
Injectable polyoxazoline grafted hyaluronic acid thermoresponsive hydrogels for biomedical applications†
Morgane
Morel
ab,
Mathieu
Madau
a,
Didier
Le Cerf
a,
Virginie
Dulong
a,
Anne-Claire
Groo
 b,
Aurélie
Malzert-Fréon
b and
Luc
Picton
*a
b,
Aurélie
Malzert-Fréon
b and
Luc
Picton
*a
aUniv Rouen Normandie, CNRS, PBS UMR6270, F-76000 Rouen, France. E-mail: luc.picton@univ-rouen.fr
bUniv Caen Normandie, CERMN, UR4258, F-14000 Caen, France
First published on 15th February 2024
Abstract
Injectable thermosensitive hydrogels based on hyaluronic acid (HA) grafted with lower critical solution temperature (LCST) polyoxazoline (copolymers of poly(isopropyl-co-butyl oxazoline)) or P(iPrOx-co-BuOx) have been elaborated with tunable solution/gel temperature transitions and gel state elastic modulus. A suitable HA-g-P(iPrOx-co-BuOx-67/33)-0.10 sample with an iPrOx/BuOx ratio of 67/33, a polymerization degree (DP) of 25, a substitution degree (DS) of 10%, and displaying thermally induced gelling character with elastic (G′) and viscous (G′′) moduli crossover points at 25 °C and a G′ at 37 °C around 80 Pa has been chosen for medical application. Hydrogels obtained with HA-g-P(iPrOx-co-BuOx-67/33)-0.10 exhibited high stability at 37 °C and excellent injectability properties with full and quick reversibility. The incorporation of a secondary network (HA), until 35 wt%, into the thermosensitive hydrogel also demonstrated very good stability and injectability.
1. Introduction
Hydrogels are tridimensional crosslinked networks soluble in water made from artificial or natural polymers. They can be created under several shapes such as coatings and films, micro and nanoparticles, etc.1–3 Hydrogels are present in a lot of applications, especially in tissue engineering,4 regenerative medicine,5 diagnostics,6 cell immobilization,7 and controlled delivery of active agents.8 Hydrogels can be classified into chemical or physical hydrogels, with chemical hydrogels being made of covalent crosslinking,9 while physical hydrogels10 are instead made of non-covalent bonds (ionic interaction, van der Waals interactions, hydrogen bonds, etc.). Physical hydrogels are preferred for injection treatments because of their reversibility, and in some cases, they can respond to external stimuli such as temperature, pH, electric field, etc.The number of studies on injectable hydrogels has been growing steadily in the biomedical field for many years. This is because injectable hydrogels offer several advantages over conventional methods of administration (intravenous, intranasal, and oral). In fact, injectable hydrogels enable the therapy to be delivered directly to the target site, thus avoiding problems of diffusion into “healthy” areas of the human body. This method also overcomes certain barriers (such as the blood–brain barrier) which limit access to certain molecules, so that the dose of delivered therapy is better controlled.11–14
For in situ injection, thermosensitive physical hydrogels that undergo a transition from the liquid state at room temperature to the gel state once they arrive at the desired site with the help of body temperature are very interesting materials as they make the injection process easier.8,15–17 They can be obtained by grafting LCST (lower critical solution temperature) polymers onto a water-soluble polymer. LCST polymers are water soluble below the LCST temperature and precipitate above the LCST temperature. This phenomenon is explained by the fact that heat causes the polymer/polymer interactions to be favorable over the polymer/solvent interactions. As a matter of fact, thermosensitive polymers lead to physical crosslinking nodes between the LCST polymer chains at a given temperature making it possible to form a gel.10,18–20
At present, there are several types of LCST polymers known, such as some polyether amines (Jeffamine®), PNIPAM, or some types of polyoxazolines (POXs).18,21–25 However, we’ll be focusing solely on polyoxazolines because, unlike Jeffamine® or PNIPAM, POXs appear more biocompatible, allow easier control of the gelation temperature, and, even more strikingly, an almost spontaneous resulting sol–gel transition.20,26–29
During this work, we chose to create an injectable thermoresponsive hydrogel thanks to hyaluronic acid (HA). Hyaluronic acid is an anionic polysaccharide that is completely biocompatible and biodegradable due to its ubiquity in the human body especially in the extracellular matrix but also in various conjunctive tissues.18,30–33 Unfortunately, hyaluronic acid doesn’t naturally form a heat-sensitive hydrogel, so we decided to graft polyoxazoline (POX) copolymers displaying an LCST behavior on it. This work follows our previous study focusing on the sol–gel transition of HA grafted with suitable POX copolymers (i.e., poly(isopropyl-co-butyl-oxazoline) or P(iPrOx-co-BuOx)).34 The thermal transition can then be perfectly controlled by adjusting the IPrOx/BuOx ratio, the degree of polymerization (DP), and the degree of substitution (DS) of the LCST polymer.34,35
Indeed, the present work exploits this synthesis route to design an easily and controlled injectable thermo-sensitive hydrogel that is stable at 37 °C with suitable, quick, and reversible gelation. In contrast to our previous study,34 we have here set the synthesis conditions to obtain these injectable heat-sensitive hydrogels compatible with biomedical use. However, in this study, no specific application or specific site of injection was sought; hence, the following study focuses on the simulated injectability of a thermosensitive and biocompatible system in the body at 37 °C.
The injectability of the hydrogel will be thoroughly evaluated. Moreover, the robustness of the hydrogel properties when inserting a secondary HA network will be assessed. The secondary HA network could then be grafted with drug molecules to devise a hydrogel-based drug delivery system.
2. Experimental
2.1 Materials
Hyaluronic acid sodium salt (Na–HA) from streptococcus equi bacterial glycosaminoglycan polysaccharide (1.2 MDa), tetrabutylammonium hydroxide (TBAOH), 2-n-isopropyl-2-oxazoline (iPrOx), 2-n-butyl-2-oxazoline (BuOx), methyl-p-toluenesulfonate, chloroform-D (CDCl3), sodium deuteroxide (NaOD), deuterium oxide (D2O), boric acid, (4-(dimethylamino)benzaldehyde) (DMAB), acetic acid 96%, hyaluronidase from bovine testes type IV-S and anhydrous acetonitrile were purchased from Sigma-Aldrich. Dimethylsulfoxide anhydrous (DMSO), sodium chloride (NaCl), sodium hydroxide pellets (NaOH), and acetone were purchased from VWR. Potassium hydroxide (KOH) and hydrochloric acid 35% were purchased from Prolabo. Potassium hydrogen phthalate was purchased from Fisher Chemicals and potassium nitrate from Wako. The phosphate buffer (0.1 M; pH 7.4) was made from sodium dihydrogen phosphate monohydrate from AppliChem and di-sodium hydrogen phosphate dihydrate from Merck-Millipore. Milli-Q water used was purified using the Milli-Q water reagent system from Millipore (MA, USA).2.2 Synthesis of HA-PiPrOx-co-PBuOx
HA-g-P(iPrOx-co-BuOx) was synthesized as shown previously.34 Briefly, Na–HA was converted into tetrabutylammonium hyaluronate (TBA-HA) in order to be soluble in a polar aprotic solvent (DMSO). The polymerization of 2-n-isopropyl-2-oxazoline (iPrOx) and 2-n-butyl-2-oxazoline (BuOx) (at a theoretical molar ratio of 68/32, and a theoretical polymerization degree (DP) of 30) has been done through living cationic ring-opening polymerization (CROP) at 70 °C during 24 h, using methyl-p-toluenesulfonate as the initiator. After that, the termination step was realized with the use of TBA-HA (dissolved in DMSO) as a termination agent for 24 h at 60 °C.35 Dialysis (with Spectra/Por®4 dialysis membrane MWCO: 12–14 kDa from Repligen), lyophilization, and acetone washing were performed to purify the product. 1H NMR (in D2O and NaOD) and rheological (in NaCl 0.9% (0.15 M)) measurements have been successful in confirming the efficiency of the grafting.2.3 Characterization methods
Hydrogel stability was then monitored by tracking elastic (G′) and viscous (G′′) moduli over time at 37 °C for 24 hours (0.1 Pa and 1 Hz).
For injectability measurements, the samples were subjected to various stresses ranging from 5 to 100 Pa for 5 min at 20 °C or injected in the geometry using different types of syringes and needles. They were then placed under a stress of 0.1 Pa for 5 min at 20 °C. Finally, the temperature was raised to 37 °C (0.1 Pa) for 10 min.
Flow measurements have been carried out with up (10′) and down (10′) applied shear rates scans between 0.1 to 1000 s−1.
