
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in light-triggered cancer immunotherapy
Jin-Kyoung
Yang
a,
Hayoon
Kwon
bc and
Sehoon
Kim
 *bc
*bc
aDepartment of Chemical Engineering, Dong-eui University, Busan, 47340, Republic of Korea. E-mail: yjk0627@deu.ac.kr
bChemical & Biological integrative Research Center, Korea Institute of Science and Technology, Seoul, 02792, Republic of Korea. E-mail: sehoonkim@kist.re.kr
cKU-KIST Graduate School of Converging Science and Technology, Korea University, Seoul, 02841, Republic of Korea
First published on 30th January 2024
Abstract
Light-triggered phototherapies, such as photodynamic therapy (PDT) and photothermal therapy (PTT), have shown strong therapeutic efficacy with minimal invasiveness and systemic toxicity, offering opportunities for tumor-specific therapies. Phototherapies not only induce direct tumor cell killing, but also trigger anti-tumor immune responses by releasing various immune-stimulating factors. In recent years, conventional phototherapies have been combined with cancer immunotherapy as synergistic therapeutic modalities to eradicate cancer by exploiting the innate and adaptive immunity. These combined photoimmunotherapies have demonstrated excellent therapeutic efficacy in preventing tumor recurrence and metastasis compared to phototherapy alone. This review covers recent advancements in combined photoimmunotherapy, including photoimmunotherapy (PIT), PDT-combined immunotherapy, and PTT-combined immunotherapy, along with their underlying anti-tumor immune response mechanisms. In addition, the challenges and future research directions for light-triggered cancer immunotherapy are discussed.
1. Introduction
Cancer is a leading cause of mortality and morbidity worldwide. Much progress and technical improvements against cancer have been made over the past few decades, contributing to a decline in cancer mortality rates. However, cancer remains a substantial burden, with more than 1.6 million newly diagnosed cases in 2017.1 Several novel anti-tumor methods, such as chemotherapy, radiotherapy, gene therapy, photothermal therapy (PTT), and photodynamic therapy (PDT), have been developed to treat and prevent cancer.2 Although these strategies have shown highly effective performances for cancer treatment, they only focus on regional therapy rather than fundamental therapy, hindering the prevention of metastasis and invasion.3 However, the single treatment approach has become insufficient in eliminating the entire tumor owing to increased resistance to therapy. Therefore, the use of long-lasting therapeutics with multiple agents is a promising strategy for cancer treatment.Immunotherapy, which exploits the innate immune system to eliminate malignant cells, has attracted considerable attention over the past few years.4–8 Cancer cells have multiple resistance mechanisms, such as local immune evasion, induced tolerance, and systemic disruption of T cell signaling, enabling them to escape immune recognition and subsequent destruction by the innate immune system.9 Furthermore, through immune editing, tumors can grow unrestrained and become clinically apparent.10 Since receiving the Nobel Prize in immunology in 2018, immunotherapy has prompted many researchers to use the immune system to protect humans against cancer. Immunotherapy against cancers include various approaches ranging from the use of IL-2 or IFN-α cytokine to stimulate the host's immune system,11 employing cell-based therapies for T cell regulation,12–15 to using antibodies against cytotoxic T lymphocyte-associated antigen 4 (CTLA-4), programmed cell-death protein 1 (PD-1) and its ligand (PD-L1) for immune checkpoint blockade.6,16–19 Immune systems activated through the abovementioned methods exhibited considerably improved anti-tumor effects and yielded increased memory effects, thereby successfully preventing tumor metastasis and recurrence. However, several recent studies have reported that immunotherapy is either ineffective or inconsistent in terms of therapeutic outcomes. Without cancer specificity, immunotherapy can induce several immune side effects, including the hyperimmunization of normal cells, autoimmune responses, organ toxicity, and increased drug resistance.20–22 Therefore, one strategy to improve the efficacy of immunotherapeutic drugs and reduce their side effects is by combining immunotherapy with region-selective therapeutic methods.
In this regard, many studies that combine immunotherapy with external therapy have been reported.23–28 Although chemotherapy, radiotherapy, and surgery can be effective in tumor regression, these methods are generally not associated with the induction of systemic tumor immunity.29 On the other hand, photoirradiation can trigger an immune-active response by itself in three major ways: (1) laser irradiation could induce tissue stress, disintegration, and ejection itself. (2) Photochemical reactions alter the chemical bonds on cell surfaces and generate reactive oxygen species (ROS) with the assistance of a photosensitizer, leading to both the death of irradiated tissues and a host immune response.
(3) Photothermal reactions that produce elevated heat from laser irradiation could effectively destroy the tumor vasculature owing to its high sensitivity to temperature.29–34 Therefore, phototherapy could be a good candidate for synergistic therapeutic effects owing to its applicability as a tumor-focused treatment, which leads to tumor regression and prevents toxic effects in healthy tissues.35–37
In this review, we focus on dual-mode therapies that combine immunotherapy and light-based therapy (photoimmunotherapy). The types of phototherapies integrated with immunotherapy for cancer treatment are summarized in Fig. 1. First, we discuss the three latest developments in photoimmunotherapy: photoimmunotherapy (PIT), photodynamic therapy (PDT)-combined immunotherapy, and photothermal therapy (PTT)-combined immunotherapy. In addition, we present the immunological effects of each phototherapy, as well as those of the combined therapies. The last section summarizes the advantages and disadvantages of photoimmunotherapy and discusses its future aspects in detail.
 | ||
| Fig. 1 Schematic representation of combinatorial treatment strategies of light-triggered immunotherapy. | ||
2. Photoimmunotherapy
Photodynamic therapy (PDT) is a minimally invasive therapeutic method used in cancer treatment.38–40 In PDT, a non-toxic photosensitive dye (photosensitizer) absorbs incident light and subsequently produces cytotoxic ROS via photochemical reactions involving the transfer of excited electrons. PDT has advantages over conventional cancer therapies such as surgery, chemotherapy, and radiotherapy owing to its minimal systemic toxicity and repeatability in the tumor region. In addition, PDT can trigger an immunostimulatory effect on the immune system.29,31,41 However, the use of PDT is limited owing to skin photosensitivity, phototoxicity, tumor selectivity, and the unfavourable distribution of photosensitizers leading to normal tissue damage.42,43 A potential method to overcome these disadvantages is to provide tumor selectivity to photosensitizers through the conjugation of antibodies, i.e., photoimmunotherapy (PIT).44 Since the lifetime and radius of action of reactive oxygen species are <0.04 μs and <0.02 μm, respectively, dimensions smaller than the size of tumor cells (10 μm), PIT can offer enhanced performance compared to the conventional PDT method.452.1 PIT
The PIT strategy, which has been studied for approximately 30 years, is based on photosensitizers conjugated to monoclonal antibodies (mAbs) that bind to particular tumor antigens in a specific manner. Therefore, its clinical effects depend on the antigen expressed on the tumor cell surface and not on normal tissues. Unfortunately, a considerable number of antigens identified in human tumors are not exclusive to tumor tissues but are also expressed in normal tissues. Despite this challenge, photoimmunotherapy (PIT) can still achieve adequate therapeutic efficacy in situations where mAbs exhibit limited accessibility to normal tissues or when the tissue remains unexposed to laser irradiation or normal daylight. Some target antigens and photosensitizer conjugates for PIT are provided in Table 1, along with their therapeutic responses.| Target antigen | Photosensitizer | Anti-tumor response | Ref. |
|---|---|---|---|
| CAMAL | Hematoporphyrin | Leukemia cells producing CAMAL were selectively killed. | 49 |
| Leu1 | Chlorin e6 | Human leukemia cells were selectively photodestructed. | 50 |
| CD4 | Pyrene liposome | Human leukemia cells were selectively destructed. | 51 |
| CA 125 | Chlorin e6 | Immunoconjugate was accumulated in ovarian tumor, but showed dark toxicity. | 52 |
| ICAM | Tin chlorin e6 | Melanoma cells were photolysed by a point rupture of the plasma membrane. | 53 |
| CA 125 | Poly-L-lysine chlorin e6 | Cationic photoimmunoconjugates showed enhanced cancer-cell uptake and phototoxicity. | 54 |
| CD44 | m-Tetrahydroxyphenylchlorin | Tumor selectivity of conjugates increased compared to free photosensitizers. | 55 |
| CD44 | Aluminium(III) phthalocyanine tetrasulfonate | Photoimmunoconjugates were more toxic to head and neck cancer cells than free photosensitizers. | 56 |
| CEA, ErbB2 | Aluminium(III) phthalocyanine tetrasulfonate | Photo-induced toxicity of conjugates to ovarian carcinoma cells was increased by using internalized mAbs. | 57 |
| CEA, ErbB2, EpCAM | Porphyrin isothiocyanates | Photo-induced toxicity of conjugates to carcinoma cells was increased by using internalized mAbs. | 58 |
| CEA | IRDye800DX | Single PIT treatment caused rapid cell death and significantly inhibited tumor growth. | 59 |
| FAP | ZnF16Pc | Ferritin nanoparticle-based PIT (nano-PIT) selectively eliminated CAFs, enhanced T cell infiltration, and increased tumor suppression. | 48 |
As described in Table 1, various antigens have been used for tumor-specific targeting. In most studies, the photosensitizing and phototoxic performances of photoimmunoconjugates depend on the ratio of mAbs to the photosensitizer. In addition, the binding region of mAbs should not be interrupted or shielded by the conjugation reaction with the photosensitizer to ensure specific targeting. Therefore, the development of high-quality photoimmunoconjugates is very important.
Generally, the conjugation between mAbs and the photosensitizer depends on the reaction of functional groups such as –NH2, –COOH, and –SH. In addition, hydrophilic moieties have been introduced into photosensitizers, which are hydrophobic, to improve their water solubility.46 Photoimmunoconjugates have been characterized to evaluate their therapeutic performances by altering the pharmacokinetics, biodistribution, specific and non-specific binding to antigens, and internalization.
Targeted PIT molecular therapeutic agents show anti-tumor effects by generating ROS that contribute to cancer cell destruction via direct cancer cell death, destruction of cell membranes or tumor vasculature, leading to tumor ischemia, and activation of the immune response.31,38,47
Recently, nanotechnology has been adopted in PIT for enhanced specific delivery of therapeutic agents to tumor regions through enhanced permeation and retention (EPR) effects, as well as active targeting via antibody–antigen conjugation. Zhen et al. reported a nanoparticle-based PIT utilizing ferritin as a photosensitizer carrier and an anti-fibroblast-activation protein (FAP) single-chain variable fragment (scFv) for targeting mAbs.48 The nano-PIT system successfully eliminated carcinoma-associated fibroblasts (CAF) from tumors but caused slight damage to healthy tissues owing to its localized specific treatment. This approach prevents tumor growth via CAF elimination. Further studies have shown that the CAF elimination-induced destruction of the extracellular matrix (ECM) in the surrounding tumors sensitizes cancer cells to immunotherapeutic effects via the infiltration of CD8+ T cells into tumors. Although PIT efficiently triggers a specific anti-cancer PDT response, its application is limited to in vitro use, and only a few studies have shown in vivo therapeutic effects. There are several reasons for these inconsistent results. First, most conventional photosensitizers absorb visible light that cannot penetrate deep tissues, thus deteriorating the light-triggered therapeutic performance of PIT. Second, the hydrophobic nature of conventional photosensitizers disrupts their conjugation to mAbs and leads to poor water solubility of the immunoconjugates, thereby reducing the targeting capacity of the conjugate. Third, the reproducibility of photoimmunoconjugate production could be affected by conjugation procedures, potentially reducing their integrity and antigen-binding capacities.58 Therefore, additional work is required to overcome these limitations to achieve successful in vivo therapeutic performance.
2.2 Near-infrared PIT (NIR-PIT)
Near-infrared (NIR, 700–1000 nm) light with deep tissue penetrability had already been broadly applied in imaging and therapy in vivo.60 Several NIR light-sensitive photosensitizers have been discovered for in vivo PDT agents.61,62 PIT has also been combined with NIR-sensitive photosensitizers to improve the anti-tumor response. Kobayashi et al. found that the NIR phthalocyanine dye IR700, conjugated to mAbs (mAb-IR700), along with NIR light irradiation, enhanced the target-selective anti-tumor performance with immune activation.63–79 As shown in Fig. 2(a) and (b), the tumor volume significantly reduced after NIR-PIT with mAb-IR700 treatment compared to the untreated control, while survival was prolonged in mice. In addition, dendritic cell (DC) maturation was confirmed using CD80 and CD86 markers after NIR-PIT-mediated cancer cell death (Fig. 2(c)). The molecular and cellular mechanisms underlying cell death induced by NIR-PIT have recently been elucidated.80 IR700 molecules (a photoactivating chemical, APC) were conjugated with three hydrophilic sulfonic acid moieties that could be released from APC after NIR irradiation and subsequent hydrolysis under electron-donor-rich conditions. This light-triggered cleavage results in the formation of less soluble phthalocyanine molecules (Fig. 3(a)). The diminished water solubility contributed to the stacking and aggregation between IR700 conjugated to mAbs and mAbs bound to target antigens on the cell membrane, which damages the target membrane proteins, subsequently depleting the cell membrane integrity (Fig. 3(b)). Next, the targeted cancer cells rapidly swell and release cytoplasmic contents into the ECM through membrane rupture. In this cell death process, ROS generation by APCs plays a minor role, although conventional PDT could partially induce phototoxic cell death.23 Danger signals released from targeted dying cells such as calreticulin, ATP, HMGB1, and heat shock proteins (HSPs), potently induce the phenotype maturation of DCs and their functional activation, characterized by an increase in CD80 and CD86 markers. In turn, the activated mature DCs cause cellular immune responses, leading to CD8+ T cell expansion, which contributes to cancer cell killing and confers a key feature of immune memory (Fig. 3(c)).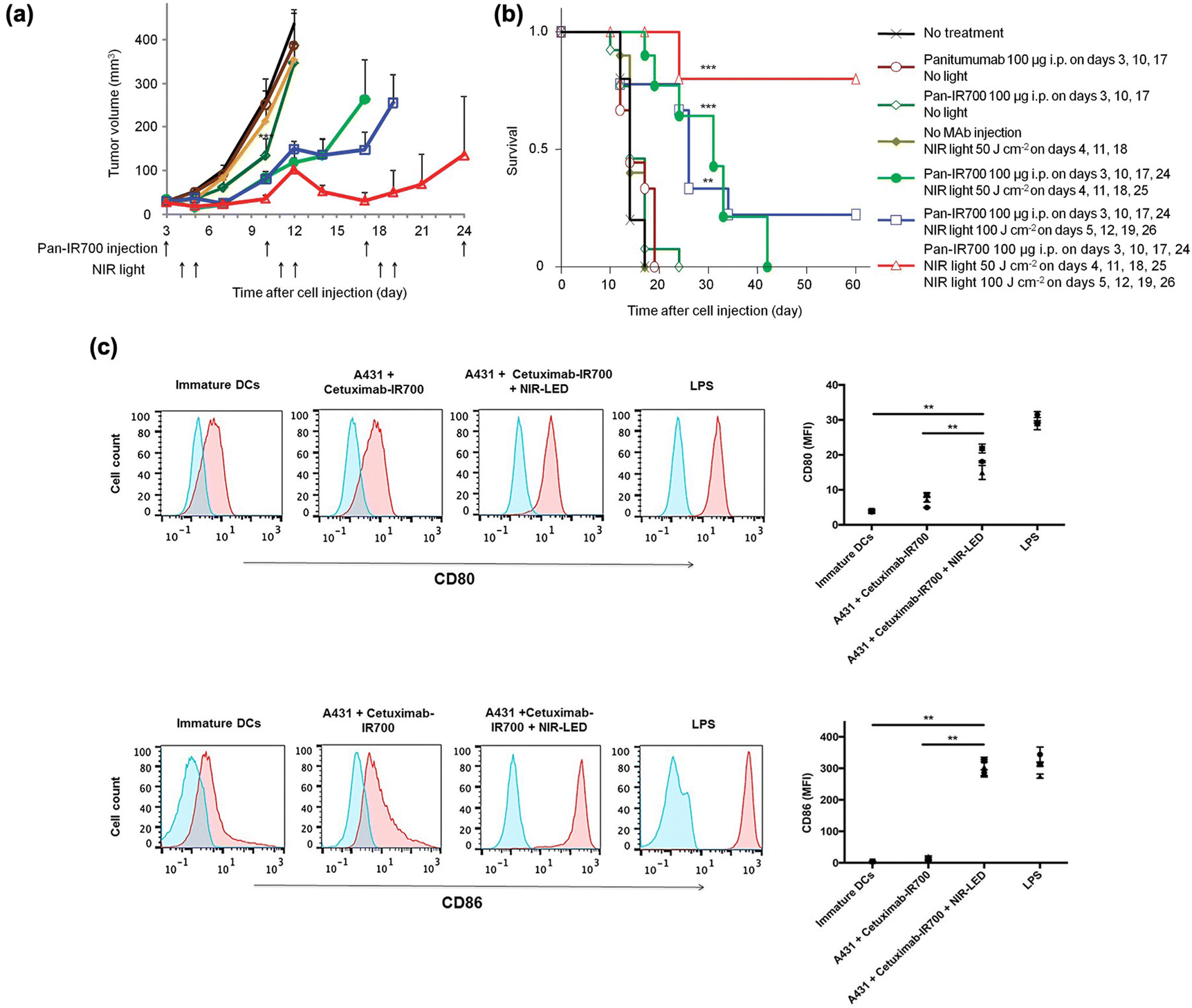 | ||
| Fig. 2 Therapeutic performance of mAb-IR700 and immune activation. (a) Tumor volume reduction and (b) survival after NIR-PIT with repeated light irradiation. (c) DC maturation after NIR-PIT. Reproduced with permission from ref. 62 and 73. Copyright©2024, American Chemical Society and Impact Journals, LLC. | ||
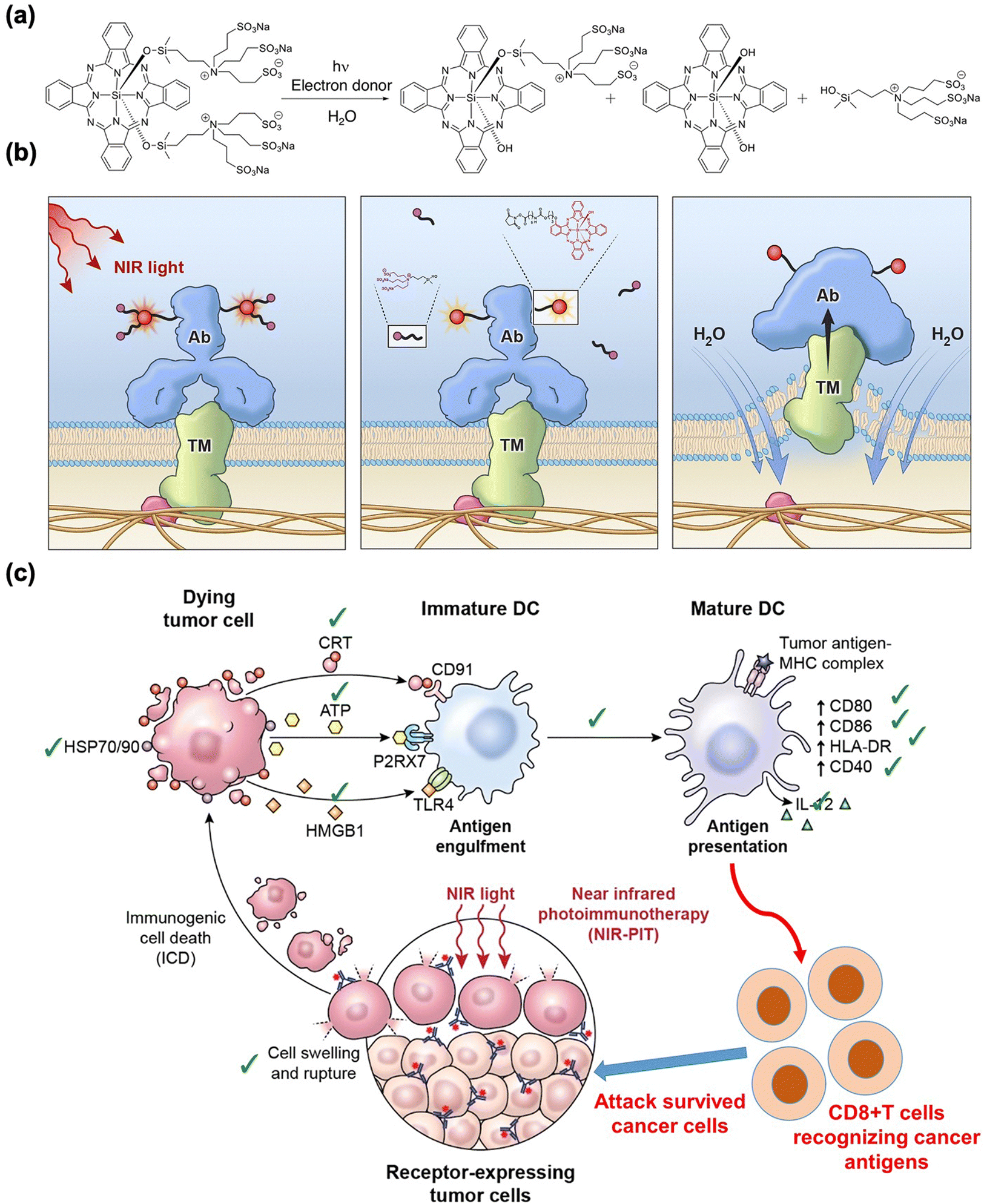 | ||
| Fig. 3 Schematic illustration of the (a) chemistry, (b) physiological action of NIR-PIT, and (c) immunogenic cell death mechanism of NIR-PIT. Reproduced with permission from ref. 26. Copyright©2024, American Chemical Society. | ||
NIR-PIT has been applied to various types of cancers with overexpressed target membrane proteins and has shown successful in vitro and in vivo performances. Based on these early results, NIR-PIT was fast-tracked for approval by the US Food and Drug Administration for the treatment of malignant head and neck cancer in 2019.
Overall, NIR-PIT is a good platform for photo-triggered cancer immunotherapy with several advantages: (i) target specificity and selective phototoxicity, (ii) deep-tissue penetration of NIR light, (iii) high NIR-PIT repeatability, and (iv) flexibility of the platform that can be applied to various different biomarkers and antibodies. Thus, NIR-PIT is a promising novel approach for phototriggered cancer immunotherapy.
3. Photodynamic therapy-combined immunotherapy
3.1 Immunogenic response of PDT
PDT directly causes both apoptotic and necrotic cell death by producing reactive singlet oxygen, whereas other cytotoxic agents usually induce apoptotic cell death.23 During necrotic and/or apoptotic cell death, an acute inflammatory response can occur by attracting leukocytes such as DCs and neutrophils into the tumor site, thus increasing antigen presentation. HSP70 plays an important role in promoting tumor antigen delivery and stimulating anti-tumor immunity.81 HSP70 is a protective protein that mitigates external stress, such as heat and inflammation, by interacting with the peptide segments of proteins. This engagement shields proteins from aggregation or misfolding, ultimately averting consequential cellular death. Notably, both HSP70 expression and APC recruitment increased at the tumor site after PDT, which leads to the activation and maturation of DCs, resulting in the cross-presentation of the antigen peptide and the activation of CD8+ effector T cells.82 Furthermore, PDT-induced acute inflammation releases various types of cytokines. IL-6, a proinflammatory cytokine, was detected in increased levels after PDT.83 IL-1β, IL-8, TNF-α, and IFN-γ contents were also elevated after PDT.84,85 These cytokines can trigger the immune response by directing the migration and activation of phagocytes and lymphocytes.Tumor-specific and memory immunity following PDT has been demonstrated in several studies. Cubeddu et al. proved that MS-2 fibrosarcoma in mice were able to resist a rechallenge with tumor cells in a tumor-specific manner after PDT, whereas immunosuppressed models were unable to inhibit the rechallenge.86 Several studies have reported that systemic anti-tumor immunity could be induced by PDT.87,88
On the other hand, the immunosuppressive properties of PDT have also been reported.89 Bowen et al. reported that hematoporphyrin derivative-mediated PDT resulted in 50% suppression of contact hypersensitivity, which is a T cell-mediated immune response.90 Several subsequent studies have also shown the immunosuppressive effect of PDT using different photosensitizers.91–94 Although the exact mechanism of the immunosuppressive effect of PDT has not been discovered yet, Hamblin et al. proposed that the local inflammation response following PDT may trigger a counteraction, leading to immune suppression through regulatory T cell (Treg) activation.89 Elevated levels of the vascular endothelial growth factor (VEGF) after PDT might also inhibit DC maturation and increase immature phenotypes.95,96
These contradictory observations may be attributed to different photosensitizers, light treatments, fluency levels, and evaluation time points. Therefore, to elicit a robust immunostimulatory effect with PDT, a combination of PDT and immunotherapy may be a good candidate for powerful cancer therapy with an intensified anti-cancer immune response.
3.2 PDT-combined immunotherapy
There have been several reports on the combination of PDT with various immunostimulatory agents, which can be divided into four broad classes.Another broadly used TLR-related immunostimulant is CpG-oligodeoxynucleotide (CpG-ODN), which triggers the host immune system by activating B cells, NK cells, monocytes, and macrophages after TLR9 recognition.117 Hamblin et al. reported a combination of CpG-ODN as an adjuvant and liposomal benzoporphyrin derivative-based PDT for metastatic mouse breast cancer therapy.118,119 The CpG adjuvant and PDT improved the therapeutic efficacy via DC maturation and activation. Ahn et al. treated cell lysates, which contain immunogenic HSP70, with CpG-ODN after PDT in a human metastatic lung cancer mouse model.120 Interestingly, injecting PDT-generated cell lysates with CpG-ODN suppressed the tumor growth and improved the immune defense through IFN-γ secretion and cytotoxic T lymphocyte (CTL) activation.
Chitosan is a biodegradable polymer of D-glucosamine and N-acetyl-D-glucosamine and is known to stimulate immune-activity by inducing cytokine release and T cell upregulation.121 A chitosan derivative, glycated chitosan (GC), has been used as an immunostimulator with photofrin-mediated PDT for mammary and lung cancer therapies.114,122 Notably, the combined GC and PDT treatment increased the survival rate by approximately 2-fold compared to PDT alone. Furthermore, another chitosan derivative, schizophyllan (SPG), increased the macrophage-1 antigen-positive innate immune cell content and prolonged the retention time of the photofrin photosensitizer, leading to an increased rate of tumor cell death and enhanced tumor cure rates in SCCVII-bearing mice.123 GC and SPG exhibited significant immunostimulatory activities; however, further studies are needed to understand the therapeutic mechanisms and improve their practical efficacy.
Bacterial-derived anti-cancer vaccines have been used as non-specific adjuvants to develop immunity against cancer.124Bacillus Calmette-Guérin (BCG) administration not only improved the curative efficacy of PDT treatment toward tumors, but also boosted the incidence of immune memory T cells in tumor-draining lymph nodes.125,126 Another bacterial-derived immuno-adjuvant, Corynebacterium parvum (CP), also exhibited a synergistic effect on PDT cancer treatment.127 The therapeutic performance of CP-PDT depends on the amount and the sequence of CP. This method is promising, but further clinical studies are required. OK-432, which kills Streptococcus pyogenes, has been tested in combination with hematoporphyrin-mediated PDT in mice bearing NR-S1 mouse squamous cell carcinoma.128 The survival time was prolonged when OK-432 was injected 3 h before light irradiation, where OK-432 activated macrophages and neutrophils, leading to tumoricidal effects. Although the exact immunostimulatory mechanism of the abovementioned bacteria-derived vaccines remains unknown, it may be attributed to multiple immunomodulatory effects that could enhance and sustain the inflammatory response, followed by anti-cancer immunity.
PDT-combined ACT involves the ex vivo manipulation of immune cells, including immature DCs, T cells, and NK cells, and their subsequent reinfusion into the PDT-treated tumor bearing host. Immature DCs efficiently acquire antigens from apoptotic and necrotic tumor cells after PDT treatment, which transforms immature DCs into effective antigen-presenting cells, mature DCs, that migrate to regional lymph nodes for the activation of T lymphocytes. In addition, mature DCs could induce activation and proliferation of NK cells, which in turn lead to the maturation of immature DCs. These combination approaches can potentiate PDT-induced immune response and increase therapeutic efficiency (Fig. 4). Golab et al. investigated the anti-tumor activity of PDT by administering immature DCs.129 Immature DCs co-cultured with PDT-treated murine colon carcinoma cells (C-26) efficiently induced anti-tumor responses through the maturation and production of IL-12, and stimulation of cytotoxic T and NK cells. In addition, intratumoral DC administration combined with PDT resulted in in vivo tumor regression.130 Most importantly, DC treatment with PDT led to the regression of contralateral untreated tumors, including multiple lung metastases. Korbelik et al. showed the therapeutic performance of mTHPC-mediated PDT combined with ACT using NK cells genetically modified to produce IL-2 (NK92MI cells) in human cervical squamous cell carcinoma and human colorectal adenocarcinoma mouse models.131 They found that the transfer of NK92MI cells substantially improved the therapeutic outcome of PDT.
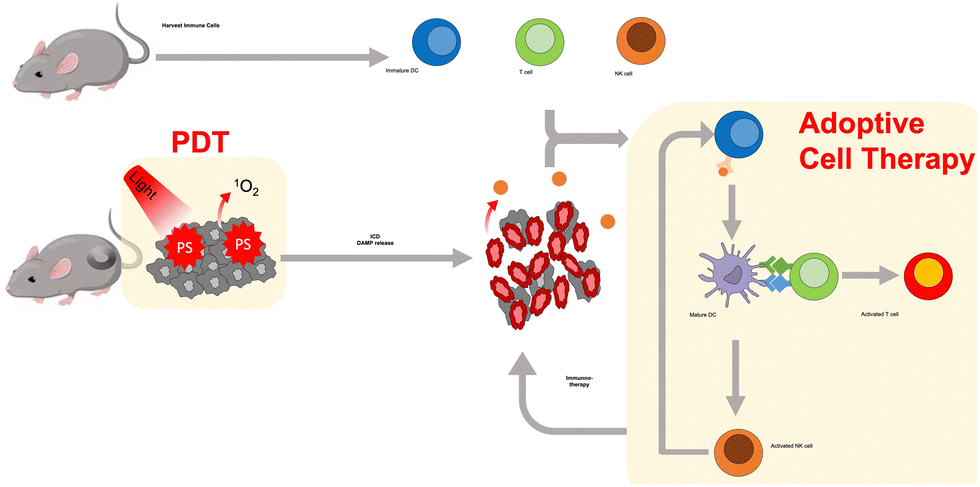 | ||
| Fig. 4 Schematic illustration of PDT-combined adoptive cell transfer. | ||
Various types of nanoparticles (NPs) have been developed to selectively deliver photosensitizers to cancer cells and activate anti-cancer immunity simultaneously. A summary of the different NPs used for PDT-combined immunotherapy is provided in Table 2. There are three major combinations of PDT with immunotherapeutic agents: (1) immunostimulators such as CpG-ODN and imiquimod (R837), (2) immune checkpoint inhibitors, and (3) indoleamine 2,3-dioxygenase (IDO) inhibitors. These approaches suppress primary and secondary tumors with memory effects induced by mature DC and activated T cells resulting from PDT-combined immunotherapy.
| NPs | Immunotherapeutic agents | Advantages | Therapeutic effects | Ref. |
|---|---|---|---|---|
| PC@GCpD(Gd) | CpG ODN | Multimodal-NPs combining PDT and immunotherapy with MRI/FI dual imaging modalities | Complete suppression of EMT6 murine mammary cancer model by maturation of DCs and activation and infiltration of T cells | 136 |
| W-TBP/CpG (MOF) | CpG ODN | Efficient delivery of CpGs through charge interaction between cationic MOF and anionic CpGs | Strong anti-tumor immunity with 97% tumor regression and abscopal effect in a bilateral TUBO mouse model | 137 |
| bMSN(CpG/Ce6)-Neoantigen | CpG ODN, neoantigen | Biodegradable NPs, image-guided therapeutic NPs | Recruitment of DCs to tumor sites by PDT, followed by neoantigen-specific CD8α+ CTL response | 142 |
| Strong synergistic effect on local and distant tumors in multiple tumor models | ||||
| NCP@pyrolipid (polymer NPs) | PD-L1 inhibitor | Chemotherapy and PDT combined NPs | Regression of primary and distant tumors in CT26 and MC38 mouse models by inducing tumor-specific T cell response | 143 |
| ZnP@pyro | PD-L1 inhibitor | Biocompatible NPs | Eradication of the primary tumors, prevention of lung metastases in the 4T1 mTNBC murine model | 144 |
| Complete inhibition of the non-irradiated distant tumors in 4T1 and TUBO mouse models | ||||
| CAT@S/Ce6-CTPP/DPEG | PD-L1 inhibitor | pH responsive mitochondria-targeting NPs | Eradication of tumors with enhanced PDT effects | 145 |
| Synergistic effect by anti-tumor immunities in 4T1 mouse model | ||||
| Ce6/MLT@SAB (serum albumin–Aluminum hydroxide) | PD-1 inhibitor | DC-activatable peptide (melittin, MLT) containing NPs | Synergistic immunogenic PDT with MLT, increase in therapeutic effects by combination with anti-PD-1 immunotherapy in the 4T1 mouse model | 146 |
| UCNP | Imiquimod (R837), CTLA-4 inhibitor | NIR-active NPs, stimulating immune responses with two immunogenic agents | Direct DC maturation by UCNP-Ce6-R837 in the CT26 mouse model, elimination of primary and secondary tumors by the long-term immune memory effect | 147 |
| Ce6-CAT/RPNPs/PEGDA | Imiquimod (R837), CLTA-4 inhibitor | Long-term tumor retention and multiple rounds of therapy by in situ polymerization | Strong anti-tumor response, abscopal effect and long-term immune memory protection in the CT26 mouse model | 148 |
| PpIX-1MT | 1-Methyltryptophan (1MT), IDO-1 inhibitor | Caspase-responsive NPs | Activation of CD8+ T cells via IDO inhibition, eradication of both primary tumor and lung metastasis in the CT26 mouse model | 149 |
| Pegylated PPa-NLG919 liposome | NLG919, IDO-1 inhibitor | MMP-2 responsive NPs, tumor-cell specific release of NLG919 | Enhanced anti-tumor efficacy in both CT26 and 4T1 mouse models by reducing intratumoral Treg infiltration and improving intratumoral infiltration of IFN-γ + T cells | 150 |
| pRNVs/HPPH/IND | Indoximod (IND), IDO inhibitor | pH-responsive NPs | ICD by PDT and vehicle, significant inhibition of both primary and distant tumors by increasing CD8+ T cell infiltration in the B16F10 mouse model | 151 |
First, immunostimulators have been used in some studies in combination with PDT. Liu et al. used graphene quantum dots containing a Cy3 photosensitizer, Gd3+ as the MR probe, and CpG-ODN (PC@GCpD(Gd)) for combined PDT immunotherapy.136 The TLR-9 adjuvant, CpG-ODN, enhanced the anti-tumor immune response through the maturation of DCs and secretion of TNF-α and IL-6, thus stimulating CD4+ and CD8+ T cell infiltration to enhance the anti-tumor immunity. Other nanomaterials, including metal–organic frameworks (MOFs) and DNA nanostructures, have been reported as PDT and CpG-ODN therapeutic agents, efficiently suppressing primary and distant tumors via DC and T cell expansion, respectively.137,138
Neoantigens have recently emerged as next-generation immunotherapeutics.139–141 Neoantigens are mutant peptide fragments of major histocompatibility complexes (MHCs) on the surface of cancer cells that are absent in normal cells. Thus, the use of neoantigens could allow for tumor- and patient-specific T cell activation-mediated cancer immunotherapy. Recently, Moon et al. developed biodegradable mesoporous silica NPs (MSNs) as nanocarriers for PDT-combined immunotherapy. As MSNs can undergo hydrolysis under aqueous conditions through nucleophilic interaction between hydroxyl groups in water and unbridged oxide on the surface of MSNs, they have been widely used as a platform for drug delivery.152 Moon et al. loaded CpG-ODN, neoantigen, photosensitizer chlorin e6, and 64Cu into MSNs for positron emission tomography-guided PDT and cancer vaccination in MC38 and B16F10 tumor mouse models (Fig. 5).142 PDT alone could reduce only primary tumors but not distant tumors, whereas the combined therapy showed a good abscopal effect through DC maturation and neoantigen-specific CD8α+ CTL responses (Fig. 5(b)–(d)).
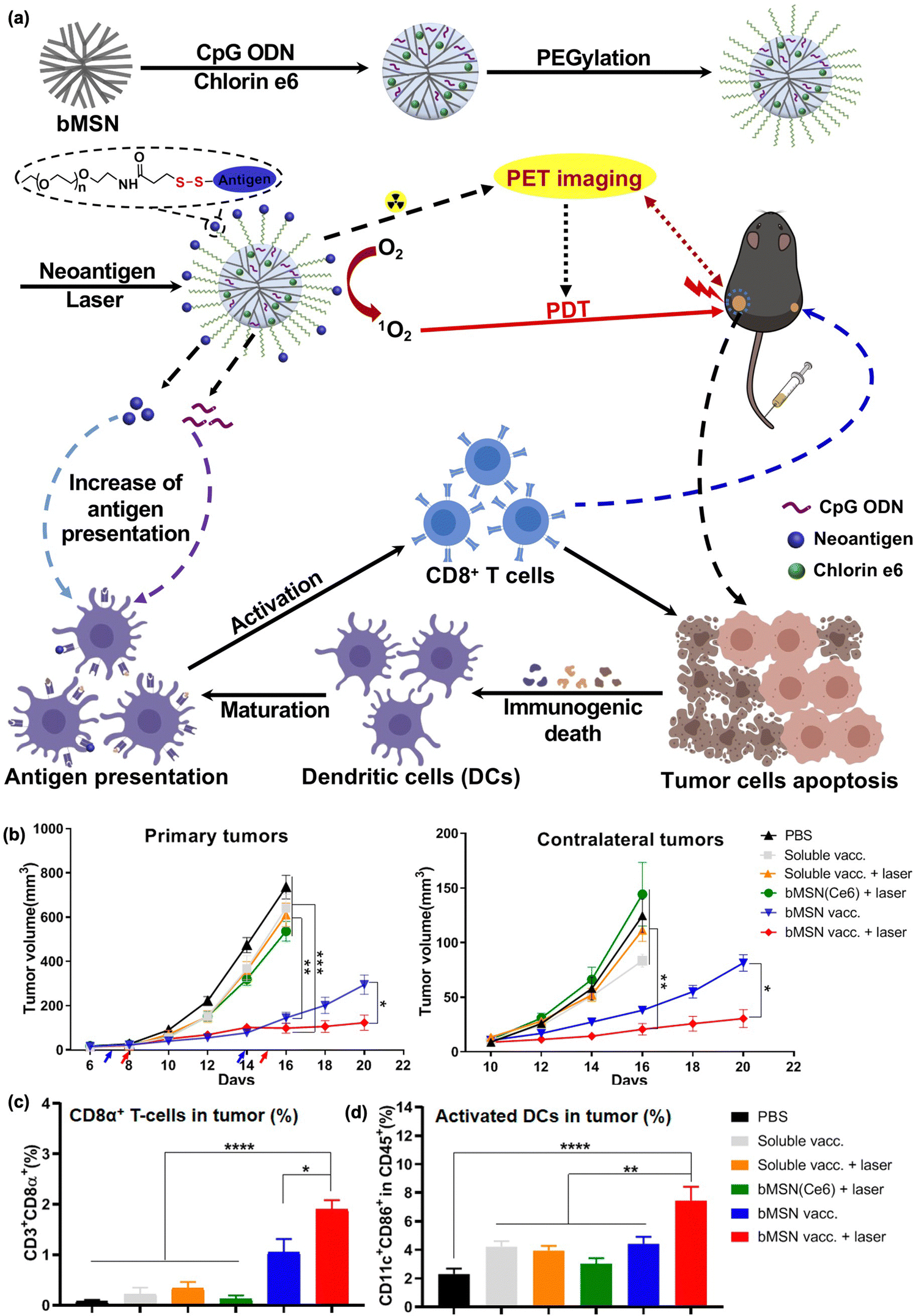 | ||
| Fig. 5 (a) Schematic illustration for the synthesis of bMSN nanovaccines and the mechanism of therapeutic action. (b) Average primary and contralateral tumor growth in each group. Percentage of (c) CD3 + CD8α+ T cells and (d) CD11c + CD86 + DCs. Reproduced with permission from ref. 142. Copyright @ 2024, American Chemical Society. | ||
Immune checkpoint inhibitors have also been used in several studies for PDT-combined immunotherapy. Lin et al. combined polymeric core–shell NPs containing a photosensitizer pyropheophorbide–lipid (pyrolipid) conjugate and PD-L1 blockade to treat MC38 and CT26 bilateral tumor models.143 PDT using pyrolipid NPs induced strong ICD in CT26 cells, which resulted in an effective vaccination against cancer cells. Although pyrolipid NPs alone effectively inhibited tumor growth under light irradiation, their combination with the PD-L1 blockade reduced tumor size by 2.9% compared to the control group. Furthermore, distant tumors that did not receive local light irradiation were almost eliminated. This abscopal effect can be attributed to the infiltration of CD8+ T cells by the PD-L1 blockade. Lin et al. also demonstrated that pyrolipid-loaded Zn-pyrophosphate (ZnP) NPs can kill tumor cells with light irradiation directly by inducing apoptosis and necrosis, followed by disrupting the tumor vasculature and increasing tumor immunogenicity.144 The anti-tumor therapeutic performance was enhanced after co-treatment with the PD-L1 blockade, as the primary and untreated distant tumors were eradicated by inducing tumor-specific cytotoxic T cell responses.153,154 These studies proved that NP-mediated PDT can be synergized with checkpoint blockade immunotherapy by activating tumor-specific innate and adoptive immune systems.
The oxygen-deficient (hypoxic) microenvironment of tumors can limit the effective use of PDT, which uses oxygen for cell death. Moreover, oxygen consumption induced by PDT further increases tumor hypoxia, which can lead to cancer recurrence and progression through the activation of various angiogenic factors, significantly impeding therapeutic outcomes. Therefore, it is of great interest to develop PDT agents that can provide oxygen to hypoxic tumor microenvironments. Liu et al. reported hydrogel-based PDT agents that were gelated in situ through light irradiation.148 Catalase (CAT)-conjugated chlorin e6 (Ce6), a photosensitizer capable of decomposing H2O2 into oxygen and water, was combined with a polymeric matrix comprising poly(ethylene glycol) double acrylate (PEGDA) and imiquimod (R837)-loaded poly(lactic-co-glycolic acid) (PLGA). After injecting this mixture into the tumor site, polymerization was initiated by light-triggered ROS generation by Ce6. The hydrogel produces oxygen by decomposing the tumor endogenous H2O2 with the gel-retained CAT, which improved the PDT efficacy substantially. Furthermore, the immune response can be activated through DC maturation by recognizing R837, a TLR7 agonist. The in situ gelation of Ce6-CAT/PEGDA resulted in ICDs, inducing ICD infiltration into tumors, while the hypoxic conditions of the tumor created a reversed immunosuppressive tumor microenvironment, enhancing the therapeutic performance of PDT and immunotherapy. Combination therapy with the CTLA-4 blockade effectively inhibited primary and distant tumor growth, with a memory effect. Increased amounts of effector memory T cells in the spleen and IFN-γ generated an immune response. Additional details of the checkpoint blockade combination therapies are provided in Table 2.
