
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient in vitro and in vivo transfection of self-amplifying mRNA with linear poly(propylenimine) and poly(ethylenimine-propylenimine) random copolymers as non-viral carriers†
Lisa
Opsomer
 a,
Somdeb
Jana
b,
Ine
Mertens
b,
Xiaole
Cui
a,
Richard
Hoogenboom
*b and
Niek N.
Sanders
*ac
a,
Somdeb
Jana
b,
Ine
Mertens
b,
Xiaole
Cui
a,
Richard
Hoogenboom
*b and
Niek N.
Sanders
*ac
aLaboratory of Gene Therapy, Department of Veterinary and Biosciences, Faculty of Veterinary Medicine, Ghent University, B-9820 Merelbeke, Belgium. E-mail: Niek.Sanders@ugent.be; Tel: +32 9 264 78 08
bSupramolecular Chemistry Group, Centre of Macromolecular Chemistry (CMaC), Department of Organic and Macromolecular Chemistry, Ghent University, 9000 Ghent, Belgium. E-mail: Richard.Hoogenboom@ugent.be; Tel: +32 9 264 4481
cCancer Research Institute (CRIG), Ghent University, B-9000 Ghent, Belgium
First published on 18th March 2024
Abstract
Messenger RNA (mRNA) based vaccines have been introduced worldwide to combat the Covid-19 pandemic. These vaccines consist of non-amplifying mRNA formulated in lipid nanoparticles (LNPs). Consequently, LNPs are considered benchmark non-viral carriers for nucleic acid delivery. However, the formulation and manufacturing of these mRNA-LNP nanoparticles are expensive and time-consuming. Therefore, we used self-amplifying mRNA (saRNA) and synthesized novel polymers as alternative non-viral carrier platform to LNPs, which enable a simple, rapid, one-pot formulation of saRNA-polyplexes. Our novel polymer-based carrier platform consists of randomly concatenated ethylenimine and propylenimine comonomers, resulting in linear, poly(ethylenimine-ran-propylenimine) (L-PEIx-ran-PPIy) copolymers with controllable degrees of polymerization. Here we demonstrate in multiple cell lines, that our saRNA-polyplexes show comparable to higher in vitro saRNA transfection efficiencies and higher cell viabilities compared to formulations with Lipofectamine MessengerMAX™ (LFMM), a commercial, lipid-based carrier considered to be the in vitro gold standard carrier. This is especially true for our in vitro best performing saRNA-polyplexes with N/P 5, which are characterised with a size below 100 nm, a positive zeta potential, a near 100% encapsulation efficiency, a high retention capacity and the ability to protect the saRNA from degradation mediated by RNase A. Furthermore, an ex vivo hemolysis assay with pig red blood cells demonstrated that the saRNA-polyplexes exhibit negligible hemolytic activity. Finally, a bioluminescence-based in vivo study was performed over a 35-day period, and showed that the polymers result in a higher and prolonged bioluminescent signal compared to naked saRNA and L-PEI based polyplexes. Moreover, the polymers show different expression profiles compared to those of LNPs, with one of our new polymers (L-PPI250) demonstrating a higher sustained expression for at least 35 days after injection.
Introduction
The COVID-19 pandemic has created momentum for messenger RNA (mRNA) technology.1 Decades of research in this field combined with the urgent need of a vaccine against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) have ensured that mRNA vaccines were the first to receive emergency use authorization by the Food and Drug Administration (FDA) and European Medical Agency.2,3 This is not surprising given that Moderna generated the first batch of a clinical-grade COVID-19 mRNA vaccine only 42 days after the publication of the Wuhan SARS-CoV-2 genomic sequence by the Chinese authorities.4 This demonstrates one of the major advantages of the mRNA platform, being its customisable and rapid development, which allows codes for new antigens or proteins to be plugged in without major changes to the production process. Other advantages of mRNA are its effectiveness in both dividing and non-dividing cells and this with a well-defined, predictable period of expression without the risk of insertional mutagenesis.5–7 These features make mRNA more predictable than DNA and viral vectors, which are also investigated for vaccine and therapeutic applications.6–8 Furthermore, mRNA molecules do not contain antibiotic resistant genes and its manufacturing process is rapid, scalable and cell-free in contrast to DNA and viral vector based vaccines.9,10These excellent properties make mRNA a good platform for vaccines, which can be seen in the high number of clinical trials of mRNA vaccines against viral-based infectious diseases, such as: dengue, influenza, HIV-1 (AIDS), respiratory syncytial virus (RSV) and rabies, but also against parasite-derived diseases, such as malaria.10–12 Moreover, the clinical use of in vitro transcribed (IVT) mRNA as a therapeutic vaccine against cancer or as a protein replacement therapy for acquired or congenital genetic disorders is also receiving significant attention.13–16 Altogether this indicates that in vitro synthesized mRNA-based vaccines and therapeutics will soon revolutionise modern medicine.17 The short expression of about a week still represents a point of improvement for mRNA, as this often requires relatively high doses to be administered at a high frequency. This mainly applies to protein replacement therapy where a long-term effect is desired.18,19 Self-amplifying mRNA (saRNA) is an advanced alternative to mRNA that potentially can overcome these limitations and is being developed for the next generation of mRNA vaccines and therapeutics.20–23 SaRNA shares many structural similarities with non-replicating mRNA as it also has a 5′ cap, an open reading frame (ORF) with the gene of interest, two UTR's that flank the ORF, and a 3′ poly(A) tail. However, it differs from non-replicating mRNA in that it also codes for a viral replicase, which is usually derived from positive sense alphaviral genomes.23 The latter enlarges the saRNA molecules to ∼9500–12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 nucleotides, while the size of non-replicating mRNA ranges between 2000 and 5000 nucleotides.24 The viral replicase consists of four non-structural proteins (nsPs) and enables the amplification of genomic and subgenomic mRNA upon reaching the host cell's cytosol. This results in an enhanced protein or antigen expression.10,23,24 Consequently, the saRNA dose to be administrated may be 24- to 64-fold lower than mRNA, depending on the administration route.22,25 Since it has been previously stated that the amount of RNA per vaccine dose has a high impact on the vaccine production cost per dose,(18) the use of saRNA vaccines could reduce these costs. Interestingly, very recently the first approved saRNA vaccine (ARCT-154, Arcturus Therapeutics & CSL) uses a 6-fold lower dose compared to the Pfizer/BioNTech and Moderna COVID-19 mRNA vaccines.26,27 However, the RNA purification method has also an important impact on the production cost and can be more challenging in case of saRNA compared to mRNA. From the available lab-scale purification methods, the cellulose-based purification is one of the few methods that removes dsRNA artefacts from IVT RNA.(28) This technique is also compatible for saRNA and results in a lower innate immune response and higher expression level compared to non-cellulose-based purified saRNA.29 Importantly, despite the self-amplifying capacity, the expression is limited in time, resulting in a temporal in vivo saRNA expression which, depending on the dose, delivery agent and the administration route, can last for up to seven weeks.19,30,31 Notably, the very recent approval of the first saRNA-based vaccine (ARCT-154) by the Japanese authorities highlights the efficacy, safety and benefit of saRNA vaccines.27
000 nucleotides, while the size of non-replicating mRNA ranges between 2000 and 5000 nucleotides.24 The viral replicase consists of four non-structural proteins (nsPs) and enables the amplification of genomic and subgenomic mRNA upon reaching the host cell's cytosol. This results in an enhanced protein or antigen expression.10,23,24 Consequently, the saRNA dose to be administrated may be 24- to 64-fold lower than mRNA, depending on the administration route.22,25 Since it has been previously stated that the amount of RNA per vaccine dose has a high impact on the vaccine production cost per dose,(18) the use of saRNA vaccines could reduce these costs. Interestingly, very recently the first approved saRNA vaccine (ARCT-154, Arcturus Therapeutics & CSL) uses a 6-fold lower dose compared to the Pfizer/BioNTech and Moderna COVID-19 mRNA vaccines.26,27 However, the RNA purification method has also an important impact on the production cost and can be more challenging in case of saRNA compared to mRNA. From the available lab-scale purification methods, the cellulose-based purification is one of the few methods that removes dsRNA artefacts from IVT RNA.(28) This technique is also compatible for saRNA and results in a lower innate immune response and higher expression level compared to non-cellulose-based purified saRNA.29 Importantly, despite the self-amplifying capacity, the expression is limited in time, resulting in a temporal in vivo saRNA expression which, depending on the dose, delivery agent and the administration route, can last for up to seven weeks.19,30,31 Notably, the very recent approval of the first saRNA-based vaccine (ARCT-154) by the Japanese authorities highlights the efficacy, safety and benefit of saRNA vaccines.27
By using non-viral carriers, RNA expression can be increased and prolonged compared to naked RNA.32 Non-viral carriers electrostatically interact and condense RNA into nanoparticles. These carriers protect RNA against enzymatic degradation and facilitate the cellular uptake and endosomal escape of the RNA cargo, which are crucial for its transfection.7 Up to now, saRNA has been formulated in lipid nanoparticles (LNPs),32 polymer-based nanoparticles (PNPs) or polyplexes33 and cationic nano emulsions.34 Currently, LNPs are the state-of-the-art non-viral carriers since they are clinically used in the COVID-19 mRNA vaccines and in Patisiran (trade name: Onpattro), a siRNA-based therapeutic.1,35 In general, LNPs consist of an ionizable amino lipid, helper lipids (cholesterol and a phospholipid) and a poly(ethylene glycol)-conjugated lipid (PEGylated lipid).36–39 They show a high delivery efficiency, but there are some limitations, such as the time-consuming,40 complex formulation process,41 the tropism to the liver42 and the presence of PEG-lipids that can trigger anti-PEG antibodies and in rare cases (severe) allergic reactions.43,44 Also, the low thermostability is an issue, requiring storage and transportation of the mRNA-LNPs at −20 °C or lower.45 From a pharmaceutical and regulatory point-of-view, it would be more favourable to have a more straightforward formulation involving fewer components and production steps. A one-step formulation of mRNA-based therapeutics and vaccines is possible by using cationic polymers containing protonatable tertiary or secondary amine groups.46,47 Compared to lipids, polymers usually interact better with nucleic acids and condense these nucleic acids into smaller nanoparticles (polyplexes).46,48 Moreover, polyplexes self-assemble in organic solvent-free, aqueous conditions by simply mixing the polymer with the nucleic acids and have a high flexibility towards chemical modifications with targeting or shielding moieties.44 Among the polymers, linear poly(ethyleneimine) (L-PEI) is one of the most efficient carriers as it shows both high in vitro and in vivo transfection efficiencies, due to its high density of protonatable secondary amino groups that result in an adequate RNA condensation and efficient endosomal escape through the so called: “proton sponge effect”,47,49 although other endosomal escape mechanisms involving phospholipid degradation have also been proposed.50,51
Despite the high transfection efficiency of L-PEI, its toxicity and related safety issues are still a major concern in clinical translation.52 This toxicity is most likely caused by the high overall positive charge of the L-PEI-based polyplexes, which can induce mitochondrial depolarization and subsequent necrotic cell death and apoptosis.47,52,53 One approach to reduce cytotoxicity but maintain transfection efficiency is to chemically modify the PEI structures,52 another one is to synthesize copolymers with lower charge density54 or copolymers that resemble endogenous structures, e.g. polyamines.47
In this work, we aimed to create a new polymeric carrier platform to overcome the drawbacks and disadvantages of LNP and L-PEI-based formulations. Therefore, we recently established a synthesis method for the preparation of linear PEI with randomly incorporated linear polypropylenimine (L-PPI) repeating units, through copolymerization of 2-ethyl-2-oxazoline with 2-isopropyl-2-oxazine followed by acidic hydrolysis of the resulting poly(2-ethyl-2-oxazoline-ran-2-isopropyl-2-oxazine) copolymers.49,55–57 For more details on the synthesis of the near-ideal random copolymers, we refer to our previous publication.49 Of these resulting linear, random PEIx–PPIy copolymers (L-PEIx-ran-PPIy), we were able to tune the PEI/PPI ratios and degrees of polymerization, resulting in controllable corresponding charge densities, and this over a wide range of molecular weights, with a narrow size distribution (see Fig. 1). It was shown previously that our L-PEIx-ran-PPIy copolymers are potent carriers for intracellular DNA delivery in vitro, and this with exceptional serum tolerance.49 In the current work we investigated if our linear, random L-PEIx-ran-PPIy copolymers with certain PEIx/PPIy ratios, and their homopolymers variants, all with a DP 250 (∼10–15 kDa) are efficient non-viral carriers for saRNA delivery (with x and y the percentages of the total amount of monomers in the polymer). Depending on the N/P ratio, the polymers were able to condensate the saRNA into very small nanoparticles. Four saRNA-based polyplexes showed higher in vitro transfection activities and cell viability compared to Lipofectamine MessengerMAX™ (LFMM). Six saRNA-based polyplexes, including the four in vitro best performing ones, were selected to further verify their in vivo saRNA delivery capacity in comparison to a state-of-the-art LNP carrier. This direct comparison of polymer-based to lipid-based carriers is very rare, but nevertheless very important to understand the differences in performances. At this point, the few studies that made the comparison are inconclusive, with some showing lower58 and others showing higher23 protein expression with polymer-based carriers compared with lipid-based carriers. Our in vivo study demonstrated that L-PEI60-ran-PPI190 and L-PPI250 show a lower, but more prolonged expression of luciferase-coding saRNA (at least until day 14 and day 35 after injection, respectively) compared to a LNP. In summary, this paper presents a new L-PEIx-ran-PPIy based polymeric carrier platform for the intracellular delivery of saRNA-based applications, with efficient in vitro transfection and great promise for in vivo translation.
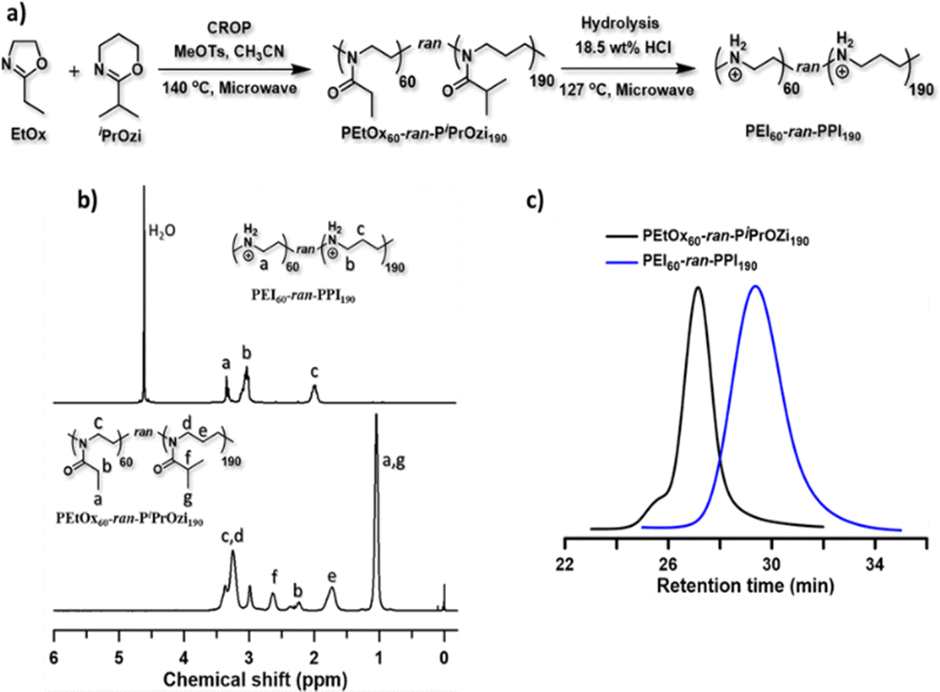 | ||
| Fig. 1 Overview copolymer synthesis and analysis. Panel (a): synthesis scheme for the preparation of L-PEI60-ran-PPI190 copolymer. Panel (b): 1H NMR spectra of PEtOx60-ran-PiPrOzi190 and L-PEI60-ran-PPI190 in CDCl3 and D2O, respectively. Panel (c): SEC traces of PEtOx60-ran-PiPrOzi190 in DMA and L-PEI60-ran-PPI190 in aqueous medium. | ||
Results and discussion
Synthesis and characterization of linear PEIx–PPIy random copolymers
With the aim of creating a cost-effective, non-toxic and efficient non-viral carrier platform, initially for saRNA-based nanoparticles, we synthesized and tested novel L-PEIx-ran-PPIy and L-PPI polymeric carriers, as alternative to the LNPs. The motivation for making these polymers was the hypothesis that the introduction of more hydrophobic comonomers (PPI) into the linear PEI structure (resulting in linear PEI–PPI copolymers and the PPI homopolymer), would lower the charge density and increase the hydrophobicity of the polymers. The log P values of the hypothetical homopolymer structures (L-PEI and L-PPI DP 1) calculated with ChemDraw software confirm this hypothesis considering hydrophobicity (Fig. S1, ESI†). Consequently, this lower charge density and increased hydrophobicity could potentially lead to polymers with higher transfection efficiency and lower cytotoxicity. Indeed, it has been reported that the charge-dense L-PEI interacts too strongly with mRNA resulting in an inefficient intracellular release of the mRNA, compared to the lipid based carrier LFMM.59 Additionally, the high cytotoxicity of PEI is known to be due to its high charge density.59,60 Introducing more hydrophobicity in linear polymers such as e.g., L-PEI and polycation poly(dimethylaminoethyl acrylate) (pDMAEA) has shown to increase efficiency and decrease cytotoxicity when an optimal balance is found.60,61 Introducing PPI comonomers is therefore expected to enhance transfection efficiency and decrease cytotoxicity. Moreover, due to the random distribution of PEI and PPI units, the structure of the L-PEIx-ran-PPIy copolymers more closely resembles the endogenous oligoamine spermine. This is beneficial, since spermine, which also consists partly out of PPI monomers, electrostatically interacts with DNA and RNA at physiological pH, as a result of its net positive charge.62 Interestingly, it is also assumed that spermine forms H-bonds to both the phosphate and the 2′-OH of RNA.62,63 Lastly, the findings of Ziebarth et al. that show that an additional methylene group (–CH2–) in polyamines provides extra distance between the amine groups, leading to much easier protonation compared to PEI, which might also be the case for the PEI–PPI copolymers.64With this motivation, a small library of linear, near-ideal random PEIx–PPIy copolymers and the corresponding homopolymers (with x:y = 0:250; 60:190; 100:150; 150:100; 190:60 and 250:0) were synthesized with low dispersity, by cationic ring-opening polymerization (CROP) of 2-ethyl-2-oxazoline (EtOx) and/or 2-isopropyl-2-oxazine (iPrOzi), followed by acidic side chain hydrolysis according to our recently developed protocol (see Fig. 1(a)).49 The degree of polymerization (DP) and molecular weight (Mn) of the L-PEIx-ran-PPIy (co)polymers were 250 and ∼13 kDa (see Table 1) as determined by 1H NMR spectroscopy (see Fig. 1(b)) and size exclusion chromatography (SEC) (see Fig. 1(c)), respectively. The aqueous SEC usually gives higher dispersity (Đ), but the unimodal traces indicate the absence of main chain hydrolysis, as expected by using a controlled microwave heating protocol for side-chain hydrolysis of the poly(2-oxazoline-ran-2-oxazine) copolymers.65 Furthermore, the SEC analysis of the precursor poly(2-oxazoline-ran-2-oxazine) copolymers revealed well-defined structures with narrow molar mass distribution and low Đ (see Table 1 and Fig. S2–S5, ESI†).65
| Precursor polymers | Final polymers | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Samples | Compositiona (mol%) [EtOx]:[iPrOZi] |
M n (theo) (kDa) | M n (SEC) (kDa) | Đ (SEC) | Samples | Compositiona (mol%) [PEI]:[PPI] |
M n (theo) (kDa) | M n (SEC) (kDa) | Đ (SEC) |
| a Exact composition of PEtOx/PiPrOzi and PEI/PPI are determined from 1H NMR analysis. b Determined by SEC in DMAc with RI detection (calculated against narrow disperse PMMA standards from PSS). c Determined by SEC in methanol–sodium acetate buffer with RI detection (calculated against narrow disperse PEG standards from PSS). | |||||||||
| PEtOx60-ran-PiPrOZi190 | 25:75 |
30.1 | 29.0 | 1.14 | PEI60-ran-PPI190 | 25:75 |
13.0 | 44.0 | 1.38 |
| PiPrOZi250 | 0:100 |
31.7 | 33.5 | 1.17 | PPI250 | 0:100 |
15.0 | 33.0 | 1.25 |
In vitro transfection efficiency and cytotoxicity of the saRNA-polyplexes
Recently, we reported on the synthesis and full characterization of a library of L-PEIx-ran-PPIy copolymers and their DNA transfection efficiency.49 In this paper, we report for the first time on the in vitro transfection efficiency, cytotoxicity, physicochemical properties and in vivo performance of saRNA-polyplexes based on a small library of L-PEIx-ran-PPIy copolymers with varying PEI/PPI monomer ratio's and a constant DP of 250. Only a limited number of polymer-based carriers are successful in delivering saRNA-based cargo in vivo, therefore this work is of great importance in the search for new and better performing non-viral carriers.66First, the ability of six L-PEIx-ran-PPIy (co)polymers, to serve as a carrier for saRNA was tested in HeLa cells. SaRNA encoding luciferase was formulated with the polymers at six different N/P ratios (N/P 40 – 20 – 10 – 5 – 1 – 0.2). The resulting saRNA-polyplexes, 36 in total, were incubated with HeLa cells and their transfection efficiency was measured and compared with saRNA formulated with Lipofectamine MessengerMAX™ (LFMM), the in vitro gold standard, lipid-based mRNA transfecting agent. Non-transfected (NTF) cells served as negative control. Twenty-four hours after administration, the saRNA-polyplexes based on L-PEI60-ran-PPI190 and L-PPI250 were the best performing polymers (see Fig. 2 and Fig. S6, ESI†). For both polymers, a N/P ratio of 5 resulted in the highest transfection efficiency with bioluminescent signals that were significantly higher compared to the N/P ratios of 40, 20, 1, 0.2 and the non-transfected cells (see Fig. 2, graphs a and d). In case of L-PPI250, the saRNA-polyplexes prepared at a N/P 5 also outperformed the N/P 10 formulation. Remarkably, at this optimal N/P ratio, both polymers resulted in a slightly, but not statistically significant, higher bioluminescent signal compared to LFMM, the commercial, lipid-based gold standard carrier. Independent repeated experiments showed the same trends and hence confirm the reproducibility of these findings (see Fig. S7, ESI†). One of these two best performing polymers, i.e., L-PPI250 has also been tested as non-viral carrier in combination with non-self-amplifying, nucleotide-modified mRNA. Interestingly, similar trends in optimal N/P ratio could be distinguished for the non-self-amplifying and self-amplifying mRNA cargo in HeLa cells (see Fig. S8, ESI†).
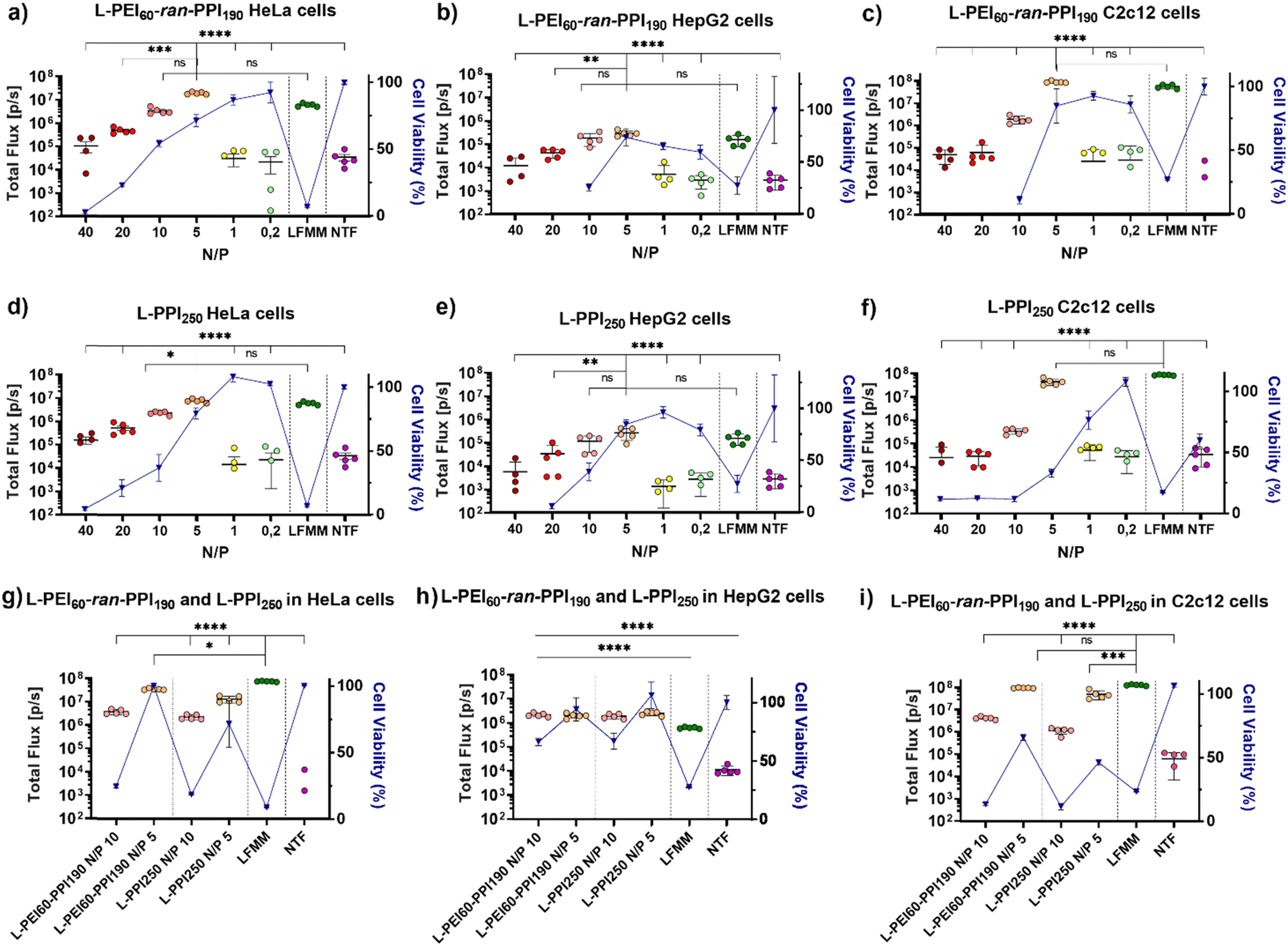 | ||
| Fig. 2 Bioluminescent signals and cell viability data of in vitro saRNA-polyplex transfections with L-PEI60-ran-PPI190 and L-PPI250, in HeLa cells, HepG2 and C2c12 cells. Cells were transfected with 500 ng saRNA per well in a 24 well-plate, saRNA was either complexed with L-PEI60-ran-PPI190 or L-PPI250 (considering six N/P ratios, N/P 40 – 0,2) or encapsulated by LFMM at a 2:1 ratio (ratio represents: ng saRNA:μL LFMM). Graphs (a)–(f) show data 24 hours after transfection, without changing medium, thus cells were 24 hours exposed to the complexes. Graphs (g)–(i) show the data obtained by replacing the nanoparticle-containing medium by full DMEM 4 hours after transfection. The data were obtained with the IVIS Lumina III (Total Flux [p/s]). Each circle represents one well of the 24 well-plate (N = 5), the line represents mean ± SEM. * indicates significance of p ≤ 0.05, ** indicates significance of p ≤ 0.01, *** indicates significance of p ≤ 0.001, **** indicates significance of p ≤ 0.0001. All means were compared to each other with one-way ANOVA, after log-transformation of the obtained data and testing for normality. Adjustment for multiple comparisons was performed using Tukey's multiple comparisons test in GraphPad Prism 8.4.3. Cell viability data (blue triangles) is based on the WST-1 assay 24 hours after initial transfection, and after 2–3 hours of incubation with the WST-1 reagent. Outliers were removed according to the IQR method and data represent mean ± SEM. Abbreviations: LFMM = Lipofectamine MessengerMAX™, NTF = non-transfected cells. | ||
Remarkably, L-PEI250 and two polymers with near equal amounts of PEI and PPI monomers (L-PEI100-PPI150 and L-PEI150-PPI100) exhibited the lowest transfection efficacy for saRNA (Fig. S6, ESI†), which highlights that even subtle changes in the structure of carriers have great impact on the carrier's efficiency and that different polymer structures are optimal for different nucleic acids, as previously we reported that for DNA transfection a 1:1 ratio of PEI and PPI was best for in vitro transfection.49
After in vivo administration, the contact time of nanoparticles with cells is usually less than 24 hours, because of their bio-distribution. Therefore, the transfection efficiency of the four-best performing saRNA-polyplexes (L-PEI60-ran-PPI190 and L-PPI250, N/P 5 and 10) was also evaluated after a shorter incubation time, i.e. 4 hours instead of 24 hours. This was conducted by replacing the medium containing the saRNA-polyplexes with full medium 4 hours after administration. Subsequently, 24 hours after initial addition of the saRNA-polyplexes, the bioluminescent signal was measured. Shortening the transfection time increased the transfection of the saRNA-LFMM formulation, while the transfection of the saRNA-polyplexes remained the same (Fig. 2(g)). Consequently, at a shorter incubation time, LFMM induced slightly, significant, better saRNA translation data compared to the polymers (p ≤ 0,001), except for L-PEI60-ran-PPI190-based saRNA-polyplexes prepared at N/P 5, which exhibited a comparable efficiency as LFMM (p ≤ 0, 05) (see Fig. 2(g)).
Next to high transfection efficiency, biocompatibility is one of the most important criteria for efficient and safe non-viral carriers. The term ‘biocompatibility’ covers many different properties, however, in vitro cytotoxicity and hemocompatibility are addressed in particular when screening novel carriers.67 The cell viability gradually decreased as a function of the N/P ratio of the saRNA-polyplexes. This was measured 24 h after addition of the nanoparticles to the HeLa cells, by using the WST-1 assay. Generally, L-PPI250 tends to be slightly more cytotoxic than L-PEI60-ran-PPI190, especially at N/P ratios above 5. SaRNA-polyplexes prepared at N/P 5, the most efficient ratio for transfection, moderately decreased cell viability (∼20–30% drop) relative to non-transfected cells, while saRNA-LFMM nanoparticles were very cytotoxic as they induced a drastic reduction in cell viability (>90% drop). Since a relatively high dose of saRNA (500 ng/50000 cells in a 24 well-plate) was used compared to other work, the saRNA-LFMM nanoparticles here might show higher cytotoxicity. SaRNA-polyplexes formulated with L-PEI60-ran-PPI190 at N/P 5 did not induce cytotoxicity in HeLa cells when the incubation time was shortened to 4 h.
In order to reinforce our findings with the HeLa cells, the translation efficiencies of the two best performing polymers, i.e. L-PEI60-ran-PPI190 and L-PPI250 were studied in humane HepG2 cells (see Fig. 2, graphs b and e) and murine C2c12 cells (see Fig. 2, graphs c and f). These cell types were chosen, as they are important target cells after intravenous and intramuscular injection of RNA-containing nanoparticles, respectively. Again, the cells were incubated for 24 h and 4 h with the saRNA-polyplexes. Generally, expression levels in C2c12 cells were comparably high to those in HeLa cells, while HepG2 cells showed lower bioluminescent signals for all tested nanoparticles. Interestingly, the overall trend in transfection efficiency of the saRNA-polyplexes as a function of N/P ratio in these cells was similar to those in HeLa cells and for both polymers the N/P ratio of 5 resulted in the highest bioluminescent signals. At this N/P ratio the saRNA translation obtained with the polymeric carriers was comparable with LFMM in both HepG2 and C2c12 cells (see Fig. 2, graphs b, c, e and f). Reducing the incubation time of the cells with the saRNA-nanoparticles from 24 h to 4 h resulted in a noticeable higher luciferase expression in HepG2 cells, most pronounced at N/P 10, with a circa 8-fold increase in bioluminescent signal. No changes were noticed in C2c12 cells.
Similar to HeLa cells, the cell viability of C2c12 and HepG2 cells dropped as a function of the N/P ratio. SaRNA-polyplexes prepared at N/P 5 induced a moderate drop in cell viability in these cells, analogous to HeLa cells, except for the L-PPI250-based saRNA-polyplexes, which were more cytotoxic in C2c12 cells. In both cell lines, the saRNA-LFMM nanoparticles were also drastically more cytotoxic than the most efficient saRNA-polyplexes. However, with the highest N/P ratios (40 and 20), the excess of polymer in the saRNA-polyplexes became too toxic in case of HepG2 and C2c12 cells (except for L-PPI250 in C2c12 cells). This resulted in rounding-up of the cells and loss of attachment to the culture plate. Consequently, these cells were washed away prior to read-out, with no detectable absorbance and hence, zero to sub-zero values for cell viability.
Shortening the incubation time form 24 h to 4 h, resulted in a higher cell viability of HepG2 cells transfected with saRNA-polyplexes at N/P 5, similar to HeLa cells, and an increase in cell viability at N/P 10. This might indicate that in HepG2 cells longer incubation times increase the cytotoxicity of the saRNA-nanoparticles. In C2c12 cells, the shorter incubation times did not clearly reduce the cytotoxicity of the saRNA-polyplexes. This indicates that our saRNA-polyplexes cause less cytotoxic effects in human cell lines compared to murine cell lines, since a broader N/P range was tolerated, except for the L-PEI60-ran-PPI190-based nanoparticles in the HepG2 cells. In summary, the transfections in these three different cell lines suggest that the transfection efficiency and cytotoxicity of our saRNA-polyplexes is cell-type and polymer-dependent, with L-PEI60-ran-PPI190 being the best performing polymer with the lowest cytotoxicity in HeLa and C2c12 cells and L-PPI250 being less cytotoxic in HepG2 cells, with no clear distinction in transfection efficiency between the polymers in this cell line.
Interestingly, higher cell viabilities do not necessarily result in higher transfection efficiencies, but could indicate that the intracellular endocytosis of the polyplexes was not successful, see polyplexes with N/P 1 and 0.2 (see Fig. 2). In some cases the cell viability was higher than 100%, and hence higher than observed for the untreated cells. This may be due to cell proliferation, cell stress, chemical reduction of the WST-1 reagent by the polymer or hormesis, a phenomenon in toxicology where compounds can be beneficial/stimulatory at low doses but toxic/inhibiting at higher doses.68 In this context, it is interesting to note that polyamines may indirectly act as scavengers of oxygen free radicals, protecting nucleic acids and other cellular components from oxidative damage which could result in higher cell viability.62
Physicochemical properties, complexation efficiency and protection towards RNase A
Transfection efficiency and biocompatibility of nanoparticles are closely related to their size and zeta potential (which is related to the surface charge). Nanoparticles with small sizes and a slightly negative to neutral zeta potential are considered optimal for in vivo applications.69 Regarding the size of RNA-based nanoparticles, a range of 6–8 nm70,71 to 150–200 nm72,73 is considered to be ideal, so that particles are retained by the glomerulus and are small enough to escape from phagocytosis by macrophages, which would remove the particles from the body as part of the mononuclear phagocyte system.73Regarding our saRNA-polyplexes based on L-PEI60-ran-PPI190 and L-PPI250, all of them show sizes larger than the minimum size range and the majority shows sizes smaller than 150 nm (mean diameter), except for L-PEI60-ran-PPI190 at N/P 40 with a size of ∼250 nm. This last result was unexpected, since we hypothesized that the more polymer we would use (i.e. the higher the N/P ratio), the more the saRNA would be condensed, the smaller the saRNA-polyplexes would be. This was true, however, only up to an N/P of 5–10 after which the saRNA-polyplexes did not become smaller anymore, but larger (N/P ≥ 20). This limit probably represents a “saRNA condensation limit” of the polymers, where polyplexes with N/P 5–10 have a maximal sRNA condensation capacity resulting in the smallest and the most monodisperse saRNA-polyplexes. The later can be interpreted from their relatively low PDI values (Table S1, ESI†). Future improvements to lower the PDI value to ≤0.2 will be necessary to make the polyplexes acceptable for clinical applications.74 Potentially, introducing more steric hindrance on the nanoparticle surface or performing polyplex formulation through a more controlled mixing process may result in monodisperse nanoparticles.43
Regarding the ZP of the saRNA-polyplexes, the majority of them have a positive charge, which a clear shift to negative starting from N/P 1 and lower (N/P ≥ 5: ∼+30 to +40 mV; N/P ≤ 1: ∼−25 to −40 mV). This shift is accompanied with a size increase, with a maximum size of ∼350–400 nm at N/P 0.2, which might indicate an incomplete saRNA-complexation (see Fig. 3). The positive charge and the relatively small size of the saRNA-polyplexes with N/P 5 can explain the higher in vitro transfection efficiency, as they might efficiently interact with the negatively charged cell membranes, enabling cellular uptake. However, only particles with a neutral to slightly negative charge are generally accepted as desirable for in vivo applications.69
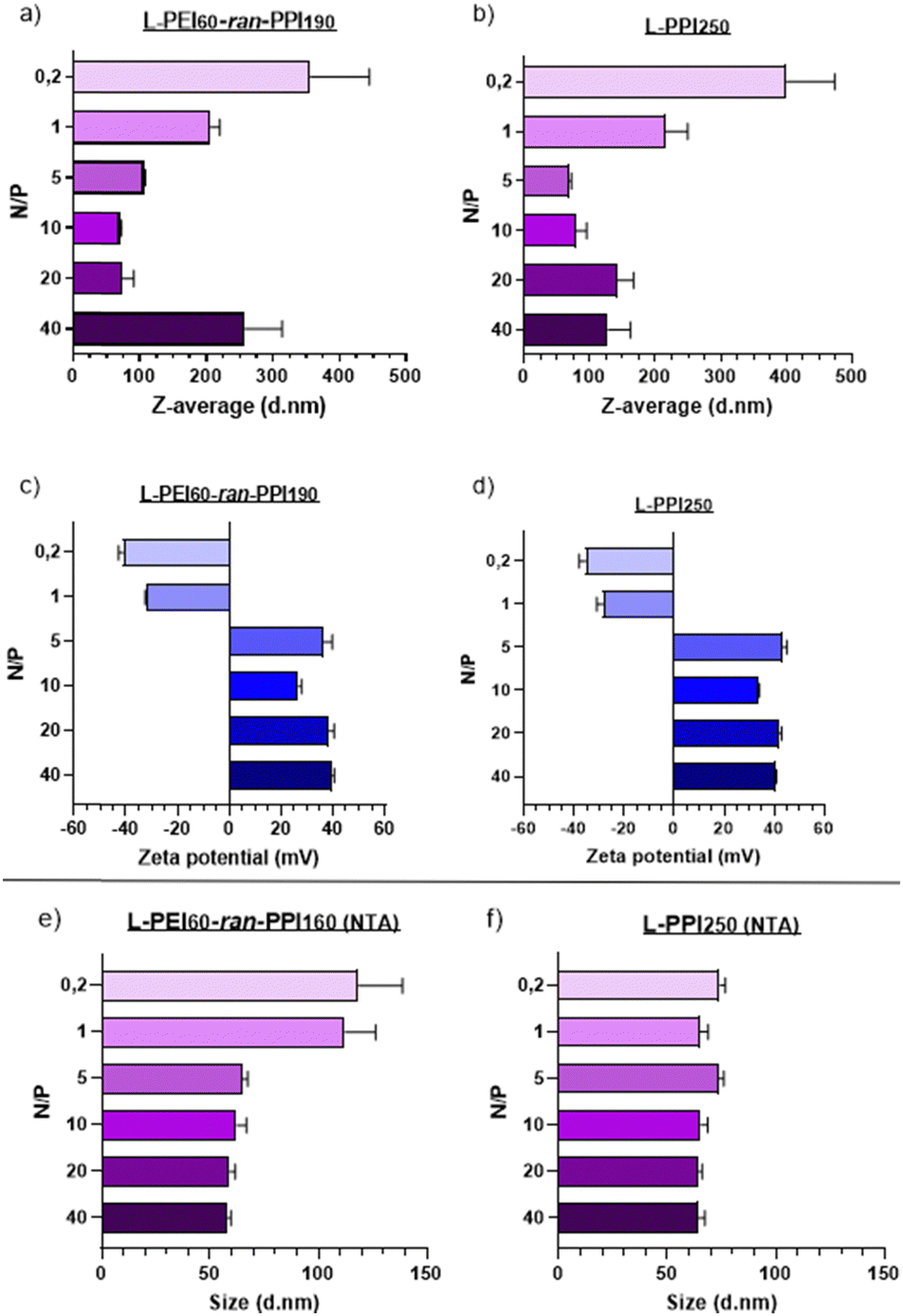 | ||
| Fig. 3 Size and zeta potential of the saRNA-based polyplexes, with L-PEI60-ran-PPI190 and L-PPI250 in NaOAC buffer. The Z-average sizes data (graphs (a) and (b), mean and SD) are based on intensity distributions of the dynamic light scattering measurements. The zeta potential data (graphs (c) and (d)) are based on laser Doppler velocimetry measurements. Data presented in these four graphs are obtained with the ZetaSizer Nano-ZS. Size data (graphs (e) and (f), mode and SE) are based on intensity distributions of the nanoparticle tracking analysis (NTA) measurements obtained with the NanoSight NS300. | ||
To determine the N/P ratio at which our saRNA-polyplexes become neutral, we made additional saRNA-polyplexes with N/P ratios 2, 3 and 4, and discovered that a N/P ratio of 2 results in approximately neutrally charged, but large (∼4 μm) saRNA-polyplexes as there is no charge stabilization (Fig. S9, ESI†). The transfection of these saRNA-polyplexes showed that N/P ratios of 5, 4 and 3 perform about equally good, with no statistically significant difference (Fig. S10, ESI†).
Additionally, to confirm these DLS-based results, nanoparticle tracking analysis (NTA) was performed, which showed sizes for all nanoparticles lower than 150 nm. In addition, the size differences between the N/P ratios were much smaller when NTA was used (see Fig. 3(e) and (f)). The mean diameter of the saRNA-polyplexes with a N/P from 5 till 40 was ∼70 nm. Only the L-PEI60-ran-PPI190 saRNA-polyplexes with a N/P of 1 and 0.2 had mean sizes above 100 nm (see Fig. 3). These size differences between DLS (ZetaSizer Nano-ZS) and NTA (NanoSight NS300) is a common observation that is most likely attributed to the tendency of DLS to skew towards larger particles sizes due to the use of intensity-weighted values.24
Ideal non-viral carriers must also maintain an optimal balance between retaining and releasing the (sa)RNA cargo, to protect it from degradation and to release it in the cytosol for efficient translation.61,75,76 Electrophoretic mobility shift assays or gel retardation assays were performed with the L-PEI60-ran-PPI190 and L-PPI250 saRNA-polyplexes (N/P 0.2-40) to assess the saRNA encapsulation capacity. These assays demonstrate that at N/P ratios above 1 all (100%) saRNA is encapsulated, resulting in absence of the free saRNA-band in the gel (see Fig. 4, 1). However, L-PEI60-ran-PPI190 and L-PPI250, prepared at N/P 1, show a partial saRNA-retainment of respectively 78% and 73%, based on the intensity of the free saRNA band. The polymers could not complex the saRNA at N/P 0.2 (see Fig. 4, 1). These results confirm that the saRNA-polyplexes with N/P 1 and 0.2 are not able to retain (all) saRNA, when they encounter an electrical field. This inefficient saRNA-complexation is in line with the less efficient in vitro transfections of these particular saRNA-polyplexes.
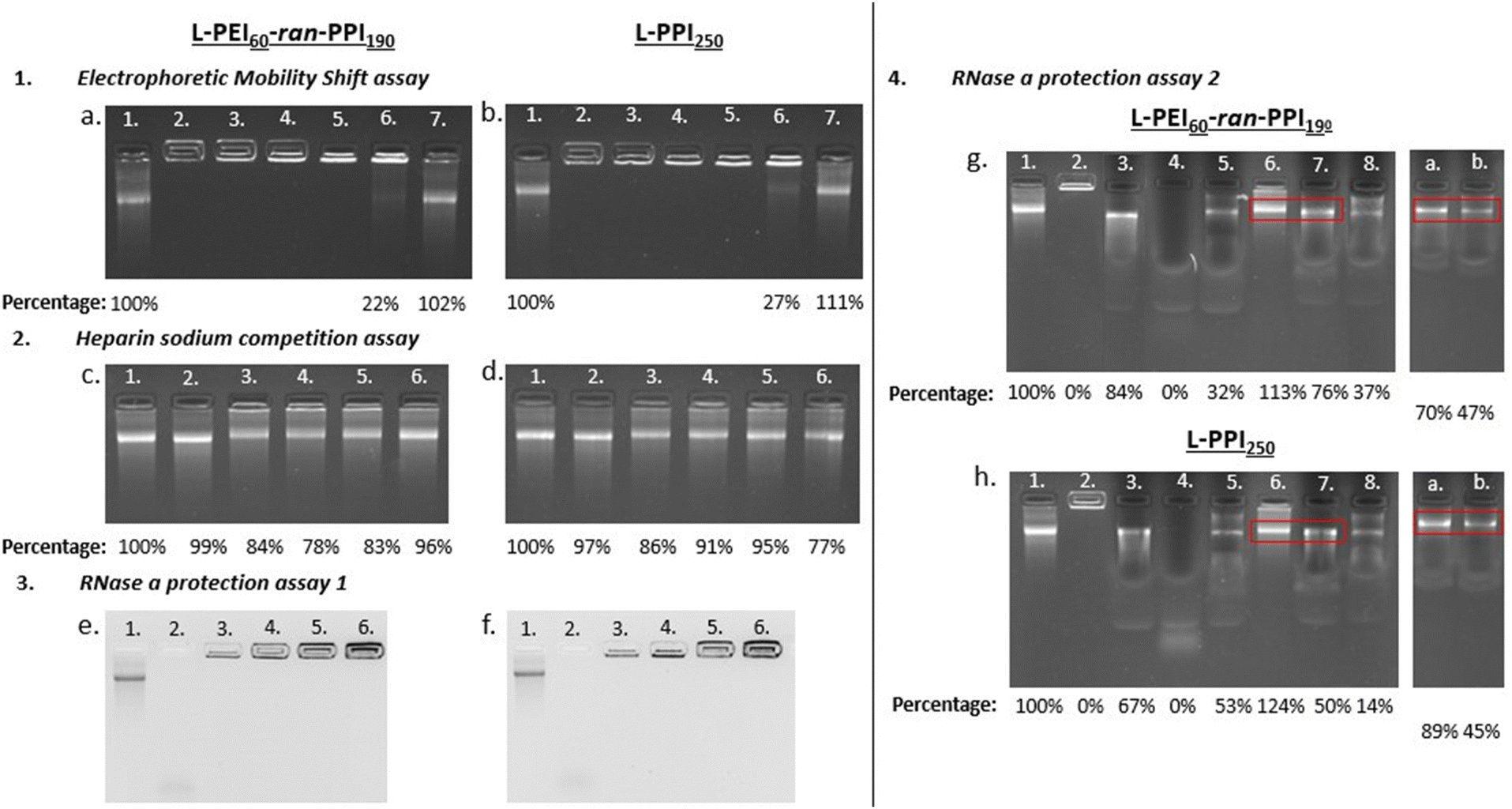 | ||
| Fig. 4 Results of the gel electrophoresis-based experiments, with left the saRNA-polyplexes based on L-PEI60-ran-PPI190 and right the ones based on L-PPI250. Fig. 1 (a) and (b) show the results of the electrophoretic mobility shift assay. Lane numbers in white correspond to “saRNA only” (lane 1) and the saRNA-polyplexes composed of 500 ng saRNA prepared at N/P ratios: 40 (lane 2) – 20 (lane 3) – 10 (lane 4) – 5 (lane 5) – 1 (lane 6) and 0,2 (lane 7). Fig. 2 (c) and (d) show the results of the heparin sodium competition assay. The lane numbers in white correspond to: “saRNA only” (lane 1), “saRNA only incubated at 37 °C for 1 hour (lane 2) and the saRNA-polyplexes incubated with 80 μg HS (lanes 3–6)”. For each condition 500 ng saRNA was used. Lane 1 shows naked saRNA stored on ice and lane 2 shows naked saRNA incubated for 1 hour at 37 °C, both serve as positive control. From left to right, lane 3 to 6 show the saRNA-polyplexes with N/P 20, 10, 5 and 1 after incubation with 80 μg HS for 1 hour at 37 °C. Fig. 3 (e) and (f) show the RNase A protection assay 1. The lane numbers in white correspond to: “saRNA only” as positive control (lane 1), naked saRNA with RNase A (lane 2) and the saRNA-polyplexes (N/P 5) with increasing amounts of saRNA and constant amounts of RNase A (1 ng) (lane 3.–6.). From left to right, lane 3 to 6 show the saRNA-polyplexes (N/P 5) with 500 ng, 1 μg, 2 μg and 5 μg saRNA, incubated with 1 ng of RNase A for 30 minutes at 37 °C. Fig. 4 (g) and (h) show the results of the RNase A protection assay 2, with the red boxes being the areas of interest, i.e. the location of the intact saRNA band. All lanes show a signal originating from 500 ng saRNA. Lane 1 shows “saRNA only” which serves as positive control, lane 2 shows the untreated saRNA-polyplex which serves as negative control. The composition of the samples (with respect of the order of the compounds) in the next lanes is as follows: RNA + SDS + RNase A (lane 3), RNA + RNase A + SDS (lane 4), saRNA-polyplex + RNase A + SDS (lane 5), saRNA-polyplex + HS (lane 6), saRNA-polyplex + HS + RNase A + SDS (lane 7) and saRNA-polyplex + RNase A + SDS + HS (lane 8). Lanes (a) and (b) originate from secondary gels, the composition of the samples is: (a) polyplex + HS and SDS + RNase A, and (b) saRNA-polyplex + RNase A + HS and SDS. Polyplexes are composed based on N/P 5. All UV-pictures show EtBr bleach agarose gels, with a detection limit of 10 ng saRNA. A quantification of the released saRNA by densiometric analysis (Fiji) is shown below the pictures as percentages relative to the “saRNA only” signal. | ||
Complementary to the gel retardation assay a RiboGreen assay was performed. However, for this assay a saRNA releasing agent is needed as the encapsulation efficiency is the percentage of fluorescent signal, derived from the RiboGreen stain complexed to the RNA, of the free/released saRNA reduced with the signal of the intact complex relative to the signal of free/released saRNA. To that end the polyanion heparin sodium (HS) was added to the saRNA-polyplexes (N/P 5) to compete with the saRNA for complexation with the polymers, in order to disassemble the saRNA-polyplexes and release the cargo. The addition of 160 μg HS/1 μg saRNA (total HS concentration: 3.2 μg μL−1) to the saRNA-polyplexes in NaOAc buffer, following an incubation of 1 hour at 37 °C showed the highest saRNA release of ∼80%. This HS concentration is at least 640 times higher compared to human blood.77 This illustrates the strong intermolecular interactions between our saRNA and our polymers, indicating the exceptional stability of our saRNA-polyplexes. Overall, this assay indicates, that at the best performing N/P ratios (5 and 10) the L-PPI250 based saRNA-polyplexes tend to release more saRNA than the complexes with the more charge dense L-PEI60-ran-PPI190 (see Fig. 4, 2c and d, lane 3–6). Interestingly, with this assay we also show that our saRNA is stable at 37 °C for at least 1 hour (see Fig. 4, 2c and d, lane 2).
After showing successful saRNA release with HS-addition, the RiboGreen assay was performed to quantify the saRNA encapsulation efficiency (EE%) of the polymers. This assay showed that L-PEI60-ran-PPI190 had an average EE% of 94.45% ± 0.13% and 94.20% ± 0.11%; and L-PPI250 an average EE% of 95.34% ± 0.09% and 95.11% ± 0.04%, at the optimal ratios for transfection (i.e. N/P 5 and 10, respectively), with no significant difference in EE% between both N/P ratios. Since our saRNA differs significantly in length from the rRNA standards of the RiboGreen kit, the EE%'s of the polymers relative to the fluorescence signal of a naked saRNA sample were also calculated. This resulted in EE%'s of ∼99% for both polymers at the tested N/P ratios. The influence of HS on the fluorescence signal was negligible.
Next, the ability of the two polymers to protect the saRNA from RNase A mediated degradation was investigated. A series of RNase A protection assays were conducted with the most efficient polyplexes (N/P 5). First, saRNA-polyplexes with increasing amounts of saRNA (0.5 to 5 μg) were challenged with a constant amount of RNase A (1 ng). After 30 minutes of incubation at 37 °C no traces of degraded saRNA could be discerned in the lanes of the polyplexes (see Fig. 4, 3e and f, lane 3–6). In contrast, non-formulated naked saRNA was heavily degraded by RNase A, which is visible by the smear at the bottom of the lane (see Fig. 4, 3e and f, lane 2).
However, this assay may result in artefacts if the degraded saRNA is retained by the polymers or if the RNase A enzyme is not able to reach the core saRNA, but only the external saRNA present on the surface of the saRNA-polyplexes. Hence, a second RNase A protection assay was performed in which the saRNA-polyplexes were first incubated with RNase A, followed by inhibition of the RNase A, and next, dissembled by HS in order to quantify the amount of released, intact saRNA. The order of these steps were changed to get a better understanding of the mechanism of action. First, several agents were tested as inhibitors of RNase A (Fig. S11 and S12, ESI†).
Addition of sodium dodecyl sulphate (SDS) to naked saRNA resulted in full (100%) protection from RNase A degradation, based on the relative intensity of the saRNA band after SDS and RNase A incubation compared to the naked “saRNA only”-band (Fig. S12, ESI†). Interestingly, HS also showed some inhibitory activity towards RNase A (see Fig. 4, 4g and h, lane 7). Finally, the amount of released, intact saRNA was compared between saRNA-polyplexes first incubated with (i) RNase A or (ii) HS + SDS, followed by incubation with (i) SDS + HS or (ii) RNase A, respectively. Together with the necessary control samples, this assay estimated that L-PEI60-ran-PPI190 and L-PPI250 protect at least ∼70% and ∼50% of saRNA from RNase A degradation, respectively, with the saRNA band intensity percentage of lane b divided by the one of lane a (see Fig. 4, 4g and h). This is in line with the results obtained by dividing the saRNA band intensity of lane 7 by that of lane 6, resulting in ∼70% and ∼40% protection with the same respective polymers (see Fig. 4, 4g and h). Thus, L-PEI60-ran-PPI190 tends to form saRNA-polyplexes that are the most protective towards saRNA degradation by RNase A. Furthermore, the enzyme is most likely able to penetrate the saRNA-polyplexes, but degraded saRNA fragments are not released or not detectable in the gel, since no saRNA smear was present in lane 7, a or b, as compared to lane 4 (see Fig. 4, 4g and h). Apart from saRNA integrity assessments after RNase A challenge by gel retardation experiments, it could potentially be of interest to perform transfections with HS treated saRNA-polyplexes (released saRNA) as well, similar to the work of Akhter S. et al.78 This may result in additional evidence that saRNA integrity is guaranteed upon condensation and release by our polymers. Despite the transfection experiments already showed this indirectly. Altogether, L-PEI60-ran-PPI190 appears to be more protective towards saRNA degradation and to result in somewhat more stable complexes compare to L-PPI250. Interestingly, this is in line with the RiboGreen data that confirmed that saRNA-polyplexes based on L-PEI60-ran-PPI190 were the hardest to disintegrate, with HS only being able to release ∼63–66% of the saRNA, compared to ∼73–75% saRNA in case of L-PPI250-based polyplexes. These results are somewhat expected as disintegration of L-PPI250-based saRNA-polyplexes is assumed to be more easy, because of the larger space between the N-atoms, which results in lower charge density and finally a presumable lower stability compared to saRNA-polyplexes based on L-PEI60-ran-PPI190, despite the anticipated stronger hydrophobic interaction in L-PPI250. Therefore, these results indicate that in these saRNA-polyplexes the charge density is of higher importance for complexation than the hydrophobic interactions. In addition, both assays showed that the saRNA-polyplexes were never completely disintegrated by addition of HS, presumably because the utilized saRNA molecule is very large in size (9664 nt), resulting in a high negative charge density, making it difficult for HS to compete for binding with the polymers (∼13 kDa). A challenge of the saRNA-polyplexes with a more representative mixture of proteins and serum components or mucus and extracellular matrix components would give us a more detailed view on their stability and saRNA protection ability in an in vivo setting.
Switch to a physiologically compatible formulation, ex vivo toxicity and first in vivo bioluminescent study
In what preceded, the saRNA-polyplexes were prepared in acidic NaOAc buffer (20 mM, pH 5.2). However, with in vivo administration and applications in mind, the saRNA-polyplexes (L-PEI60-ran-PPI190 and L-PPI250, N/P 10 and 5) were now formulated in HEPES buffer (pH 7.4), since in vivo use of an acidic buffer is considered inappropriate. First, the influence of the pH difference on the size and ZP of these saRNA-polyplexes were measured. We hypothesized that this would increase the size and decrease the ZP, as a higher pH would result in a less efficient protonation of the polymers. Consequently, the reduced amount of positive charges available for electrostatic interactions would result in larger sized saRNA-polyplexes as the saRNA would be less condensed. However, the opposite was true, saRNA-polyplexes in HEPES buffer showed no significant differences in size or ZP compared to the ones formulated in NaOAc (Fig. S13 and S14, ESI†). Again, the sizes measured with NTA tended to be slightly smaller. In a neutral environment, the polymer charge density may be lower, leading to a greater contribution of hydrophobic-based interactions to the saRNA-polyplex formation compared to an acidic environment, where charge-based electrostatic interactions may predominate.79,80 Alternatively, it is possible that at pH 7.4 there are still sufficient positive charge to induce a maximal condensation of the saRNA.Subsequently, the in vitro transfection efficiency of the saRNA-polyplexes formulated in HEPES buffer were compared with saRNA-polyplexes formulated in NaOAc buffer. For this, L-PEI60-ran-PPI190 based saRNA-polyplexes (N/P 5) were prepared in both buffers and transfected in HeLa cells with OptiMEM medium. Remarkably, the nanoparticles formulated in HEPES buffer outperformed the nanoparticles formulated in the acidic buffer with a ∼8-fold increase in the bioluminescent signal. Besides a neutral pH, the nanoparticles must also be resistant to environments with high serum to be suitable for in vivo applications. Hence, the nanoparticles formulated in NaOAc were also transfected in serum containing medium, being full DMEM (10% FBS, 1% P/S). Interestingly, this did not reduce the transfection efficiency of the L-PEI60-ran-PPI190 based saRNA-polyplexes, and they even performed equivalent to the OptiMEM condition (see red dots Fig. 5). This illustrates that L-PEI60-ran-PPI190 is able to transfect the saRNA in serum-containing medium. Finally, replacing the OptiMEM medium containing the nanoparticles 4 hours after transfection with full DMEM medium (shorter transfection time) has no effect on the transfection efficiency (see pink dots Fig. 5).
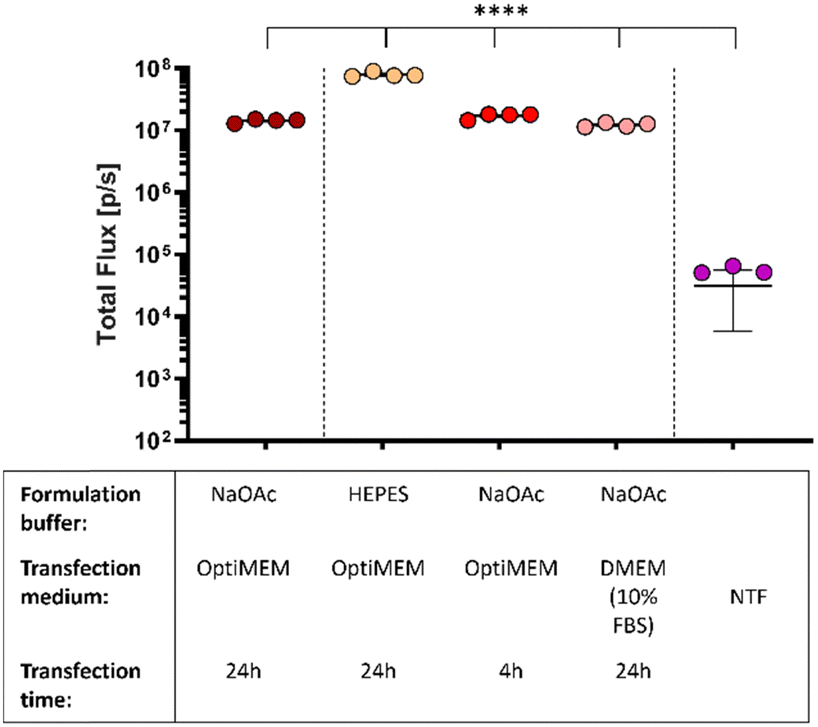 | ||
| Fig. 5 Bioluminescent signals of in vitro saRNA-polyplex transfections with L-PEI60-ran-PPI190 in HeLa cells. Cells (exposed to OptiMEM or full DMEM) were transfected with L-PEI60-ran-PPI190-based saRNA-polyplexes (N/P 5) containing 500 ng saRNA per well (24 well-plate). Polyplexes were formulated in HEPES (20 mM, pH 7.4; orange dots) or NaOAc (20 mM, pH 5.2). The graph shows data 24 hours after transfection, medium was not changed except for the pink dots, here OptiMEM medium was replaced by full DMEM/10% FBS 4 hours after transfection. Data was obtained with the IVIS Lumina III (Total Flux [p/s]). Each circle represents one well of the 24 well-plate (N = 4), the line represents mean ± SEM. *indicates significance of p ≤ 0.05, **indicates significance of p ≤ 0.01, ***indicates significance of p ≤ 0.001, ****indicates significance of p ≤ 0.0001. All conditions were compared to each other with one-way ANOVA, after log-transformation of the obtained data and testing for normality. Adjustment for multiple comparisons was performed using Tukey's multiple comparisons test. Abbreviations: NTF = non-transfected cells. | ||
Next, the in vivo biocompatibility of the most promising saRNA-polyplexes was investigated with a haemolysis assay performed at physiological pH. The saRNA-polyplexes were prepared in HBG (20 mM HEPES, pH 7.4 with 5% glucose) buffer, according to N/P ratios of 1, 5 and 10, and were incubated with fresh pig red blood cells (RBCs) at two different concentrations, i.e. 20 μg and 20 ng complexed saRNA per 5.37 × 109 RBCs per mL. The highest saRNA/RBC ratio reflected the in situ situation immediately after IV administration (“administration phase”), while the lowest ratio represented the situation in which nanoparticles had already been distributed throughout the whole circulatory system (“distribution phase”). After 1 hour of incubation, a significant haemolysis (35%) was noticed with the L-PEI60-ran-PPI190-based polyplexes with N/P 10 in the administration phase (see Fig. 6(a)). In the distribution phase, the same saRNA-polyplex resulted in a very low haemolytic activity of 0.15% (see Fig. 6(b)). Since, an in vitro haemolytic activity of less than 10% is considered to be non-haemolytic and percentages above 25% to be haemolytic,81 saRNA-polyplexes with N/P 5 and 1 can be recognized as safe in both the “administration” (20 μg saRNA) and “distribution” (20 ng saRNA) phases, for both L-PEI60-ran-PPI190 and L-PPI250. An explanation for the higher haemolysis with increasing N/P ratio is most likely the presence of higher amounts of free cationic polymer at higher N/P ratios. Indeed, previous studies have demonstrated that polyplexes exhibit a dynamic equilibrium between free polymer and polymer in the polyplexes,82 and that the actual N/P ratio within polyplexes does not usually exceed 2, despite their typical formulation at N/P ratios of 10 or greater.61,83,84 Consequently, this implies that at high N/P ratios substantially higher amounts of free cationic polymer are present, which potentially can cause toxicity.
 | ||
| Fig. 6 Ex vivo haemolysis (%) of the polyplexes composed of saRNA complexed with L-PEI60-ran-PPI190 or L-PPI250. Panel (a) shows the “administration phase” where RBCs were incubated with 20 μg saRNA formulated in polyplexes, graph (b) presents the “distribution phase” where 20 ng saRNA-polyplexes were used. RBCs were derived from fresh pig blood. Spectrophotometric read-out was performed with the TECAN plate-reader at 600 nm. Each box represents the minimum and maximum spectrophotometric values based on the absorbance of 4 wells per condition (n = 4), compared to the negative (HBG) and positive (Triton X-100) control. *indicates significance of p ≤ 0.05, **indicates significance of p ≤ 0.01, ***indicates significance of p ≤ 0.001, ****indicates significance of p ≤ 0.0001. All means were compared to each other with one-way ANOVA, after testing for normality. Adjustment for multiple comparisons was performed using Tukey's multiple comparisons test in GraphPad Prism 8.4.3. | ||
In conclusion, although no clear trend could be derived from the cell viability assays as to which polymer is the least toxic, the haemolysis assay points out that the polymer with the highest PPI content (L-PPI250) exhibits the lowest toxicity towards the erythrocytes. This is in line with the hypothesis that a lower charge density reduces cytotoxicity.60–62 The cytotoxicity assay and the haemolysis assay both demonstrated that saRNA-polyplexes prepared at N/P ratio 5 and 1 induce less toxicity compared to N/P of 10.
Finally, when the saRNA-polyplexes could be considered safe for in vivo administration, the in vivo efficiency of the novel L-PEI60-ran-PPI190 (co)polymers as carriers for saRNA delivery (N/P 1 and 5) was investigated and compared with a D-Lin-MC3-DMA-based saRNA-LNP (MC3-LNP), during a preliminary study (see Fig. 7, graph (a) and panel (b)). This study was conducted using luciferase encoding saRNA and in vivo optical imaging. We chose to use the MC3-LNP as reference, since this ionizable lipid was commercially available and is proven safe, as it is used in the approved siRNA-drug Patisiran. This preliminary study shows that, compared to the MC3-LNP, the L-PEI60-ran-PPI190-based saRNA-polyplexes (N/P 1 and 5) are less efficient with regard to peak luciferase expression during the first 3 days. However, from that day on, the MC3-LNP tends to maintain an expression plateau up to day 7, which was not statistically significant higher compared to the polymer carrier, starting from day 4 after injection. After this plateau, the expression gradually decreases to background level. The expression profile of L-PEI60-ran-PPI190 shows a slow increase up to day 7 and exceeds the bioluminescent signal of the MC3-LNP at day 14. Remarkably, from that day on, a constant signal persists at least until day 21 after injection.
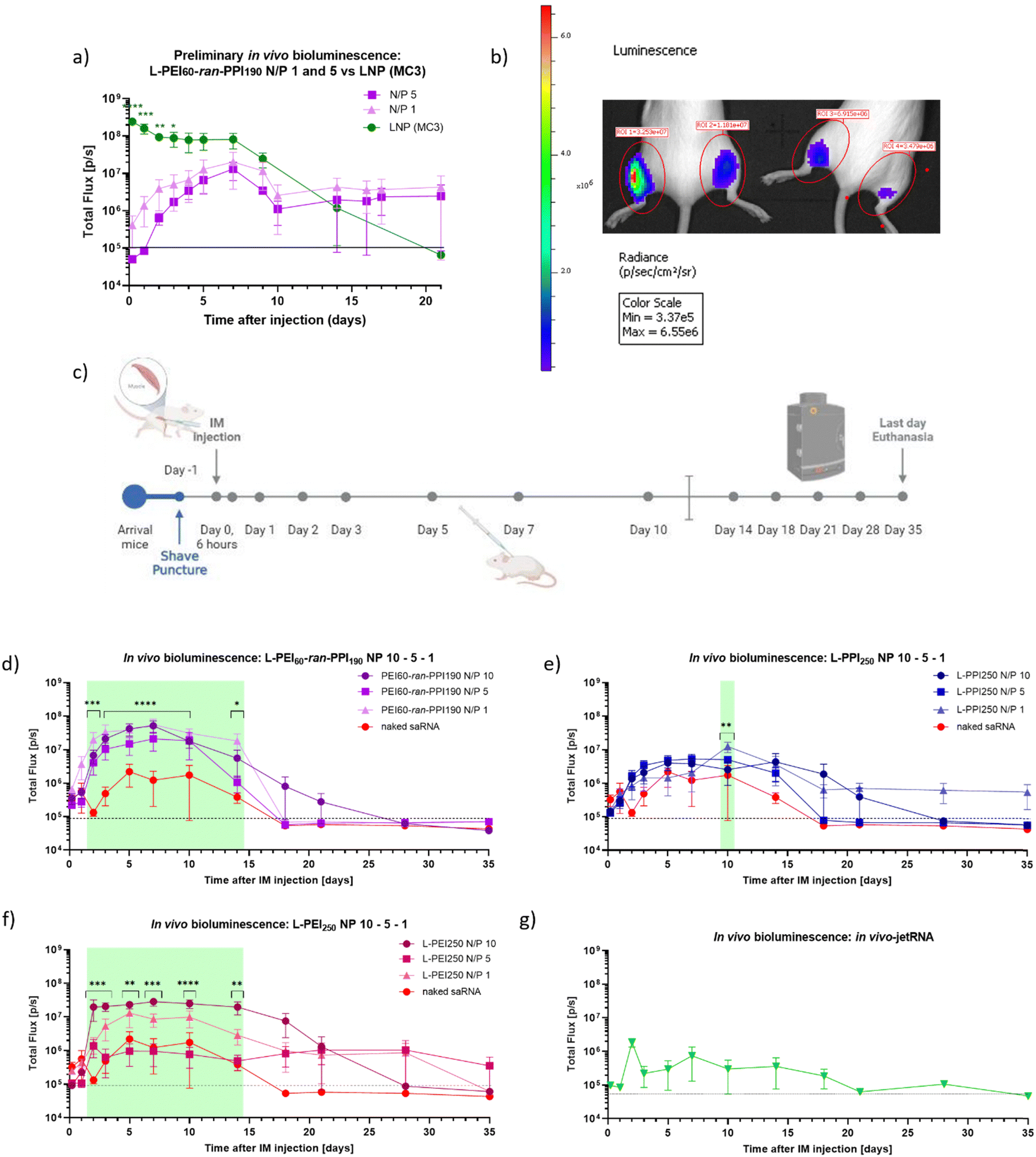 | ||
| Fig. 7 Timeline and results (preliminary) in vivo bioluminescence study in BALB/cJRj mice. Graph (a) and picture (b) show the bioluminescent signals (Total Flux [p/s]) of a preliminary study, during which 1 μg firefly luciferase coding saRNA was administered (IM) in the hindlimbs of three 8 week-old BALB/cJRj mice (N = 3). Graph (a) the saRNA was either complexed with L-PEI60-ran-PPI190 (N/P 1 or 5) (purple) or with lipids (LNP, based on the D-Lin-MC3-DMA ionizable lipid, N/P 10) (Oda, #119). Datapoints represent the average of the signals measured from the front and the back. Panel (b) shows the IVIS picture of two mice 7 days after injection of saRNA with L-PEI60-ran-PPI190 (N/P 1 (left leg) or N/P 5 (right leg)). Panel (c) demonstrates the timeline of the in vivo bioluminescence study with six mice/“treatment”. Mice were again IM injected with 1 μg saRNA per leg (N = 6). Graphs (d)–(f) show the bioluminescent signals (Total Flux [p/s]) after injection of naked saRNA (‘red’) compared to the signals after injection of saRNA complexed with L-PEI60-ran-PPI190 (purple), L-PPI250 (blue) and L-PEI250 (pink) (N/P 10, 5 and 1), respectively. The green panels represent the days of significant differences between groups (for more details: ESI†). Graph (g) shows data obtained after IM injection of saRNA complexed with In vivo-jetRNA™, a commercial mRNA delivery agent. Datapoints represent the average signals measured from the back. All data was obtained with the IVIS Lumina III, 12 minutes after SC injection of D-luciferin and anesthesia via isoflurane aerosol. The dashed black line represents background signal. * indicates significance of p ≤ 0.05, ** indicates significance of p ≤ 0.01, *** indicates significance of p ≤ 0.001, **** indicates significance of p ≤ 0.0001. All means were compared to each other with one-way ANOVA (a) (each day separately) or with two-way ANOVA (b) (matched values over time), after log-transformation of the obtained data and testing for normality. Adjustment for multiple comparisons was performed using Tukey's multiple comparisons test in GraphPad Prism 8.4.3. | ||
Based on these promising results, a similar, more extensive in vivo bioluminescence study was performed to compare the in vivo efficiency of L-PEI60-ran-PPI190 and L-PPI250 (N/P 1, 5 and 10). Here, naked saRNA was included as negative control and L-PEI250 and in vivo-JetRNA (the only commercial available in vivo mRNA carrier at the moment of the study) as positive controls (see Fig. 7, panel (c) and graphs (d)–(g)). All considered carriers show statistically significant higher bioluminescent signals compared to naked saRNA, except in vivo-JetRNA™, despite following the manufacturers protocol (see Fig. 7, graphs (d)–(g)). The latter, lipid-based, commercial in vivo carrier was used, in the absence of a commercially available LNP for saRNA. Only a relatively low signal from day 1 until day 18 could be distinguished after IM administration of saRNA-nanoparticles formulated with this in vivo carrier. Potentially, this is the first time in vivo-JetRNA™ was used in combination with saRNA, and more specifically cellulose-purified saRNA. This purification method was performed since it is crucial in removing dsRNA, which otherwise causes detrimental cytokine production following in vivo administration of saRNA.28 Naked saRNA results in a low, but relatively stable expression between day 4 and 14, next the signal steadily decreases until reaching background signal on day 18.
Regarding the saRNA-polyplexes, this second in vivo study revealed two trends. First, the relatively high in vivo transfection efficiency of L-PEI60-ran-PPI190 is confirmed here, with ∼20–200 times higher bioluminescent signals compared to naked saRNA (Fig. 7, graph (d)). In addition, L-PEI60-ran-PPI190-based saRNA-polyplexes result in higher luciferase expression profiles compared to the L-PPI250-based saRNA-polyplexes, which is in line with the in vitro transfection data. In case of L-PEI60-ran-PPI190, saRNA-polyplexes with N/P 10 show statistically significant (p ≤ 0.0001) higher signals compared to naked saRNA, starting from day 3 until day 10 after IM administration, while L-PPI250-based saRNA-polyplexes with N/P 1 only showed minor statistically significant (p = 0.0010) higher signals compared to naked saRNA 10 days after IM injection. Noteworthy, L-PEI60-ran-PPI190-based polyplexes prepared at N/P 1, show high in vivo efficiency which is in contrast with the in vitro results. Second, unlike the MC3-LNP (from the first in vivo study), the saRNA-polyplexes induce a gradual increase in saRNA expression and reach plateau expression after 3–4 days, that persists for about 10–14 days (in case of L-PPI250 and L-PEI250), which is a significantly longer plateau period compared to the MC3-LNP. This highlights the sustained expression profile of L-PPI250 in particular, with the longest relevant expression that lasted at least for 35 days after IM injection, when formulated with saRNA according to a N/P ratio of 1.
The area under the curve (AUC) of the bioluminescence data from the second study was also calculated, these data consider the total/overall saRNA expression over the 35-day period. This data suggests that L-PEI60-ran-PPI190 causes the overall greatest saRNA expression when complexed with saRNA in a N/P ratio of 1, as it shows a statistical significant (p ≤ 0.0001) higher AUC compared to 8 of the other conditions (see Fig. 8). This is remarkable given the absence of saRNA expression in in vitro transfections. Furthermore, the data of the same polymer with N/P 10 shows comparable results, and the N/P 5 results in the lowest AUC within these saRNA-polyplexes, which is also in contrast with the in vitro data. The AUC of the three L-PPI250-based saRNA-polyplexes are very similar to each other. Linear-PEI250 at N/P 5 results in the smallest, although greater AUC compared to naked saRNA. The ultimate smallest AUC value was obtained by In vivo-JetRNA™.
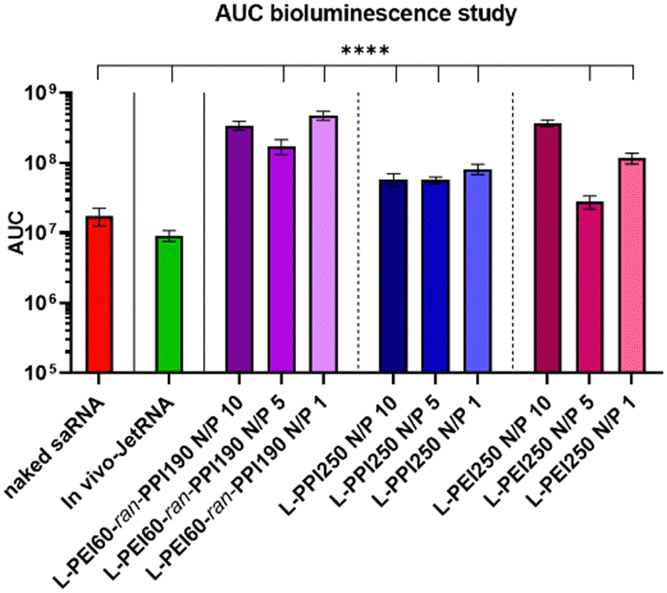 | ||
| Fig. 8 Area Under Curve (AUC) in vivo bioluminescence. Data was obtained with the IVIS Lumina III (Total Flux [p/s]). * indicates significance of p ≤ 0.05, ** indicates significance of p ≤ 0.01, *** indicates significance of p ≤ 0.001, **** indicates significance of p ≤ 0.0001. All means were compared to each other with one-way ANOVA after testing for normality. Adjustment for multiple comparisons was performed using Tukey's multiple comparisons test in GraphPad Prism 8.4.3. | ||
Conclusions
Only a limited number of polymer-based carriers are successful in delivering saRNA, so this research is of great importance in the search for new and more efficient polymers.66 In this study, CROP of 2-oxazolines and 2-oxazines followed by acidic hydrolysis was used to synthesize L-PEIx-ran-PPIy random copolymers and the corresponding homopolymers with a molecular weight of ∼15 kDa and DP250. The polymers were tested as non-viral carriers for saRNA revealing that particularly two polymers, i.e. L-PEI60-ran-PPI190 and L-PPI250, outperformed the in vitro gold standard, lipid-based carrier Lipofectamine MessengerMAX™, both with regard to transfection efficiency as well as cell viability. Moreover, the saRNA-polyplexes prepared at N/P ratio 5 did not induce cytotoxicity when transfected in humane cell lines (HeLa and HepG2) during a short period of time. These saRNA-polyplexes were fully characterized and had a small size (∼70–100 nm), a positive zeta potential (∼40 mV), a complete complexation/binding of the saRNA, a partial protection against high doses of RNase and a good hemocompatibility. Together, this indicates their high potential as in vitro carriers.The in vivo study taught us that saRNA-polyplexes based on the L-PEI60-ran-PPI190 copolymer were the most efficient, however, they are still less efficient than Dlin-MC3-DMA LNPs with regard to peak luciferase expression. Interestingly, the saRNA-polyplexes, and especially these based on the L-PPI250 polymer trend to induce a longer saRNA expression compared to (MC3-) LNPs. The somewhat lower saRNA expression obtained with our polyplexes relative to saRNA-LNPs does not necessarily mean that our polymers should not be considered for e.g., vaccine purposes, since the linear relationship between protein expression and immunogenicity is not necessarily true.23
Remarkably, saRNA-polyplexes with N/P 10 and N/P 1, of which the latter did not result in relevant in vitro transfection efficiencies, demonstrated the highest luciferase expression and. Therefore, these N/P ratios are considered the most interesting for in vivo applications. Based on this and since the best in vitro performing saRNA-polyplexes with N/P 5 showed the lowest in vivo saRNA expression within all polymer groups, in vivo postulations based on in vitro data are not always true.
In our future studies we plan to include other administration routes and polymers with different molecular weights and architectures. Additionally, more in-depth metabolization, toxicity and stability studies based on e.g., ROS production, complement cascade activation and long-term storage at sub-zero, refrigerator and room temperatures (with or without cryoprotectants and freeze-drying) are planned to gain more understanding about the behaviour and application potential of our (co)polymer-based carrier platform.
Experimental
Materials
All reagents were obtained from commercial sources and used without further purification unless stated otherwise. 2-Ethyl-2-oxazoline was kindly provided by Polymer Chemistry Innovations and distilled over barium oxide under reduced pressure prior to use. Acetonitrile and dichloromethane were dried in a solvent purification system (Pure Solv EN, Innovative Technology). Dried acetonitrile was further distilled (from barium oxide in the presence of ninhydrin) before use as a polymerization solvent. 37% Hydrochloric acid (HCl), anhydrous magnesium sulphate and Spectra/Por 6 dialysis membrane (3.5 kDa molecular weight cut-off) were obtained from fisher scientific. All other chemicals, including isobutyronitrile (99%), 3-amino-1-propanol (>99%), dichloromethane (99%), zinc acetate dihydrate (≥98%), methyl tosylate (98%, distilled over barium oxide and stored under argon), deuterated chloroform (CDCl3; 99.8%), deuterated water (D2O; 99.9%) and benzophenone (99%) were purchased from Sigma-Aldrich.Synthesis of 2-isopropyl-2-oxazine (iPrOzi)
The monomer, 2-isopropyl-2-oxazine (iPrOzi) was synthesized by a standard procedure85 in which isobutyronitrile (200 mL; 2.238 mol; 1 equiv.) and zinc acetate dehydrate (catalyst; 14.67 g; 66.85 mmol; 0.03 equiv.) were heated to 127 °C under reflux conditions. 3-Amino-1-propanol (190 mL; 2.495 mol; 1.12 equiv.) was then added into the reaction mixture dropwise and the resulting mixture was refluxed at 127 °C. After a reaction time of ∼72 h, the reaction mixture was allowed to cool to room temperature, and 450 mL dichloromethane (DCM) was added. The organic phase was washed with water (3 × 350 mL) and with brine (1 × 350 mL). The organic layer was then dried over magnesium sulfate and evaporated under reduced pressure resulting in crude 2-isopropyl-2-oxazine (iPrOzi). The iPrOzi monomer was further purified by repeated distillation over BaO and a final distillation over metallic sodium/benzophenone. The resulting colourless viscous liquid monomer was stored under argon atmosphere (yield: ∼50%). The monomer structure and purity were confirmed via1H NMR spectroscopy.
1H NMR (400 MHz; CDCl3) δ (ppm): 4.10 (2H, t, O![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif)
![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2), 3.36 (2H, t, N2), 2.32 (1H, m, CH3CH3), 1.83 (2H, m, OCH22CH2N), 1.08 (6H, d, C3CHC3).
2), 3.36 (2H, t, N2), 2.32 (1H, m, CH3CH3), 1.83 (2H, m, OCH22CH2N), 1.08 (6H, d, C3CHC3).
Synthesis of poly(2-isopropyl-2-oxazine) [PiPrOzi(DP250)] homopolymer
The cationic ring-opening polymerization (CROP) technique was used to synthesize the polymer. The polymerization was performed in a capped microwave vial in a single-mode microwave Biotage Initiator Sixty (IR temperature sensor) (Biotage, Uppsala, Sweden). The sample was prepared in a VIGOR Sci-Lab SG 1200/750 glovebox system with obtained purity levels below 1 ppm, both for water and oxygen content. iPrOzi monomer (1.5 mL; 11.45 mmol), MeOTs initiator (7 μL; 0.046 mmol) and 3 mL CH3CN were added in the 2-5 mL glass microwave vial and capped inside the glove box. The theoretical molar ratios of monomer and initiator was as follows: [iPrOzi]/[MeOTs] = 250:1. The vial was removed from the glovebox and placed in the microwave reactor to be heated at 140 °C for 4.30 h. Subsequently, the polymerization was terminated by the addition of a potassium hydroxide solution (0.5 M in methanol, 0.1 mL). The resulting polymer was isolated by precipitation in cold diethyl ether. The precipitate was filtered, washed three times with cold diethyl ether and dried in a vacuum oven at 40 °C. The white amorphous solid polymer was analysed through 1H NMR spectroscopy and size-exclusion chromatography (SEC). The latter resulted in a molecular weight (Mn) of 33.5 kDa and dispersity (Đ) of 1.17.
1H NMR (300 MHz; CDCl3) δ (ppm): 3.37 (4H, b, N2CH22), 2.70 (1H, b, NCOC(CH3)2), 1.80 (2H, b, NCH2C2CH2), 1.07 (6H, b, NCOCH(C3)2).
Synthesis of poly(2-ethyl-2-oxazoline)-random-poly(2-isopropyl-2-oxazine) (L-PEtOx60-ran-PiPrOzi190) copolymers
:60:1. The polymerization was terminated by the addition of a potassium hydroxide solution (0.5 M in methanol, 0.1 mL). Dichloromethane was added to dilute the polymerization mixture and the copolymer was isolated by triple precipitation/reprecipitation from cold diethyl ether and was dried in a vacuum oven at 40 °C. The copolymer was further purified by dialysis against distilled water for one day using a membrane (molecular weight cut-off of 3.5 kDa) and was obtained as white amorphous/powdery solid by evaporating the water in a lyophilizer (yield ∼70%). The copolymer was characterized through 1H NMR spectroscopy and the compositions of the copolymer was determined/confirmed from the integration of the appropriate signals (signals at 2.75 and 2.25 ppm for the PiPrOzi and PEtOx segments, respectively) in the 1H NMR spectrum (see Fig. 1). Molecular weight (Mn) and dispersity (Đ) of the polymer was analysed from SEC and were found to be 29.0 kDa and 1.14, respectively.
1H NMR (300 MHz; CDCl3) δ (ppm): 3.37 (8H, b, NC2CH2C2N and NC2C2N), 2.72 (2H, b, NCOC(CH3)2), 2.25 (2H, b, NCOC2CH3), 1.79 (2H, b, NCH2C2CH2), 1.06 (9H, b, NCOCH(C3)2 and NCOCH2C3).
Synthesis of poly(propyleneimine) [PPI(250)] homopolymer
The precursor ([PiPrOzi(DP250)]) homopolymer (∼300 mg) was dissolved in aqueous hydrochloric acid (∼18 wt%, 15 mL) and heated for 6 h at 127 °C in a microwave reactor. The solvents and acids were evaporated under reduced pressure at high temperature (∼80 °C). The crude product was then dissolved in distilled water and purified by dialysis against distilled water using a dialysis membrane (molecular weight cut-off of 3.5 kDa) and recovered by lyophilizing to obtain the PPI(250) copolymer as white solid (∼175 mg; yield 80%). The PPI(250) was characterized through 1H NMR spectroscopy and aqueous SEC (Mn = 44 kDA, and Đ = 1.38).
1H NMR (300 MHz; D2O) δ (ppm): 3.12 (4H, b, NHC2CH2C2), 2.07 (2H, b, NCH2C2CH2).
Synthesis of poly(ethylenimine)-random-poly(propyleneimine) (L-PEI60-ran-PPI190) copolymers
2CH2C2) of the PPI units and at δ(a) 3.34 ppm for the methylene protons (NHC2C2) of PEI units (see Fig. 1). The appropriate DP of PEI and PPI units was then determined from the ratio of integrated intensities of these two well-resolved signals and was found to be 60 and 190, respectively. The copolymer was also analysed through SEC (in aqueous medium) resulting in a Mn of 33 kDa, and Đ of 1.25.
1H NMR (300 MHz; D2O) δ (ppm): 3.34 (4H, b, NHC2C2 from PEI) 3.03 (4H, b, NHC2CH2C2 from PPI), 1.98 (2H, b, NCH2C2CH2 from PPI).
Characterization of polymers
A Bruker Avance 300 MHz Ultrashield or Bruker Avance II 400 MHz were used to measure 1H-nuclear magnetic resonance (1H NMR) spectra at room temperature to characterize the monomer and (co)polymers. The chemical shifts (δ) are given in parts per million (ppm) relative to tetramethylsilane. The compositions of the L-PEtOx60-ran-PiPrOzi190 and L-PEI60-ran-PPI190 copolymers were also determined from the integration of the appropriate signals in the 1H NMR spectra (see Fig. 1). Size-exclusion chromatography (SEC) was used to determine the molar masses (Mw: weight-averaged molar mass, Mn: number-averaged molar mass) and the dispersity (Đ = Mw/Mn) of the prepared polymers. The molecular weights of PEtOx60-ran-PiPrOzi190 and PiPrOzi250 copolymers were determined by SEC using an Agilent 1260-series HPLC system equipped with a 1260 online degasser, a 1260 ISO-pump, a 1260 automatic liquid sampler,14 a thermostatic column compartment (TCC) set at 50 °C equipped with two PLgel 5 μm mixed-D columns (7.5 mm × 300 mm) and a precolumn in series, a 1260 diode array detector (DAD) and a 1260 refractive index detector (RID). The used eluent was N,N-dimethyl acetamide (DMA) containing 50 mM of LiCl at a flow rate of 0.500 mL min−1. The spectra were analyzed using the Agilent Chemstation software with the GPC add on. Molar mass values and molar mass distribution, i.e. dispersity (Đ) values were calculated against narrow disperse poly(methyl methacrylate) (PMMA) standards from PSS. SEC system with aqueous medium as eluent was also employed to measure the molar mass distributions of L-PEIx-ran-PPIy and L-PEI250 as the polymers are insoluble in DMA. This was performed on an Agilent 1260-series HPLC system equipped with an online PSS degasser, a 1260 ISO-pump, a 1260 automatic liquid sampler (ALS), a 1261 thermostatic column compartment (TCC) at 35 °C equipped with two PSS Novema Max 5 μm columns and a precolumn in series, a 1262 diode array detector (DAD) and a 1290 refractive index detector (RID). The used eluent was methanol–sodium acetate buffer containing 0.1 M NaNO3 at a flow rate of 0.500 mL min−1. The spectra were analyzed using the Agilent Chemstation software with the GPC add on. Molar mass and Đ values were calculated against PEG standards from PSS. The polymerizations of EtOx and iPrOzi were performed in glovebox-filled capped vials in a single mode microwave Biotage initiator sixty (IR temperature sensor). Lyophilisation was performed on a Martin Christ freeze-dryer, model Alpha 2–4 LSC plus.SaRNA synthesis and silica purification
Luciferase coding saRNA was produced by in vitro transcription (IVT). The pTK160 plasmid,19 containing a T7 promotor followed by the template, was isolated using the MidiPrep Spin Plus Kit (Qiagen, Hilden, Germany, #12943) and digested with the I-SceI enzyme (New England Biolabs, Ipswich, Massachusetts, USA, #R0694S) to produce linear template. Digested DNA was purified using the Wizard DNA Clean-up system (Promega, Madison, Wisconsin, USA, #A9281) and 1 μg was loaded on 1% agarose gels to confirm complete digestion. IVT was performed using the MEGAScript T7 Transcription Kit (Thermo Fisher Scientific, Waltham, Massachusetts, USA, #AM1334), according to the manufacturer's instructions. Next, the pDNA template was degraded by TURBO DNase™, provided in the MEGAScript T7 kit, according to manufacturer's protocol. The RNA was purified using the RNeasy Mini Kit (Qiagen, Hilden, Germany, #74104), capped enzymatically using the ScriptCap Cap 1 Capping System (CellScript, Madison, Wisconsin, USA, #C-SCCS1710) and purified again. The concentration and quality of RNA preparations was determined spectrophotometrically using a NanoDrop ND-1000 (Thermo Fisher Scientific, Waltham, Massachusetts, USA) and RNA integrity was analyzed by agarose gel electrophoresis on a 1% gel.Cellulose-based purification of saRNA
For the in vivo experiments, the luciferase encoding saRNA was purified by the earlier described cellulose-based purification,29 after IVT, capping and silica based purification. This purification method was used to remove dsRNA by-product and was performed as described previously.29Polymer-based nanoparticle (polyplex) formulation
A polymer solution was established by dissolving the polymers in 20 mM NaOAc (pH 5.2) at a concentration of 2 mg mL−1 this solution was aliquoted and stored at −20 °C. Freeze–thaw cycles were limited to five. SaRNA-polyplexes were formulated in one step by adding an equal volume of the saRNA solution to the polymer solution and pipetting 8 times up and down (both solutions diluted in either 20 mM NaOAc, pH 5.2 or in 20 mM HEPES, pH 7.4), followed by a 30 minute incubation at room temperature. The saRNA concentration was kept constant (25 ng saRNA per μL polyplex) and a polymer dilution series was constructed according to the desired N/P ratio's (N: mole of cationic nitrogen atoms in the polymer structure; and P: mole of anionic phosphates in the saRNA molecule). For in vivo administration, at the day of injection the saRNA solution and the polymer solutions were formulated in a 20 mM HEPES buffered glucose (5% w/v) (HBG) solution according to the desired N/P ratio.LNP formulation
MC3-LNPs were formulated by adding an ethanolic lipid solution to an aqueous saRNA solution (7.5 mM NaOAc buffer, pH 4.5) under rapid mixing with vigorous stirring. The ethanolic solution contained the ionizable lipid D-Lin-MC3-DMA, cholesterol, DOPE and DMG-PEG 2000 at a molar ratio of 50:38.5:10:1.5, and the lipids were added to the saRNA according to a N/P ratio of 10. Subsequently, the saRNA-MC3-LNPs were subjected to overnight dialysis in a dialysis cassette (Thermo Scientific, Rockford, U.S.A) against DPBS (1×, pH 7.4, no calcium, no magnesium) according to manufacturer's guidelines, to remove ethanol. The saRNA-MC3-LNP formulations were then adjusted to 20 ng μL−1 of saRNA.
Cell culture, in vitro transfection of saRNA-polyplexes and bioluminescent imaging
HeLa cells (a kind gift from prof. dr Daisy Vanrompay), C2C12 cells and HepG2 cells (ATCC, Virginia, US) were cultured in Dulbecco's Modified Eagle Medium (DMEM; Thermo Fisher Scientific, Waltham, Massachusetts, USA), containing 10% heat inactivated fetal bovine serum (FBS) (Biowest, Nuaillé, France) and 1% penicillin/streptomycin (P/S) (Thermo Fisher Scientific, Waltham, Massachusetts, USA). One day before transfection, 5 × 104/500 μL HeLa cells, 4 × 104 C2C12 cells or 1 × 105 HepG2 cells were seeded in each well of 24-well plates to obtain 70–80% confluency. Cells were washed with 1× DPBS and transfected with saRNA-polyplexes (500 ng saRNA) in Opti-MEM or full DMEM (i.e., with 10%FBS and 1% P/S). After 4 hours the medium containing the complexes was either replaced or not by full DMEM medium. Twenty-four hours after transfection, the medium was replaced, cells were washed with 1× DPBS and trypsinized. During the 10 minute trypsinization, cells were incubated at 37 °C and afterwards neutralized with full DMEM medium. Part of the neutralized cell suspension (36%) was transferred to a black 96-well plate. A D-luciferin solution (50 mg ml−1; 1/10 dilution) was added to each well with transfected cells, and left to incubate at 37 °C for 10 minutes. Subsequently, the bioluminescent signal was measured using the IVIS lumina III (Xenogen Corporation, Alameda, California, US).Cell viability assay
Cell viability after transfection experiments was determined using the Cell Proliferation Reagent WST-1 (2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt) (Abcam, Cambridge, UK). After trypsinization, 36% of the volume was transferred to a clear 96 well-plate and WST-1 solution was added according to the manufacturer's instructions (1/10 dilution). After 30 minutes of incubation at 37 °C, the absorbance at 450 nm (660 nm reference) was determined using the EZ Read 400 microplate reader (Biochrom, Cambridge, UK). The measurement was repeated after 1 hour, 2 hours, 3 hours and 4 hours of incubation. To correct for the background signal, the WST-1 reagent was added to DMEM only. Cells from non-transfected (NTF) wells were used as positive control. Viability graphs show the percentage of cell viability (%) (see formula below) based on the measured absorbance before reaching saturation.Calculation:
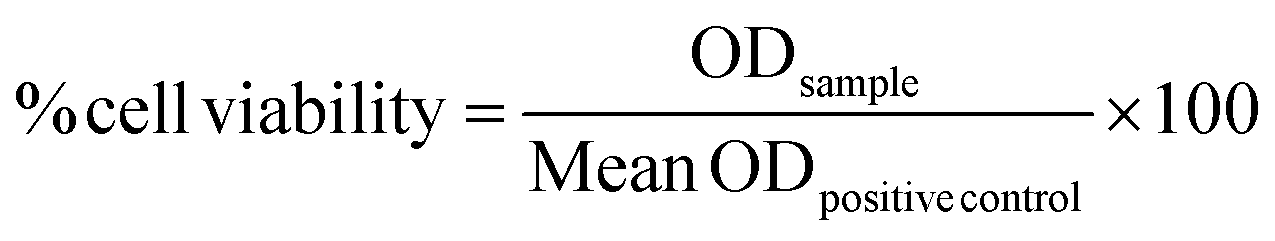
Particle size and zeta-potential analysis
The particle size (hydrodynamic diameter (DH), Z-average) and zeta potential (ZP) of the saRNA-polylexes were measured using the Zetasizer Nano-ZS (Malvern Instruments, Malvern, UK). For the size measurements, 400 μL samples were made in either NaOAc buffer (pH 5.2, 20 mM) or HEPES buffer (pH 7.4, 20 nM) (25 ng μL−1 saRNA) and loaded into a disposable semi-micro polystyrene cuvette (BRAND, Wertheim, Germany). These measurements are based on the principle of Dynamic Light Scattering (DLS) using a laser beam of 632.8 nm and a detector at a scattering angle of 93°.ZP measurements were performed with samples at a concentration of 10 ng μL−1 saRNA loaded in a folded capillary cell (polystyrene) cuvette (Malvern Panalytical, Malvern, UK). Both size and ZP measurements were conducted at 25 °C, with the following settings: dispersant ‘water’, with viscosity 0.8872 cP, refractive index of 1.33, and dielectric constant of 78.5.
The size of the saRNA-polyplexes was also determined with a NanoSight NS300 (Malvern Panalytical, Malvern, UK). To that end the saRNA-polyplex samples were diluted with NaOAc (20 mM, pH 5.2) or HEPES (20 mM, pH 7.4) to concentrations ranging from ∼10–40 particles per frame or ∼ 2–7 × 1011 particles per mL, corresponding to concentrations of 0.25–0.5 μg mL−1 saRNA. The samples were loaded with a syringe pump (speed 50), irradiated by a 488-nm laser and visualised by a high-sensitivity sCMOS camera. The resulting experiment recordings were analysed using Nanoparticle Tracking Analysis (NTA′)[1] 3.4 Build 3.4.003 software (Malvern Instruments) after capture in script control mode (3 recordings of 60 s per measurement). In total ∼1500 frames were created per sample to determine the size of the saRNA-polyplexes.
Electrophoretic mobility shift assay (EMSA), heparin competition assay and RNase a protection assay
An electrophoretic mobility shift assay (EMSA) or gel retardation assay was performed to assess the complexation status of the saRNA-polyplexes. To that end, the saRNA-polyplexes were formulated in NaOAc buffer as described earlier (see Polyplex Formulation). Next, 1% (w/v) agarose gels containing 1% (v/v) bleach (NaClO) were made in tris–borate–EDTA (TBE) buffer (0.045 M tris–borate and 0.001 M EDTA, pH 8.3). After homogenization, the mixtures were heated to melt the agarose and then cooled before adding ethidium bromide (EtBr) (10 mg mL−1) to a final 0,02% concentration. Next, the gels were poured into the mold and allowed to cool and set for at least 15 minutes at room temperature. Before loading the saRNA-polyplexes (500 ng saRNA), (10×) loading buffer (50% (w/v) sucrose in 20 mM HEPES buffer pH 7.4) was added to allow for RNA sedimentation in the wells. RNase-free water was added to all samples and to obtain identical total sample volumes. A 1 kb+ DNA-ladder (Thermo Fisher Scientific, Massachusetts, USA) and/or free saRNA, made with the same stock solution, were also loaded on the gels to identify the location of the free saRNA band. Absence of this band represents retention of the saRNA by the polymer. After loading the gel was run at 100 V for 30–35 minutes. Afterwards, the gel was exposed to UV light and images were taken and analysed with Fiji software.To assess the strength of the saRNA-polyplexes, a Heparin competition assay was performed. To that end, 20 μl saRNA-polyplexes containing 500 ng saRNA were incubated during 1 hour at 37 °C with 1 μL of a 80 mg mL−1 heparin sodium (HS) dissolved in RNase-free H2O. After incubation the samples were analysed by agarose gel electrophoresis as described above.
The level of protection of the saRNA against nuclease degradation was investigated using in-house RNase A Protection Assay. During this assay saRNA and saRNA-polyplexes were exposed to RNase A (1 μL, 1 ng μL−1) (Thermo Fisher Scientific, Waltham, Massachusetts, USA) for 30 minutes at 37 °C, before or after the addition of 80 μg HS and/or 15% (w/v) SDS. In this experiment 15% SDS dissolved in RNase-free water was used as RNase A inhibitor. Subsequently, the samples were loaded on an agarose gel, run and analysed as described above.
RiboGreen assay
The amount of unbound or loosely surface bound saRNA and the amount of released saRNA after HS exposure was also quantified by using the Quant-iT™ Ribogreen RNA reagent and kit (Invitrogen, Massachusetts, US). This also allowed us to calculate the encapsulation efficiency (%EE) of the two best performing polymers (L-PEI60-ran-PPI190 and L-PPI250). For this assay, saRNA-polyplexes with a N/P ratio of 10 and 5 were formulated in 20 mM NaOAc buffer (pH 5.2), as described previously. The HS mediated saRNA release from the polymers was established by incubating the saRNA-polyplexes (20 μL, 500 ng saRNA) with HS (80 μg) for 1 hour at 37 °C (see Heparin Competition Assay). Next, all samples were 1/25 diluted in 1× TE buffer (10 mM tris–HCl, 1 mM EDTA, pH 7.5) to obtain RNA concentrations in the range of the high-range assay described in the manufacturer's guideline (“the high-range assay allows quantitation of 20 ng mL−1–1 μg mL−1”). A few minutes before the measurement, the 200-fold diluted RiboGreen reagent was added 1:1 to the samples in a black 96 well-plate which then was incubated in the dark for 2–5 minutes. Finally, the fluorescence intensity was measured from above by a Tecan Infinite 200 PRO plate reader (Tecan, Männedorf, Switzerland) with an excitation wavelength of 485 nm and an emission wavelength of 535 nm. A “saRNA only” sample, made out of the same saRNA stock and containing an equal saRNA mass as the saRNA-polyplexes, and a “heparin only” sample were also included to determine the fluorescence of 100% free saRNA and to assess the influence of heparin on the fluorescent detection, respectively. All measurements, except the rRNA standards curve samples (n = 2), were performed in triplicates (n = 3).
The following equation was used to calculate the EE%:
| (AHS − Ac)/AHS × 100% |
Haemolysis assay
Red blood cells (RBCs) were isolated from fresh heparinized pig blood (kindly gifted by Prof. Devriendt, Ghent University, EC no. EC2017/121EC). After 1:1 dilution in 20 mM HEPES containing 5% (w/v) glucose (HEPES buffered glucose or HBG, pH 7.4), the blood was added in a 4/3 ratio to the Ficoll-Paque PLUS density gradient media (VWR, Pennsylvania, US) and centrifugated at 800 rcf for 10 minutes at 20 °C. The plasma along with the buffy coat were discarded and the erythrocytes were washed several times in 3 volumes of HBG followed by centrifugation at 500 rcf for 10 minutes until the supernatant was clear. After microscopically checking the morphology of the RBCs, they were resuspended in PBS at a concentration of 5.37 × 109 RBC per mL. SaRNA-polyplexes were made in the same way as mentioned before (see Polyplex Formulation) but with 20 μg or 20 ng saRNA per saRNA-polyplex sample. Polymers (L-PEI60-ran-PPI190 and L-PPI250) were added according to the desired saRNA quantity and N/P ratio. Next, the saRNA-polyplexes were added to the RBC suspension to obtain a final 1:20 dilution. Triton X-100 (4% v/v) (VWR, Pennsylvania, US), 1× DPBS, and HBG were prepared and added to the RBCs in the same way as the samples and served as positive (100% hemolysis) and negative controls (0% hemolysis), respectively. Since previous experiments (data not shown) demonstrated that HBG is less toxic to RBCs than NaOAc buffer (20 mM, pH 5.2), the latter buffer was not included here. Moreover, this study showed that HBG is even less toxic than DPBS, so the hemolysis% was calculated using the values derived for HBG as negative control.
After shaking the samples at 650 rpm for 1 h at 37 °C by using the ThermoMixer™ (Eppendorf, Hamburg, Germany) and centrifugation at 500 rcf for 10 minutes, four times 200 μL supernatant from each tube was transferred to a clear 96-well plate and the absorbance was measured at 600 nm using the Tecan Infinite® 200 PRO plate reader. This wavelength was chosen because it resulted in interpretable data for Triton X-100 samples, which served as a positive control.
The following equation was used to calculate the percentage of haemolysis:
| (A − A0)/(A100 − A0) × 100% |
In vivo bioluminescence study
Female 6–8 weeks old BALB/cJRj mice were purchased from Janvier Laboratories (Paris, France) 10–14 days before the start of the in vivo study. They were housed in individually ventilated cages connected to an IVC rack with controlled temperature and humidity. All cages were equipped with bedding material (wood chips), nesting material (paper towel and toilet rolls) and a red mouse house. Mice feed and water were provided ad libitum. During the preliminary study, mice were intramuscularly (IM) injected in both (unshaved) hindlegs with L-PEI60-ran-PPI190 based saRNA-polyplexes prepared in HBG at either N/P 1 or 5 and containing 1 μg saRNA (25 ng saRNA per μL). As benchmark we injected 1 μg saRNA encapsulated in MC3-LNPs (N/P 10, 20 ng saRNA per μL).For the subsequent larger in vivo study, 69 mice were randomly assigned to a cage, punctured in the ear and housed in groups of 6 or 9 in each cage. The mice were IM injected (on day 0) in both shaved hindlegs (shaved on day-1) with L-PEI60-ran-PPI190 and L-PPI250 based saRNA-polyplexes prepared in HBG at either N/P 1, 5 or 10 and containing 1 μg saRNA. Naked saRNA was used as negative control, In vivo-jetRNA™ (Polyplus) formulated with saRNA (according to manufacturer's protocol) and L-PEI250 were used as positive controls. All saRNA, in both studies, was purified using the cellulose-based purification protocol, as described earlier (see Cellulose-based purification of saRNA). All bioluminescent signals were measured with the IVIS Lumina III, 12 minutes after subcutaneous injection of D-luciferin (15 μg μL−1) when mice were anaesthetised with isoflurane aerosol. The IVIS Lumina III was used to measure all bioluminescent signals 12 minutes after subcutaneous D-luciferin (15 μg μL−1) injection, while mice were anaesthetized with isoflurane aerosol (Zoetis, Louvain-La-Neuve, Belgium) (5% for induction and 2% for maintenance). Measurements were performed: 6 hours after injection and at day 1, 2, 3, 5, 7, 10, 14, 18, 21, 28 and 35. Finally, mice were euthanized after sedation with isoflurane via cervical dislocation.
Mice experiments were approved by the ethics committee of the Faculty of Veterinary Medicine, Ghent University (EC no. EC2021/047).
Statistical analysis
All statistical analyses were executed using GraphPad Prism 8.4.3. First, normality and log-normality of data was checked by the Shapiro-Wilk test, and visually confirmed via QQ plots. Data obtained with the IVIS Lumina III were log-transformed and checked for normality again. Normally distributed data of three and more groups were tested to each other with the one-way or two-way ANOVA (analysis of variance) test, followed by Tukey's test for multiple comparisons. p-Values below 0.05 were considered statistically significant and indicated as follows: * indicates significance of p ≤ 0.05, ** indicates significance of p ≤ 0.01, *** indicates significance of p ≤ 0.001, **** indicates significance of p ≤ 0.0001.Notes
Part of the results described in this article have been used in the patent application WO2018002382A1.Author contributions
L. O. established the methodology and was responsible for the investigation, involving all in vitro and in vivo experiments performed with the saRNA-polyplexes. Next, she performed the statistical analysis, created the visuals and, wrote everything down in this research article. S. J. and I. M. performed polymer synthesis, purification and analysis, S. J. described this in this research article and provided the visuals, X. C. formulated the MC3-LNPs and assisted in the preliminary in vivo study. R. H. and N. S. were responsible for the conceptualisation of the polymer design and, funding acquisition, they provided supervision and reviewed the writing of this article. L. O., S. J., R. H. and N. S. contributed to manuscript editing and provided constructive feedback and all authors provided final approval.Conflicts of interest
The authors declare the following competing financial interest(s): the following statement has been included in the manuscript: R. H. is one of the founders of Avroxa BVBA that commercializes poly(2-oxazoline)s as Ultroxa. R. H. is listed as one of the inventors of patent application WO2018002382A1 that is based on parts of this work. The other authors have no conflicts to declare.Acknowledgements
The authors want to thank Prof. Dr. Daisy Vanrompay (UGent, Department of Animal Sciences and Aquatic Ecology) for the donation of HeLa cells, and Prof. Dr. Bert Devriendt (UGent, Department of Translational Physiology, Infectiology and Public Health) and his research members for the drawing of the pig blood. This work was supported by BOF.24Y.2020.0010.01., BOFBAS2018002801, F2021/IOF-Equip/034, F2020 IOF StarTT 077, grated to N. S. and R. H. Both, R. H. and N. S. thank Ghent University for continuous financial support.References
- R. Verbeke, I. Lentacker, S. C. De Smedt and H. Dewitte, The dawn of mRNA vaccines: The COVID-19 case, J. Controlled Release, 2021, 333, 511–520 CrossRef CAS PubMed.
- R. M. Schiffelers, A. Ansari, J. Xu, Q. Zhou, Q. Tang and G. Storm, et al., Cancer siRNA therapy by tumor selective delivery with ligand-targeted sterically stabilized nanoparticle, Nucleic Acids Res., 2004, 32(19), e149 CrossRef PubMed.
- FDA Takes Key Action in Fight Against COVID-19 By Issuing Emergency Use Authorization for First COVID-19 Vaccine 2020, https://www.fda.gov/news-events/press-announcements/fda-takes-key-action-fight-against-covid-19-issuing-emergency-use-authorization-first-covid-19.
- MODERNA SHIPS MRNA VACCINE AGAINST NOVEL CORONAVIRUS (MRNA-1273) FOR PHASE 1 STUDY, https://investors.modernatx.com/news/news-details/2020/Moderna-Ships-mRNA-Vaccine-Against-Novel-Coronavirus-mRNA-1273-for-Phase-1-Study/default.aspx2020.
- S. Mc Cafferty, J. De Temmerman, T. Kitada, J. R. Becraft, R. Weiss and D. J. Irvine, et al., In Vivo Validation of a Reversible Small Molecule-Based Switch for Synthetic Self-Amplifying mRNA Regulation, Mol. Ther., 2021, 29(3), 1164–1173 CrossRef CAS PubMed.
- M. Ranzani, S. Annunziato, D. J. Adams and E. Montini, Cancer gene discovery: exploiting insertional mutagenesis, Mol. Cancer Res., 2013, 11(10), 1141–1158 CrossRef CAS PubMed.
- Z. F. Zhong, S. Mc Cafferty, F. Combes, H. Huysmans, J. De Temmerman and A. Gitsels, et al., mRNA therapeutics deliver a hopeful message, Nano Today, 2018, 23, 16–39 CrossRef CAS.
- N. Pardi, M. J. Hogan, F. W. Porter and D. Weissman, mRNA vaccines – a new era in vaccinology, Nat. Rev. Drug Discovery, 2018, 17(4), 261–279 CrossRef CAS PubMed.
- A. Wadhwa, A. Aljabbari, A. Lokras, C. Foged and A. Thakur, Opportunities and Challenges in the Delivery of mRNA-based Vaccines, Pharmaceutics, 2020, 12, 102 CrossRef CAS PubMed.
- K. Bloom, F. van den Berg and P. Arbuthnot, Self-amplifying RNA vaccines for infectious diseases, Gene Ther., 2021, 28(3–4), 117–129 CrossRef CAS PubMed.
- M. Zhang, J. Sun, M. Li and X. Jin, Modified mRNA-LNP Vaccines Confer Protection against Experimental DENV-2 Infection in Mice, Mol. Ther., 2020, 18, 702–712 CAS.
- K. L. Mallory, J. A. Taylor, X. Zou, I. N. Waghela, C. G. Schneider and M. Q. Sibilo, et al., Messenger RNA expressing PfCSP induces functional, protective immune responses against malaria in mice, npj Vaccines, 2021, 6(1), 84 CrossRef CAS PubMed.
- H. Wang, Q. Meng, J. Qian, M. Li, C. Gu and Y. Yang, Review: RNA-based diagnostic markers discovery and therapeutic targets development in cancer, Pharmacol. Ther., 2022, 234, 108123 CrossRef CAS PubMed.
- P. S. Kowalski, A. Rudra, L. Miao and D. G. Anderson, Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery, Mol. Ther., 2019, 27(4), 710–728 CrossRef CAS PubMed.
- C. Wang, Y. Zhang and Y. Dong, Lipid Nanoparticle-mRNA Formulations for Therapeutic Applications, Acc. Chem. Res., 2021, 54(23), 4283–4293 CrossRef CAS PubMed.
- D. Papukashvili, N. Rcheulishvili, C. Liu, Y. Ji, Y. He and P. G. Wang, Self-Amplifying RNA Approach for Protein Replacement Therapy, Int. J. Mol. Sci., 2022, 23, 12884 CrossRef CAS PubMed.
- J. B. Sandbrink and R. J. Shattock, RNA Vaccines: A Suitable Platform for Tackling Emerging Pandemics?, Front. Immunol., 2020, 11, 608460 CrossRef CAS PubMed.
- Z. Kis, C. Kontoravdi, R. Shattock and N. I. Shah, Resources, Production Scales and Time Required for Producing RNA Vaccines for the Global Pandemic Demand, Vaccines, 2021, 9, 3 CrossRef CAS.
- H. Huysmans, Z. Zhong, J. De Temmerman, B. L. Mui, Y. K. Tam and S. Mc Cafferty, et al., Expression Kinetics and Innate Immune Response after Electroporation and LNP-Mediated Delivery of a Self-Amplifying mRNA in the Skin, Mol. Ther.–Nucleic Acids, 2019, 17, 867–878 CrossRef CAS.
- T. E. Wagner, J. R. Becraft, K. Bodner, B. Teague, X. Zhang and A. Woo, et al., Small-molecule-based regulation of RNA-delivered circuits in mammalian cells, Nat. Chem. Biol., 2018, 14(11), 1043–1050 CrossRef CAS PubMed.
- O. Andries, T. Kitada, K. Bodner, N. N. Sanders and R. Weiss, Synthetic biology devices and circuits for RNA-based ‘smart vaccines’: a propositional review, Expert Rev. Vaccines, 2015, 14(2), 313–331 CrossRef CAS PubMed.
- A. B. Vogel, L. Lambert, E. Kinnear, D. Busse, S. Erbar and K. C. Reuter, et al., Self-Amplifying RNA Vaccines Give Equivalent Protection against Influenza to mRNA Vaccines but at Much Lower Doses, Mol. Ther., 2018, 26(2), 446–455 CrossRef CAS PubMed.
- A. K. Blakney, P. F. McKay, K. Hu, K. Samnuan, N. Jain and A. Brown, et al., Polymeric and lipid nanoparticles for delivery of self-amplifying RNA vaccines, J. Controlled Release, 2021, 338, 201–210 CrossRef CAS PubMed.
- A. K. Blakney, Y. Zhu, P. F. McKay, C. R. Bouton, J. Yeow and J. Tang, et al., Big Is Beautiful: Enhanced saRNA Delivery and Immunogenicity by a Higher Molecular Weight, Bioreducible, Cationic Polymer, ACS Nano, 2020, 14(5), 5711–5727 CrossRef CAS PubMed.
- F. W. Johanning, R. M. Conry, A. F. LoBuglio, M. Wright, L. A. Sumerel, M. J. Pike and D. T. Curiel, A Sindbis virus mRNA polynucleotide vector achieves prolonged and high level heterologous gene expression in vivo, Nucleic Acids Res., 1995, 23(9), 1495–1501 CrossRef CAS PubMed.
- Y. Oda, Y. Kumagai, M. Kanai, Y. Iwama, I. Okura and T. Minamida, et al., Immunogenicity and safety of a booster dose of a self-amplifying RNA COVID-19 vaccine (ARCT-154) versus BNT162b2 mRNA COVID-19 vaccine: a double-blind, multicentre, randomised, controlled, phase 3, non-inferiority trial, Lancet Infect. Dis., 2023, 24, 351–360 CrossRef PubMed.
- E. Dolgin Self-copying RNA vaccine wins first full approval: what's next? 2023, https://www.nature.com/articles/d41586-023-03859-w.
- M. Baiersdorfer, G. Boros, H. Muramatsu, A. Mahiny, I. Vlatkovic, U. Sahin and K. Kariko, A Facile Method for the Removal of dsRNA Contaminant from In Vitro-Transcribed mRNA, Mol. Ther.–Nucleic Acids, 2019, 15, 26–35 CrossRef PubMed.
- Z. Zhong, S. McCafferty, L. Opsomer, H. Wang, H. Huysmans and J. De Temmerman, et al., Corticosteroids and cellulose purification improve, respectively, the in vivo translation and vaccination efficacy of sa-mRNAs, Mol. Ther., 2021, 29(4), 1370–1381 CrossRef CAS PubMed.
- B. Leyman, H. Huysmans, S. Mc Cafferty, F. Combes, E. Cox, B. Devriendt and N. N. Sanders, Comparison of the Expression Kinetics and Immunostimulatory Activity of Replicating mRNA, Nonreplicating mRNA, and pDNA after Intradermal Electroporation in Pigs, Mol. Pharmaceutics, 2018, 15(2), 377–384 CrossRef CAS.
- H. Huysmans, J. De Temmerman, Z. Zhong, S. Mc Cafferty, F. Combes, F. Haesebrouck and N. N. Sanders, Improving the Repeatability and Efficacy of Intradermal Electroporated Self-Replicating mRNA, Mol. Ther.–Nucleic Acids, 2019, 17, 388–395 CrossRef CAS PubMed.
- A. J. Geall, A. Verma, G. R. Otten, C. A. Shaw, A. Hekele and K. Banerjee, et al., Nonviral delivery of self-amplifying RNA vaccines, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(36), 14604–14609 CrossRef CAS PubMed.
- T. Demoulins, P. Milona, P. C. Englezou, T. Ebensen, K. Schulze and R. Suter, et al., Polyethylenimine-based polyplex delivery of self-replicating RNA vaccines, Nanomedicine, 2016, 12(3), 711–722 CrossRef CAS PubMed.
- L. A. Brito, M. Chan, C. A. Shaw, A. Hekele, T. Carsillo and M. Schaefer, et al., A cationic nanoemulsion for the delivery of next-generation RNA vaccines, Mol. Ther., 2014, 22(12), 2118–2129 CrossRef CAS PubMed.
- M. Jayaraman, S. M. Ansell, B. L. Mui, Y. K. Tam, J. Chen and X. Du, et al., Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo, Angew. Chem., Int. Ed., 2012, 51(34), 8529–8533 CrossRef CAS PubMed.
- Y. Eygeris, S. Patel, A. Jozic and G. Sahay, Deconvoluting Lipid Nanoparticle Structure for Messenger RNA Delivery, Nano Lett., 2020, 20(6), 4543–4549 CrossRef CAS PubMed.
- C. B. Roces, G. Lou, N. Jain, S. Abraham, A. Thomas, G. W. Halbert and Y. Perrie, Manufacturing Considerations for the Development of Lipid Nanoparticles Using Microfluidics, Pharmaceutics, 2020, 12, 1095 CrossRef CAS PubMed.
- H. Song, S. L. Hart and Z. Du, Assembly strategy of liposome and polymer systems for siRNA delivery, Int. J. Pharm., 2021, 592, 120033 CrossRef CAS.
- I. Ermilova and J. Swenson, DOPC versus DOPE as a helper lipid for gene-therapies: molecular dynamics simulations with DLin-MC3-DMA, Phys. Chem. Chem. Phys., 2020, 22(48), 28256–28268 RSC.
- R. L. Ball, P. Bajaj and K. A. Whitehead, Achieving long-term stability of lipid nanoparticles: examining the effect of pH, temperature, and lyophilization, Int. J. Nanomed., 2017, 12, 305–315 CrossRef CAS PubMed.
- S. T. LoPresti, M. L. Arral, N. Chaudhary and K. A. Whitehead, The replacement of helper lipids with charged alternatives in lipid nanoparticles facilitates targeted mRNA delivery to the spleen and lungs, J. Controlled Release, 2022, 345, 819–831 CrossRef CAS PubMed.
- R. N. Kularatne, R. M. Crist and S. T. Stern, The Future of Tissue-Targeted Lipid Nanoparticle-Mediated Nucleic Acid Delivery, Pharmaceuticals, 2022, 15, 897 CrossRef CAS PubMed.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Poly(ethylene glycol) in drug delivery: pros and cons as well as potential alternatives, Angew. Chem., Int. Ed., 2010, 49(36), 6288–6308 CrossRef CAS PubMed.
- W. Yang, L. Mixich, E. Boonstra and H. Cabral, Polymer-Based mRNA Delivery Strategies for Advanced Therapies, Adv. Healthcare Mater., 2023, e2202688 CrossRef PubMed.
- E. Oude Blenke, E. Ornskov, C. Schoneich, G. A. Nilsson, D. B. Volkin and E. Mastrobattista, et al., The Storage and In-Use Stability of mRNA Vaccines and Therapeutics: Not A Cold Case, J. Pharm. Sci., 2023, 112(2), 386–403 CrossRef CAS PubMed.
- D. Rafael, F. Andrade, A. Arranja and A. Luis, Videira M. Lipoplexes and Polyplexes: Gene Delivery Applications, 2015 Search PubMed.
- A. Hall, U. Lachelt, J. Bartek, E. Wagner and S. M. Moghimi, Polyplex Evolution: Understanding Biology, Optimizing Performance, Mol. Ther., 2017, 25(7), 1476–1490 CrossRef CAS PubMed.
- B. B. Mendes, J. Conniot, A. Avital, D. Yao, X. Jiang and X. Zhou, et al., Nanodelivery of nucleic acids, Nat. Rev. Methods Primers, 2022, 2, 24 CrossRef CAS PubMed.
- M. R. Elzes, I. Mertens, O. Sedlacek, B. Verbraeken, A. C. A. Doensen and M. A. Mees, et al., Linear Poly(ethylenimine-propylenimine) Random Copolymers for Gene Delivery: From Polymer Synthesis to Efficient Transfection with High Serum Tolerance, Biomacromolecules, 2022, 23(6), 2459–2470 CrossRef CAS PubMed.
- B. D. Monnery, M. Wright, R. Cavill, R. Hoogenboom, S. Shaunak, J. H. G. Steinke and M. Thanou, Cytotoxicity of polycations: Relationship of molecular weight and the hydrolytic theory of the mechanism of toxicity, Int. J. Pharm., 2017, 521(1–2), 249–258 CrossRef CAS PubMed.
- B. D. Monnery, Polycation-Mediated Transfection: Mechanisms of Internalization and Intracellular Trafficking, Biomacromolecules, 2021, 22(10), 4060–4083 CrossRef CAS PubMed.
- H. L. Weien Yuan, Polymer-based nanocarriers for therapeutic nucleic acids delivery, in Micro and Nano Technologies. Nanostructures for Drug Delivery, ed. A. M. G. Ecaterina Andronescu, Elsevier, 2017, ch. 14, pp. 445–460 Search PubMed.
- G. Grandinetti, N. P. Ingle and T. M. Reineke, Interaction of poly(ethylenimine)-DNA polyplexes with mitochondria: implications for a mechanism of cytotoxicity, Mol. Pharmaceutics, 2011, 8(5), 1709–1719 CrossRef CAS PubMed.
- D. Il, Development of poly(ethyleneimine) grafted amphiphilic copolymers: Evaluation of their cytotoxicity and ability to complex DNA, J. Bioact. Compat. Polym., 2021, 36(6), 447–463 CrossRef.
- M. Mees, E. Haladjova, D. Momekova, G. Momekov, P. S. Shestakova and C. B. Tsvetanov, et al., Partially Hydrolyzed Poly(n-propyl-2-oxazoline): Synthesis, Aqueous Solution Properties, and Preparation of Gene Delivery Systems, Biomacromolecules, 2016, 17(11), 3580–3590 CrossRef CAS PubMed.
- V. de la Rosa, Fast and accurate partial hydrolysis of poly(2-ethyl-2-oxazoline) into tailored linear polyethylenimine copolymers, Polym. Chem., 2014, 5(17), 4957–4964 RSC.
- O. Sedlacek, K. Lava, B. Verbraeken, S. Kasmi, B. G. De Geest and R. Hoogenboom, Unexpected Reactivity Switch in the Statistical Copolymerization of 2-Oxazolines and 2-Oxazines Enabling the One-Step Synthesis of Amphiphilic Gradient Copolymers, J. Am. Chem. Soc., 2019, 141(24), 9617–9622 CrossRef CAS PubMed.
- J. Rejman, G. Tavernier, N. Bavarsad, J. Demeester and S. C. De Smedt, mRNA transfection of cervical carcinoma and mesenchymal stem cells mediated by cationic carriers, J. Controlled Release, 2010, 147(3), 385–391 CrossRef CAS PubMed.
- H. Zhang, S. C. De Smedt and K. Remaut, Fluorescence Correlation Spectroscopy to find the critical balance between extracellular association and intracellular dissociation of mRNA complexes, Acta Biomater., 2018, 75, 358–370 CrossRef CAS PubMed.
- R. K. Oskuee, A. Dehshahri, W. T. Shier and M. Ramezani, Alkylcarboxylate grafting to polyethylenimine: a simple approach to producing a DNA nanocarrier with low toxicity, J. Gene Med., 2009, 11(10), 921–932 CrossRef CAS PubMed.
- P. Gurnani, A. K. Blakney, R. Terracciano, J. E. Petch, A. J. Blok and C. R. Bouton, et al., The In Vitro, Ex Vivo, and In Vivo Effect of Polymer Hydrophobicity on Charge-Reversible Vectors for Self-Amplifying RNA, Biomacromolecules, 2020, 21(8), 3242–3253 CrossRef CAS PubMed.
- H. L. Lightfoot and J. Hall, Endogenous polyamine function-the RNA perspective, Nucleic Acids Res., 2014, 42(18), 11275–11290 CrossRef CAS PubMed.
- P. H. Bolton and D. R. Kearns, Hydrogen bonding interactions of polyamines with the 2′ OH of RNA, Nucleic Acids Res., 1978, 5(4), 1315–1324 CrossRef CAS PubMed.
- J. D. Ziebarth and Y. Wang, Understanding the protonation behavior of linear polyethylenimine in solutions through Monte Carlo simulations, Biomacromolecules, 2010, 11(1), 29–38 CrossRef CAS PubMed.
- L. Tauhardt, K. Kempe, K. Knop, E. Altuntas, M. Jäger and S. Schubert, et al., Linear Polyethyleneimine: Optimized Synthesis and Characterization - On the Way to “Pharmagrade” Batches, Macromol. Chem. Phys., 2011, 212(17), 1918–1924 CrossRef CAS.
- A. K. Blakney, S. Ip and A. J. Geall, An Update on Self-Amplifying mRNA Vaccine Development, Vaccines, 2021,(2), 9 Search PubMed.
- D. Fischer, Y. Li, B. Ahlemeyer, J. Krieglstein and T. Kissel, In vitro cytotoxicity testing of polycations: influence of polymer structure on cell viability and hemolysis, Biomaterials, 2003, 24(7), 1121–1131 CrossRef CAS PubMed.
- H. Sun, E. J. Calabrese, Z. Lin, B. Lian and X. Zhang, Similarities between the Yin/Yang Doctrine and Hormesis in Toxicology and Pharmacology, Trends Pharmacol. Sci., 2020, 41(8), 544–556 CrossRef CAS PubMed.
- E. Blanco, H. Shen and M. Ferrari, Principles of nanoparticle design for overcoming biological barriers to drug delivery, Nat. Biotechnol., 2015, 33(9), 941–951 CrossRef CAS PubMed.
- H. Kobayashi, R. Watanabe and P. L. Choyke, Improving conventional enhanced permeability and retention (EPR) effects; what is the appropriate target?, Theranostics, 2013, 4(1), 81–89 CrossRef PubMed.
- B. Du, X. Jiang, A. Das, Q. Zhou, M. Yu, R. Jin and J. Zheng, Glomerular barrier behaves as an atomically precise bandpass filter in a sub-nanometre regime, Nat. Nanotechnol., 2017, 12(11), 1096–1102 CrossRef CAS PubMed.
- D. Manzanares and V. Cena, Endocytosis: The Nanoparticle and Submicron Nanocompounds Gateway into the Cell, Pharmaceutics, 2020, 12, 371 CrossRef CAS PubMed.
- G. Song, J. S. Petschauer, A. J. Madden and W. C. Zamboni, Nanoparticles and the mononuclear phagocyte system: pharmacokinetics and applications for inflammatory diseases, Curr. Rheumatol. Rev., 2014, 10(1), 22–34 CrossRef CAS PubMed.
- M. Danaei, M. Dehghankhold, S. Ataei, F. Hasanzadeh Davarani, R. Javanmard and A. Dokhani, et al., Impact of Particle Size and Polydispersity Index on the Clinical Applications of Lipidic Nanocarrier Systems, Pharmaceutics, 2018, 10, 57 CrossRef PubMed.
- M. I. Syga, E. Nicoli, E. Kohler and V. P. Shastri, Albumin Incorporation in Polyethylenimine-DNA Polyplexes Influences Transfection Efficiency, Biomacromolecules, 2016, 17(1), 200–207 CrossRef CAS PubMed.
- H. J. Kim, S. Ogura, T. Otabe, R. Kamegawa, M. Sato, K. Kataoka and K. Miyata, Fine-Tuning of Hydrophobicity in Amphiphilic Polyaspartamide Derivatives for Rapid and Transient Expression of Messenger RNA Directed Toward Genome Engineering in Brain, ACS Cent. Sci., 2019, 5(11), 1866–1875 CrossRef CAS PubMed.
- G. Choijilsuren, R. S. Jhou, S. F. Chou, C. J. Chang, H. I. Yang and Y. Y. Chen, et al., Heparin at physiological concentration can enhance PEG-free in vitro infection with human hepatitis B virus, Sci. Rep., 2017, 7(1), 14461 CrossRef PubMed.
- S. Akhter, M. Berchel, P. A. Jaffres, P. Midoux and C. Pichon, mRNA Lipoplexes with Cationic and Ionizable alpha-Amino-lipophosphonates: Membrane Fusion, Transfection, mRNA Translation and Conformation, Pharmaceutics, 2022, 14, 581 CrossRef CAS PubMed.
- F. Xiao, Z. Chen, Z. Wei and L. Tian, Hydrophobic Interaction: A Promising Driving Force for the Biomedical Applications of Nucleic Acids, Adv. Sci., 2020, 7(16), 2001048 CrossRef CAS PubMed.
- S. M. Sarett, T. A. Werfel, I. Chandra, M. A. Jackson, T. E. Kavanaugh and M. E. Hattaway, et al., Hydrophobic interactions between polymeric carrier and palmitic acid-conjugated siRNA improve PEGylated polyplex stability and enhance in vivo pharmacokinetics and tumor gene silencing, Biomaterials, 2016, 97, 122–132 CrossRef CAS PubMed.
- K. Amin and R. M. Dannenfelser, In vitro hemolysis: guidance for the pharmaceutical scientist, J. Pharm. Sci., 2006, 95(6), 1173–1176 CrossRef CAS PubMed.
- R. Riera, N. Feiner-Gracia, C. Fornaguera, A. Cascante, S. Borros and L. Albertazzi, Tracking the DNA complexation state of pBAE polyplexes in cells with super resolution microscopy, Nanoscale, 2019, 11(38), 17869–17877 RSC.
- P. L. Ma, M. D. Buschmann and F. M. Winnik, Complete physicochemical characterization of DNA/chitosan complexes by multiple detection using asymmetrical flow field-flow fractionation, Anal. Chem., 2010, 82(23), 9636–9643 CrossRef CAS PubMed.
- P. L. Ma, M. D. Buschmann and F. M. Winnik, One-step analysis of DNA/chitosan complexes by field-flow fractionation reveals particle size and free chitosan content, Biomacromolecules, 2010, 11(3), 549–554 CrossRef CAS PubMed.
- M. M. Bloksma, R. M. Paulus, H. P. van Kuringen, F. van der Woerdt, H. M. Lambermont-Thijs, U. S. Schubert and R. Hoogenboom, Thermoresponsive poly(2-oxazine)s, Macromol. Rapid Commun., 2012, 33(1), 92–96 CrossRef CAS PubMed.
- B. Layek, M. K. Haldar, G. Sharma, L. Lipp, S. Mallik and J. Singh, Hexanoic acid and polyethylene glycol double grafted amphiphilic chitosan for enhanced gene delivery: influence of hydrophobic and hydrophilic substitution degree, Mol. Pharmaceutics, 2014, 11(3), 982–994 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tb03003b |
| This journal is © The Royal Society of Chemistry 2024 |