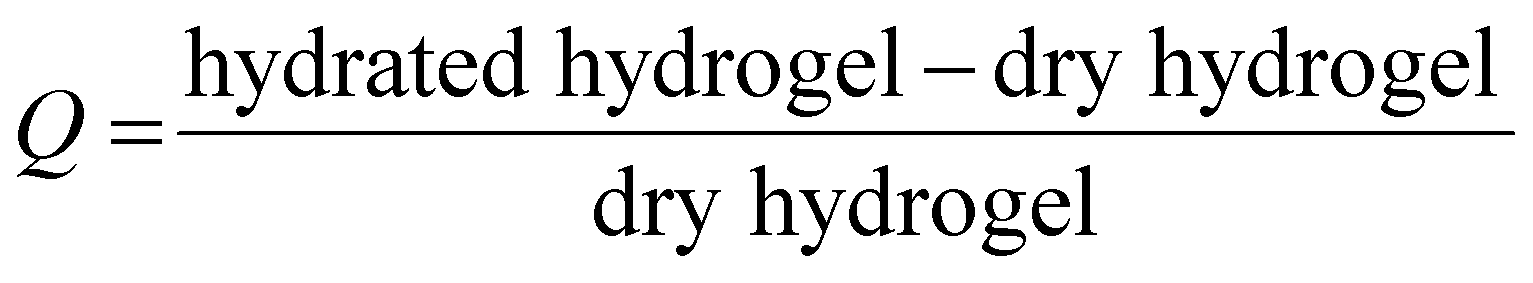 | (1) |
3. Results and discussions
3.1 Synthesis of HA-g-P(iPrOx-co-BuOx)
HA-g-P(iPrOx-co-BuOx) was prepared from the CROP (cationic ring-opening polymerization) reaction between 2-n-isopropyl-2-oxazoline (iPrOx), 2-n-butyl-2-oxazoline (BuOx), and tetrabutylammonium hyaluronate as shown on the reaction mechanism in Fig. 1.34,35 The 1H NMR characterization for HA-g-P(iPrOx-co-BuOx) and P(iPrOx-co-BuOx) is shown in the ESI† (S1 and S2). The DS, DP, and the IPrOx/BuOx ratio are determined as explained in our previous work.34 The substitution degree (DS) was determined according to eqn (2) (more information in the ESI†).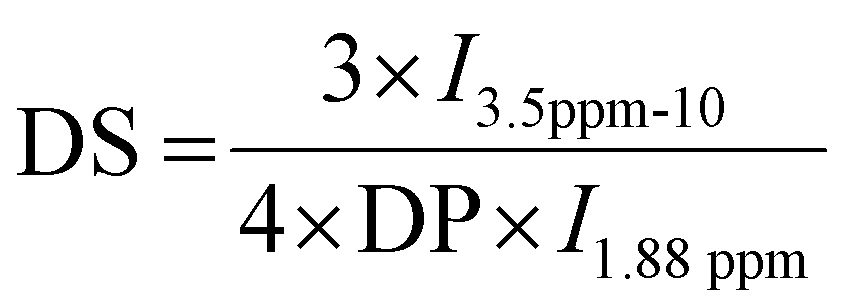 | (2) |
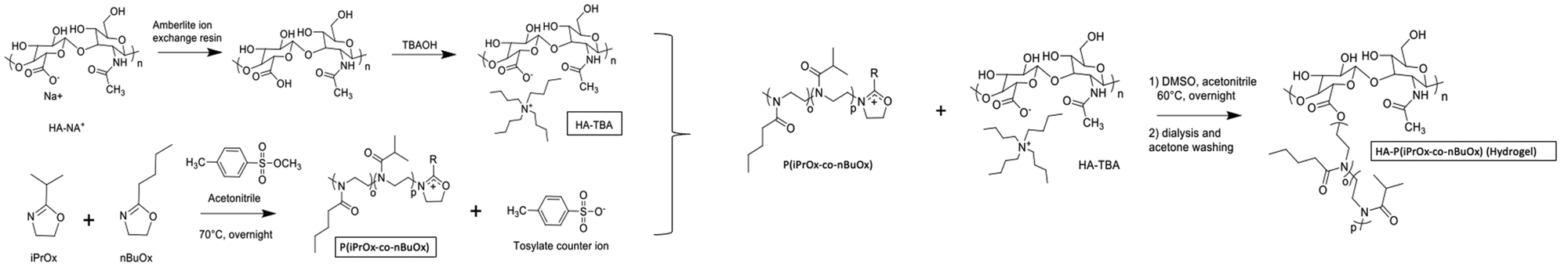 | ||
| Fig. 1 Reaction mechanism for grafting the P(iPrOX-co-BuOx) copolymer onto hyaluronic acid. | ||
Ideal conditions for hydrogel injection/gelation were achieved at an experimental iPrOx/BuOx ratio of 67/33 (±1/1), a DS of 10 (±3), and a DP of 25 (±5).
Rheological measurement of this sample has been done to check its gelation ability when heated to 37 °C (body temperature). As shown in Fig. 2 (see part 2.3.2 for conditions), it can be observed that below 20 °C, G′′ is largely above G′ indicating the viscous state of the system, which is therefore expected to be easily injectable. In this range both moduli decrease with the temperature according to thermal agitation, this behavior is typical of water-soluble polymer dynamic. After 20 °C, both moduli increase with the temperature as the grafted POX chains start to associate themselves (explained by their LCST character). The associative behavior is reinforced with the increase of the temperature. Thus, and logically, G′ increases more than G′′ leading to a critical temperature, sometimes called transition temperature (Tsol/gel), that corresponds to the crossover of both moduli. Finally, the elastic modulus seems to reach a plateau (∼100 Pa) from about 35 °C. It is largely higher than the G′′ (more than 1 decade), which means that the elastic behavior of the material is predominant over its viscous behavior.
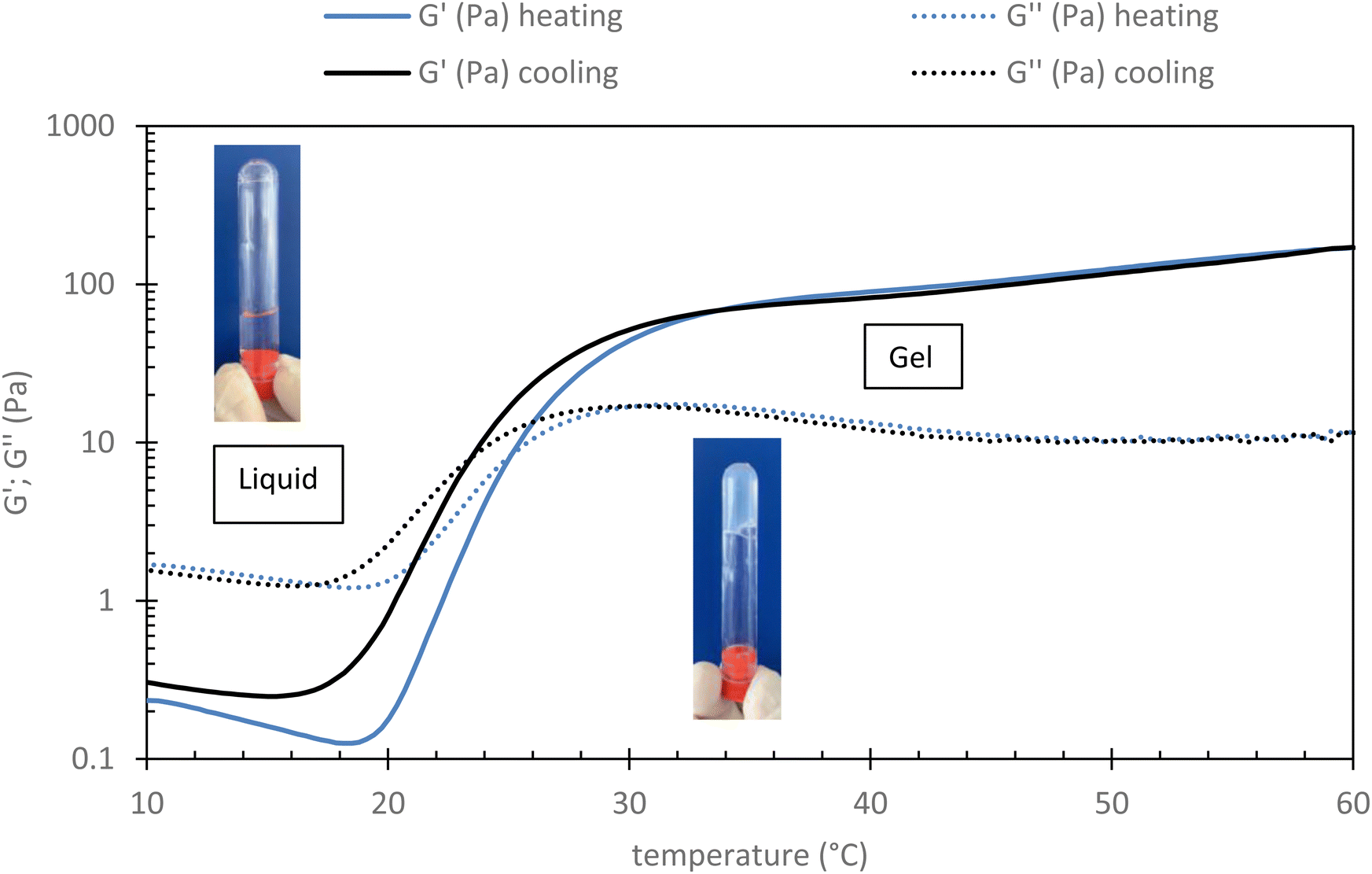 | ||
| Fig. 2 Evolution of rheological profile of HA-g-P(iPrOx-co-BuOx-67/33)-0.10 015 g L−1 in NaCl 0.15 M in function of temperature (shear stress: 0.1 Pa; frequency: 1 Hz and rate: 0.5 °C min−1). | ||
This result seems to indicate that this sample is well adapted for human body medical sol/gel application in terms of the temperature of final gelation (∼37 °C) and of the magnitude of the elastic modulus.
3.2 Hydrogel stability at 37 °C
To ensure that the hydrogel remains in the gel state at 37 °C, a 24-hour rheological analysis was set up in oscillatory mode (measurement of G′ and G′′ moduli for 24 h at 37 °C, 0.1 Pa, and 1 Hz).As shown in Fig. 3, it can be seen that the elastic modulus G′ remains higher than the viscous modulus G′′ for at least 15 h at 37 °C, above this time, solvent evaporation probably occurs leading to an increase in both moduli. This confirms that the hydrogel remains stable at 37 °C for at least 15 h (maximum operating conditions). One can notice a very slight diminishing of both moduli over time that could be explained by a possible reorganization in the associations, without changing the main behavior.
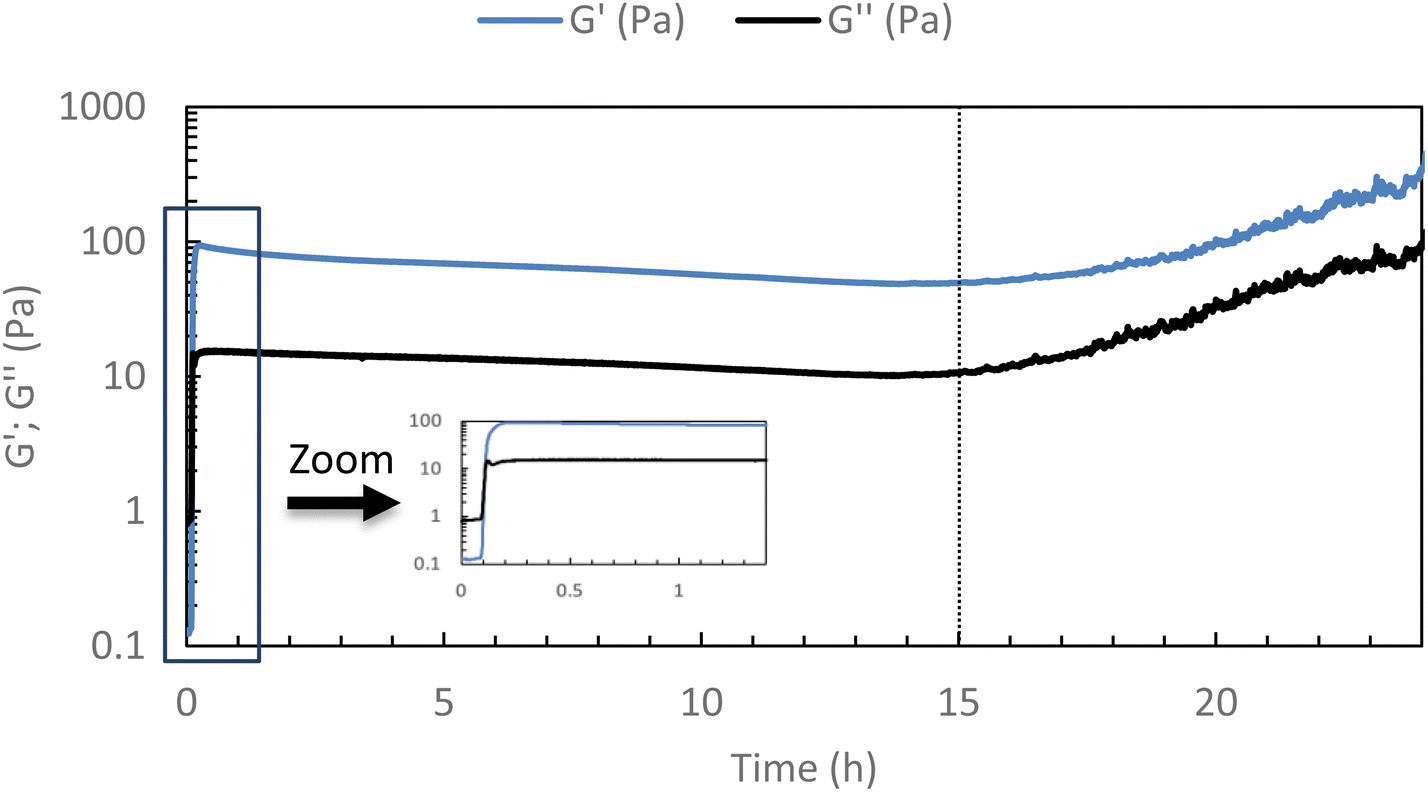 | ||
| Fig. 3 Stability studies of HA-g-P(iPrOx-co-BuOx-67/33)-0.10 at 37 °C (15 g L−1 in NaCl 0.15 M) for 24 h (0.1 Pa and 1 Hz). | ||
A second study on the same sample (i.e. HA-g-P(iPrOx-co-BuOx-67/33)-0.10) was designed to confirm whether the hydrogel remained stable in an excess of solvent at 37 °C. The hydrogel was immersed in an excess of a 0.15 M NaCl solution. Then, supernatant samples were taken off every day to check if some polymer was extracted from the gel toward the supernatant. These samples were analyzed by TOC to follow the variation in carbon content in the supernatant as a function of time for 5 days, and thus to trace the release polymer concentration in the supernatant, as shown in Fig. 4(A).
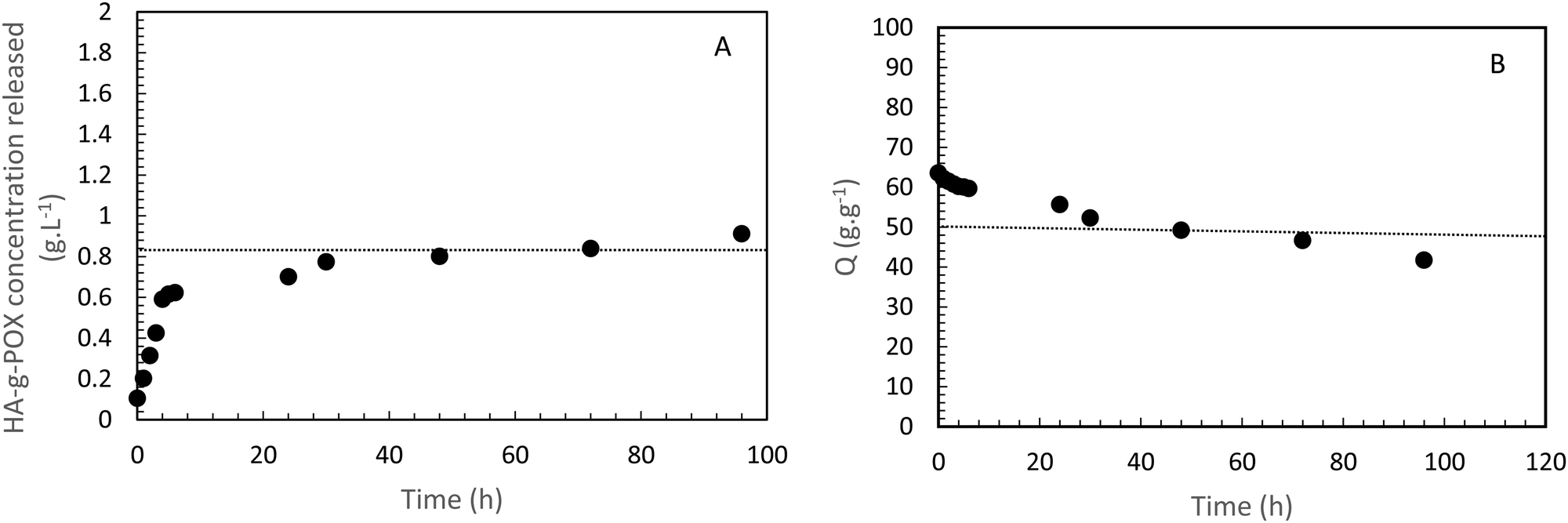 | ||
| Fig. 4 HA-g-P(iPrOx-co-BuOx-67/33)-0.10 concentration released in the supernatant over time at 37 °C. B-Swelling rate (Q) of HA-POX over time at 37 °C. | ||
Regarding this sample, it appears that only 0.9 g L−1 has been released after 5 days which represents less than 6% of the initial amount of polymer in the gel. This result clearly indicates that the gel is strongly stable in an excess of solvent at the body temperature. The burst effect observed during the first 6 hours can be explained by the diffusion of polymers which should not be correctly crosslinked with a major part of hydrogel in the supernatant.
To confirm these results, a third study was carried out to control the swelling rate of the hydrogel over time in order to prove that the hydrogel remains in the gel state for at least 5 days even with the presence of a solvent excess applied to its surface. As can be seen in Fig. 4(B), 80% of the initial swelling is conserved after 5 days. The swelling loss observed seems to be correlated to the loss of polymer (6%) observed during the TOC analysis described above. Finally, over the experimental time of 15 hours induced by rheological limits, our swelling and polymer release results confirm that the gel state is globally preserved over 5 days.
3.3 Enzymatic degradation
The enzymatic degradation preliminary study of the hydrogel in the presence of hyaluronidase (Fig. 5(A)) shows that the reducing ends groups (N-acetylglucosamine or NAG) concentration is higher when the HA-g-POX degradation is done at 20 °C than at 37 °C after 72 h (1.3 mmol L−1 at 20 °C vs. 0.3 mmol L−1 at 37 °C). More importantly, the HA-g-POX degradation at 37 °C reaches a plateau after one hour, whereas HA-g-POX degradation at 20 °C increases over time. This plateau may be explained by the fact that, at 37 °C, the hydrogel's polyoxazoline grafts limit access to the glycosidic β (1–4) bond of hyaluronic acid, in addition to the gel structure, which greatly slows down the enzyme.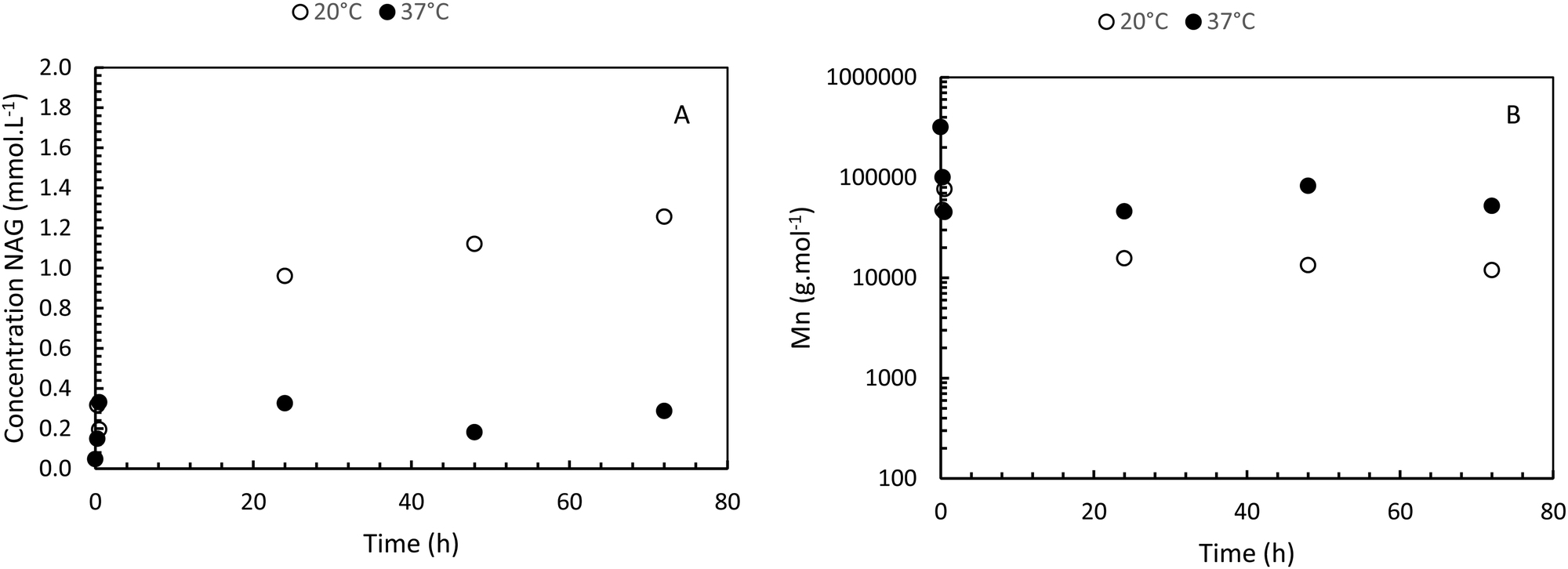 | ||
| Fig. 5 (A) Enzymatic degradation of HA-g-P(iPrOx-co-BuOx-67/33)-0.10 in presence of hyaluronidase (100 μL at 1 mg mL−1 for 10 mL hydrogel at 15 g L−1) over time at 20 °C and 37 °C. (B) Molar mass evolution of HA-g-P(iPrOx-co-BuOx-67/33)-0.10 over time in presence of hyaluronidase at 20 °C and 37 °C. | ||
To better visualize the phenomenon, the evolution of the number average molar mass (Mn, determined using eqn (3)) was plotted as a function of time for the hydrolysis undergone by the gel.
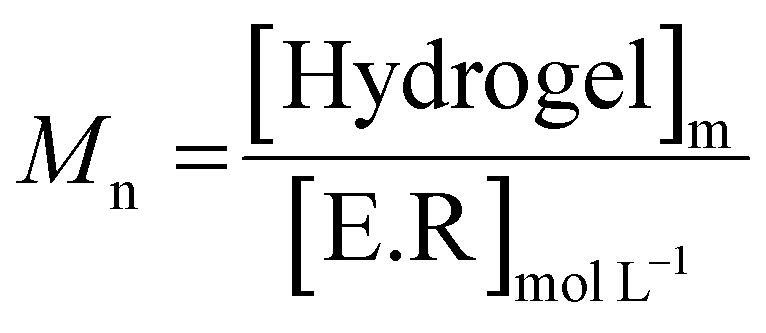 | (3) |
As shown in Fig. 5(B), for the solution at 20 °C, the molar masses decrease to reach an Mn of around 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 after 5 days, unlike the gel at 37 °C, which degrades to around 52000 g mol−1. If we compare this result with the literature,39 the total degradation of native hyaluronic acid leads to a Mn of about 1600 g mol−1, that means about 4 disaccharide units.
000 g mol−1 after 5 days, unlike the gel at 37 °C, which degrades to around 52000 g mol−1. If we compare this result with the literature,39 the total degradation of native hyaluronic acid leads to a Mn of about 1600 g mol−1, that means about 4 disaccharide units.
Thus, our results show that HAase leads to a degradation of HA-g-POx which is largely less important as compared to native HA. This is due to the presence of the grafted POx groups which limit the accessibility of the enzyme. Moreover, at the gel state, the degradation is more limited than at the sol state due probably to the hindering of the network. As reported in part 3.2 this slight enzymatic degradation at the gel state does not lead to the destructuration of the network. These results are very encouraging for future applications.
3.4 Hydrogel injectability
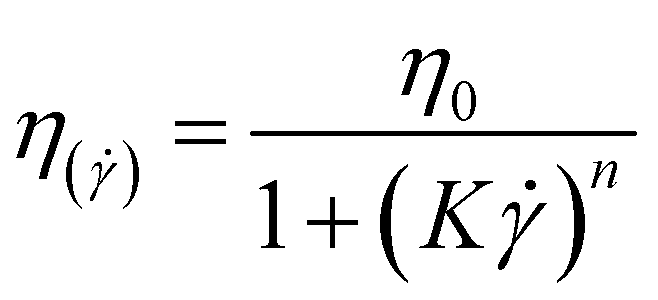 | (4) |
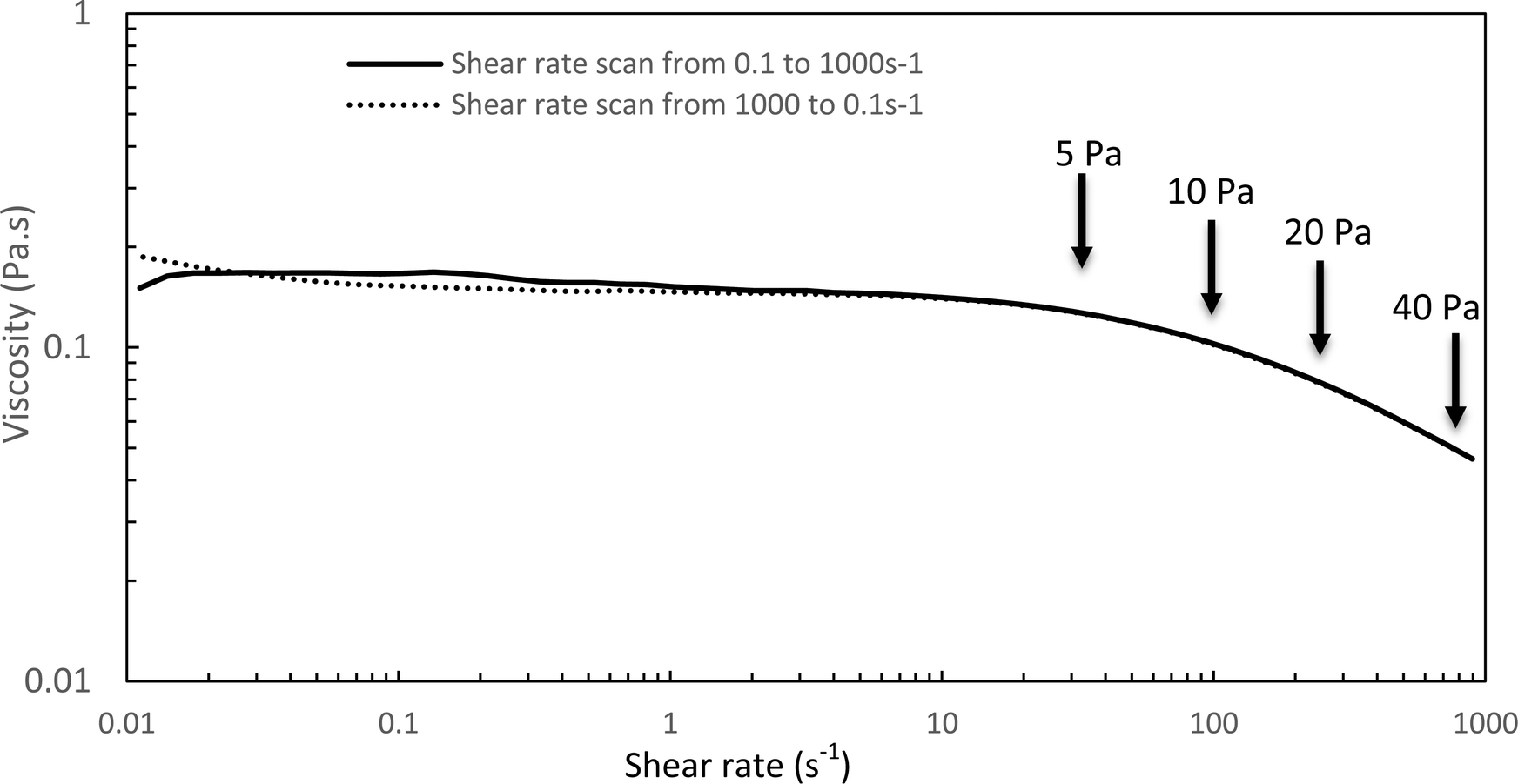 | ||
| Fig. 6 Flow curve for HA-g-P(iPrOx-co-BuOx-67/33)-0.10 at 20 °C; 15 g L−1. | ||
The Williamson model well fits the experimental flow curve of the solution (R2 = 0.9996) and gives the following result: η0 = 155 mPa s−1; K = 5 ms; n = −0.77. This behavior is a good indication and prognostic of potential easy injectability.
In the following, G′ and G′′ (at 1 Hz) are measured under various conditions of stress and temperature. Step 0 gives the original elastic state of the hydrogel at 37 °C in the linearity domain (low stress: 0.1 Pa, 1 Hz). Then, the hydrogel was subjected to various stresses at 20 °C ranging from 5 to 100 Pa during 5 min (step 1 in Fig. 7). Thus, this step simulates the applied stress when the hydrogel is injected via a syringe with a needle or capillary. These stresses were reported with their corresponding viscosities in Fig. 6 (flow curve), which evidences that the tested stresses correspond to the shear thinning behavior range. The hydrogel is then left to rest at 20 °C under very low stress (0.1 Pa) for 5 minutes (step 2 in Fig. 7). This stage mimics the moment when the gel exits the syringe/needle and enters the human body (with no stress applied and not heated at 37 °C yet). Finally, during step 3 (Fig. 7), the geometry is heated to 37 °C to mimic the introduction in the human body in the same conditions as step 0. In a second stage (Fig. 8), the same analysis was carried out, but instead of applying stress to the hydrogel via the rheometer, it was injected into the device geometry using different syringes, needles, micropipettes, and capillaries (cf. S3 ESI† for capillaries experiments).
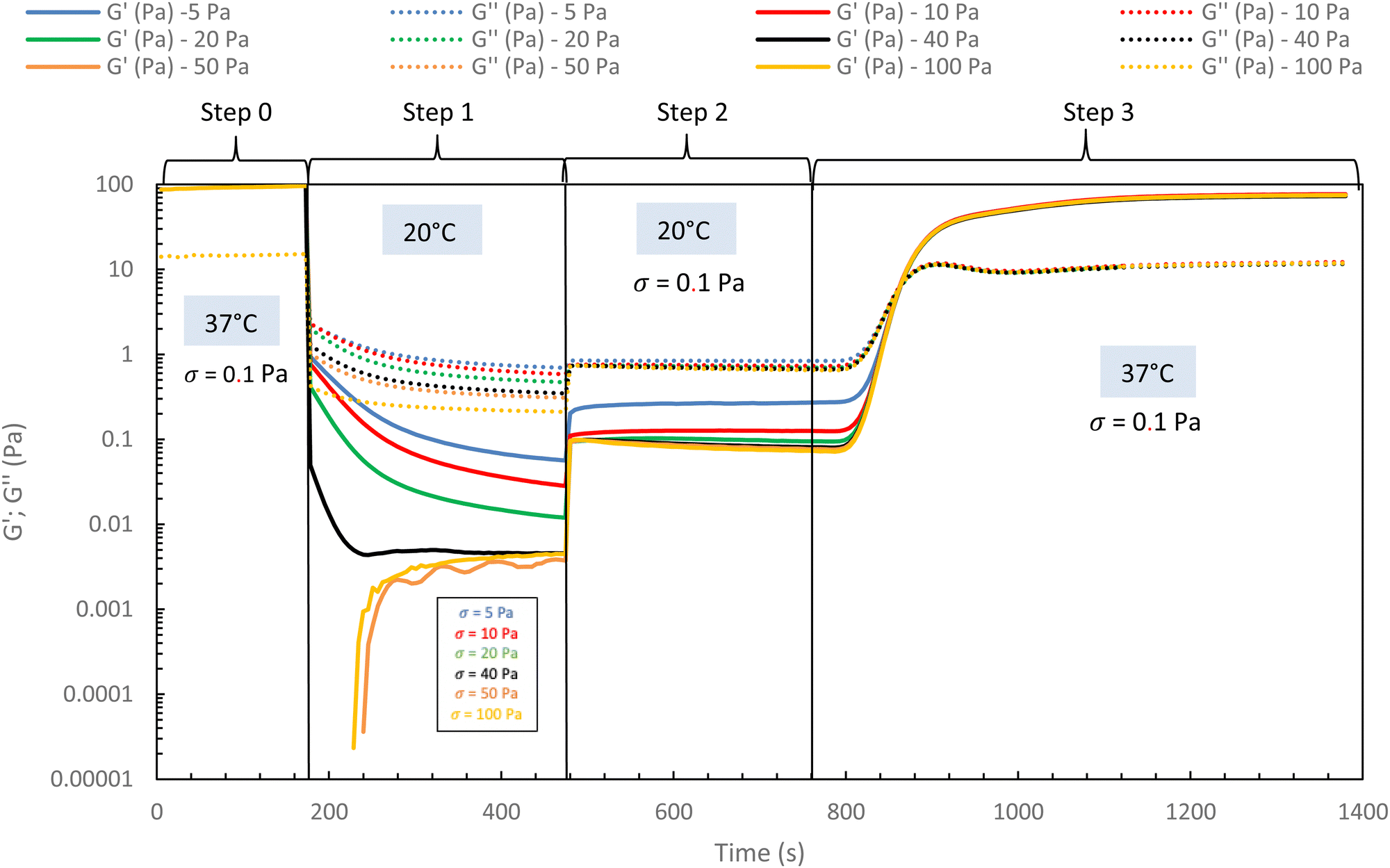 | ||
| Fig. 7 Rheological injection simulation for HA-g-(PiPrOx-co-PBuOx-67/33)-0.10 via different stress apply at 20 °C to mimic the injection phenomenon (15 g L−1 in NaCl 0.15 M). | ||
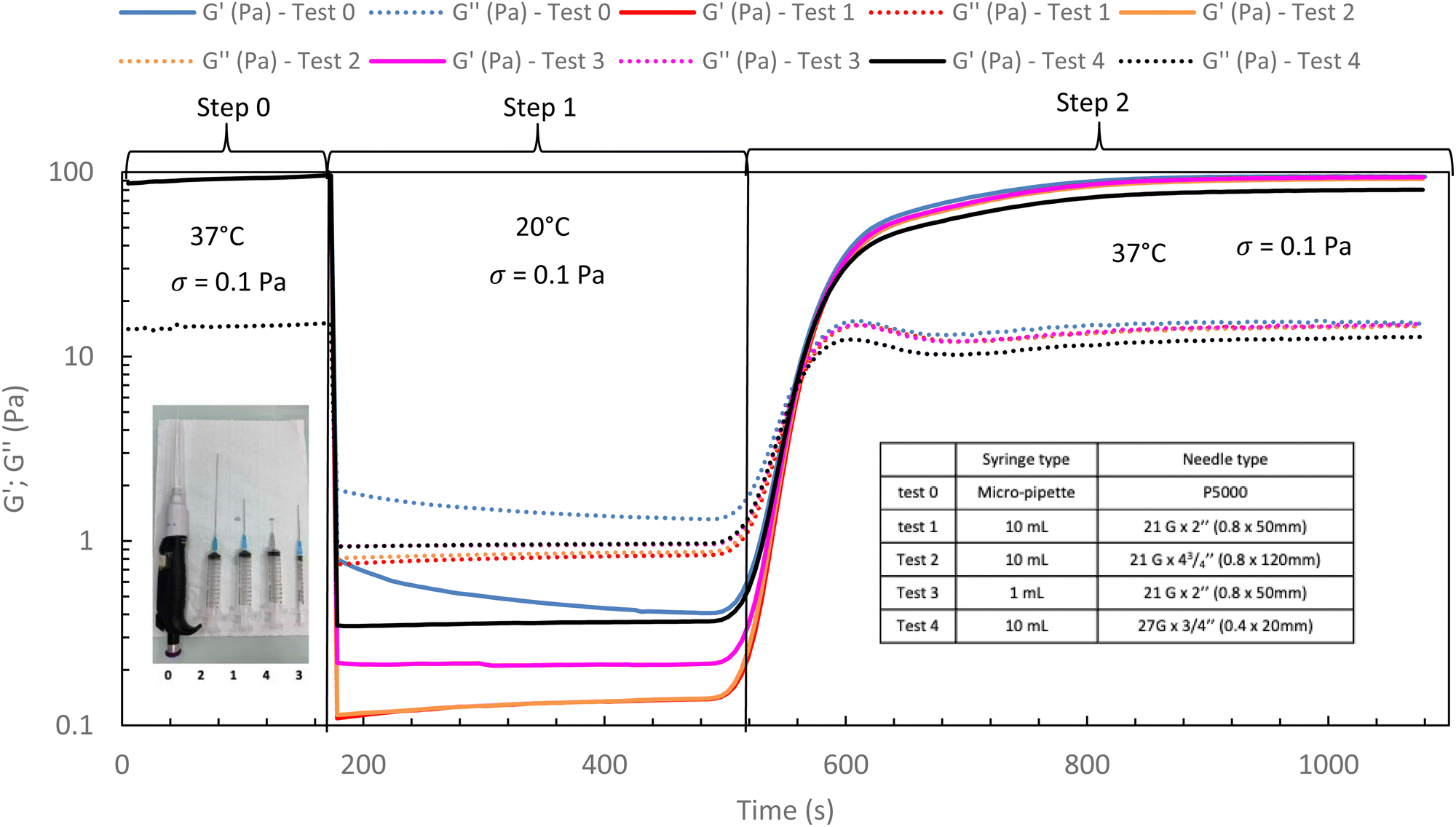 | ||
| Fig. 8 Rheological in situ injection via different syringes and needles for HA-PiPrOx-co-PBuOx-66/34-0.10 at 20 °C to mimic the injection phenomenon (15 g L−1 in NaCl 0.15 M). | ||
As shown in Fig. 7 and 8, whatever the applied stress and whatever the used injection method, the hydrogel will regain its initial properties at 37 °C very quickly. The time frame between the temperature set and the beginning of the modulus increase is about 150 s. It can be easily explained and has to be subtracted to the time of gelation. Indeed, the time needed by the Peltier temperature control to reach the set temperature inside the geometry (double wall concentric cylinder) was found to be 290 s. This time becomes largely lower if another geometry with less metal envelope, such as a cone-plate geometry, is used. Otherwise, these results also indicate that injections do not cause irreversible damage to the polymer and do not affect the thermo-gelling behavior. Thus, we confirm that such thermosensitive hydrogels are perfectly injectable, whatever the injection used method.
Excepted for both 50 and 100 Pa which are not strong enough to completely break the structure of the hydrogel at 37 °C, the results of Fig. 9 show that the high shear stresses lead to a decrease in moduli due to the disorganization of the physically associated network. This becomes noticeable from 150 Pa with G′ lower than G′′ (that means a liquid and viscous state). Whatever the applied stress, once the stress is removed, the hydrogel returns very quickly to its initial gelled state without irreversible damage.
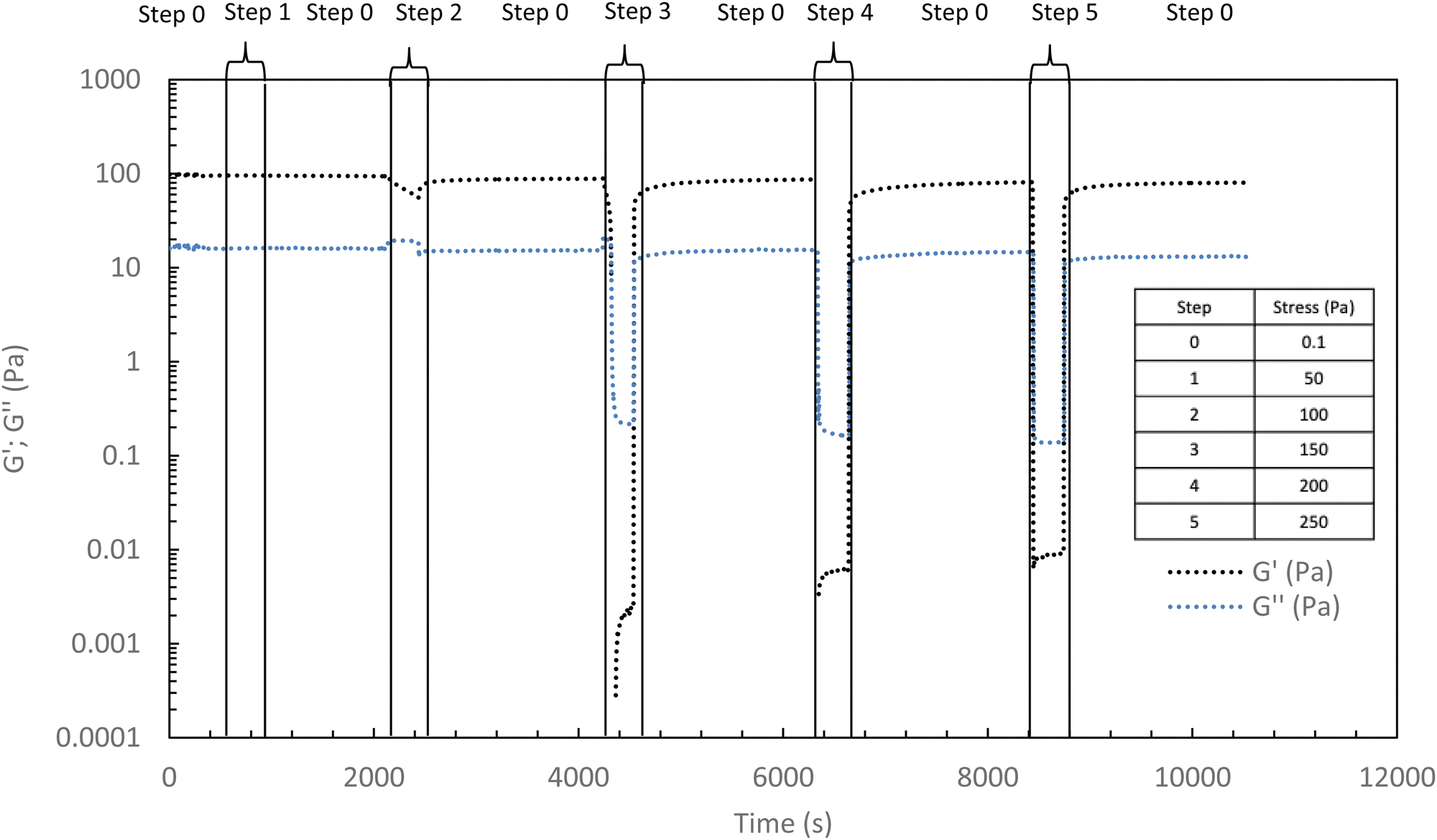 | ||
| Fig. 9 Elastic and viscous moduli during different stresses applications (ranging from 50 to 250 Pa) to the hydrogel (HA-PiPrOx-co-PBuOx-66/34-0.10, 15 g L−1 in NaCl 0.15 M) at 37 °C (1 Hz) with a rest time (0.1 Pa, 1 Hz) between stresses. | ||
3.5 Incorporation of a secondary network (semi-interpenetrating network (IPN))
For future applications, we could be led to add new properties to the hydrogel, such as contrast agents, drug delivery molecules, and others. Thus, it appears to be easier to modify a new native HA than an already thermo-sensitive HA. Indeed, the chosen strategy for a potential medical application will be to graft the various molecules onto new native HA chains and blend them into the thermosensitive hydrogel network. It must then be ensured that the incorporation of a secondary network based on native HA does not prevent the thermosensitive hydrogel from gelling.To confirm this strategy, a 15 g L−1 solution of the heat-sensitive HA-g-P(iPrOx-co-BuOx-67/33)-0.10 was mixed with 15 g L−1 native HA solution at different volume ratios at 4 °C and the gelation of the resulting mixture were then monitored. To begin, a mixture with a volume ratio of 75/25 thermosensitive/native HA was studied (Fig. 10). One can notice a slight decrease in G′ modulus for the hydrogel/HA mixture compared with the hydrogel alone. This drop in modulus can be explained by the secondary hyaluronic acid network which will prevent certain physical cross-linking nodes from forming that leads to a weakened 3D network. Nevertheless, the respective gelation temperature of the mixture and the corresponding thermosensitive HA remain identical, which is a promising feature for future research.
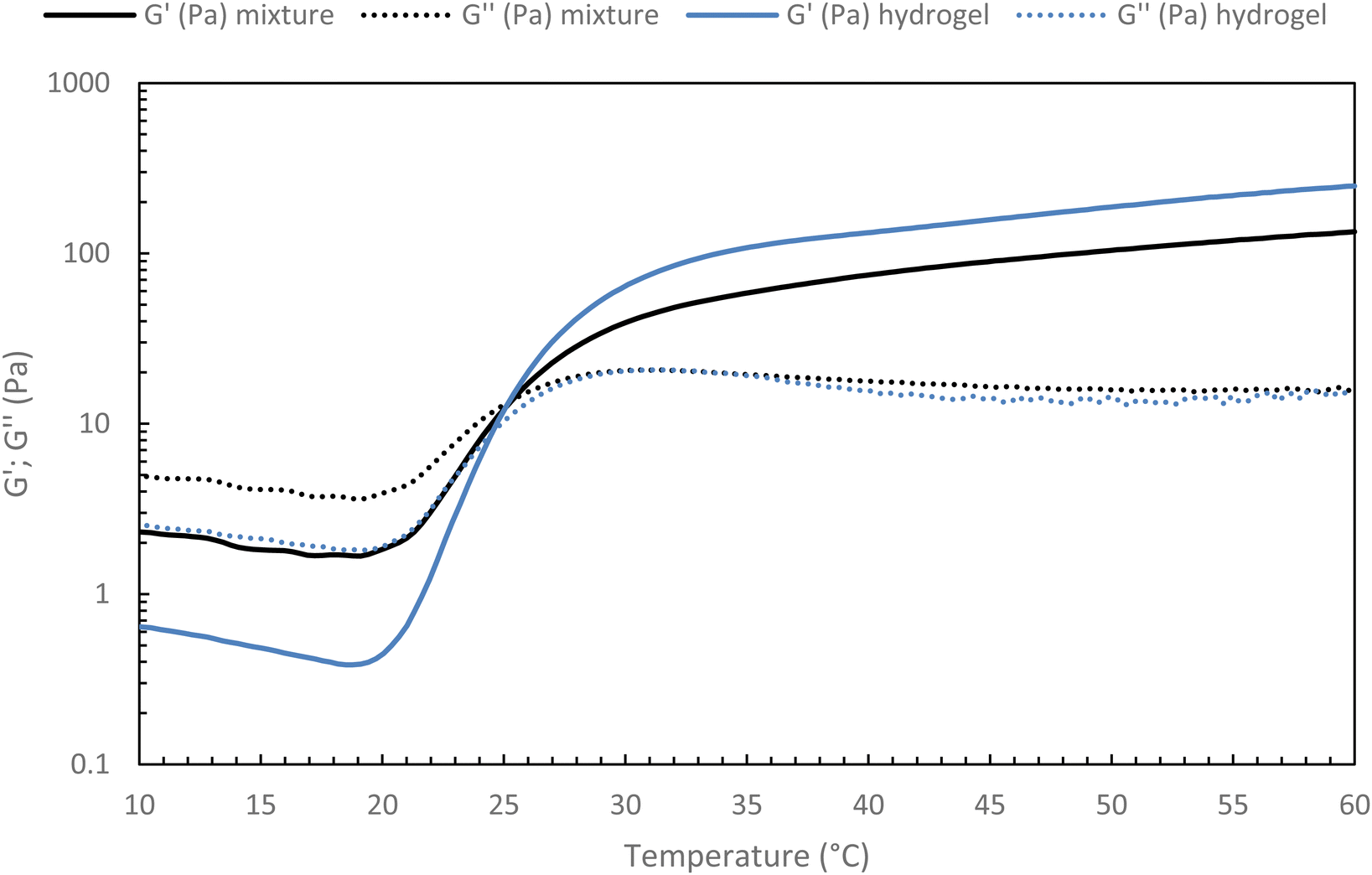 | ||
| Fig. 10 Temperature ramp for hydrogel alone (HA-g-(PiPrOx-co-PBuOx-67/33)-0.10) and hydrogel/HA mixture (75/25 – v/v) at 15 g L−1 in NaCl 0.15 M (0.1 Pa and 1 Hz; 0.5 °C min−1). | ||
Then, the native HA content in the mixture ratio was increased to find out whether a limit should not be exceeded to preserve the gelation of the mixture. To this end, different HA-g-P(iPrOx-co-BuOx-67/33)-0.10/native HA ratios were tested, and the results are presented in Fig. 11.
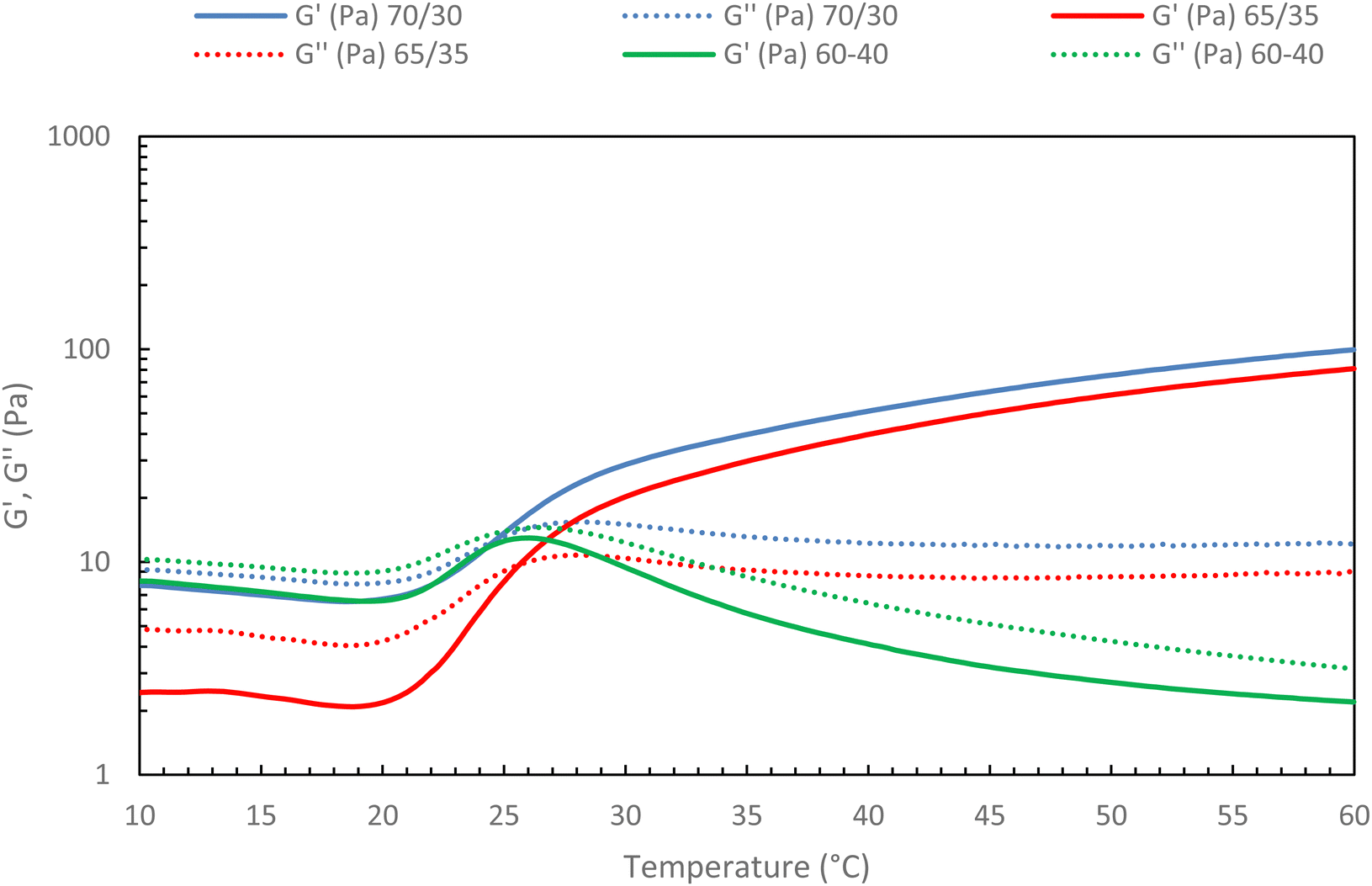 | ||
| Fig. 11 Rheological ramp of different mixture between HA-g-P(iPrOx-co-BuOx-67/33)-0.10 and native HA (15 g L−1 in NaCl 0.15 M; 0.5 °C min−1, 0.1 Pa and 1 Hz). | ||
The first thing to note is that the more secondary network is added to the mixture, the more G′ decreases. This confirms that the secondary network will prevent certain cross-linking nodes from forming as was hypothesized above. Indeed, the more secondary network is added, the more difficult it becomes for the polyoxazoline chains to associate with each other to form the gel, up until a critical point is reached where gelation becomes impossible. Indeed, the absence of a crossover point between G′ and G′′ moduli is evidenced for a 60/40 ratio (green curves) in Fig. 11, with G′′ being higher than G′ throughout the temperature range. This behavior indicates the absence of gelation in the system, implying that it will be impossible to add a secondary network in proportions greater than 35% without losing the thermosensitive character of the mixture at 15 g L−1. This is an encouraging result, as it indicates that the incorporation of hyaluronic acid modified with molecules of interest should be feasible for future studies.
Studies of stability (S6 supporting data, ESI†), flow behavior (S7 supporting data, ESI†), and injectability (S8 supporting data, ESI†) of the 75/25 ratio mixtures are perfectly similar in terms of interpretation to the pure thermosensitive systems discussed above.
Conclusions
Following on from our previous work,34 the thermoresponsive HA-g-P(iPrOx-co-BuOx) copolymer was designed to be ideally suited to an injectable biomedical application. We obtained a grafting of the P(iPrOx-co-BuOx) copolymer (67/33 molar ratio; DP: 25) with a grafting rate of around 10%. This grafting results in a reversible thermosensitive hydrogel (at 15 g L−1) with a G′, G′′ crossover point of around 25 °C (for a G′ at 37 °C of around 80 Pa ± 20). We have shown that this hydrogel is perfectly stable at 37 °C for at least 5 days. We have also demonstrated that the gel is maintained when submitted to HAase action for 4 days, even if some chain cuts occur. These are very encouraging results for future applications in the human body, but further studies will be necessary notably in vivo for which other variables could occur. In addition, the slight shear thinning behavior of the solution at 20 °C led to comfortable injectability conditions. Nevertheless, in the gel state, injectability measurements showed high resistance, requiring the hydrogel to be subjected to high stresses leading to the breaking of the 3D network. However, once the stress is removed, the gel network reforms very quickly without causing irreversible damage to the polysaccharide chains. These good results prompted us to try the incorporation of a secondary network (HA) inside the heat-sensitive hydrogel, with the aim of grafting therapeutic molecules onto it at a later stage. The results obtained showed that a secondary network could be added to the hydrogel in a maximum proportion of 35 wt%. The mixed gel showed a slight loss of G′ modulus at 37 °C but remained stable at 37 °C for at least 3 days and had the same injection ability as the hydrogel alone. These results are very encouraging and allow us to envisage the incorporation of therapeutic molecules in the future.Author contributions
Morgane Morel: investigation, writing the original draft. Mathieu Madau: supervision – writing – review & editing. Virginie Dulong: supervision – writing – review & editing. Anne-Claire Groo: supervision – writing – review & editing. Aurélie Mazert-Fréon: supervision – writing – review & editing. Didier Le Cerf: supervision – writing – review & editing. Luc Picton: supervision – writing – review & editing.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors would like to thank the Normandy region for their financial contribution to the RIN TREMPLIN NeuroOncoChimie project (ERDF funding). The authors would also like to thank Koceila Boundaoui for his help with injectability studies at 37 °C.References
- C. Zhao, L. Zhou, M. Chiao and W. Yang, Adv. Colloid Interface Sci., 2020, 285, 102280 CrossRef PubMed.
- I. Tokarev and S. Minko, Soft Matter, 2009, 5, 511–524 RSC.
- M. Hamidi, A. Azadi and P. Rafiei, Adv. Drug Delivery Rev., 2008, 60, 1638–1649 CrossRef CAS PubMed.
- S. Mantha, S. Pillai, P. Khayambashi, A. Upadhyay, Y. Zhang, O. Tao, H. M. Pham and S. D. Tran, Materials, 2019, 12(20), 3326 CrossRef PubMed.
- M. C. Catoira, L. Fusaro, D. Di Francesco, M. Ramella and F. Boccafoschi, J. Mater. Sci.: Mater. Med., 2019, 30, 115 CrossRef PubMed.
- M. Guenther, G. Gerlach, T. Wallmersperger, M. N. Avula, S. H. Cho, X. Xie, B. V. Devener, F. Solzbacher, P. Tathireddy, J. J. Magda, C. Scholz, R. Obeid and T. Armstrong, Adv. Sci. Technol., 2013, 85, 47–52 CAS.
- A. C. Jen, M. C. Wake and A. G. Mikos, Biotechnol. Bioeng., 1996, 50, 357–364 CrossRef CAS PubMed.
- T. R. Hoare and D. S. Kohane, Polymer, 2008, 49, 1993–2007 CrossRef CAS.
- J. Lou and D. J. Mooney, Nat. Rev. Chem., 2022, 6, 726–744 CrossRef CAS PubMed.
- S. Graham, P. F. Marina and A. Blencowe, Carbohydr. Polym., 2019, 207, 143–159 CrossRef CAS PubMed.
- L. Yu and J. Ding, Chem. Soc. Rev., 2008, 37, 1473–1481 RSC.
- D. J. Overstreet, D. Dutta, S. E. Stabenfeldt and B. L. Vernon, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 881–903 CrossRef CAS.
- E. Hasanzadeh, A. Seifalian, A. Mellati, J. Saremi, S. Asadpour, S. E. Enderami, H. Nekounam and N. Mahmoodi, Mater. Today Bio, 2023, 20, 100614 CrossRef CAS PubMed.
- C. Mo, R. Luo and Y. Chen, Macromol. Rapid Commun., 2022, 43, 2200007 CrossRef CAS PubMed.
- M. Norouzi, J. Firouzi, N. Sodeifi, M. Ebrahimi and D. W. Miller, Int. J. Pharm., 2021, 598, 120316 CrossRef CAS PubMed.
- J. Basso, A. Miranda, S. Nunes, T. Cova, J. Sousa, C. Vitorino and A. Pais, Gels, 2018, 4, 62 CrossRef PubMed.
- J. Zhang, C. Chen, A. Li, W. Jing, P. Sun, X. Huang, Y. Liu, S. Zhang, W. Du, R. Zhang, Y. Liu, A. Gong, J. Wu and X. Jiang, Nat. Nanotechnol., 2021, 16, 538–548 CrossRef CAS PubMed.
- M. D’Este, M. Alini and D. Eglin, Carbohydr. Polym., 2012, 90, 1378–1385 CrossRef PubMed.
- G. Mocanu, D. Mihaï, V. Dulong, L. Picton and D. Le Cerf, Carbohydr. Polym., 2012, 87, 1440–1446 CrossRef CAS.
- R. Hoogenboom and H. Schlaad, Polym. Chem., 2016, 8, 24–40 RSC.
- D. J. Overstreet, R. Y. McLemore, B. D. Doan, A. Farag and B. L. Vernon, Soft Mater., 2013, 11, 294–304 CrossRef CAS.
- V. Dulong, G. Mocanu, L. Picton and D. Le Cerf, Carbohydr. Polym., 2012, 87, 1522–1531 CrossRef CAS.
- P. M. Niang, Z. Huang, V. Dulong, Z. Souguir, D. Le Cerf and L. Picton, Carbohydr. Polym., 2016, 139, 67–74 CrossRef CAS PubMed.
- G. Mocanu, Z. Souguir, L. Picton and D. Le Cerf, Carbohydr. Polym., 2012, 89, 578–585 CrossRef CAS PubMed.
- X. Xu, Y. Liu, W. Fu, M. Yao, Z. Ding, J. Xuan, D. Li, S. Wang, Y. Xia and M. Cao, Polymers, 2020, 12, 580 CrossRef CAS PubMed.
- T. X. Viegas, M. D. Bentley, J. M. Harris, Z. Fang, K. Yoon, B. Dizman, R. Weimer, A. Mero, G. Pasut and F. M. Veronese, Bioconjugate Chem., 2011, 22, 976–986 CrossRef CAS PubMed.
- A. L. Fisher, J. M. H. Schollick, D. G. A. L. Aarts and M. C. Grossel, RSC Adv., 2016, 6, 66438–66443 RSC.
- A. Bogomolova, S. K. Filippov, L. Starovoytova, B. Angelov, P. Konarev, O. Sedlacek, M. Hruby and P. Stepanek, J. Phys. Chem. B, 2014, 118, 4940–4950 CrossRef CAS PubMed.
- A. Amirova, S. Rodchenko, M. Kurlykin, A. Tenkovtsev, I. Krasnou, A. Krumme and A. Filippov, Polymers, 2020, 12, 698 CrossRef CAS PubMed.
- A. Borzacchiello, L. Russo, B. M. Malle, K. Schwach-Abdellaoui and L. Ambrosio, BioMed Res. Int., 2015, 2015, e871218 Search PubMed.
- J. A. Burdick and G. D. Prestwich, Adv. Mater., 2011, 23, H41–H56 CrossRef CAS PubMed.
- S. Federico, U. Nöchel, C. Löwenberg, A. Lendlein and A. T. Neffe, Acta Biomater., 2016, 38, 1–10 CrossRef CAS PubMed.
- I. S. Bayer, Molecules, 2020, 25, 2649 CrossRef CAS PubMed.
- M. Madau, G. Morandi, V. Lapinte, D. Le Cerf, V. Dulong and L. Picton, Polymer, 2022, 244, 124643 CrossRef CAS.
- M. Madau, G. Morandi, C. Rihouey, V. Lapinte, H. Oulyadi, D. L. E. Cerf, V. Dulong and L. Picton, Polymer, 2021, 230, 124059 CrossRef CAS.
- G. Alonci, R. Mocchi, S. Sommatis, M. C. Capillo, E. Liga, A. Janowska, L. Nachbaur and N. Zerbinati, Pharmaceutics, 2021, 13, 1194 CrossRef CAS PubMed.
- Y. Li, X. Zhang, X. Liu, W. Pan, N. Li and B. Tang, Chem. Sci., 2021, 12, 3130–3145 RSC.
- J. L. Reissig, J. L. Strominger and L. F. Leloir, J. Biol. Chem., 1955, 217, 959–966 CrossRef CAS PubMed.
- H. Duan, M. Donovan, F. Hernandez, C. Di Primo, E. Garanger, X. Schultze and S. Lecommandoux, Angew. Chem., Int. Ed., 2020, 59, 13591–13596 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tb02108d |
| This journal is © The Royal Society of Chemistry 2024 |