
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effective design of PEGylated polyion complex (PIC) nanoparticles for enhancing PIC internalisation in cells utilising block copolymer combinations with mismatched ionic chain lengths†
Fadlina
Aulia
a,
Hiroaki
Matsuba
a,
Shoya
Adachi
a,
Takumi
Yamada
a,
Ikuhiko
Nakase
g,
Teruki
Nii
b,
Takeshi
Mori
 bc,
Yoshiki
Katayama
bcdef and
Akihiro
Kishimura
*bcd
bc,
Yoshiki
Katayama
bcdef and
Akihiro
Kishimura
*bcd
aGraduate School of Systems Life Sciences, Kyushu University, 744 Moto-oka, Nishi-ku, Fukuoka 819-0395, Japan
bDepartment of Applied Chemistry, Faculty of Engineering, Kyushu University, 744 Moto-oka, Nishi-ku, Fukuoka 819-0395, Japan. E-mail: kishimura.akihiro.776@m.kyushu-u.ac.jp
cCenter for Future Chemistry, Kyushu University, 744 Moto-oka, Nishi-ku, Fukuoka 819-0395, Japan
dCenter for Molecular Systems, Kyushu University, 744 Moto-oka, Nishi-ku, Fukuoka 819-0395, Japan
eCenter for Advanced Medical Open Innovation, Kyushu University, 744 Moto-oka, Nishi-ku, Fukuoka 819-0395, Japan
fDepartment of Biomedical Engineering, Chung Yuan Christian University, 200 Chung Pei Rd., Chung Li, Taiwan 32023, ROC
gDepartment of Biological Chemistry, Graduate School of Science, Osaka Metropolitan University, 1-1, Gakuen-cho, Naka-ku, Sakai-shi, Osaka 599-8531, Japan
First published on 20th January 2024
Abstract
In nanomedicine, PEGylation of nanomaterials poses a dilemma since it inhibits their interaction with target cells and enables their retention in target tissues despite its biocompatibility and nonspecific internalisation suppression. PEGylated polypeptide-based polyion complexes (PICs) are fabricated via the self-assembly of PEGylated aniomers and homocatiomers based on electrostatic interactions. We propose that various parameters like block copolymer design and PIC domain characteristics can enhance the cell–PEGylated PIC interactions. Remarkably, the properties of the PIC domain were tuned by the matched/mismatched ionomer chain lengths, PIC domain crosslinking degree, chemical modification of cationic species after crosslinking, PIC morphologies (vesicles/micelles) and polyethylene glycol (PEG) chain lengths. Cellular internalisation of the prepared PICs was evaluated using HeLa cells. Consequently, mismatched ionomer chain lengths and vesicle morphology enhanced cell–PIC interactions, and the states of ion pairing, particularly cationic residues, affected the internalisation behaviours of PICs via acetylation or guanidinylation of amino groups on catiomers. This treatment attenuated the cell–PIC interactions, possibly because of reduced interaction of PICs with negatively charged species on the cell-surface, glycosaminoglycans. Moreover, morphology and PEG length were correlated with PIC internalisation, in which PICs with longer and denser PEG were internalised less effectively. Cell line dependency was tested using RAW 264.7 macrophage cells; PIC recognition could be maintained after capping amino groups on catiomers, indicating that the remaining anionic groups were still effectively recognised by the scavenger receptors of macrophages. Our strategy for tuning the physicochemical properties of the PEGylated PIC nanocarriers is promising for overcoming the PEG issue.
Introduction
The introduction of polyethylene glycol (PEG) into a polymer or the surface of nanomaterials, termed PEGylation, has become a gold standard to enhance the biocompatibility, anti-fouling activity, and colloidal stability of such materials as well as to reduce their immunogenicity and possibility of enzyme digestion.1 PEGylation can prolong the blood retention time of nanomaterials and promote their accumulation in tumour sites via the enhanced permeability and retention (EPR) effect. The effect of prolonged circulation can be achieved by reducing cell interaction, as reported for several PEGylated nanoparticles, PEGylated liposomes, and several inorganic-sourced nanoparticles, such as iron, silver and gold.2,3 However, biomacromolecular drugs, such as nucleic acids and proteins, must be delivered inside the target cells to show their therapeutic effects. Based on an in vitro study reported by Mishra et al.,4 PEGylation of polycation–plasmid DNA complexes suppressed their cellular uptake and then reduced their protein expression to less than half of that of the non-PEGylated variants. Several solutions have been suggested to overcome these problems, such as installing targeting ligands on the PEG surface to enhance cellular uptake and environmentally responsive cleavable PEGylation.5,6 However, to realise a more versatile and facile application of PEGylated nanocarriers, the correlation between their physicochemical properties and cellular internalisation must be clarified in the presence of PEG without cumbersome modifications. In other words, the cellular internalisation of nanocarriers without PEG detachment or ligand introduction should provide insights into an effective design strategy to overcome the PEG dilemma. Oh et al. discussed how the PEGylated gold nanoparticle size affected its cellular uptake, but in the presence of a peptide ligand.7 Li et al. studied the endocytosis behavior of PEGylated nanocarriers using dissipative particle dynamics (DPD) simulations but from the view point of PEG properties.8 Pelaz et al. investigated the effect of PEG-coated nanoparticles on the cellular uptake from the viewpoint of protein adsorption.3 As no specific studies discussed the aspect of materials design, particularly the physicochemical nature of nanoparticles themselves, we attempted to focus on tuning the nanocarrier physicochemical properties, followed by PEG characteristics.Our goal is to identify parameters related to the properties of nanocarriers that can promote their cell internalisation and compromise the impact of PEG on the nanocarriers. To this end, we decided to use PEGylated polyion complexes (PICs), which are prepared from PEGylated polyelectrolytes under aqueous conditions via electrostatic complexation, to provide different types of nanoarchitectures designed for drug delivery systems (Scheme 1). The most typical example is the PIC micelle, first reported as an assembly of oppositely charged block copolymers with a PEG segment in an aqueous environment.9 Since PIC micelles are characterised by their sizes of several tens of nanometres and protective hydrophilic and biocompatible shell and drug-loadable core, they can show prolonged blood circulation time and enhanced permeability and retention (EPR) effect, which are favourable properties for a drug delivery vehicle.10,11 Many studies reported the successful loading of plasmid DNA, mRNA, oligonucleotides, and proteins into PIC micelles.10–15 Thus, they can be a promising carrier for developing non-viral gene delivery systems for vaccines and oligonucleotide therapeutics for cancer therapy.10,15 A relatively new PIC nanoarchitecture is the PICsome, a nanosized unilamellar vesicular PIC with a hollow structure.16 PICsomes can be prepared from PEGylated polyelectrolytes by tuning the chemical design of PEGylated polymers, such as the PEG fraction17,18 and charged species,19,20 which allow effective encapsulation of neutral and charged macromolecules, such as dextran,21 enzymes,22,23 and inorganic nanoparticles,24,25 and incorporate oligonucleotides in their PIC membrane.26,27 Compared to lipid-based vesicles, PICsomes are featured by their unique semipermeable PIC membrane, which is useful for retaining macromolecular cargoes and transporting small molecular weight compounds, e.g., drugs and substrates for enzymes, across their membrane. Therefore, PICsomes can function in vivo as nanoreactors and drug reservoirs.22,23,28–30 Besides, the PIC domain's physicochemical properties, particularly stability and semipermeability, can be modulated by crosslinking31,32 and the choice of side chain functionalities of the polyelectrolytes.28,33 Crosslinked PICsomes show size-dependent disposition into organs and tumour sites in vivo.31 Thus, PICs can provide a promising platform to obtain nano-vehicles with systematic design based on a series of chemically tuned components. This platform is helpful for clarifying the correlation between the physicochemical properties of nanocarriers and their ability to interact with cells and further internalisation.
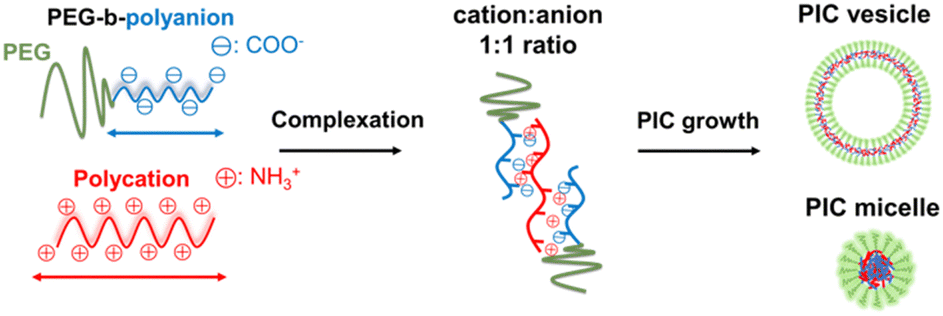 | ||
| Scheme 1 Illustration of typical PIC nanoarchitecture formation. | ||
In this study, we demonstrate that fine-tuning the physicochemical properties of PICs is a promising solution for overcoming the PEG dilemma. A series of PEG-based block ionomers were synthesised by varying their charged segment and PEG lengths to fabricate various types of PIC nanoparticles and thus evaluate their internalisation in living cells. To modulate the properties of the PIC domains, combinations of polymers with mismatched ionomer lengths were examined to enhance the cellular internalisation. In addition, the importance of PIC domain crosslinking and its fine-tuning, such as cationic residue modification, which modulates PIC interactions with the cell surface, was revealed. To the best of our knowledge, this study is the first that sought to reveal the correlations between the physicochemical properties of PEGylated PICs and their cellular internalisation. The convenient design and tunable characteristics of PICs might provide key insights into biomaterials development and applications to overcome the PEG dilemma.
Experimental
Materials
β-Benzyl-L-aspartate N-carboxy-anhydride (BLA-NCA) and ε-trifluoroacetyl-L-lysine N-carboxy-anhydride (L-Lys(TFA)-NCA) were obtained from Chuo Kaseihin Co. Inc. (Tokyo, Japan). α-Methoxy-ω-amino poly(ethylene glycol) (MeO–PEG–NH2) (number average molecular weight, Mn = 2000, 5000) was purchased from NOF Co. Ltd (Tokyo, Japan) and purified by ion-exchange chromatography using CM Sephadex C-50 from Sigma-Aldrich (Missouri, USA). n-Butylamine (special grade) was purchased from Wako Pure Chemical Co., Ltd (Osaka, Japan), refluxed for 3 h, and distilled over CaH2 at 78 °C under a N2 atmosphere. Acetonitrile, dimethyl sulfoxide (DMSO), methanol, n-hexane, chloroform (CHCl3) (super dehydrated), benzene (super dehydrated), dichloromethane (super dehydrated), and N,N-dimethylformamide (DMF) (super dehydrated) were also purchased from Wako Pure Chemical Industries and used as received. Di-isopropyl ethyl amine (DIPEA) and deuterium oxide (D2O) were purchased from Sigma-Aldrich. The Cy3 NHS ester was purchased from LumiProbe (Maryland, OH, USA). 1-Ethyl-3-(3-(dimethylamino)propyl)carbodiimide hydrochloride (EDC) was purchased from Tokyo Chemical Industry Co., Ltd (Tokyo, Japan). Calcein was purchased from Wako Pure Chemical Co., Ltd. 3,5-Dimethyl-1-guanylpyrazole nitrate (DMGP) was purchased from Sigma-Aldrich. Acetic anhydride and 2,4,6-trinitrobenzenesulphonic acid sodium salt dihydrate were purchased from Wako Pure Chemical Industries, Ltd. Copper grids (150 mesh) coated with a thin Formvar film and reinforced with a carbon coating were purchased from JEOL (Tokyo, Japan).Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1 at λ = 570 nm). For further particle fabrication, the labelling ratio was adjusted to 10% of the total polyanion chains by mixing with a non-labelled polymer solution.
000; Scharlab, Barcelona, Spain). The results are summarised in Table 1.
000 for vesicles, 100000 for micelles; VivaSpin 6, Sartorius AG, Gottingen, Germany) by exchanging the solvent with UPW three times at 4 °C using gravity force.
000 M−1 cm−1 at λ = 570 nm). For further particle fabrication, the labelling ratio was adjusted to 10% of the total polyanion chains by mixing with a non-labelled polymer solution.
000; Scharlab, Barcelona, Spain). The results are summarised in Table 1.
000 for vesicles, 100000 for micelles; VivaSpin 6, Sartorius AG, Gottingen, Germany) by exchanging the solvent with UPW three times at 4 °C using gravity force.
| PIC type | Polyanion (DP) | Polycation (DP) | f PEG (%) | Average diameter (nm) | Polydispersity index (PdI) | ζ-potential (mV) | Crosslinking degree of PICs (%) | Amine modification rate (%) |
|---|---|---|---|---|---|---|---|---|
| a f PEG: PEG fraction after complexation, defined by the fraction of PEG in a total PIC. See eqn (S3) in the ESI. | ||||||||
| Vesicle | PEG2k–PAsp(56) | Homo–PLL(100) | 10 | 127 | 0.03 | −1.5 | None (N) | |
| 159 | 0.13 | −9.9 | 50 | 50 (for acetylation, A) | ||||
| 106 | 0.12 | −1.8 | 20 (for guanidinylation, G) | |||||
| 162 | 0.08 | −0.2 | 80 | — | ||||
| PEG2k–PAsp(69) | Homo–PLL(64) | 9 | 180 | 0.07 | −1.8 | 80 | — | |
| PEG2k–PAsp(93) | Homo–PLL(64) | 7 | 200 | 0.2 | −1.2 | 80 | — | |
| 2k-Micelle | PEG2k–PAsp(56) | PEG2k-PLL(81) | 14 | 29 | 0.03 | −2.1 | 50 | — |
| 5k-Micelle | PEG5k–PAsp(70) | homo–PLL(100) | 19 | 42 | 0.03 | −3.6 | 50 | — |
| Crosslinking degree [%] = 100 − (detected amino groups, %) |
000) by gravity force. A buffer solution of DMGP (40 mg mL−1) was then added to the PIC solution with 50 equiv. of residual amines in the PIC particles. After shaking for 12 h at 40 °C, the resulting mixture was purified by ultrafiltration (VivaSpin 6, MWCO: 300000) by gravity force using UPW. Thereafter, the modification rate was evaluated using the same method as described in the ‘Determination of the crosslinking degree’ section by quantifying residual amines.
000 U mL−1, streptomycin 10000 μg mL−1 and amphotericin B 25 μg mL−1) (Nacalai Tesque Inc., Kyoto, Japan). CHO-K1 (wild-type) and CHO-A745 (glycosaminoglycan-deficient cells) were cultured in Ham's F-12K (Kaighn's Modification) (Fujifilm Wako Pure Chemical Corporation, Osaka, Japan) as a basic medium supplemented with 10% FBS and 1% antibiotic–antimycotic mixed stock solution under 5% CO2 at 37 °C. Human colorectal adenocarcinoma (Caco2) cells were cultured in phenol red-containing DMEM (Nacalai Tesque Inc.) supplemented with 10% FBS (GIBCO), 1% antibiotic–antimycotic mixed stock solution (Nacalai Tesque Inc.) and 1% MEM non-essential amino acid solution (Nacalai Tesque Inc.). Mice dendritic cells (DC 2.4) were cultured in Roswell Park Memorial Institute-1640 (RPMI-1640) medium (Nacalai Tesque Inc.) supplemented with FBS (GIBCO), 1% antibiotic–antimycotic mixed stock solution (Nacalai Tesque Inc.) and 2-mercaptoethanol (final conc. 50 μM) (Fujifilm; Wako Pure Chemical Corporation). Cell passage was performed after suspending the cells for 5 min in enzyme-free cell dissociation solution (Merck, Frankfurt, Germany), followed by centrifugation at 1000 rpm for 3 min to obtain a cell pellet. The cells were resuspended in the cell culture medium, seeded at 2 × 105 cells in 10 mL of the medium on a 10 mm culture dish, and allowed to grow to 80% confluency.
Results and discussion
Polymer synthesis and characterisation
Polypeptide-based materials have been generally used for polyelectrolyte complexation because of their feasible polymerisation and stable secondary and tertiary structures.37 A combination of carboxyl groups from PAsp as an anion source and amine groups from PLL as a cation source was selected because both ionic residues exhibited high ionisation rates at neutral pH.38 Catiomers and aniomers were prepared as homo- and block copolymers using PEGs with different molecular weights of 2000 (PEG2k) and 5000 (PEG5k). The DP was designed to be between 60 and 100 repeating units for homo- and PEG-block copolymers. The DP was determined by NMR spectroscopy, and Đ of the polymers was determined by SEC (Table 1 and Fig. S1–S7 (NMR) and Fig. S9–S15 (SEC) in the ESI†).PIC fabrication and characterisation
In this study, we fabricated PICs with an initial polymer concentration of 1 mg mL−1 for all cases and a charge ratio under neutral conditions (anionic–cationic ratio = 1:1) (Scheme 1). All PIC samples were chemically crosslinked for biological evaluation. Crosslinking was performed using EDC to crosslink –NH2 on a catiomer side chain and –COOH on an aniomer side chain to form an amide bond, which can increase particle robustness.18 The resulting PICs were purified and subjected to a TNBS assay for crosslinking degree determination to obtain PICs with crosslinking degrees of 50% and 80%. The three different morphologies are listed in Table 2: vesicles, micelles from PEG2k–PAsp and PEG2k–PLL (2k-micelle), and micelles from PEG5k–PAsp and homo–PLL (5k-micelle). The morphologies of the resulting particles were confirmed using TEM (Fig. S16, ESI†). Zeta potential measurements indicated that all PICs had almost neutral values.
Correlation between the physicochemical properties of PICs and their cellular internalisation
:1; thus, the overall charges were assumed to be neutral regardless of the chain length. DLS measurement indicated that PIC vesicles had a monodisperse distribution (Fig. S23, ESI†). Their cellular internalisation was evaluated using HeLa cells (human cervical cancer cells) by two methods: microscopic imaging by CLSM to observe internalised particles and FCM quantification to quantify the total fluorescence of particles on the cell surface and inside cells.
Consequently, mismatched pairings of PEG–PAsp and homo–PLL with the DP (56, 100) and (93, 64), respectively, exhibited a higher cellular internalisation than matched pairing with the DP (69, 64) (Fig. 1). Furthermore, there was no significant difference between (56, 100) and (93, 64), indicating no preference for longer polymer charge signs. Notably, cellular internalisation was found to be similar for PIC (56, 100) and (93, 64), despite the difference in the weight fraction of PEG (fPEG) values. In all cases, PICs were found inside the cells and were possibly internalised by endocytosis. Nevertheless, by the calcein leakage assay of PIC (56, 100), endosomal escape cannot be confirmed, which is supported by the overlapping fluorescence signal of calcein and PIC entrapped in endosomes (Fig. S25, ESI†). Overall, these results suggested that mismatched pairing in PICs can enhance their cellular internalisation. Presumably, this trend can be explained by the existence of dynamic and transient unpaired ionic residues, that is, a frustrated state in proximity to the interfacial region of the PIC domain and PEG palisades. Since the length of the polymer might contribute to cellular internalisation, that is, shorter chain lengths might suppress the internalisation to a certain extent, we focused on PICs with mismatched chain lengths with slightly shorter PAsp and longer PLL for further evaluation in the remainder of this study.
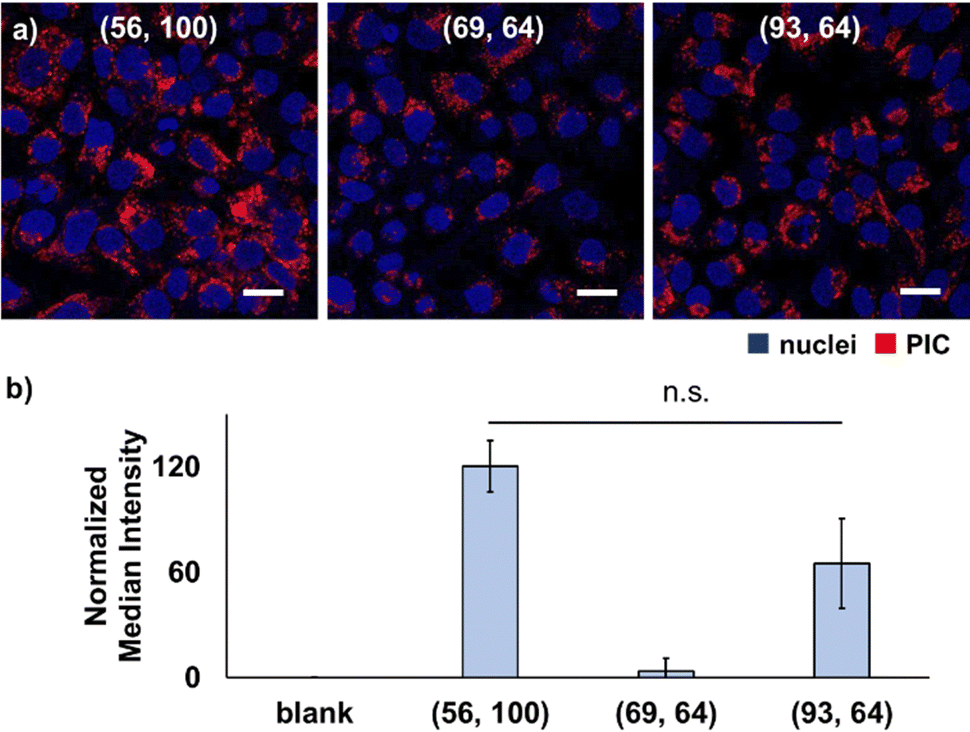 | ||
| Fig. 1 Cellular internalisation of vesicles with different chain length combinations. (a) CLSM images of HeLa cells after 24-h incubation with the 80%-CL PIC with different chain length combinations. Blue, Hoechst 33342-(nuclei); red, Cy3-labelled PEG2k-PAsp (PICs). (b) Fluorescence quantification of cells after 24 h-incubation detected by FCM (means ± standard deviation). Chain lengths are shown as (the DP of the aniomer, the DP of the catiomer). | ||
Next, to test the effect of another type of cationic residue and guanidium group, guanidinylation was performed on the primary amino groups of the catiomers. In previous reports, guanidinylation was performed against the catiomer before PIC formation, and the resulting PIC particles showed improved stability for the application.33 However, direct comparison with regular PEG–PAsp/PLL PICsomes is difficult because the preparation conditions of guanidinylated polymer-based PICs were different from those of conventional PICsomes without guanidinium functionalities. In this study, guanidinylation was conducted for ready-made PIC particles, which allowed us to use the same platform particles and evaluate the effect of ionic residues more directly. The introduction of guanidinium groups into the PIC domain resulted in the formation of slightly smaller particles with similar ζ-potentials, possibly because of the stronger binding of –COO− and –NH-(C=NH2+)-NH2via salt bridge formation (Table 2).
Both modifications resulted in the decreased cellular internalisation by HeLa cells after 24 h of incubation (Fig. 2a and d). Capped cationic residues may impede the interaction between the acetylated vesicles and the cell surface. In the case of guanidinylated vesicles, the introduction of a guanidium group inside the PIC domain enhances the interaction between the catiomer and aniomer, stabilising the PIC domain to exhibit less interaction with cells. Considering these results, the cell–PIC interaction was mainly ascribed to the interaction between dynamic cationic residues and certain cell surface components, presumably negatively charged components, via electrostatic interactions. In other words, the presence of negatively charged materials on the cell surface and positively charged materials with high mobility in the PIC domain is essential for particle recognition on the cell surface and further cellular internalisation. Therefore, the states of the PIC domain are considered to be recognisable by the cell surface, indicating that the modulation of the states of the PIC domain is a promising strategy for managing the interaction of nanoparticles with living cells.
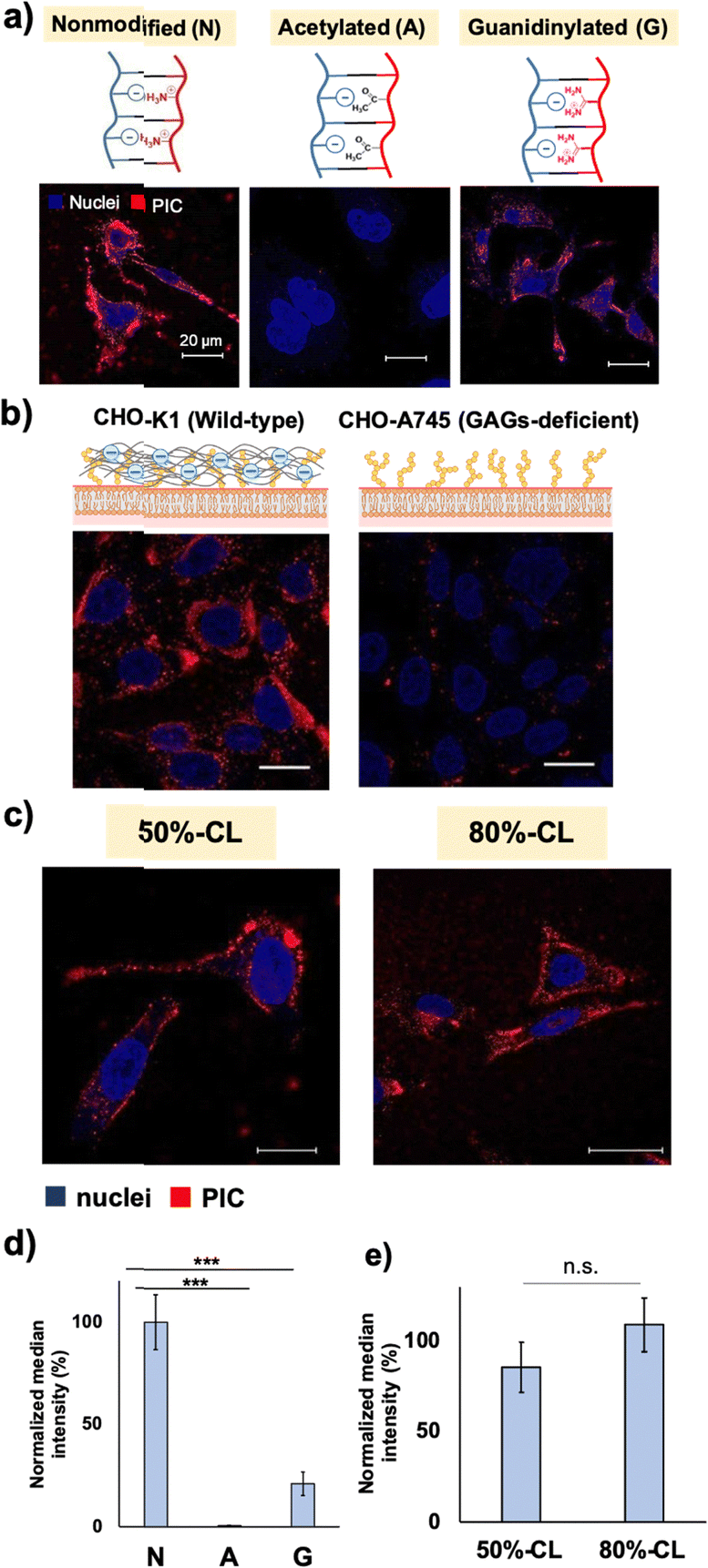 | ||
| Fig. 2 Effect of the charge state difference on the cell–PIC interaction. (a) CLSM images of HeLa cells treated with non-modified (N), acetylated (A) and guanidinylated (G) PIC vesicles after 24 h of incubation. (b) CLSM images of the effect of cell-surface glycosaminoglycans (GAGs) on the cell–PIC interaction using wild-type CHO-K1 (left) and GAG-deficient CHO-A745 (right). Red, 50%-CL vesicle; blue, Hoechst-stained nuclei (scale bar: 20 μm). (c) Fluorescence microscopic evaluation of the crosslinking degree dependency of the cell–PIC interaction using 50%- and 80%-CL vesicles (scale bar: 20 μm). (d) Quantification results of cellular internalisation of non-modified/modified PICs by FCM (y-axis was normalised to the N-vesicle) and (e) that between 50%- and 80%-CL vesicles by FCM (y-axis was normalised to the 80%-CL vesicle). | ||
First, the evaluation focused on vesicle deformability, which was expected to correlate with the degree of crosslinking owing to the restriction of polypeptide chain movement in the PIC domain. We estimated that the difference in deformability could be visualised using AFM, particularly by measuring the vesicle-spreading area on the mica substrate. The vesicle spreading area can be interpreted as the contact area on the cell surface upon recognition by cells. A series of vesicles with different degrees of crosslinking, namely 50%- and 80%-CL, were prepared (Table 2). Notably, PIC vesicles with a crosslinking degree of <50% could be obtained; however, particle loss occurred during the purification process; this loss may be attributed to nonspecific adsorption onto the filtration membrane. In addition, vesicles without crosslinking were not prepared because non-crosslinked PICs are unstable and their behaviour is unpredictable. After the crosslinking treatment, the number of residual amine groups was determined using the TNBS assay to determine the degree of crosslinking of the particles (Scheme S1 and eqn (S2), ESI†). The degree of crosslinking was obtained from the remaining amine ratio, which was calculated using the amount of remaining amine in the complex quantified by TNBS assay using a calibration curve for PLL (Fig. S19 and S20, ESI†). The remaining amine ratio was calculated as the ratio of the amount of remaining amine and the total amount of amine existing before crosslinking, which was obtained from the amount of polyanions under the assumption of equimolar charge ratio in the PIC (see Fig. S21 and S22 for polymer calibration curves, ESI†).
AFM images showed higher deformability for 50%-CL vesicles than that for 80%-CL vesicles, based on information on the spreading area on the mica surface (Fig. 3). However, no significant difference was observed between the vesicles in terms of cellular internalisation (Fig. 2c and 2e). Thus, the difference in deformability in this range may not be significant for determining the difference in cellular internalisation. This result has several interpretations. First, vesicle–mica interaction and subsequent deformation of vesicles occur more explicitly under in situ conditions than under cell culture conditions, under which proteins and other biological components inhibit the effective interaction of PICs with the cell surface. Second, although the charge density of mica (0.02–0.5 e− nm−2, as reported by Pashley, 1981 and Israelachvili and Adams, 1978)41,42 is comparable to that of the HeLa cell surface (−12 mC m−2, which is equal to 0.072 e− nm−2),43 the real cell surface is not as hard and flat as the mica surface. Specifically, the cells possess a three-dimensional proteoglycan structure that forces the PICs to behave differently. Finally, PICs with different deformabilities did not differ significantly in their interactions with the cell surface. Thus, in the present experimental design, such deformation of the cell surface was not the primary factor for the vesicles. Instead, dynamic cationic charges can be a dominant factor in cell–PIC interactions. In addition, PIC vesicles with 20–50% non-crosslinked cations exhibited similar interaction capabilities, regardless of the difference in the residual number of cationic charges in the PIC domain.
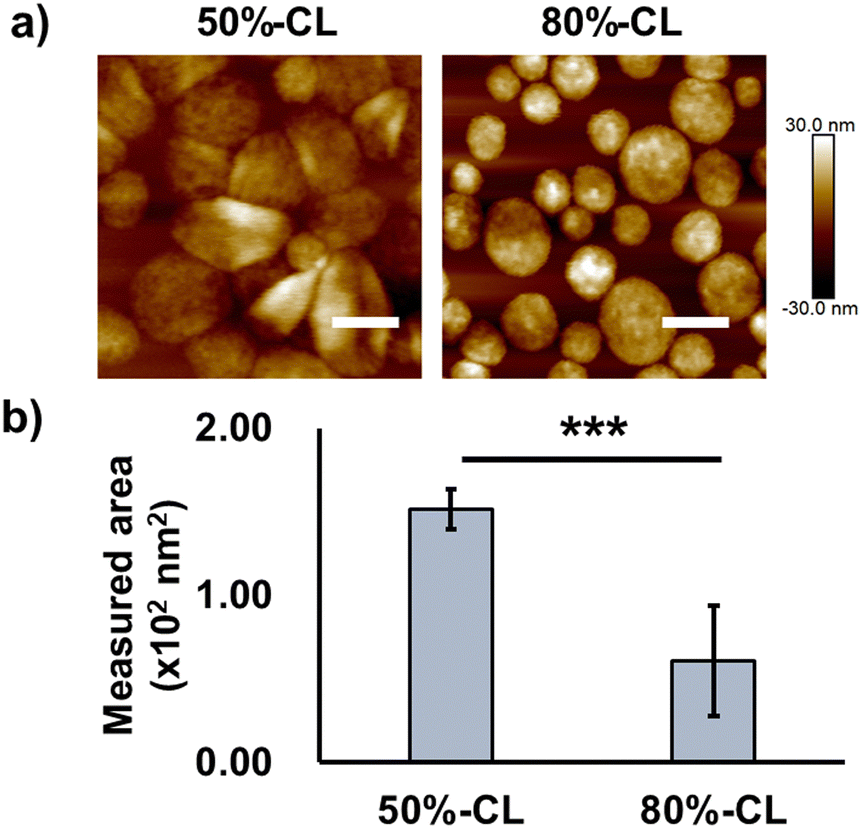 | ||
| Fig. 3 Deformable properties of PIC vesicles with different crosslinking rates. (a) AFM results of vesicles with different crosslinking rates (scale bar: 200 nm) measured under wet conditions (PBS buffer) on the mica surface; (b) the area of particle deformation (analysed using ImageJ, quantified from a minimum of 20 particles and shown as means ± standard deviation). | ||
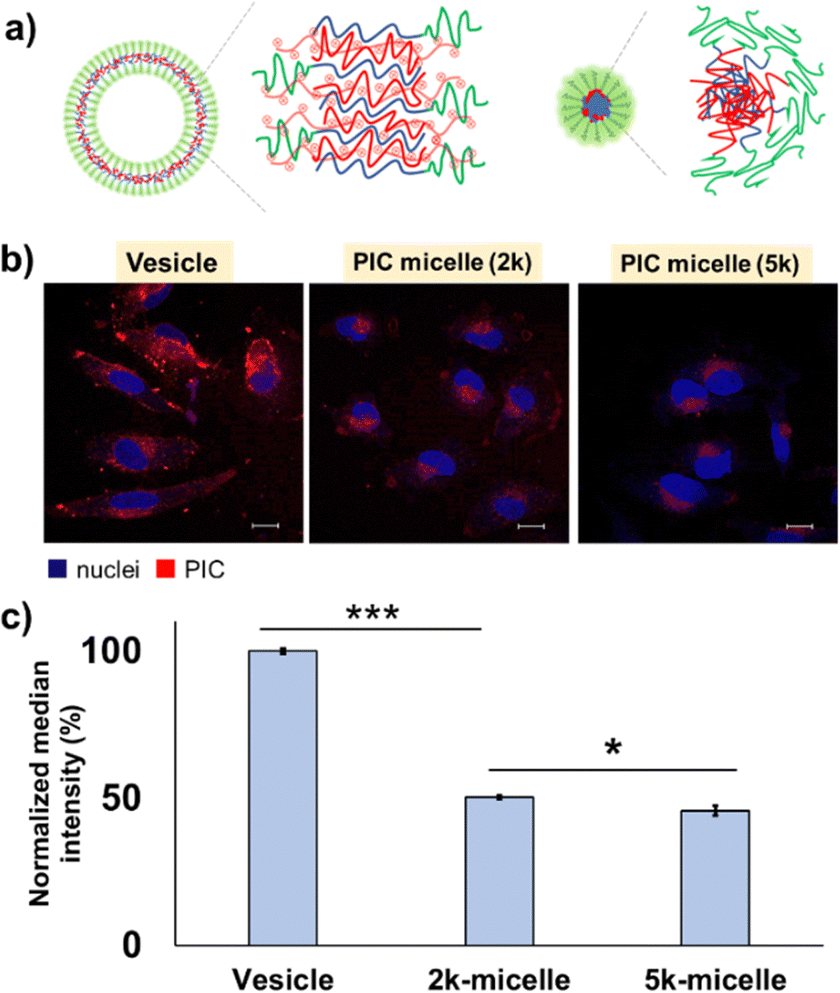 | ||
| Fig. 4 Morphology dependency of the cell–PIC interaction. (a) Schematic illustration of the domain structure difference between vesicles (left) and micelles (right); (b) CLSM observation of PIC vesicles, 2k-micelles and 5k-micelles after 24 h of incubation with HeLa cells (scale bar: 20 μm; blue, Hoechst-stained nuclei; red, PIC); and (c) their quantification results after 24 h of incubation with HeLa cells by FCM (y-axis is normalised against vesicles). | ||
The PIC domain structure and fPEG may be key to determining the trend for the interaction with cells. While the PIC micelle possesses a core–shell structure with higher fPEG (≥∼14%), the PIC vesicle has a hollow structure with lower fPEG (≤∼10%), which can provide a more accessible two-dimensional lamellar structure of PIC with a large area at its interface against a PEG layer. The unilamellar structure of the PIC vesicle was confirmed by cryo-TEM (Fig. S18, ESI†). The thickness of the PIC membrane was estimated to be ∼12.0 ± 2.5 nm, which is close to the previously reported value.16 Moreover, as shown in the AFM results, the hollow PIC architecture exhibited a deformable nature, which may result in a remarkable difference in the interaction with the cells. Presumably, the deformable vesicle architecture may promote the exposure of its PIC domain to the exterior, in contrast to the zero-dimensional architecture of the PIC micelle. However, we could not complete the deformability measurement of 2k-micelles owing to the less adherent properties of micelles on the mica surface. This behaviour can be ascribed to the effective coverage of PEG chains on the rigid PIC core.
It is noteworthy that PIC vesicles formed aggregates on the surface of most cells (Fig. S26, ESI†), while less aggregate formation was observed for 2k- and 5k-micelles; in fact, 10% and 8% of the cells (N ∼ 100) showed aggregate formation on their surface, respectively (data not shown). This indicates that the exposed PIC domains of vesicles may cause secondary aggregation on the cell surface, which can inhibit further cell internalization and show long-term retention on the cell surface. Besides domain structure and deformability, the size effect might play a certain role in cellular uptake. As an example of a similar trend, in the case of polystyrene nanoparticles, the highest cellular uptake efficiency is found for the particles with the size of 100 nm, while the particles with the size less than 50 nm exhibited less potency owing to a point at which the size no longer has a significant impact on cellular uptake.45 Similar to this report, PIC vesicles with the size of 100–200 nm showed better internalisation than micelles with the size <50 nm, presumably owing to their higher surface area and deformable nature. However, further investigation is needed to prove this point.
In addition, to inspect the effect of PEG length, we used PICs with the same morphology and similar fPEG. In fact, 2k- and 5k-micelles with close fPEG values were selected for this purpose (Table 2). Due to similar fPEG values, we expected that exposure of the PIC domain might be close. After 24 h of incubation with HeLa cells, the results showed that the 2k-micelle exhibited a slightly higher cellular internalisation than the 5k-micelle (Fig. 4c). Detailed analysis was performed using images obtained by CLSM (Fig. 4b and Fig. S27, ESI†). Results show that cellular internalisation is promoted for the 2k-micelle. This might be explained by the particle number. Probably, the greater number of particles in the 2k-micelle is due to its smaller particle size compared to that of the 5k-micelle, which increased the cell–PIC interaction probability. The subcellular distribution of PIC micelles seems similar, that is, most particles were found in the region between cell and nuclear membranes in a dot-like pattern, supported by the line profiling analysis (Fig. S27, ESI†). Presumably, all PIC micelles would be endocytosed and not escape from the endosome. At this stage, the difference in cellular uptake efficiency can also be explained by the longer PEG chain length for suppressing interaction with cells and higher fPEG value. Nevertheless, the PEG chain length effect can be found to be the size and morphology determinant factor, which should be considered for the design of particles. Moreover, different subcellular distributions are confirmed for the PIC vesicles, in which vesicular PICs were more prominently found in the cell membrane region, indicating that PIC vesicles show higher cell surface adsorption and retention capability (Fig. S26 and S27, ESI†). This result seems consistent with the deformable nature of vesicles as discussed above. Thus, in terms of cellular uptake behaviour, the image-based analysis showed a similar trend to the FCM results, PIC vesicles > 2k-micelles > 5k-micelles. Overall, all PICs are considered to be endocytosed in HeLa cells judging from their size, ≤100 nm,46 but further investigation is required.
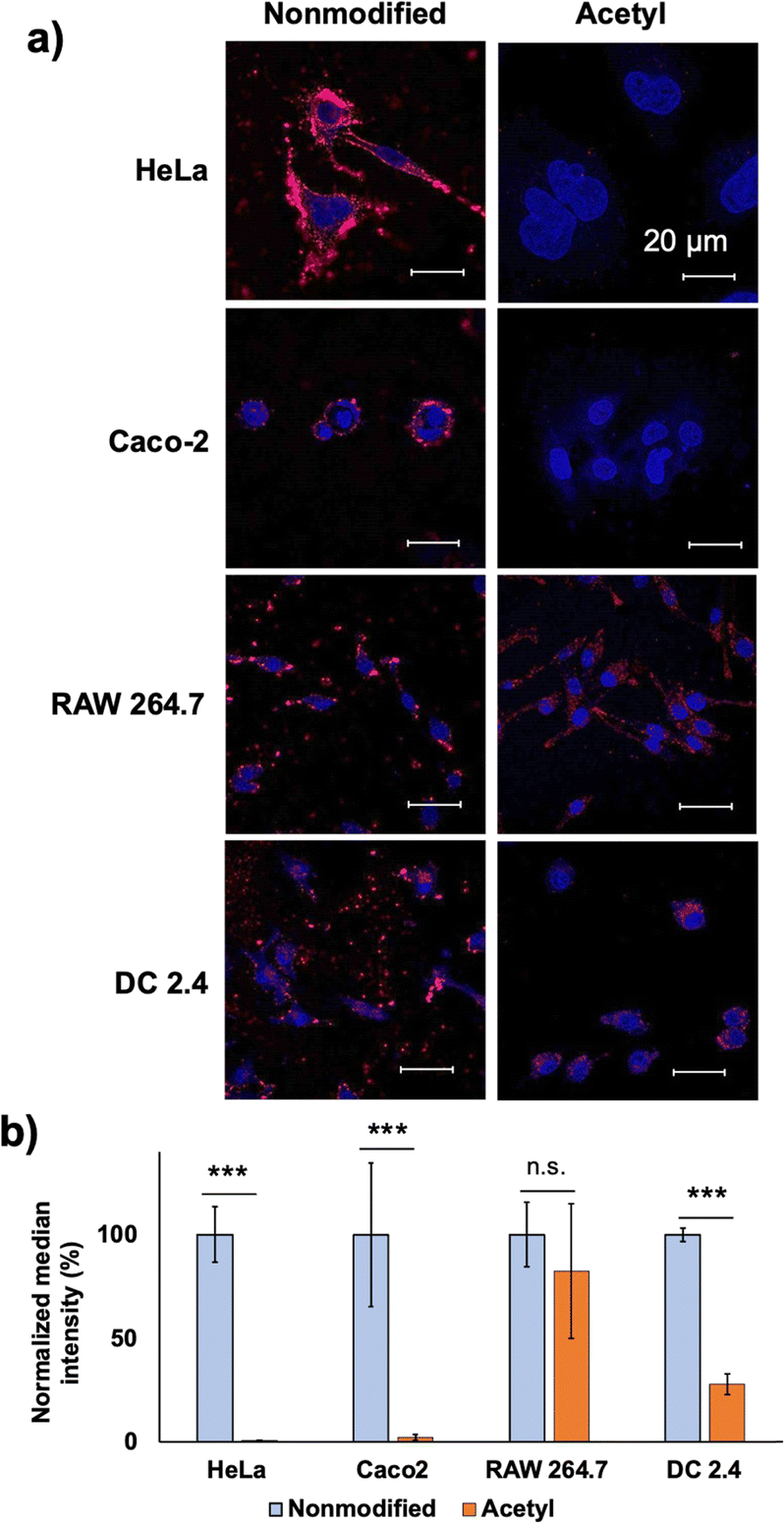 | ||
| Fig. 5 Cell-line dependency of the cell–PIC interaction. (a) CLSM observations of 50% crosslinked vesicles with and without acetylation (scale bar: 200 nm; blue, Hoechst-stained nuclei; red, PIC) and (b) quantification by FCM. The y-axis was normalised against HeLa results. | ||
Conclusion
In summary, we clarified several design aspects of PEGylated PICs to overcome the PEG dilemma by systematically tuning their properties; particularly, vesicle morphology with a mismatched catiomer–aniomer length enabled enhanced cell interaction capability. The PEG block length and crosslinking degree made a minor contribution to the cell–PIC interaction modulation. Remarkably, chemical modification of the PIC domain, particularly acetylation of the remaining amino groups, sharply inhibited the cell–PIC interaction with non-phagocytic cells, while retaining the interaction with phagocytic RAW264.7 cells. In the future, the insights of this study could be applied to the rational design of PEGylated PIC nanoparticles, where a balance between their stability, biocompatibility, and cellular internalisation can be achieved. Thus, our strategy is beneficial for the development of PEGylated nanocarriers without involving the PEG dilemma.Author contributions
F. Aulia: conceptualisation, methodology, validation, investigation, writing – original draft, visualization; H.Matsuba: conceptualization, methodology, investigation, visualization; S. Adachi: methodology, investigation, visualization; T. Yamada: methodology, investigation, visualization; I. Nakase: cell line providence (CHO-A745), editing, validation; T. Nii: validation; T. Mori: validation; Y. Katayama: validation; and A. Kishimura: conceptualisation, methodology, investigation, writing – reviewing and editing, validation, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported in part by KAKENHI (Grant No. JP18H03534 and JP22H02202 (from JSPS) and JP22H05429 (from MEXT) to A. K.), and a Monbukagakusho Scholarship, MEXT (to F. A.). The authors are grateful to the Nanotechnology Platform Program and Advanced Research Infrastructure for Materials and Nanotechnology in Japan (ARIM) of MEXT of Kyushu University and the University of Tokyo for their valuable support with the TEM analysis. We thank Drs M. Tanaka, D. Murakami and S. Shiomoto (Kyushu University) for their assistance with the AFM measurements, as well as Drs M. Goto, N. Kamiya and R. Wakabayashi (Kyushu University) for their assistance with the CLSM measurements.References
- J. M. Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214–221 CrossRef CAS PubMed
.
- T. M. Allen, G. A. Austin, A. Chonn, L. Lin and K. C. Lee, Biochim. Biophys. Acta, 1991, 1061, 56–64 CrossRef CAS PubMed
- B. Pelaz, P. del Pino, P. Maffre, R. Hartmann, M. Gallego, S. Rivera-Fernández, J. M. de la Fuente, G. U. Nienhaus and W. J. Parak, ACS Nano, 2015, 9(7), 6996–7008 CrossRef CAS PubMed
- S. Mishra, P. Webster and M. E. Davis, Eur. J. Cell Biol., 2004, 83(3), 97–111 CrossRef CAS PubMed
- H. Hatakeyama, H. Akita and H. Harashima, Biol. Pharm. Bull., 2013, 36(6), 892–899 CrossRef CAS PubMed
- Y. Fang, J. Xue, S. Gao, A. L. D. Yand, H. Jiang, Y. He and K. Shi, Drug Delivery, 2017, 24(2), 22–32 CrossRef CAS PubMed
- E. Oh, J. B. Delehanty, K. E. Sapsford, K. Susumu, R. Goswami, J. B. Blanco-Canosa, P. E. Dawson, J. Granek, M. Shoff, Q. Zhang, P. L. Goering, A. Huston and I. L. Medintz, ACS Nano, 2011, 5(8), 6434–6448 CrossRef CAS PubMed
- Y. Li, M. Kröger and W. K. Liu, Biomaterials, 2014, 35(30), 8467–8478 CrossRef CAS PubMed
- A. Harada and K. Kataoka, Macromolecules, 1995, 28, 5294–5299 CrossRef CAS
- H. Cabral, K. Miyata, K. Osada and K. Kataoka, Chem. Rev., 2018, 118(14), 6844–6892 CrossRef CAS PubMed
- J. Sun and Z. Li, Macromolecules, 2020, 53(20), 8737–8740 CrossRef CAS
- K. Itaka, K. Yamauchi, A. Harada, K. Nakamura, K. H. Kawaguchi and K. Kataoka, Biomaterials, 2003, 24(34), 4495–4506 CrossRef CAS PubMed
- K. Kataoka, H. Togawa, A. Harada, K. Yasugi, T. Matsumoto and S. Katayose, Macromolecules, 1996, 29(26), 8556–8557 CrossRef CAS
- A. Harada and K. Kataoka, Macromolecules, 1998, 31(2), 288–294 CrossRef CAS
- L. Zhao, M. Skwarczynski and I. Toth, ACS Biomater. Sci. Eng., 2019, 5(10), 4937–4950 CrossRef CAS PubMed
- Y. Anraku, A. Kishimura, M. Oba, Y. Yamasaki and K. Kataoka, J. Am. Chem. Soc., 2010, 132, 1631–1636 CrossRef CAS PubMed
- S. Chuanoi, A. Kishimura, W. Dong, Y. Anraku, Y. Yamasaki and K. Kataoka, Polym. J., 2014, 46(7), 130–135 CrossRef CAS
- W. Dong, A. Kishimura, Y. Anraku, S. Chuanoi and K. Kataoka, J. Am. Chem. Soc., 2009, 131, 3804–3805 CrossRef CAS PubMed
- A. Ahmad, T. Nii, T. Mori, Y. Katayama, M. Toyofuku and A. Kishimura, Macromol. Rapid Commun., 2022, 2200316 CrossRef CAS PubMed
- K. Naoyama, K. T. Mori, Y. Katayama and A. Kishimura, Macromol. Rapid Commun., 2016, 37, 1087–1093 CrossRef CAS PubMed
- A. Koide, A. Kishimura, K. Osada, W. D. Jang, Y. Yamasaki and K. Kataoka, J. Am. Chem. Soc., 2006, 128(18), 5988–5989 CrossRef CAS PubMed
- Y. Anraku, A. Kishimura, M. Kamiya, S. Tanaka, T. Nomoto, K. Toh, Y. Matsumoto, S. Fukushima, D. Sueyoshi, M. R. Kano, Y. Urano, N. Nishiyama and K. Kataoka, Angew. Chem., Int. Ed., 2016, 55, 560–565 CrossRef CAS PubMed
- D. Sueyoshi, Y. Anraku, T. Komatsu, Y. Urano and K. Kataoka, Biomacromolecules, 2017, 18(4), 1189–1196 CrossRef CAS PubMed
- D. Kokuryo, Y. Anraku, A. Kishimura, S. Tanaka, M. R. Kano, J. Kershaw, N. Nishiyama, T. Saga, I. Aoki and K. Kataoka, J. Controlled Release, 2013, 169, 220–227 CrossRef CAS PubMed
- W. Kawamura, Y. Miura, D. Kokuryo, K. Toh, N. Yamada, T. Nomoto, Y. Matsumoto, D. Sueyoshi, X. Liu, I. Aoki, M. R. Kano, N. Nishiyama, T. Saga, A. Kishimura and K. Kataoka, Sci. Technol. Adv. Mater., 2015, 16, 035004 CrossRef PubMed
- B. S. Kim, S. Chuanoi, T. Suma, Y. Anraku, K. Hayashi, M. Naito, H. J. Kim, I. C. Kwon, K. Miyata, A. Kishimura and K. Kataoka, J. Am. Chem. Soc., 2019, 141(8), 3699–3709 CrossRef CAS PubMed
- B. S. Kim, M. Naito, H. Chaya, M. Hori, K. Hayashi, H. S. Min, Y. Yi, H. J. Kim, T. Nagata, Y. Anraku, A. Kishimura, K. Kataoka and K. Miyata, Biomacromolecules, 2020, 21(10), 4365–4376 CrossRef CAS PubMed
- S. Chuanoi, Y. Anraku, M. Hori, A. Kishimura and K. Kataoka, Biomacromolecules, 2014, 15(7), 2389–2397 CrossRef CAS PubMed
- A. Goto, H. Yen, Y. Anraku, S. Fukushima, P. Lai, M. Kato, A. Kishimura and K. Kataoka, ACS Biomater. Sci. Eng., 2017, 3(5), 807–815 CrossRef CAS PubMed
- A. Goto, Y. Anraku, S. Fukushima and A. Kishimura, Polymers, 2023, 15, 1368 CrossRef CAS PubMed
- Y. Anraku, A. Kishimura, A. Kobayashi, M. Oba and K. Kataoka, Chem. Commun., 2011, 47, 6054–6056 RSC
- O. F. Mutaf, Y. Anraku, A. Kishimura and K. Kataoka, Polymer, 2017, 133, 1–7 CrossRef CAS
- M. Hori, H. Cabral, K. Toh, A. Kishimura and K. Kataoka, Biomacromolecules, 2018, 19, 4113–4121 CrossRef CAS PubMed
- M. Nakanishi, J. S. Park, W. D. Jang, M. Oba and K. Kataoka, React. Funct. Polym., 2007, 67, 1361–1372 CrossRef CAS
- Y. Liu, T. Maruyama, B. KC, T. Mori, Y. Katayama and A. Kishimura, Chem. Lett., 2021, 50, 1034–1037 CrossRef CAS
- O. F. Mutaf, A. Kishimura, Y. Mochida, A. Kim and A. K. Kataoka, Macromol. Rapid Commun., 2015, 36, 1958–1964 CrossRef CAS PubMed
- A. Carlsen and S. Lecommandoux, Curr. Opin. Colloid Interface Sci., 2009, 14, 329–339 CrossRef CAS
- A. Harada and K. Kataoka, Science, 1999, 283(5398), 65–67 CrossRef CAS PubMed
- R. Raman, V. Sasisekharahn and R. Sasisekharan, Chem. Biol., 2005, 12(3), 267–277 CrossRef CAS PubMed
- J. D. Esko, T. E. Stewart and W. H. Taylor, Proc. Natl. Acad. Sci. U. S. A., 1985, 82(10), 3197–3201 CrossRef CAS PubMed
- R. M. Pashley, J. Colloid Interface Sci., 1981, 80(1), 153–162 CrossRef CAS
- J. N. Israelachvili and G. E. Adams, J. Chem. Soc., Faraday Trans. 1, 1978, 74, 975–1001 RSC
- L. Ouyang, R. Shaik, R. Xu, G. Zhang and J. Zhe, Cells, 2021, 10(6), 1519 CrossRef CAS PubMed
- F. Alexis, E. Pridgen, L. K. Molnar and O. C. Farokhzad, Mol. Pharmaceutics, 2008, 5(4), 505–515 CrossRef CAS PubMed
- K. Y. Win and S. S. Feng, Biomaterials, 2005, 26(15), 2713–2722 CrossRef CAS PubMed
- J. Rejman, V. Oberle, I. S. Zuhorn and D. Hoekstra, Biochem. J., 2004, 377(1), 159–169 CrossRef CAS PubMed
- S. Gordon, Cell, 2002, 111, 927–930 CrossRef CAS PubMed
- E. Fröhlich, Int. J. Nanomed., 2012, 7, 5577–5591 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tb02049e |
| This journal is © The Royal Society of Chemistry 2024 |