Indoleamine 2,3-dioxygenase (IDO1 and IDO2) is an intracellular enzyme that catalyzes the conversion of tryptophan into kynurenine.155 Kynurenine produced by IDO is a potent suppressor of CD8+ effector T and NK cells, while inducing the activation of Treg and myeloid-derived suppressor cells (MDSCs).156,157 In addition, IDO is known to be resistant to immune checkpoint inhibitors.158 Therefore, many IDO inhibitors have been developed for effective cancer immunotherapy and are currently undergoing preclinical and clinical trials. Recently, various IDO inhibitors have been used in combination with PDT. Zhang et al. reported primary and lung metastasis therapies based on PDT combined with the IDO1 inhibitor 1-methyltryptophan (1MT).149 As shown in Fig. 6(a) and (b), the PpIX photosensitizer was linked to 1MT by bridging the casepase-3 substrate peptide (PpIX-1MT). After light irradiation, the photosensitizer produces reactive oxygen species that induce the apoptotic death of cancer cells, followed by an increase in caspase-3 activation and the production of tumor antigens. Subsequently, 1MT is released through caspase-3 substrate cleavage, thereby activating the CD8+ T cells and immune responses. Upon irradiation, PpIX-1MT yielded a higher ratio of CD3 + CD8+ effective T cells to CD3 + CD4+ Tregs than the other groups after IDO blockade (Fig. 6(c)). In addition, immune-activated factors such as TNF-α, IFN- γ, IL-17, CD-8, CD-86, and granzyme-B were upregulated with PpIX-1MT under light irradiation, while immune-suppressive IL-10 was downregulated in both primary and lung tumors (Fig. 6(d)). Other studies have shown the therapeutic potential in different tumor models or the abscopal effect of PDT combined with IDO inhibition, highlighting the potential of IDO inhibitors for highly efficient photodynamic immunotherapy.150,151,159
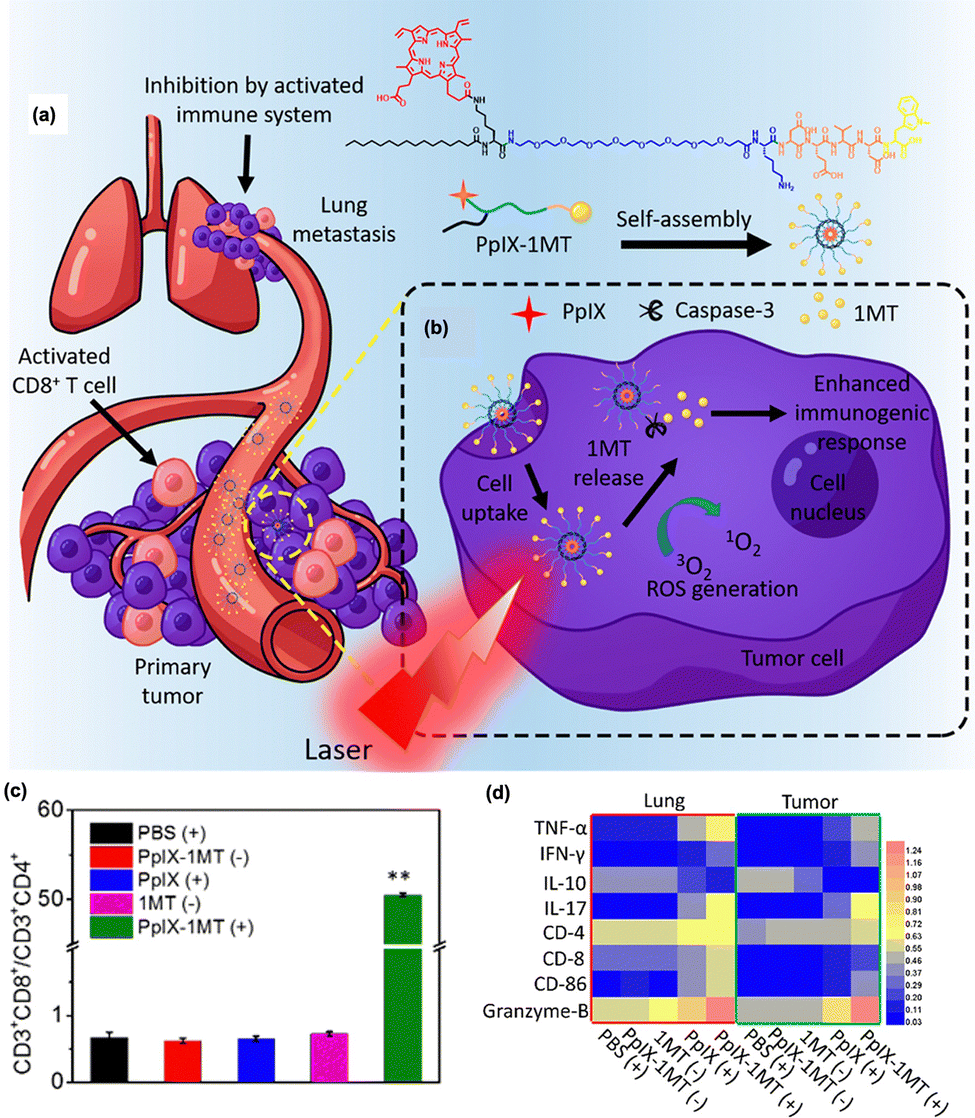 | ||
| Fig. 6 (a) Structure of PpIX-1MT and (b) therapeutic mechanism involving a series of cascade activations to inhibit primary tumor and lung metastasis. (c) Ratio of CD3 + CD8+ T cells to CD3 + CD4+ T cells after treatment, showing higher CD8+ effector T cell contents and lower CD4+ regulatory T cell contents due to the IDO inhibitor. (d) Relative values of the immune-associated factors determined by western blot within the tumor and lung. “(+)” and “(−)” represent the sample with and without light irradiation. Reproduced with permission from ref. 149. Copyright@ 2024, American Chemical Society. | ||
As described above, the combination of PDT with immunotherapy has been actively studied to achieve enhanced therapeutic outcomes. It is noteworthy that PDT, presented as an alternative for advanced cancer patients, has demonstrated biosafety, as evidenced by the FDA approval of the photosensitizer photofrin. Consequently, the combination of clinically pertinent photosensitizer-based PDT with immunotherapy holds the potential to yield clinical advantages, leading to efficacious therapeutic outcomes. One limitation of PDT-related therapy is that light cannot pass through deep tissue, which restricts the use of PDT in tumors under the skin or on the lining of internal organs or cavities.160 Moreover, PDT is less effective in treating large tumors, as light sources cannot cover the all parts of the tumors.160,161 For these reasons, PDT is usually considered as a local treatment. To overcome these limitations, it is important to combine PDT with immunotherapy to stimulate the immune system and improve the natural ability of the immune system to enhance the effectiveness of PDT.
4. Photothermal therapy-combined immunotherapy
4.1 Immunogenic response of photothermal therapy
Photothermal therapy (PTT) is another photo-triggered therapeutic method that uses materials that can absorb and convert NIR light energy to thermal energy, leading to cancer cell death with hyperthermia. Conventional PTT, necessitating hyperthermia exceeding 50 °C, frequently results in undesired damage to the surrounding healthy tissues or recruited immune cells. In contrast, the emerging mild-temperature PTT, with a temperature range from 38 °C to 43 °C, shows great potential with much lower phototoxicity to neighboring normal tissues.162 Heat ablation mainly changes the cell membrane integrity, resulting in the dysfunction of the cell cytoskeleton, thereby leading to cell necrosis and apoptosis and the release of tumor-associated antigens in the central zone where the laser is directly irradiated.163 In the surrounding peripheral zone, the induction of heat shock proteins (HSPs) plays a major role in stimulating the anti-tumor immune response.164,165 The HSP content, especially HSP70 and HSP72, on the cancer cell surface increases in response to stress, i.e., the unfolded protein response (UPR), after heat treatment, followed by recruitment of NK cells as well as DC maturation and activation of tumor cell-killing cytotoxic CD8+ T cells. In addition, HSP is also released from cancer cells as HSP alone, HSP-tumor antigen complexes, and HSP exosomes, all of which can induce activation and attract DCs and subsequently prime cytotoxic T cells by interacting with various surface receptors of APCs, including TLR, CD40, and CD91.166 In essence, the localized photothermal effect can induce an anti-tumor immune response through the expression and release of HSP, NK cell recognition, and activation of DCs and cytotoxic T cells.4.2 Photothermal therapy-combined immunotherapy
Since its development by Chen et. al in 1997, PTT-combined immunotherapy has been known as a laser immunotherapy method, which involves using a laser dye for photothermal destruction and an immunoadjuvant for immune stimulation.167 In this initial report, indocyanine green (ICG) dye was used as a PTT agent in combination with glycated chitosan (GC) as an immunoadjuvant. ICG-GC co-administration successfully eradicated tumors and increased resistance to tumor rechallenge by enhancing the immune response, which resulted from PTT-induced tumor antigen exposure, subsequent recognition of the exposed tumor antigen by GC, and an enhanced immunological effect on the treated and metastatic tumors.30,167,168 They also combined ICG-mediated PTT with imiquimod as an immunoadjuvant for clinical studies of metastatic melanoma treatment, and showed ICG–imiquimod therapy to be a safe and useful method with promising therapeutic outcomes.169 This pioneering work demonstrated that PTT-combined immunotherapy is potentially a fruitful arena for effective cancer therapy.Recently, several studies combining PTT with immunotherapy studies have been conducted using nanotechnology (Table 3). The combination of immunoadjuvants, such as GC, GM-CSF, CpG-ODN, ovalbumin (OVA) and PTT have shown enhanced anti-cancer activities.170–174 Xing et al. combined GC immunostimulators and carbon nanotube (CNT)-mediated PTT to treat murine mammary carcinoma.170 CNTs are NIR-absorbing carbon materials that exhibit dimensional advantages across cell membranes without any cytotoxicity.175 After the application of this combined method, GC delivered into tumor cells serves as both pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), resulting in an increase in the immunogenicity of tumors and APC activation. Both primary and secondary tumors were thoroughly eradicated.
| NPs | Immunotherapeutic agents | Advantages | Therapeutic effects | Ref. |
|---|---|---|---|---|
| Carbon nanotubes | GC | Ease of cell internalization and protein delivery, NIR active optical property | Eradication of both primary and secondary tumors in EMT6 mouse models with higher DAMPs and PAMPs and enhanced immunogenecity | 170 |
| Cationic graphene oxide | CpG-ODN | Ease of negative CpG ODN delivery | Synergistic therapeutic effects in the CT26 mouse model | 171 |
| Chitosan-coated hollow CuS NPs | CpG-ODN | Biodegradable and NIR absorbing NPs | Suppression of primary and distant tumors in the EMT6 mouse model by DC maturation and CD8+ T cell activation, and subsequent immune response | 172 |
| Au NPs-CpG-ODN hydrogel | CpG-ODN | Facile loading and release of CpG-ODN | Suppression of primary tumor in the EG7-OVA mouse model via enhanced HSP70 expression and immune response | 173 |
| OVA-ICG mixture nanovaccine | OVA | Simple fabrication, high antigen loading efficiency | Suppression of primary tumor and rechallenging in the B16 mouse model | 174 |
| PLGA-ICG-R837 | Imiquimod, CTLA-4 inhibitor | Biodegradable polymer | Suppression of secondary tumor and rechallenging in 4T1 and CT26 mice models by DC maturation and blocking action of Treg cells | 176 |
| Au nanorods-R837 | Imiquimod, PD-1 inhibitor | Biocompatible NPs | Suppression of primary tumor and rechallenging by enhanced CD8+ T cell infiltration | 177 |
| Fe3O4-R837 | Imiquimod, PD-L1 inhibitor | Magnetic targeting and MR imaging active NPs | Suppression of primary and secondary tumors through DC maturation | 178 |
| Inhibition of metastasis by NK cells, B cells, CD8+ T cells, and CD4+ T cells in distant tumors | ||||
| NGL919/IR780 micelles | IDO inhibitor (NGL919) | — | Suppression of primary and secondary tumors | 179 |
| Upregulation of CD8+ T cells and downregulation of Treg cells | ||||
| rGO | IDO inhibitor (epacadostat), PD-L1 inhibitor | High loading efficiency of the IDO inhibitor | Inhibition of primary and secondary tumor growth in the CT26 mouse model | 180 |
| Increase in CD45+ leukocytes, CD4+ T cells, CD8+ T cells and decrease in Treg cell infiltration in distant tumors |
Meaningful results were reported by Lu et al., in which hollow CuS NPs and CpG-ODNs were used as photothermal and immunostimulating agents, respectively.172 Hollow CuS NPs broke down after laser irradiation, before reassembling and transforming into polymeric structures, leading to enhanced tumor retention and therapeutic efficacy. PTT induced tumor cell death and the release of tumor antigens, whereas the CpG immunoadjuvant triggered the activation of NK cells and DCs. Furthermore, IFN-γ production in the tumor and spleen was significantly increased, resulting in successful anti-tumor immunity. Hollow CuS- and CpG-ODN-mediated photothermal immunotherapy elicits effective systemic anti-tumor immune responses that can eradicate primary and secondary tumors.
Although various types of nanomaterial-based vaccines have been developed for combined immunotherapy, their clinical use is limited owing to their low antigen loading efficiency, low yield, complex fabrication steps, and high cytotoxicity. Sun et al. reported a simple synthetic method to prepare natural carriers loaded with ICG in high loading and high yields.174 OVA, which is immunogenic, was used as a model antigen. OVA-ICG NPs were prepared by simply mixing these two components, resulting in a high antigen-loading efficiency of over 80%, high yield, high reproducibility, and excellent biocompatibility. OVA-ICG induced high levels of MHC-II, CD80, and CD83 expression, indicating the maturation of DCs. Subsequently, matured DCs activate CD8+ cytotoxic T cells and initiate an immune response to eliminate primary tumors and prevent tumor rechallenge. These results demonstrated that PTT-based long-term anti-cancer efficacy could be supported by a combination of immunostimulating agents or adjuvants.
More rigorous and promising therapeutic studies have been conducted by combining PTT with certain types of immune checkpoint blockades.176 Liu et al. reported that PLGA-coated NPs containing ICG and imiquimod (R837) (PLGA-ICG-R837) achieved vaccine-like anti-tumor immunity and showed synergistic effects when PLGA-ICG-R837 was combined with a CTLA-4 blockade (Fig. 7(a)). After administration of NPs and tumor ablation, DC maturation was significantly increased by tumor destruction post-PTT, as well as by the immune-stimulating effect of the adjuvant imiquimod (Fig. 7(b)). In addition, immune activation was observed in secondary tumors via a substantial increase in CD8+ CTL and CD4+ effector T cells by the CTLA-4 blockade, which reduced the number of Tregs (Fig. 7(c)). Based on these mechanisms, this strategy successfully inhibited secondary tumor growth and rechallenge after the primary tumor treatment (Fig. 7(d)).
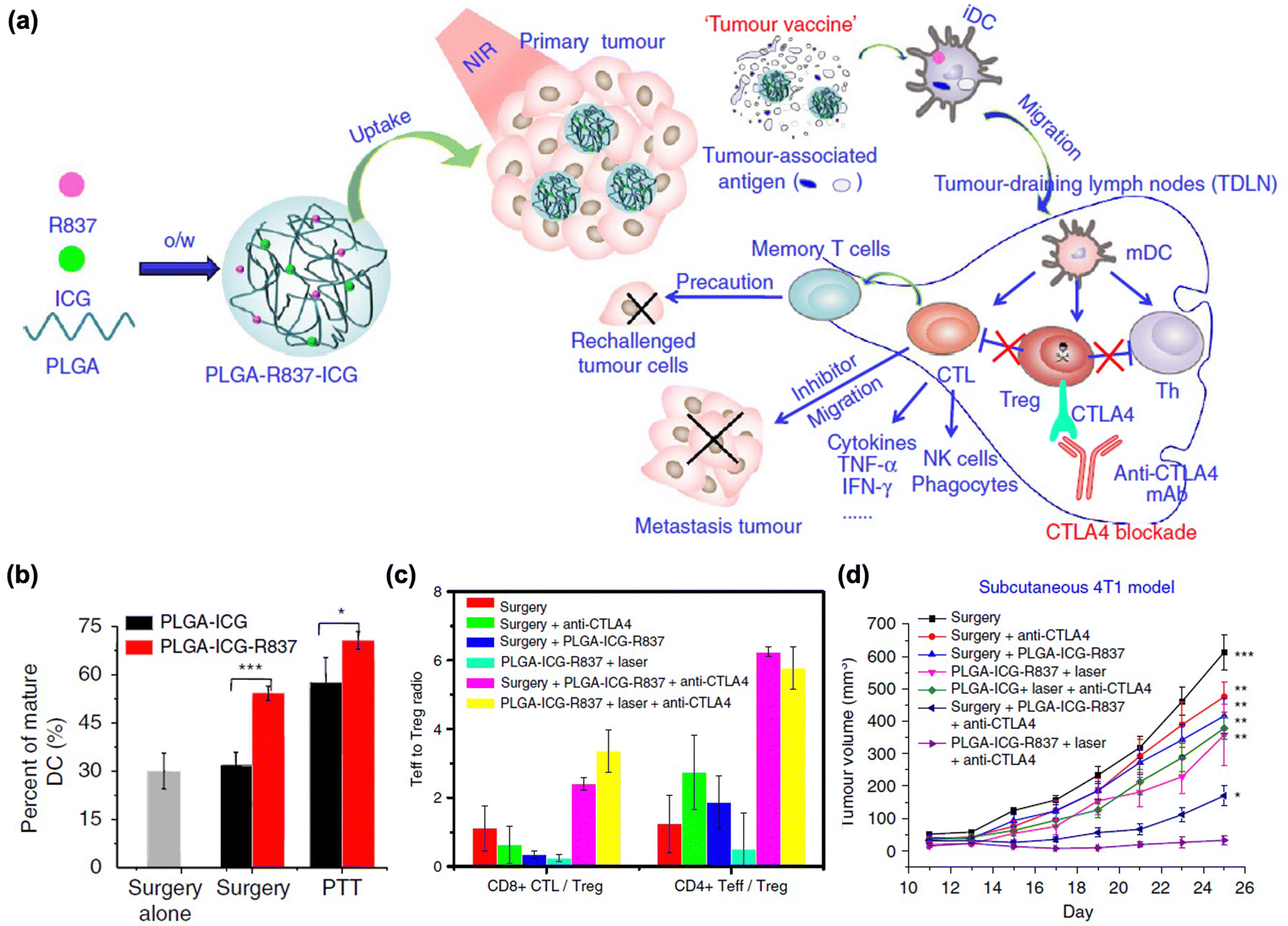 | ||
| Fig. 7 (a) The mechanism of anti-tumour immune responses induced by PLGA-ICG-R837-based PTT in combination with the checkpoint-blockade. (b) DC maturation after PLGA-ICG-R837 treatment. (c) CD8+ CTL: Treg ratios and CD4+ effector T cells: Treg ratios in the secondary tumours upon various treatments to remove the first tumours. (d) Tumour growth curves of different groups of mice. Reproduced with permission from ref. 176. Copyright @ 2024, Nature Publishing Group. | ||
Recently, the regulation of T cell metabolism has attracted considerable attention as a target of immunotherapy.181 As mild heating resulting from local light irradiation could upregulate tumor cell protective and T cell suppressive proteins related to T cell metabolism after PTT, inhibiting these T cell metabolite proteins could enhance the immune response to PTT. In this regard, IDO inhibition has been utilized to further improve the therapeutic effect in combination with PTT.179,182–185 Ma et al. used an IDO inhibitor (epacadostat)-loaded PEG-coated rGO in combination with a PD-L1 blockade inhibitor (Fig. 8(a)).180 Here, they observed that tumor ablation led to the maturation of the DCs. Furthermore, with the aid of an IDO inhibitor, this method showed higher IFN-γ, CD4+ T cell and CD8+ T cell contents, while inhibiting the Treg function. In addition, these immune responses were significantly augmented in distant tumors in combination with PD-L1 blockade therapy (Fig. 8(b)–(f)).
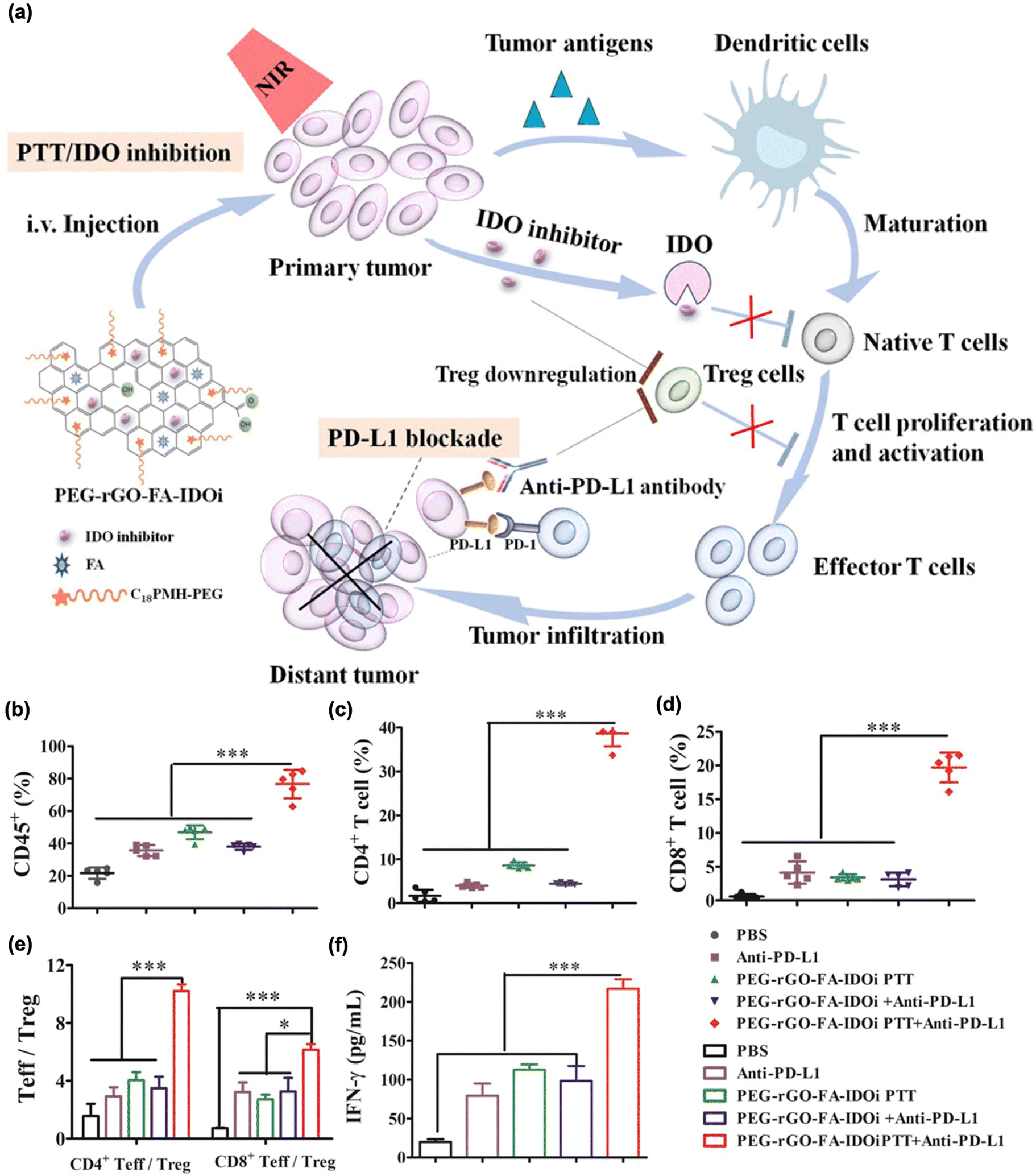 | ||
| Fig. 8 (a) The mechanism of anti-tumour immune responses induced by PEG-rGO-FA-IDOi-based PTT in combination with checkpoint-blockades. Percentage of (b) CD45+ leukocytes, (c) CD3 + CD4+ T cells, (d) CD3 + CD8+ T cells, (e) CD3 + CD4+ Foxp+ effector T cells (CD4+ Teff): CD3 + CD4+ Foxp- regulator T cells (Treg) ratios and CD3 + CD8+ T cells: Treg ratios in distant tumors. (f) Production of INF-γ in serum was determined through ELISA. Reproduced with permission from ref. 180. Copyright © 2024, American Chemical Society. | ||
Overall, PTT provides anti-tumor immunity by killing primary tumor cells and releasing tumor antigens. However, long-term therapy has been limited to inducing memory effects to prevent tumor metastasis. In particular, mild local heating of tumors owing to the narrow or shallow penetration depth of NIR lasers results in not only a temporary immune response but also an increase in some proteins that can block T cell expansion. Therefore, combining PTT with immunotherapeutic agents such as checkpoint blockers, immunostimulators, and adjuvants is a promising strategy for effective anti-cancer therapy with long-term memory effects to prevent tumor rechallenge and metastasis.
5. PDT–PTT-combined immunotherapy
Integrated trimodal therapy, encompassing PDT, PTT, and immunotherapy, has been reported to enhance the anti-cancer efficacy. Given the previously discussed ability of PDT and PTT to stimulate immune responses via ICD, the synergistic combination of PTT and PDT is likely to activate long-term immunity more effectively compared to either PTT or PDT alone.186–188 Meng et al. proposed nanoparticles consisting of highly conjugated and donor–acceptor alternating molecular photosensitizer BTAC6.186 These BTAC6 NPs exhibited superb NIR absorption with enhanced intersystem crossing (ISC), resulting in efficient energy conversion into cytotoxic ROS and heat. From these properties, BTAC6 NPs activated immune cells, promoting therapeutic efficacy even in hypoxic tumors, and restricting tumor reccurence. Another recent article showed PDT/PTT/immunotherapy designed by Cui's group.189 The authors prepared a trimodal therapeutic nanoplatform by loading CaCO3-coated gold nanostars with the Ce6 photosensitizer (GNS@CaCO3/Ce6) into human NK cells. The three parts, namely, GNS for PTT, Ce6 for PDT, and NK cells for adoptive cellular immunotherapy, significantly inhibited primary tumor growth via increased secretion of cytokines including TNF-α, IFN-γ, IL-18, granzyme A, perforin, FASL, and TRAIL. Shi et al. reported Cu9S5-based nanoparticles decorated with CpG (CSPM@CpG).190 This strategy can enhance CpG uptake by NP penetration and subsequent production of immune cytokines, leading to the stimulation of the immune response. Liu et al. developed low molecular weight hyaluronic acid (HA) modified black phosphorous (BP) nanoparticles (HA-BP) as trimodal therapeutics.191 In tumor microenvironments, there are two types of tumor associated microphages (TAMs), anti-tumor M1 macrophages and pro-tumor M2 macrophages.192 It has been proven that low molecular HA can induce the conversion of TAMs to M1 macrophages as well as reverse TAMs from M2 into M1 macrophages to promote anti-tumor therapeutic efficacy.193,194 HA-BP showed good photothermal and photodynamic activity, and great potential in inducing M1 phenotype from TAMs rather than M2. Li's group constructed core–shell nanoparticles that contained IR780 and 2,2′-azobis[2-(2-imidazolin-2-yl)propane]-dihydrochloride (AIPH) encapsulated within a matrix metalloproteinase (MMP)-sensitive PLGVRGC-anti-PD-L1 peptide shell (Fig. 9(a)).195 IR780, AIPH, and peptide serve as the PTT agent, oxygen-independent PDT agent, and anti-PD-L1 precursor for immune checkpoint blockades, respectively. IR780/AIPH with MMP-sensitive PLGVRGC-anti-PD-L1 peptide significantly inhibited primary and distant tumors compared with the control groups (Fig. 9(b) and (c)). This nanohybrid showed effective PTT-induced oxygen-independent free radical generation even under hypoxic primary tumor conditions, which led to ICD. Moreover, the anti-PD-L1 peptide, released subsequent to MMP-selective cleavage, functions as an antagonist against PD-L1 on cancer cells. This action further augments immune activation by facilitating the infiltration and activation of CD8+ T cells. Notably, the outcomes from the 4T1 xenograft mouse model showcase significant inhibition of both primary and distant tumors. These findings suggest that the proposed combination strategy holds substantial promise as an approach for PTT/PDT/immunotherapy in hypoxic tumors.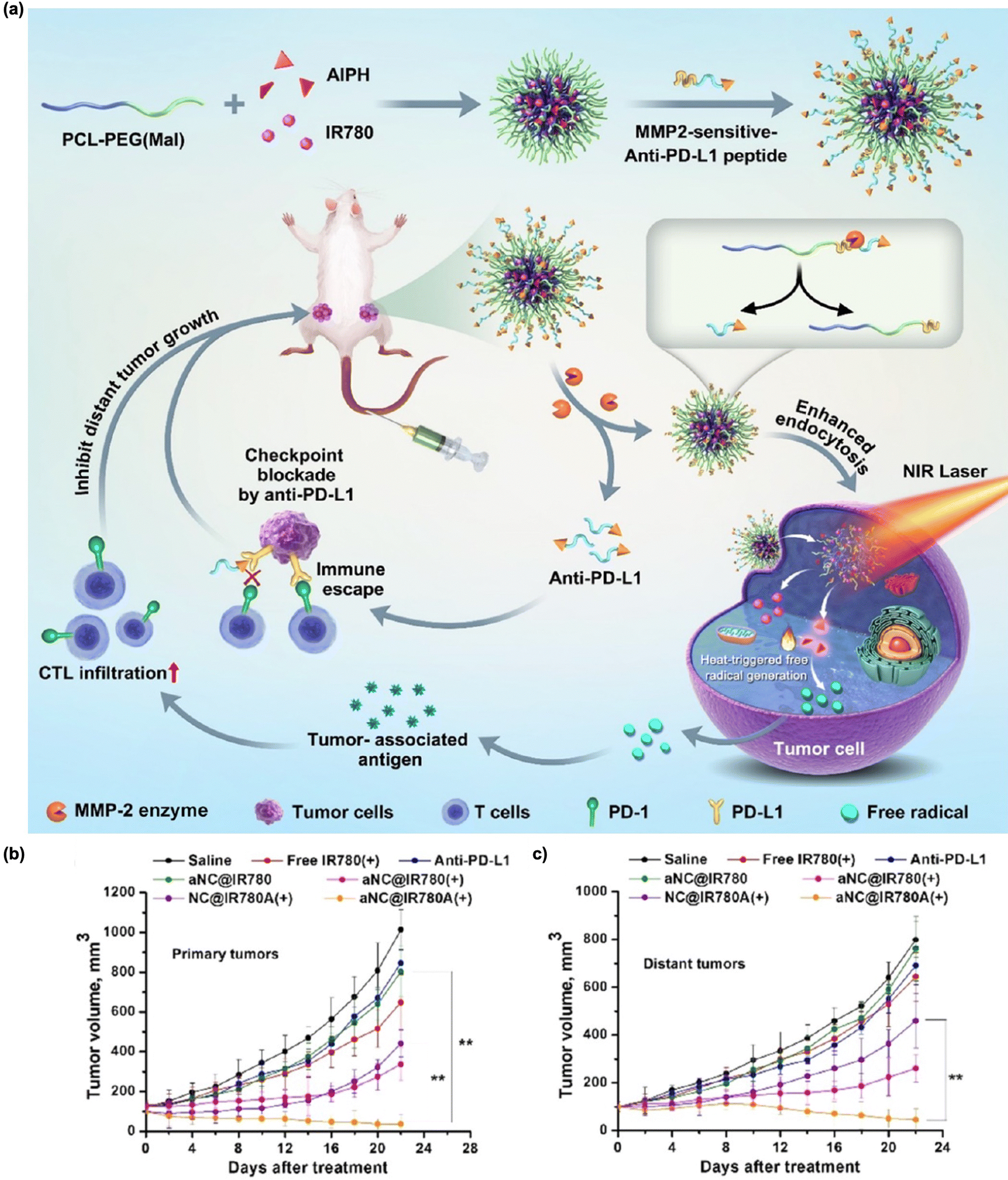 | ||
| Fig. 9 (a) Schematic illustration of IR780/AIPH nanocomposites encapsulated within an anti-PD-L1 peptide selectively cleaved by MMP for the treatment of primary hypoxic tumors and distant tumors. Average primary (b) and distant (c) tumor growth of each group. Reproduced with permission from ref. 195. Copyright©2024, American Chemical Society. | ||
Beyond the combination of PTT/PDT/immunotherapy, various studies are underway to explore the integration of PTT and PDT with other therapeutic modalities to achieve more effective cancer treatment and immunotherapy.23,196–198
6. Conclusions and perspectives
Phototherapy, PDT, and PTT can trigger anti-tumor immune responses during direct ROS- or heat-mediated cancer cell death. However, additional treatment modalities are required to achieve long-term therapeutic efficacy. Cancer immunotherapy activates the host immune system to kill primary tumors, thereby preventing tumor recurrence and metastasis through a long-term memory immune response. Hence, the combination of phototherapy and immunotherapy may be a good candidate for effective cancer treatment. Particularly, photoimmunotherapy (PIT) and NIR-PIT can provide disease-specific therapeutic outcomes by activating the immune system. With the aid of immunostimulators and checkpoint inhibitors, enhanced anti-tumor immunity and therapeutic efficacy against primary and distant tumors have been achieved using PDT and PTT in various tumor models. In particular, NP-based combination therapy provides advantages in specific functions, such as tumor-specific delivery, multimodality, high loading of therapeutics, and stimuli-responsive release of cargo.Although light-triggered cancer immunotherapy has shown high treatment efficacy and synergistic effects, several issues and challenges remain to be addressed. First, most of the current studies use multiple components or treatments for each therapy. The therapeutic components do not have the same formula when combined with an immune checkpoint inhibitor. Several steps are required to achieve therapeutic outcomes and incorrect delivery or unwanted side reactions remain issues to be addressed. Therefore, it is necessary to design a photoimmunotherapeutic platform in which each therapeutic element is well arranged and released under tumor-specific stimuli, leading to an appropriate cascade immune-stimulating reaction. Second, the challenge of deep tissue penetration should be addressed because phototherapies are limited to treating only superficial cancers owing to the low penetration depth of the light source. There have been a few recent studies on enhancing the phototherapy efficiency with improved tissue penetration.199 Gao et al. reported the treatment of orthotopic malignant glioblastoma using NIR (NIR-II) photoactive nanomaterials.200 NIR-II light ranging from 1000 to 1700 nm is superior to NIR-I light for deep tissue phototherapy. Meng et al. demonstrated the direct transarterial administration of MoS2-based PTT agents to efficiently eliminate orthotopic liver tumors.201 However, further studies on deep-tissue phototherapy are necessary. Third, the mechanism of immune activation through combination therapy is yet to be fully understood. Multiple complex immune responses against cancer must be established in future clinical studies. Finally, phototherapeutic elements and nanoplatforms for combination therapy should be optimized for therapeutic efficacy, stability, biodistribution, and safety in further clinical applications.
Author contributions
Writing – original draft preparation, writing, and preparing the final version of the manuscript, J. K. Yang; literature analysis and collection of references, H. Kwon; writing – review and editing and supervision, S. H. Kim. All the authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the grants received from the National Research Foundation of Korea (2021R1A2C2005418, and 2022M3H4A1A03076638) and the KIST intramural program. This work was supported by the Dong-eui University grant (202301350001).References
- R. L. Siegel, K. D. Miller, S. A. Fedewa, D. J. Ahnen, R. G. Meester, A. Barzi and A. Jemal, Ca-Cancer J. Clin., 2017, 67, 177–193 CrossRef PubMed.
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2016, 66, 7–30 CrossRef PubMed.
- S. Valastyan and R. A. Weinberg, Cell, 2011, 147, 275–292 CrossRef CAS PubMed.
- S. A. Rosenberg, J. C. Yang and N. P. Restifo, Nat. Med., 2004, 10, 909–915 CrossRef CAS PubMed.
- I. Mellman, G. Coukos and G. Dranoff, Nature, 2011, 480, 480–489 CrossRef CAS PubMed.
- A. Ribas and J. D. Wolchok, Science, 2018, 359, 1350–1355 CrossRef CAS PubMed.
- H. Wang and D. J. Mooney, Nat. Mater., 2018, 17, 761–772 CrossRef CAS PubMed.
- S. Farkona, E. P. Diamandis and I. M. Blasutig, BMC Med., 2016, 14, 73 CrossRef PubMed.
- D. S. Chen and I. Mellman, Immunity, 2013, 39, 1–10 CrossRef CAS PubMed.
- J. S. O’Donnell, M. W. Teng and M. J. Smyth, Nat. Rev. Clin. Oncol., 2019, 16, 151–167 CrossRef PubMed.
- P. Sharma, K. Wagner, J. D. Wolchok and J. P. Allison, Nat. Rev. Cancer, 2011, 11, 805–812 CrossRef CAS PubMed.
- K. Yaddanapudi, R. A. Mitchell and J. W. Eaton, OncoImmunology, 2013, 2, e23403 CrossRef PubMed.
- D. E. Gilham, J. Anderson, J. S. Bridgeman, R. E. Hawkins, M. A. Exley, H. Stauss, J. Maher, M. Pule, A. K. Sewell and G. Bendle, Hum. Gene Ther., 2015, 26, 276–285 CrossRef CAS PubMed.
- S. A. Rosenberg, N. P. Restifo, J. C. Yang, R. A. Morgan and M. E. Dudley, Nat. Rev. Cancer, 2008, 8, 299–308 CrossRef CAS PubMed.
- X. Qian, X. Wang and H. Jin, J. Immunol. Res., 2014, 2014, 525913 Search PubMed.
- D. J. Byun, J. D. Wolchok, L. M. Rosenberg and M. Girotra, Nat. Rev. Endocrinol., 2017, 13, 195 CrossRef CAS PubMed.
- F. S. Hodi, S. J. O’Day, D. F. McDermott, R. W. Weber, J. A. Sosman, J. B. Haanen, R. Gonzalez, C. Robert, D. Schadendorf and J. C. Hassel, N. Engl. J. Med., 2010, 363, 711–723 CrossRef CAS PubMed.
- J. F. Grosso and M. N. Jure-Kunkel, Cancer Immunity Archive, 2013, 13, 5 Search PubMed.
- J. R. Brahmer, S. S. Tykodi, L. Q. Chow, W.-J. Hwu, S. L. Topalian, P. Hwu, C. G. Drake, L. H. Camacho, J. Kauh and K. Odunsi, N. Engl. J. Med., 2012, 366, 2455–2465 CrossRef CAS PubMed.
- M. Sade-Feldman, Y. J. Jiao, J. H. Chen, M. S. Rooney, M. Barzily-Rokni, J.-P. Eliane, S. L. Bjorgaard, M. R. Hammond, H. Vitzthum and S. M. Blackmon, Nat. Commun., 2017, 8, 1–11 CrossRef CAS PubMed.
- M. Pollack, A. Betof, H. Dearden, K. Rapazzo, I. Valentine, A. Brohl, K. Ancell, G. Long, A. Menzies and Z. Eroglu, Ann. Oncol., 2018, 29, 250–255 CrossRef CAS PubMed.
- R. Costa, R. B. Costa, S. M. Talamantes, I. Helenoswki, B. A. Carneiro, Y. K. Chae, W. J. Gradishar, R. Kurzrock and F. J. Giles, Oncotarget, 2017, 8, 67782 CrossRef PubMed.
- W. Sang, Z. Zhang, Y. Dai and X. Chen, Chem. Soc. Rev., 2019, 48, 3771–3810 RSC.
- S. K. Rajendrakumar, S. Uthaman, C.-S. Cho and I.-K. Park, Biomacromolecules, 2018, 19, 1869–1887 CrossRef CAS PubMed.
- X. Hou, Y. Tao, Y. Pang, X. Li, G. Jiang and Y. Liu, Int. J. Cancer, 2018, 143, 3050–3060 CrossRef CAS PubMed.
- H. Kobayashi and P. L. Choyke, Acc. Chem. Res., 2019, 52, 2332–2339 CrossRef CAS PubMed.
- H. S. Hwang, H. Shin, J. Han and K. Na, J. Pharm. Invest., 2018, 48, 143–151 CrossRef CAS PubMed.
- J. Nam, S. Son, K. S. Park, W. Zou, L. D. Shea and J. J. Moon, Nat. Rev. Mater., 2019, 4, 398–414 CrossRef.
- M. Korbelik, J. Clin. Laser. Med. Sur., 1996, 14, 329–334 CrossRef CAS PubMed.
- W. R. Chen, W. G. Zhu, J. R. Dynlacht, H. Liu and R. E. Nordquist, Int. J. Cancer, 1999, 81, 808–812 CrossRef CAS.
- A. P. Castano, P. Mroz and M. R. Hamblin, Nat. Rev. Cancer, 2006, 6, 535–545 CrossRef CAS PubMed.
- J. Nam, S. Son, L. J. Ochyl, R. Kuai, A. Schwendeman and J. J. Moon, Nat. Commun., 2018, 9, 1–13 CrossRef CAS PubMed.
- J.-L. Boulnois, Lasers Med. Sci., 1986, 1, 47–66 CrossRef.
- S. L. Jacques, Surg. Clin. North Am., 1992, 72, 531–558 CrossRef CAS PubMed.
- N.-C. Jung, H. J. Kim, M.-S. Kang, J.-H. Lee, J.-Y. Song, H. G. Seo, Y.-S. Bae and D.-S. Lim, Cancer Lett., 2012, 324, 58–65 CrossRef CAS PubMed.
- M. Mitsunaga, M. Ogawa, N. Kosaka, L. T. Rosenblum, P. L. Choyke and H. Kobayashi, Nat. Med., 2011, 17, 1685 CrossRef CAS PubMed.
- C. Du, X. Wu, M. He, Y. Zhang, R. Zhang and C.-M. Dong, J. Mater. Chem. B, 2021, 9, 1478–1490 RSC.
- D. E. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS PubMed.
- S. B. Brown, E. A. Brown and I. Walker, Lancet Oncol., 2004, 5, 497–508 CrossRef CAS PubMed.
- J. Moan and Q. Peng, Anticancer Res., 2003, 23, 3591–3600 Search PubMed.
- F. H. van Duijnhoven, R. I. Aalbers, J. P. Rovers, O. T. Terpstra and P. J. Kuppen, Immunobiology, 2003, 207, 105–113 CrossRef CAS PubMed.
- Y. N. Konan, R. Gurny and E. Allémann, J. Photochem. Photobiol., B, 2002, 66, 89–106 CrossRef CAS PubMed.
- M. Ochsner, J. Photochem. Photobiol., B, 1997, 39, 1–18 CrossRef CAS PubMed.
- G. Van Dongen, G. Visser and M. Vrouenraets, Adv. Drug Delivery Rev., 2004, 56, 31–52 CrossRef CAS PubMed.
- P. R. Ogilby, Chem. Soc. Rev., 2010, 39, 3181–3209 RSC.
- D. Kobzev, O. Semenova, A. Tatarets, A. Bazylevich, G. Gellerman and L. Patsenker, Dyes Pigm., 2023, 212, 111101 CrossRef CAS.
- E. Buytaert, M. Dewaele and P. Agostinis, Biochim. Biophys. Acta, Rev. Cancer, 2007, 1776, 86–107 CrossRef CAS PubMed.
- Z. Zhen, W. Tang, M. Wang, S. Zhou, H. Wang, Z. Wu, Z. Hao, Z. Li, L. Liu and J. Xie, Nano Lett., 2017, 17, 862–869 CrossRef CAS PubMed.
- D. Mew, V. Lum, C. Wat, G. Towers, C. C. Sun, R. Walter, W. Wright, M. Berns and J. Levy, Cancer Res., 1985, 45, 4380–4386 CAS.
- A. R. Oseroff, D. Ohuoha, T. Hasan, J. C. Bommer and M. L. Yarmush, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 8744–8748 CrossRef CAS PubMed.
- S. Yemul, C. Berger, M. Katz, A. Estabrook, R. Edelson and H. Bayley, Cancer Immunol. Immunother., 1990, 30, 317–322 CrossRef CAS PubMed.
- B. Goff, U. Hermanto, J. Rumbaugh, J. Blake, M. Bamberg and T. Hasan, Br. J. Cancer, 1994, 70, 474–480 CrossRef CAS PubMed.
- W. P. Thorpe, M. Toner, R. M. Ezzell, R. G. Tompkins and M. L. Yarmush, Biophys. J., 1995, 68, 2198–2206 CrossRef CAS PubMed.
- M. R. Hamblin, J. L. Miller and T. Hasan, Cancer Res., 1996, 56, 5205–5210 CAS.
- M. B. Vrouenraets, G. W. Visser, F. A. Stewart, M. Stigter, H. Oppelaar, P. E. Postmus, G. B. Snow and G. A. Van Dongen, Cancer Res., 1999, 59, 1505–1513 CAS.
- M. B. Vrouenraets, G. W. Visser, M. Stigter, H. Oppelaar, G. B. Snow and G. A. Van Dongen, Cancer Res., 2001, 61, 1970–1975 CAS.
- M. Carcenac, M. Dorvillius, V. Garambois, F. Glaussel, C. Larroque, R. Langlois, N. Hynes, J. Van Lier and A. Pelegrin, Br. J. Cancer, 2001, 85, 1787–1793 CrossRef CAS PubMed.
- R. Hudson, M. Carcenac, K. Smith, L. Madden, O. Clarke, A. Pelegrin, J. Greenman and R. Boyle, Br. J. Cancer, 2005, 92, 1442–1449 CrossRef CAS PubMed.
- N. Shirasu, H. Yamada, H. Shibaguchi, M. Kuroki and M. Kuroki, Int. J. Cancer, 2014, 135, 2697–2710 CrossRef CAS PubMed.
- R. Weissleder and V. Ntziachristos, Nat. Med., 2003, 9, 123–128 CrossRef CAS PubMed.
- S. Luo, E. Zhang, Y. Su, T. Cheng and C. Shi, Biomaterials, 2011, 32, 7127–7138 CrossRef CAS PubMed.
- E. Lukyanets, Near-Infrared Dyes for High Technology Applications, Springer, 1998, pp.307–324 Search PubMed.
- M. Mitsunaga, M. Ogawa, N. Kosaka, L. T. Rosenblum, P. L. Choyke and H. Kobayashi, Nat. Med., 2011, 17, 1685–1691 CrossRef CAS PubMed.
- M. Mitsunaga, T. Nakajima, K. Sano, P. L. Choyke and H. Kobayashi, Bioconjugate Chem., 2012, 23, 604–609 CrossRef PubMed.
- T. Nakajima, K. Sano, P. L. Choyke and H. Kobayashi, Theranostics, 2013, 3, 357 CrossRef CAS PubMed.
- M. Mitsunaga, T. Nakajima, K. Sano, G. Kramer-Marek, P. L. Choyke and H. Kobayashi, BMC Cancer, 2012, 12, 1–8 CrossRef PubMed.
- K. Sato, T. Nagaya, Y. Nakamura, T. Harada, P. L. Choyke and H. Kobayashi, Oncotarget, 2015, 6, 19747 CrossRef PubMed.
- K. Sato, T. Nagaya, M. Mitsunaga, P. L. Choyke and H. Kobayashi, Cancer Lett., 2015, 365, 112–121 CrossRef CAS PubMed.
- K. Sato, H. Hanaoka, R. Watanabe, T. Nakajima, P. L. Choyke and H. Kobayashi, Mol. Cancer Ther., 2015, 14, 141–150 CrossRef CAS PubMed.
- T. Nagaya, K. Sato, T. Harada, Y. Nakamura, P. L. Choyke and H. Kobayashi, PLoS One, 2015, 10, e0136829 CrossRef PubMed.
- K. Ito, M. Mitsunaga, T. Nishimura, H. Kobayashi and H. Tajiri, Oncotarget, 2016, 7, 14143 CrossRef PubMed.
- J. Jin, B. Krishnamachary, Y. Mironchik, H. Kobayashi and Z. M. Bhujwalla, Sci. Rep., 2016, 6, 27871 CrossRef CAS PubMed.
- K. Ito, M. Mitsunaga, T. Nishimura, M. Saruta, T. Iwamoto, H. Kobayashi and H. Tajiri, Bioconjugate Chem., 2017, 28, 1458–1469 CrossRef CAS PubMed.
- M. Ogawa, Y. Tomita, Y. Nakamura, M.-J. Lee, S. Lee, S. Tomita, T. Nagaya, K. Sato, T. Yamauchi and H. Iwai, Oncotarget, 2017, 8, 10425 CrossRef PubMed.
- Q. Tang, T. Nagaya, Y. Liu, J. Lin, K. Sato, H. Kobayashi and Y. Chen, J. Controlled Release, 2017, 260, 154–163 CrossRef CAS PubMed.
- F. Ogata, T. Nagaya, Y. Nakamura, K. Sato, S. Okuyama, Y. Maruoka, P. L. Choyke and H. Kobayashi, Oncotarget, 2017, 8, 35069 CrossRef PubMed.
- T. Nagaya, A. P. Gorka, R. R. Nani, S. Okuyama, F. Ogata, Y. Maruoka, P. L. Choyke, M. J. Schnermann and H. Kobayashi, Mol. Cancer Ther., 2018, 17, 661–670 CrossRef CAS PubMed.
- Y. Maruoka, T. Nagaya, K. Sato, F. Ogata, S. Okuyama, P. L. Choyke and H. Kobayashi, Mol. Pharmaceutics, 2018, 15, 3634–3641 CrossRef CAS PubMed.
- T. Nagaya, J. Friedman, Y. Maruoka, F. Ogata, S. Okuyama, P. E. Clavijo, P. L. Choyke, C. Allen and H. Kobayashi, Cancer Immunol. Res., 2019, 7, 401–413 CrossRef CAS PubMed.
- K. Sato, K. Ando, S. Okuyama, S. Moriguchi, T. Ogura, S. Totoki, H. Hanaoka, T. Nagaya, R. Kokawa and H. Takakura, ACS Cent. Sci., 2018, 4, 1559–1569 CrossRef CAS PubMed.
- M. Korbelik, J. Sun and I. Cecic, Cancer Res., 2005, 65, 1018–1026 CrossRef CAS PubMed.
- S. Todryk, A. A. Melcher, N. Hardwick, E. Linardakis, A. Bateman, M. P. Colombo, A. Stoppacciaro and R. G. Vile, J. Immunol., 1999, 163, 1398–1408 CrossRef CAS.
- G. Kick, G. Messer, A. Goetz, G. Plewig and P. Kind, Cancer Res., 1995, 55, 2373–2379 CAS.
- W. J. de Vree, M. C. Essers, J. F. Koster and W. Sluiter, Cancer Res., 1997, 57, 2555–2558 CAS.
- P. Mroz, A. Szokalska, M. X. Wu and M. R. Hamblin, PLoS One, 2010, 5, e15194 CrossRef CAS PubMed.
- G. Cantl, D. Lattuada, A. Nicolin, P. Taroni, G. Valentinl and R. Cubeddu, Anti-Cancer Drugs, 1994, 5, 443–447 CrossRef PubMed.
- S. O. Gollnick and C. M. Brackett, Immunol. Res., 2010, 46, 216–226 CrossRef CAS PubMed.
- P. Mroz, J. T. Hashmi, Y.-Y. Huang, N. Lange and M. R. Hamblin, Expert Rev. Clin. Immunol., 2011, 7, 75–91 CrossRef CAS PubMed.
- P. Mroz and M. R. Hamblin, Photochem. Photobiol. Sci., 2011, 10, 751–758 CrossRef CAS PubMed.
- C. A. Elmets and K. D. Bowen, Cancer Res., 1986, 46, 1608–1611 CAS.
- D. A. Musser and R. J. Fiel, Photochem. Photobiol., 1991, 53, 119–123 CrossRef CAS PubMed.
- D. A. Musser, S. H. Camacho, P. A. Manderscheid and A. R. Oseroff, Photochem. Photobiol., 1999, 69, 222–225 CrossRef CAS PubMed.
- G. O. Simkin, D. E. King, J. G. Levy, A. H. Chan and D. W. Hunt, Immunopharmacology, 1997, 37, 221–230 CrossRef CAS PubMed.
- S. O. Gollnick, D. A. Musser, A. R. Oseroff, L. Vaughan, B. Owczarczak and B. W. Henderson, Photochem. Photobiol., 2001, 74, 811–816 CrossRef CAS PubMed.
- N. Solban, S. K. Pål, S. K. Alok, C. K. Sung and T. Hasan, Cancer Res., 2006, 66, 5633–5640 CrossRef CAS PubMed.
- S. Laxmanan, S. W. Robertson, E. Wang, J. S. Lau, D. M. Briscoe and D. Mukhopadhyay, Biochem. Biophys. Res. Commun., 2005, 334, 193–198 CrossRef CAS PubMed.
- R. Mittler, Trends Plant Sci., 2017, 22, 11–19 CrossRef CAS PubMed.
- D. A. Bellnier, J. Photochem. Photobiol., B, 1991, 8, 203–210 CrossRef CAS PubMed.
- D. A. Bellnier, S. O. Gollnick, S. H. Camacho, W. R. Greco and R. T. Cheney, Cancer Res., 2003, 63, 7584–7590 CAS.
- G. Krosl, M. Korbelik, J. Krosl and G. J. Dougherty, Cancer Res., 1996, 56, 3281–3286 CAS.
- J. Goląb, G. Wilczyński, R. Zagożdżon, T. Stoklosa, A. Dąbrowska, J. Rybczyńska, M. Wąsik, E. Machaj, T. Oldak and K. Kozar, Br. J. Cancer, 2000, 82, 1485–1491 CrossRef PubMed.
- A. Stoppacciaro, C. Melani, M. Parenza, A. Mastracchio, C. Bassi, C. Baroni, G. Parmiani and M. P. Colombo, J. Exp. Med., 1993, 178, 151–161 CrossRef CAS PubMed.
- M. Korbelik and P. Cooper, Br. J. Cancer, 2007, 96, 67–72 CrossRef CAS PubMed.
- T. W. Dubensky Jr and S. G. Reed, 2010.
- H. HogenEsch, Front. Immunol., 2013, 3, 406 Search PubMed.
- B. A. Beutler, Blood, 2009, 113, 1399–1407 CrossRef CAS PubMed.
- C. Olive, Expert Rev. Vaccines, 2012, 11, 237–256 CrossRef CAS PubMed.
- X.-L. Wang, H.-W. Wang, M.-X. Guo and Z. Huang, Photodiagn. Photodyn. Ther., 2007, 4, 88–93 CrossRef CAS PubMed.
- M. Schön and M. Schön, Br. J. Dermatol., 2007, 157, 8–13 CrossRef PubMed.
- E. Sotiriou, A. Lallas, Z. Apalla and D. Ioannides, Photodermatol., Photoimmunol. Photomed., 2011, 27, 164–166 CrossRef PubMed.
- U. Winters, S. Daayana, J. T. Lear, A. E. Tomlinson, E. Elkord, P. L. Stern and H. C. Kitchener, Clin. Cancer Res., 2008, 14, 5292–5299 CrossRef CAS PubMed.
- T.-K. Lo, Y.-C. Chen, W.-T. Liu and T.-W. Wong, Photodiagn. Photodyn. Ther., 2022, 40, 103128 CrossRef CAS PubMed.
- D. Kim, J. Byun, J. Park, Y. Lee, G. Shim and Y.-K. Oh, Biomater. Sci., 2020, 8, 1106–1116 RSC.
- Q. Zeng, J. Yang, J. Ji, P. Wang, L. Zhang, G. Yan, Y. Wu, Q. Chen, J. Liu and G. Zhang, OncoImmunology, 2022, 11, 2061396 CrossRef PubMed.
- A. Nikkels, C. Pierard-Franchimont, N. Nikkels-Tassoudji, R. Bourguignon and G. Pierard, Acta Clin. Belg., 2005, 60, 227–234 CrossRef CAS PubMed.
- B. J. Osiecka, K. Jurczyszyn and P. Ziółkowski, Med. Sci. Monit., 2012, 18, P15 CrossRef PubMed.
- D. M. Klinman, Nat. Rev. Immunol., 2004, 4, 249–259 CrossRef CAS PubMed.
- A. P. Castano and M. R. Hamblin, 2006.
- Y. Xia, G. K. Gupta, A. P. Castano, P. Mroz, P. Avci and M. R. Hamblin, J. Biophotonics, 2014, 7, 897–905 CrossRef CAS PubMed.
- S. M. Bae, Y. W. Kim, S. Y. Kwak, Y. W. Kim, D. Y. Ro, J. C. Shin, C. H. Park, S. J. Han, C. H. Oh and C. K. Kim, Cancer Sci., 2007, 98, 747–752 CrossRef CAS PubMed.
- I. Van der Lubben, J. Verhoef, G. Borchard and H. Junginger, Adv. Drug Delivery Rev., 2001, 52, 139–144 CrossRef CAS PubMed.
- W. R. Chen, M. Korbelik, K. E. Battels, H. Liu, J. Sun and R. E. Nordquist, Photochem. Photobiol., 2005, 81, 190–195 CAS.
- G. Krosl and M. Korbelik, Cancer Lett., 1994, 84, 43–49 CrossRef CAS PubMed.
- J. A. Chabalgoity, G. Dougan, P. Mastroeni and R. J. Aspinall, Expert Rev. Vaccines, 2002, 1, 495–505 CrossRef CAS PubMed.
- M. Korbelik and I. Cecic, J. Photochem. Photobiol., B, 1998, 44, 151–158 CrossRef CAS PubMed.
- M. Korbelik, J. Sun and J. J. Posakony, Photochem. Photobiol., 2001, 73, 403–409 CrossRef CAS.
- R. C. Myers, B. H. Lau, D. Y. Kunihira, R. R. Torrey, J. L. Woolley and J. Tosk, Urology, 1989, 33, 230–235 CrossRef CAS.
- M. Uehara, K. Sano, Z.-L. Wang, J. Sekine, H. Ikeda and T. Inokuchi, Cancer Immunol. Immunother., 2000, 49, 401–409 CrossRef CAS PubMed.
- A. Jalili, M. Makowski, T. Świtaj, D. Nowis, G. M. Wilczyński, E. Wilczek, M. Chorąży-Massalska, A. Radzikowska, W. Maśliński and Ł. Biały, Clin. Cancer Res., 2004, 10, 4498–4508 CrossRef CAS PubMed.
- H. Saji, W. Song, K. Furumoto, H. Kato and E. G. Engleman, Clin. Cancer Res., 2006, 12, 2568–2574 CrossRef CAS.
- M. Korbelik and J. Sun, Int. J. Cancer, 2001, 93, 269–274 CrossRef CAS.
- V. Petrova, M. Annicchiarico-Petruzzelli, G. Melino and I. Amelio, Oncogenesis, 2018, 7, 1–13 CrossRef CAS PubMed.
- P. Darvin, S. M. Toor, V. S. Nair and E. Elkord, Exp. Mol. Med., 2018, 50, 1–11 CrossRef PubMed.
- J. W. Kleinovink, M. F. Fransen, C. W. Löwik and F. Ossendorp, Cancer Immunol. Res., 2017, 5, 832–838 CrossRef CAS PubMed.
- P. Gurung, J. Lim, R. Shrestha and Y.-W. Kim, Sci. Rep., 2023, 13, 4647 CrossRef CAS PubMed.
- C. Wu, X. Guan, J. Xu, Y. Zhang, Q. Liu, Y. Tian, S. Li, X. Qin, H. Yang and Y. Liu, Biomaterials, 2019, 205, 106–119 CrossRef CAS PubMed.
- K. Ni, T. Luo, G. Lan, A. Culbert, Y. Song, T. Wu, X. Jiang and W. Lin, Angew. Chem., Int. Ed., 2020, 132, 1124–1128 CrossRef.
- D. Wang, J. Liu, J. Duan, Y. Ma, H. Gao, Z. Zhang, J. Liu, J. Shi and K. Zhang, ACS Appl. Mater. Interfaces, 2022, 14, 44183–44198 CrossRef CAS PubMed.
- M. Yarchoan, B. A. Johnson III, E. R. Lutz, D. A. Laheru and E. M. Jaffee, Nat. Rev. Cancer, 2017, 17, 209 CrossRef CAS PubMed.
- P. A. Ott, Z. Hu, D. B. Keskin, S. A. Shukla, J. Sun, D. J. Bozym, W. Zhang, A. Luoma, A. Giobbie-Hurder and L. Peter, Nature, 2017, 547, 217–221 CrossRef CAS.
- T. N. Schumacher and R. D. Schreiber, Science, 2015, 348, 69–74 CrossRef CAS PubMed.
- C. Xu, J. Nam, H. Hong, Y. Xu and J. J. Moon, ACS Nano, 2019, 13, 12148–12161 CrossRef CAS PubMed.
- C. He, X. Duan, N. Guo, C. Chan, C. Poon, R. R. Weichselbaum and W. Lin, Nat. Commun., 2016, 7, 1–12 Search PubMed.
- X. Duan, C. Chan, N. Guo, W. Han, R. R. Weichselbaum and W. Lin, J. Am. Chem. Soc., 2016, 138, 16686–16695 CrossRef CAS PubMed.
- G. Yang, L. Xu, J. Xu, R. Zhang, G. Song, Y. Chao, L. Feng, F. Han, Z. Dong and B. Li, Nano Lett., 2018, 18, 2475–2484 CrossRef CAS PubMed.
- H. Liu, Y. Hu, Y. Sun, C. Wan, Z. Zhang, X. Dai, Z. Lin, Q. He, Z. Yang and P. Huang, ACS Nano, 2019, 13, 12638–12652 CrossRef CAS PubMed.
- J. Xu, L. Xu, C. Wang, R. Yang, Q. Zhuang, X. Han, Z. Dong, W. Zhu, R. Peng and Z. Liu, ACS Nano, 2017, 11, 4463–4474 CrossRef CAS PubMed.
- Z. Meng, X. Zhou, J. Xu, X. Han, Z. Dong, H. Wang, Y. Zhang, J. She, L. Xu and C. Wang, Adv. Mater., 2019, 31, 1900927 CrossRef PubMed.
- W. Song, J. Kuang, C.-X. Li, M. Zhang, D. Zheng, X. Zeng, C. Liu and X.-Z. Zhang, ACS Nano, 2018, 12, 1978–1989 CrossRef CAS PubMed.
- A. Gao, B. Chen, J. Gao, F. Zhou, M. Saeed, B. Hou, Y. Li and H. Yu, Nano Lett., 2019, 20, 353–362 CrossRef PubMed.
- W. Yang, F. Zhang, H. Deng, L. Lin, S. Wang, F. Kang, G. Yu, J. Lau, R. Tian and M. Zhang, ACS Nano, 2019, 14, 620–631 CrossRef PubMed.
- Y. Hu, S. Bai, X. Wu, S. Tan and Y. He, Ceram. Int., 2021, 47, 31031–31041 CrossRef CAS.
- S. Kim, S. A. Kim, G.-H. Nam, Y. Hong, G. B. Kim, Y. Choi, S. Lee, Y. Cho, M. Kwon, C. Jeong, S. Kim and I.-S. Kim, J. Immunother. Cancer, 2021, 9, e001481 CrossRef PubMed.
- H. Wang, Z. Gao, D. Jiao, Y. Zhang, J. Zhang, T. Wang, Y. Huang, D. Zheng, J. Hou and D. Ding, Adv. Funct. Mater., 2023, 33, 2214499 CrossRef CAS.
- G. C. Prendergast, C. Smith, S. Thomas, L. Mandik-Nayak, L. Laury-Kleintop, R. Metz and A. J. Muller, Cancer Immunol. Immunother., 2014, 63, 721–735 CrossRef CAS PubMed.
- D. H. Munn and A. L. Mellor, J. Clin. Invest., 2007, 117, 1147–1154 CrossRef CAS PubMed.
- D. H. Munn, E. Shafizadeh, J. T. Attwood, I. Bondarev, A. Pashine and A. L. Mellor, J. Exp. Med., 1999, 189, 1363–1372 CrossRef CAS PubMed.
- R. B. Holmgaard, D. Zamarin, D. H. Munn, J. D. Wolchok and J. P. Allison, J. Exp. Med., 2013, 210, 1389–1402 CrossRef CAS PubMed.
- C. Wu, J. Xu, Z. Xie, H. Huang, N. Li, X. Wei, T. Li, H. Yang, S. Li and X. Qin, Biomater. Sci., 2021, 9, 8019–8031 RSC.
- M. B. Vrouenraets, G. Visser, G. B. Snow and G. Van Dongena, Anticancer Res., 2003, 23, 505 CAS.
- M. A. M. Capella and L. S. Capella, J. Biomed. Sci., 2003, 10, 361–366 CrossRef CAS PubMed.
- K. Yang, S. Zhao, B. Li, B. Wang, M. Lan and X. Song, Coord. Chem. Rev., 2022, 454, 214330 CrossRef CAS.
- K. F. Chu and D. E. Dupuy, Nat. Rev. Cancer, 2014, 14, 199–208 CrossRef CAS PubMed.
- J. J. Skitzki, E. A. Repasky and S. S. Evans, Curr. Opin. Invest. Drugs, 2009, 10, 550 CAS.
- B. Frey, E.-M. Weiss, Y. Rubner, R. Wunderlich, O. J. Ott, R. Sauer, R. Fietkau and U. S. Gaipl, Int. J. Hyperthermia, 2012, 28, 528–542 CrossRef CAS PubMed.
- J. Boulanger, D. Faulds, E. Eddy and C. Lingwood, J. Cell. Physiol., 1995, 165, 7–17 CrossRef CAS PubMed.
- W. R. Chen, R. L. Adams, R. Carubelli and R. E. Nordquist, Cancer Lett., 1997, 115, 25–30 CrossRef CAS PubMed.
- W. R. Chen, H. Liu, J. W. Ritchey, K. E. Bartels, M. D. Lucroy and R. E. Nordquist, Cancer Res., 2002, 62, 4295–4299 CAS.
- X. Li, M. F. Naylor, H. Le, R. E. Nordquist, T. K. Teague, C. A. Howard, C. Murray and W. R. Chen, Cancer Biol. Ther., 2010, 10, 1081–1087 CrossRef PubMed.
- F. Zhou, S. Wu, S. Song, W. R. Chen, D. E. Resasco and D. Xing, Biomaterials, 2012, 33, 3235–3242 CrossRef CAS PubMed.
- Y. Tao, E. Ju, J. Ren and X. Qu, Biomaterials, 2014, 35, 9963–9971 CrossRef CAS PubMed.
- L. Guo, D. D. Yan, D. Yang, Y. Li, X. Wang, O. Zalewski, B. Yan and W. Lu, ACS Nano, 2014, 8, 5670–5681 CrossRef CAS PubMed.
- T. Yata, Y. Takahashi, M. Tan, H. Nakatsuji, S. Ohtsuki, T. Murakami, H. Imahori, Y. Umeki, T. Shiomi and Y. Takakura, Biomaterials, 2017, 146, 136–145 CrossRef CAS PubMed.
- J. Pan, Y. Wang, C. Zhang, X. Wang, H. Wang, J. Wang, Y. Yuan, X. Wang, X. Zhang and C. Yu, Adv. Mater., 2018, 30, 1704408 CrossRef PubMed.
- H. K. Moon, S. H. Lee and H. C. Choi, ACS Nano, 2009, 3, 3707–3713 CrossRef CAS PubMed.
- Q. Chen, L. Xu, C. Liang, C. Wang, R. Peng and Z. Liu, Nat. Commun., 2016, 7, 1–13 Search PubMed.
- B. Zhou, J. Song, M. Wang, X. Wang, J. Wang, E. W. Howard, F. Zhou, J. Qu and W. R. Chen, Nanoscale, 2018, 10, 21640–21647 RSC.
- R. Ge, C. Liu, X. Zhang, W. Wang, B. Li, J. Liu, Y. Liu, H. Sun, D. Zhang and Y. Hou, ACS Appl. Mater. Interfaces, 2018, 10, 20342–20355 CrossRef CAS PubMed.
- J. Peng, Y. Xiao, W. Li, Q. Yang, L. Tan, Y. Jia, Y. Qu and Z. Qian, Adv. Sci., 2018, 5, 1700891 CrossRef PubMed.
- M. Yan, Y. Liu, X. Zhu, X. Wang, L. Liu, H. Sun, C. Wang, D. Kong and G. Ma, ACS Appl. Mater. Interfaces, 2018, 11, 1876–1885 CrossRef PubMed.
- C.-H. Chang and E. L. Pearce, Nat. Immunol., 2016, 17, 364–368 CrossRef CAS PubMed.
- C. Zhang, K. Zhao, W. Bu, D. Ni, Y. Liu, J. Feng and J. Shi, Angew. Chem., Int. Ed., 2015, 54, 1770–1774 CrossRef CAS PubMed.
- Y. Yang, W. Zhu, Z. Dong, Y. Chao, L. Xu, M. Chen and Z. Liu, Adv. Mater., 2017, 29, 1703588 CrossRef PubMed.
- P. Huang, Y. Yang, W. Wang, Z. Li, N. Gao, H. Chen and X. Zeng, Biomaterials, 2023, 299, 122157 CrossRef CAS PubMed.
- L. Zhou, B. Feng, H. Wang, D. Wang and Y. Li, Nano Today, 2022, 44, 101466 CrossRef CAS.
- X. Meng, Y. Han, S. Wang, X. Wang, Z. Zhang, S. Yao, X. Wan, Z. Liu, Z. Ge and L. Li, Nano Today, 2023, 53, 102030 CrossRef CAS.
- Y. Zhou, S. Liu, C. Hu, L. Cai and M. Pang, J. Mater. Chem. B, 2020, 8, 5451–5459 RSC.
- J. Li, D. Cui, J. Huang, S. He, Z. Yang, Y. Zhang, Y. Luo and K. Pu, Angew. Chem., Int. Ed., 2019, 58, 12680–12687 CrossRef CAS PubMed.
- B. Liu, W. Cao, J. Cheng, S. Fan, S. Pan, L. Wang, J. Niu, Y. Pan, Y. Liu and X. Sun, Cancer Biol. Med., 2019, 16, 756 CrossRef CAS PubMed.
- L. Zhou, L. Chen, X. Hu, Y. Lu, W. Liu, Y. Sun, T. Yao, C. Dong and S. Shi, Commun. Biol., 2020, 3, 343 CrossRef CAS PubMed.
- X. Zhang, J. Tang, C. Li, Y. Lu, L. Cheng and J. Liu, Bioact. Mater., 2021, 6, 472–489 CAS.
- S. K. Jeong, J. S. Kim, C. G. Lee, Y.-S. Park, S. D. Kim, S. O. Yoon, D. H. Han, K. Y. Lee, M. H. Jeong and W. S. Jo, Immunobiology, 2017, 222, 55–65 CrossRef CAS PubMed.
- J. E. Rayahin, J. S. Buhrman, Y. Zhang, T. J. Koh and R. A. Gemeinhart, ACS Biomater. Sci. Eng., 2015, 1, 481–493 CrossRef CAS PubMed.
- M. Song, T. Liu, C. Shi, X. Zhang and X. Chen, ACS Nano, 2016, 10, 633–647 CrossRef CAS PubMed.
- X. Gu, B. Wu, G. Feng, Z. Chen, F. Ren, X. Chen, W. Hong and W. Li, Mol. Pharmaceutics, 2023, 20, 4007–4020 CrossRef CAS PubMed.
- M. He, Y. Song, W. Xu, X. Zhang and C. M. Dong, Adv. Funct. Mater., 2023, 2304216 CrossRef CAS.
- B. Ji, M. Wei and B. Yang, Theranostics, 2022, 12, 434 CrossRef CAS PubMed.
- M. Chang, Z. Hou, M. Wang, C. Li and J. Lin, Adv. Mater., 2021, 33, 2004788 CrossRef CAS PubMed.
- Q. Liu, J. Tian, Y. Tian, Q. Sun, D. Sun, F. Wang, H. Xu, G. Ying, J. Wang and A. K. Yetisen, ACS Nano, 2021, 15, 515–525 CrossRef CAS PubMed.
- H. Zhang, T. Wang, H. Liu, F. Ren, W. Qiu, Q. Sun, F. Yan, H. Zheng, Z. Li and M. Gao, Nanoscale, 2019, 11, 7600–7608 RSC.
- L. Tan, S. Wang, K. Xu, T. Liu, P. Liang, M. Niu, C. Fu, H. Shao, J. Yu and T. Ma, Small, 2016, 12, 2046–2055 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |