
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent progress and perspectives of liquid organic hydrogen carrier electrochemistry for energy applications
Jinyao
Tang
 ,
Rongxuan
Xie
,
Parsa
Pishva
,
Xiaochen
Shen
,
Yanlin
Zhu
and
Zhenmeng
Peng
*
,
Rongxuan
Xie
,
Parsa
Pishva
,
Xiaochen
Shen
,
Yanlin
Zhu
and
Zhenmeng
Peng
*
Department of Chemical Engineering, University of South Carolina, Columbia, SC 29208, USA. E-mail: zmpeng@sc.edu
First published on 22nd May 2024
Abstract
Amidst the global pursuit of clean and sustainable energy, the transition towards a hydrogen economy holds immense promise, yet is encumbered by significant storage challenges. Liquid organic hydrogen carrier (LOHC) electrochemistry emerges as a promising solution to enable efficient and sustainable hydrogen storage, meanwhile broadening the horizons of LOHCs for use as clean, renewable, and intense energy carriers to store and generate electricity. In this perspective, we embark on a review of recent trends and progress in LOHC redox electrochemistry, properties, and applications. We categorize electrochemically active, regenerable LOHCs into alcohols, amines, aromatic compounds, aminoxyl species, and others, based on their functional groups, with a keen emphasis on their electrochemical properties. Our examination of electrochemically redoxable LOHCs, with a particular focus on secondary alcohols such as isopropanol and cyclohexanol, primary amines such as ethylamine, aromatic compounds such as quinones, and aminoxyl species such as TEMPO, unveils their vast potential across diverse applications including ambient hydrogen storage, regenerative fuel cells for electricity generation, and rechargeable batteries and flow batteries for intensive electricity storage. Through this exploration, we discern promising avenues for advancing sustainable hydrogen and energy storage solutions by harnessing the diverse capabilities of different LOHC categories, thus paving the way towards a greener and more resilient energy landscape.
1. Introduction
The global demand for sustainable energy solutions has prompted a transition towards cleaner energy sources such as hydrogen. However, the challenges associated with hydrogen storage and transportation hinder its widespread application. To address these challenges, liquid organic hydrogen carriers (LOHCs) have emerged as a promising alternative, offering efficient and sustainable hydrogen storage and extending energy storage possibilities even beyond hydrogen storage.LOHCs offer significant advantages for hydrogen storage and energy applications by facilitating the release and uptake of hydrogen through chemical reactions.1 Previous research efforts mainly focused on LOHC thermal dehydrogenation/hydrogenation processes for completing a hydrogen storage cycle. However, the thermal chemistry approach poses challenges for reaction conditions, typically requiring elevated pressure and temperature (10–50 bar and 100–250 °C for hydrogenation, atmospheric pressure and 150–400 °C for endothermic dehydrogenation).2 In contrast, LOHC electrochemistry presents compelling perks over its thermal chemistry. The utilization of electrode potentials overcomes the thermodynamics of endothermic reactions and promotes kinetics, allowing operations at ambient pressure and temperature. Electrification of LOHC chemistry can not only enhance energy efficiency but also enable the direct use of LOHCs in electrical energy storage devices, representing a significant improvement over conventional thermal methods. Moreover, the working principle of LOHC electrochemical cells helps minimize the need for hydrogen separation after release, enhancing the overall efficiency.3
Certain alcohols such as isopropanol (IPA)4–6 and cyclohexanol7 have attracted significant research interest as LOHCs due to their ease of acquisition from industrial processes. These alcohols can undergo dehydrogenation to yield carbonyl derivatives as H-lean LOHCs (LOHC−), which can subsequently be rehydrogenated to H-rich LOHCs (LOHC+), establishing the basis for an alcohol-based LOHC economy. Besides alcohols, some amines8–10 and aromatic compounds11,12 serve as potential LOHC candidates. Noteworthily, ethylamine has been demonstrated as a promising LOHC, boasting an impressive 8.9 wt% hydrogen storage content and exhibiting highly reversible merit.10 In a broader sense, any chemicals configured with H-lean form and H-rich counterparts, existing in a liquid phase during use, qualify as LOHCs. For instance, the N-hydroxyphthalimide (NHPI) redox couple switches between the hydroxylamine and aminoxyl radical. This redox couple undergoes a one-proton transfer reaction without the need for catalysts and is typically dissolved in water or other solvents for flow batteries.13,14 Therefore, this category extends to various chemicals acting as energy sources, enabling hydrogen storage, contributing to electricity generation through fuel cells, and functioning as the anolyte or catholyte in flow batteries. With the field of sustainable energy storage continuously evolving, there is growing research interest in LOHCs and diverse unrecognized LOHC categories. As recent reviews predominantly concentrate on the application of LOHCs solely in fuel cells, it becomes imperative to summarize recent advancements and trends in the broader field of LOHCs. This includes highlighting the emerging role of LOHCs in hydrogen storage, flow batteries and other innovative energy storage technologies, as well as identifying future research directions in LOHC development.
In this perspective, our focus is directed towards electrochemically active and regenerable LOHCs. We classify these compounds according to their distinct functional groups and delve into their electrochemical characteristics. Furthermore, the perspective offers insights into the diverse applications of LOHC electrochemistry, encompassing hydrogen storage, regenerative fuel cells, batteries, and flow batteries. It culminates by proposing avenues for future research aimed at enhancing electrochemical properties of LOHCs and facilitating their widespread integration into sustainable energy solutions.
2. LOHC redox electrochemistry
Electrochemically active and regenerable LOHCs represent a class of compounds which can undergo redox electrochemistry, either with or without a catalyst. The LOHC electrochemical redox reactions under acidic and basic conditions can be depicted with eqn (1) and (2), respectively.15,16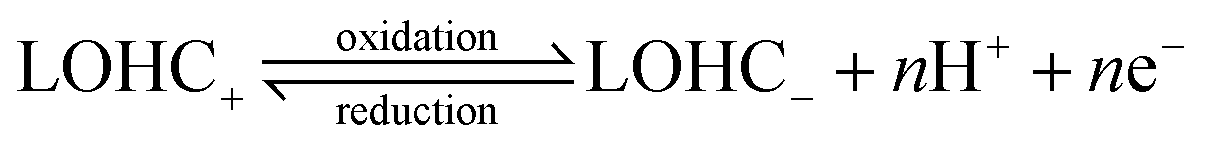 | (1) |
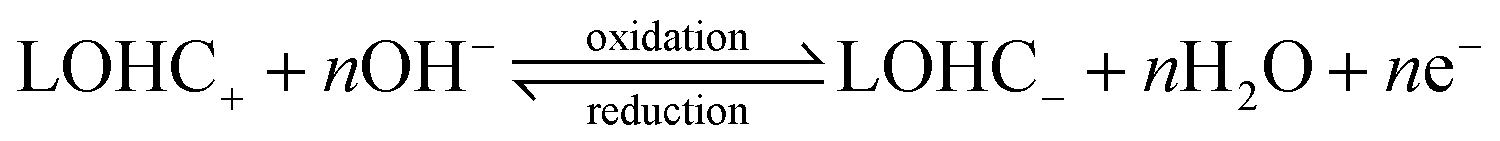 | (2) |
In brief, LOHC+ undergoes electrochemical oxidation above its thermodynamic redox potential, resulting in dehydrogenation to LOHC−. When the electrode potential is below the redox potential, the reverse process, i.e., electrochemical reduction of LOHC−, occurs that leads to its hydrogenation back into LOHC+. Table 1 summarizes some representative LOHC chemicals that have been investigated for their electrochemical properties.
| LOHC+/LOHC− | E 0 (V vs. RHE) | H wt% | Boiling point (°C) | Water solubility (/100 mL) |
|---|---|---|---|---|
| Ethylamine/acetonitrile | 0.13 (ref. 10) | 8.9 | 16.6/82.0 | Miscible/miscible |
| Isopropanol/acetone | 0.12 (ref. 29) | 3.3 | 82.6/56.1 | Miscible/miscible |
| Cyclohexanol/cyclohexanone | 0.15 (ref. 29) | 2.0 | 161.8/155.6 | 3.6 g/8.6 g |
| Methylcyclohexane/toluene | 0.15 (ref. 30) | 6.1 | 101/110.6 | 1.4 mg/51.9 mg |
| 9,10-Dihydroxyanthracene-2,7-disulfonic acid/anthraquinone-2,7-disulfonic acid (AQDS) | 0.2 (ref. 31) | 0.5 | −/− | Soluble/>1 M |
| 1-Hydroxy-2,2,6,6-tetramethylpiperidine (TEMPOH)/2,2,6,6-tetramethylpiperidine N-oxyl (TEMPO) | −0.38 vs. NHE22 | 0.6 | 208.8/193 | Soluble/1.0 g |
| N-Hydroxyphthalimide (NHPI)/phthalimide N-oxyl (PINO) | 1.30 vs. NHE32 | 0.6 | −/− | Slightly soluble/<100 mg |
2.1. Fundamentals of LOHC electrochemistry
The underlying principles of LOHC electrochemistry stem from straightforward reaction pathways that can be triggered by low overpotentials. These reactions primarily involve facile activation and cleavage of H-bonds, such as O–H, N–H and C–H bonds. As a result, the activation energy (Ea) associated with LOHC electrochemistry typically falls within a low to moderate range. For instance, the Ea of ethylamine oxidation was measured to be 52.4 kJ mol−1 at 0.5 V vs. RHE, and the Ea of acetonitrile reduction was 11.6 kJ mol−1.10 The relatively low E0 values of LOHC pairs, as shown in Table 1, facilitate these reactions, when coupled with hydrogen electrochemistry reactions, to be driven by minimal energy input, an important feature to enable efficient hydrogen storage.The LOHC electrochemistry can follow various mechanisms, primarily governed by catalysts and reaction conditions. Scheme 1a depicts a typical reaction pathway for LOHC electrooxidation on transition metal catalysts.17 The absorbed reactant forms bonds with the metal catalyst during electrooxidation, and the rate determining step (RDS) is the desorption of the product. Another reaction pathway for LOHC electrooxidation proposed by Fleischmann et al. is the indirect oxidation mechanism as shown in Scheme 1b.18–20 In this mechanism, catalysts such as NiOOH act as chemical oxidizing agents, initiating a reaction with the amine/alcohol via a rate-limiting hydrogen atom transfer (HAT) step. This step transfers a hydrogen radical from the α-carbon of the alcohol/amine to NiOOH, reducing it to Ni(OH)2. This regeneration occurs rapidly, even at low overpotentials, and the overall rate of oxidation remains unaffected by the applied potential, mainly governed by the chemical HAT step. For LOHCs such as anthraquinone (AQ), the reaction pathway depends on the metal ions in the electrolytes.21 It undergoes a two-step single-electron reductive reaction in metal ion electrolytes with a low charge-to-radius ratio. Initially, AQ forms a radical anion [AQ]˙−, which pairs with metal ions to create stable [AQ–Mn+](n−1)+ molecules (Scheme 1c). Spin-density analysis reveals delocalization of spin densities for certain metal ions, facilitating dissociation from the AQ crystal and formation of red species in the electrolyte. Additionally, metal ions such as Al3+, Mg2+, and Zn2+ with larger charge-to-radius ratios form unstable radical species due to higher spin densities localized on the metal centers. A proton-assisted one-step two-electron reductive reaction mechanism is proposed for certain aqueous solutions (Scheme 1c). High binding energies between Mn+ ions and [AQ]2− anions indicate favorable metathesis reactions between AQ–H2 and metal ions, leading to the formation of AQ metal products (AQ–M). Another example is aminoxyl radicals, which exhibit distinctive redox properties (Scheme 1d).22 Aminoxyl radicals (1) undergo a reversible one-electron oxidation to form oxoammonium species (2) and a quasi-reversible one-electron/one-proton reduction to hydroxylamines (3).
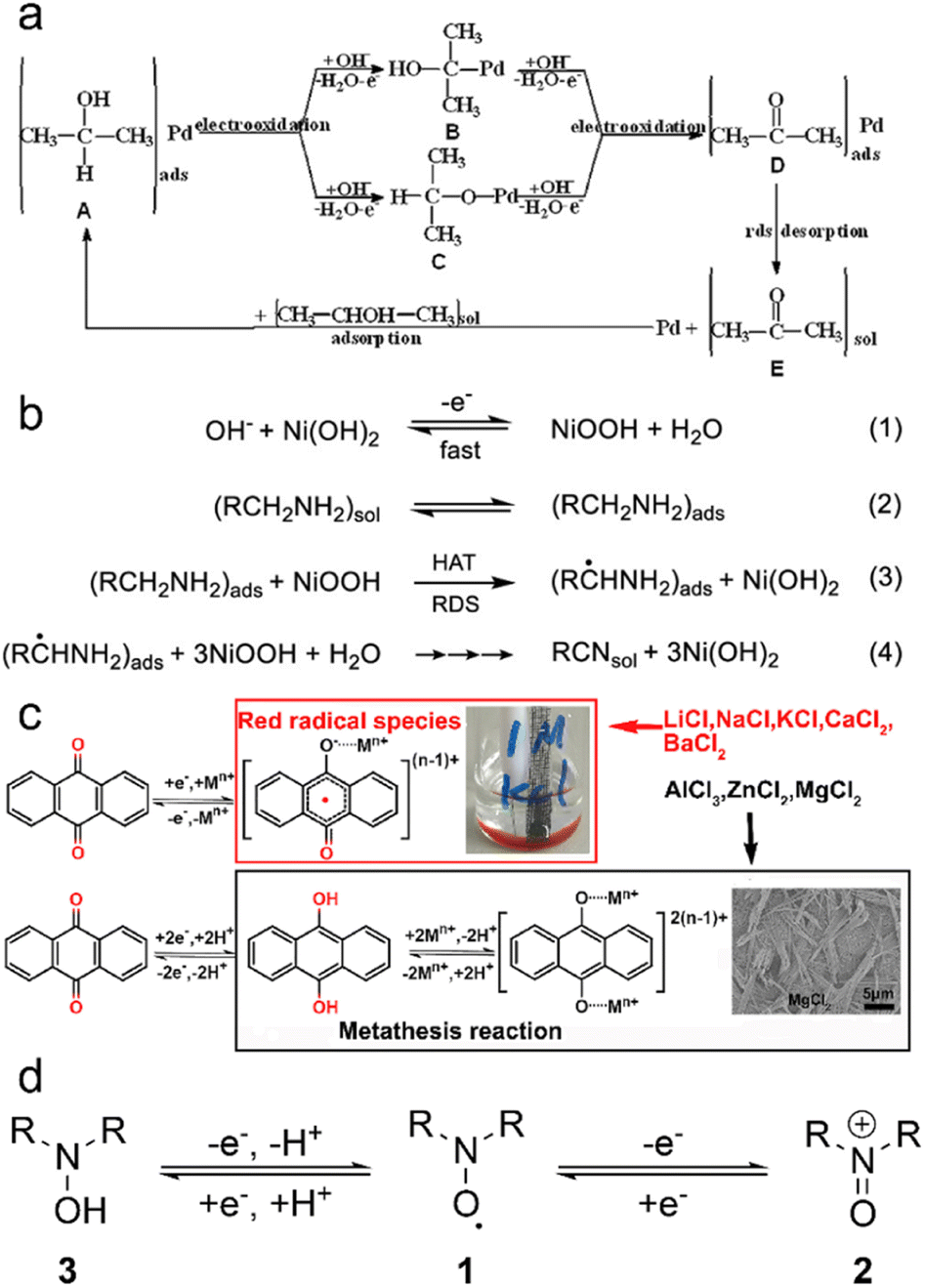 | ||
| Scheme 1 Reaction pathways of LOHCs. (a) The schematic reaction pathway for 2-propanol electrooxidation on a Pd/Ni-foam electrode in an alkaline medium with acetone as the sole product. Reproduced with permission from ref. 17. Copyright 2011 Elsevier. (b) Proposed mechanism for the indirect oxidation of amines at NiOOH electrodes in alkaline aqueous media. Reproduced with permission from ref. 18. Copyright 2022 American Chemical Society. (c) Proposed redox mechanism of AQ in different aqueous metal ion electrolytes. Reproduced with permission from ref. 21. Copyright 2020 Elsevier. (d) Simplified redox chemistry of aminoxyl radicals. Reproduced with permission from ref. 22. Copyright 2018 American Chemical Society. | ||
2.2. Categories of LOHCs
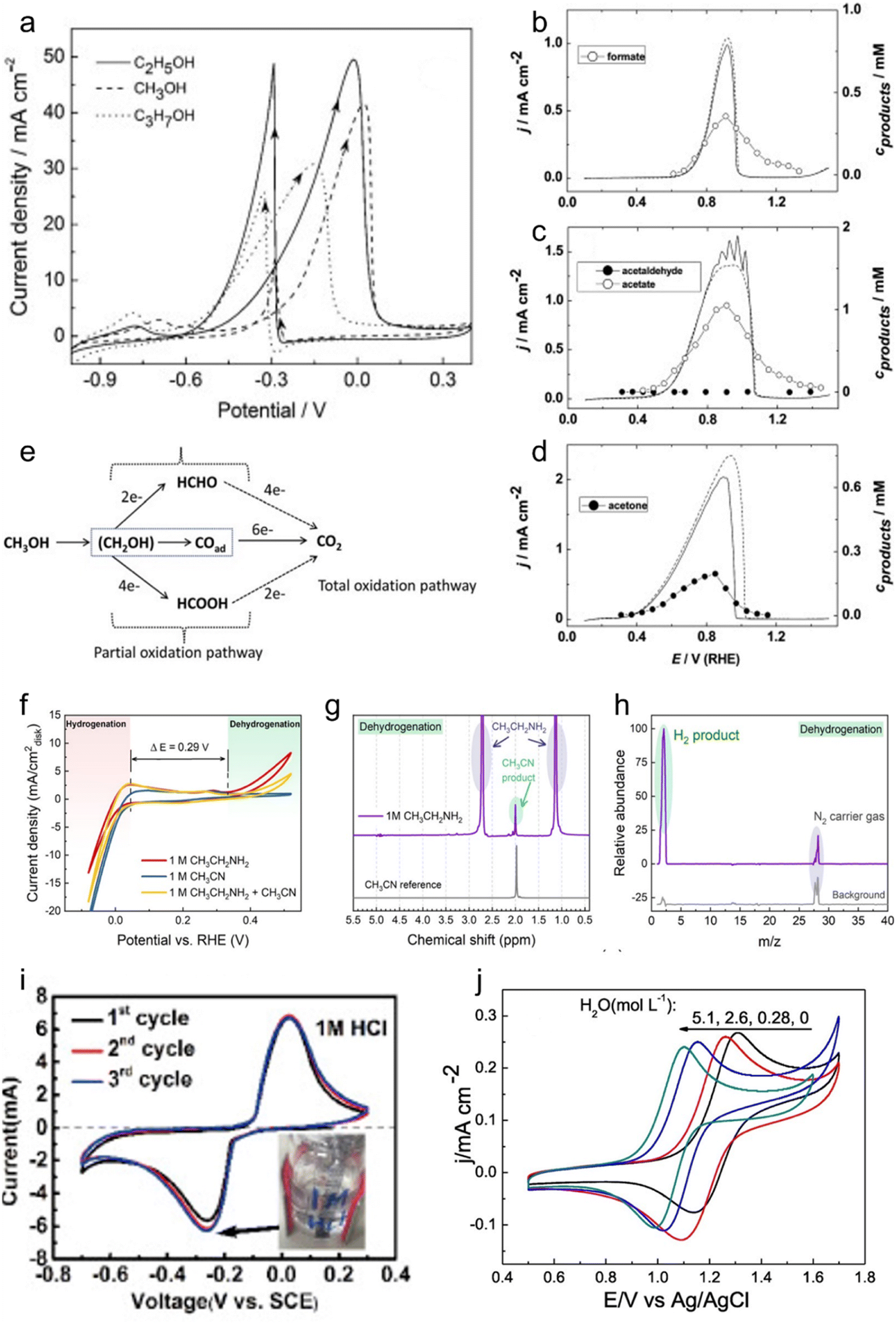 | ||
| Fig. 1 Electrochemistry of LOHCs. (a) Pd/TiO2-MWCNTs catalysts in 0.5 M NaOH + 1.0 M CH3OH (C2H5OH or C3H7OH) at a scanning rate of 50 mV s−1. Adapted with permission from ref. 25. Copyright 2015 Elsevier. Potential sweeps and formed oxidation products of (b) methanol, (c) ethanol and (d) iso-propanol on a palladium electrode. The solid lines represent the potential sweep with sample collection and the dashed ones without. Reproduced with permission from ref. 26. Copyright 2011 Elsevier. (e) Schematic of the parallel pathways for the electrocatalytic oxidation of methanol. Reproduced with permission from ref. 27. Copyright 2022 Elsevier. (f) CV of a commercial Pt black catalyst-loaded working electrode in 1 M NaOH aqueous solution with the addition of 1 M ethylamine, 1 M acetonitrile, and 1 M ethylamine + 1 M acetonitrile. (g) 1H NMR spectrum of the liquid product from ethylamine dehydrogenation obtained by applying 0.7 V vs. RHE for 10 h, with acetonitrile as a reference. (h) Mass spectrum of the gas product from the counter electrode in the CH3CH2NH2 dehydrogenation experiment. Reproduced with permission from ref. 10. Copyright 2021 American Chemical Society. (i) CV curves of AQ at 5 mV s−1 in different 1 M HCl. Reproduced with permission from ref. 21. Copyright 2020 Elsevier. (j) CVs of 2 mmol L−1 NHPI at 20 mV s−1 with increasing amounts of H2O, c[H2O] (mol L−1): 0 (black), 0.28 (red), 2.6 (blue), and 5.1 (dark cyan). Reproduced with permission from ref. 33. Copyright 2020 Elsevier. | ||
IPA, classified as a secondary alcohol, emerges as a more promising LOHC due to its distinctive electrochemical and physical attributes. The reversible nature inherent in secondary alcohols renders them highly suitable for LOHC electrochemistry. Specifically, IPA electro-oxidation produces sole liquid product acetone (ACE) that can be selectively reduced back to IPA,28 enhancing its competitiveness as an LOHC (Fig. 1d). The reported low onset potential for IPA oxidation, approximately 0.2 V vs. RHE using a Pd catalyst, and excellent selectivity towards the ACE product underscore its electrochemical viability, ensuring a controlled and efficient process.29 Moreover, the physical properties of IPA as well as ACE, including low cost, low toxicity, 3.3 wt% hydrogen storage capacity, moderate boiling point, and miscibility with water, contribute to its status as a promising LOHC candidate under ambient conditions. However, catalyst poisoning due to strong ACE adsorption was reported in previous studies, which led to a decay in the IPA electro-oxidation activity.4 Addressing the catalyst stability problem is thus crucial for IPA oxidation to ACE. Further research endeavors are warranted to identify advanced catalysts capable of catalyzing selective IPA oxidation and meanwhile enhancing both reaction efficiency and catalyst stability.
Some other secondary alcohols also exhibit remarkable electro-redox activity and selectivity. For instance, cyclohexanol electro-oxidation shows a low onset potential of about 0.2 V vs. RHE when using a Pt catalyst and produces cyclohexanone as the only LOHC− product. Notably, Pt can also effectively catalyze electrochemical reduction of cyclohexanone, which regenerates cyclohexanol.29 The high selectivity appears to be a distinct advantage of secondary alcohols, as compared to primary ones. This interesting characteristic, coupled with their physical merits and electrochemical properties, positions secondary alcohols as promising candidates for efficient and reversible LOHC systems in hydrogen storage and energy applications.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 in product distribution in a solid oxide fuel cell (SOFC) at 420 °C.30 Imada et al. investigated toluene electrochemical hydrogenation and reported the interesting catalytic properties of Rhx/Pt/C that are 2.7 times as high compared to pure Pt.34 Quinones, representing another typical class of aromatic compounds as possible LOHCs, possess interesting attributes due to their inherent ability to undergo redox electrochemistry without the need for catalysts. Moreover, their redox potential can be finely tuned by introducing various modifications to the aromatic ring.35 Yan et al. investigated the rocking-chair electrochemical behavior of 9,10-anthraquinone (AQ) with different ions in the electrolyte (Fig. 1i), revealing the highly reversible nature of AQ's redox electrochemistry.21 Notably, it was proposed that AQ undergoes a two-step redox process with a poor reversibility in neutral or alkaline aqueous electrolytes and undergoes a reversible one-step two-electron redox process facilitated by protons in acidic electrolytes. In general, aromatic compound-based LOHCs hold potential to be applied, given their interesting redox electrochemistry properties and even requiring no catalyst. It needs to be noted that this category of LOHCs typically has a low water solubility that may restrict their applications. Incorporating water-solubilizing groups through modifications can enhance the solubility of such LOHCs, thereby improving their performance.
:6 in product distribution in a solid oxide fuel cell (SOFC) at 420 °C.30 Imada et al. investigated toluene electrochemical hydrogenation and reported the interesting catalytic properties of Rhx/Pt/C that are 2.7 times as high compared to pure Pt.34 Quinones, representing another typical class of aromatic compounds as possible LOHCs, possess interesting attributes due to their inherent ability to undergo redox electrochemistry without the need for catalysts. Moreover, their redox potential can be finely tuned by introducing various modifications to the aromatic ring.35 Yan et al. investigated the rocking-chair electrochemical behavior of 9,10-anthraquinone (AQ) with different ions in the electrolyte (Fig. 1i), revealing the highly reversible nature of AQ's redox electrochemistry.21 Notably, it was proposed that AQ undergoes a two-step redox process with a poor reversibility in neutral or alkaline aqueous electrolytes and undergoes a reversible one-step two-electron redox process facilitated by protons in acidic electrolytes. In general, aromatic compound-based LOHCs hold potential to be applied, given their interesting redox electrochemistry properties and even requiring no catalyst. It needs to be noted that this category of LOHCs typically has a low water solubility that may restrict their applications. Incorporating water-solubilizing groups through modifications can enhance the solubility of such LOHCs, thereby improving their performance.
2.3. Electrocatalysts for LOHC redox electrochemistry
Electrocatalysts play a crucial role in the electrochemical redox processes of alcohol/amine-based and certain aromatic compound-based LOHCs by lowering overpotentials and accelerating kinetics. As of now, the design of these electrocatalysts predominantly centers on transition metal-based nanomaterials, known for their intriguing catalytic properties in a broad range of redox reaction systems.Catalyst selection for different LOHCs can be guided by theories such as d-band (εd) theory37 or descriptors such as the work function38 to predict the performance, which can then be validated experimentally. For instance, integrating non-noble metals such as Cu with Pt alters the electronic structure of Pt through ensemble and ligand effects.39,40 Additionally, less noble Cu provides OH− species at lower potentials41 and effectively adjusts the d-band center of Pt, preventing its oxidation42 and thereby enhancing the LOHC+ oxidation activity. Furthermore, engineering the morphology and geometry of the Pt–Cu alloy catalyst can lead to higher performance, achieved through techniques such as morphological tailoring and polyhedral structures.43,44
3. Applications of LOHC redox electrochemistry
The LOHC redox electrochemistry, when coupled with other electrochemical reactions, can be utilized to fulfil a variety of applications, particularly hydrogen storage and electrical energy generation and storage applications. Different LOHCs possess their own unique traits that would render them well-suited for specific applications. For instance, ethylamine stands out as an excellent choice for onboard hydrogen storage and electric energy generation, thanks to its impressive hydrogen capacity. Conversely, IPA demonstrates excellent suitability for stationary hydrogen and energy storage applications due to its low cost and low toxicity. LOHCs that have a good hydrogen capacity and do not require catalysts may well suit batteries and flow batteries for energy storage. Table 2 summarizes the applications of selected LOHCs and their respective performance.| LOHC+ | Catalyst | Application | OCV (V) | Max power density (mW cm−2) | Specific capacity | Temperature (°C) | Stability | Reference |
|---|---|---|---|---|---|---|---|---|
| Isopropanol | Pt–Ru | Fuel cell | 0.782 | 254 | — | 100 | — | 4 |
| Cyclohexanol | Pt/C | Fuel cell | 0.93 | 74.7 | — | 80 | — | 7 |
| Ethanol | Pd–(Ni–Zn)/C | Fuel cell | 0.80 | 170 | — | 80 | 217 h | 46 |
| Quinone | — | Fuel cell | 0.85 | ∼215 | — | 80 | >8 h | 31 |
| 1,2-Dihydrobenzoquinone-3,5-disulfonic acid (BQDS) | — | Fuel cell | 0.86 | 122 | — | RT | 50 cycles | 47 |
| PEG-substituted anthraquinones (PEGAQs) | — | Flow battery | 1.0 | 220 | 25.2 W h L−1 | RT | 220 cycles/>18 days | 48 |
| Methylene blue (MB) | — | Battery | 0.9 | 4.706 | 1324 μA h cm−2 | RT | 8000 cycles | 49 |
| N-Hydroxyphthalimide (NHPI) | — | Battery | 1.15 | — | 270 mA h L−1 | 0 | >20 cycles | 32 |
| 4-Carboxylic-7-sulfonate fluorenol (4C7SFL–OH) | — | Flow battery | 1.10 | — | ∼80% theoretical capacity | RT | 780 cycles | 50 |
| Isopropanol | Pt/CC | Battery | — | — | 525 mA h g−1 | RT | 50 cycles | 29 |
| Cyclohexanol | Pt/CC | Battery | 0.96 | — | 374 mA h g−1 | RT | 250 cycles | 29 |
| (2,2,6,6-Tetramethylpiperidin-1-yl)oxyl (TEMPO) | — | Fuel cell | 0.7 | 90 | — | 30 | 200 min | 51 |
| 3,3′-(Phenazine-1,8-diyl)dipropionic acid (1,8-PFP) | — | Flow battery | 1.15 | 94 | ∼135C | 45 | 53 days | 52 |
3.1. Criteria for LOHCs in various applications
The applications of LOHCs span various domains, including hydrogen storage, regenerative LOHC fuel cells, rechargeable LOHC batteries, and LOHC flow batteries. While rapid and reversible LOHC redox electrochemistry is universally desired, different applications have different priorities and thus place varying emphasis on the specific requirements of LOHCs for effective implementation.In hydrogen storage applications, paramount attention is directed towards storage efficiency and capacity. Attaining a low E0cell for LOHC electrolysis is vital to ensure an effective hydrogen uptake and release cycle. Additionally, a high hydrogen storage density, both in terms of LOHC mass and volume, holds significant importance, especially in onboard hydrogen storage applications. Conversely, for stationary, large-scale applications, stringent demands on mass and volume are somewhat relaxed, with cost and safety emerging as predominant factors.
Regenerative LOHC fuel cells are engineered for emission-free, efficient electricity generation through the coupling of LOHC electro-oxidation and oxygen reduction reactions. Therefore, greater emphasis is placed on fuel cell energy and power density. LOHCs with lower E0 values facilitate higher E0cell values and increased power output. Furthermore, a higher hydrogen density in LOHCs results in more available hydrogen carriers for utilization, thereby enhancing electrical energy generation.
In battery applications, the primary considerations for LOHCs revolve around reversibility and specific capacity. Given the need for in situ charge and discharge within a single device, LOHC pairs must demonstrate high stability and selectivity between LOHC+ and LOHC− chemicals to deter capacity decay. Furthermore, specific capacity holds particular significance, particularly in portable applications where energy density is a primary concern. Therefore, a high hydrogen carrier density of LOCHs would be preferred.
In the realm of flow batteries, primarily utilized for stationary energy storage, more emphasis is placed on reversibility and rechargeability rather than specific capacity. Flow batteries favor LOHCs that do not necessitate catalysts, aligning with their intended purpose of providing large-scale and cost-effective energy storage solutions. Consequently, low-cost LOHCs devoid of catalysts emerge as good options for flow battery applications.
3.2. Hydrogen storage
Hydrogen storage represents the foremost application of LOHC electrochemistry. In a pivotal contribution, Wu et al. introduced a framework for a sustainable hydrogen economy, centering on the electrochemical ethylamine/acetonitrile redox method for hydrogen storage (Fig. 2a).10 Employing a custom-made 10 cm2 electrochemical cell, they probed the H2 uptake and release processes, effectively simulating a complete H2 storage cycle. The electrochemical cell exhibited commendable durability in H2 uptake through acetonitrile electrochemical hydrogenation, maintaining notable current levels and showcasing an augmented H2 uptake rate at higher cell voltages. The efficacy of H2 release was underscored by continuous H2 generation with nearly 100% faradaic efficiency (FE). Energy consumption analysis validated the method's efficiency, particularly at slower rates, highlighting its potential for sustainable hydrogen storage (Fig. 2b). Notably, the electrochemical ethylamine/acetonitrile redox method outperformed existing approaches by its capability to achieve an impressive 8.9 wt% hydrogen storage capacity under ambient conditions, aligning with the 2025 Department of Energy (DOE) onboard hydrogen storage target. The cell durability was assessed through chronoamperometry experiments conducted at various voltages. Initially, the current exhibited a sharp decline within the first few minutes, followed by a more stable behavior for the remainder of the experiment at constant voltage, indicating favorable cell durability. However, when the voltage was intermittently toggled on and off to simulate startup and shutdown operations, the current decayed more rapidly. Moreover, as the voltage increased, the decay rate intensified. This phenomenon is likely attributed to the gradual deactivation of the Pt black catalyst under these stability test conditions. The discovery of a more robust catalyst is imperative to improve the stability.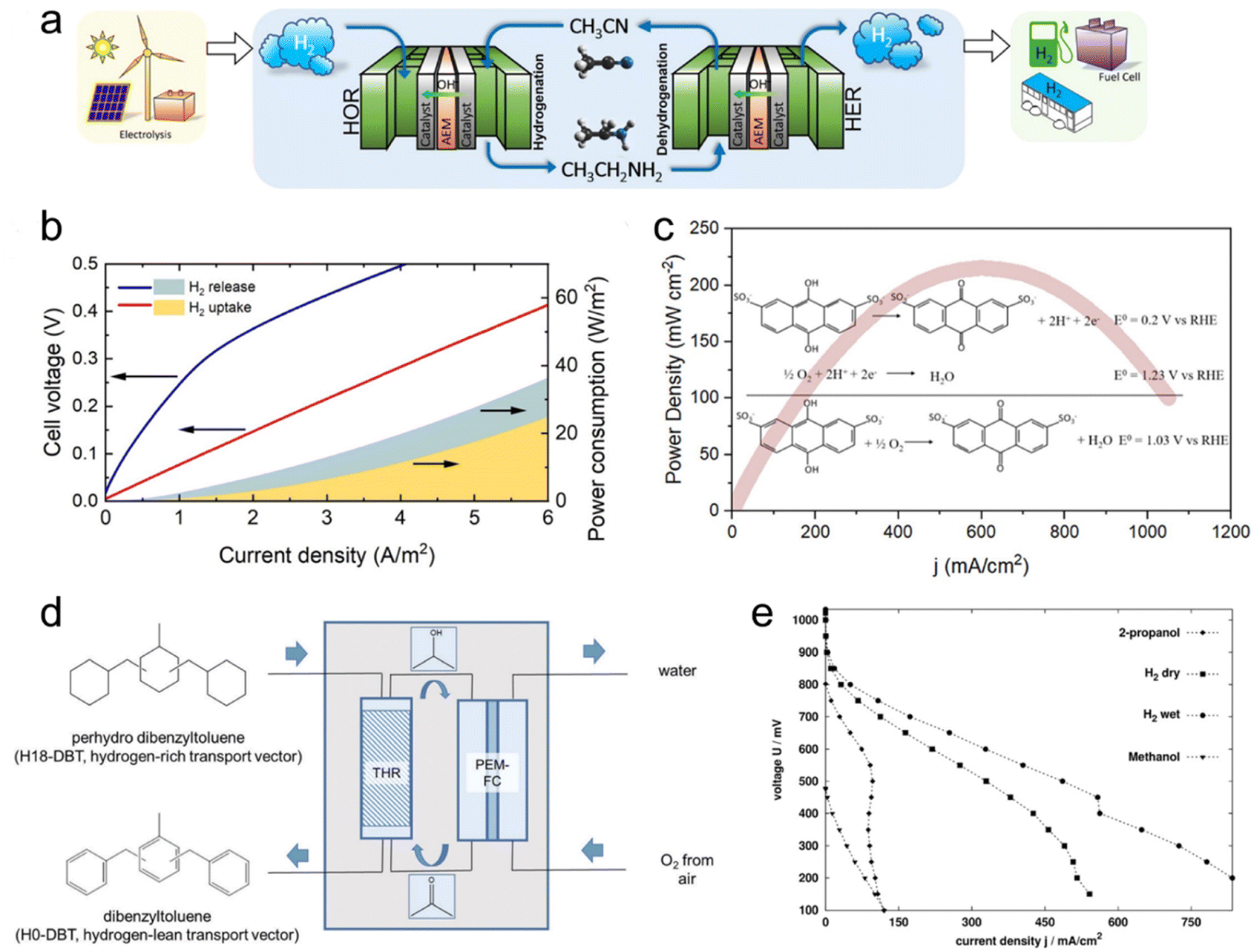 | ||
| Fig. 2 LOHC electrochemistry for hydrogen storage and regenerative fuel cells. (a) Schematic drawing of a full-cell system that realizes a complete cycle of hydrogen storage under ambient conditions by coupling the electrochemical ethylamine/acetonitrile redox method with the HER/HOR, with hydrogen production from renewable energy and hydrogen applications included to illustrate a sustainable hydrogen economy. (b) Full-cell with ethylamine/acetonitrile I–V curves for H2 uptake and release steps, and corresponding power consumption. Reproduced with permission from ref. 10. Copyright 2021 American Chemical Society. (c) Power density curves of a direct quinone fuel cell. Reproduced with permission from ref. 31. Copyright 2023 American Chemical Society. (d) Conversion of H18-DBT-bound hydrogen to electricity in a sequence of transfer hydrogenation of ACE to IPA in a transfer hydrogenation reactor (THR) followed by use of the formed 2-propanol as organic fuel in a subsequent PEM fuel cell. (e) Current–voltage curves of different fuels (IPA, methanol, and hydrogen (dry and wet)). Reproduced with permission from ref. 3. Copyright 2019 Royal Society of Chemistry. | ||
In a follow-up study, Li et al. focused on the electrolysis of ethylamine and showcased a remarkable H2 production rate of 309 μmol h−1 with an impressive 97% FE at room temperature.9 Moreover, the process yielded acetonitrile with a 100% FE at a cell voltage of 0.6 V. This signifies a notable advancement in ethylamine electrolysis for H2 release, demonstrating both speed control and excellent selectivity under ambient conditions. The stability of ethylamine electrolysis was evaluated via chronoamperometry, revealing rapid current decay from 81 mA cm−2 to 9 mA cm−2 within 15 minutes, indicating catalyst deactivation. In situ attenuated total reflectance Fourier-transform infrared (ATR-FTIR) characterization showed stronger adsorption of primary amine-related peaks over time, suggesting intermediate accumulation on the catalyst surface. A regeneration cycling experiment involving a negative voltage sweep effectively recovered initial current density, with subsequent linear sweep voltammetry indicating a 14% drop in final current density compared to an 80% drop without treatment. FTIR spectra demonstrated desorption of accumulated species with negative voltage treatment, regenerating active sites and alleviating performance degradation.
3.3. Regenerative LOHC fuel cells
The electrochemical redox properties of LOHCs lay the foundation for their direct utilization as regenerative fuels in fuel cells for electrical energy generation. Unlike traditional liquid fuel cells, such as direct methanol fuel cells (DMFCs), which encounter issues related to their complete oxidation to emit CO2, the electro-oxidation of LOHC+s produces their corresponding LOHC− that can be facilely hydrogenated to regenerate the original LOHC+s, forming a closed loop and having no emissions.Taking regenerative IPA fuel cells as an example, IPA reacts with air to generate electricity and meanwhile produces ACE and water as products. Then ACE can be hydrogenated back to IPA on-board, by coupling ACE electro-reduction and water oxidation reactions. Alternatively, the produced ACE can be collected and centralized to regenerate IPA for re-use. In a study by Sievi et al., they presented a concept using perhydro-dibenzyltoluene (H18-DBT) as the hydrogen transport vector to reduce ACE to IPA. Subsequently, they employed IPA as the fuel in a proton exchange membrane fuel cell (PEMFC) for electricity generation (Fig. 2d).3 The initial validation confirmed the selective formation of IPA from ACE with H18-DBT as the hydrogen source and Pt/C as the catalyst, demonstrating the feasibility of the reaction under relatively mild conditions (190 °C). In a semi-continuous experiment under ambient pressure conditions with a 0.3 wt% Pt/AlOx catalyst at 170 °C, they achieved a 13.6% yield of IPA after 1 h, affirming the viability of acetone hydrogenation with the assistance of H18-DBT. The performance of the IPA fuel cell was then evaluated with a commercial DMFC setup with a PtRu/C anode and compared with other established fuel cells such as hydrogen and methanol. Fig. 2e illustrates the corresponding I–V curves of the fuel cells, indicating that the performance of the IPA fuel cell is three to four times lower than that of hydrogen but significantly better than that of methanol. Importantly, this process requires no external heat input and demonstrates promising potential for over 50% efficiency, surpassing traditional dehydrogenation/fuel cell approaches.
Quinone was demonstrated as another candidate for the regenerative fuel cell application, offering a cost-effective alternative by eliminating the need for catalysts. Yurko et al. reported an anthraquinone-2,7-disulfonic acid (AQDS) fuel cell, which delivered three times peak power density of a DMFC without the application of any catalyst at the anode (Fig. 2c).31 The cell demonstrated reversibility when switched into electrolyzer mode, with the oxidized AQDS solution being completely regenerated within 25 min. The chemical stability of AQDS was evaluated over 35 cycles, revealing a slight decrease in total charge passed during AQDS oxidation cycles, possibly indicating crossover or self-discharge mechanisms. Material degradation from acidic conditions may promote AQDS disproportionation, while cathode degradation and poisoning by leaked AQDS could further impact performance. Enhancing material strength and selecting improved membranes can reduce crossover and enhance stability.
3.4. Rechargeable LOHC batteries
With the capacity for in situ regeneration, LOHC redox electrochemistry can be utilized in rechargeable battery application, with LOHCs being integrated for use as active electrode materials. Building upon the approach of hydrogen gas batteries, Tang et al. introduced a new concept employing LOHCs as anodes instead of H2 gas in batteries.29 Their study focused on two LOHC redox couples, i.e., cyclohexanol (CHOL)/cyclohexanone (CHON) and IPA/ACE, and investigated their electrochemical redox properties under half-cell conditions in a 1 M KOH electrolyte. CV and H1NMR analysis confirmed the reversible interconversion between CHOL and CHON and between IPA and ACE. Coin cells employing LOHC-loaded Pt/CC as the anode and Ni(OH)2/NF as the cathode exhibited promising performance (Fig. 3a). The CHOL cell achieved a maximum specific capacity of 374 mA h g−1 (Fig. 3b), while the IPA cell reached 525 mA h g−1. Notably, both cells showed high cell voltage comparable to hydrogen gas batteries, while exhibiting significantly higher specific capacities (Fig. 3c). Cycling tests revealed fair cyclability for the CHOL cell and a rapid decay in discharge capacity for the IPA cell (Fig. 3d), attributed to catalyst deactivation caused by poisoning. Further electrochemical impedance spectroscopy (EIS) and distribution of relaxation times (DRT) analysis provided insights into the mechanistic aspects of LOHC batteries, indicating slower reaction kinetics for both CHOL/CHON and IPA/ACE anode reactions compared to Li-ion batteries. The study highlighted the potential of LOHCs for energy storage applications in rechargeable batteries, with opportunities for improving catalyst stability and performance in the future.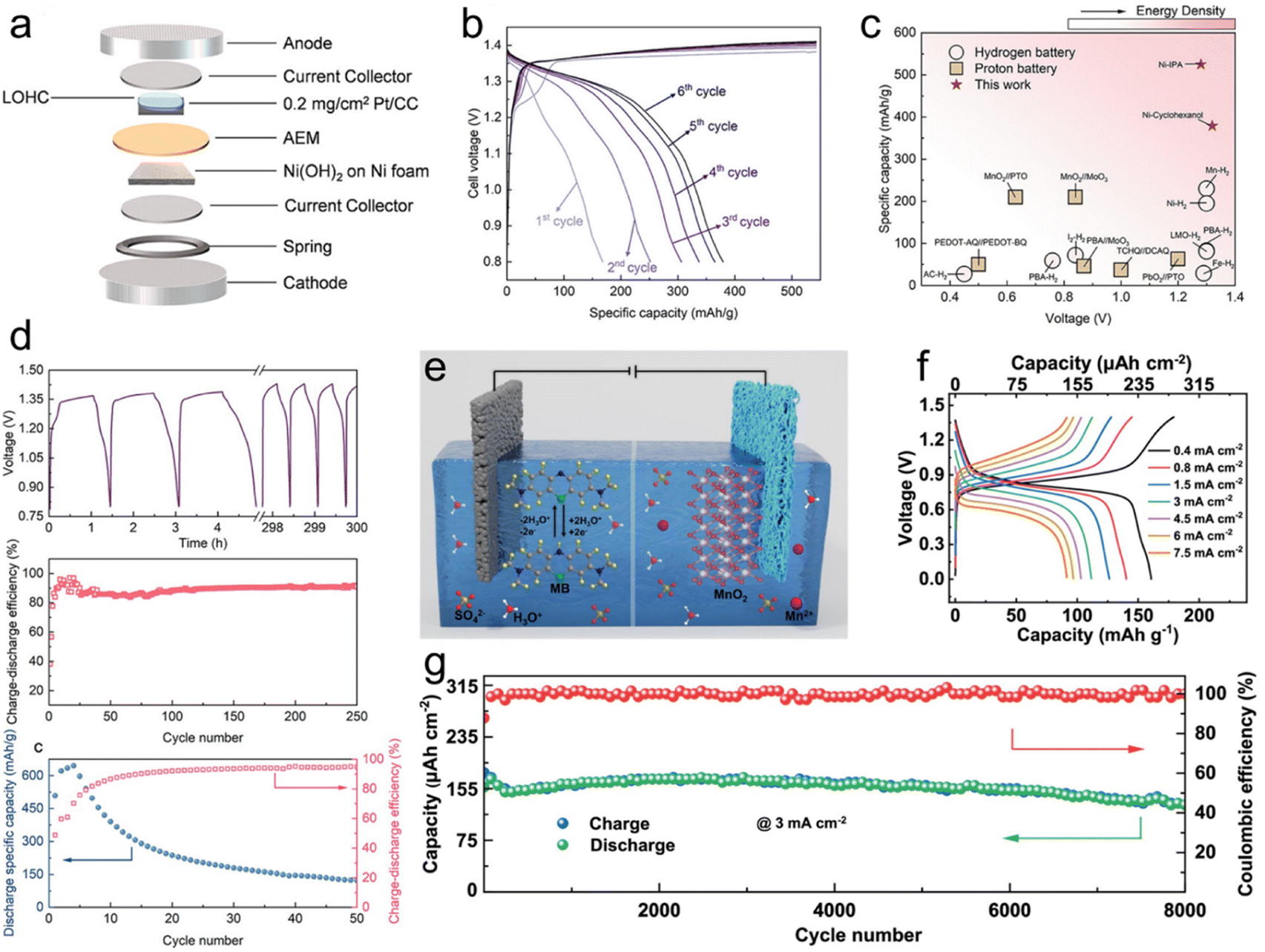 | ||
| Fig. 3 LOHC electrochemistry of rechargeable batteries. (a) Schematic drawing of the assembly of a LOHC coin cell. (b). GCD profiles of CHON/CHOL coin cells at 1C from 0.8 to 1.43 V from the 1st cycle to the 6th cycle. (c) Comparison of the voltage and specific capacity of ACE/IPA and CHON/CHOL to other hydrogen batteries and proton batteries. (d) Cycling life and efficiency of the Ni(OH)2/NF-LOHC/Pt/CC batteries. Reproduced from ref. 29. Copyright 2023 American Chemical Society. (e) Schematic illustration of the operation mechanism of MB//MnO2 hydronium-ion batteries. (f) GCD curves of MB//MnO2 hydronium-ion batteries at various current densities. (g) Cycle stability of MB//MnO2 hydronium-ion batteries tested at a current density of 3 mA cm−2. Reproduced with permission from ref. 49. Copyright 2024 Elsevier. | ||
In another study, a hydronium-ion battery employing a soluble methylene blue (MB) anode and a MnO2@graphite felt cathode was introduced (Fig. 3e).49 The anode undergoes a –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N/–C–N–H functional group transition, demonstrating a maximum capacity of 1324 μA h cm−2 at 2 mA cm−2 and exceptional cycle stability with 93% retention over 7500 cycles. In situ and ex situ characterization studies reveal MB's highly reversible interfacial redox reaction and robust ring skeleton stability. When coupled with the MnO2@graphite felt cathode, the MB//MnO2 batteries achieved an energy density of 198 μW h cm−2 (Fig. 3f) and outstanding long cycle stability exceeding 8000 cycles (Fig. 3g). These findings paved the way for high-stability LOHC hydronium-ion batteries, leveraging the advantages of high conductivity, economic benefits, and environmental friendliness.
N/–C–N–H functional group transition, demonstrating a maximum capacity of 1324 μA h cm−2 at 2 mA cm−2 and exceptional cycle stability with 93% retention over 7500 cycles. In situ and ex situ characterization studies reveal MB's highly reversible interfacial redox reaction and robust ring skeleton stability. When coupled with the MnO2@graphite felt cathode, the MB//MnO2 batteries achieved an energy density of 198 μW h cm−2 (Fig. 3f) and outstanding long cycle stability exceeding 8000 cycles (Fig. 3g). These findings paved the way for high-stability LOHC hydronium-ion batteries, leveraging the advantages of high conductivity, economic benefits, and environmental friendliness.
3.5. LOHC flow batteries
Aqueous flow batteries are gaining recognition for their safety, cost-effectiveness, and scalability, positioning them as a promising option for next-generation energy storage and conversion. The growing popularity of LOHC flow batteries stems from their structural diversity, tailorability, and potential for low cost.13,53Fig. 4a depicts the reactions for benzoquinone, phenazine, and naphthalenediimide in acidic aqueous electrolytes, highlighting their ability to undergo deprotonation, a working principle shared among LOHCs. These organic compounds can be strategically paired with other organic or inorganic redox couples to form a flow battery to store electricity.54 For example, Jin et al. exploited the potential of a water-miscible quinone, specifically 1,8-bis(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)anthracene-9,10-dione (AQ-1,8-3E-OH), by constructing a full cell using concentrated AQ-1,8-3E-OH negolyte and 0.31 M ferrocyanide posolyte. The negolyte achieved a record volumetric capacity of 80.4 A h L−1, with a theoretical limit of 120.1 A h L−1 when AQ-1,8-3E-OH transitions into a liquid at temperatures exceeding 35 °C. Polarization experiments showed higher OCV across all state-of-charge (SOC) levels compared to a low-concentration cell (Fig. 4b and c). Though the high-concentration cell exhibited increased viscosity that impacts mass-transfer rates, a peak power density of 0.17 W cm−2 was maintained. Cycling experiments demonstrated a capacity fade rate of 0.5% per day or 0.043% per cycle over 220 cycles, with a current efficiency averaging 99.90% (Fig. 4d). They attributed the capacity fade solely to the decomposition of the active material in the negolyte by identifying the decomposition products, which may be controlled by adjusting the electrolyte environment and inert atmosphere. To enhance energy density, the study paired the 1.5 M negolyte with a more concentrated posolyte, achieving a peak power density of 0.22 W cm−2 and accessing 94% of theoretical capacity. These findings hint at the potential of LOHCs for high-stability hydronium-ion batteries with remarkable energy density.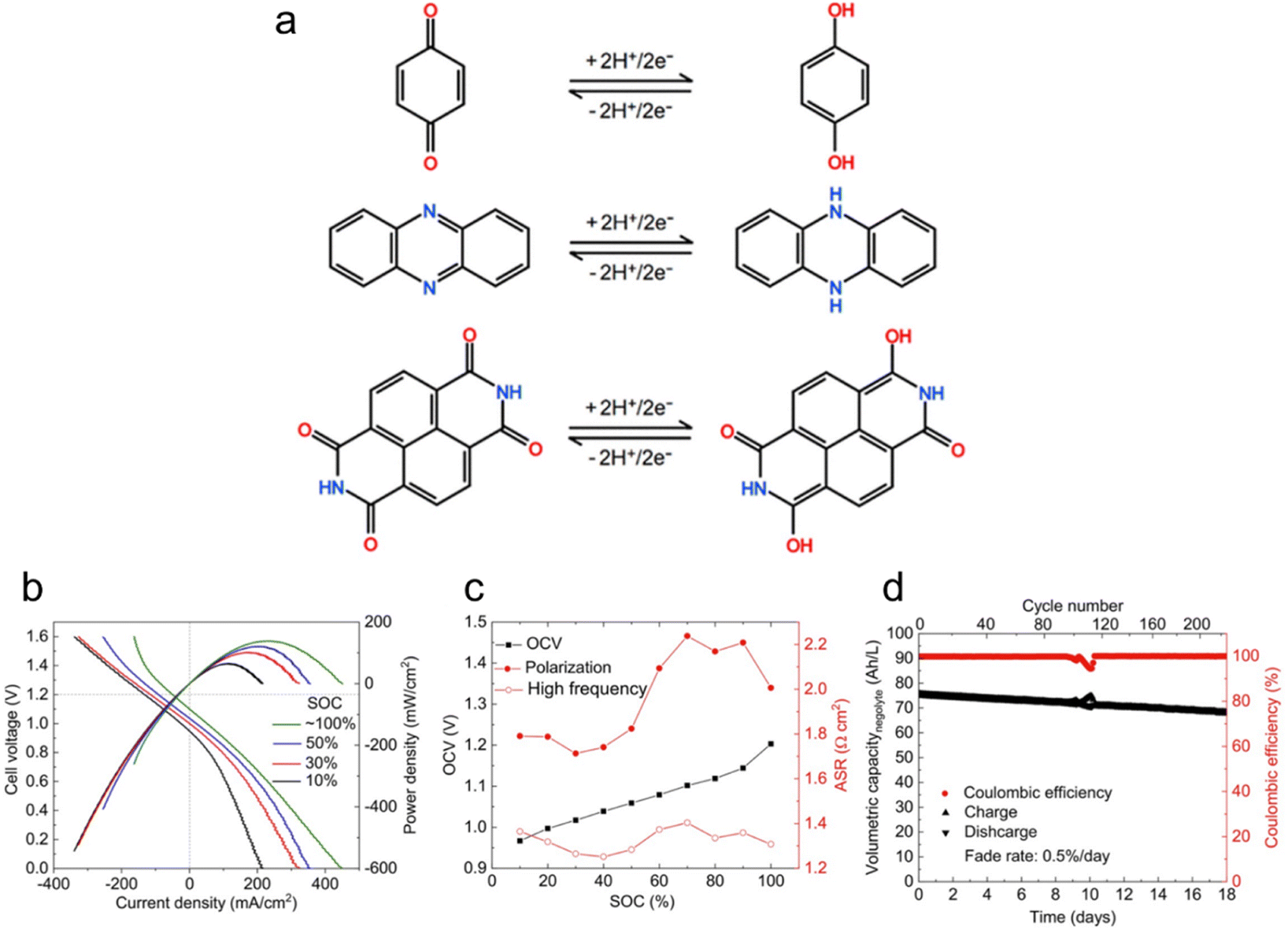 | ||
| Fig. 4 LOHC electrochemistry for flow batteries. (a) Schematic illustration of a two-electron transfer for benzoquinone, phenazine, and naphthalenediimide in acidic aqueous electrolytes. Reproduced with permission from ref. 53. Copyright 2020 American Chemical Society. (b) Cell voltage and power density versus current density at room temperature at 10%, 30%, 50%, and ∼100% SOC. Electrolytes comprised 7 mL of 1.5 M AQ-1,8-3E-OH (negolyte) in 1 M KCl and 150 mL of 0.31 M potassium ferrocyanide and 0.31 M potassium ferricyanide (posolyte) in 1 M KCl. (c) OCV, high frequency, and polarization ASR versus SOC. (d) Current efficiency and charge–discharge capacities versus time and the cycle number. Reproduced with permission from ref. 48. Copyright 2019 American Chemical Society. | ||
4. Outlook
In this perspective, our exploration of LOHC redox electrochemistry involved categorizing LOHCs based on their distinct functional groups, alongside an introduction and summary of their electrochemical properties and applications, particularly for hydrogen storage and electrical energy storage and generation. We mainly focused on organic molecules, either in liquid form or dissolved in liquid when utilized as carriers that can reversibly take up and release hydrogen through electrochemical redox reactions. This would include previously underexplored candidates such as PINO and phenazine. Previous studies have clearly demonstrated great potential and unique advantages of LOHC redox electrochemistry in different applications, for instance, the capability to take up and release H2 for its storage under ambient conditions, the merit of enabling regenerative fuel cells that generate clean electricity without emission, and the feasibility of constructing energy-intensive rechargeable batteries and flow batteries for electricity storage.4.1. Challenges at the fundamental level
Meanwhile, there are remaining research challenges that are hindering understanding and application of LOHC redox electrochemistry. At the fundamental level, gaining a deeper knowledge of LOHC redox electrochemistry mechanisms is imperative. Advanced approaches such as density functional theory (DFT) and in situ spectroscopy can unveil intricate details, assisting in elucidating intermediate species and reaction pathways, understanding reaction reversibility, kinetics, selectivity and stability properties, and identifying governing factors to guide catalyst discovery. These are especially crucial in the case of LOHC electrochemical reactions that require a catalyst, in which catalyst selectivity and stability often pose a significant challenge. Research efforts need to prioritize the discovery of robust catalysts to address issues such as side reactions and poisoning to enhance selectivity and stability performance. Furthermore, establishing structure–property relationships is essential in catalyst research to identify superior catalysts with high activity, selectivity, and stability.4.2. Challenges at the application level
At the application level, other physiochemical properties of a LOHC, including but not limited to gravimetric and volumetric hydrogen capacity, cost, safety, eco-friendliness, storage and operating conditions, solubility, etc., are also important factors to consider besides its electrochemical redox properties. It is challenging to obtain a LOHC with ideal properties from every perspective. Prioritization and trade-offs are often needed to apply LOHC electrochemistry for specific applications. Different categories of LOHCs, owing to their characteristics, offer distinct advantages tailored to various applications. For onboard hydrogen storage and electrical energy generation that typically demand intense energy, LOHCs with high gravimetric and volumetric hydrogen capacities, such as ethylamine, render them promising candidates. For stationary hydrogen storage and energy storage and generation scenarios that impose fewer mass and volume restrictions, LOHCs with low cost and eco-friendliness and meanwhile decent hydrogen capacity, such as isopropanol, serve as promising candidates. Those that require inexpensive or even no catalyst materials would be even more ideal candidates for such applications, paving the way for cost-effective systems that eliminate dependency on precious metals.4.3. Potential future directions
The integration of LOHC redox electrochemistry with existing electrochemical redox reactions has been emerging to transform LOHCs as clean energy carriers, beyond their hydrogen carrier role, for electricity storage and generation using regenerative fuel cells, rechargeable batteries and flow batteries. LOHCs hold a great promise for these energy applications with advantages such as low cost, high charge capacity, and safe operation. The research and development in this field is still at the rudimentary stage. Therefore, extensive studies, at both fundamental and applied levels, are crucial for deepening our understanding and performance control of LOHC-based regenerative fuel cells, rechargeable batteries and flow batteries. For example, in the realm of flow batteries, the use of redox couples such as TEMPO as active materials for the electrolyte often leads to higher costs associated with the flow battery strategy. Therefore, there is substantial potential for cost reduction in flow batteries by adopting readily available LOHCs such as IPA and ethylamine as active materials, sourced directly from industry.Overall, the continuous exploration of LOHC electrochemistry and its diverse applications, propelled by the quest for sustainability and enhanced efficiency, signifies it as a dynamic and burgeoning field. With their multifaceted potential, LOHC electrochemical technologies stand at the forefront of innovation, poised to redefine the landscape of hydrogen and energy storage solutions for the future. As researchers delve deeper into the intricacies of LOHC systems, uncovering novel electrochemistry mechanisms and unlocking new avenues for practical implementation, the significance of this field is expected to continue growing exponentially. Indeed, the trajectory of LOHC research points towards a transformative impact on the energy sector, promising to revolutionize how we store, transport, and utilize renewable energy in a sustainable and environmentally conscious manner.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the support of the US National Science Foundation (NSF and 1955452) and University of South Carolina.References
- P. C. Rao and M. Yoon, Energies, 2020, 13, 6040 CrossRef CAS.
- E. Rakić, M. Grilc and B. Likozar, Chem. Eng. J., 2023, 144836 CrossRef.
- G. Sievi, D. Geburtig, T. Skeledzic, A. Bösmann, P. Preuster, O. Brummel, F. Waidhas, M. A. Montero, P. Khanipour and I. Katsounaros, Energy Environ. Sci., 2019, 12, 2305–2314 RSC.
- P. Hauenstein, D. Seeberger, P. Wasserscheid and S. Thiele, Electrochem. Commun., 2020, 118, 106786 CrossRef CAS.
- F. Waidhas, S. Haschke, P. Khanipour, L. Fromm, A. Görling, J. Bachmann, I. Katsounaros, K. J. Mayrhofer, O. Brummel and J. r. Libuda, ACS Catal., 2020, 10, 6831–6842 CrossRef CAS.
- R. Bayat, A. El Attar, M. Akin, M. Bekmezci, M. El Rhazi and F. Sen, Int. J. Hydrogen Energy, 2024, 51, 1577–1586 CrossRef CAS.
- J. Li, J. Tang, P. Pishva and Z. Peng, SSRN Electron. J., 2023, 4548072 Search PubMed.
- D. Zhang, J. Chen, Z. Hao, L. Jiao, Q. Ge, W.-F. Fu and X.-J. Lv, Chem Catal., 2021, 1, 393–406 CrossRef CAS.
- J. Li, J. Tang, D. Wu, L. Yao and Z. Peng, Int. J. Hydrogen Energy, 2023, 48, 36286–36294 CrossRef CAS.
- D. Wu, J. Li, L. Yao, R. Xie and Z. Peng, ACS Appl. Mater. Interfaces, 2021, 13, 55292–55298 CrossRef CAS PubMed.
- K. Müller, R. Aslam, A. Fischer, K. Stark, P. Wasserscheid and W. Arlt, Int. J. Hydrogen Energy, 2016, 41, 22097–22103 CrossRef.
- L. Shi, S. Qi, J. Qu, T. Che, C. Yi and B. Yang, Int. J. Hydrogen Energy, 2019, 44, 5345–5354 CrossRef CAS.
- C. G. Cannon, P. A. Klusener, N. P. Brandon and A. R. Kucernak, ChemSusChem, 2023, 16, e202300303 CrossRef CAS PubMed.
- C. Yang, L. A. Farmer, D. A. Pratt, S. Maldonado and C. R. Stephenson, J. Appl. Chem. Sci., 2021, 143, 10324–10332 CAS.
- H. Shuang, H. Chen, F. Wu, J. Li, C. Cheng, H. Li and J. Fu, Fuel, 2020, 275, 117896 CrossRef CAS.
- J. Cho, B. Kim, S. Venkateshalu, D. Y. Chung, K. Lee and S.-I. Choi, J. Appl. Chem. Sci., 2023, 145, 16951–16965 CAS.
- Y. Cheng, Y. Liu, D. Cao, G. Wang and Y. Gao, J. Power Sources, 2011, 196, 3124–3128 CrossRef CAS.
- M. T. Bender and K.-S. Choi, JACS Au, 2022, 2, 1169–1180 CrossRef CAS PubMed.
- M. Fleischmann, K. Korinek and D. Pletcher, J. Electroanal. Chem. Interfacial Electrochem., 1971, 31, 39–49 CrossRef CAS.
- M. Fleischmann, K. Korinek and D. Pletcher, J. Chem. Soc., Perkin Trans. 2, 1972, 1396–1403 RSC.
- L. Yan, C. Zhao, Y. Sha, Z. Li, T. Liu, M. Ling, S. Zhou and C. Liang, Nano Energy, 2020, 73, 104766 CrossRef CAS.
- J. E. Nutting, M. Rafiee and S. S. Stahl, Chem. Rev., 2018, 118, 4834–4885 CrossRef CAS PubMed.
- F. Vigier, S. Rousseau, C. Coutanceau, J.-M. Leger and C. Lamy, Top. Catal., 2006, 40, 111–121 CrossRef CAS.
- M. J. Farias, W. Cheuquepán, A. A. Tanaka and J. M. Feliu, ACS Catal., 2019, 10, 543–555 CrossRef.
- H. An, L. Pan, H. Cui, D. Zhou, B. Wang, J. Zhai, Q. Li and Y. Pan, J. Electroanal. Chem., 2015, 741, 56–63 CrossRef CAS.
- A. Santasalo-Aarnio, Y. Kwon, E. Ahlberg, K. Kontturi, T. Kallio and M. T. Koper, Electrochem. Commun., 2011, 13, 466–469 CrossRef CAS.
- A. Toghan, M. Khairy, M. M. Mohamed and A. A. Amer, J. Mater. Res. Technol., 2022, 16, 362–372 CrossRef CAS.
- C. Li, A. M. Sallee, X. Zhang and S. Kumar, Energies, 2018, 11, 2691 CrossRef.
- J. Tang, J. Li, P. Pishva, R. Xie and Z. Peng, ACS Energy Lett., 2023, 8, 3727–3732 CrossRef CAS.
- A. Fukunaga, A. Kato, Y. Hara and T. Matsumoto, Appl. Energy, 2023, 348, 121469 CrossRef CAS.
- Y. Yurko and L. Elbaz, J. Appl. Chem. Sci., 2023, 145, 2653–2660 CAS.
- Y. Tian, K.-H. Wu, L. Cao, W. H. Saputera, R. Amal and D.-W. Wang, Chem. Commun., 2019, 55, 2154–2157 RSC.
- Y. Li, Y. Wei and W. Zhang, J. Electroanal. Chem., 2020, 870, 114251 CrossRef CAS.
- T. Imada, M. Chiku, E. Higuchi and H. Inoue, ACS Catal., 2020, 10, 13718–13728 CrossRef CAS.
- M. T. Huynh, C. W. Anson, A. C. Cavell, S. S. Stahl and S. Hammes-Schiffer, J. Appl. Chem. Sci., 2016, 138, 15903–15910 CAS.
- C. Kim, Y. Lee, K. Kim and U. Lee, Catalysts, 2022, 12, 1113 CrossRef CAS.
- B. Hammer and J. K. Nørskov, Surf. Sci., 1995, 343, 211–220 CrossRef CAS.
- X. Shen, Y. Pan, B. Liu, J. Yang, J. Zeng and Z. Peng, Phys. Chem. Chem. Phys., 2017, 19, 12628–12632 RSC.
- R. G. dos Reis and F. Colmati, J. Solid State Electrochem., 2016, 20, 2559–2567 CrossRef CAS.
- Y. Liao, G. Yu, Y. Zhang, T. Guo, F. Chang and C.-J. Zhong, J. Phys. Chem. C, 2016, 120, 10476–10484 CrossRef CAS.
- J. Maya-Cornejo, R. Carrera-Cerritos, D. Sebastián, J. Ledesma-García, L. Arriaga, A. Aricò and V. Baglio, Int. J. Hydrogen Energy, 2017, 42, 27919–27928 CrossRef CAS.
- K. Wang, R. Sriphathoorat, S. Luo, M. Tang, H. Du and P. K. Shen, J. Mater. Chem. A, 2016, 4, 13425–13430 RSC.
- G. Zhang, Z. Yang, W. Zhang, H. Hu, C. Wang, C. Huang and Y. Wang, Nanoscale, 2016, 8, 3075–3084 RSC.
- T. Liu, K. Wang, Q. Yuan, Z. Shen, Y. Wang, Q. Zhang and X. Wang, Nanoscale, 2017, 9, 2963–2968 RSC.
- C. Tang, C. Wei, Y. Fang, B. Liu, X. Song, Z. Bian, X. Yin, H. Wang, Z. Liu and G. Wang, Nat. Commun., 2024, 15, 3233 CrossRef CAS PubMed.
- C. Bianchini, V. Bambagioni, J. Filippi, A. Marchionni, F. Vizza, P. Bert and A. Tampucci, Electrochem. Commun., 2009, 11, 1077–1080 CrossRef CAS.
- J. Rubio-Garcia, A. Kucernak, A. Parra-Puerto, R. Liu and B. Chakrabarti, J. Mater. Chem. A, 2020, 8, 3933–3941 RSC.
- S. Jin, Y. Jing, D. G. Kwabi, Y. Ji, L. Tong, D. De Porcellinis, M.-A. Goulet, D. A. Pollack, R. G. Gordon and M. J. Aziz, ACS Energy Lett., 2019, 4, 1342–1348 CrossRef CAS.
- S. An, L. Hu, X. Li, S. Zhao, M. Shi and C. Yan, Energy Storage Mater., 2024, 64, 103076 CrossRef.
- R. Feng, X. Zhang, V. Murugesan, A. Hollas, Y. Chen, Y. Shao, E. Walter, N. P. Wellala, L. Yan and K. M. Rosso, Science, 2021, 372, 836–840 CrossRef CAS PubMed.
- S.-B. Han, D.-H. Kwak, H. S. Park, J.-Y. Park, K.-B. Ma, J.-E. Won, D.-H. Kim, M.-C. Kim and K.-W. Park, J. Power Sources, 2018, 393, 32–36 CrossRef CAS.
- J. Xu, S. Pang, X. Wang, P. Wang and Y. Ji, Joule, 2021, 5, 2437–2449 CrossRef CAS.
- J. Cao, J. Tian, J. Xu and Y. Wang, Energy Fuels, 2020, 34, 13384–13411 CrossRef CAS.
- C. Wang, Y. Wang, M. Tao, B. Yu, K. Zhang, J. Wei, Y. Liu, P. Zhang, G. Ding and Z. Tie, ACS Appl. Energy Mater., 2022, 5, 10379–10384 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |