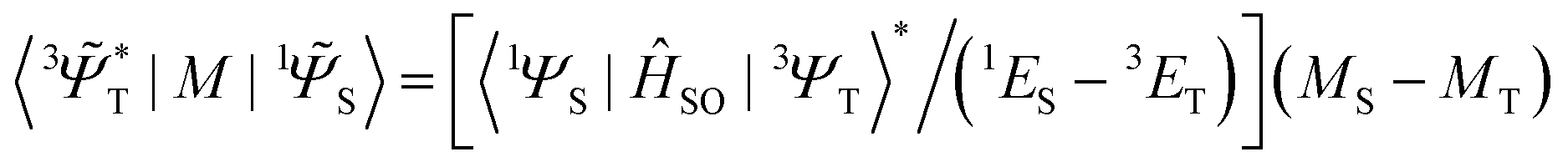 | (9) |
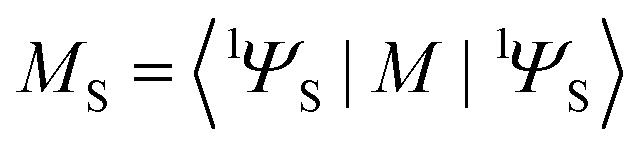 is the molecular ground state dipole moment. Besides this T1–S0 state mixing, eqn (7) and (8), all other closely lying Tn and Sm states can participate in similar SOC perturbations;11 this will lead to the T1 → S0 intensity borrowing from the EDTM-allowed S0–Sm and T1–Tn transitions. The S0 → Sm transitions are usually well-known from the absorption spectra; thus, the RTP properties are often discussed in terms of their S–S intensity borrowing. However, direct ab initio calculations often indicate the importance of the contributions from the T1–Tn transitions to the RTP rate constant.6,12
is the molecular ground state dipole moment. Besides this T1–S0 state mixing, eqn (7) and (8), all other closely lying Tn and Sm states can participate in similar SOC perturbations;11 this will lead to the T1 → S0 intensity borrowing from the EDTM-allowed S0–Sm and T1–Tn transitions. The S0 → Sm transitions are usually well-known from the absorption spectra; thus, the RTP properties are often discussed in terms of their S–S intensity borrowing. However, direct ab initio calculations often indicate the importance of the contributions from the T1–Tn transitions to the RTP rate constant.6,12
The larger the SOC ME and the smaller the energy gaps in eqn (7)–(9), the more intense the RTP intensity and the kp rate. Adachi et al. have proposed that the T1–S1 energy gap can effectively regulate the TADF when the small T1–S1 gap is comparable with the SOC ME integral.13 In such case, the T1 ← S1 reverse intersystem crossing (rISC) could be very fast and compete with the RTP, thus providing the TADF radiation.4,13 The temperature dependence of the rISC rate and RTP intensity really indicates the TADF importance for specially synthesized chromophores with a charge transfer nature of the T1–S1 states and very low energy gaps.12
Comparison of singlet oxygen and organic phosphorescence
The singlet oxygen lifetime in solvents is comparable with that of organic RTP. The singlet oxygen emission O2(1Δg) → O2(3Σ−g) ranging 1270–1060 nm represents the reverse phosphorescence according to the IUPAC definition6 (since it is a transition between states of different multiplicity, S1 → T0). Moreover, it is observed as weak RTP in solvents and matrices.6,12 It is easy to show that SOC ME between triplet O2(3Σ−g) and singlet O2(1Δg) states is equal to zero and the singlet oxygen relaxation is strictly forbidden in the electric dipole approach.6,12 Both the states O2(3Σ−g) and O2(1Δg) differ by the orbital angular momentum projection (ΔΛ = 2); this selection rule strictly forbids their SOC mixing12 and EDTM activity. The O2(1Δg) state radiative rate constant kp in a free O2 molecule is determined by a weak magnetic-dipole transition and its lifetime exceeds an hour. In contrast, the non-radiative ISC rate O2(1Δg) → O2(3Σ−g) in organic solvents is rather fast and provides a τΔ lifetime in the μs11 range. The radiative lifetime of oxygen phosphorescence 1Δg → 3Σ−g is much longer in any solvent, even though this emission acquires an electric-dipole nature in the condensed phase.6 The organic RTP quenching by O2 is often accompanied by the generation of singlet oxygen. The observed τΔ lifetime strongly depends on SOC between O2(3Σ−g) and O2(1Σ+g) states and on charge-transfer interactions with the nearest environment.6 One can see that the RTP quenching problem by environmental temperature-factors and the τΔ lifetime theory11 both depend on the fine tuning between intramolecular and intermolecular SOC effects including spin–vibronic interactions and exciton and phonon dynamics in the vicinity of the RTP emitter. Because of this, the corresponding lifetimes are strongly reduced depending on the environment. Besides the kp rate constant, the origin of RTP depends on other parameters connected with knr, kISC, and krISC and other photophysical processes rates which will be discussed below.3. Blue RTP emitters
The first sources of blue lasting emission for colour TV pictures were developed with the implementation of noble metal atoms. The internal atomic SOC ςA constants for heavy metals (A = Pt, Ir, and Au) exceed those for the first-row elements by two orders of magnitude; thus, the SOC ME integrals (6)–(9) in such metal–organic complexes with pronounced “metal-to-ligand charge transfer effects” provide strong mixing of S–T states and efficient RTP with short lifetime.6 The kp rate constant of RTP depends on the square of the SOC ME value; thus, the kp rate is four orders of magnitude higher for metal complexes. The development of organic RTP materials containing no metal atoms that emit light in the short-wavelength region of the electromagnetic spectrum (with emission wavelengths up to 475 nm) is one of the most complicated tasks, because of the low kp rate. After the discovery of the TADF phenomenon, researchers have encountered several problems related to the blue TADF emitters. For a long time, researchers could not overcome the problem of 20% external quantum efficiency for OLEDs based on blue TADF emitters. Such devices suffered from short operation lifetimes and colour aging that were caused by lower photostability of blue TADF-emitters compared to their green and red counterparts.13 The major problem with blue RTP materials is that phosphorescence is usually red-shifted in comparison to fluorescence because the triplet-excited states usually have lower energy compared to the S1 excited state, following the molecular analog of the Hund rule.6 The S1–T1 energy gap of the ψi → ψu excitation is determined by the 2(iu|iu) expression, being twice the exchange integral between the HOMO and LUMO, eqn (1). In order to achieve blue RTP, the organic compounds have to possess triplet excitation energy levels of ca. 3 eV. High-lying triplets have a tendency to populate singlet excited states via reverse intersystem crossing resulting in TADF.4 To avoid complete triplet upconversion and to afford RTP for a single compound kISC should be larger than krISC. The non-radiative loss of triplet excitation states can be prevented by embedding a luminophore into a rigid polymer matrix to reduce the non-radiative channels of energy dissipation or by crystallization of the luminophore.14The latter concept was successfully implemented by Han et al.15 They reported N-phenyl-substituted 9,10-dihydro-9,9-dimethylacridine B1 (Fig. 5 and Table 1), which exhibits persistent luminescence in the blue area of the spectrum. Compared to its methyl and methoxy-containing analogues (G1 and G2, Fig. 11 and Table 2) that showed green RTP, fluorescence and RTP maxima of B1 were blue-shifted to 351 nm and 475 nm, respectively. The long-lived RTP of B1 was explained by the peculiarities of the crystal structure. When two phenyl rings of triphenylamine are intramolecularly locked via an sp3-hybridized carbon atom to afford N-phenyl-9,10-dihydro-9,9-dimethylacridine, the target compound exhibits a twisted conformation with a torsion angle of ca. 90° between acridine and the unlocked phenyl ring. DFT calculations revealed rather weak overlap between the HOMO and the LUMO of B1 that leads to a low ΔEST gap, thereby promoting intersystem crossing.15 X-ray analysis performed for B1 single crystals revealed the presence of intermolecular interactions of different types such as C⋯H and C⋯NH, whereas no π–π stacking was observed. In the case of methyl and methoxy-containing counterparts such interactions are more abundant, which sufficiently prolong the RTP lifetime compared to B1.
| Compound | λ FL, nm | τ PL, ns | FLQY, % | λ RTP, nm | τ RTP, ms | PHQY, % | Host | Ref. |
|---|---|---|---|---|---|---|---|---|
| λ FL – the wavelength of fluorescence maxima, τPL – fluorescence lifetime, FLQY – fluorescence quantum yield, λRTP – the wavelength of room temperature phosphorescence maxima, τRTP – RTP lifetime, PHQY – phosphorescence quantum yield, and gr. – ground. | ||||||||
| B1 | 351 | — | 13.47 | 475 | 30 | 2.26 | — | 15 |
| B2 | 387 | 0.89 | 4.2 | 460 | 542 | 11.7 | PMMA | 16 |
| B3-1 | 373 | 3.82 | 0.9 | 467 | 3.5 | 9.1 | — | 17 |
| B3-2 | 378 | 1.62 | 3 | 467 | 2.7 | 6 | — | 17 |
| B3 gr. | 334 | 1.6 | 7.6 | 467 | 2.1 | 1.4 | — | 17 |
| B4 | 392 | — | — | 403 | 51.96 | — | — | 18 |
| B5 | 338 | — | — | 471 | 1116.10 | — | — | 18 |
| B6 | 379 | — | — | 409 | 60.75 | — | — | 18 |
| B7 | 351 | — | — | 471 | 249.43 | — | — | 18 |
| B8 | — | — | — | 465 | 580 | — | — | 19 |
| B9 | 390, 410 | 4.46 | — | 434, 464 | 760 | — | PVA | 20 |
| B10 | 405 | 5.44 | — | 434, 464, 494 | 87 | — | PVA | 20 |
| Compound | λ FL, nm | τ PL, ns | FLQY, % | λ RTP, nm | τ RTP, ms | PHQY, % | Host | Ref. |
|---|---|---|---|---|---|---|---|---|
| λ FL – the wavelength of fluorescence maxima, τPL – fluorescence lifetime, FLQY – fluorescence quantum yield, λRTP – the wavelength of room temperature phosphorescence maxima, τRTP – RTP lifetime, and PHQY – phosphorescence quantum yield.a Under ambient conditions.b At a pressure of 4.15 GPa.c After 30 s of 365 nm UV irradiation, and cr. – crystal. | ||||||||
| G1 | 361 | — | 20.8 | 529 | 28 | 2.0 | — | 15 |
| G2 | 386 | — | 83.5 | 508 | 18 | — | — | 15 |
| G3 | 363 | 4.73 | — | 489 | 517 | 7.7 | PVA | 22 |
| G4 | 491 | 10.19 | — | 502 | 229 | 19.6 | PVA | 22 |
| G5 | 495 | 12.57 | — | 494 | 222 | 10.7 | PVA | 22 |
| G6 | — | — | — | 515 | 32 | 25.6 | DIPHOS | 23 |
| G7 | 447 | — | — | 510a | 6.92 | 4.80 | — | 26 |
| G7 | — | — | — | 567b | 4.24 | — | — | 26 |
| G8 | 453 | 1.08 | 3.8 | 500 | 1.62 | 44.3 | PS | 27 |
| G8cr | 483 | 0.79 | 2.1 | 543 | 0.016 | 2.9 | — | 27 |
| G9 | 441 | 1.35 | 11.8 | 487 | 10.38 | 10.1 | PS | 27 |
| G9cr | 472 | 1.21 | 7.6 | 529 | 0.61 | 21.7 | — | 27 |
| G10 | 424 | 1.91 | 0.5 | 497 | 3.09 | 66.7 | PS | 37 |
| G11 | — | — | — | 536 | 0.67 | 7.7 | — | 28 |
| G12 | — | — | — | — | — | 0 | — | 28 |
| G13 | 393 | 1.79 | 21.3 | 486, 514 | 730, 747 | 10.6 | PMMA | 29 |
| G14 | 380 | 1.34 | 14.2 | 485, 518 | 822, 760 | 11.6 | PMMA | 29 |
| G15 | 358 | 3.28 | 29.4 | 510, 545 | 340, 284 | 7.4 | PMMA | 29 |
| G16 | 387 | 1.08 | 7.5 | 477 | 818 | 20.2 | PMMA | 16 |
| G17 | 409 | 1.97 | 8.5 | 480 | 380 | 15.6 | PMMA | 16 |
| G18 | 450 | 2.37 | 7.3 | 499 | 87 | 16.8 | PMMA | 16 |
| G19 | 430 | 7.1 | 16.3 | 476 | 96 | — | — | 30 |
| G20 | 455 | 3.26 | 5.9 | 466, 539 | 125, 136 | — | — | 30 |
| G21 | 423 | 1.03 | 2.9 | — | — | — | — | 30 |
| G22 | — | 6.63 | 3.01 | 516 | 98 | 0.37 | OPPh3 | 30 |
| G23 | — | 4.95 | 4.45 | 510 | 980 | 3.33 | OPPh3 | 30 |
| G24 | — | 6.01 | 5.09 | 515 | 334 | 1.13 | OPPh3 | 30 |
| G25 | 360 | 2.66 | 3.29 | 508 | 339 | 0.73 | OPPh3 | 30 |
| G26 | 393 | 2.07 | 6.35 | 512 | 907 | 4.48 | OPPh3 | 30 |
| G27 | 397 | 2.29 | 2.18 | 516 | 464 | 0.24 | OPPh3 | 30 |
| G28 | 440 | 3.24 | — | 516 | 32a | 11a | PMMA | 32 |
| G29 | 440 | 3.23 | — | 516 | 25a | 20a | PMMA | 32 |
| G30 | 440 | 3.15 | — | 516 | 30a | 22c | PMMA | 32 |
| G31 | 563 | 10.71 | 30.2 | 530 | 1.06, 5.48 | 3.8 | PMMA | 33 |
| 2 × 104 | ||||||||
| G31 | 590 | 25.96 | 14.7 | 532 | 2.03, 19.53 | 3.3 | DPEPO | 33 |
| 3.36 × 103 | ||||||||
| G32 | 558 | 11.33 | 25.5 | 530 | 1.1, 5.5 | 2.5 | PMMA | 33 |
| 12.82 | ||||||||
| G32 | 584 | 23.51 | 16.4 | 532 | 4.05, 23.07 | 3.6 | DPEPO | 33 |
| 3.69 × 104 | ||||||||
| G33 | 453 | — | 3 | 551 | 16.6 | 54 | Zeonex | 34 |
| G34 | 456 | 11.81 | — | 533 | 0.001 | — | Zeonex | 35 |
| G35 | — | — | — | 545 | 80.4 | — | H35 | 36 |
| G36 | — | — | — | 550 | 3.85 | — | H36 | 36 |
| G37 | — | — | — | 550 | 59.44 | — | H37 | 36 |
Deng et al. managed to achieve blue RTP by incorporating the phenyl fragment into the sulfone-locked triphenylamine heteroaromatic core.16 Thorough investigations disclosed that the introduction of the phenyl fragment results in the rearrangement of its excited state energy levels thus providing more efficient ISC and strengthened SOC between the triplet excited states and first singlet excited state S1. Embedding 5 wt% of compound B2 (Fig. 5 and Table 1) into a PMMA matrix resulted in the cessation of the non-radiative relaxation of triplex excitons. Steady-state PL spectra revealed a peak at 387 nm, which was attributed to fluorescence whereas RTP peaked at 460 nm with a τRTP of 542 ms. CIE colour coordinates of blue afterglow were (0.16, 0.27).
Xie et al. reported phase-dependent RTP of very simple bromine-containing biphenyl derivative B3 ((Fig. 5 and Table 1)).17 The authors managed to obtain two crystalline forms of compound B3. High-quality crystals of B3-1 were obtained by vacuum gradient sublimation and low-quality crystals of B3-2 were grown by quick solvent evaporation. The PL spectra of two crystalline forms revealed two peaks around 370 nm (τ of 3.82 ns) and 470 nm (τ of 3.5 ms) that were attributed to fluorescence and RTP. Interestingly, the ratios between the intensities of fluorescence and phosphorescence were different and strongly dependent on the quality of the crystal. The RTP of crystal B3-1 was much more expressed than fluorescence whereas lower-quality crystalline form B3-2 displayed the opposite behaviour. When compound B3 was ground in a mortar, its RTP almost disappeared confirming that crystallization is the key factor in initiation of RTP in B3 (Fig. 3). The phosphorescence quantum yields were 9.1%, 6% and 1.4% for B3-1, B3-2 and the ground sample of B3, respectively. In the case of lower quality crystals and a thoroughly ground sample, the crystalline lattice is damaged to some degree as well as specific molecular arrangement, leading to less efficient RTP.
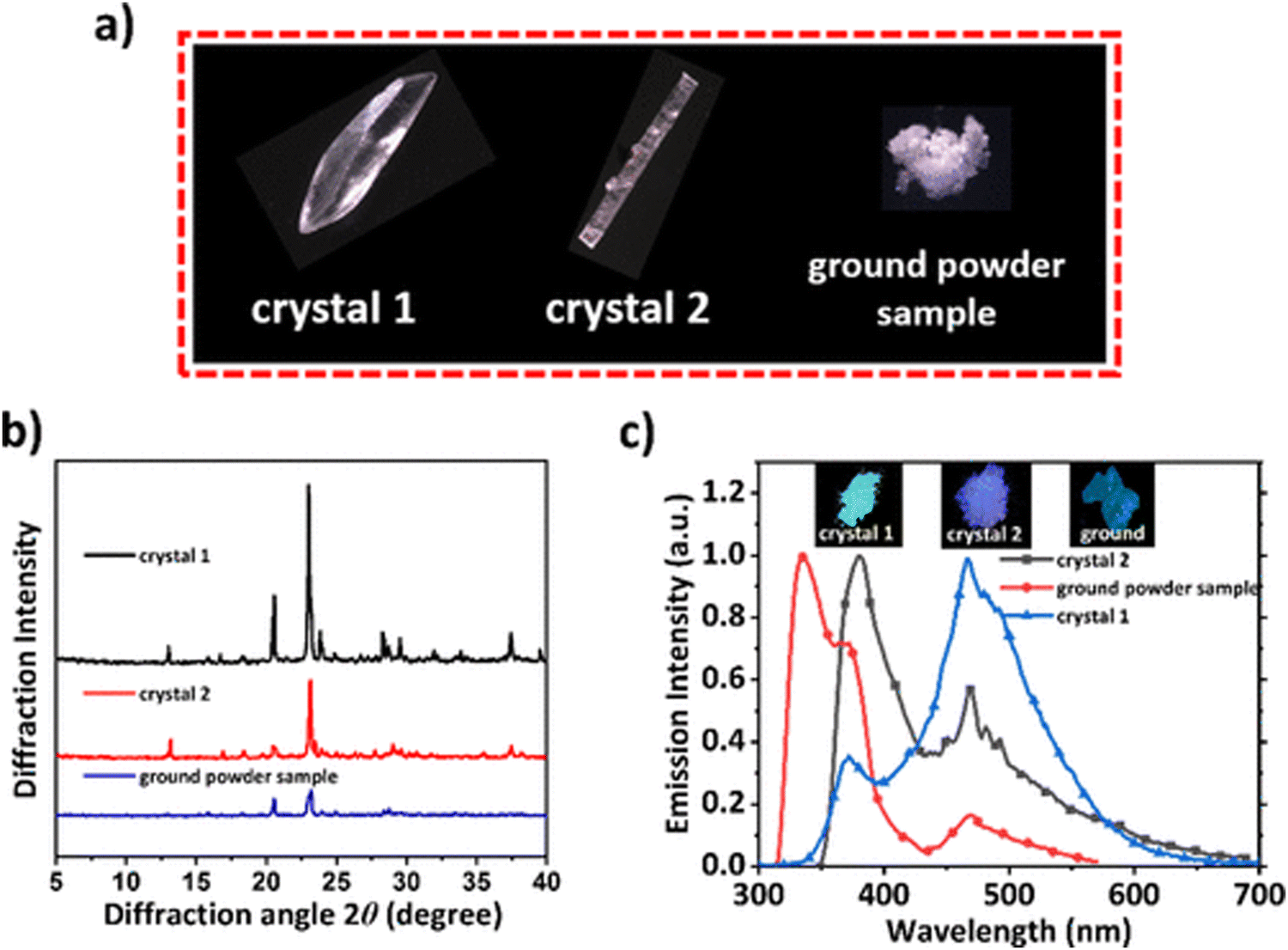 | ||
| Fig. 3 (a) Photographs of B3 different crystalline samples under a microscope; (b) PXRD patterns of B3-1, B3-2, and ground powder samples; and (c) PL spectra of B3 in different solid states. Adapted with permission from ref. 17. Copyright 2023 American Chemical Society. | ||
Cong and co-workers18 recently studied the possibility of initiating blue RTP in the phenothiazine-5,5-dioxide moiety by the attachment of cycloalkane fragments with different sizes of the rings. Cycloalkyl groups have superiority above alkyl ones because the rotation of C–C single bonds is restricted in cyclic structures which is favourable for RTP. Having this in mind, the authors18 synthesized a family of compounds with cycloalkyl rings varying from cyclopropyl to cycloheptyl and acyclic analogous compounds. All the compounds were obtained through a two-step synthetic pathway including the acylation of phenothiazine with the corresponding cycloalkyl or alkyl moiety and the following oxidation of phenothiazine to phenothiazine-5,5-dioxide. In the crystalline state, cyclopropyl- and cyclobutyl-containing materials demonstrated green RTP, whereas cyclopentyl–cycloheptyl containing ones B4–B6 ((Fig. 5 and Table 1)) afforded blue afterglow, visible by the naked eye after turning off the excitation source. The most efficient blue afterglow was observed for compound B5 having a cyclohexyl fragment linked with a phenothiazine-5,5-dioxide moiety, with a τRTP of 1116.1 ms and a RTP intensity maximum wavelength of 471 nm. This is reported to be the longest RTP lifetime among phenothiazine-5,5-dioxide derivatives. Among aliphatic analogues, only the butyl-containing compound B7 (Fig. 5 and Table 1) showed blue RTP also peaking at 471 nm but with a sufficiently shortened lifetime of 249.43 ms.18 After grinding the crystals to powder all the materials demonstrated much weaker afterglow with shortened decay lifetimes, which proves that RTP properties of this family are promoted by multiple intermolecular interactions in the crystalline phase.
Miao et al. found that under certain conditions, blue RTP can be initiated in widely used melamine B8 (Fig. 5 and Table 1).19 Excitation of the crystalline powder of B8 with 310 nm resulted in blue RTP with the intensity maximum at 465 nm and a τRTP of 580 ms. The decrease in the excitation energy to 365 nm resulted in green RTP peaking at 490 nm. The authors19 explained such a dynamic shift in the phosphorescence colour by the formation of H-aggregates between adjacent melamine molecules. Blue RTP of melamine originated from triplet excited-state radiative transition, and the excited S1 exciton was converted into a T1 exciton via intersystem crossing. Next, the T1 exciton was trapped by the H-aggregates and stabilized by the strong aggregation. Then, the captured T1 excitons were slowly released and returned to S0 to emit green RTP.
Although carbazole is the most widely used electroactive organic material, the indolocarbazole family remains scarcely investigated due to the solubility problems of such compounds. However, there is a high probability that they are RTP-active due to their high crystallinity and the presence of two amino groups that can readily form intermolecular non-covalent bonds, thus preventing the non-radiative loss of triplets. It was recently proved that all existing indolocarbazole isomers are indeed RTP-active when doped into a PVA matrix, and two of them (B9 and B10, Fig. 5 and Table 1) were found to exhibit bright blue afterglow.20 Compounds B9 and B10 revealed biluminescent behaviours with fluorescence maxima located at 390–410 nm and RTP peaking up to 494 nm. The symmetrical isomer B9 demonstrated a τRTP of 0.76 s, which is one of the highest values among organic blue RTP emitters. In order to verify that RTP of the investigated compounds is promoted by guest–host intermolecular interactions, the NMR spectra for B9, PVA and B9/PVA mixture were recorded. For pure PVA, the –OH proton peak ranged from 4.36 to 4.76 ppm, but in the case of B9-PVA, it appeared to be shifted by 0.16 ppm. For pure B9, the signal from the –NH group was shifted to 0.16 ppm, whereas the signals of the protons of phenyl rings appeared almost at the same place (shift of 0.01 ppm).20 This confirms the existence of intermolecular hydrogen bonding between the B9 guest and the PVA host matrix (Fig. 4).
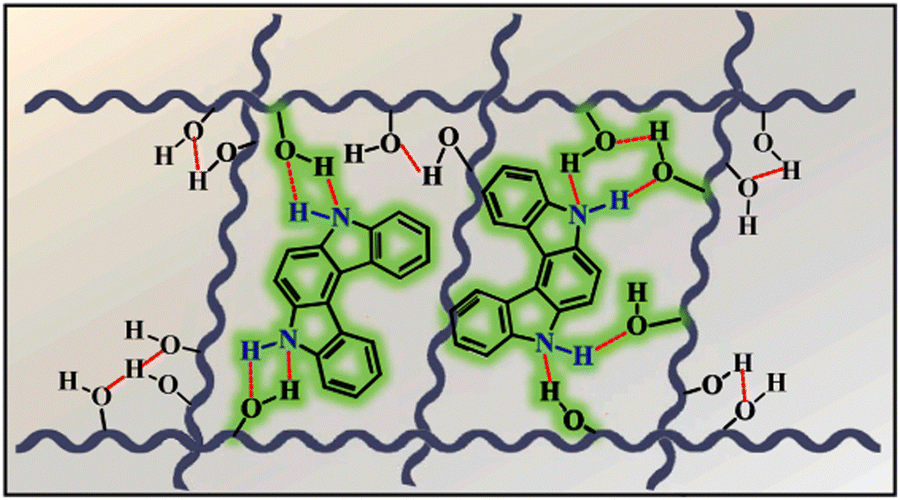 | ||
| Fig. 4 Schematic illustration of intermolecular hydrogen bonding between B9 and PVA. Reprinted from M. Jian, Z. Song, X. Chen, J. Zhao, B. Xu and Z. Chi, Afterglows from indolocarbazole families, Chem. Eng. J., 429, 132346,20 Copyright 2023, with permission from Elsevier. | ||
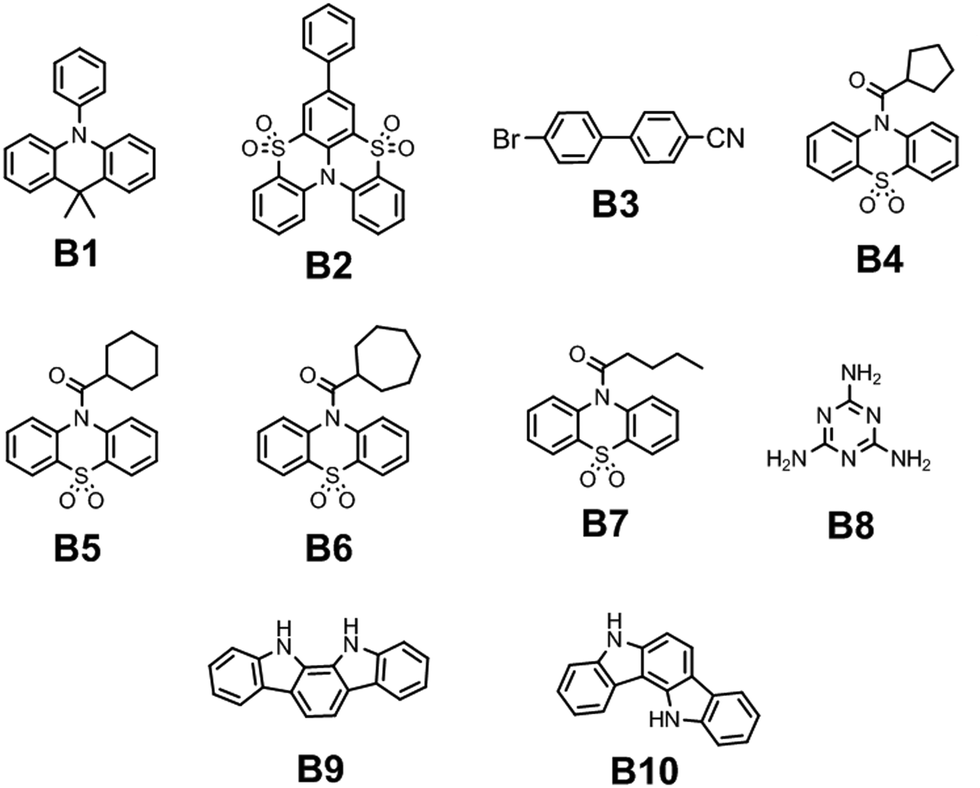 | ||
| Fig. 5 Molecular structures of blue RTP-agents B1–B10. | ||
4. Green RTP emitters
Since the human eye is most sensitive to yellowish-green emission in the daylight, green RTP emitters are desirable for bioimaging applications that require high contrast and resolution.21 Green RTP emitters might attract considerable attention for the development of OLEDs with high efficiency and stability. For example, they can be used for data encryption and decryption by exploiting their long-lived afterglow.Eventually, even very small organic molecules without heavy atoms can exhibit green RTP. Han et al.15 found that intramolecular rigidification on triphenylamine phenyl rotors can evoke efficient RTP in the former. Locking of two phenyl rings of triphenylamine via an sp3-hypridized carbon atom diminishes the rotations around C–N bonds, thus opening up a way for triplet excitons to relax radiatively. The authors15 synthesized three derivatives of 9,10-dihydro-9,9-dimethylacridine by Buchwald–Hartwig cross-coupling: B1 (mentioned in chapter 1, Fig. 5 and Table 1), G1 and G2 (Fig. 11 and Table 2). Compound G1 which has a methyl group in the para-position of its unlocked phenyl ring demonstrated green RTP peaking at 529 nm (τRTP of 28 ms). It showed a PHQY of 2%. Changing the substituent from methyl- to methoxy- in G2 results in a hypsochromic shift of the RTP peak to 508 nm and a shortening of τRTP to 18 ms. At first glance, RTP in the former compound should be more expressed due to the presence of an oxygen atom that promotes intermolecular interactions because of the electronic n-lone pair activity. The authors15 explained such opposite RTP-behaviour by more compact and ordered molecular packing in G1 crystals that suppressed non-radiative transitions of G1 in comparison to G2.
A family of phenanthrolines, exhibiting RTP, were synthesized and investigated by Sun et al.22 The suppression of molecular rotations was achieved by doping phenanthrolines G3, G4, and G5 (Fig. 11 and Table 2) into a rigid PVA matrix. Such systems create abundant H-bonding interactions that are key to restrain the non-radiative decay of the triplet excited states. The unique property of such host–guest systems is that they can be excited from UV to visible-light to afford RTP. Compound G4 that contains one aminogroup demonstrated the best performance among the whole family (τRTP of 229 ms and a PHQY of 19.6%). It was revealed that the RTP properties of phenanthroline derivatives strongly depend on the number of incorporated aminogroups. Apparently, in the case of unsubstituted phenanthroline G3 doping into a PVA matrix, it did not provide the necessary amount of intermolecular interactions to facilitate RTP, and NH2-abundant G5 has plenty of N–H groups not attached to PVA that dissipate energy non-radiatively, resulting in a decrease of efficiency of RTP.
Another example of efficient RTP of low-molar-mass organic compounds was recently reported by Li and co-workers.23 They synthesized a tetracyclic compound G6 (Scheme 1, Fig. 11 and Table 2) that exhibited no RTP in the solid state but became RTP-active when molecularly dispersed in the newly developed phosphorus-containing rigid host (DIPHOS). The new host not only enabled more effective exploitation of triplet excitons of the guest G6, but also the ethyl chain between two phosphorus atoms provided good compatibility with surrounding guest molecules. Moreover, two phosphorus atoms of the host promote the heavy atom effect, resulting in enhanced intersystem crossing. The authors23 managed to grow guest–host co-crystals by the evaporation of the guest–host ethanol solution with a molar ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 correspondingly. Compound G6 itself exhibited deep-blue fluorescence in the solid state while the host was almost nonluminous with an extremely low photoluminescence quantum yield of 0.6% in the solid state. The resulting cocrystal was excited at 350 nm, which was the excitation wavelength of the guest G6. The host could not be excited with such irradiation. The G6-DIPHOS co-crystal revealed sky-blue emission at a steady state and persistent green afterglow (515 nm) with a PHQY of 25.6% and a τRTP of 32 ms.
:1 correspondingly. Compound G6 itself exhibited deep-blue fluorescence in the solid state while the host was almost nonluminous with an extremely low photoluminescence quantum yield of 0.6% in the solid state. The resulting cocrystal was excited at 350 nm, which was the excitation wavelength of the guest G6. The host could not be excited with such irradiation. The G6-DIPHOS co-crystal revealed sky-blue emission at a steady state and persistent green afterglow (515 nm) with a PHQY of 25.6% and a τRTP of 32 ms.
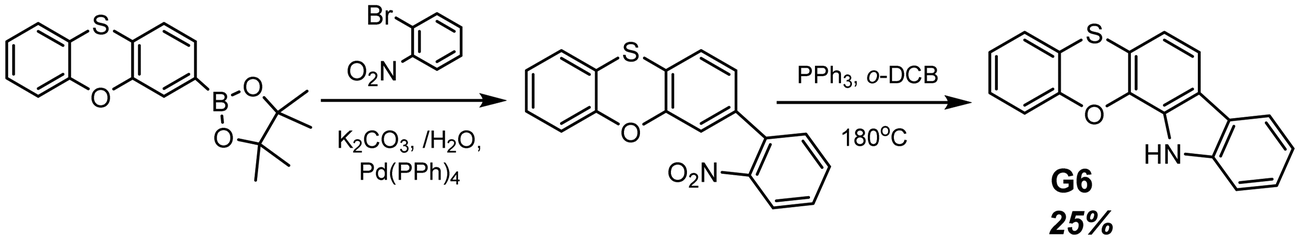 | ||
| Scheme 1 Synthetic route for compound G6. Adapted from the literature.23 | ||
To date, the vast majority of luminescent materials exhibiting piezochromism are attributed to purely fluorescent compounds.24,25 However, RTP often coexists with fluorescence which makes it impossible to study RTP piezochromism separately. Yang et al.26 recently reported pressure-dependent RTP properties of green organic phosphor selenanthrene G7 (Fig. 11 and Table 2) that exhibited almost no coexisting fluorescence under ambient conditions and therefore is a perfect candidate for studying the influence of pressure on RTP. The isotopic hydrostatic pressure produced by a diamond anvil cell was applied to crystals of G7. With increasing pressure, the RTP of G7 revealed two-step transformation. At the first step the pressure was increased from 1 atm to 4.15 GPa and RTP of G7 profoundly increased, simultaneously shifting from green to greenish-yellow. The authors26 observed the sharp shrinkage of the emission spectrum having a vibronic structure together with the appearance of a single peak at 567 nm when the pressure was increased to 4.15 GPa. At the second step, when the pressure was increased over 4.15 GPa, the emission of G7 decreased drastically with a redshift from greenish-yellow to orange and completely ceased under 9.21 GPa. The authors26 concluded that pressure-dependent RTP reinforcement in selenanthrene occurred due to the more folded geometry along with more intensive intermolecular interactions under high pressure, which results in an improved rate of triplet radiative transition. The shorter the intermolecular distances and the stronger the out-of-plane distortions under pressure, the more efficient the π–σ mixing; the orbital angular momentum matrix elements 〈σ||π*〉 of the SOC operator (6) increase and the influence of the heavy Se atoms get more pronounced.6 This results in the strengthening of the SOC ME and the EDTM (9), increasing the kp rate constant.
Selenium is a promising element for the organic halogen-free RTP agents due to its large atomic radius that results in large SOC. However, due to the high cost of selenium-containing starting materials and complex synthetic routes, such materials are not widespread. Wang et al.27 developed a selenium-containing donor–acceptor compound G8 (Scheme 2, Fig. 11 and Table 2) with the 9H-selenoxanthen-9-one moiety as an electron acceptor and 9-phenyl-9H-carbazole as an electron donor unit. They synthesized its sulfur-containing counterpart G9 (Scheme 2, Fig. 11 and Table 2) bearing 9H-thioxanthene-9 as an electron-accepting moiety to brighten RTP. The presence of both a selenium heavy atom and a carbonyl group with oxygen lone electron pair promotes RTP by strengthening SOC which results in more effective non-radiative singlet–triplet transition. A photophysical investigation of 1 wt% of compounds G8 and G9 embedded into the polystyrene (PS) matrix revealed that both compounds exhibit a biluminescence behaviour. In the case of the molecular dispersion of compound G8 in PS, the film exhibited bright green emission peaking at 500 nm and a shoulder at 453 nm under vacuum at room temperature. The investigation of the sample at 77 K with a delay time of 1 ms revealed complete disappearance of the mentioned shoulder, suggesting its fluorescent origin. The sulfur-containing counterpart G215 demonstrated the opposite behaviour. Its fluorescence peak at 441 nm (emission lifetime of 1.35 ns) was more intensive than the phosphorescence shoulder with emission maxima at 487 nm (τRTP of 10.38 ms). Noteworthily, the film of the molecular dispersion of G8 in PS exhibited a high phosphorescence quantum yield of 44.3%, which is one of the highest among selenium containing RTP-materials and four times higher than that of the film of the molecular dispersion of G9 in PS (10.1%). The authors27 calculated the radiative rate constants of fluorescence (kFr) and phosphorescence (kPr), the intersystem crossing rate constant (kISC), and the sum of the non-radiative constant of phosphorescence (kPr) and the quenching rate of T1 (kq) of the films of G8 and G9 doped in PS. The theoretical calculations revealed that when sulphur is replaced with a heavier selenium atom, it results in a 1.5-fold increase in kISC and an increase in kPr by two orders of magnitude.
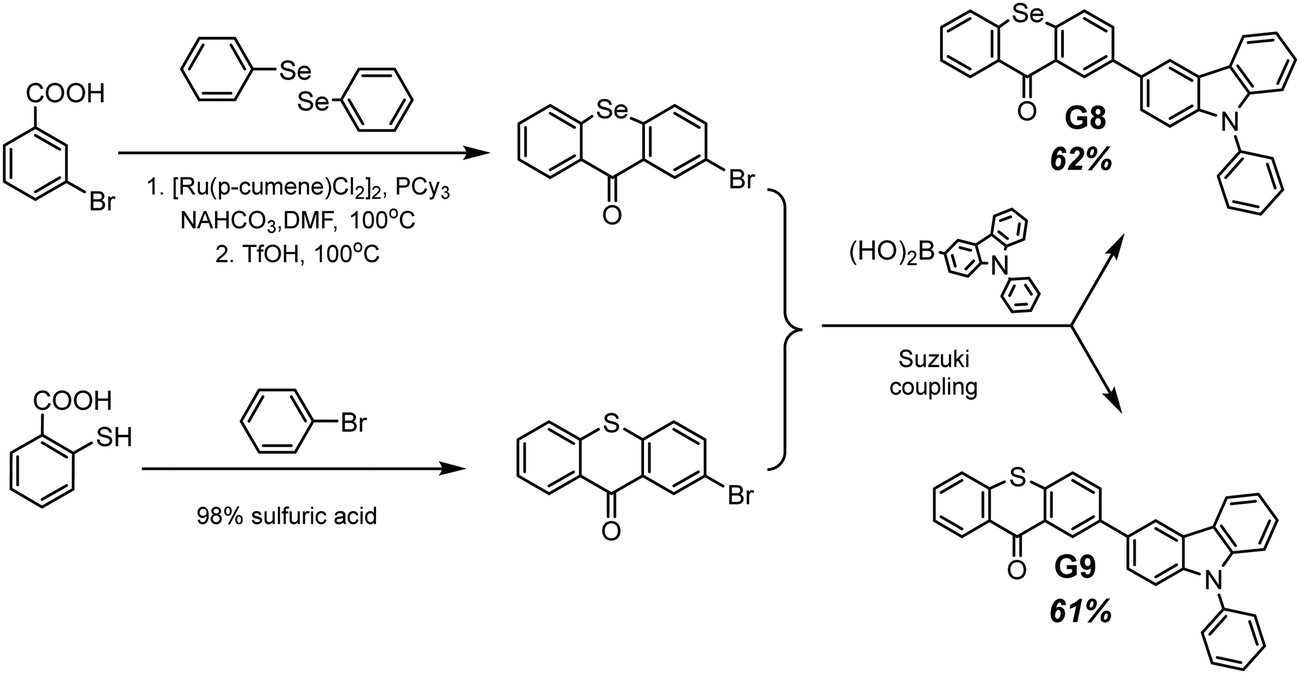 | ||
| Scheme 2 Synthetic route of obtaining compounds G8–G9. Adapted from the literature.27 | ||
For most organic RTP-materials, crystallization enhances phosphorescent emission; however, the powder of compound G8 exhibited a PHQY of only of 2.9%. Calculated kPr + kq for G8 crystals was 200-fold higher than that of the film of the molecular dispersion in PS; therefore, in a crystalline state, the non-radiative loss of triplet excitons increased drastically, resulting in low PHQY.
The same group of researchers27 investigated the effects of the structural isomerism on the RTP properties of compound G8. They synthesized three donor–acceptor isomers of G8, varying by the linking topology of the phenylcarbazole electron donor and selenoxanthenone electron acceptor. The film of compound G10 (Fig. 11 and Table 2) with a phenylcarbazole moiety at C-3 position of the 9H-selenoxanthen-9-one ring being doped in a PS matrix has demonstrated an RTP intensity maximum at 497 nm (τRTP of 3.09 ms) and with PHQY reaching 66.7%, which is the highest value among the selenium-containining RTP materials. The isomer with a phenylcarbazole moiety at the C-4 position of the 9H-selenoxanthen-9-one ring exhibited blue RTP peaking at 468 nm with a PHQY of 27.3%, while the isomer with C-1 substituted position of 9H-selenoxanthen-9-one exhibited no RTP. It showed only green fluorescence.
Another illustrious example of the advantageous co-presence of a selenium heavy atom and a ketogroup in one molecule to maximize SOC for initiating RTP was reported in ref. 28. The simple (10H-phenoselenazin-10-yl)(phenyl)methanone G11 (Fig. 11 and Table 2), where a phenoselenazine moiety is bonded with benzoyl fragment via an N atom, and its sulfur containing counterpart G12 (Fig. 11 and Table 2) were synthesized. Compound G11 did not exhibit fluorescence in a tetrahydrofuran solution at room temperature. This indicated that singlet excitons were converted into triplet excitons with almost 100% efficiency due to the enhanced SOC. Fig. 6a depicts NTO of G12 in T1 state. Both the HONTO and LUNTO are localized on the phenoselenazine fragment. Therefore, it can be outlined that the locally excited phenoselenazine unit vastly contributes to the phosphorescence of G11 and the contribution of charge transfer is rather negligible. Under excitation with 365 nm the crystal of G11 emitted bright light whereas the crystal of G12 demonstrated faint luminescence (Fig. 6b and c). PHQYs of the crystals of G11 and G12 were 7.7% and close to 0%, respectively.
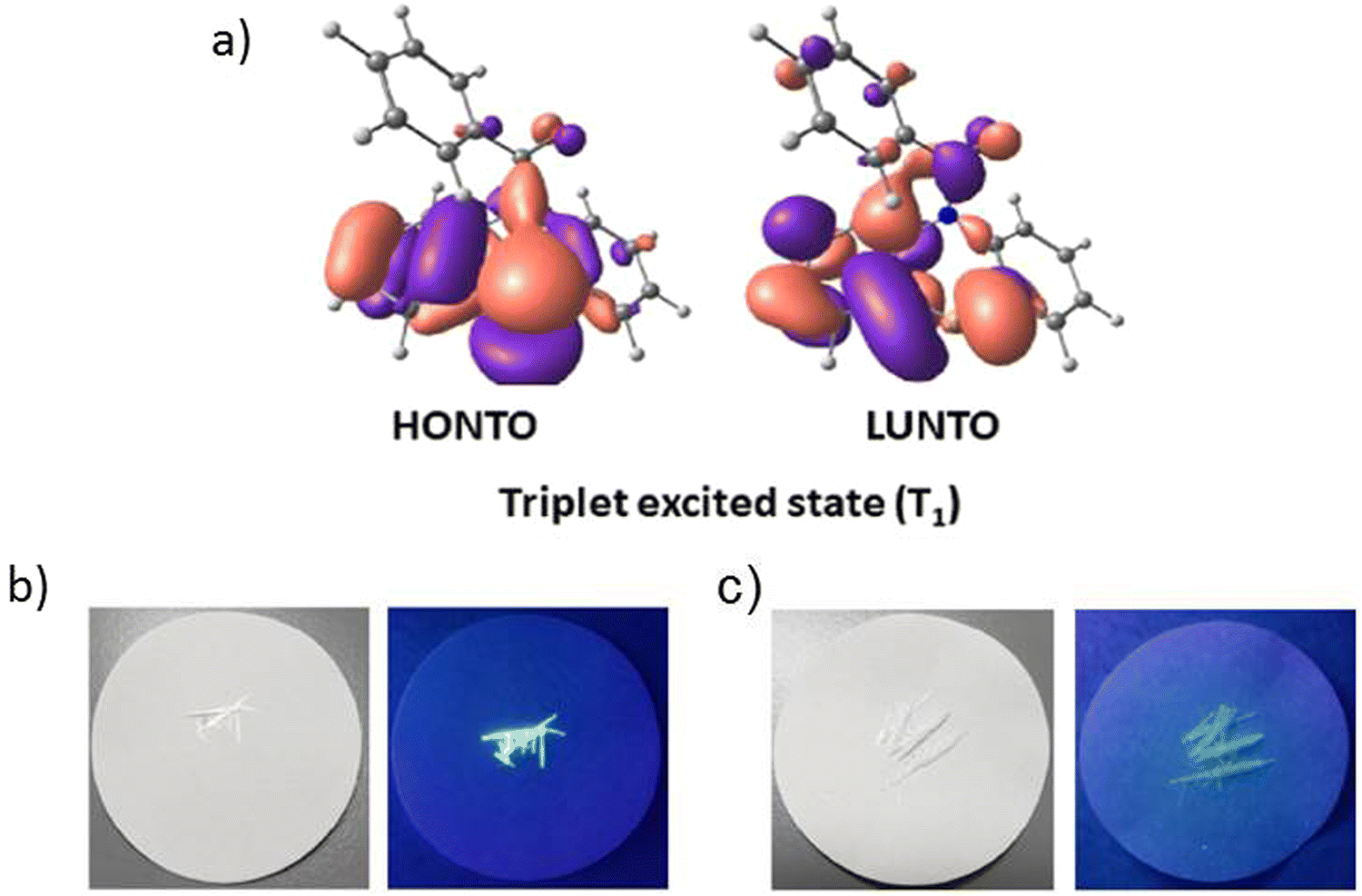 | ||
| Fig. 6 (a) Natural transition orbitals of G12 at T1 state; (b) and (c) photographs of G12 and G11 under daylight and under 365 nm excitation. Reprinted from D. R. Lee, J. Park, J. Y. Lee, Metal and halogen-free purely organic room temperature phosphorescence material using heavy atom effect of phenoselenazine, Org. Electron., 106, 106534,28 Copyright 2023, with permission from Elsevier. | ||
Derivatives of the benzothiazino-phenothiazine tetraoxide (BTPO) core were reported as a new category of organic emitters.29 Functionalization of BTPO with different substituents enabled tuning the origin of luminescence from TADF to RTP. The BTPO skeleton was functionalized with the different moieties such as p-cyanophenyl-, carbazolyl- and triphenylamino to obtain compounds G13–G15 (Fig. 11 and Table 2) in 82%. 77% and 78% yields correspondingly. Investigation of photophysical properties of the target compounds doped into a rigid PMMA matrix revealed that all of them exhibited biluminescence with obvious RTP. The RTP/PL ratio for G14 was almost unity, whereas those for G13 and G15 were reduced to less than 0.5. The PHQYs were estimated to be in the range from 7.4% to 11.6%. These values are among the highest values for organic luminogens with phosphorescence lifetimes above 0.1 s. These results signify that decoration of the BTPO core with various peripheral substituents allow tuning the phosphorescence lifetime and quantum yield. The film of 5 wt% BTPO doped into the PMMA matrix exhibited no phosphorescence under ambient conditions under vacuum.29 The S1 and T1 values were calculated to be 3.31 eV and 3.15 eV correspondingly, resulting in a low ΔEST of 0.16 eV. The molecular dispersions of compounds G13–G15 in PMMA showed larger ΔEST values of 0.59, 0.7 and 0.44 eV, respectively. To verify the necessity of introduction of a phenyl ring into the BTPO core to afford efficient RTP, the authors also investigated BTPO with an extra phenyl group as a reference compound. The 5% solid solution of the reference material in PMMA showed dual emission, with a lifetime of prompt emission of 1.27 ns peaking at 391 nm, and phosphorescence peaking at 465 nm and 493 nm with the corresponding lifetimes of 658 ms and 657 ms. Thus, the triplet energy could be significantly decreased, and efficient phosphorescence could be realized by introduction of an additional aromatic substituent to the BTPO core.
BTPO serving as a favourable RTP-synthone was also used in the work.16 BTPO was functionalized with the same substituents but at the C-7 position. Compounds G16–G18 (Fig. 11 and Table 2) showed much brighter RTPs than compounds G13–G15 maintaining long phosphorescence lifetimes. The 5 wt% molecular dispersions of the compounds in PMMA showed RTP with obvious vibronic features of the spectra, except for the dispersion of triphenylamine-containing G18 that revealed one emission peak. The possible explanation of this phenomenon is a more expressed CT-character of G18 than those of G16 and G17. Singlet–triplet energy gaps, calculated from the fluorescence and phosphorescence onsets, were in the range of 0.28–0.6 eV, allowing efficient ISC. The cyano-containing derivative G16 was found to exhibit a high PHQY of 20.2% together with an ultralong phosphorescence lifetime of 818 ms. This value stands among the best results for green organic phosphors dispersed in a polymer matrix.
Planar tricoordinate boron derivatives G19–G21 (Fig. 11 and Table 2) that are isostructural with the abovementioned BTPO derivatives, were recently reported.30 Target compounds were obtained through lithiation reaction using n-BuLi at −70 °C and following borination with BBr3 at −40 °C (Scheme 3). In a crystalline state, the compounds under UV irradiation emit blue, cyan and deep-blue light with corresponding maxima at 430, 455 and 423 nm, respectively. Upon ceasing UV irradiation, luminogens G19 and G20 maintain green afterglow with single maxima at 476 for G19 and double maxima at 466 and 539 for G20. Compound G21, although having a bromine atom, did not exhibit any emission from triplet states. This is due to a specific molecular packing. The incorporation of two bulky tert-butyl groups into a boron-centred core was performed in order to regulate the packing of the molecule and amplify intermolecular interactions (compound G21 in Scheme 3). However, surprisingly such a molecular design failed to afford RTP. Further investigation of the G21 crystal structure revealed antiparralel π-stacking that prohibits the radiative transition of H-aggregates and leads to the annihilation of triplet excited states. After doping into the PMMA host, luminogens G19 and G20 maintained the RTP properties. RTP maxima of the film of the molecular dispersion of G19 in PMMA was blue-shifted to 415 nm whereas that of the film of the molecular dispersion of G20 in PMMA remained at ca. 472 nm. For compound G21, even embedding into the rigid polymer matrix did not unlock the triplet radiative pathway.
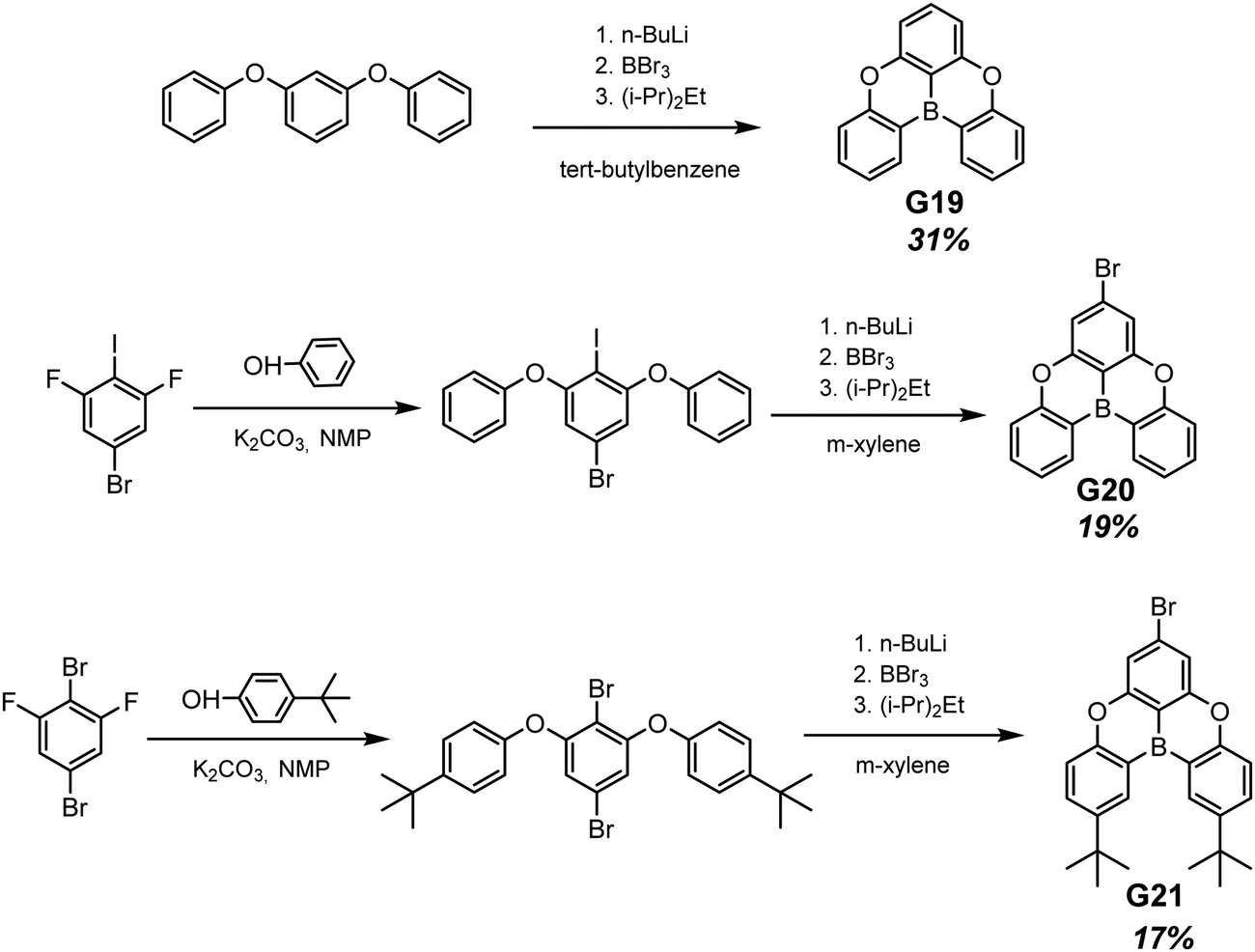 | ||
| Scheme 3 Synthetic routes for compounds G19–G21. Adapted from the literature.30 | ||
The luminescence properties of the crystals of G19–G21 were studied under hydrostatic pressure with the aid of a diamond anvil cell. When the pressure was increased from 0 atm to 10.9 GPa, the fluorescent peak of G19 was slightly shifted to a longer wavelength area from 427 nm to 433 nm alongwith a sharp decrease in the intensity. The intensity of the phosphorescent band around 470–550 nm was gradually amplified and also exhibited a red-shift. As it was mentioned above in the case of selenanthrene G7, high pressure shortens the intermolecular distance, diminishing molecular vibration by reducing free molecular volume, and facilitates the overlap of electronic orbitals between neighbouring molecules.26 The short intermolecular distances and high MO overlap between molecules lead to an increase in the σ–π intramolecular mixing and stronger SOC perturbations.6 The processes occurring are not favourable for fluorescence but promote RTP through the enhancement of SOC and acceleration of ISC. The more intense SOC of G20 because of the internal heavy atom effect of bromine resulted in a more expressed enhancement of phosphorescence under high pressure. With increasing pressure, the fluorescence of the crystal of G20 also diminished sharply and red shifted from 429 nm to 437 nm. The phosphorescence intensity even outperformed fluorescence at a pressure higher than 3.1 GPa, reached its full intensity at 7.9 GPa (538 nm) and became completely quenched at 10.6 GPa. Surprisingly, high pressure released the forbidden phosphorescence in G21 that reached its maximum at 3.0 GPa (442 nm), and gradually decreased at 9.9 GPa.
A guest–host strategy to evoke RTP in non-phosphorescent sulfone-containing homologs was proposed.31 A family of compounds G22–G27 (Fig. 11 and Table 2) exhibit efficient green afterglow when doped into triphenylphosphine oxide (OPPh3) as a host. A strong afterglow was noticed from the co-crystal sample of G23 (PHQY of 3.33% and τPHOS of 980 ms) whereas G22 and G24 revealed rather weak afterglow. To elucidate the reason of different RTP efficiency in three similar compounds in this family of compounds the authors of ref. 31 studied the photophysical properties of the solid samples of the guests and the host separately. The emission band of OPPh3 was found to be significantly overlapped with the absorption bands of guest molecules that indicated a good match of their energy levels. From the output of single crystal analysis the energy levels of singlet and triplet excited states were calculated using the TD-DFT method. With the B3LYP functional and 6-31G basis set the energy of the S1 state for OPPh3 was calculated to be of 5.60 eV. The Sn states of guests with energy ±0.3 eV were enumerated. The energy transfer from OPPh3 host to a guest could occur easily in such energy frame. It was found that compound G23 had four energy levels (5.31 eV, 5.70 eV, 5.77 eV and 5.85 eV) that could match with the S1 energy level of the host, resulting in energy transfer, whereas G22 and G24 exhibited only two (5.55 eV and 5.79 eV) and three (5.35 eV, 5.50 eV and 5.65 eV) matchable energy levels correspondingly (Fig. 7). The authors31 also observed that after grinding of the guest–host samples RTP gradually rose both in intensity and the lifetime. It was explained by the decrease of the distance between the guest and host with the help of mechanical force, thus facilitating energy transfer.
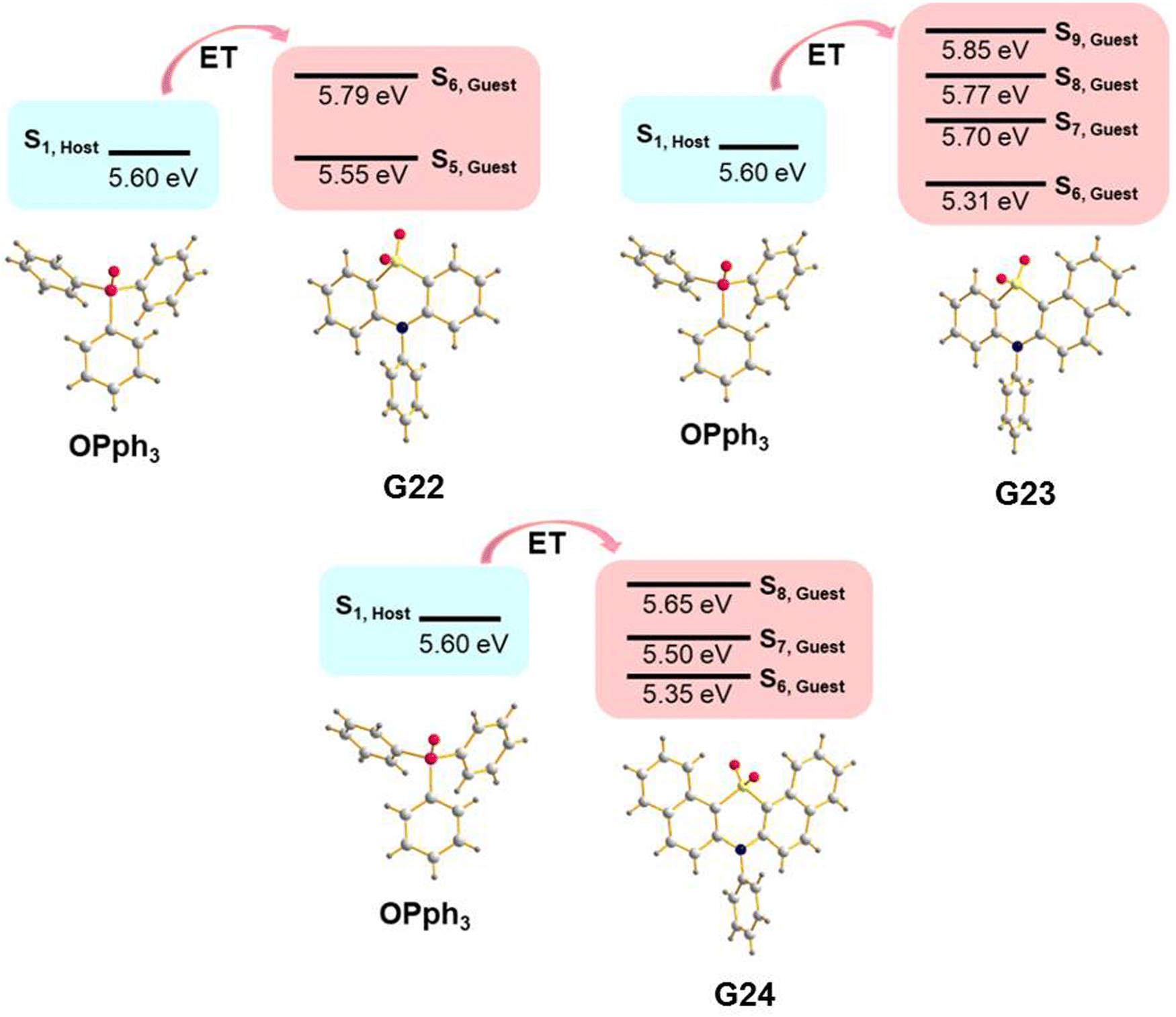 | ||
| Fig. 7 Illustration of host–guest energy transfer for the molecular dispersions of G22–G24 in OPPh3. Reproduced from ref. 31 with permission from John Wiley and Sons, Copyright 2023. | ||
Pyridine containing analogues of G22–G24, namely, G25–G27 (Fig. 11 and Table 2) were obtained via similar synthetic pathways. When they were embedded into the OPPh3 host, green afterglow was also observed and was also amplified by grinding. When the films of the molecular dispersions of pyridine-containing compounds G25–G27 in OPPh3 films interacted with acid, their RTP was switched off as a consequence of structural change in guest molecules (Fig. 8).
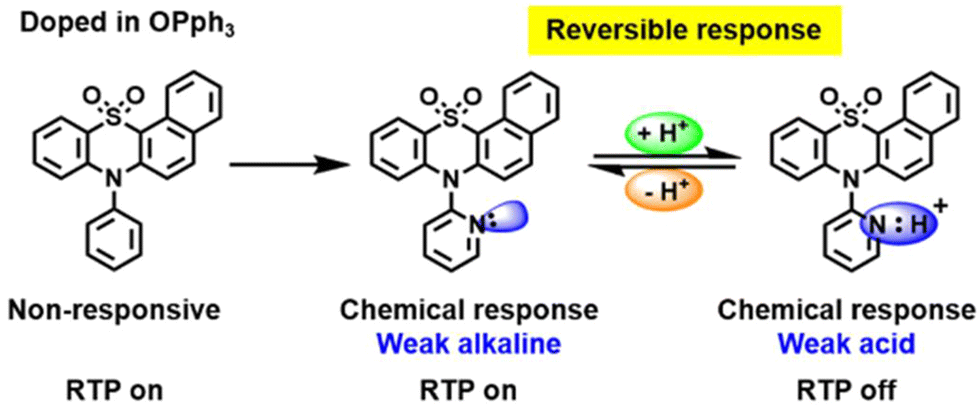 | ||
| Fig. 8 Acid–base stimulus response scheme for compound G26. Reproduced from ref. 31 with permission from John Wiley and Sons, Copyright 2023. | ||
Three very simple phenothiazine-based phosphors G28–G30 (Fig. 11 and Table 2) varying by the number of phenothiazine moieties were reported by Wang et al.32 The films of compounds G28–G30 doped into the PMMA host matrix exhibited weak photoinduced RTP at initial time. The authors explained the RTP photoinduction by O2 consunption under UV irradiation since the triplet oxygen can quench the triplet excitons of the luminophores.11 RTP of compound G30 that contains three phenothiazine fragments is almost negligible under initial ambient conditions, but enhances gradually with the increase in the irradiation time (λirr of 365 nm). Expressed RTP emerges after 5 s and reaches its maximum after 25 s of irradiation with a PHQY of 22% and a τRTP of 30 ms. The film returns back to a non-RTP state after heating the sample at 60 °C for 1 min or maintaining it for 30 min under ambient conditions. Compounds G28 and G29 also revealed reversible photoinduced phosphorescence with PHQYs of 11% and 20% respectively.
Other phenothiazine-containing conjugates with a quinoline acceptor fragment—G31, G32 (Fig. 11 and Table 2)—were reported by Xie et al.33 The special value of this work is that it demonstrated very deep theoretical insight into photophysical processes of abovementioned compounds. The authors33 confirmed that three excited states 1CT, 3CT, and 3LE contribute to the rISC process resulting in TADF and green RTP. For both the luminophores (G31 and G32) the intermediate 3CT was observed when they were molecularly dispersed in a polar matrix, that leads to TADF. RTP bands that were found to be blue-shifted with respect to charge-transfer peaks, originated from phenothiazine donor 3LE, which is higher in energy than CT.
The phenothiazine-triazatruxene derivative G33 (Fig. 11 and Table 2) demonstrated conformer-induced green RTP.34 Triazatruxene core was decorated with three N-ethylphenothiazine fragments via C-2 position of the former. Luminescence properties of the films with 1% solid solutions of the compound in rigid Zeonex matrix were studied. Under vacuum, the investigated film demonstrated two peaks: one at 453 nm which was attributed to fluorescence and another one at 551 nm that increased the intensity of the fluorescence band by 19 times (τPHOS = 16.6 ms) was recognized as RTP band. The film demonstrated a PHQY of 54% that is among the highest values for green RTP-agents to date. The authors of ref. 34 also investigated the photophysical properties of 5% and 10% solid solutions of G35 in Zeonex, but none of the samples revealed such an impressive RTP performance. The corresponding PLQYs were 33% and 22.5% and the RTP/PL intensities ratio were 11 and 7.5, respectively (Fig. 9). The authors34 suggested the possibility of the existence of various types of conformations for G32 that differ by the tilts of phenothiazine fragments. As a result, many triplet states with close, but different energies can exist and provide multichannel ISC leading to RTP.
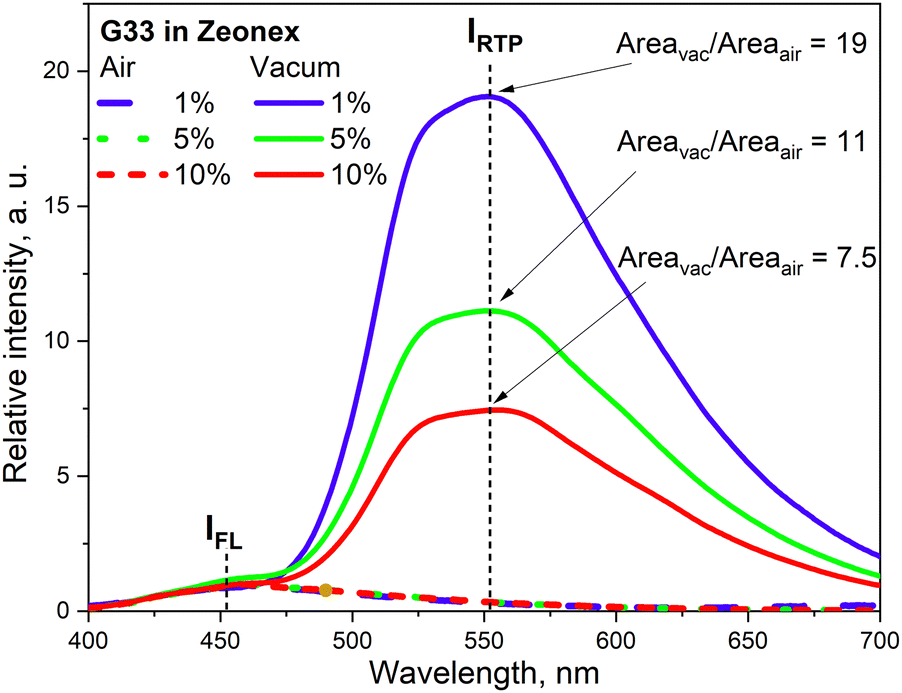 | ||
| Fig. 9 RTP vs. fluorescence ratio for the different concentrations of G34 in Zeonex. Reprinted from E. Skuodis, K. Leitonas, A. Panchenko, L. Volyniuk, J. Simokaitienė, R. Keruckienė, D. Volyniuk, B. F. Minaev and J. V. Gražulevičius, Very sensitive probes for quantitative and organoleptic detection of oxygen based on conformer-induced room-temperature phosphorescence enhancement of the derivative of triazatruxene and phenothiazine, Sens. Actuators, B, 373, 132727,34 Copyright 2023, with permission from Elsevier. | ||
Phenothiazine-containing phosphor G34 (Fig. 11 and Table 2) was obtained via Ullmann coupling between 4,4′-diodo-2,2′-bis(trifluoromethyl)biphenyl and phenothiazine in 59% yield (Scheme 4).35 Two trifluoromethyl groups were employed with the aim of intensifying intermolecular interactions, boosting thermal stability and enhancing ISC and obtaining efficient radiative simultaneous decay of singlet and triplet excited states. Under vacuum, 1 wt% of compound G34 dispersed in a Zeonex rigid matrix revealed dual emission with fluorescence peaking at 456 nm and RTP peaking at 533 nm (τPHOS of 1 μs). The ratio between the RTP intensity and fluorescence intensity of evacuated and air-equilibrated samples was 3.42 (Fig. 10).
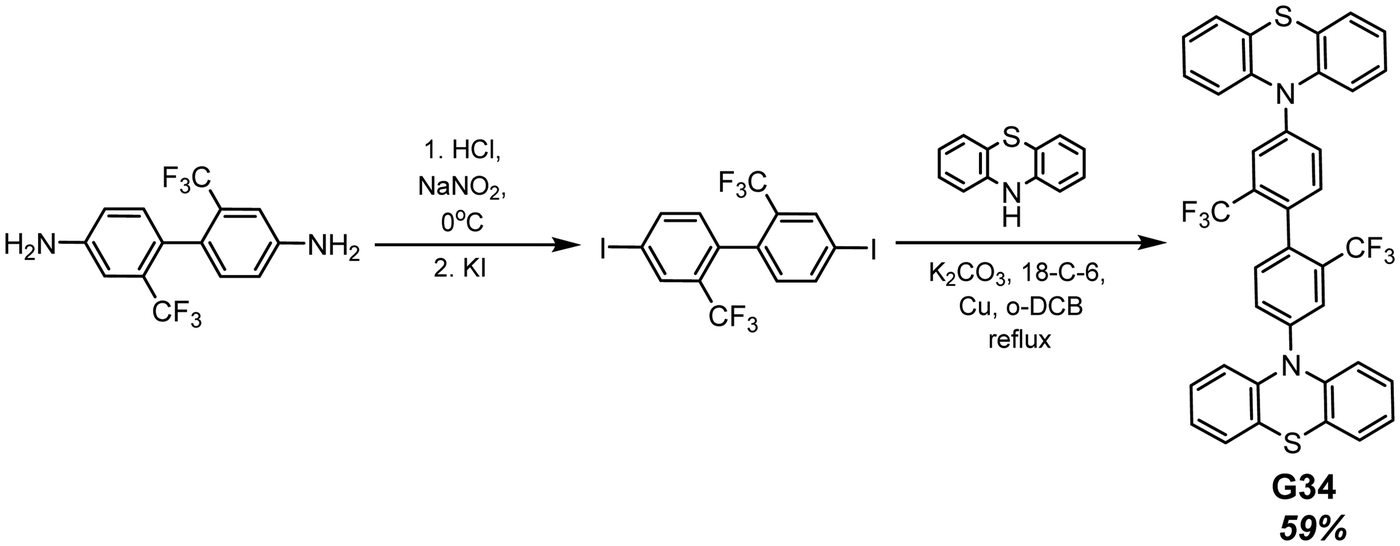 | ||
| Scheme 4 Synthesis of compound G34. Adapted from the literature.35 | ||
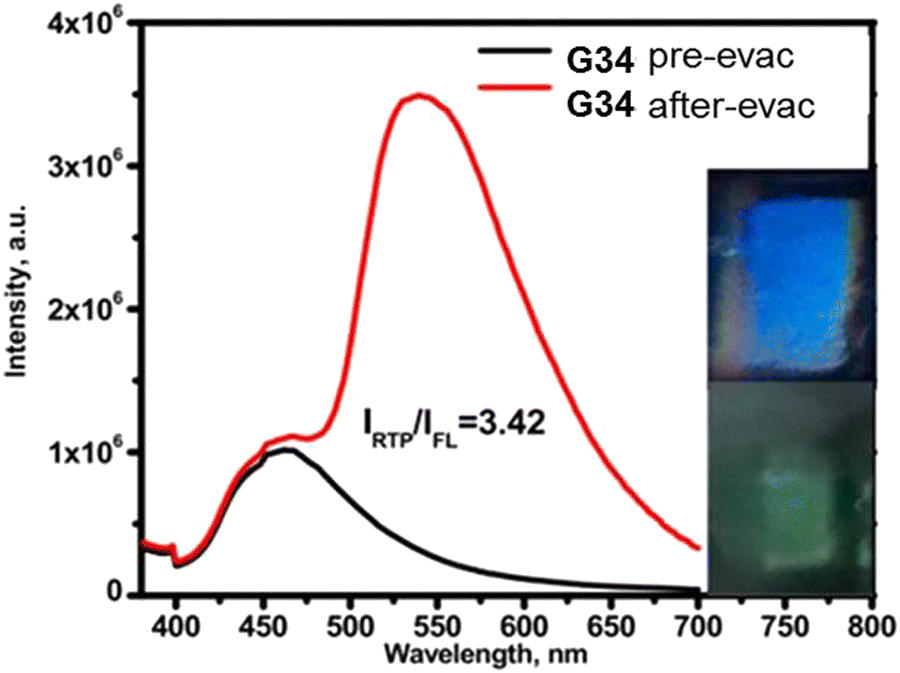 | ||
| Fig. 10 Photoluminescence spectra of the film of the molecular dispersion of compound G34 in Zeonex before and after evacuation. Reproduced from R. Keruckiene, N. Kusas, L. Dvylys, E. Skuodis, V. E. Matulis, E. G. Ragoyja, D. A. Lyakhov, I. Klymenko and J. V. Grazulevicius, Derivatives of Bis(trifluoromethyl)biphenyl and Various Donor Moieties Exhibiting Dual State Emission, J. Luminescence, 241, 118502,35 Copyright 2023, with permission from Elsevier. | ||
 | ||
| Fig. 11 Molecular structures of green RTP-agents G1–G37. | ||
Multi-twisted organic compounds that have many rotational bonds possess great potential to be used in molecular machines, sensors etc. However, the abundance of rotational freedom that results in non-radiative energy loss makes it hard to achieve RTP for such molecules. Shen et al.36 introduced an elegant solution for this problem by engineering appropriate hosts for highly twisted compounds. They designed and synthesized three asterisk-like sulfur containing luminophores G35–G37 (Fig. 11 and Table 2) and proposed an individual host for each compound to afford efficient RTP. Hosts H35–H37 (Fig. 11 and Table 2) were designed taking into account the structural peculiarities of the guest compounds to ensure not only restriction of molecular motions but also compatibility of energy levels. All the guest compounds did not exhibit RTP after being doped into the PVA matrix, confirming that RTP of the guest–host solid mixtures is driven by energy transfer rather than by restriction of rotational freedom. All the guest–host solid mixtures exhibited RTP peaking at ca. 550 nm with a τRTP of 80.4 ms for G35–H35, 3.85 ms for G36–H36 and 59.44 for G37–H37. The authors36 attributed the short RTP of the G36–H36 solid mixture to the fast triplet–triplet energy transfer from the H36 host to the G36 guest, which was further proven by theoretical calculations, revealing that ΔETHOST − TGUEST was only 0.18 eV.
5. Orange/red RTP agents
The field of organic red light-emitting materials is thriving at this point in time since there is still a high demand for such materials to be utilized in optical sensors, displays, lighting devices, security probes and night vision devices.38,39 Organic red-emitting materials are exclusively used in medical applications, i.e., bioimaging, drug tracking, and especially, tumour diagnosis and treatment.40 Emitters with a wavelength shorter than 580 nm cannot be applied for achieving the abovementioned goals because their high photon energies can damage live tissues instead of penetrating them.41 Moreover, organic compounds have obvious advantages over their inorganic counterparts and metal complexes such as low toxicity, good biocompatibility, the vast variety of possible materials and cost efficiency of the synthesis. However, in terms of luminescence lifetimes and quantum yields, the inorganic orange/red emitters are still superior.42 Fluorescence organic materials are not sufficiently efficient because of the limitation imposed by spin statistics. In addition, the lifetimes of fluorescence are usually in the range of nanoseconds. Room-temperature phosphorescence, i.e., emissive relaxation from the triplet excited state to the ground state at a temperature higher than 290 K has considerable advantages against fluorescence both in terms of efficiencies and lifetimes.While developing red-emissive organic compounds, researchers have encountered several challenges:
(1) in order to generate emission in the long-wavelength region of the spectrum, a compound has to have a low optical bandgap;
(2) in order to harvest energy not only from singlet excitons but also from triplet excited states, a compound has to have high-lying triplet energy levels. Generally, extension of the π-conjugation system results in a shift in emission to longer wavelengths, but it also increases the number of rotational bonds that results in lower emission quantum yield. Highly planar and rigid π-conjugated compounds such as pyrene and coronene derivatives have low-lying triplet energy levels which make it hard for these compounds to harvest energy from them via ISC;43
(3) An additional problem arises when it comes to RTP quenching. For triplet excitons to relax radiatively, non-emissive pathways of energy dissipation have to be blocked. It can be easily achieved via dispersing small amounts of the compound into a rigid host matrix, but it is almost impossible to observe RTP of liquid solutions, where molecular motions are intensive. Therefore, in cases when triplet energy levels of compounds are low, the major challenges in achieving red RTP refer to the enhancement of the ISC rate and suppression of the non-radiative decay of excitons.
A simple and efficient method of initiating RTP in the long-wavelength region of the spectrum is to functionalize the compounds that are used as acceptor units in fluorescent/TADF donor–acceptor-type organic emitters with heavy halogen atoms to promote intersystem crossing and populate triplet excitons. The 2,1,3-benzothiadiazole (BTD) moiety, a very common electron-accepting constituent of organic fluorescent red/NIR emitters is RTP-inactive when unsubstituted, while its RTP properties can be easily evoked by introducing heavy halogen atoms.
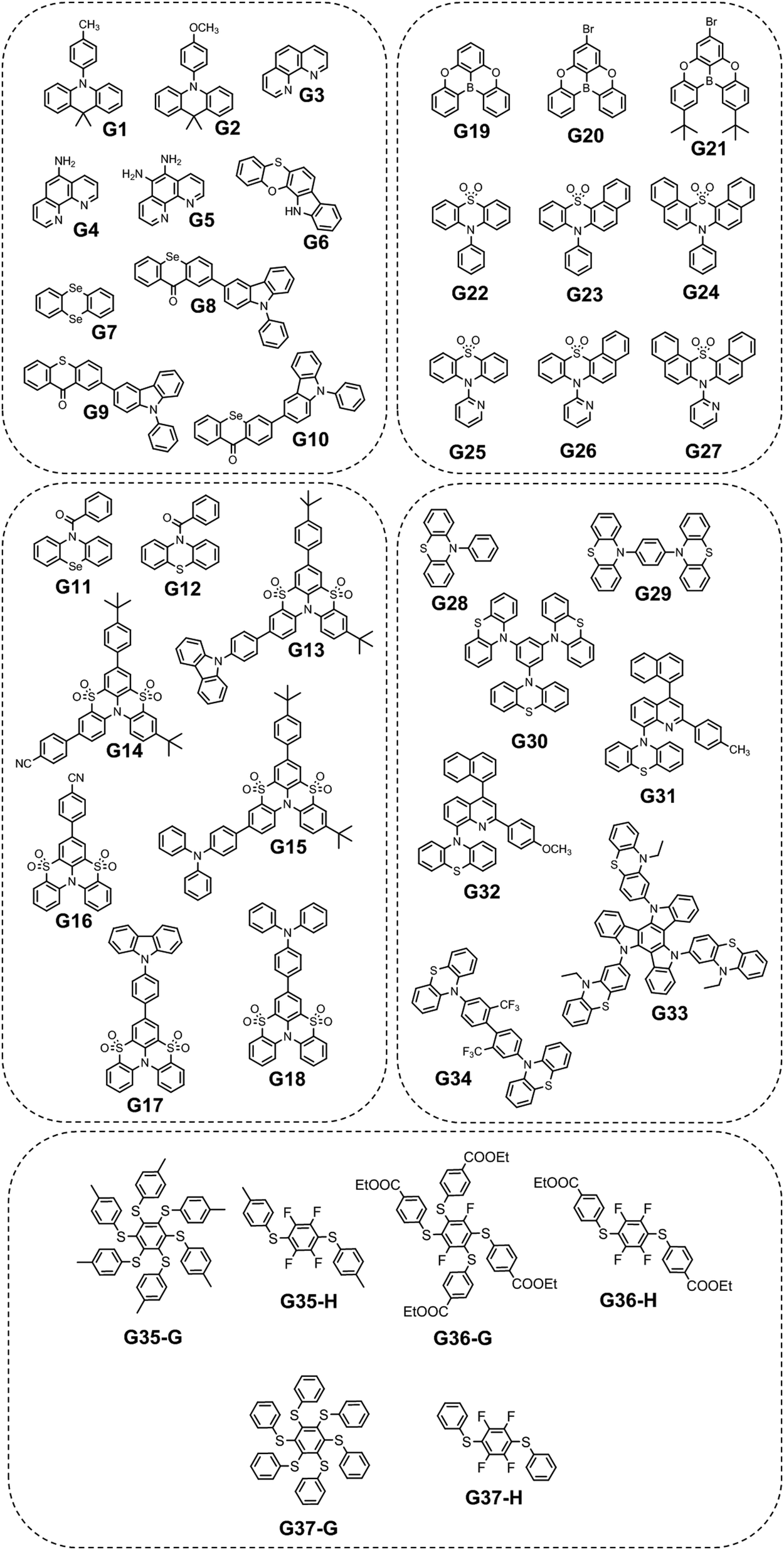
Long-wavelength RTP of bihalogenated derivatives of BTD was previously observed.44,45 Recently, Ishi-e et al. studied the impact of methoxy-groups and halogen heavy atom co-presence on RTP of BTD derivatives.46 Compounds R1–R4 (Fig. 17 and Table 3) were synthesized starting from o-dimentoxybenzene in moderate yields (Scheme 5). First, the nitration of o-dimethoxybenzene was performed by heating it with fuming nitric acid to afford 1,2-dimethoxy-4,5-dinitrobenzene. Then the nitro groups were reduced to amino groups using palladium on the carbon catalyst and ammonium acetate at room temperature. Dimethoxy-2,1,3-benzothiadiazole R1 was synthesized by treating the amino derivative with pyridine and thionylchloride. Bromination of R1 using bromine resulted in the formation of compound R2 with 43% yield. Condensation of 4,5-dimethoxybenzene-1,2-diamine with glyoxal afforded compound R3 that was transformed into R4 after treating it with bromine.
| Compound | λ FL, nm | TPL, ns | FLQY, % | λ RTP, nm | τ RTP, ms | PHQY, % | Host | Ref. |
|---|---|---|---|---|---|---|---|---|
| λ FL – the wavelength of fluorescence maxima, τPL – fluorescence lifetime, FLQY – fluorescence quantum yield, λRTP – the wavelength of room temperature phosphorescence maxima, τRTP – RTP lifetime, and PHQY – phosphorescence quantum yield.a After UV irradiation for 40 s.b Under ambient conditions.c Under vacuum.d Excitation wavelength = 450 nm.e Excitation wavelength = 400 nm.f After photoactivation, cr. – crystal, and cr. pw. – crystalline powder. | ||||||||
| R1 | 448 | 2.32 | 0.4 | 648 | 0.585 | 3.7 | — | 46 |
| R1 | 426 | — | — | 642 | — | — | PMMA | 46 |
| R2 | 422 | 2.34 | 0.4 | 629 | 0.233 | 1.8 | — | 46 |
| R3 | 426 | 23.1 | 1 | 559 | 0.110 | 1.6 | — | 46 |
| R4 | 402 | 0.48 | 2.4 | 555 | 0.073 | 0.5 | — | 46 |
| R5 | — | 5 | — | 575 | 1.9 | 0.21 | — | 47 |
| R6 | 510 | 0.29 | 0.19 | 605 | 1.57 | 13.43 | pIA | 48 |
| R7 | 512 | 0.31 | 2.34 | 611 | 830 | 15.96 | pIA | 48 |
| R8 | 610 | 25.54 | — | 625 | 0.029 | — | — | 49 |
| R9 | 620 | 9.25 | — | 636 | 0.027 | — | — | 49 |
| R10 | 622 | 3.03 | — | 628 | 0.017 | — | — | 49 |
| R11 r-cr. | 433 | — | — | 615 | 2.24 | 4.56 | — | 50 |
| R11 g-cr. | 559 | 1.79 | — | — | — | — | — | 50 |
| R11 g-cr. | — | — | — | 580 | 0.685 | — | PVK | 50 |
| R11 g-cr. | — | — | — | 580 | 0.006 | 13.9 | PMMAa | 50 |
| R12-B | 398 | 3.3, 8.08 × 105 | 1.4 | 546 | 2.1 | 6.7 | — | 52 |
| R12-Y | 398 | 4.5, 8.42 × 105 | 75.2 | — | — | — | — | 52 |
| R13 | — | — | — | 549 | 280 | 5 | — | 53 |
| R14 | — | — | — | 570,620 | 140 | 11 | — | 53 |
| R15 | 635 | 7.2 × 105 | 8.2 | 789 | 0.72 | 1.8 | PVA | 54 |
| R16 | 637 | 5.8 × 105 | 13.9 | 793 | 0.78 | 2.6 | PVA | 54 |
| R17 | 665 | 2.7 × 105 | 15 | 819 | 0.27 | 3 | PVA | 54 |
| R18 | 400 | 2.70 | — | 576 | 10.5 | 26b, 28.7c | — | 56 |
| R18 | 400 | 2.11 | — | 480 | 1.4 | 18, 54.7 | PMMA 1% | 56 |
| R18 | — | — | — | 650 | 5.2 | 17.5, 23.5 | PMMA 50% | 56 |
| R19 | 400 | 2.75 | — | 585 | 0.61 | 48b, 68.2c | - | 56 |
| R19 | 400 | 2.57 | — | 487 | 0.006 | 2, 2.4 | PMMA 1% | 56 |
| R19 | — | — | — | 650 | 0.32 | 30,45.9 | PMMA 50% | 56 |
| R20cr | — | — | — | 655 | 0.087 | 3.4d/7.8e | — | 55 |
|
R20
cr.pw.
|
— | — | — | 652 | 0.010 | 1.8d/5e | — | 55 |
| R20 film | — | — | — | 649 | 0.007 | 0.8d/2.8e | — | 55 |
| R21 cr. | — | — | — | 664 | 0.011 | 11.8d/15.1e | — | 55 |
|
R20
cr.pw.
|
— | — | — | 664 | 0.012 | 6.8d/10.8e | — | 55 |
| R20 film | — | — | — | 662 | 0.01 | 3.8d/4.5e | — | 55 |
| R22 | 403 | — | 27.6 | 541 | 403 | 4.2 | o-Phthalic acid | 58 |
| R22 | 425 | — | — | 569 | 373 | 4.6 | m-Phthalic acid | 58 |
| R22 | 451 | — | — | 649 | 337 | 1.8 | p-Phthalic acid | 58 |
| R23 | 443 | 2.86 | 0.3 | 573 | 79 | — | — | 59 |
| R24 | 412, 432 | 18.4, 18.55 | — | 552, 600 | 423, 414 | 0.3 | — | 60 |
| R25 | 411, 431 | 13.74, 13. 68 | — | 552, 596, 650 | 753, 731, 715 | 0.7 | — | 60 |
| R26 | 411, 432 | 5. 36, 5. 43 | — | 553, 602, 664 | 383, 335, 313 | 7 | — | 60 |
| R27 | 415, 441, 466 | 9.10, 8.25, 7.59 | — | 555, 604 | 6.7, 4.4 | 0.03 | — | 60 |
| R28 | — | — | — | 560 | 170 | 2.3 | — | 62 |
| R28 | 560 | 420f | 3.8f | — | 62 | |||
| R29 | — | — | — | 530 | 40 | 2.3 | — | 62 |
| R29 | 530 | 760f | 5.1f | — | 62 | |||
| R30 | — | — | — | 531 | 190 | 1.5 | — | 62 |
| R30 | 531 | 460f | 3.9f | — | 62 | |||
| R31 | 415 | 1.43 | 14.2 | 600, 657 | 327, 324 | 9.2 | BP | 64 |
| R32 | 424 | 2.03 | 13.4 | 623, 680 | 215, 210 | 8 | BP | 64 |
| R33 | 440 | 1.98 | 15.2 | 643, 697 | 201, 198 | 6.3 | BP | 64 |
| R34 | 471 | 2.19 | 12.3 | 657, 713 | 180, 175 | 5.4 | BP | 64 |
| R35 | 483 | 2.32 | 16.1 | 681, 732 | 106, 102 | 4.2 | BP | 64 |
| R36 | — | — | — | 679, 736 | 0.032 | 11 | — | 65 |
| R37 | 474 | 0.3 | — | 654, 702 | 0.018 | 1.2 | — | 65 |
| R38 | 448 | — | — | 540 | 299.3 | 8.3 | BP | 70 |
| R39 | 417 | — | — | 540 | 120.3 | 3.6 | BP | 70 |
| R40 | 418 | — | — | 541 | 244.7 | 11.2 | BP | 70 |
| R41 | 468 | — | — | 551 | 4.4 | 6.6 | BP | 70 |
| R42 | 440 | — | — | 544 | 26.8 | 4.1 | BP | 70 |
| R43 | 467 | — | — | 553 | 240.4 | 18.6 | BP | 70 |
| R44 | 418 | — | — | 548 | 165.7 | 13.4 | BP | 70 |
| R45 | 422 | — | — | 544 | 93.0 | 7.2 | BP | 70 |
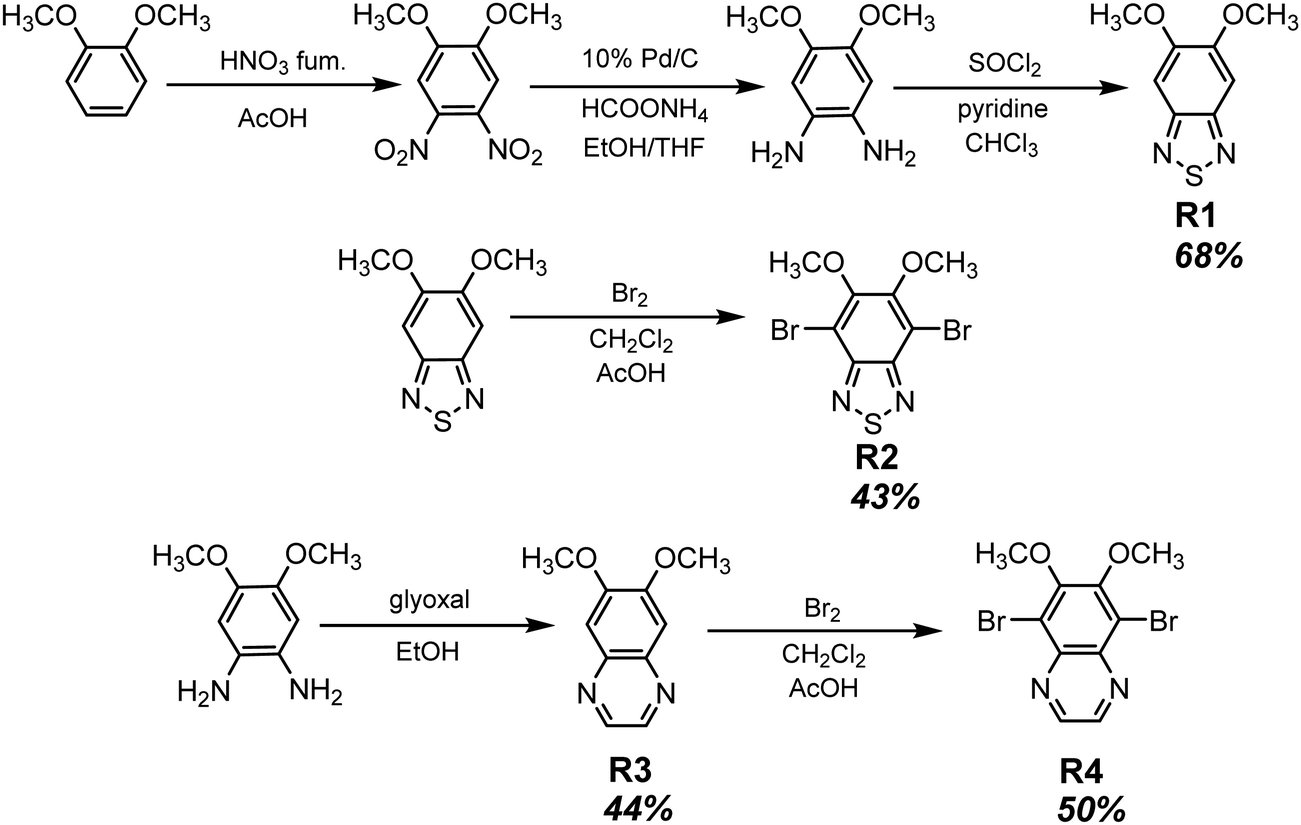 | ||
| Scheme 5 Synthetic routes for compounds R1–R4. Adapted from the literature.46 | ||
In the solid state, the compounds emitted blue light with fluorescence maxima ranging from 402 nm to 448 nm. The observed phosphorescence under ambient conditions ranged from 555 to 648 nm. The highest PHQY (3.7%) as well as the longest τRTP (585 μs at 648 nm) were observed for compound R1 having two methoxy groups incorporated into the benzothiadiazole core. The analogous compound R2 with fluorine atoms instead of methoxy groups exhibited phosphorescence in a shorter wavelength region peaking at 629 nm, with a reduced PHQY (1.8%) and τRTP (233 μs), pointing out that methoxy-groups are superior to fluorine atoms in amplification of RTP of 4,7-dibromo-2,1,3-benzothiadiazole. The authors46 also examined the photophysical properties of compound R4 which is similar to R1 but being without bromine atoms. Based on the significantly reduced PHQY (0.5%) and τRTP (73 μs) of the former, they came to the conclusion that the co-presence of both methoxy groups and bromine heavy atoms is important for phosphorescence of BTD derivatives. To shed light on the triplet harvesting origin of the BDT derivatives, the authors46 investigated compound R1 in more detail. By doping 1% R1 into the rigid PMMA matrix, RTP was observed with intensity maximum at 642 nm. The shape of the spectrum was very similar to that observed for the crystalline sample. The similarity of the RTP properties of R1 in the crystalline state, where molecular packing is tight and in the PMMA matrix where intermolecular interactions are minimized, indicates that RTP of BDT derivatives arises not only from the formation of intermolecular aggregates but also from inherent monomeric characteristics.
Compound R5 (Fig. 17 and Table 3) that has thionyl units fused with a quinoline ring was obtained via Suzuki–Miyaura coupling between 3,4-dibromoisoquinoline and 2-methylbenzo[b]thiophene pinacol boronic ester and next methylation with subsequent anionic exchange in 84% yield.47 Steady state investigations of the film of R5 revealed green fluorescence with a lifetime of 5 ns. The PL spectrum of the film of R5 with a delay of 120 μs revealed the disappearance of fluorescence and appearance of red-shifted emission with the maximum at 575 and a lifetime of 1.9 ms, which was recognized as RTP. The authors of ref. 47 supposed that the formation of a boron complex was the key factor that helped in achieving RTP for R5. The cation–anion interactions between the aromatic unit and the [BF4]− anion apparently isolated the emissive thiophene-quinoline core from molecular oxygen and promoted harvesting the energy from triplets. To verify the assumption, a non-complexed precursor of R5 was investigated and no RTP was observed. Quantum chemical simulations of collision complexes between O2 and organic chromophore models6,12 support this assumption.
Stabilization of sulfur-containing boronic complexes with an appropriate rigid host matrix can not only improve RTP but also make it pH-sensitive. Xiao et al.48 designed and synthesized compounds R6 and R7 (Fig. 17 and Table 3) in a three-step synthetic pathway (Fig. 12a and c). The formation of the corresponding boronic complexes was achieved by a treatment of intermediates possessing a free NH-group with boron trifluoride diethyletherate in DCM. Both resulting compounds were RTP-inactive in the crystalline form; however, red RTP was observed after doping of 1 wt% of R6 and R7 into a p-iodoaniline (p-IPhNH2) matrix. X-ray analysis revealed the built-in halogen bonds between the cyano-group of R6 and the iodine atom of p-IPhNH2 host molecules and between the iodine atoms of R7 guest molecules and the amino-group of the p-IPhNH2 host. The molecules of the guests became strongly restricted in motion that allow triplet excited states to relax radiatively. After treatment with hydrochloric acid the concurrent formation of complexes p-IPhNH2·HCl takes place, thus disrupting the iodine-NH2 guest–host bonding in the R7-p-IPhNH2 system and consequently turning off the red RTP (Fig. 12d). In the case of RTP, persistent luminescence of the assembled R6-p-IPhNH2, where intermolecular bonding does not involve NH2-groups, remained unchanged (Fig. 12b).
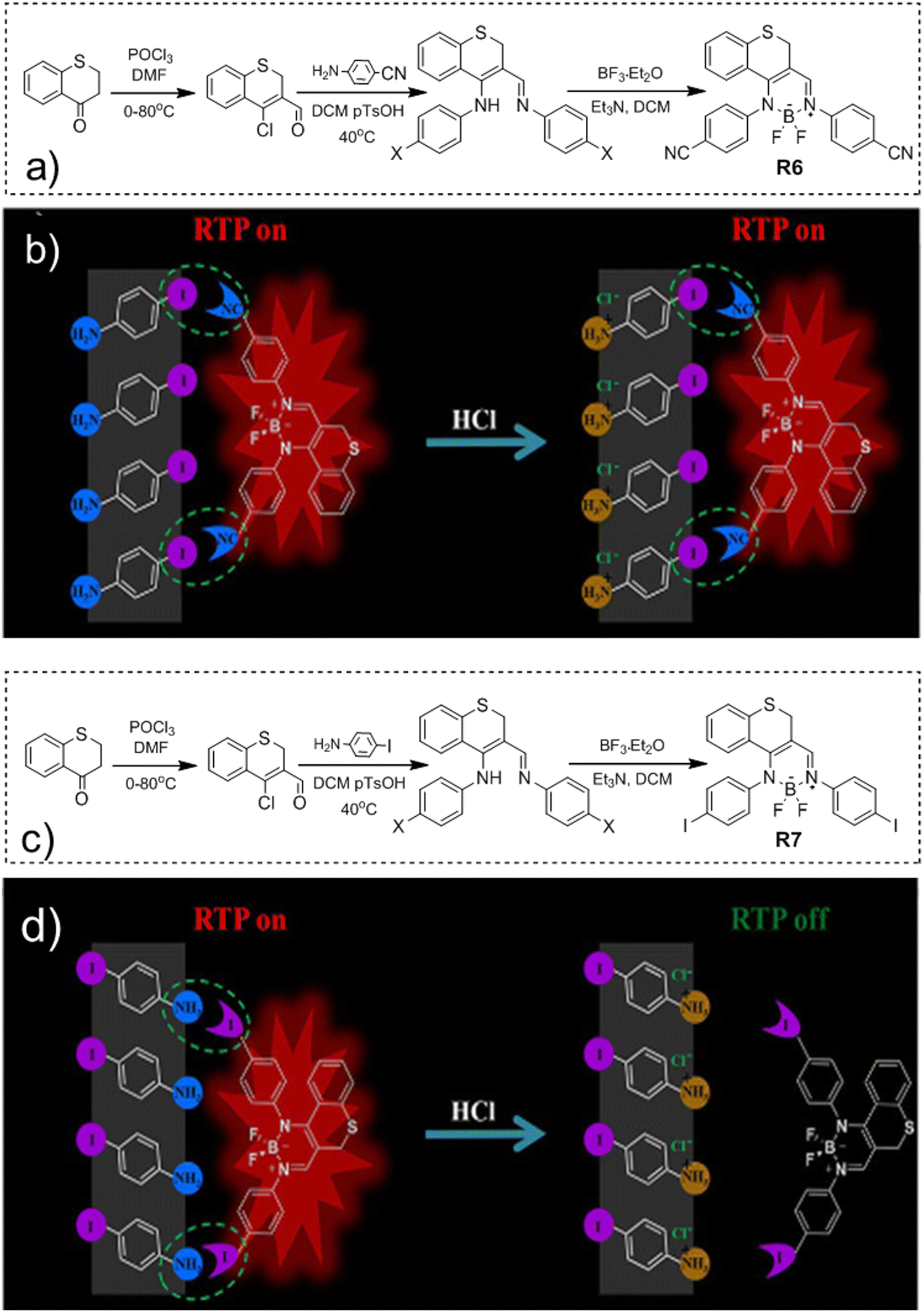 | ||
| Fig. 12 (a) Synthetic route for the synthesis of compound R6, (b) schematic mechanism of HCl-sensitive behaviour of R6, (c) synthetic route for the synthesis of compound R7, and (d) schematic mechanism of HCl-sensitive behaviour of R7. Reproduced from ref. 48 with permission from John Wiley and Sons, Copyright 2023. | ||
Three boron-containing organic complexes that show RTP in aqueous media were reported by Wang et al.49 Difluoroboron β-diketonate derivatives R8–R10 (Fig. 17 and Table 3) were obtained via two-step synthesis from acetyl-carbazole intermediates and the corresponding naphthalic acid methyl esters. All the complexes in an aqueous solution exhibited fluorescence and RTP upon excitation with visible light (470 nm). The longest living RTP (29 μs) was observed for halogen-free compound R8. The mostly red-shifted phosphorescence peaking at 636 nm was observed for R9 that has a naphthyl fragment substituted with a bromine atom. The authors49 managed to achieve RTP of R8–R10 even under NIR excitation. The compounds exhibited maximum emission by femtosecond laser excitation in the wavelength range of 780–820 nm. The examination of the photophysical properties of dilute THF solutions of the compounds revealed that the corresponding luminescence maxima were significantly blue-shifted. The phosphorescence intensity maxima observed at 77 K also were blue-shifted with respect to those observed at room temperature. It was assumed that RTP of all these compounds may arise from some form of aggregates rather than from monomeric species.49 The single-crystals of R9 and R10 were obtained and it was revealed that the neighbouring molecules are in close proximity to each other. On the basis of the obtained crystallographic data quantum chemical calculations were performed for the monomeric and dimeric structures of R9 and R10. The calculated energy gap between the lowest singlet and triplet excited state of the dimer was substantially lower than those of the monomeric forms (Fig. 13). Therefore, in the case of dimers with lower energies ISC was promoted and efficient intermolecular interactions were enabled, which resulted in efficient RTP. These lower energy dimers acted as the ISC channels between S and T states, resulting in the decrease of ΔEST. Thus, they enabled multiple intermolecular interactions which resulted in the appearance of efficient RTP in aqueous media.
 | ||
| Fig. 13 Molecular structures of compounds R8–R10 and Jablonski diagram illustrating the origin of RTP from their dimers. Adapted with permission from ref. 49. Copyright 2023, American Chemical Society. | ||
Huand et al. decided to decorate the phenanthrene fragment with bromine and cyano groups with the aim of not only facilitating ISC but also obtaining tighter molecular packing by enchancing intermolecular contacts.50 The luminophore R11 (Fig. 17 and Table 3) was obtained from 2-(4-bromophenyl)acetonitrile in two steps (Scheme 6). The steady state and time-resolved luminescence spectra of R11 in the crystalline form exhibited intensity maxima at 615 nm with a PHQY of 4.56% and a luminescence lifetime of 2.24 ms indicating that the nature of luminescence was RTP. When the crystal of R11, namely, R11-r was left under ambient conditions for 24 hours, its colour changed from red to yellow (R11-g), along with the emission colour altering to green. Furthermore, the steady-state spectra recorded showed that the mentioned green emission (559 nm) had nanosecond-range lifetimes, pointing out its fluorescence origin. These authors decided to evacuate the R11-r crystal for 25 min and obtained the same change in colour and emission. When the the R11-g crystal was annealed for 6 min with DMF vapor, the red emission was restored, while green fluorescence significantly shrinked in intensity. Further experiments proved that the process was reversible and can be repeated more than 10 times. The crystal of non-brominated dicyanophenanthrene showed no red phosphorescence itself, proving that the bromine heavy atoms are essential to initiate phosphorescence. However, it was reported that DMF can decompose into N,N-dimethylamine upon heating.51 Therefore, it was speculated that the substitution product of R11 by N,N-dimethylamine, namely N-R11 may be present in the R11-r crystal. To prove the assumption, N-R11 was synthesized according to the route reproduced in Scheme 6. After that, a co-crystal of N-R11 and R11 was grown and investigated. It emitted in the very same way as compound R11-r, losing RTP after evacuation and restoring it after fumigation with DMF. X-ray analysis revealed that in the case of R11-r red phosphorescence occurred from the host–guest system cluster of R11 and N-R11, in which DMF stabilized the crystal lattice by providing abundant intermolecular interactions.
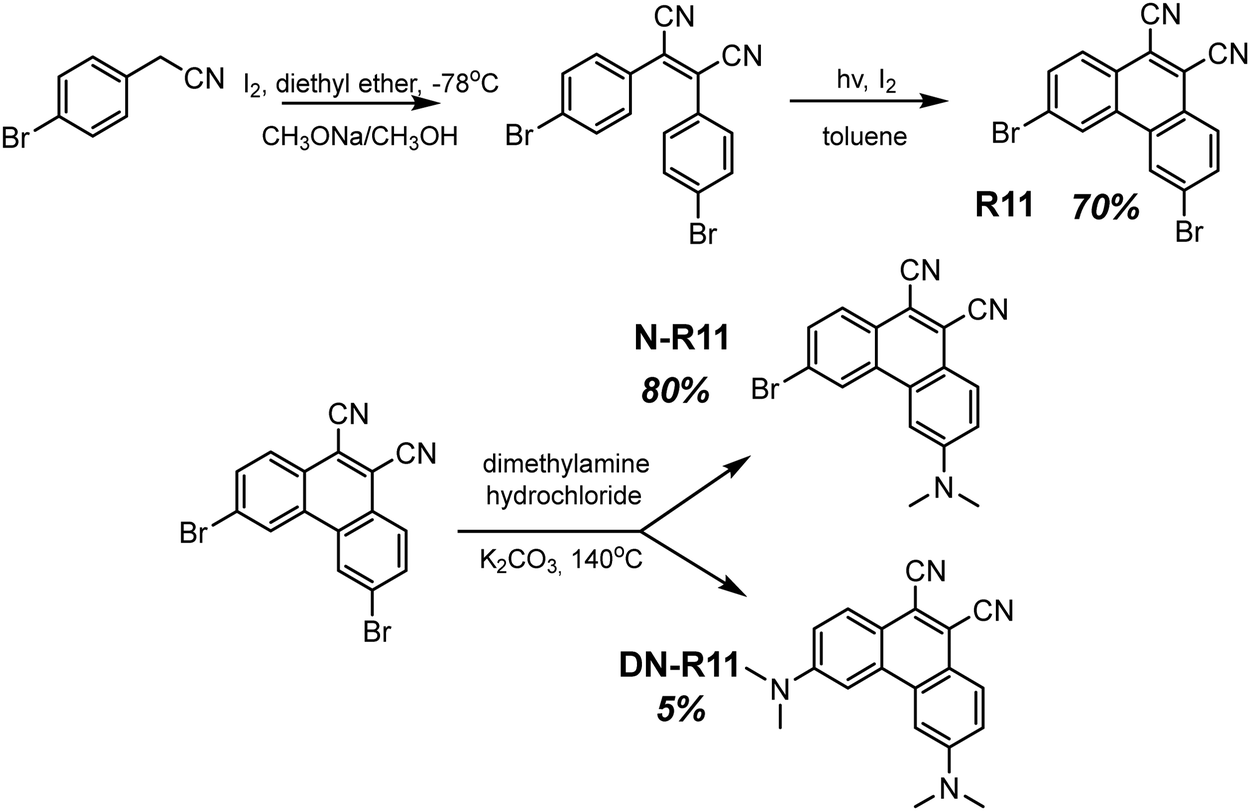 | ||
| Scheme 6 Synthetic route for obtaining compounds R11. Adapted from the litetature.50 | ||
Although crystallization is quite a facile strategy to promote RTP by tightening molecular packing, for a single compound with polymorphism one crystalline form may demonstrate RTP while another may be RTP-inactive. The work of Wen et al. is an illustrious example.52 Compound R12 (Fig. 17 and Table 3) that has a structure very similar to common organic RTP luminophores was synthesized in one step from iminostilbene and p-chlorobenzoyl chloride in 68% yield. Two types of crystals were obtained by different methods: R12-B crystals were obtained by sublimation and R12-Y crystals were obtained by slow evaporation of the R12 DCM solution. The R12-B crystals showed a rare highly efficient near-ultraviolet emission with the intensity maximum at 398 nm, accompanied with a very high PLQY of 75.2% The PL decay of the R12-B crystal exhibited prompt (4.5 ns) and delayed (842.0 μs) components. The R12-Y crystals exhibited yellowish emission with a PLQY of 8.1%. Two bands contributed to the resulting emission: a weak band at 398 nm (τPF of 3.3 ns, τDF of 808.3 μs) and a major band peaking at 546 nm (τ of 2.1 ms) that had obvious RTP character. Single crystal X-ray analysis was performed for both types of crystals and it was revealed that R12-B crystals exhibited a donor iminostylbene fragment rotation by 9.55° with respect to the acceptor fragment. The corresponding angle for R12-Y crystals was 18.9° (Fig. 14). Thus, molecular packing and the degree of intermolecular interaction were different. Based on the difference in the crystal structure, theoretical and experimental investigations were further performed to elucidate the emission mechanism of the crystals of R12. Singlet–triplet energy splittings were calculated to be too large, enabling rISC, therefore, TADF as a mechanism of blue emission in both crystal forms was excluded. The delayed component in blue emission originated from triplet–triplet annihilation (TTA), which was proved experimentally by recording the photoluminescence spectra of crystals under different excitation powers. Additionally, participation of upper triplet state T2 was confirmed for R12-Y crystals by recording their PL spectra at different temperatures. A new emission band appeared at ca. 438 nm at 80 K with a long lifetime of 71.1 ms. Thus, it was demonstrated that this new emission peak originated from the high-lying excited state T2.
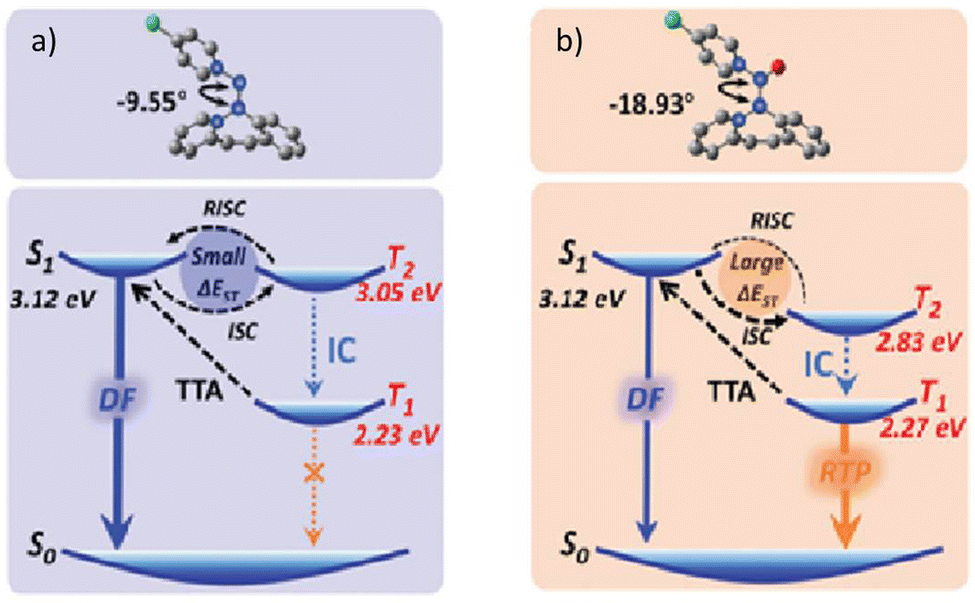 | ||
| Fig. 14 Geometries and schematically drawn mechanisms of delayed fluorescence in R12-B (a) and R12-Y (b) types of crystals. Reproduced from ref. 52 with permission from Royal Society of Chemistry, Copyright 2023. | ||
It was further predicted by calculations52 that in the case of R12-B, when the donor–acceptor twist angle is small, only TTA is achieved through the rISC between the T2–S1 process due to the small ΔES1–T2. With an increase in the twist angle, the energy level of the T2 state is lowered and therefore RTP is achieved for R12-Y crystals from the T1 excited state via a more competitive internal conversion (T2–T1) process in comparison to the slower rISC (T2–S1) process. The tighter molecular packing contributed to the RTP-dominated emission of the R12-Y crystal in addition to more effective ISC for triplet exciton population.
The effect of incorporation of a long alkyl chain into the RTP-luminophore on molecular packing was studied by Fateminia et al.53 A soft alkoxy spacer group was introduced between the moieties of, already reported by that time, RTP agent R13 (Fig. 17 and Table 3) aiming to prolong the emission lifetimes and enhance PHQY. A butoxy spacer helped to separate carbazole and the 4-bromobenzophenone groups, thus affording a new luminogen R14 (Fig. 17 and Table 3) with a more versatile conformational behaviour. Compound R14 was obtained from p-bromobenzoyl chloride in 90% yield. X-ray analysis revealed that the distance between carbazole and bromobenzophenone units in compound R14 was shortened, compared to those observed in R13. That indicated stronger intermolecular interactions and more efficient RTP. Due to the crystal packing differences, even the effect of the bromine heavy atom was different. The C–Br bond in the crystal of R13 is far from the carbazolyl fragment. It was arranged nearly parallel to it, weakening the effect of the heavy halogen atom on the carbazolyl unit and resulting in weaker RTP. Taking into account of the observed long phosphorescence lifetime of 0.14 s for R14, it can be characterized as one of the most efficient red-emissive persistent RTP materials reported so far.
By incorporating multiple bromine atoms into the well-known organic acid–base indicator phenolsulfonephthaleine, Sun et al.54 recently managed to achieve near-infrared RTP. They doped the R15–R17 derivatives with different amounts of bromine atoms into a rigid PVA matrix. It was reported earlier55 that the R15–R17 derivatives were highly sensitive to the acidity of the media and undergo structural transformation in acidic and neutral aqueous solutions (Fig. 15). In acidic media, sulfogroups of compounds R15–R17 were found in a close-ring structural form while in neutral and alkaline media, the anionic form prevailed, resulting in ring opening. The authors54 prepared two types of the films of the molecular dispersions of R15–17 in PVA. The films of the first type were prepared under acidic conditions and the films of the other one were fabricated under neutral conditions. The films, fabricated under acidic conditions, were light-yellow, whereas those fabricated under neutral conditions were dark-blue, manifesting that compounds R15–R17 appeared in the different forms in the differently prepared films. Photophysical investigation revealed that the neutral films exhibited TADF with the intensity maxima at 600–700 nm and RTP peaking above 750 nm. Eventually, the film of the molecular dispersion of R17 in PVA exhibited the reddest RTP peaking at 819 nm with τRTP of 0.27 ms. The films prepared under acidic conditions exhibited only red fluorescence and no RTP was observed.
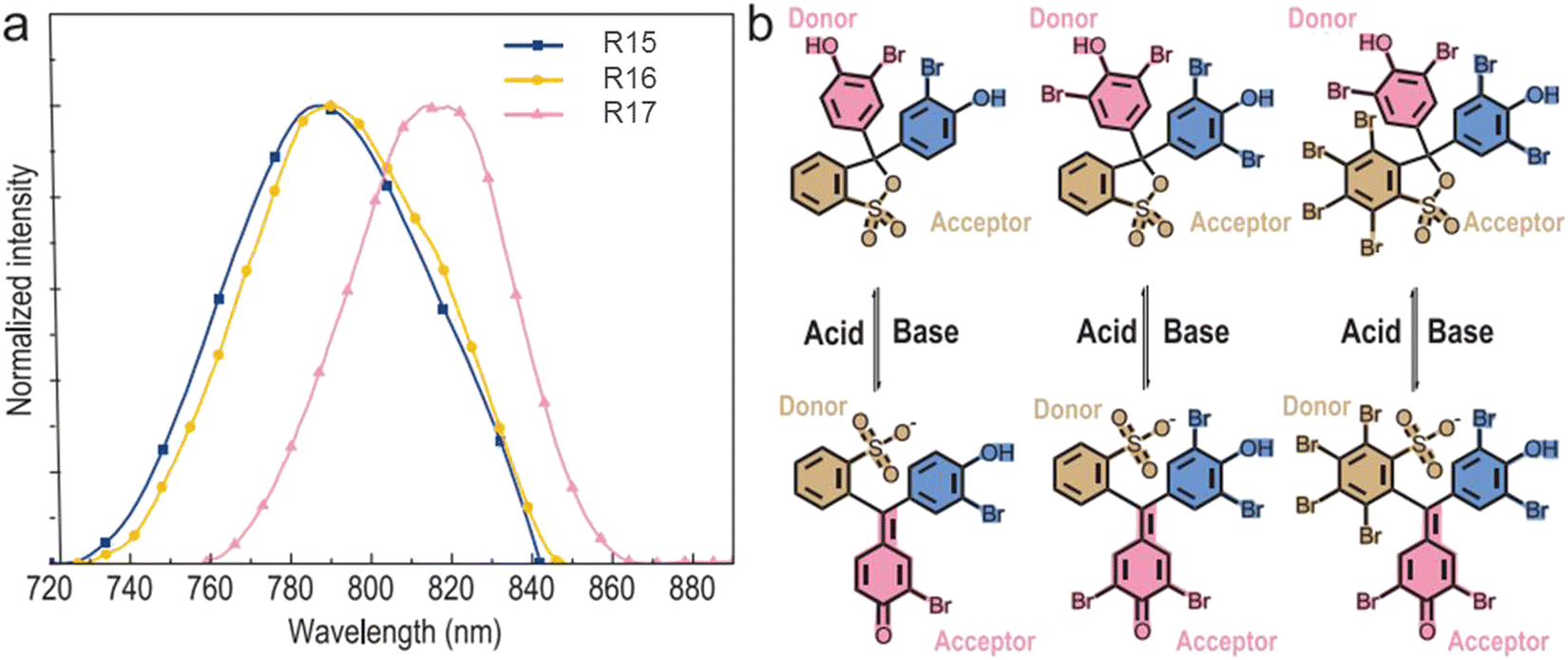 | ||
| Fig. 15 (a) The RTP profiles of molecular mixtures of compounds R15–R17 with PVA; (b) structural transformation of R15–R17 in an aqueous solution with differing pH values. Reproduced from ref. 54 with permission from Oxford University Press. Copyright 2023. | ||
Aromatic imides, such as naphthalene monoimide and phthalimide derivatives are well-known to exhibit orange-red RTP even in the presence of oxygen. Garain et al.56 reported the derivatives of arylene diimide R18 and R19 (Fig. 17 and Table 3) functionalized with heavy halogen atoms not only as RTP agents in the long-wavelength spectral region but also as very rare examples of air-persistent RTP. In order to harvest triplet excitons by diminishing the non-radiative energy dissipation the authors56 dispersed 1 wt% of R18 and R19 in a PMMA rigid medium. A strong cyan emission was witnessed for the films of the dispersions of both R18 and R19 with the corresponding maxima at 480 (τRTP = 1.4 ms) and 487 nm (τRTP = 6.25 μs). In air, the corresponding PHQY values were 18% and 2%, but in a vacuum the R18 containing film exhibited a remarkably high PHQY of 54%. Interestingly, while increasing the ratio of PMMA to 50%, triplet excitons did not become quenched by the intensified molecular motions but the RTP maxima were shifted to orange-red region peaking at 576 (τRTP = 5.2 ms) nm and 585 nm (τRTP = 0.32 ms) for R18 and R19 (Fig. 16). The authors56 assumed that the presence of abundant non-covalent interactions in the aggregated state helps to stabilize the triplet energy. This assumption was further supported by the investigation of the photophysical properties of the single crystals of R18 and R19 that exhibited orange-red RTP with maxima at 576 and 585 nm for R18 and R19, indicating the similarity to the phenomena observed for 50% molecular dispersions of the compounds in PMMA. Both the crystals of R18 and R19 demonstrated high PHQYs of 26% and 48% in air, which are among the highest PHQY values reported for the orange-red RTP emitters.
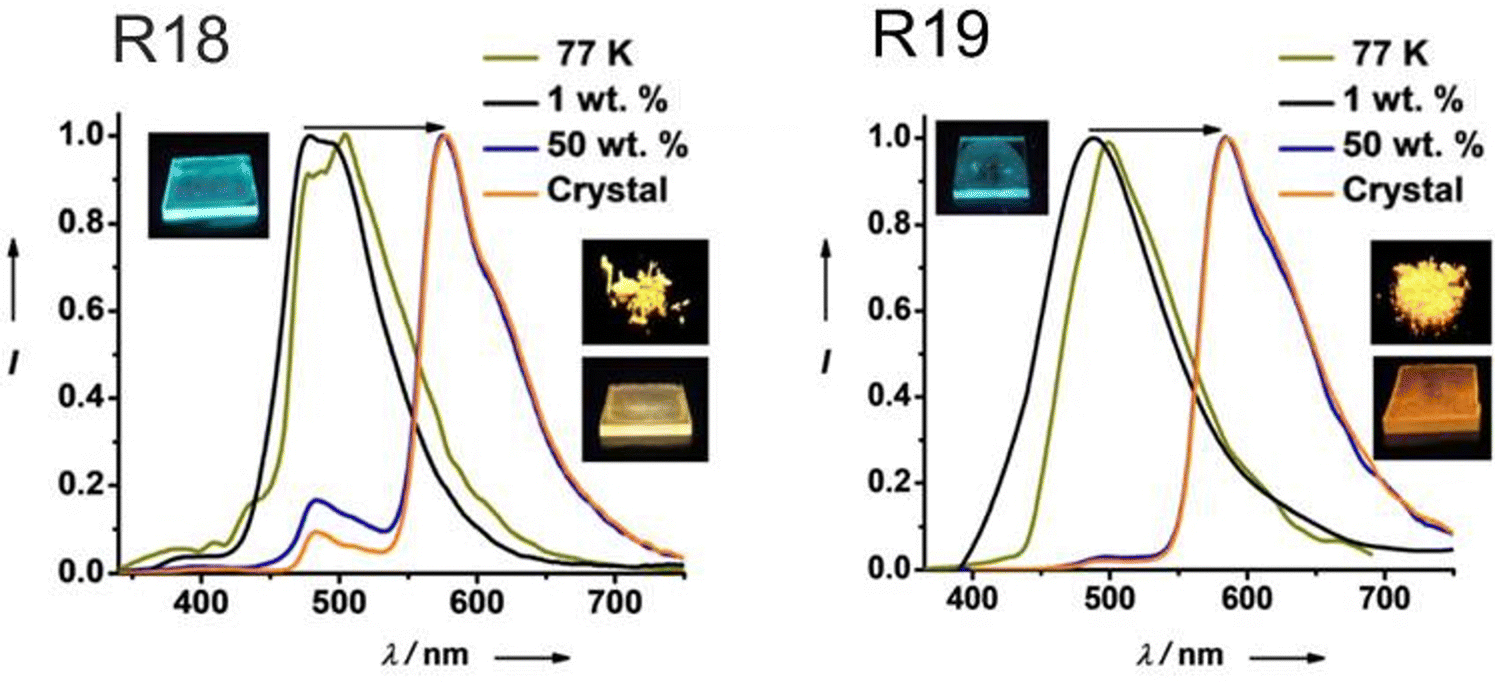 | ||
| Fig. 16 Phosphorescence spectra of the solutions of R18 and R19 in frozen THF recorded at 77 K of the films of molecular dispersions in PMMA with the concentrations of the phosphors of 1 wt% and 50 wt% and of the crystals. Reproduced from ref. 56 with permission from John Wiley and Sons. Copyright 2023. | ||
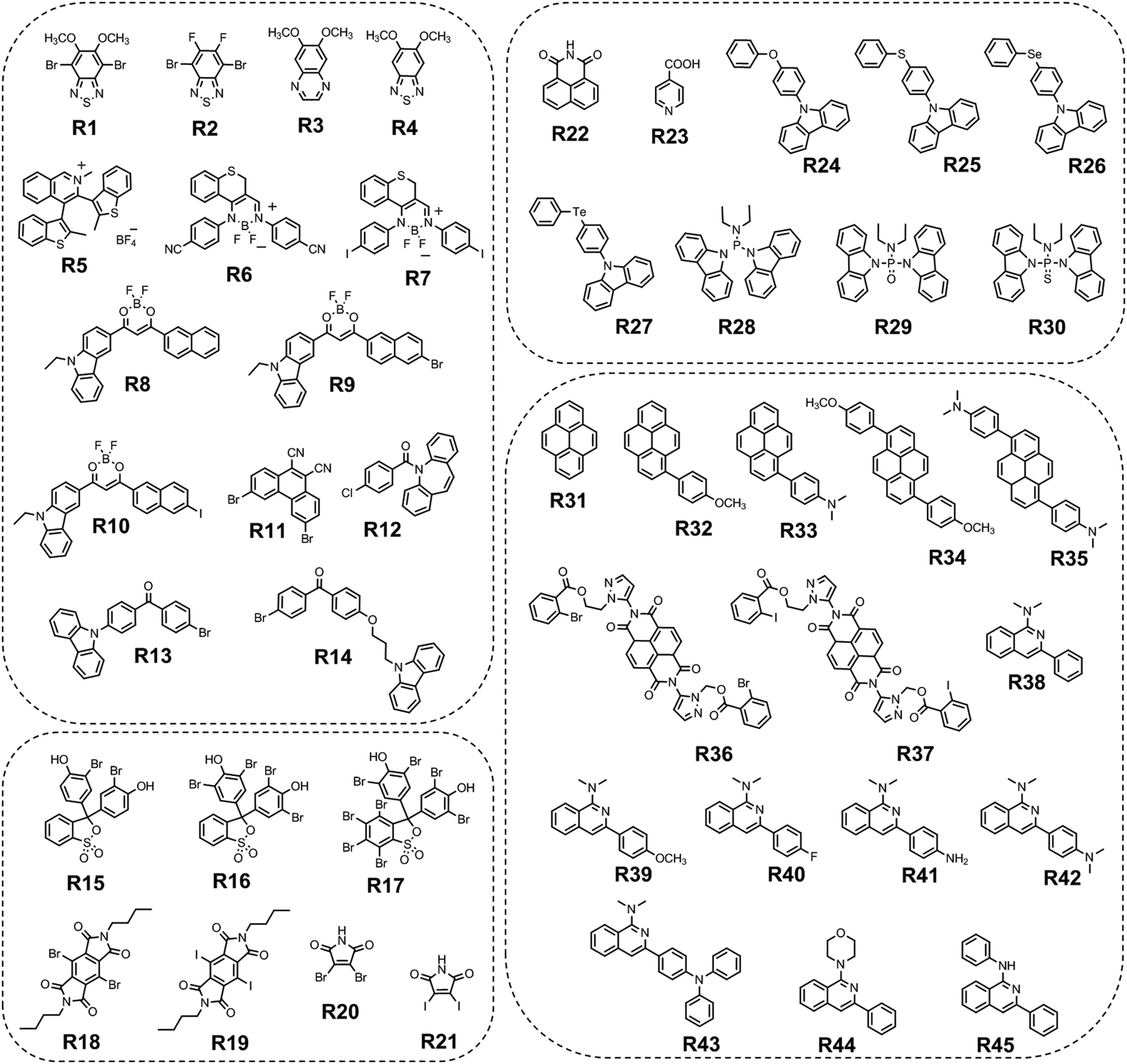 | ||
| Fig. 17 Molecular structures of red RTP-agents R1–R45. | ||
A limited number of organic crystals exhibiting RTP persistent under high humidity conditions have been reported.57 Interaction with water is destructive for RTP since water molecules easily destroy the original intermolecular bonding in crystals and form hydrogen bonds. Furthermore, molecular oxygen present in water quenches triplet excitons. Wang et al. reported halogenated maleimide derivatives R20 and R21 (Fig. 17 and Table 3) as efficient RTP-agents in the near infrared region.55 Their RTP demonstrated unique resistivity to water. Yellow crystals of R20 and R21 emitted bright red light under a UV lamp. Upon excitation at 450 nm their emission spectra revealed broad unstructured peaks ca. 650 nm. The emission lifetimes were found to be 87 μs and 11 μs for R20 and R21, respectively. For compound R20, the excitation spectrum revealed four excitation peaks at 385 nm, 405 nm, 450 nm, and 530 nm. Upon all these excitations, compound R21 demonstrated very similar phosphorescence decay profiles with constant lifetimes, indicating that RTP of R20 originates from one emission centre. The authors of ref. 55 also studied the impact of morphology of the solid samples on RTP of R20 and R21, fabricating their thin films with a thickness of ca. 465 nm. Due to the low thickness, some light penetrated the surface before getting absorbed by it. The films demonstrated photophysical behaviour very similar to that of the crystalline samples confirming that red RTP of R20 and R21 is stable and independent of the morphology of the samples. PHQYs of the films were lower than those of the crystals. This observation can be attributed to the degradation of the crystalline lattice to some extent that facilitates the molecular motions and leads to nonradiative energy losses. In order to establish whether the RTP properties of maleimides R20 and R21 originate from the single molecules or from aggregates, a single crystal X-ray analysis was performed. It was revealed that the molecules of both R20 and R21 are packed in a quite stable 2D-layered structure, that is stabilized by abundant intermolecular interactions. No π–π stacking was observed that indicated the absence of J-type and H-type aggregates and proved that the RTP properties of R20 and R21 originated from single molecules. Amazingly, after soaking the R20 crystal in water for one hour, the phosphorescence remained almost unchanged. The authors of ref. 55 explained such a high water stability by a stiff layered structure stabilized by halogen bonds, preventing the diffusion of water into the crystalline lattice.
Wang et al.58 succeeded in evoking RTP from 1,8-naphthalimide R22 (Fig. 17 and Table 3) by doping it into isomeric dicarboxylic acids. Planar and rigid R22 is reported to be an efficient fluorescent material without any hints of delayed emission at room temperature due to its large singlet–triplet energy splitting. The authors58 considered the aromatic dicarboxylic acids as favourable hosts for R22 to activate RTP because of their high PHQYs, long phosphorescence lifetimes and high singlet and triplet energy levels which ensure efficient energy transfer to the R22 guest. Additionally, plentiful intermolecular interactions can be formed between the carboxylic groups of the aromatic acid and the imidyl group of R22 that results in a stable molecular framework. Guest–host films of R22 and ortho-, iso- and terephthalic acid were prepared. All the films exhibited a bright yellow afterglow after ceasing the UV irradiation. The best RTP-performance was demonstrated by the film of the molecular mixture of R22 isophthalic acid with a τRTP of 1.169 s and a PHQY of 5.9%. The authors of ref. 58 also investigated the impact of guest concentration on RTP. After increasing the molar ratio of the R22 guest from 0.02 to 1 mol%, the relative RTP intensity significantly decreased. That could be caused by triplet quenching. A concentration lower than 0.02 mol% resulted in inefficient energy transfer that again led to inefficient phosphorescence.
The essential role of intermolecular interactions in evoking RTP in organic systems was proved by Chen et al.59 The isonicotinic acid R23 (Fig. 17 and Table 3) was found to be an excellent RTP agent being in the crystalline state but lost its RTP properties when dispersed in the rigid PMMA matrix. Under excitation two luminescence bands were observed for the crystals of isonicotinic acid R23. The fluorescence band peaked at 443 nm while the RTP band peaked at 573 nm (τRTP = 79 ms). After doping isonicotinic acid into the PMMA matrix it did not exhibit RTP. This observation suggested that RTP of R23 crystals was initiated by hydrogen bond interactions between the carboxyl group and the pyridine nitrogen atom.
Xu et al. studied the impact on RTP of heavy chalcogen atoms by combining a chalcogen-linked diphenyl fragment with the carbazolyl donor unit (R24–R27, Fig. 17 and Table 3).60 The simplicity of the structure made it possible to tune the RTP properties by incorporating chalcogen atoms with different sizes something that made it possible to study the mechanism of RTP at a single-molecular level. All the compounds in the crystalline state demonstrated structured fluorescence peaking in the range of 400–470 nm. For selenium-containing R26 an additional band appeared at 525–675 nm. After switching off the excitation source, all the samples emitted yellow light with maxima at 525–675 nm. The similarity of emission profiles of the compounds indicates that they had similar T1 energies. Nevertheless, the PHQYs and τRTP were different: 0.3% and 423 ms for R24; 0.7% and 753 ms for R25; 7% and 380 ms for R26 and 0.03% and 7 ms for R27. The phosphor R27 exhibited one of the longest phosphorescence lifetimes among the reported tellurium-containing compounds. A single crystal X-ray analysis revealed that molecular conformations as well as the crystal packing were similar for all the compounds, indicating that differences in the RTP-behaviour were predetermined by the individual chalcogen atom. In the case of compounds R24–R25 the low values of spin–orbit coupling were predicted indicating a rather weak ISC for these compounds. In contrast, compounds R26–R27, featuring heavier selenium and tellurium atoms, have high spin orbit coupling indicating that an electron can be easily moved from S1 to Tn. R27 exhibited the largest SOC constant that resulted in the shortest phosphorescence lifetime among the compounds of the series.
Tao et al.61 synthesized three photoresponsive organic luminophores containing phosphorus atoms R28–R30 (Fig. 17 and Table 3). Steady-state luminescence spectra revealed orange ultralong organic phosphorescence for the powders of compounds R28 and R30. Considerably shorter RTP with a lifetime of 40 ms was measured for the sulfur-containing compound R30. When the powders of all three compounds were irradiated with 365 nm UV light, their phosphorescence lifetimes as well as phosphorescence quantum yields profoundly increased. A most significant change in PHQY by a factor of 18.5 was observed for compound R29.62 Pyrene is widely used in the design and synthesis of electroactive materials due to the high population of triplet excitons.63 Xiao et al.64 recently proposed a facile method of activating RTP in pure pyrene R31 (Fig. 17 and Table 3) as well as in its mono- and di-substituted derivatives R32–R35 (Fig. 17 and Table 3) by doping them into a benzophenone host matrix. Highly conjugated pyrene-based guests had T1 energy levels sufficiently low to afford RTP in the red/NIR area. The authors64 chose benzophenone as a host with the aim of promoting exciton transfer from the guest to the host and to diminish their non-radiative transitions by suppressing molecular motions. Resulting RTP was observed in the range of 600–732 nm together with long τRTP ranging from 102 ms to 324 ms. Through this smart guest–host architecture, the authors64 managed to achieve both long τRTP and emission in the NIR area, which is a perfect combination for biomedical applications. The stability of guest–host films containing R31 and R33 was studied. After soaking in water for 24 h, the films still had a strong red afterglow. Their phosphorescence remained almost unchanged after keeping under ambient conditions for a month.
Near-infrared RTP derivatives of naphthalene diimide (NDI) were reported by Ono et al.65 Similarly, to its hydrocarbon relative pyrene, NDI has low-lying triplet energy levels that result in large ΔEST.66 Efficient RTP was realized in metalorganic complexes where NDI acted as a ligand. The intermediate NDI-derivative was synthesized in 58% yield by the reaction between 1,4,5,8-naphthalenetetracarboxylicdianhydride and 5-amino-1-(2-hydroxyethyl)pyrazole.65 Compounds R36 and R37 (Fig. 17 and Table 3) were obtained via coupling of an intermediate with the corresponding halobenzoates. The NDI-derivative with a 3-chlorodenzoate unit exhibited only fluorescence while a compound bearing a 3-bromobenzoate unit (R36) afforded both green fluorescence and orange RTP. Surprisingly, two out of three iodine-containing compounds that have an iodine atom introduced at C-3 and C-4 positions of the benzene ring exhibited no RTP while compound R37 that has the C-2 position occupied by iodine demonstated bright NIR RTP with a lifetime of 11 μs. At first glance, there is a trend in RTP performance of the series determined by the heavy atom effect that promotes ISC. While the chlorine-containing derivative showed no RTP, the bromine containing compound showed both RTP and fluorescence. To clear up why there was no RTP observed for the two iodine-containing isomers, the authors65 performed single X-ray analysis for the compounds containing iodine atoms. It was revealed that the compounds formed open structures originating from a flexible linker at the NDI core and halobenzoate fragments. In the case of compound R37, the NDI core and 2-iodobenzoate packed closely and formed face-to-face intermolecular stacking. The authors65 assumed that such packing promoted CT interaction between molecules, which resulted in RTP. This assumption was confirmed by quantum chemical calculations.
Isoquinoline derivatives are widely used in organic optoelectonics due to their high photostabitity and a great possibitity of structural modifications.67–69 Chen et al.70 reported isoquinoline-based compounds R38–R45 (Fig. 17 and Table 3) which exhibited persistent yellow afterglow at room temperature when doped in a benzophenone matrix. They suggested a simple and cost-effective method of obtaining isoquinoline derivatives using copper catalysts such as copper(I) iodide. Compounds R38–R45 containing different peripheral moieties were doped into the benzophenone host (molar ratio 1:100). A persistent yellow afterglow was observed for all the films after ceasing the excitation source at 418–468 nm but with very different lifetimes. Phosphorescense lifetimes and PHQYs were lower for compounds R39, R41, and R42, indicating that functionalizing the isoquinoline core with electron-donating groups results in a lower RTP performance. Triphenylamine-containing compound R43 exhibited the highest PHQY and one of the longest τRTP among the whole family.
6. White RTP agents
The fabrication of white solid-state light-emitting devices is a challenging task.71 A conventional strategy to generate white emission is to appropriately adjust emissive layers with complementary blue/orange or primary blue/green/red colours of emission in a multi-layered device.72 This approach has a series of drawbacks, including unavoidable phase separation that appears as a consequence of a multi-layered structure and results in unstable luminescence. An intuitive strategy to overcome the issue is to fabricate a device that consists of one white light-emitting layer. This means the necessity of obtaining of a combination of several emission bands in a single molecule. As long as white light is composed of emission with different wavelengths, it is practically impossible to reach an RGB spectrum with the same excitation wavelength and with the same photoluminescence lifetime using one compound. If three different types of chromophores (i.e., blue, green and red) are combined in a single molecule, the resulting emission arises from the red moiety. For the single molecule, the emissive spectral coverage of 400–700 nm (the spectral coverage of white light) is not consistent with Kasha's rule that states that the photons can only be emitted from the lowest excited state.73 Various approaches are explored with an attempt to achieve single-molecule white emission, e.g., aggregation-induced emission enhancement (AIEE), molecular excited-state intramolecular proton transfer (ESIPT), excimer and electroplex formation.74 In particular, harvesting of triplet emission imposes a vast contribution to the above-listed phenomena. A balanced white spectrum can be achieved either from blue fluorescent material by carefully adjusting the suitable host that induces orange afterglow or from the co-existence of blue fluorescence and complementary orange RTP in a single molecule. The latter seems to be more challenging but advantageous from a technological perspective. It is possible to outline a number of general strategies for the design of purely organic molecules with dual excited state emission to afford white light:(1) The singlet excited state should not be converted into a triplet excited state ultimately, because in the opposite case we will have only phosphorescence. Therefore, ISC should be rather balanced, not too rapid, to achieve both fluorescence and phosphorescence because for white light, we are interested in both.
(2) Singlet–triplet energy splitting has to be ca. 0.2 eV. Too large splittings will not be efficient in evoking triplet excited states, while too small splittings may initiate thermally activated delayed fluorescence.
(3) The structure of the emitter has to be sterically hindered to suppress the non-radiative energy dissipation.
The first statement is essential for white light generation whereas the 2nd and the 3rd ones are general rules for the initiation of RTP in pure organic compounds.
Along the path of searching for organic white-emitting materials, Yang et al.26 came across an interesting finding. By studying the pressure effect on RTP, they discovered that the well-known yellow RTP-agent thianthrene is an uncommon white emitter.26 The authors26 compared the pressure-responsive emission of thianthrene W1 with that of selenanthrene G7 (Fig. 11 and Table 2). Both thianthrene and selenanthrene are RTP-active due to the presence of heavy atoms and a folded structure that facilitates spin–orbit coupling. However, in the case of heavier selenanthrene, SOC is strengthened and only pure RTP was observed, whereas thianthrene W1 showed dual-emission, i.e., minor blue fluorescence and the dominating yellow RTP (Fig. 18a). Isotropic hydrostatic pressure was applied to the powder of W1 with the aid of a diamond anvil cell. As the pressure was increased, more molecular interactions occurred which effectively suppressed restrictions of molecular motions and non-radiative quenching of triplet excitons. Interestingly, for thianthrene W1, high pressure did not close up a fluorescence radiative way by shortening the intermolecular distances and promoting ISC, but intensified it to the point comparable with RTP (Fig. 18b). The authors26 explained such unusual enhancement by the prevalence of nonradiative molecular motions over the ISC process. Consequently, both RTP and fluorescence were enhanced profoundly, which resulted in the change of the emission colour from yellow to white (Fig. 18c). The most intense white emission was achieved at a pressure of 4.55 GPa. At a higher pressure the emission shrunk drastically.
 | ||
| Fig. 18 (a) Emission spectra of the crystal of W1 observed under ambient conditions, (b) pressure-responsive luminescence spectra of the single crystal of W1; and (c) luminescence photographs of the single crystals of W1 under different pressures. Reproduced from ref. 26 with permission from Royal Society of Chemistry, Copyright 2023. | ||
Two unique examples of the smallest single-molecular white emitters were reported by Zheng et al. in 2021.75 One of the first attempts to develop tiny single-molecular white emitters was made by Zhang and co-workers, who reported derivatives of triazole with intrinsic broadband white light emission in 2017.76 However, the white emission of the mentioned derivatives did not include RTP but originated from supramolecular aggregates and involved several processes, like excited-state intramolecular proton transfer (ESIPT), synergetic emission from a monomer and excimer/exciplex, aggregation-induced emission (AIE), delayed fluorescence, and simultaneous emission from singlet and triplet states of a single monomer.
Rigid and planar compounds W2 and W3 (Fig. 20 and Table 4) both of which feature electron-rich sulfur atoms were obtained from commercially available thiophene-3,4-dicarboxylic acid with yields of 68.3% and 85%, respectively.
| Compound | λ FL, nm | τ PL, ns | FLQY, % | λ RTP, nm | τ RTP, ms | PHQY, % | Host | CIE coord. | CRI index | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| λ FL – the wavelength of fluorescence maxima, τPL – fluorescence lifetime, FLQY – fluorescence quantum yield, λRTP – the wavelength of room temperature phosphorescence maxima, τRTP – RTP lifetime, PHQY – phosphorescence quantum yield, CIE – Commission Internationale d’Eclaraige, CRI – colour rendering index, and n/m – not mentioned.a Estimated for the fabricated device. | ||||||||||
| W1 | n/m | n/m | n/m | n/m | n/m | n/m | n/m | n/m | — | 26 |
| W2 | — | — | — | 585 | 39 | — | — | (0.26, 0.30) | 85a | 75 |
| W3 | — | — | — | 587 | 44 | — | — | (0.32, 0.42) | — | 75 |
| W4 | 386 | 0.4 | — | 519 | 35.6 | — | — | (0.24, 0.26) | — | 77 |
| W5 | 388 | 0.7 | — | 549 | 0.007 | — | — | (0.27, 0.27) | — | 77 |
| W6 | 455 | 1.75, 6730 | — | 571 | 4.16 | — | — | (0.33, 0.33) | 78 | |
| W7 | 393 | 2.71 | 31.0 | 546 | 626 | 1.7 | PVA | — | — | 79 |
| W7 film | — | — | — | 543 | 328 | 2.5 | — | — | — | 79 |
| W8 | 413 | 2.70 | 3.2 | 562 | 480 | 5.5 | PVA | — | — | 79 |
| W8 film | — | — | — | 546 | 356 | 5 | — | (0.33, 0.34) | 87a | 79 |
| W9 | 422 | 2.07 | 0.5 | 547 | 142 | 1.6 | PVA | — | — | 79 |
| W9 film | — | — | — | 546 | 150 | 0.5 | — | — | — | 79 |
| W10 | 455 | 89 × 106 | 50.68 | 542, 592 | 280 | 10.92 | — | (0.30, 0.35) | — | 80 |
| W11 | 402 | 1.53 | 2.9 | 547 | 134 | 7.4 | — | (0.33, 0.34) | — | 81 |
| W12 | 405 | 0.7 | 1.6 | 544 | 219 | 11.3 | — | — | — | 81 |
| W13 | 410 | 3.4 | 6.34 | 567 | 204 | 2.85 | — | (0.27, 0.26) | — | 82 |
| W14 | 431 | 3.4 | 3.6 | 546 | 79 | 5.5 | — | (0.30, 0.31) | — | 83 |
| W15 | 448, 475, 550 | 4.7, 5.6, 4.5 | — | 575 | 370 | — | — | (0.28, 0.31) | — | 84 |
| W16 | 467 | — | 42 | 564 | 13.67 | 42 | Zeonex | (0.32, 0.39) | 79 | 85 |
Compound W3 possesses the unique ability of emitting white light both in solution and in crystalline states. The origin of white emission in DMF is the emission enhancement caused by the formation of aggregates with different sizes, while emission in the solid state exhibits orange RTP. Investigation of the photophysical properties of DMF solutions of W3 in DMF led to the conclusion that the luminescence intensity of W3 depends on the concentration and increases considerably with the increase in the concentration from 5 × 10−3 M to 0.25 M.75 In addition, the change in the colour of emission from blue to green, and finally to yellow-white was observed. A conclusion was made that compounds W2 and W3 are AIE-active. However, their AIE-activity is different from that of the conventional AIE agents, rich in rotational fragments. The lack of the latter in W2 and W3 allowed the authors75 to deduce that the AIE mechanism of their emission does not refer to a restriction of molecular rotations but to space-conjugation of the aggregates. At a concentration of W3 in DMF of 0.25 M the excitation at 340 nm afforded a wide white emission with CIE coordinates of (0.30, 0.36). In the case of compound W2 the intermolecular interaction of its aggregates at high concentrations was found to be weaker than that of W3 which was explained by the absence of a nitrogen heteroatom. As a result, white emission of its DMF solution was not observed. The crystals of W2 and W3 were easily grown from toluene and methanol solutions. They showed yellow RTP after turning off the 365 nm light source. The crystals of W2 showed emission intensity maximum at 585 nm and a lifetime of 39 ms, while the RTP of the crystal of W3 peaked at 587 nm with a lifetime of 44 ms. Their photoluminescence spectra covered the whole visible light region. The crystal of W2 demonstrated cold white luminescence with CIE coordinates of (0.26, 0.30), while that of W3 displayed white light with CIE coordinates of (0.32, 0.42). On the basis of theoretical calculations, the authors75 concluded that RTP of the crystals of W2 and W3 originated from a certain kind of specific aggregate and that it had no obvious excitation wavelength dependence.
Two efficient dual-emissive compounds W4 and W5 (Fig. 20 and Table 4) were synthesized by Xu et al.77 Butterfly-type luminogens W4 and W5 were obtained via a Suzuki cross-coupling reaction from the corresponding arylbromide and dibenzo[b,d]thiophen-4-yl-boronic acid in 76% and 78% yields, respectively. Their solutions exhibited only blue fluorescence, but in the solid state, both compounds were found to exhibit biluminescence that included orange RTP with the CIE coordinates of (0.24, 0.26) and (0.27, 0.27), respectively. The authors77 observed that phosphorescence of the crystals of both the compounds shrank significantly after applying a mechanical force. Fluorescence also underwent minor red shifts from 385 nm to 392 nm in the case of the crystal of W4 and from 400 nm to 414 nm in the case of W5. Thus, their emission is easily tuneable from white to deep blue even with a minor mechanical treatment or hand grinding. Based on the single crystal analysis the authors77 concluded that mechanochromism appeared as a result of a transition from the crystalline phase to the amorphous one that destroyed intermolecular interactions thus opening up a way for triplet excitons to relax nonradiatively. By fuming the films of both the compounds with dichloromethane vapor, the yellow phosphoresce of W4 and W5 was revitalized again and the emission colors were restored to white. Interestingly, the authors77 also discovered that thermal annealing also caused a change in the emission colour of W5 from white to blue with CIE coordinates of (0.18, 0.11). Emission of the annealed sample was found to return back to the white colour after the fumigation with DCM vapour.
One more white-emitting dibenzothiophene derivative W6 (Fig. 20 and Table 4) was reported by Zhang et al.78 They attached pyrimidine to dibenzothiophene via Suzuki coupling between 4-chloropyrimidine hydrochloride and dibenzo[b,d]thiophen-4-yl-boronic acid. The PL spectra of crystalline powder of W6 displayed dual emission with the peaks at 455 nm (1.75 ns, 6.73 μs) and 571 nm (1.16 ms). Similarly to W4 and W5,77 the emission of compound W6 also exhibited stimuli responsiveness. The fluorescence peak shifted after grinding, and the phosphorescence peak significantly decreased. The authors78 managed to grow yellow-green crystals of W6, namely W6-G by slow evaporation of the hexane/DCM mixture. The PL spectrum of W6-G demonstrated a dominant peak of phosphorescence at 550 nm, and a minor peak of fluorescence at 443 nm. When slightly ground in a mortar, the W6-G crystals transformed into a W6-Y type of crystal along with an RTP colour shifting from yellow-greenish to yellow (565 nm). After continuing grinding, the W6-Y crystals underwent another transformation into W6-W powder, and its emission peaks were further red-shifted to 454 nm and 574 nm, with almost equal intensities (Fig. 19). The CIE coordinates of W6-W were calculated to be (0.33, 0.33), confirming its pure white light emission. The process was reversible. It could be repeated several times.
 | ||
| Fig. 19 PL spectra (a) and powder X-ray diffraction (b) of compound W6 in different solid states. Reprinted from Y. Sun, H. Qu, J. Zhang, X. Duan and X. Zhang, Room temperature phosphorescence, thermally activated delayed fluorescence and multicolor mechanochromic luminescence of emitters through molecular interaction and conformational modulations, Tetrahedron, 110, 132692,78 Copyright 2023, with permission from Elsevier. | ||
 | ||
| Fig. 20 Molecular structures of white RTP-agents W1–W16. | ||
Crystalline materials usually suffer from poor processability and reproducibility. In particular, the preparation of their thin films is always a critical and challenging task, while thin films with desirable optical properties are highly desired for many important practical applications. Shu et al.79 studied the impact of heavy chalcogen atoms on RTP of three asterisk-shaped molecules W7–W9 (Fig. 20 and Table 4). The heavy atom effect and large steric hindrance led to the appearance of RTP not only in crystalline forms, but also in neat and host-containing films. The compounds were obtained via a nucleophilic aromatic substitution reaction between carbazole and the corresponding tetrafluorinated chalcogene-containing intermediate in the moderate yields of 60–75%. The most effective kISC in the series was observed for compound W9 containing the selenium atom. Unlike most organic RTP materials, all three compounds demonstrate RTP even in neat films peaking at 543, 546, and 546 nm, respectively. This is due to high steric hindrance of star-shaped structures that suppress non-radiative electronic transitions. The neat film of compound W8 showed the longest lived RTP with τPHOS of 356 ms. This was the highest τPHOS value among all reported low-molar-mass organic molecules. Although compound W7–W9 exhibited complementary blue-yellow emissions, the authors79 managed to generate white emission only from compound W8. This compound doped into DPEPO was used for the preparation of an emitting layer of a solution processed WOLED with white electroluminescence and CIE colour coordinates of (0.36, 0.38).
Liu et al.80 reported RTP-containing white afterglow of the molecule containing no heavy halogen or chalcogen atoms. Compound W10 (Fig. 20 and Table 4) that consists of a pyridine ring, to which two carbazole moieties are linked at the meta-positions and a cyano group linked at the ortho-position, was easily obtained via nucleophilic substitution between carbazole and 4-cyano-3,5-difluoropyridine. The presence of two carbazolyl donor units led to a decrease in ΔEST and a promotion of ISC and rISC. Under excitation at 365 nm, the crystal of compound W10 exhibited blue emission with white afterglow. The white emission of W10 consisted of TADF (λmax = 455 nm, τ = 89 ms), distinguishable thermally activated delayed phosphorescence (λmax = 483 nm) and organic ultralong phosphorescence from the stabilized triplet state  (λPHOS = 542 nm, 592 nm, and τRTP = 240 ms). This is the first and only example of a molecule's white afterglow which appears from multi-mode triplet emissions.
(λPHOS = 542 nm, 592 nm, and τRTP = 240 ms). This is the first and only example of a molecule's white afterglow which appears from multi-mode triplet emissions.
RTP has a tendency to cease when the temperature of the environment is increased. The molecular packing tightens as temperature decreases, leading to the enhancement of intermolecular interactions. When the temperature increases, molecular movement becomes more intensive, resulting in weaker intermolecular interactions. Bi et al.81 reported two isomeric compounds W11 and W12 (Fig. 20 and Table 4) that are similar to W10 in structure but have a bromine heavy atom instead of one carbazole moiety. The compounds were reported to be the first examples of materials with stable RTP at elevated temperatures. The introduction of bromine helps not only to enhance the RTP efficiency but also to increase the thermal stability and most importantly resistance of RTP of W11 and W12 to the elevated temperatures. The authors81 explained this observation by the co-presence of bromine atoms and cyano groups, facilitating abundant intermolecular interactions. The solid powders of both the compounds W11 and W12 demonstrated biluminescence with fluorescence maxima in the deep-blue region of the spectrum (at 402 nm for W11, 405 nm for W12), and an RTP peaking in the 523–683 nm range for W11 and in the 520–689 nm range for W12. Both compounds still exhibited double emission in the crystalline state. Compared to the emission of the solid powders, the fluorescence maxima did not change, but τFL and PHQY significantly increased. At the same time, FLQY and τRTP were found to be significantly diminished as they were competing processes. The isomers W11 and W12 exhibited temperature-dependent emission both as solid powders and in the crystalline state. For a balanced fluorescence-RTP intensity ratio with the help of temperature changing, almost pure white light with the CIE coordinates of (0.33, 0.34), could be achieved for compound W11. In addition, a significant RTP peak was still present at 380 K (τRTP of 4 ms and a PHQYof 3.9%). To the best of our knowledge, these are the first compounds reported in which RTP shows resistance to such temperatures. In addition to the temperature-dependent luminescence, the authors81 revealed the dependence of the emission on the intensity of the excitation light for both compounds. With the constant growth of excitation intensity, the intensity of fluorescence increased two to three times faster than that of phosphorescence resulting in a modulated multicolour luminescence that includes pure white light (CIE coordinates 0.33, 0.33).
An illustrious example of pure organic RTP of the different morphological forms is compound W13 (Fig. 20 and Table 4), designed and synthesized by Du et al.82 Luminophore W13 consists of an electron donating 9,9-dimethyl-9,10-dihydroacridine moiety, an electron accepting 4-phenylpyridine unit and a phenyl linker between them. The authors82 managed to obtain two crystalline forms of W13: W13-B that was afforded by the evaporation of the solvents from the solution of W13 in a DCM–acetonitrile mixture and W13-W that was obtained by diffusing hexane vapour into a DCM solution of W13. The PL spectrum of W13-B crystals revealed a blue fluorescence peak at 402 nm with a τFL of 3 ns. The time resolved luminescence spectrometry revealed yellow RTP peaking at 567 nm with a τRTP of 118 ms. Despite the presence of two complementary emission colours, the authors in ref. 82 failed to obtain white emission from the corresponding crystalline form due to its low PHQY of only 1.07%. Meanwhile, the W13-W crystal revealed single-molecular white emission that consisted of blue fluorescence at 410 nm (τFL of 3.4 ns) and orange RTP at 567 nm (τRTP of 204 ms) with a PHQY of 2.85%. Consequently, white light with CIE coordinates of (0.27, 0.26) was afforded. The authors82 performed single X-ray analysis for each polymorph. The molecules in both crystals were characterized by similar dihedral angles between the donor fragment and the phenyl linker, but the dihedral angle between the pyridine acceptor and benzene ring was smaller in the case of W13-B crystals. This provides more efficient intramolecular charge transfer in the W13-B polymorph. Additionally, W3-W crystals were found to form more abundant intermolecular interactions than W13-B crystals. Surprisingly, the film of compound W13 doped in the rigid PMMA matrix also afforded white emission with CIE coordinates of (0.26,0.30) that consisted of two fluorescence components with maxima around 430 nm and 540 nm and an RTP component peaking around 500 nm. The sample was characterized by a PLQY of 40.2%.
Ishi-I et al.83 demonstrated that if a compound exists in two polymorph forms and both of them exhibit dual fluorescence-RTP behaviour, reversible switching can be achieved with the aid of external stimuli. They synthesized a bromine-containing carbazole–pyrimidine conjugate W14 with a yield of 76% (Fig. 20 and Table 4), which was found to crystallize in two different polymorphs.83 White crystals of W14-A were obtained by the evaporation of the solvent from the chloroform solution of W14, while yellow crystals of W14-B were grown from a hexane/chloroform mixture. Upon excitation, the W14-B crystalline form demonstrated very weak RTP peaking at 546 nm (τRTP of 8 ms), and fluorescence peaking at 476 nm. Meanwhile, the W14-A crystals showed blue fluorescence with the maximum at 431 nm and intensive yellow afterglow (λRTP of 546 nm and τRTP of 77 ms). Almost pure white emission, (0.30, 0.31), could be generated by heating W14-A crystals above their melting point (210 °C) and then cooling to room temperature. The obtained crystalline form W14-W demonstrated enhanced RTP with PHQY raised to 5.5% together with the decrease of FLQY to 3.6%. The process was reversible and could be repeated. The authors83 concluded that the heating–cooling cycle of the sample W14-A helped to increase its crystallinity that promoted ISC, and therefore in the obtained W14-W crystalline form, the phosphorescence intensity increased whereas the fluorescence decreased.
The unique excitation-dependent luminescence was observed by Ma et al.84 from the simple terpyridine derivative W15 (Fig. 20 and Table 4). After being excited at 254–330 nm, the W15 film demonstrated two emission peaks at 372 nm and 550 nm. After an increase in the excitation wavelength to 365 nm, a slight decrease in intensity of the peak at 550 nm was observed, but the appearance of three additional peaks at 448 nm and 475 nm and 575 was noticed. The last peak was characterized by a luminescence lifetime of 0.73 s and was recognized as RTP. The resulting emission had CIE coordinates of (0.28, 0.31) which can be described as cold white light. After a further increase in the excitation wavelength to 380 nm, the emission peaks at 448, 475, and 550 nm increased in intensity, but the white emission was still observable (CIE coordinates of (0.30, 0.37)). After increase of the excitation wavelength to 420 nm, the peaks at 448 and 475 nm still increased and a minor peak at 505 nm emerged, while the peak at 550 nm almost disappeared.
Our group recently reported high-quality white emission from a single heavy-atom-free molecule.85 State-of-the-art monomolecular white emission with a PLQY of 84%, a CRI of 79, a TC of ca. 5100 K, and CIE 1931 colour coordinates of (0.32, 0.39) close to that of the natural white colour (0.33, 0.33) was observed for compound W16 (Fig. 20 and Table 4). The concept of monomolecular white emission was proved after the investigation of twelve compounds containing thianthrene and benzoyl moieties with different arrangements of halogen atoms. All the compounds were obtained in good yields via Fridel–Crafts acylation of thianthrene with differently substituted benzoyl chlorides. The structural peculiarities of the compounds are responsible for their efficient fluorescence and RTP. Taking into account the results of X-ray analyses and the theoretical study, the high PLQY values of the thianthrene derivatives are attributed to the absence of π–π intermolecular interactions and the low amount of weak van der Waals intermolecular stacking due to the strong bending of the heterocyclic thianthrene moiety and due to the high dihedral angle between thianthrene and benzoyl moieties. The valence ![[double bond splayed right]](https://www.rsc.org/images/entities/char_e00a.gif) C–S–C angle of the thianthrene fragment based on a 1,4-dithiin structural moiety is ca. 102°. In the case of the investigated compounds, the theoretically calculated dihedral bending angle between the two planes of the 1,4-dithiin structural moiety via the S⋯S axis was 140.6°. Such dihedral bending of the rigid heterocyclic thianthrene can prevent the formation of π–π intermolecular interactions. Among all twelve compounds, only the W16 derivative with a fluorine atom in the para-position to the carbonyl fragment was found to exhibit almost pure white emission. Such emissive properties originate from well-balanced rates of radiative transition from singlet states (3.24 × 107 s−1) and intersystem crossing (3.09 × 107 s−1).
C–S–C angle of the thianthrene fragment based on a 1,4-dithiin structural moiety is ca. 102°. In the case of the investigated compounds, the theoretically calculated dihedral bending angle between the two planes of the 1,4-dithiin structural moiety via the S⋯S axis was 140.6°. Such dihedral bending of the rigid heterocyclic thianthrene can prevent the formation of π–π intermolecular interactions. Among all twelve compounds, only the W16 derivative with a fluorine atom in the para-position to the carbonyl fragment was found to exhibit almost pure white emission. Such emissive properties originate from well-balanced rates of radiative transition from singlet states (3.24 × 107 s−1) and intersystem crossing (3.09 × 107 s−1).
7. Applications of organic triplet emitters
Organic RTP materials have been used in chemical sensors, anticounterfeiting, molecular oxygen and temperature sensors, for in vivo bioimaging, lighting applications, multicolor displays, chemical qubits, optical memory, luminescence printing, 3D printing, information storage in programmable luminescent tags, quantum computers, microfluidic devices, and optoelectronic devices, to mention some examples.86–88 Multifunctional applications of organic RTP compounds are discussed in many research articles.89–91 The most widespread applications of organic RTP materials are discussed in review articles.92–97 Therefore, we will highlight only the recent records and the unique applications of organic RTP materials.From the point of view of colour purity and efficiency, very attractive applications of organic RTP-agents have been recently reported by Zou et al.98 The authors developed high-colour-purity afterglow materials and demonstrated their potential applications in afterglow displays, information encryption, identification, etc. (Fig. 21a–e). For the development of high-colour-purity afterglow materials, phosphorescent chromophores 4,4′-biphenol (BP), 1,1′-bi-2-naphthol (BNP), and 1,8-naphthalimide (NPI) were used as as electron-donating moieties. Rhodamine derivatives (BCA, BPH, R123, RdB, and R101) were used as electron-accepting compounds with narrow-band fluorescence spectra. Both donor and acceptor compounds were dispersed in the polyvinyl alcohol (PVA) matrix. The rigidity of this matrix enabled the prevention of non-radiative transitions of organic RTP-agents and fluorescent derivatives of rhodamine. The efficient Förster resonance energy transfer was observed in donor–acceptor molecular mixtures owing to large emission-absorption spectral overlap of the co-doped donor and acceptor in the PVA polymeric matrix (Fig. 21f). Such systems showed long-lived afterglow at room temperature. The authors98 investigated a series of narrowband polymeric afterglow materials with high colour purity. By optimization of the donor–acceptor pairs, colours of narrowband polymeric afterglow materials were tuned from green to red. The resulting organic afterglow materials exhibited narrow full width at half-maximum (FWHM) approaching 23 nm, and a long lifetime of 721.22 ms at room temperature (Fig. 21g).
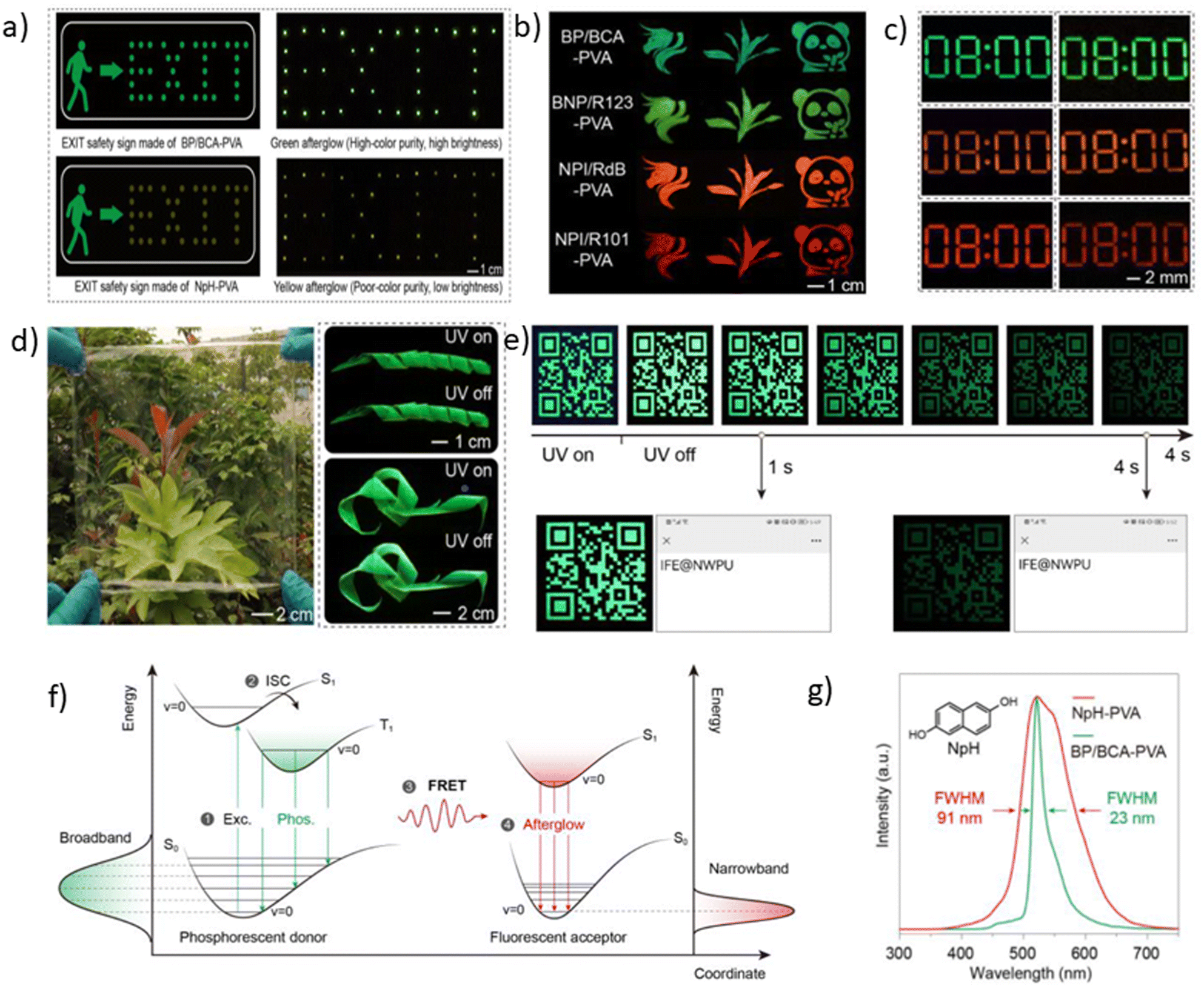 | ||
| Fig. 21 Examples of applications of narrowband polymeric afterglow materials based on electron-donating RTP-agents for safety sign afterglow displays (a); afterglow photographs with multicolor patterns (b); miniature display patterns in different colors (c); and large-area transparent and flexible film applications under on/off UV excitation at a wavelength of 254 nm (d). QR code before and after switching off the UV excitation at 310 nm (e). Schematic visualisation of the emission mechanism of narrowband polymeric afterglow materials based on electron-donating RTP-agents (f) and delayed photoluminescence spectra of narrowband polymeric afterglow materials and reference films BP/BCA-PVA and NpH-PVA recorded under ambient conditions (g). Reproduced from ref. 98 with permission from John Wiley and Sons, Copyright 2023. | ||
A record external quantum efficiency of 15.8% for host-free OLEDs exhibiting aggregation-induced organic room temperature electrophosphorescence was reported by Xu et al.99 The device efficiency is superior in comparison to that of OLEDs based on previously reported organic RTP emitters.100,101 The authors99 designed donor-oxygen-acceptor-type compounds containing an acridine moiety as a donor, a triazine unit as an acceptor and oxygen as the bridge between the acridine and triazine moieties. They claimed that such a molecular structure caused effective ISC resulting in efficient RTP. The neat emitting layers exhibited PLQYs of up to 77.5% under an inert atmosphere. The donor-oxygen-acceptor type RTP compounds were used as the hosts for multiple resonance emitters facilitating the fabrication of pure colour OLEDs with a maximum EQE of 26.4% and narrow electroluminescence spectra approaching FWHMs of 26 nm. In our group, white host-free OLEDs based on organic RTP emitters were developed102 (Fig. 22a). White electroluminescence of the fabricated devices resulted from the overlapping of blue fluorescence and orange RTP of 2-(3,7-di-tert-butyl-phenothiazin-10-yl)thianthrene (PTZ-tBu-TA).102 The maximum external quantum efficiencies of the devices did not exceed 1% because of the low PLQY of the neat films of PTZ-tBu-TA. Meannwhile, the PMMA hosting of PTZ-tBu-TA leads to intensive green RTP with high oxygen sensitivity described by a high Stern–Volmer constant of 21 × 10−4 ppm−1 (Fig. 22b).
 | ||
| Fig. 22 Electroluminescence (EL) spectra of host-free devices based on the organic RTP emitter PTZ-tBu-TA (a) and PL spectra of PTZ-tBu-TA dispersed in the PMMA matrix at different concentrations of oxygen (b). Reprinted from K. Leitonas, A. Tomkeviciene, G. Baratte, A. Dabuliene, S. M. Punniyakoti, D. Volyniuk and J. Vidas Grazulevicius, oxygen sensing properties of thianthrene and phenothiazine derivatives exhibiting room-temperature phosphorescence: Effect of substitution of phenothiazine moieties, Sens. Actuators, B, 345, 130369,102 Copyright 2023, with permission from Elsevier. | ||
Reineke et al.103 used 4,4-dithianthrene-1-yl-benzophenone (BP-2TA) exhibiting RTP with PHQY up to 20% for the fabrication of photonic devices, such as programmable luminescent labels or tags. BP-2TA showed much better performance in programmable luminescent labels in comparison to Ir(III) and Pt(II) complexes (Fig. 23a). The advantages of BP-2TA are mainly attributed to the ratio of ΦRTP and ΦF (P2F = ΦRTP/ΦF), which shows a better contrast of the tags than those of tags based on organometallic complexes. Compound BP-2TA demonstrates a P2F value of 21. The same group104 proposed an interesting application of BP-2TA as the active material of organic thin-film wavelength sensors for accelerating wavelength tracking of inorganic light-emitting diodes under heating. Such applications of BP-2TA were also possible due to its P2F parameter, resulting in the production of mostly triplet excited states due to efficient ISC under absorption of irradiation.
 | ||
| Fig. 23 Programmable luminescent tags with readability under continuous-wave illumination (a) and molecular structure of PRT fluorophores used for their fabrication: NPB, Ir(MDQ)2(acac), PtOEP, and BP-2TA (b). Reproduced from ref. 103 with permission from John Wiley and Sons, Copyright 2023. | ||
RTP is detectable not only under optical and electrical excitations but also under X-ray excitation, resulting in various applications of organic RTP materials such as radiation detection, X-ray imaging, radiotherapy, non-destructive testing, and X-ray astronomy.105,106 (Fig. 24a). Gan et al.105,106 obtained amorphous copolymers through the copolymerization of bromine-containing RTP-agents and acrylic acid for organic phosphorescent scintillation and the related emissive processes under X-ray irradiation (Fig. 24b and c). The developed polymeric scintillators were able to efficiently respond to X-rays and have a respectable PHQY of up to 51.4% at room temperature. To achieve simultaneous X-ray absorption and ISC, heavy atoms such as bromine and iodine are used in the design of X-ray detectable organic RTP materials (Fig. 24d and e).
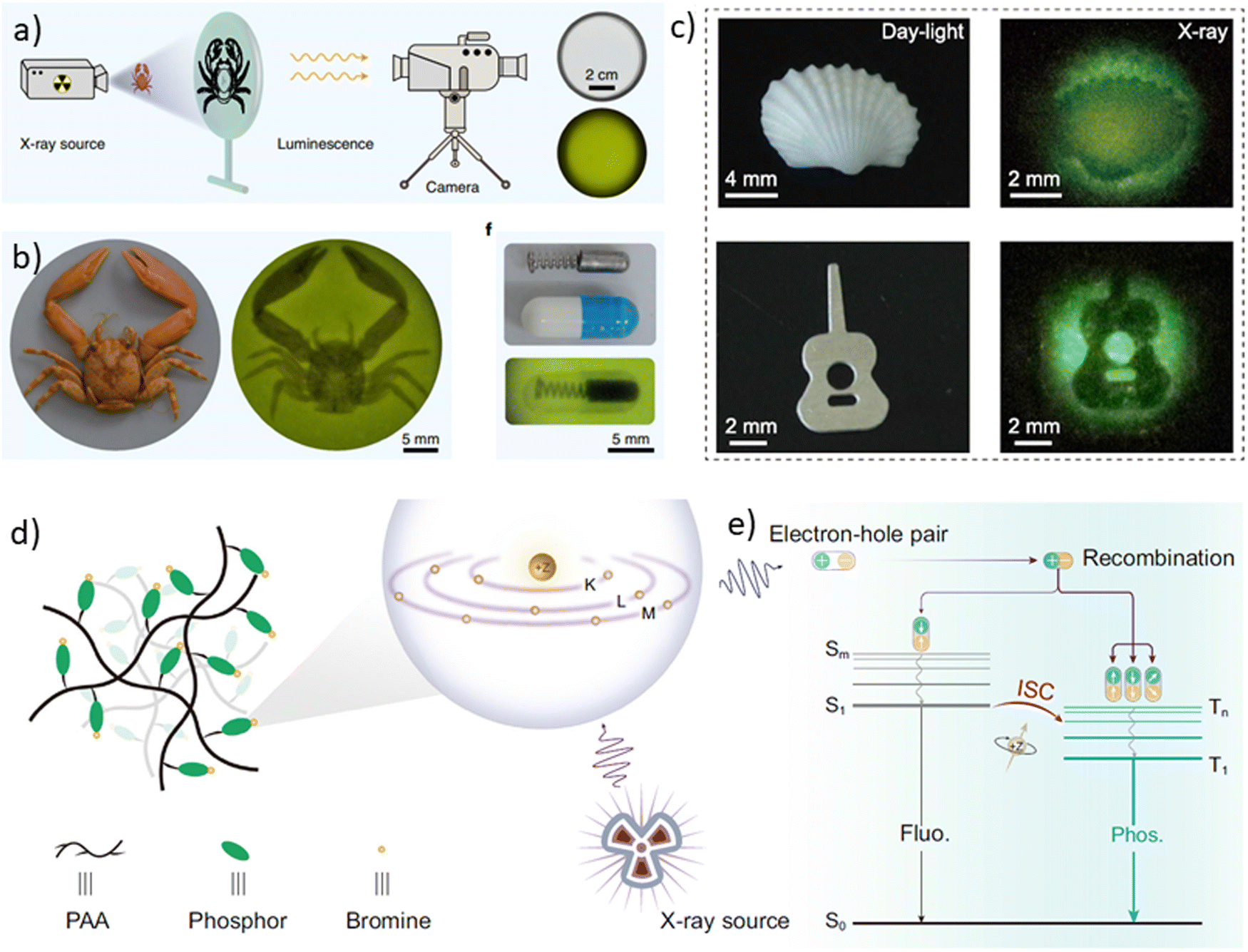 | ||
| Fig. 24 Schematic visualization of a radiography set up (a), examples of the aplication of RTP-agent-based X-ray detectors containing RTP-agents with heavy atoms such as bromine or iodine (b) and (c), design strategy of X-ray detecteble organic RTP materials (d) and schematic mechanism of X-ray-irradiated RTP (e). Reproduced from ref. 105 and 106 with permission from Springer Nature. Copyright 2023. | ||
At the end of this review, we have to mention that energy transfer from the singlet oxygen to solvent vibrations is an important temperature dependent process.6,12,107 Since the RTP quenching by O2 is accompanied by the singlet oxygen generation and quenching, all the temperature dependence studies should be considered in cooperation. In this respect, the role of high-frequency C–H, N–H and O–H vibrations could be considered since they determine the most active energy-accepting vibrational modes in the singlet oxygen quenching by solvents. The same vibrations could be important for the organic RTP quenching by matrixes and solvents (especially for orange and red RTP). This aspect of the RTP problem needs additional analysis.12,107
We also want to stress an important SOC peculiarity, which is almost forgotten in the vast modern literature on RTP problems. The quantum-chemical modelling of intermolecular interactions in host–guest (H–G) organic crystals indicates that at the typical H–G distances, even weak overlap between H–G molecular orbitals can induce the σ–π mixing inside the electronic shell of the planar phosphorescent guest molecule, which is sufficient for increasing SOC ME and notably diminishing the phosphorescence lifetime.108,109 This internal magnetic effect is due to the appearance of one-centre contributions to the SOC integrals 〈Sππ*|HSO|Tππ*〉.108 High pressure and tight interactions in the polymer matrix led to shorter contacts and higher intermolecular overlap of the H–G orbitals, which increases the σ–π intramolecular mixing and induces stronger SOC perturbations inside the G chromophore responsible for the RTP.6,108,109
8. Conclusion and outlook
Organic RTP materials offer numerous benefits, such as energy-saving and environmental benefits. They can harvest emissive triplets and do not contain any hazardous elements. These benefits allow the prediction of the usage of organic RTP materials in many fields of human activities including sensing and X-ray detection as well as lighting and photonic information technologies. However, in order to expand the fields of application of organic RTP materials, their efficiency, stability, and simplicity, there are still challenges that must be resolved.Although fluorophores that have a phosphorescence quantum yield higher than 50% have been developed, many organic RTP materials have low efficiency because of weak spin–orbit coupling and high non-radiative decay rates of triplet states. Even the best examples exhibit phosphorescence quantum yields significantly lower than 100%. There are various methods to improve the efficiency of RTP luminophores, especially in the solid-state. For example, a polymer can interact with the molecules of a luminophore through specific non-covalent interactions, thereby facilitating the vibronic confinement of the luminophores. The most frequently exploited non-covalent perturbations are host–guest and charge-transfer interactions, hydrogen bonding, isolation from oxygen, and suppression of internal vibrational movements. The latter type of perturbation can shift vibration frequency and destroy resonances that are typically associated with free luminophores. All confinements produced by polymer matrices or by other hosts introduce perturbations to the electronic–vibrational coupling and thus a subtle regulation of non-radiative relaxation processes. At a higher pressure and tighter interactions in the polymer matrices the shorter contacts and the higher intermolecular MO overlap occur, which increases the σ–π intramolecular mixing and leads to stronger SOC perturbations inside the RTP chromophore.
Significant attention should be paid to the role of a host matrix in ensuring/improving/tuning the RTP properties of a guest compound. The main task of hosts for RTP luminophores is to provide an environment rigid enough to suppress molecular vibrations and to stabilize triplet excitons. For this reason, rigid polymers such as Zeonex, PS, PMMA, etc. are employed as hosts. This approach seems to be the simplest and the most easily available one.
More advanced technologies exploit hosting of RTP agents in supramolecular systems such as cucurbituril and cyclodextrin that act as cavities into which the guest molecules can fit. However, the role of a host matrix for organic RTP guests is not limited by the rigidification only. Engineering of host–guest systems to initiate energy/charge transfer seems to be the least studied. While the roles of hosts that promote charge/energy transfer to fluorescent and TADF emitters are widely investigated and discussed, the host–guest interactions in organic RTP systems are studied to considerably less extent. It is established that triplet–triplet energy transfer (Dexter energy transfer determined by exchange interaction, eqn (1)) usually occurs when host and guest molecules have closely located triplet energy levels. Charge transfer in host–guest RTP systems is provided by closely located 1CT and 3CT excited states. Such systems are very scarce, because in this case rISC can easily prevail above ISC, thus initiating TADF instead of RTP; therefore, deeper theoretical insights as well as more experimental data are needed.
There are numerous problems related to organic RTP materials which have to be addressed in the future. For instance, these materials have limited RTP colours, mainly in the deep-blue to blue region due to colour purity issues arising from the broad emission spectra and overlap of fluorescence and phosphorescence. For lighting applications, efficient white emissions of organic RTP emitters with colour coordinates close to that of natural white (0.33, 0.33) have to be developed. In several instances, it is possible to achieve white emission of organic RTP emitters by applying external stimuli such as pressure or oxygen quenching.
Most organic RTP materials are prone to being quenched by molecular oxygen and moisture, which leads to the deactivation of the triplet states and a reduction in both phosphorescence lifetime and intensity.105 In order to improve the efficiency of organic RTP materials, it is important to develop hosts with low oxygen and water permeability and effective encapsulation methods. On the other hand, utilizing intricate fabrication techniques such as crystallization, polymerization, or rigid matrix formation to achieve efficient RTP emission can result in poor processability and reproducibility.
Low stability and durability are also issues associated with organic RTP materials. Many RTP materials are prone to degradation under ambient conditions or UV excitation, which can affect their phosphorescence efficiency and lifetime. The mechanism of RTP of organic materials is still not fully understood since it can be affected by several factors such as molecular structure, intermolecular interactions, energy transfer, and exciplex formation. Extensive research has still to be conducted on RTP compounds to gain a better understanding of how their structure and properties are related.
In this article, we reviewed the fluorescence and phosphoresence quantum yields, lifetimes, colour purity, and the wavelengths of intensity maxima of organic RTP materials. However, it is difficult to directly compare the photophysical parameters of the reported compounds since they were investigated under different conditions (in either an amorphous or a crystalline phase, in hosts or in neat films, and in air or in an inert atmosphere). To date, there has been no unified standard or method for the estimation and comparison of RTP of different organic materials. The terms phosphorescence, long pristine luminescence, long-lived afterglow, ultralong organic phosphorescence, ultralong organic luminescence, and room temperature phosphorescence are often used for the characterization of the same phenomenon.
Poor water solubility and biocompatibility of many hydrophobic and insoluble in water RTP materials limits their applications in aqueous environments and biological systems. Many RTP materials have low RTP efficiency and stability in solutions due to the collisional quenching, molecular motions, and oxygen diffusion. Some RTP compounds have low photostability and photobleaching resistance due to the photochemical degradation or oxidation of the chromophores under prolonged or intense irradiation. This can reduce their phosphorescence intensity and lifetime over time. These factors can reduce the intersystem crossing rate and increase the non-radiative decay rate of the triplet state. In addition, certain RTP materials possess cytotoxic or phototoxic properties that can cause damage to living cells or tissues, limiting bioimaging applications.
Although organic RTP materials have shown promise in chemical and physical sensors, such as optical sensors of oxygen and temperature, many of these materials exhibit a weak or non-existent response to external stimuli such as pH, metal ions, and biomolecules. This limitation reduces their potential applications in sensing and imaging. Moreover, some RTP materials are characterized by poor reversibility or reproducibility of their response, which affects the accuracy and reliability of the devices. Many RTP materials have similar molecular structures or design strategies, which limit their diversity and functionality.
We anticipate that the efficiency, stability, and cost-efficiency of the synthesis of organic RTP materials will be improved, enabling the discovery of chemical and physical approaches to better meet the requirements of both the known and forthcoming applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has received funding from the Research Council of Lithuania (LMTLT), agreement no. S-MIP-23-50, from the Ministry of Science and Education of Ukraine (project 0122U000760), the Swedish Wenner-Gren Foundations (project no. GFU 2022-0036) and the Swedish Natural Science Research Council (Dnr 2022-03405).References
- R. C. Sharma, R. Nandal, N. Tanwar, R. Yadav, J. Bhardwaj and A. Verma, J. Phys.: Conf. Ser., 2023, 2426, 012008 CrossRef.
- K. J. Yu, Z. Yan, M. Han and J. A. Rogers, npj Flexible Electron., 2017, 1, 1–14 CrossRef CAS.
- M. A. Baldo, D. F. O’Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS.
- J. Gibson, A. P. Monkman and T. J. Penfold, ChemPhysChem, 2016, 17, 2956–2961 CrossRef CAS PubMed.
- Kenry, C. Chen and B. Liu, Nat. Commun., 2019, 10, 1–15 CrossRef CAS PubMed.
- G. Baryshnikov, B. Minaev and H. Ågren, Chem. Rev., 2017, 117, 6500–6537 CrossRef CAS PubMed.
- Y. Gong, J. Yang, M. Fang and Z. Li, Cell Rep. Phys. Sci., 2022, 3, 100663 CrossRef CAS.
- Y. Patil, C. Demangeat and L. Favereau, Chirality, 2023, 35(7), 390–410 CrossRef CAS PubMed.
- M. Singh, K. Liu, S. Qu, H. Ma, H. Shi, Z. An and W. Huang, Adv. Opt. Mater., 2021, 9, 2002197 CrossRef CAS.
- H. E. Hackney and D. F. Perepichka, Aggregate, 2022, 3, e123 CrossRef CAS.
- B. F. Minaev, Chem. Phys., 2017, 483–484, 84–95 CrossRef CAS.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- A. Monkman, ACS Appl. Mater. Interfaces, 2022, 14(18), 20463–20467 CrossRef CAS PubMed.
- C. R. Wang, Y. Y. Gong, W. Z. Yuan and Y. M. Zhang, Chin. Chem. Lett., 2016, 27, 1184–1192 CrossRef CAS.
- M. Han, Z. Xu, J. Lu, Y. Xie, Q. Li and Z. Li, Mater. Chem. Front., 2021, 6, 33–39 RSC.
- L. Deng, Z. Ma, J. Zhou, L. Chen, J. Wang, X. Qiao, D. Hu, D. Ma, J. Peng and Y. Ma, Chem. Eng. J., 2022, 449, 137834 CrossRef CAS.
- N. Xie, H. Yu, J. Wang, Z. Li, J. Wei and Y. Wang, J. Phys. Chem. C, 2021, 125, 27489–27496 CrossRef CAS.
- Z. Cong, M. Han, Y. Fan, Y. Fan, K. Chang, L. Xiao, Y. Zhang, X. Zhen, Q. Li and Z. Li, Mater. Chem. Front., 2022, 6, 1606–1614 RSC.
- Y. Miao, L. Ma, J. Lv, S. Liu, W. Yang and J. Li, Anal. Chem., 2021, 93, 4075–4083 CrossRef CAS PubMed.
- M. Jian, Z. Song, X. Chen, J. Zhao, B. Xu and Z. Chi, Chem. Eng. J., 2022, 429, 132346 CrossRef CAS.
- Y. Zhang, H. Li, M. Yang, W. Dai, J. Shi, B. Tong, Z. Cai, Z. Wang, Y. Dong and X. Yu, Chem. Commun., 2023, 59, 5329–5342 RSC.
- W. Sun, B. Shi, Z. Xia and C. Lü, Mater. Today Chem., 2023, 27, 101297 CrossRef CAS.
- M. Li, X. Cai, Z. Chen, K. Liu, W. Qiu, W. Xie, L. Wang and S. J. Su, Chem. Sci., 2021, 12, 13580–13587 RSC.
- B. He, Z. Chang, Y. Jiang, X. Xu, P. Lu, H. S. Kwok, J. Zhou, H. Qiu, Z. Zhao and B. Z. Tang, Dyes Pigm., 2014, 106, 87–93 CrossRef CAS.
- Y. Dai, H. Liu, T. Geng, R. Duan, X. Li, Y. Liu, W. Liu, B. G. He, L. Sui, K. Wang, B. Zou, B. Yang and Y. Qi, J. Mater. Chem. C, 2023, 11, 4892–4898 RSC.
- Z. Yang, Z. Fu, H. Liu, M. Wu, N. Li, K. Wang, S.-T. Zhang, B. Zou and B. Yang, Chem. Sci., 2023, 14, 2640–2645 RSC.
- S. Wang, H. Shu, X. Han, X. Wu, H. Tong and L. Wang, J. Mater. Chem. C, 2021, 9, 9907–9913 RSC.
- D. R. Lee, J. Park and J. Y. Lee, Org. Electron., 2022, 106, 106534 CrossRef CAS.
- Z. Ma, Z. Yang, L. Mu, L. Deng, L. Chen, B. Wang, X. Qiao, D. Hu, B. Yang, D. Ma, J. Peng and Y. Ma, Chem. Sci., 2021, 12, 14808–14814 RSC.
- L. Tu, Y. Fan, C. Bi, L. Xiao, Y. Li, A. Li, W. Che, Y. Xie, Y. Zhang, S. Xu, W. Xu, Q. Li and Z. Li, Sci. China: Chem., 2023, 66, 816–825 CAS.
- Y. Tian, J. Yang, Z. Liu, M. Gao, X. Li, W. Che, M. Fang and Z. Li, Angew. Chem., Int. Ed., 2021, 60, 20259–20263 CrossRef CAS PubMed.
- Y. Wang, J. Yang, M. Fang, Y. Gong, J. Ren, L. Tu, B. Zhong Tang, Z. Li, Y. Wang, J. Yang, M. Fang, Y. Gong, J. Ren, L. Tu, B. Z. Tang and Z. Li, Adv. Funct. Mater., 2021, 31, 2101719 CrossRef CAS.
- N. Acharya, M. Hasan, S. Dey, S.-C. Lo, E. B. Namdas and D. Ray, Adv. Photonics Res., 2021, 2, 2000201 CrossRef CAS.
- E. Skuodis, K. Leitonas, A. Panchenko, L. Volyniuk, J. Simokaitienė, R. Keruckienė, D. Volyniuk, B. F. Minaev and J. V. Gražulevičius, Sens. Actuators, B, 2022, 373, 132727 CrossRef CAS.
- R. Keruckiene, N. Kusas, L. Dvylys, E. Skuodis, V. E. Matulis, E. G. Ragoyja, D. A. Lyakhov, I. Klymenko and J. V. Grazulevicius, J. Lumin., 2022, 241, 118502 CrossRef CAS.
- S. Shen, G. V. Baryshnikov, Q. Xie, B. Wu, M. Lv, H. Sun, Z. Li, H. Ågren, J. Chen and L. Zhu, Chem. Sci., 2023, 14, 970–978 RSC.
- S. Wang, Z. Cheng, X. Han, H. Shu, X. Wu, H. Tong and L. Wang, J. Mater. Chem. C, 2022, 10, 5141–5146 RSC.
- J. H. Kim, J. H. Yun and J. Y. Lee, Adv. Opt. Mater., 2018, 6(18), 1800255 CrossRef.
- L. Tu, Y. Xie, Z. Li and B. Tang, SmartMat, 2021, 2, 326–346 CrossRef CAS.
- Z. Liu, W. Shi, G. Hong, W. Chen, B. Song, X. Peng, X. Xiong and F. Song, J. Controlled Release, 2019, 310, 1–10 CrossRef CAS PubMed.
- A. Amaro-Ortiz, B. Yan and J. A. D’Orazio, Molecules, 2014, 19, 6202 CrossRef PubMed.
- Q. Zhang, X. Wang and Y. Wang, Inorg. Chem. Front., 2020, 7, 1034–1045 RSC.
- L. Li, Q. Zhang, C. Han, X. Sun, T. Xing, M. Wang, M. Wu, N. Weeranoppanant and W. Wu, J. Phys. Chem. C, 2023, 127, 12586–12592 CrossRef CAS.
- G. D. Gutierrez, G. T. Sazama, T. Wu, M. A. Baldo and T. M. Swager, J. Org. Chem., 2016, 81, 4789–4796 CrossRef CAS PubMed.
- H. Shi, L. Zou, K. Huang, H. Wang, C. Sun, S. Wang, H. Ma, Y. He, J. Wang, H. Yu, W. Yao, Z. An, Q. Zhao and W. Huang, ACS Appl. Mater. Interfaces, 2019, 11, 18103–18110 CrossRef CAS PubMed.
- T. Ishi-I, R. Kichise, I. S. Park, T. Yasuda and T. Matsumoto, J. Mater. Chem. C, 2023, 11, 3003–3009 RSC.
- Z. Xu, Q. T. Liu, X. Wang, Q. Liu, D. Hean, K. C. Chou and M. O. Wolf, Chem. Sci., 2020, 11, 2729–2734 RSC.
- L. Xiao, Y. Wu, Z. Yu, Z. Xu, J. Li, Y. Liu, J. Yao and H. Fu, Chem. – Eur. J., 2018, 24, 1801–1805 CrossRef CAS PubMed.
- X. F. Wang, H. Xiao, P. Z. Chen, Q. Z. Yang, B. Chen, C. H. Tung, Y. Z. Chen and L. Z. Wu, J. Am. Chem. Soc., 2019, 141, 5045–5050 CrossRef CAS PubMed.
- Y. Huang, X. Zheng, Z. Yao, W. Lv, S. Xiang, Q. Ling and Z. Lin, Chem. Eng. J., 2022, 444, 136629 CrossRef CAS.
- L. Dziewit, M. Dmowski, J. Baj and D. Bartosik, Appl. Environ. Microbiol., 2010, 76, 1861–1869 CrossRef CAS PubMed.
- Y. Wen, S. Xiao, H. Liu, X. Tian, J. De, T. Lu, Z. Yang, D. Zou, Y. Lv, S. T. Zhang, Q. Su and B. Yang, J. Mater. Chem. C, 2021, 9, 17511–17517 RSC.
- S. M. A. Fateminia, Z. Mao, S. Xu, Z. Yang, Z. Chi and B. Liu, Angew. Chem., Int. Ed., 2017, 56, 12160–12164 CrossRef CAS PubMed.
- S. Sun, L. Ma, J. Wang, X. Ma and H. Tian, Natl. Sci. Rev., 2022, 9(2), nwab085 CrossRef CAS PubMed.
- S. Wang, J. Wang, Q. Huang, X. Zheng, Z. Yao, S. Xiang, Q. Ling and Z. Lin, ACS Appl. Mater. Interfaces, 2022, 14, 14703–14711 CrossRef CAS PubMed.
- S. Garain, S. Kuila, B. C. Garain, M. Kataria, A. Borah, S. K. Pati and S. J. George, Angew. Chem., Int. Ed., 2021, 60, 12323–12327 CrossRef CAS PubMed.
- H. Sun and L. Zhu, Aggregate, 2023, 4, e253 CrossRef CAS.
- X. Wang, Z. Wang, H. Feng, C. Lin, H. Shi, Z. An, Z. M. Su and F. S. Liang, Chem. Commun., 2022, 58, 3641–3644 RSC.
- Y. Chen, Y. Xie and Z. Li, J. Phys. Chem. Lett., 2022, 13, 1652–1659 CrossRef CAS PubMed.
- L. Xu, G. Li, T. Xu, W. Zhang, S. Zhang, S. Yin, Z. An and G. He, Chem. Commun., 2018, 54, 9226–9229 RSC.
- Y. Tao, C. Liu, Y. Xiang, Z. Wang, X. Xue, P. Li, H. Li, G. Xie, W. Huang and R. Chen, J. Am. Chem. Soc., 2022, 144, 6946–6953 CrossRef CAS PubMed.
- Y. Tao, C. Liu, Y. Xiang, Z. Wang, X. Xue, P. Li, H. Li, G. Xie, W. Huang and R. Chen, J. Am. Chem. Soc., 2022, 144, 6946–6953 CrossRef CAS PubMed.
- T. M. Figueira-Duarte and K. Müllen, Chem. Rev., 2011, 111, 7260–7314 CrossRef CAS PubMed.
- F. Xiao, H. Gao, Y. Lei, W. Dai, M. Liu, X. Zheng, Z. Cai, X. Huang, H. Wu and D. Ding, Nat. Commun., 2022, 13, 1–10 Search PubMed.
- T. Ono, K. Kimura, M. Ihara, Y. Yamanaka, M. Sasaki, H. Mori and Y. Hisaeda, Chem. – Eur. J., 2021, 27, 9535–9541 CrossRef CAS PubMed.
- A. Nowak-Król, K. Shoyama, M. Stolte and F. Würthner, Chem. Commun., 2018, 54, 13763–13772 RSC.
- Y. Li, X. Li, T. Yu, W. Su, Y. Wang, Y. Zhao and H. Zhang, Dyes Pigm., 2020, 172, 107803 CrossRef.
- Ł. Balewski, F. S
![[a with combining cedilla]](https://www.rsc.org/images/entities/char_0061_0327.gif) czewski, M. Gdaniec, A. Kornicka, K. Cicha and A. Jalińska, Molecules, 2019, 24, 4070 CrossRef PubMed.
czewski, M. Gdaniec, A. Kornicka, K. Cicha and A. Jalińska, Molecules, 2019, 24, 4070 CrossRef PubMed. - J. Wang, G. Zhang, Z. Liu, X. Gu, Y. Yan, C. Zhang, Z. Xu, Y. Zhao, H. Fu and D. Zhang, Tetrahedron, 2013, 69, 2687–2692 CrossRef CAS.
- L. Chen, W. Xiong, Y. Ma, J. Y. Ge, N. Lv, X. Wu, J. Chen and Z. Chen, Chem. – Eur. J., 2023, 29, e202202909 CrossRef CAS PubMed.
- S. Reineke, M. Thomschke, B. Lüssem and K. Leo, Rev. Mod. Phys., 2013, 85, 1245–1293 CrossRef CAS.
- H. Liu, Y. Fu, B. Z. Tang and Z. Zhao, Nat. Commun., 2022, 13, 1–11 Search PubMed.
- J. C. Del Valle and J. Catalán, Phys. Chem. Chem. Phys., 2019, 21, 10061–10069 RSC.
- V. Anand, R. Mishra and Y. Barot, Dyes Pigm., 2021, 191, 109390 CrossRef CAS.
- X. Zheng, Y. Huang, D. Xiao, S. Yang, Z. Lin and Q. Ling, Mater. Chem. Front., 2021, 5, 6960–6968 RSC.
- N.-N. Zhang, C. Sun, X.-M. Jiang, X.-S. Xing, Y. Yan, L.-Z. Cai, M.-S. Wang, G.-C. Guo and R. Li, Chem. Commun., 2017, 53, 9269 RSC.
- B. Xu, H. Wu, J. Chen, Z. Yang, Z. Yang, Y. C. Wu, Y. Zhang, C. Jin, P. Y. Lu, Z. Chi, S. Liu, J. Xu and M. Aldred, Chem. Sci., 2017, 8, 1909–1914 RSC.
- Y. Sun, H. Qu, J. Zhang, X. Duan and X. Zhang, Tetrahedron, 2022, 110, 132692 CrossRef CAS.
- H. Shu, L. Chen, X. Wu, T. Wang, S. Wang, H. Tong and L. Wang, J. Mater. Chem. C, 2022, 10, 1833–1838 RSC.
- J. Liu, Z. Ma, Z. Li, Y. Liu, X. Fu, H. Jiang, Z. Ma and X. Jia, J. Mater. Chem. C, 2021, 9, 3257–3263 RSC.
- X. Bi, Y. Shi, T. Peng, S. Yue, F. Wang, L. Zheng, Q.-E. Cao, X. Bi, Y. Shi, S. Yue, L. Zheng, Q. Cao, T. Peng and F. Wang, Adv. Funct. Mater., 2021, 31, 2101312 CrossRef CAS.
- M. Du, Y. Shi, Q. Zhou, Z. Yin, L. Chen, Y. Shu, G. Y. Sun, G. Zhang, Q. Peng and D. Zhang, Adv. Sci., 2022, 9(5), 2104539 CrossRef CAS PubMed.
- T. Ishi-I, H. Tanaka, I. S. Park, T. Yasuda, S. I. Kato, M. Ito, H. Hiyoshi and T. Matsumoto, Chem. Commun., 2020, 56, 4051–4054 RSC.
- X. Ma, L. Jia, B. Yang, J. Li, W. Huang, D. Wu and W. Y. Wong, J. Mater. Chem. C, 2021, 9, 727–735 RSC.
- L. Volyniuk, D. Gudeika, A. A. Panchenko, B. F. Minaev, M. Mahmoudi, J. Simokaitiene, A. Bucinskas, D. Volyniuk and J. V. Grazulevicius, ACS Sustainable Chem. Eng., 2023, 11, 16914–16925 CrossRef CAS.
- H. Thomas, D. L. Pastoetter, M. Gmelch, T. Achenbach, A. Schlögl, M. Louis, X. Feng, S. Reineke, H. Thomas, M. Gmelch, T. Achenbach, A. Schlögl, M. Louis, S. Reineke, D. L. Pastoetter and X. Feng, Adv. Mater., 2020, 32, 2000880 CrossRef CAS PubMed.
- M. Louis, H. Thomas, M. Gmelch, F. Fries, A. Haft, J. Lindenthal and S. Reineke, Adv. Opt. Mater., 2020, 8, 2000427 CrossRef CAS.
- Z. Yang, S. Zhao, X. Zhang, M. Liu, H. Liu and B. Yang, Front. Chem., 2022, 9, 810304 CrossRef PubMed.
- Y. Li, G. V. Baryshnikov, F. Siddique, P. Wei, H. Wu and T. Yi, Angew. Chem., Int. Ed., 2022, 61, e202213051 CrossRef CAS PubMed.
- H. Liu, W. Ye, Y. Mu, H. Ma, A. Lv, S. Han, H. Shi, J. Li, Z. An, G. Wang, W. Huang, H. Liu, Y. Mu, S. Han, J. Li, G. Wang, W. Ye, H. Ma, A. Lv, H. Shi, Z. An and W. Huang, Adv. Mater., 2022, 34, 2107612 CrossRef CAS PubMed.
- X. Yao, H. Ma, X. Wang, H. Wang, Q. Wang, X. Zou, Z. Song, W. Jia, Y. Li, Y. Mao, M. Singh, W. Ye, J. Liang, Y. Zhang, Z. Liu, Y. He, J. Li, Z. Zhou, Z. Zhao, Y. Zhang, G. Niu, C. Yin, S. Zhang, H. Shi, W. Huang and Z. An, Nat. Commun., 2022, 13, 1–8 Search PubMed.
- J. Wang, X. Y. Lou, Y. Wang, J. Tang and Y. W. Yang, Macromol. Rapid Commun., 2021, 42, 2100021 CrossRef CAS PubMed.
- Z. Wang, X. Cheng, Y. Xie, S. Liu, M. Dong, J. Zhao, F. Liang, Z. An and W. Huang, CCS Chem., 2023, 5, 292–309 CrossRef CAS.
- X. Wei, J. Yang, L. Hu, Y. Cao, J. Lai, F. Cao, J. Gu and X. Cao, J. Mater. Chem. C, 2021, 9, 4425–4443 RSC.
- L. Ma and X. Ma, Sci. China: Chem., 2022, 66, 304–314 CrossRef.
- M. Ji and X. Ma, Ind. Chem. Mater., 2023, 1, 582–594 RSC.
- S. Guo, W. Dai, X. Chen, Y. Lei, J. Shi, B. Tong, Z. Cai and Y. Dong, ACS Mater. Lett., 2021, 3, 379–397 CrossRef CAS.
- X. Zou, N. Gan, M. Dong, W. Huo, A. Lv, X. Yao, C. Yin, Z. Wang, Y. Zhang, H. Chen, H. Ma, L. Gu, Z. An and W. Huang, Adv. Mater., 2023, 35, 2210489 CrossRef CAS PubMed.
- L. Xu, Y. Mo, N. Su, C. Shi, N. Sun, Y. Zhang, L. Duan, Z. H. Lu and J. Ding, Nat. Commun., 2023, 14, 1–9 Search PubMed.
- H. F. Higginbotham, M. Okazaki, P. De Silva, S. Minakata, Y. Takeda and P. Data, ACS Appl. Mater. Interfaces, 2021, 13, 2899–2907 CrossRef CAS PubMed.
- T. Hosono, N. O. Decarli, P. Z. Crocomo, T. Goya, L. E. de Sousa, N. Tohnai, S. Minakata, P. de Silva, P. Data and Y. Takeda, J. Mater. Chem. C, 2022, 10, 4905–4913 RSC.
- K. Leitonas, A. Tomkeviciene, G. Baratte, A. Dabuliene, S. M. Punniyakoti, D. Volyniuk and J. V. Grazulevicius, Sens. Actuators, B, 2021, 345, 130369 CrossRef CAS.
- M. Gmelch, T. Achenbach, A. Tomkeviciene, S. Reineke, M. Gmelch, T. Achenbach and S. Reineke, Adv. Sci., 2021, 8, 2102104 CrossRef CAS PubMed.
- A. Kirch, T. Bärschneider, T. Achenbach, F. Fries, M. Gmelch, R. Werberger, C. Guhrenz, A. Tomkevičienė, J. Benduhn, A. Eychmüller, K. Leo, S. Reineke, A. Kirch, T. Bärschneider, T. Achenbach, F. Fries, M. Gmelch, R. Werberger, A. Tomkevičienė, J. Benduhn, K. Leo, S. Reineke, C. Guhrenz and A. Eychmüller, Adv. Mater., 2022, 34, 2205015 CrossRef CAS PubMed.
- X. Wang, H. Shi, H. Ma, W. Ye, L. Song, J. Zan, X. Yao, X. Ou, G. Yang, Z. Zhao, M. Singh, C. Lin, H. Wang, W. Jia, Q. Wang, J. Zhi, C. Dong, X. Jiang, Y. Tang, X. Xie, Y. Yang, J. Wang, Q. Chen, Y. Wang, H. Yang, G. Zhang, Z. An, X. Liu and W. Huang, Nat. Photonics, 2021, 15, 187–192 CrossRef CAS.
- N. Gan, X. Zou, M. Dong, Y. Wang, X. Wang, A. Lv, Z. Song, Y. Zhang, W. Gong, Z. Zhao, Z. Wang, Z. Zhou, H. Ma, X. Liu, Q. Chen, H. Shi, H. Yang, L. Gu, Z. An and W. Huang, Nat. Commun., 2022, 13, 1–8 Search PubMed.
- M. Bregnhøj, M. Westberg, B. F. Minaev and P. R. Ogilby, Acc. Chem. Res., 2017, 50, 1920–1927 CrossRef PubMed.
- B. F. Minaev and A. F. Terpugova, Soviet Phys. J., 1969, 12, 1260–1263 CrossRef.
- H. Wu, L. Gu, G. V. Baryshnikov, H. Wang, B. F. Minaev, H. Ågren and Y. Zhao, ACS Appl. Mater. Interfaces, 2020, 12, 20765–20774 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |