
Immobilization of metal active centers in reticular framework materials for photocatalytic energy conversion
Shuo
Wang
 a,
Wei
Guo
*b,
Chao
Li
a,
Bo
Weng
c,
Shuai
Liu
e,
Ning
Han
*b,
Jinlin
Long
*a and
Honglei
Zhang
*d
a,
Wei
Guo
*b,
Chao
Li
a,
Bo
Weng
c,
Shuai
Liu
e,
Ning
Han
*b,
Jinlin
Long
*a and
Honglei
Zhang
*d
aState Key Laboratory of Photocatalysis on Energy and Environment, College of Chemistry, Fuzhou University, Fuzhou 350116, China. E-mail: jllong@fzu.edu.cn
bDepartment of Materials Engineering, KU Leuven, Leuven 3001, Belgium. E-mail: wei.guo1@kuleuven.be; ninghan51@126.com
cCAS Key Laboratory of Urban Pollutant Conversion, Institute of Urban Environment Chinese Academy of Sciences, 1799 Jimei Road, Xiamen 361021, China
dNottingham Ningbo China Beacons of Excellence Research and Innovation Institute, Department of Chemical and Environmental Engineering, University of Nottingham Ningbo China, Ningbo 315100, China. E-mail: honglei-zhang@nottingham.edu.cn
eSchool of Mechatronics and Energy Engineering, NingboTech University, Ningbo 315100, China
First published on 30th April 2024
Abstract
To enhance the economy and society's sustainability, prioritizing innovative, green energy conversion methods is essential. The conversion of solar energy into chemical energy through photocatalysis represents a viable solution in the pursuit of sustainable energy resources despite low solar-to-energy conversion efficiency. In recent years, the immobilization of metal active centers in nanospace has been considered an important strategy to improve photocatalytic performance. Metal–organic frameworks (MOFs), covalent organic frameworks (COFs) and hydrogen-bonded organic frameworks (HOFs), as exciting reticular framework materials, have attracted much attention due to their advantages of large specific surface area, high porosity and tunable functionalization sites, which are considered promising carriers for immobilizing metal sites. This paper introduces the structural features and advantages of each framework in photocatalysis, highlighting their potential applications in water splitting (H2 or O2 evolution) and CO2 reduction. The review underscores the outstanding performance of metal-active sites within these frameworks. Finally, the current limitations and challenges of the reticular framework materials in photocatalysis are pointed out, and an outlook for future development is provided, which aims to inspire continued advancements in green energy solutions for a sustainable future.
 Honglei Zhang | Dr Honglei Zhang is an Assistant Professor/Investigator in Renewable Energy & Energy Storage at The Nottingham Ningbo China Beacons of Excellence Research and Innovation Institute (CBI). He completed his PhD in Chemical & Environmental Engineering in 2016, under the mentorship of Professor George Chen and Professor Tao Wu. His research primarily focuses on the synthesis, in situ characterization, and practical application of innovative photo/electrocatalysts for CO2 reduction and hydrogen production, which are critical processes in the field of renewable energy and environmental sustainability. Prior to joining CBI, he was a postdoctoral fellow and subsequently research fellow with the Hong Kong Polytechnic University. |
1. Introduction
Over the past few decades, the conflict between the substantial energy consumption for rapid economic growth and environmental degradation has become increasingly prominent.1–5 To address the issue and reduce the consumption of non-sustainable energy resources, there has been a growing interest in developing alternative production methods and systems that utilize renewable and sustainable energy sources. Among them, photocatalysis, recognized for its prowess in solar-to-chemical energy conversion, is extensively explored as a solution for energy conservation and environmental pollution mitigation.6–9 Its advantages, including renewability, cost-effectiveness, and minimal side effects, position it as a promising avenue for addressing these pressing challenges.Photocatalysts, pivotal in photocatalytic technology, face a daunting challenge in achieving comprehensive control and optimization of performance, spanning efficiency, activity, and selectivity.10 Photocatalytic efficiency, an important indicator to evaluate their performance, is contingent on the optical properties of photocatalysts. The cornerstone for exceptional photocatalytic performance lies in enhancing quantum efficiency through bandgap structure tuning. Catalytic activity, closely linked to the number of catalytic sites, substrate adsorption, and product desorption, is influenced by morphologies, specific surface areas, and intrinsic or post-modified active centers. Selectivity hinges on the chemical properties, with redox behavior at catalytic sites being crucial. Modulating product type and concentration according to demand is integral to realizing high-value chemicals. Various photocatalytic systems, including traditional inorganic materials like metal oxides and sulfides,11,12 have been explored for efficient energy conversion. However, inorganic materials face limitations such as wide band gaps, weak light absorption, and poor chemical stability.13–15 These challenges, coupled with difficulty in rational modulation at the molecular level and dependence on noble metals, restrict the broad application of inorganic materials. Molecular catalysts allow pre-designed organic ligands but often compromise stability for increased activity and selectivity.16–18 Additionally, controlling the assembly form of molecular catalysts in different solutions poses a significant challenge in studying the conformational relationships of photocatalysts. Hence, there is an urgent need for the discovery and study of a multifunctional photocatalytic material that seamlessly combines various properties.
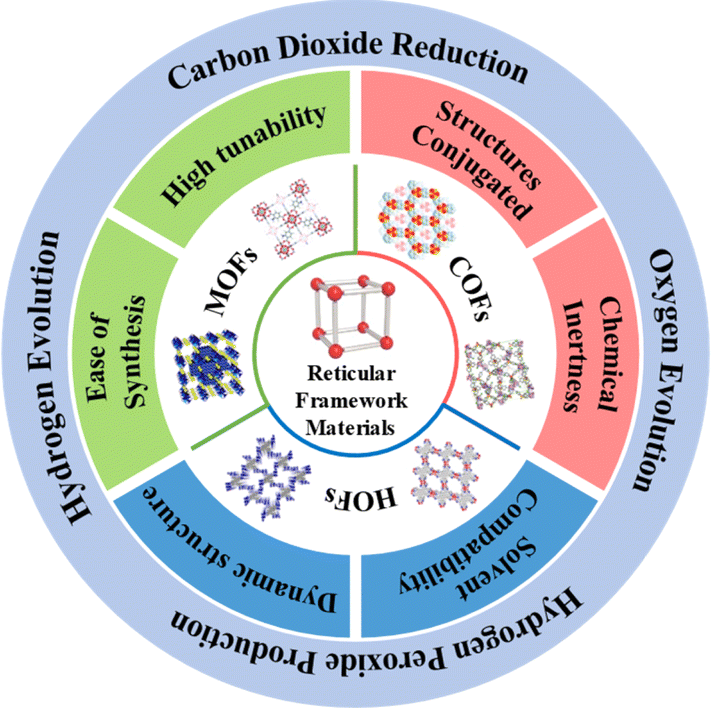
Reticular chemistry extends molecular principles to construct precise structures through chemical bonds, facilitating the prediction and design of diverse periodically expanding structures. This approach enables the creation of various reticulated materials,19,20 including discrete solids (metal–organic polyhedra, covalent organic polyhedra) and framework materials (Fig. 1) like metal–organic frameworks (MOFs), covalent organic frameworks (COFs), and hydrogen-bonded organic frameworks (HOFs).21–23 Specifically, MOFs are a class of crystalline porous skeletal materials with periodic network structures, which are formed by self-assembly of organic ligands and metal ions via coordination bonds. Generally, metal ions act as the connecting points and organic ligands support to form spatial 3D extensions with three-dimensional pore structures. COFs are a new type of crystalline organic porous polymers based on covalent bonding connections, which are usually composed of lightweight elements (C, O, N, B, etc.), while HOFs, as another ordered framework materials, are constructed only by self-assembly of organic building units by non-covalent intermolecular forces (hydrogen bonding, π–π stacking, etc.). Reticular framework materials have emerged as prominent photocatalysts due to their tunable pore sizes and exceptional specific surface areas, enhancing reactivity by enabling substrate diffusion within restricted nano spaces during catalysis. The pre-designable structure further enhances their performance for diverse photocatalytic applications.24–26 While most reviews focus on the overall design or organic unit importance, there is a limited exploration of the role of metal sites in these frameworks. Modulating original metal sites in MOFs or incorporating metal sites in COFs and HOFs addresses the vulnerability of homogeneous metal molecular catalysts,27,28 optimizing kinetic and thermodynamic processes for enhanced photocatalytic performance. The robust topology and tailorable pore structure of reticular framework photocatalysts allow effective immobilization of metal active sites, influencing the feasibility and performance of the photocatalytic system.
 | ||
| Fig. 1 Schematic representation of reticular framework materials. | ||
This review focuses on the progress in photocatalytic energy conversion using reticular framework materials (MOFs, COFs, HOFs) as ideal carriers for metal active sites. We will examine the structural features and unique advantages of each framework, emphasizing applications in water splitting (H2 or O2 evolution) and CO2 reduction. The pivotal role of metal sites in influencing the photocatalytic performance will be highlighted. Finally, challenges and opportunities associated with reticular framework materials containing metal sites in the realm of photocatalytic energy conversion will be discussed.
2. Classification of reticular framework materials and respective advantages
2.1 Metal–organic frameworks (MOFs)
MOFs, as a novel periodic reticular framework material, have emerged as ideal candidates for multiphase photocatalysis (Fig. 2a).29–31 The substantial specific surface area and high porosity of MOFs facilitate the accessibility of active sites, fostering the generation of charge–hole pairs.32,33 Their structural customizability allows efficient band gap engineering and the incorporation of additional catalytic centers. The confined catalytically active sites within MOFs serve as unique microreactors, enhancing substrate adsorption and activation through host–guest interactions.34,35 Furthermore, MOFs organize reactants strategically, minimizing entropy loss and reducing transition state energy, thereby facilitating photocatalytic reactions.36,37 The periodic order and site isolation of catalytic columns in MOFs eliminate deactivation pathways, extending the functional lifetime of catalytic centers. This characteristic enables a thorough exploration of reaction mechanisms. Currently, efforts focus on improving MOFs' photocatalytic prowess by fine-tuning the type, number, and coordination environment of metal sites, attracting significant scientific attention.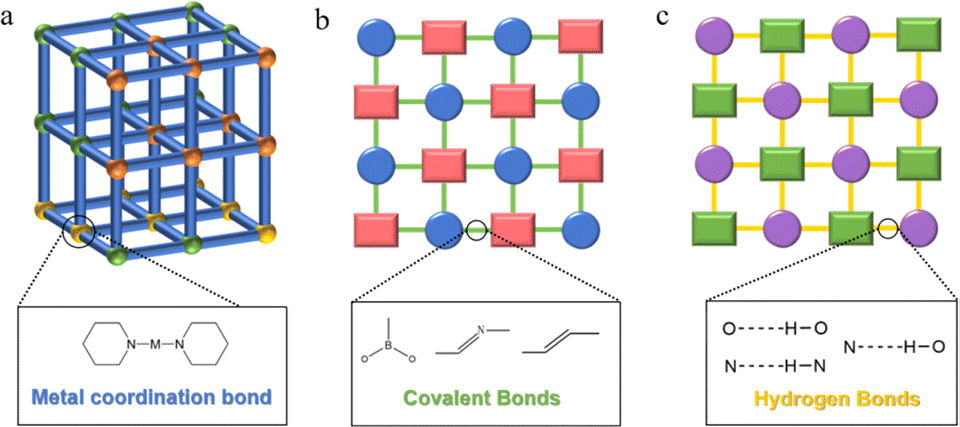 | ||
| Fig. 2 Structures of MOFs (a), COFs (b) and HOFs (c) constructed by organic monomers. | ||
Typically, there exist two main forms of introducing metal catalyzed active sites in MOFs.38 Firstly, metal-conjugated nodes with unsaturated coordination environments are utilized as the catalytic sites. The metal nodes in MOFs are usually accompanied by coordinated water molecules or other solvent molecules, which can be easily removed and do not disrupt the framework structure of the MOFs, allowing the accessible unsaturated metal centers that can be used as catalytic sites for reactions with substrate molecules. Secondly, the metal ions in the inorganic clusters can be replaced by different metal ions without compromising the structure. Thus, some MOFs can contain several kinds of metal ions, which can provide opportunities for versatility and tailoring the properties of the materials to specific applications. The possible synergistic effect due to the presence of multiple metals facilitates the realization of tandem or cascade reactions in the field of multiphase catalysis, making them suitable for a wider range of applications.
2.2 Covalent organic frameworks (COFs)
COFs, a subset of reticular organic framework materials, arise from the integration of organic building units linked by covalent bonds (Fig. 2b),39–42 mirroring biopolymer formation. Non-covalent interactions, predominantly hydrogen bonding, and hydrophobic interactions, complement covalent bonds formed through polycondensation reactions. The distinctive fusion of covalent and noncovalent interactions in COFs sets them apart from other polymers and porous materials, offering a unique structure, properties, and functionality.43,44 This distinctiveness positions COFs as an appealing platform across various fields. Notably, COFs feature pre-designed framework structures and inherent pore sizes, expanding their design versatility. Specifically, COFs excel in integrating desired architectural units or active sites into periodically ordered structures, creating permanent pores with atomic precision in sizes, shapes, and environments.45,46Since COFs were first reported by Yaghi and co-workers in 2005,47 COFs have aroused excitement among researchers over the past nearly two decades. Extensive efforts have focused on COFs, emphasizing structural design, precise synthesis, and specific modifications across various disciplines. With inherent permanent porosity, rigid framework structures, and long-range ordering, COFs emerge as ideal candidates for multiphase catalysis in photocatalytic systems. Their design flexibility enhances properties crucial for photocatalysis, ensuring optimal light absorption, superior electron–hole separation, and efficient transfer. The substantial specific surface area and porous structure facilitate the exposure of catalytic sites and the mass transfer.48,49 Notably, COFs exhibit remarkable chemical and thermodynamic stability under harsh photocatalytic conditions due to their robust covalent bonding.50 These intrinsic properties position COFs with significant potential to rival or even surpass conventional inorganic semiconductors in photocatalytic applications, driving comprehensive exploration within the scientific community.
2.3 Hydrogen-bonded organic frameworks (HOFs)
HOFs, a novel reticular framework material, stand apart from MOFs and COFs as they are stabilized by non-covalent hydrogen bonding (Fig. 2c).51–53 Their distinguishing features include high crystallinity, sustainability, and utility, attributed to the flexibility and spontaneity of hydrogen bonding. Various topologies and monomer designs contribute to the fabrication of highly crystalline HOFs, enhancing structural analysis accuracy through techniques like powder or single-crystal X-ray diffraction. This exceptional crystallinity facilitates a thorough exploration of conformational relationships in photocatalysis.54The interest in assembling crystalline HOFs using hydrogen bonding was ignited in the 2010s, and several HOFs with permanent pores have subsequently been constructed using hydrogen bonding motifs,55–57 such as pyridine, diaminotriazine, benzimidazolone, pyrazole, and carboxyl groups. The topology of HOFs is connected by weak hydrogen bonds, which leads to structural instability compared to MOFs and COFs. Indeed, several routes through π–π interactions, electrostatic interactions, and van der Waals force-assisted hydrogen-bonded assemblies have recently been used to fabricate stable HOFs.58,59 However, weak hydrogen bonding connection also has its unique advantages; for example, HOFs can usually be assembled under milder conditions, which facilitates the confinement of metal units by in situ encapsulation. Moreover, the flexibility and reversibility of hydrogen bonding led to solution processability and simple regeneration of HOFs, which helps to realize their sustainable utilization under practical conditions.
3. Photocatalytic applications
3.1 Photocatalytic hydrogen evolution
Under the context of growing global energy demand, hydrogen (H2) is an ideal alternative energy source to fossil fuels to achieve sustainable development, thanks to its high energy density (about three times that of gasoline), non-toxicity, renewability, and clean emission product. H2 is considered an ideal energy carrier for the future, despite the risk of fire in the air and the remaining limitations of safety challenges for its consumption, storage, and transport. Currently, photocatalytic water splitting for H2 production is one of the most promising strategies.60 Semiconductor materials capable of efficiently realizing this process usually have distinct properties, such as a wide range of light absorption, high efficiency of electron–hole pair separation, long lifetime of the photoexcited electrons, minimal potential barriers to photoexcited charge migration, etc. Porous reticular framework materials have received increasing attention due to their ability to provide conventional solid-state platforms with porous structures, consecutive repeating units with few defects, countless active sites, and different metal centers as options. Therefore, such materials have the potential to be effective photocatalysts for H2 evolution to overcome the limitations of the conventional semiconductors in this field.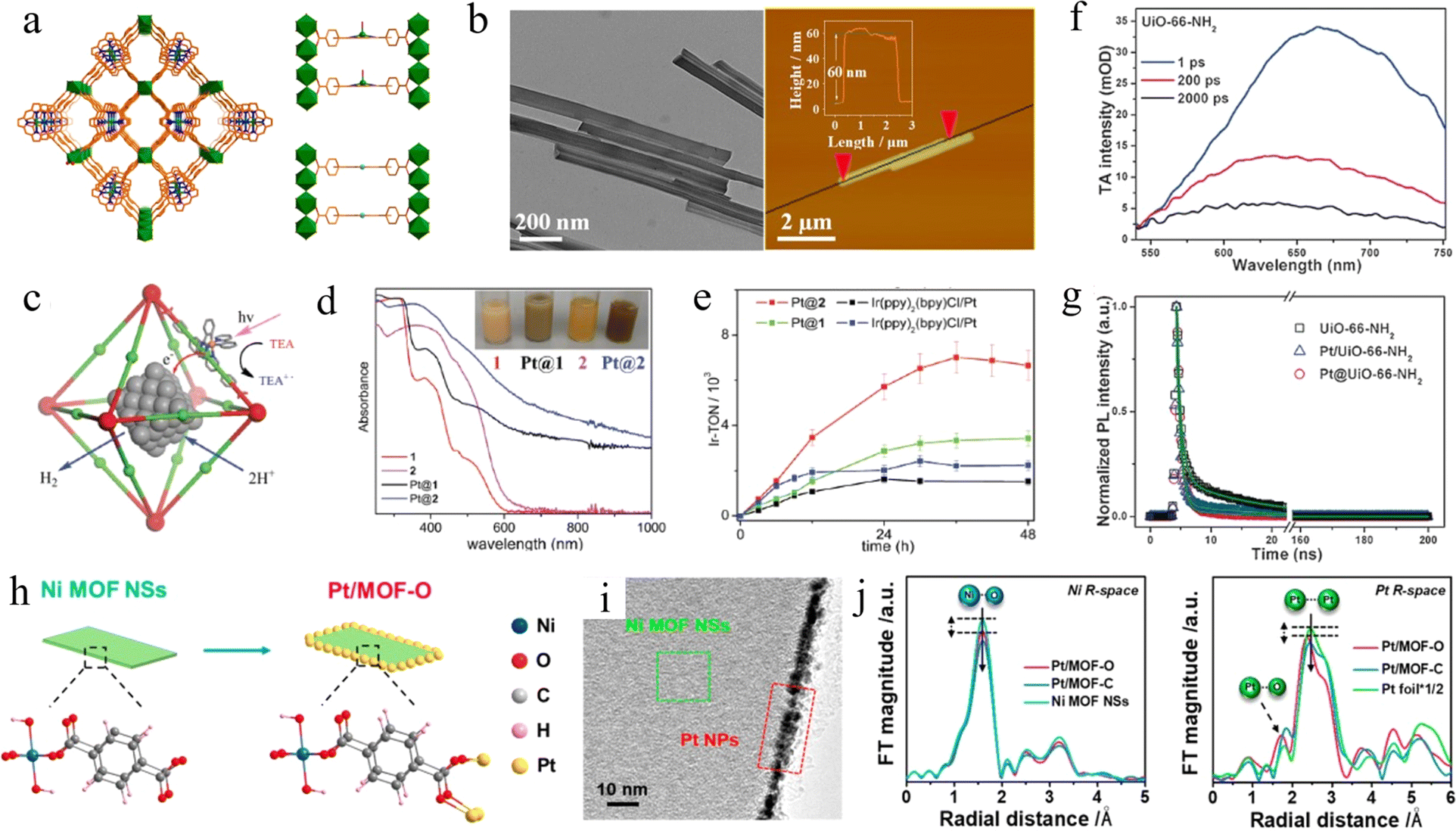 | ||
| Fig. 3 (a) Views of the 3D network of USTC-8(In) and partial structures in USTC-8(In) and USTC-8(M) (M = Cu, Co, Ni). Adapted with permission from ref. 65. Copyright 2018, American Chemical Society. (b) The TEM image and AFM topography of Ni-TBAPy-NB nanobelts. Adapted with permission from ref. 68. Copyright 2022, American Chemical Society. (c) Schematic of the synergistic photocatalytic H2 evolution process using Pt NP-incorporated MOF frameworks. (d) Diffuse reflectance spectra of 1, Pt@1, 2, and Pt. A photograph of the suspensions of these samples is shown in the inset. (e) Time-dependent H2 evolution curves of as-prepared samples. Adapted with permission from ref. 70. Copyright 2012, American Chemical Society. (f) TA spectra of UiO-66-NH2. (g) Time-resolved PL decay profiles for UiO-66-NH2, Pt@UiO-66-NH2, and Pt/UiO-66-NH2, respectively. Adapted with permission from ref. 72. Copyright 2016, Wiley-VCH. (h) Schematic illustration of the structure of Pt/MOF-O. (i) HRTEM images of Pt/MOF-O. (j) EXAFS spectra and Pt L3 edge EXAFS spectra of Pt/MOF-O, Pt/MOF-C and Ni MOF NSs. Adapted with permission from ref. 73. Copyright 2021, American Chemical Society. | ||
Doping metals as mediators in MOFs also can promote charge migration and improve photocatalytic performance, which is inspired by the enhanced performance of metal-doped semiconductors. MOFs with Ti replacing part of Zr, NH2-Uio-66 (Zr/Ti), were prepared by the post-synthetic exchange (PSE) method by Li et al.67 The results showed that the photocatalytic H2 evolution rate of NH2-Uio-66(Zr/Ti) increased 1.5 times compared with NH2-Uio-66(Zr), reaching 3.5 mmol mol−1. DFT studies revealed that the probability of photogenerated electron transfer to the Ti4+ center was significantly higher than that to the Zr4+, resulting in the formation of the (Ti3+/Zr4+)6O4(OH)4 excited state. Ti3+ played the role of an electron donor in which electrons are provided to Zr4+ to form Ti4+–O–Zr3+. Such an electron transfer mechanism helps to improve the transfer of excited interfacial charge to Zr–O clusters for enhanced visible-light photocatalysis. Zhang et al. reported a novel 2D p-type MOF nanoribbon (Ni-TBAPy-NB) photocatalyst with an average nanobelt thickness of approximately 60 nm (Fig. 3b). The MOFs consisted of 1,3,6,8-tetrakis(p-benzoic acid)pyrene (H4TBAPy) ligand as the light-harvesting center and [Ni3O16] clusters as the catalytic center.68 The in situ EPR experiments yielded a g value of 2.005 belonging to paramagnetic Ni(I) species, revealing the reduction of Ni2+ to Ni+ in the photocatalytic process. The reduced Ni(I) active center generated by the light-induced ligand-to-metal charge transfer (LMCT) process can serve as a reduction site for the adsorption and activation of water under visible-light irradiation, which subsequently enables the photocleavage of water to H2 in the absence of any co-catalysts. Aziz et al. demonstrated that the doping metal center also can affect the band gap structure.69 A narrowing of the bandgap was achieved by replacing Al with Fe in the part of the metal center, which resulted in an improved charge separation capability.
However, despite pristine MOFs exhibiting H2 evolution performance, poor conductivity and difficulties in electron transfer kinetics limit further improvement. Therefore, metal nanoparticles (NPs) are often employed as a co-catalyst in the photocatalytic H2 evolution from MOFs. Lin and co-workers successfully encapsulated Pt nanoparticles with diameters of 2–3 nm and 5–6 nm in the cavities of MOFs (Fig. 3c), to investigate the relationship between the size of Pt nanoparticles deposited in MOFs and the photocatalytic performance.70 As shown in Fig. 3e, the TON of the Pt@MOF photocatalytic H2 precipitation was about 5-fold higher than that of the control samples, which can be up to 7000, attributed to the rapid transfer of electrons from the photo-reduced Ir center to the trapped Pt NPs. Furthermore, the expanded absorption range of visible light (Fig. 3d) also played a crucial role in enhancing the performance. Similarly, Su et al. successfully prepared a series of spatially isolated metal single-atom catalysts (SACs) by binding metal single atoms (M-SAs) in the pores of porphyrin-type MOFs.71 The prepared MOFs exhibited excellent sustained efficiency in photocatalytic H2 evolution with a TON as high as 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 713 over 32 h and a start/sustained turnover frequency (TOF) greater than 1200/600 h−1 based on M-SAs under visible light irradiation. Mechanistic studies showed that the tight binding of catalytically active Pt-SAs with a Pd-porphyrin photosensitizer led to enhanced chemical binding and stabilization effects, which can accelerate the electron–hole separation and charge transfer in the pore space, thus largely promoting the photocatalytic water splitting process.
713 over 32 h and a start/sustained turnover frequency (TOF) greater than 1200/600 h−1 based on M-SAs under visible light irradiation. Mechanistic studies showed that the tight binding of catalytically active Pt-SAs with a Pd-porphyrin photosensitizer led to enhanced chemical binding and stabilization effects, which can accelerate the electron–hole separation and charge transfer in the pore space, thus largely promoting the photocatalytic water splitting process.
In addition, the photocatalytic efficiency is closely related to the position of Pt relative to the MOF. Jiang and co-workers incorporated Pt nanoparticles inside UiO-66-NH2 (Pt@UiO-66-NH2) and supported Pt nanoparticles on UiO-66-NH2 (Pt/UiO-66-NH2) with a diameter of 3 nm by two different methods, respectively.72 Pt@UiO-66-NH2 showed better photocatalytic activity than Pt/UiO-66-NH2, which is attributed to the fact that it greatly shortens the distance from the electrons to the catalytic center and promotes the separation of electrons and holes. The TA spectra of UiO-66-NH2 (Fig. 3f) and corresponding time-resolved PL (Fig. 3g) revealed that the lifetimes of UiO-66-NH2, Pt/UiO-66-NH2 and Pt@UiO-66-NH2 were 10.28 ± 0.06, 7.26 ± 0.04 and 2.86 ± 0.02 ns. The shortened lifetimes proved that the introduction of Pt NPs inhibited the recombination of photogenerated carriers in the MOFs due to the opened new channel of electron transfer. Recently, Huang et al. demonstrated that the oriented growth of ultrasmall noble metal nanoparticles (e.g., Pt, Pd, Ag, and Au) on MOFs could be achieved by precisely regulating the reduction kinetics of metal ions (Fig. 3i).73 In particular, when methanol and ethanol were used as reducing agents, the rapid reduction process of metal ions led to the random distribution on the surface of MOFs. In contrast, the oriented growth of metal particles on the edges of MOFs was achieved when ethylene glycol and diethylene glycol were chosen, due to their inhibition of the reduction kinetics, which can be seen in Fig. 3h. Ni–O, Pt–Pt, and Pt–O bonds can be clearly observed in the extended X-ray absorption fine structure (EXAFS) spectrum (Fig. 3j), which further provided strong evidence for the existence of Pt. Moreover, detailed experiments indicated that electrons may be transferred from Pt atoms to O atoms, leading to low electron density of Pt and high electron density of O in Pt/MOF-O. As a result, elevated activity of photocatalytic H2 evolution was exhibited under both acidic and alkaline conditions.
Porphyrins and their derivatives are conjugated π-electron macrocycles with unique physical and chemical properties. In principle, the incorporation of different metal ions into the porphyrin can rationally adjust the photophysical and electronic properties, thus affecting the photocatalytic activity of the corresponding COFs. Considering this, Wang et al. designed and synthesized four COFs with similar structures, which are named MPor-DETH-COF (M = H2, Co, Ni, Zn).74 Interestingly, these COFs exhibited tunable photocatalytic H2 evolution ability, in the order of CoPor-DETH-COF < H2Por-DETH-COF < NiPor-DETH-COF < ZnPor-DETH-COF. The difference in photocatalytic activity was attributed to the effect of different metal ions on the carrier dynamics in the COFs. In particular, the migration of photogenerated electrons relies on metal-to-metal channels rather than specific metal centers under photoexcitation of the porphyrin, and the photogenerated holes are transferred mainly via the pathway of macrocycle-on-macrocycle. As seen in Fig. 4a, for the metal-free H2Por-DETH-COF, the migration of both electrons and holes takes place via the macrocycle-on-macrocycle pathway, which undoubtedly increases charge recombination and leads to poorer photocatalytic activity. However, for CoPor-DETH-COF and NiPor-DETH-COF, the promoted photoactivity can be attributed to the ligand-to-metal charge transfer (LMCT) process, which is partially inhibited with the increase of d electrons (Ni2+ for 3d8), resulting in improved hole transfer ability and enhanced charge separation efficiency. Finally, the LMCT process was prohibited in Zn2+ ions with 3d10 configuration, resulting in a long-time charge separation state. Therefore, ZnPorDETH-COF showed the best photocatalytic H2 evolution activity under the same conditions.
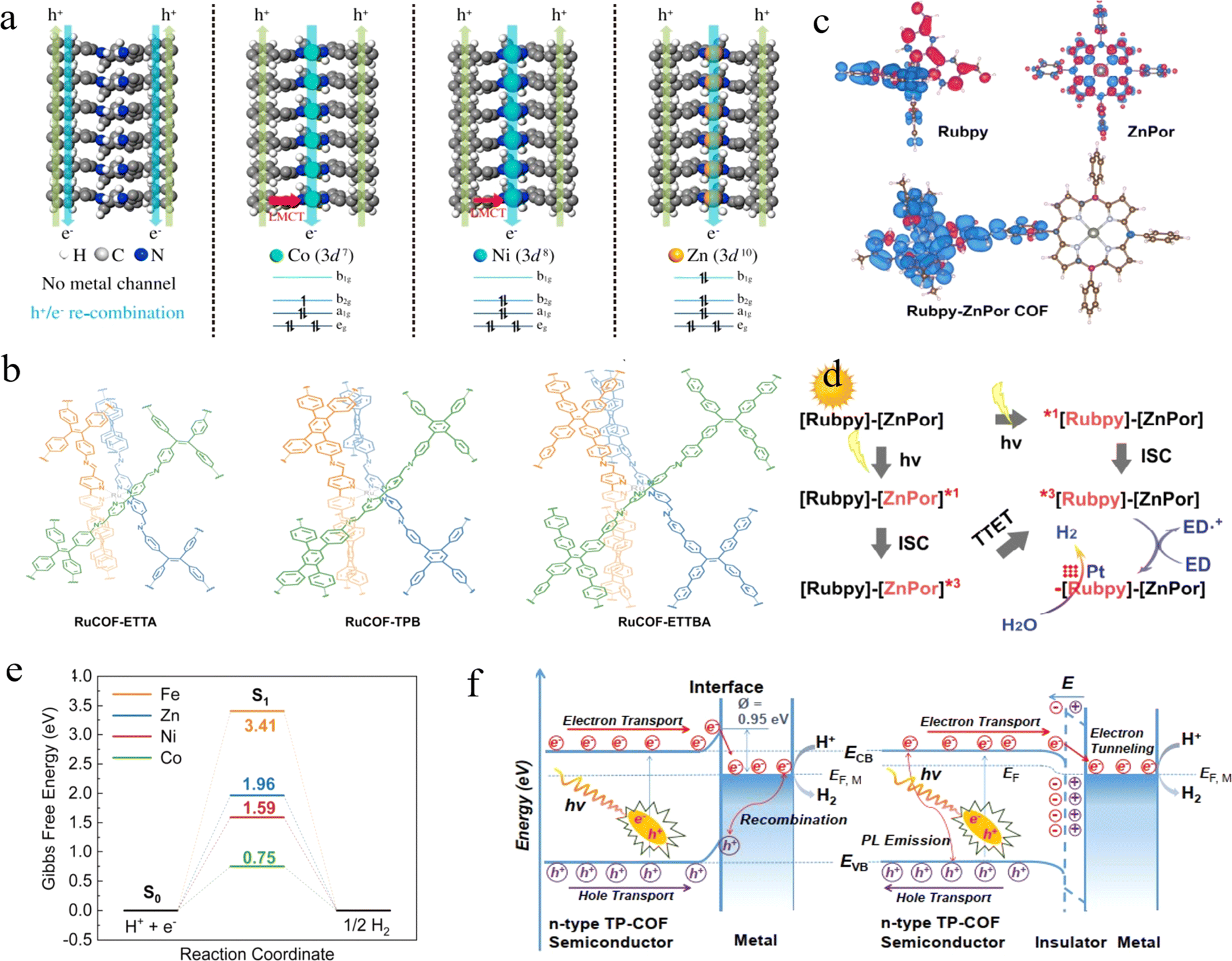 | ||
| Fig. 4 (a) The schematic illustrations of the hole–electron transport processes in MPor-DETH-COFs. Adapted with permission from ref. 74. Copyright 2021, Springer Nature. (b) Illustration of the synthetic procedures of RuCOFs. Adapted with permission from ref. 76. Copyright 2022, Wiley-VCH. (c) Triplet spin density surfaces for Rubpy-ZnPor COF. (d) Proposed photochemical process for H2 evolution. Adapted with permission from ref. 77. Copyright 2023, Wiley-VCH. (e) Gibbs Free energy diagrams for hydrogen evolution on the salen metal site in Zn-salen-COF and M/Zn-salen-COF (M = Fe, Co, Ni). Adapted with permission from ref. 78. Copyright 2023, Wiley-VCH. (f) Mott–Schottky Pt/TP-COF (left) and Pt-PVP-TP-COF MIS (right) photocatalyst. Adapted with permission from ref. 79. Copyright 2019, Wiley-VCH. | ||
In response to the frequent leakage of transition metal ions during the photocatalytic reactions, bipyridine ligand metals are a good choice. Guo et al. used a solvothermal method to anchor Ni(II) ions in bipyridine-containing COFs.75 In contrast to the traditional method of anchoring Ni(II) ions in COFs under mild conditions, this method can result in torsion of the 2,2′-Bpy part of the single bond, leading to a planar conformation coordinated to Ni(II). The COF–Ni2+ complexes allowed the unique transfer mode of metal-to-ligand charge as well as a broadened absorption range of visible light. Based on this, the H2 evolution rate of COF-Ni2+ was nearly 2.5 times higher than that of pristine COFs, and the H2 evolution can also be observed upon 700 nm light irradiation. On the other hand, Gu and co-workers extended bipyridines to 3D COFs.76 Three different COFs, RuCOF-ETTA, RuCOF-TPB, and RuCOF-ETT-BA (Fig. 4b), were synthesized based on the Ru(II)tris(2,2′-bipyridine) unit. Interestingly, each RuCOF contained three isomorphic COFs that were interlocked with the Ru center as the registration point. The homogeneous distribution of Ru units resulted in excellent chemical stability, strong light harvesting ability, and outstanding H2 evolution (20308 μmol g−1 h−1) of the RuCOFs. Moreover, Lan et al. prepared a series of bis-photosensitised COFs with strong visible light absorption by organically combining two conventional photosensitizers, pyridine ruthenium/iron (Ru(bpy)/Fe(bpy)) and porphyrin/metal porphyrin complexes (2Por/ZnPor).77 DFT calculations (Fig. 4c and d) demonstrated that both Rubpy and ZnPor can be excited and subsequently undergo a two-way electron transfer process, and this dual-excitation behavior lays the foundation for achieving fast photoelectron transfer and efficient photocatalytic water splitting to H2.
Furthermore, salen as an efficient molecular catalyst for H2 evolution integrated with the light collector in a photocatalyst is an effective strategy for solar light conversion. As an example, Deng and co-workers reported a series of rigid M/Zn-salen-COFs (M = Zn, Fe, Co, and Ni), which were synthesized from salen-M molecular catalysts with pyrene as the light trapping unit.78 Among them, Co/Zn-salen-COF exhibited the most efficient rate of H2 evolution, reaching 1378 μmol g−1 h−1. The presence of salen-Co increased the separation of photogenerated charge in the COFs and reduced the transfer resistance of interfacial charge, so salen-Co in Co/Zn-salen-COF exhibited a lower Gibbs free energy (Fig. 4e). Furthermore, a binuclear Cu-salphen COF was designed by Mak et al. to achieve an H2 evolution rate of 36.99 mmol g−1 h−1 under visible light irradiation without the decoration of Pt.79 The independent Cu sites with high loading can not only effectively increase the probability of photogenerated charge into the reaction center on the surface of the photocatalyst, but also prevent the recombination of photogenerated carriers. In addition, the authors exfoliated COFs into ultrathin nanosheets to overcome the problems of low active site utilization and slow ion diffusion prevalent in large-volume COFs. That also improved the photocatalytic efficiency thanks to the accelerated charge transfer kinetics greatly.
In the actual process of photocatalytic H2 evolution, metal Pt is often involved in COF photocatalytic H2 evolution as a proton reduction co-catalyst due to its excellent properties. Pt acts as an electron collector throughout the photocatalytic system, with the COFs forming Schottky junctions at the contact interface. In other words, the COFs act as photosensitizers to provide photogenerated electrons for proton reduction on the co-catalyst. Therefore, it is crucial to promote the electron transfer from COFs to the co-catalyst to enhance the photocatalytic H2 evolution. Zhang et al. anchored single-atom Pt to the pore walls of β-ketamine-bonded COFs (TpPa-1-COF).80 The advanced AC HAADF-STEM and XAFS techniques demonstrated that the atomically dispersed Pt existed in the form of a six-coordinated C3N–Pt–Cl2. The good dispersion of single-atom Pt as photocatalytic active sites lays the foundation for the achievement of excellent photocatalytic H2 evolution activity, which was increased 3.9 and 48 times compared to TpPa-1 COFs with Pt nanoparticles and parent TpPa-1, respectively. In addition, the presence of single-atom Pt facilitated effective photogenerated charge separation and lowered the energy barrier for the formation of H*.
Surpassing the conventional Schottky-type photocatalysts, Long's group cleverly designed a novel metal–insulator–semiconductor (MIS) photosystem (Pt-PVP-TP-COF) based on the electrostatic self-assembly of polyvinylpyrrolidone (PVP) insulator-covered Pt NPs with hydrophilic imine-linked TP-COFs.81 As shown in Fig. 4f, the photoelectrons can tunnel through the thin insulating layer (typically∼1.0 nm) to the collector when the MIS heterostructure is irradiated, where the H2 evolution reaction occurs. The electrostatic field formed perpendicular to the COFs between the TP-COF and the insulating layer provided the impetus for charge tunneling for the extraction of hot π-electrons from the photoexcited COF semiconductor. In normal Schottky-type photocatalysts, both excitons can be transferred to Pt, leading to non-radiative recombination and significant reduction of hot carriers and hole oxidation. In contrast, the MIS photosystem hindered the transfer of photogenerated holes to Pt, which can improve the separation efficiency. The combined enhancement of the photoexcitation, charge separation, and oxidation rates of holes accumulated in the VB of the TP-COF resulted in a maximum H2 evolution rate of 8.42 mmol g−1 h−1 and the highest apparent quantum efficiency of 0.4% at 475 nm.
3.2 Photocatalytic oxygen evolution
Photocatalytic O2 evolution is another half of the water-splitting reaction. Combining the two half-reactions of photocatalytic hydrogen and oxygen production, the target of light-to-energy can be achieved. However, the photocatalytic O2 evolution is often accompanied by slow kinetics because it involves multi-electron transfer processes and high overpotential requirements, especially the high energy barrier required for O–O bond formation. Therefore, the progress of reticular frameworks in the field of photocatalytic O2 evolution has been relatively slow compared to the photocatalytic H2 evolution, and only a few cases have been reported.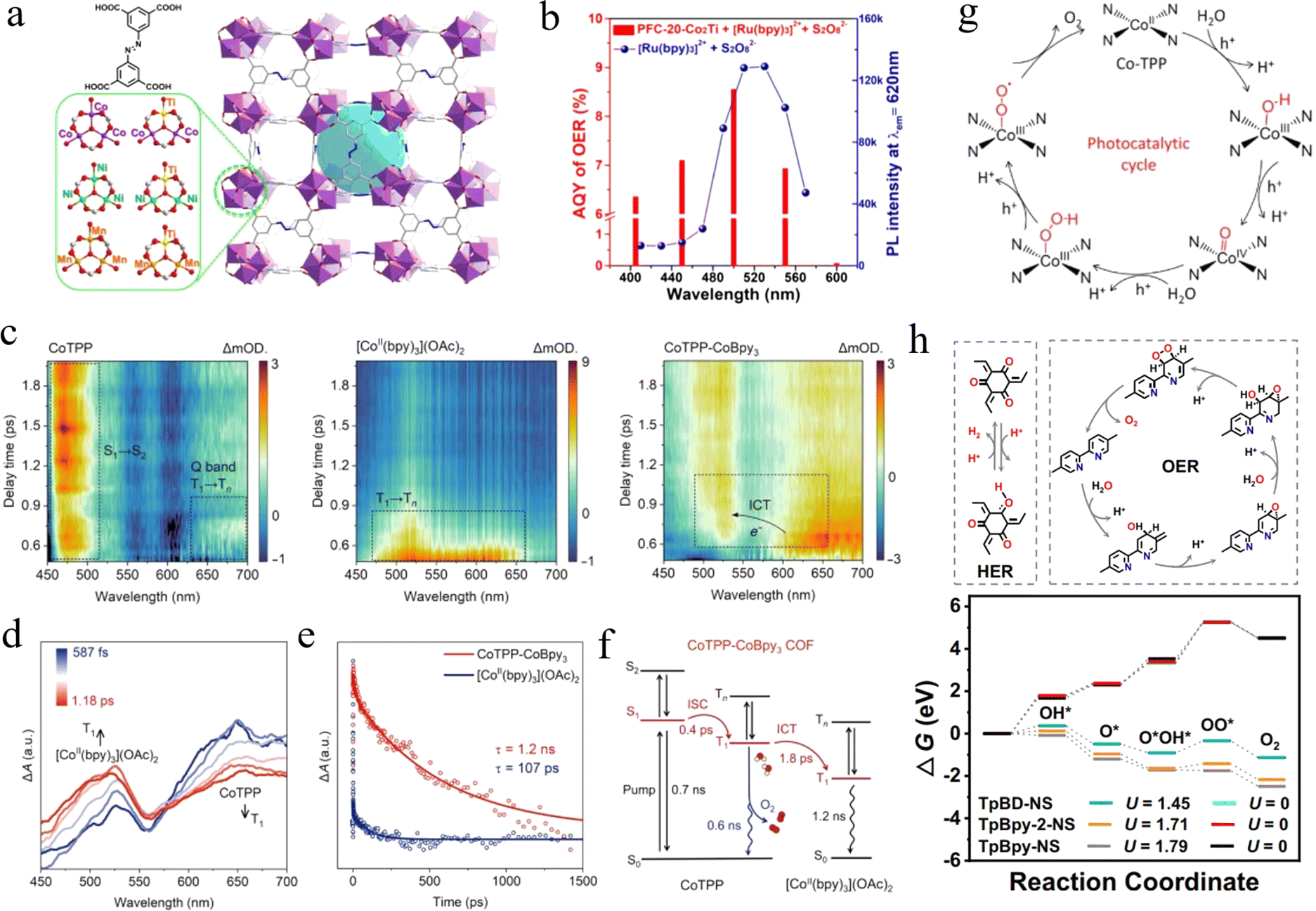 | ||
| Fig. 5 (a) The different metal clusters in the PFC-20 series and the crystal structure of PFC-20. (b) Comparison between AQYs of O2 evolution and PL intensity of [Ru(bpy)3]2+-S2O82− solution under monochromatic-light irradiation. Adapted with permission from ref. 84. Copyright 2022, Chinese Chemical Society. (c) The fs-TA spectra of CoTPP, [CoII(bpy)3](OAc)2, and CoTPP-CoBpy3. (d) ICT from CoTPP to [CoII(bpy)3](OAc)2. (e) The decay trace of ESA signal at 500 nm in CoTPP-CoBpy3 and [CoII(bpy)3](OAc)2. (f) Electron transfer pathway including ISC and ICT. (g) Proposed water oxidation mechanism in CoTPP active sites. Adapted with permission from ref. 84. Copyright 2024, American Association for the Advancement of Science. (h) The possible process of the HER on the Tp segment and the OER via a dual-site process on the Bpy segment in TpBpy-NS, and the comparison of calculated Gibbs free energy change for C2d paths at pH = 7. Adapted with permission from ref. 87. Copyright 2023, Springer Nature. | ||
In addition, the suitable energy band structure is the groundwork for achieving efficient overall water splitting, while the highly exposed active site is an important part of improving photocatalytic activity. Based on this, Yang et al. revealed the catalytic activities of several COFs for H2 and O2 evolution reactions by first-principles calculations, which provided useful guidance for the design of COFs that can be used for visible-light-driven overall water splitting.88 Twelve COFs were designed by assembling nitrogen-containing bonds (including imine, nitrogen, and azo groups) and four phenyl building blocks (including benzene, triphenylamine, 1,3,5-triphenyl benzene, and 2,4,6-triphenyl-1,3,5-triazine). The results showed that 9 of these COFs satisfy both the potential requirements for photocatalytic H2 and O2 production, and only three TA-based COFs can undergo the H2 evolution reaction in theory. In addition, the fact that I-TST COFs can produce H2 and O2 under visible light irradiation was further demonstrated experimentally. The approach of theory-guided experimental provided a new way to design efficient COF-based photocatalysts for overall water splitting under visible light.
3.3 Photocatalytic carbon dioxide conversion
Carbon dioxide (CO2) is considered one of the most dominant greenhouse gases, with approximately 60% of the global warming effect attributed to CO2 emissions. At the same time, it is also an abundant, cheap, non-toxic, and bio-renewable carbon resource. Therefore, the conversion of CO2 into other useful chemicals is an effective way to curb problems of global warming and energy shortage. Thermodynamically, abundant energy is required due to the large enthalpy (523 kJ mol−1) of the double bond in O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO. Recently, the economic solar-driven decomposition of the bond between C and O has been gaining attention. Reticular framework materials have great advantages as photocatalysts due to their high capacity of catalytic sites, semiconducting properties, and tunneling morphology. In addition, the uniform permeable structure and high specific surface area of the reticular framework materials provide an ideal platform for the introduction of metal sites into the frameworks, which lays a solid foundation for achieving more efficient photocatalytic CO2 reduction.
CO. Recently, the economic solar-driven decomposition of the bond between C and O has been gaining attention. Reticular framework materials have great advantages as photocatalysts due to their high capacity of catalytic sites, semiconducting properties, and tunneling morphology. In addition, the uniform permeable structure and high specific surface area of the reticular framework materials provide an ideal platform for the introduction of metal sites into the frameworks, which lays a solid foundation for achieving more efficient photocatalytic CO2 reduction.
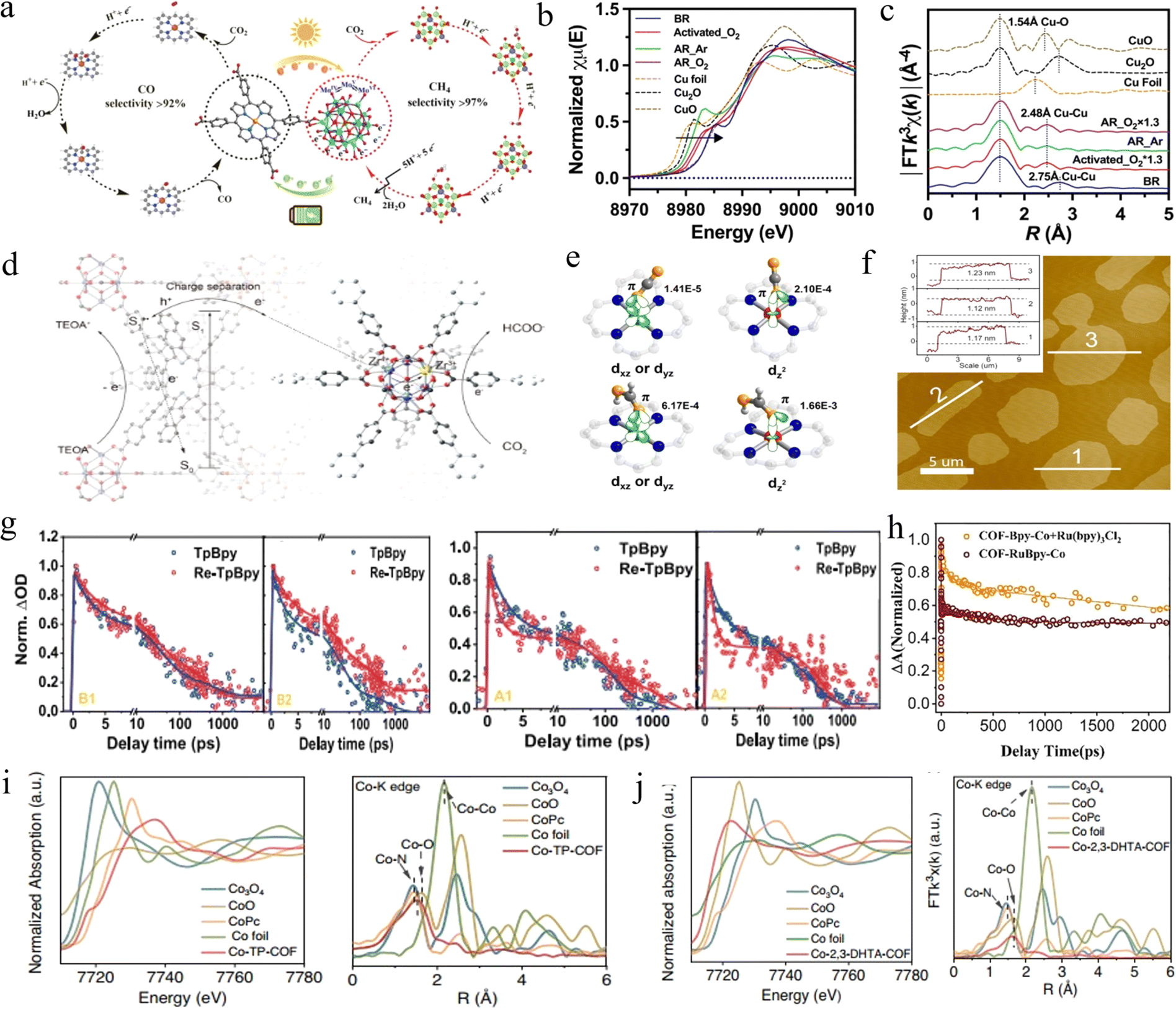 | ||
| Fig. 6 (a) Proposed reaction mechanism in PCR and ECR reactions for Fe-POMOF, respectively. Adapted with permission from ref. 94. Copyright 2022, American Association for the Advancement of Science. (b) Copper K-edge XANES spectra of pristine and (c) Fourier transformed K3-weighted χ(k) function of EXAFS for different samples. Adapted with permission from ref. 98. Copyright 2023, Wiley-VCH. (d) Schematic illustration of the NIR-light-induced electron transfer pathway fromπ-conjugated systems to Zr clusters in TNP-MOF for CO2 reduction. Adapted with permission from ref. 99. Copyright 2022, American Chemical Society. (e) Different coupling modes of CO2 and HCOOH interact with the Co site at different spin states. Adapted with permission from ref. 103. Copyright 2020, American Chemical Society. (f) AFM image and the height profiles along the marked white line of COF-367 NSs. Adapted with permission from ref. 106. Copyright 2019, American Chemical Society. (g) TA kinetics of two samples at various emission wavelengths. Adapted with permission from ref. 109. Copyright 2018, American Chemical Society. (h) Corresponding normalized TA kinetic curves probed at 450 nm. Adapted with permission from ref. 119. Copyright 2023, Elsevier. (i) Normalized XANES spectra and FT-EXAFS spectra at the Co K-edge of Co-2,3-DHTA-COF, Co3O4, CoO, CoPc and Co foil samples. (j) Normalized XANES spectra and FT-EXAFS spectra for discriminating the radial distance and k-space resolution of Co-TP-COF. Adapted with permission from ref. 114. Copyright 2023, Springer Nature. | ||
Similarly, Ti-based MOFs have attracted great interest in the photocatalytic field due to the photoinduced transition from Ti4+ to Ti3+.95 Li et al. prepared NH2-MIL-125(Ti) MOFs with the satisfactory ability to reduce CO2 to HCOOH under visible light.96 NH2 in the prepared MOFs mainly acted as an adsorbent, whereas the Ti4+ center accepted the photogenerated electrons from the NH2-functionalized ligands, then reduced to Ti3+. Subsequently, Ti3+ reacted with the excess electrons with CO2 for the generation of HCOOH. Cu-based MOFs also exhibit similar properties. Typically, Cu has higher activity in the oxidized state than metallic copper, thus pre-oxidation of Cu-MOFs before photocatalysis is the most common method. However, this method suffers from a drawback in that maintaining the oxidized state of Cu sites is difficult during CO2 reduction, which leads to a decrease in photocatalytic capacity.97 To address this, Sheng et al. achieved photocatalytic CO2 reduction to CO and CH4 by Cu-MOFs in the presence of O2 levels (20% v/v) in air, using water as the hole scavenger.98 The results of structural analyses (Fig. 6b and c) showed that the hydroxylated Cu generated by Cu sites during pre-oxidation under aerobic conditions could maintain a stable structure during CO2 reduction, whereas it undergoes structural changes and loses the reducing ability under anaerobic conditions. This resulted in a 5-fold higher CO2 reduction rate in the presence of O2 than under anaerobic conditions, which opened the possibility of CO2 reduction in an air atmosphere.
Another wide strategy is the integration of large π-conjugated organic units into MOFs, such as porphyrin. As an example, mesoporous MOFs with six macrocyclic π-conjugated units that progressively increased in number were synthesized.99 The MOFs containing the porphyrin unit (TNP-MOF) were able to convert CO2 to HCOOH at an extremely high rate of 6630 μmol g−1 h−1 under near-infrared light irradiation. The richness of the linker's electron density can broaden the photo-absorption range of the MOFs. Moreover, as shown in Fig. 6d, the transfer pathway of photogenerated electrons from the conjugated linkers to the metal clusters led to rapid charge separation. These combined factors resulted in an unprecedentedly high photocatalytic performance.
In response to the synergistic effect of individual metal sites on the CO2 reduction reaction, dual-metal-site pair catalysts have been considered as an effective strategy for inducing highly selective products in multi-step catalytic processes.100 In particular, the synergistic effect of these dual-metal-site pairs on the bonding of C1 intermediates provides a viable way to enhance the induction of specific reaction pathways in the catalysis of CO2-to-CH4. Based on this, recently, Zhong et al. merged Cu and Ni sites into a MOF (i.e., MOF-808) in the form of a single-site, which exhibited dynamic adaptive behavior to accommodate mutated C1 intermediates, leading to the photocatalytic CO2-to-CH4 process and achieving a yield of 158.7 μmol g−1 h−1.101 The theoretical calculations showed that the spatial configuration of the Cu/Ni MOF is dynamic during CO2 photoreduction, just as the bonding with substrates and intermediates induces suitable conformations in a natural enzyme system. This adaptive behavior played a crucial role in stabilizing the various C1 intermediates, thus inhibiting the desorption of undesired by-products, which is thought to be responsible for the highly selective photoreduction of CO2 to CH4.
The advantages of Re(I) complexes as photocatalysts for CO2 reduction are as follows: high product selectivity and outstanding CO2 reduction efficiency.108 Therefore, it is promising to support Bpy-Re in COFs. At first, Huang et al. revealed three key intermediates in charge separation, induction period, and CO2 reduction decisive step in the photocatalytic process of COFs containing Re complexes for the first time.109 The photogenerated electrons in this system can be transferred to the Re complexes via intramolecular charge transfer, which achieves better activity than homogeneous Re catalysts. Then, Re-TpBpy was used as an example to study the excitation kinetics and charge transfer process of such catalysts by Zheng and co-workers.110 Femtosecond transient visible (fs-TA) and time-resolved infrared (fs-TRIR) revealed the whole process. As shown in Fig. 6g, the excited electrons were in the position of the Bpy conduction band when the energy of photons was close to the Re-TpBpy band gap, then injected into the Re site within sub-picoseconds. However, these electrons recombined with photogenerated holes in 13 ps. In contrast, when the COFs are excited with high energy, the hot electrons were injected into the high-energy orbital of Re in 2 ps, and then bounced back to the Bpy fragment within 24 ps, while the hot holes slowly (340 ps) relax to the energy level of HOMO. At this point, the long lifetime of excitation electrons in the Re sites provided favorable conditions for two-electron mediated photocatalytic CO2 reduction to CO. Furthermore, Han et al. achieved both CO2 reduction and H2O oxidation by sp2 COF constructed by using the rhenium complex as the reduction site and the triazine ring as the oxidation site.111 The excellent charge separation ability of COF-Bpy-Re allowed the photogenerated electrons and holes to concentrate on the two unit sites in a periodic arrangement, respectively. This resulted in a CO yield of 190.6 μmol g−1 h−1 and an O2 evolution rate of 90.2 μmol g−1 h−1. Similarly, Bpy-Cu and Bpy-Ni were integrated into the COFs, which also achieved the same purpose of improving the efficiency of the photocatalytic CO2 reduction reaction.112 This is because of the enhanced adsorption of CO2, the expanded light absorption, and the improved electron separation efficiency of COFs.
Moreover, 1,2-acenaphthenequinone has likewise been used to anchor monatomic metal sites. Hou et al. utilized it to introduce different single atomic metal sites (e.g., Fe, Co, Ni, Zn, Cu, Mn, and Ru) to the frameworks of triazine COFs with the metal–N–Cl structure.113 It is worth noting that the CO2 conversion rate of Fe SAS/TrCOF as the representative catalyst was 26 times higher than that of the original Tr-COF, which was up to 980.3 μmol g−1 h−1. The superior photocatalytic performance could be due to the synergistic interaction of the atomically dispersed metal sites with the COF framework, which not only lowered the reaction energy barriers for the formation of the *COOH intermediates but also facilitated the adsorption of CO2 while also facilitating the desorption of CO.
The coordination environment of the metal site is also an important factor affecting the performance of COF photocatalysts except for different organic units. Wang et al. took advantage of the controllable functional groups in the precursor module and the strong coordination of Co(II) to spontaneously form Co-COFs,114 which can be based on the pre-designed hydroxyl positions in the aldehyde module by a programmable method to provide a precise coordination microenvironment. The EXAFS results in Fig. 6i and j provide ample evidence for this. In this research, two Co-COFs with different ways of oxygen coordination were designed. The results showed that the as-prepared Co–COF with Co–O4 coordination after adjusting the coordination environment, reached a CO yield of up to 18000 μmol g−1 h−1 and a selectivity of up to 95.7% under visible light irradiation. In situ characterization and theoretical calculations jointly demonstrated that the pre-designed Co–O4 sites significantly promoted the migration efficiency of charge carriers in the Co–COF matrix and inhibited the recombination of photogenerated electron–hole pairs during the photocatalytic process.
Similar to MOFs, the simultaneous integration of dual photosensitive units into the COF backbone not only further extends the range of light absorption, but also enables more efficient intermolecular multi-electron transfer towards the catalytic centers across the π-conjugated COFs,115,116 which could potentially alleviate the rapid quenching and diffusion limitations of photoexcited electrons in physical hybrid systems.117 Wang et al. used [Rel(bpy)(CO)3Cl] and protoporphyrin as building blocks to synthesize imine-conjugated H2PReBpy-COF.118 The self-properties of porphyrin and the extended π-conjugated plane of the 2D COFs resulted in excellent light absorption, efficient charge separation, and fast interfacial charge transfer, making H2PReBpy-COF a preeminent photocatalyst for CO2 reduction. On the other hand, Cao et al. utilized [Ru(bpy)3]2+ (bpy = 2,2′ bipyridine) and activated cobalt porphyrin (Co-Por) to prepare COF-RuBpy-Co.119 The efficient photoelectron transfer from [Ru(bpy)3]2+ to Co-Por enabled a CO generation rate of 547 μmol g−1 h−1, which was a 1.4-fold improvement over both physical mixtures. It is worth noting that the fs-TA results provided evidence for a faster transfer of photogenerated charge (44.2 ps, Fig. 6h) on the large π-conjugated system.
Density Functional Theory (DFT) calculations play an important role in exploring and understanding potential catalytic mechanisms, as well as guiding experimental design.120,121 A series of COFs with bimetallic active sites were designed to readily aggregate to form nanoparticles due to the higher surface energy of the isolated metal atoms. The possible intermediates, free energy, reaction pathways, and overpotentials of the CO2RR were considered and calculated by DFT. Moreover, with the descriptor identification study, the Bader charge associated with the Pauling electronegativity of the embedded bimetallic atoms was found to be an important factor affecting the catalytic activity.
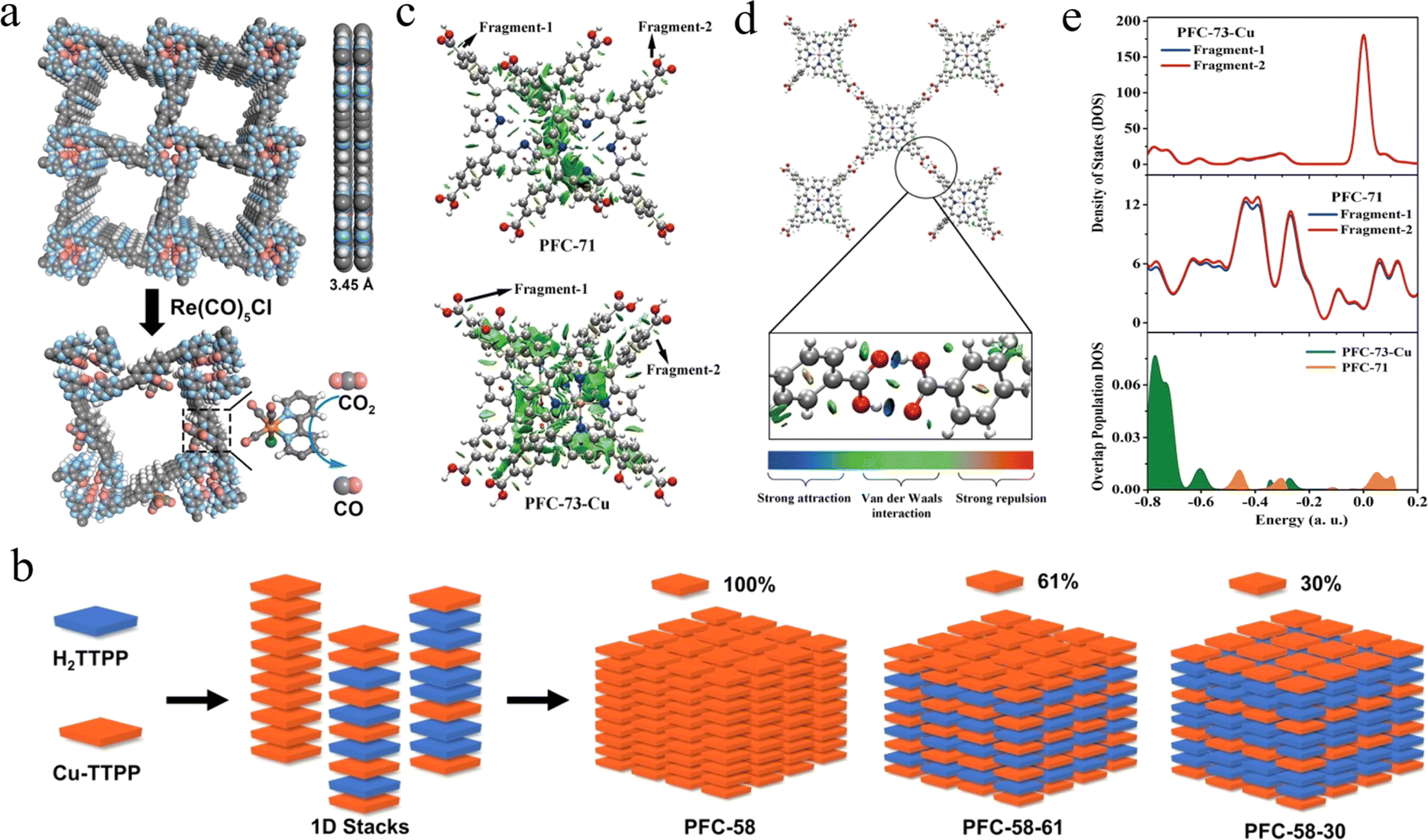 | ||
| Fig. 7 (a) AA packing mode of HOF-25 and a proposed structure of HOF-25-Re for CO2 reduction. Adapted with permission from ref. 122. Copyright 2021, Wiley-VCH. (b) Schematic diagram of the structures of PFC-58 series HOFs with different metalloporphyrin contents. Adapted with permission from ref. 123. Copyright 2022, Wiley-VCH. (c) Interlayer NCI isosurfaces for PFC-71 dimer and PFC-73-Cu dimer show a larger overlap area of PFC-73-Cu than PFC-71. (d) PFC-71 and PFC-73-Cu show similar in-plane NCI isosurfaces with a strong hydrogen bond interaction (only PFC-73-Cu is depicted as a representative). (e) Comparison of DOS and the corresponding OPDOS of PFC-71 and PFC73-Cu dimer. Adapted with permission from ref. 125. Copyright 2021, Wiley-VCH. | ||
Drawing on the fact that in homogeneous porphyrin systems, the metalloporphyrin center is usually considered to be one of the most important determinants of the robustness and activity of the materials, changing the metal center leads to drastic changes in the electronic structure of the macrocycle, axial bonding/interactions, and peripheral substitution geometries.124 Based on this, Liu et al. obtained metalloporphyrin HOFs with different non-covalent interactions, orbital overlap, and molecular geometries but the same topology by introducing different transition metals into the porphyrin center.125 The results in Fig. 7c–e demonstrate that the metalated porphyrins dramatically altered the electrostatic potential, density of states, and thus the non-covalent interactions between the interlayer porphyrin fragments. Subsequently, effective catalysis of the photoreduction of CO2 to CO was achieved. The photocatalytic performance of CO2 was heavily dependent on the type of chelating metal at the porphyrin center, which provided an effective guide for the design of porphyrin HOFs in the field of photocatalytic CO2 reduction.
3.4 Hydrogen peroxide production
Hydrogen peroxide (H2O2) is an environmentally friendly oxidizing agent with a wide range of applications in bleaching, disinfection, semiconductor manufacturing and chemical synthesis.126 At the same time, H2O2 is a potential energy carrier that can be used directly in fuel cells to generate electricity. In addition, the energy density of aqueous H2O2 is comparable to that of compressed H2, and H2O2 is more convenient than H2 in terms of storage, transport, and portability, making it a promising alternative to H2 as a clean energy source. For all the above reasons, the global H2O2 market is expected to grow to 5.7 million tonnes by 2027. Currently, more than 95% of industrial H2O2 is produced by anthraquinone oxidation, an energy-intensive, multi-step method to generate a large amount of waste.127 Therefore, the direct production of H2O2 through a photocatalytic reaction using H2O and O2 is a potentially viable and green sustainable approach.128,129The partial replacement of metal–oxygen clusters by other metal ions or the incorporation of new metal ions can lead to new physicochemical properties of MOFs.130 In particular, hydrophobicity, superior charge separation efficiency and longer lifetime of the photoexcited state can be effectively achieved in MOFs. As an example, the hydrophobic MOF MIL-125-Rn (n = 4 and 7) was firstly synthesized by Yamashita's group,131 but the drastically reduced specific surface area was not conducive to further improvement of the H2O2 production. On this basis, the authors alkylated the Ti cluster of MIL-125-NH2 by using octadecyl phosphonic acid (OPA).132 Unlike before, the as-prepared hydrophobic OPA/MIL-125-NH2 maintained the original large surface area due to the alkylation reaction only occurring on the outermost surface of the pristine MOFs with open coordination sites. The yield of H2O2 was again improved 3 times compared to MIL-125-Rn. The increased photocatalytic activity was attributed to the faster diffusion of reactants and products as well as the slower decomposition of H2O2 in OPA/MIL-125-NH2. Wang et al. loaded Pd single atoms onto defect-rich UiO-66-NH2 and subsequently processed gas permeable membranes (Pd/UiO-66-NH2).133 Photocatalytic generation of H2O2 at the gas–solid interface was achieved using humid O2 (Fig. 8a). The defect sites favored light absorption and interaction with metal single atoms, while the positively charged Pd single atoms both enhanced H2O2 generation and inhibited H2O2 decomposition. The H2O2 generation rate on the Pd/UiO-66-NH2 membrane was 10.4 mmol g−1 h−1 in the gas–membrane–gas mode, which was 4.9 times higher than that on the Pd/UiO-66-NH2 particles. Furthermore, the bimetallic PMOF-RuFe(OH) was prepared by immobilizing mono-iron hydroxyl sites in MOFs by Schröder et al.134 The photocatalytic processes were triggered by photogenerated electron transfer from the excited state PS* of [Ru(bpy)2(bpydc)]* to generate the electron-rich PW9V3IV/V. Then it drove the photoreduction of O2 to H2O2via proton-coupled (from H2O) electron transfer (from the reduced PW9VIV/V3). Intriguingly, the bimetallic catalyst PMOF-RuFe(OH) can rapidly convert the in situ formed H2O2 to ·OH radicals facilitated by the {Fe–OH} fraction and subsequently achieved the conversion of CH4 to CH3OH with 100% selectivity in the presence of H2O and O2. The restricted mono-iron hydroxyl sites in MOFs were converted to CH3OH by the formation of the [Fe–OH⋯CH4] intermediate to CH4, thus lowering the barrier to C–H bond activation. The PMOF-RuFe(OH) framework provided a stable platform for such a H2O2-coupled reaction, which could achieve higher value in energy conversion.
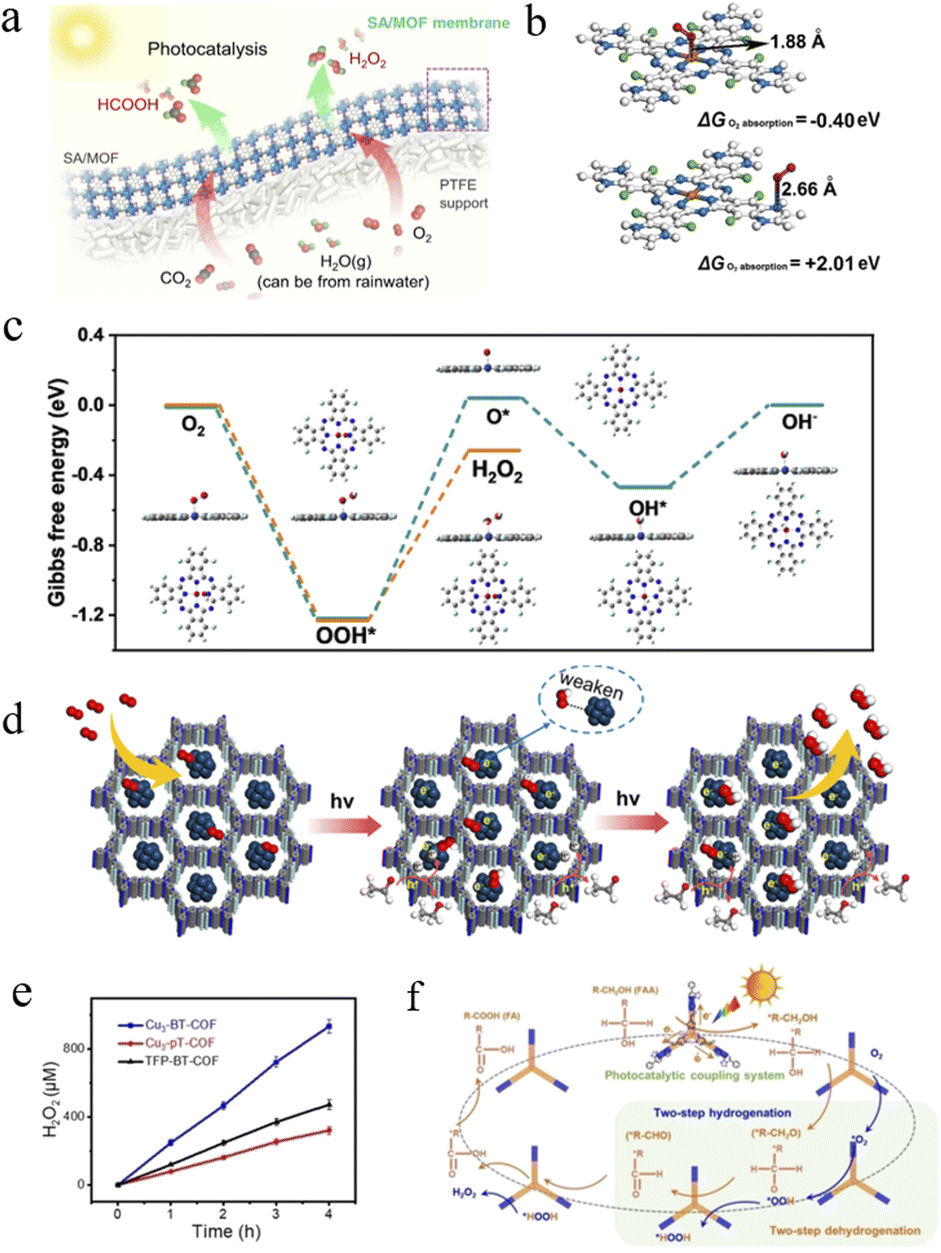 | ||
| Fig. 8 (a) Schematic illustration of the SA/MOF membranes. Adapted with permission from ref. 133. Copyright 2021, Springer Nature. (b) Calculated oxygen adsorption energies on the Co atom and N atom of CoPc-BTM-COF. (c) Free energy diagrams for 2e− (orange line) and 4e− (cyan line) ORR processes on CoPc with optimized configurations for each step. Adapted with permission from ref. 135. Copyright 2022, American Chemical Society. (d) Schematic diagram of the photocatalytic H2O2 mechanism on TAPT-TFPA@Pd ICs. Adapted with permission from ref. 136. Copyright 2023, American Chemical Society. (e) Photocatalytic activity of Cu3-BT-COF, Cu3-pT-COF and TFP-BT-COF for H2O2 photosynthesis. (f) Schematic diagram of the proposed reaction mechanism for H2O2 photosynthesis. Adapted with permission from ref. 137. Copyright 2023, Wiley-VCH. | ||
For COFs, achieving a highly active and selective 2e− oxygen reduction reaction remains the key to efficient H2O2 production. Jinag et al. obtained two piperazine-conjugated COFs by nucleophilic substitution reactions with 1,2,4,5-benzenetetramine (BTM) or 3,3′-diaminobenzidine (DAB), namely, CoPc-BTM-COF and CoPcDAB-COF.135 Both COFs exhibited excellent light absorption capacity in the visible range and enhanced photo-induced charge separation and migration efficiencies, resulting in an H2O2 yield of 2096 μmol g−1 h−1 and an apparent quantum yield of 7.2% at 630 nm. As shown in Fig. 8b, the oxygen adsorption energy on the Co atom of CoPc-BTM-COF was −0.40 eV with a 1.88 Å Co⋯O, which was much smaller than that of the N site (+2.01 eV, N⋯O distance of 2.66 Å), suggesting that the Co atom in CoPc-BTM-COF has the active site nature of the ORR. In addition, DFT demonstrated the good selectivity of CoPc-COFs for H2O2 production by the 2e− ORR, as the activation barrier of CoPc for the 2e− ORR was 0.97 eV compared to 1.27 eV for the 4e− ORR, resulting in faster kinetics of the reaction of the 2e− ORR to CoPc-COFs (Fig. 8c).
Palladium metal-isolated clusters act as an intermediate between nanomaterials and single atoms, which exhibit excellent activity and selectivity in the photocatalysis of H2O2 production due to their highly overlapping electronic orbitals. It is an effective way to optimize their poor stability by confining them in COFs. It can be clearly seen that, as shown in Fig. 8d, Guo et al. strongly confined Pd clusters in fluorinated COFs by a strong metal–carrier interaction (MSI),136 significantly improving the stability (over 100 h) and activity (2143 μmol g−1 h−1) of H2O2 production, which can be attributed to the fact that the strong electronegativity of fluorine in COFs can not only enhance MSI, but also optimize the d-band center of Pd.
The combination of H2O2 photosynthesis and bio-value addition not only maximizes energy efficiency but also enables the production of value-added products. To this end, Lan et al. prepared a series of redox molecular COFs with Cu-containing units (Cu3-BT-COF, Cu3-pT-COF, and TFP-BT-COF) and investigated the coupling of H2O2 photosynthesis to the preparation of furoic acid (FA) from the photo-oxidation of furfuryl alcohol (FFA).137 Cu3-BT-COF, constructed by the covalent linkage of Cu3 with the thiazolyl group (BT), was a newly designed COF with nanosheet morphology (∼1.25 nm), which exhibited the best performance for the photocatalytic coupling reaction. Among them, the FA generation efficiency was up to 575 mM g−1 (∼100% conversion and >99% selectivity), while the H2O2 generation rate was up to 187000 μM g−1, which was much higher than that of Cu3-pT-COF, TFP-BT-COF, and their monomers, as shown in Fig. 8e. And as seen in Fig. 8f, the covalent linkage between Cu3 and BT in the Cu3-BT-COF enabled the visible-light-driven electrons to be efficiently transferred from Cu3 to the BT portion, resulting in the generation of photoexcited electrons (on BT) and holes (on Cu3) that can be used for H2O2 production and FFA photo-oxidation reactions, respectively. Significantly, the system was highly versatile and can be extended to other alcohol photo-oxidation systems, such as H2O2 photosynthesis coupled with 2-thiophene methanol photooxidation. This opens the way for work on COFs for H2O2 photosynthesis with biomass value addition.
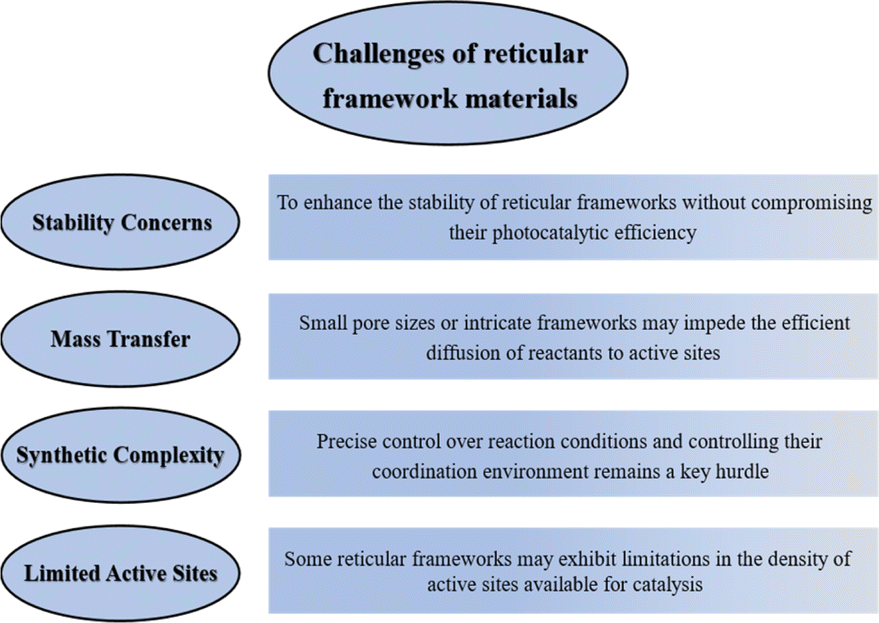
4. Outlooks and challenges
In conclusion, the integration of reticular framework materials- MOFs, COFs, and HOFs-into photocatalysis has sparked unprecedented interest, offering immense potential for sustainable and clean energy conversion. The unique characteristics (Table 1) of reticular frameworks promise enhanced photocatalyst efficiency, addressing existing challenges and propelling the field forward. These materials provide a versatile platform for designing catalysts with improved selectivity and activity. Looking ahead, the outlook for reticular frameworks containing metal sites in energy-related photocatalysis is promising. Ongoing research aims to optimize the design, structure, and composition for enhanced efficiency and selectivity. The customization of metal sites, pore structures, and surface properties holds potential for materials tailored to specific catalytic processes. Emphasizing sustainability, these materials can contribute to eco-friendly energy conversion processes and promote a greener future. The development of multifunctional reticular frameworks for integrated and efficient energy conversion systems is anticipated. Collaboration across disciplines is crucial for innovative breakthroughs and potential commercialization. Reticular framework materials containing metal sites stand poised to revolutionize energy-related applications through efficient and sustainable photocatalysis processes, marking a significant stride toward a cleaner and greener future. However, as illustrated in Fig. 9 and Table 1, the field is not without its current limitations and challenges that warrant attention and further research.| Reticular framework materials | |
|---|---|
| Advantages | Disadvantages |
| Large specific surface area | Synthetic complexity |
| Adjustable hole size | Relatively expensive |
| Easy to functionalize | The structure of multi-component frames is difficult to confirm |
| Structural design in advance | Sacrificial agents are usually required in photocatalytic reactions |
 | ||
| Fig. 9 Current limitations and challenges of reticular framework materials. | ||
1. Stability concerns: many reticular frameworks, particularly certain MOFs, may face challenges related to stability under the rigorous conditions of photocatalysis. Exposure to high temperatures, corrosive environments, or extended reaction times can lead to framework degradation, affecting both catalytic activity and long-term performance. Developing strategies to enhance the stability of reticular frameworks without compromising their photocatalytic efficiency remains a critical challenge. The exploration of novel materials or modification techniques to mitigate stability issues is an area requiring dedicated investigation, such as, the structure of MOFs should be carefully designed to avoid easily destructive groups so that the frameworks are difficult to destroy under extreme conditions.
2. Mass transfer limitations: the inherently porous nature of reticular frameworks, while advantageous for providing a high surface area, can pose challenges related to mass transfer limitations. In some instances, small pore sizes or intricate frameworks may impede the efficient diffusion of reactants to active sites, influencing overall reaction rates. Addressing mass transfer limitations necessitates innovative designs or post-synthetic modifications that maintain the structural integrity of the framework while optimizing pore size and accessibility. Developing frameworks with larger pores or hierarchically structured materials, or adding metal sites or functional groups with greater adsorption capacity for transition states to enhance mass transfer accessibility, may be potential solutions.
3. Synthetic complexity: the synthesis of certain reticular frameworks, especially COFs and HOFs, can be intricate and time-consuming. Precise control over reaction conditions, extended reaction times, and the need for specialized equipment contribute to the challenges associated with large-scale production. Furthermore, precise control over the synthesis of these frameworks with specific metal sites is essential but can be challenging. Achieving uniform distribution of metal sites and controlling their coordination environment remains a key hurdle. Modulation of the microenvironment of the metal sites through the use of different ligands, followed by more efficient photogenerated charge segregation and migration is worthy of further investigation.
4. Limited active sites: some reticular frameworks may exhibit limitations in the density of active sites available for catalysis. This can impact overall photocatalytic efficiency and may require optimization for specific reactions, which is a benefit for achieving more efficient photocatalytic systems and high selectivity for desired products.
In conclusion, the potential of reticular framework materials in advancing photocatalysis is considerable. However, addressing the current limitations and challenges is imperative for the progression of reticular framework materials containing metal sites in energy-related photocatalysis. Continuous research and innovation are essential to surmount these obstacles and fully unleash the capabilities of these materials in sustainable energy technologies.
Author contributions
The conceptualization was carried out by S. W., W. G., J. L., and H. Z.; the formal analysis was conducted by C. L., B. W, S. L., N. H., and H. Z.; the original draft preparation was done by S. W., N. H. and H. Z.; the review and editing were carried out by W. G., S. L., J. L., and H. Z.; the supervision was done by W. G., J. L., and H. Z.; the funding was provided by H. Z.; all authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the Youth Foundation of the National Natural Science Foundation of China (No. 52203300), Zhejiang Provincial Key Laboratory (No. 2020E10018), 2023 Ningbo Natural Science Funding Commonweal Programme Major Project (No. 2023S019), and the Zhejiang Provincial Natural Science Funding Major Project (No. LDT23E06011E06).Notes and references
- Y. Zhang, H. Liu, F. Gao, X. Tan, Y. Cai, B. Hu, Q. Huang, M. Fang and X. Wang, EnergyChem, 2022, 4, 100078 CrossRef CAS.
- S. Liu, M. Wang, Y. He, Q. Cheng, T. Qian and C. Yan, Coord. Chem. Rev., 2023, 475, 214882 CrossRef CAS.
- J. Di, J. Xia, H. Li, S. Guo and S. Dai, Nano Energy, 2017, 41, 172–192 CrossRef CAS.
- N. Han, Y. Wang and B. L. Su, Natl. Sci. Rev., 2024, nwae068 CrossRef PubMed.
- J. Ran, M. Jaroniec and S. Z. Qiao, Adv. Mater., 2018, 30, 1704649 CrossRef PubMed.
- T. He and Y. Zhao, Angew. Chem., Int. Ed., 2023, 62, e202303086 CrossRef CAS PubMed.
- H. Huang, J. Zhao, B. Weng, F. Lai, M. Zhang, J. Hofkens, M. Roeffaers, J. Steele and J. Long, Angew. Chem., Int. Ed., 2022, 61, e202204563 CrossRef CAS PubMed.
- J. Li, X. Lv, B. Weng, M. Roeffaers and H. Jia, Chem. Eng. J., 2023, 461, 142022 CrossRef CAS.
- C. Wang, H. Zhang, F. Lai, Z. Xie, Y. Ng, B. Weng, X. Wu and Y. Liao, J. Energy Chem., 2023, 83, 341–362 CrossRef CAS.
- Y. Z. Chen, R. Zhang, L. Jiao and H. L. Jiang, Coord. Chem. Rev., 2018, 362, 1–23 CrossRef CAS.
- N. Han, W. Zhang, W. Guo, H. Pan, B. Jiang, L. Xing, H. Tian, G. Wang, X. Zhang and J. Fransaer, Nano-Micro Lett., 2023, 15, 185 CrossRef CAS PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. B. Chorkendorff, J. K. Norskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- X. Meng, L. Liu, S. Ouyang, H. Xu, D. Wang, N. Zhao and J. Ye, Adv. Mater., 2016, 28, 6781–6803 CrossRef CAS.
- H. Dong, M. Xiao, S. Yu, H. Wu, Y. Wang, J. Sung, G. Chen and C. Li, ACS Catal., 2020, 10, 458–462 CrossRef CAS.
- S. Gong, M. Hou, Y. Niu, X. Teng, X. Liu, M. Xu, C. Xu, V. K.-M. Au and Z. Chen, Chem. Eng. J., 2022, 427, 131717 CrossRef CAS.
- C. Costentin, S. Drouet, M. Robert and J.-M. Saveant, Science, 2012, 338, 90–94 CrossRef CAS PubMed.
- A. J. Morris, G. J. Meyer and E. Fujita, Acc. Chem. Res., 2009, 42, 1983–1994 CrossRef CAS.
- C. Costentin, G. Passard, M. Robert and J.-M. Saveant, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14990–14994 CrossRef CAS.
- D. W. Sun, L. Huang, H. Pu and J. Ma, Chem. Soc. Rev., 2021, 50, 1070–1110 RSC.
- R. Freund, S. Canossa, S. M. Cohen, W. Yan, H. Deng, V. Guillerm, M. Eddaoudi, D. G. Madden, D. Fairen-Jimenez, H. Lyu, L. K. Macreadie, Z. Ji, Y. Zhang, B. Wang, F. Haase, C. Woell, O. Zaremba, J. Andreo, S. Wuttke and C. S. Diercks, Angew. Chem., Int. Ed., 2021, 60, 23946–23974 CrossRef CAS PubMed.
- X. Gong, Y. Shu, Z. Jiang, L. Lu, X. Xu, C. Wang and H. Deng, Angew. Chem., Int. Ed., 2020, 59, 5326–5331 CrossRef CAS PubMed.
- S. Wu, C. Li, Y. Wang, Y. Zhuang, Y. Pan, N. Wen, S. Wang, Z. Zhang, Z. Ding, R. Yuan, W. Dai, X. Fu and J. Long, Angew. Chem., Int. Ed., 2023, 62, e202309026 CrossRef CAS.
- P. Tholen, C. A. Peeples, M. M. Ayhan, L. Wagner, H. Thomas, P. Imbrasas, Y. Zorlu, C. Baretzky, S. Reineke, G. Hanna and G. Yuecesan, Small, 2022, 18, 2204578 CrossRef CAS.
- Z. Chen, P. Li, R. Anderson, X. Wang, X. Zhang, L. Robison, L. R. Redfern, S. Moribe, T. Islamoglu, D. A. Gomez-Gualdron, T. Yildirim, J. F. Stoddart and O. K. Farha, Science, 2020, 368, 297 CrossRef CAS.
- D. E. Jaramillo, H. Z. H. Jiang, H. A. Evans, R. Chakraborty, H. Furukawa, C. M. Brown, M. Head-Gordon and J. R. Long, J. Am. Chem. Soc., 2021, 143, 6248–6256 CrossRef CAS PubMed.
- W. K. Han, Y. Liu, X. Yan, Y. Jiang, J. Zhang and Z. G. Gu, Angew. Chem., Int. Ed., 2022, 61, e202208791 CrossRef CAS PubMed.
- H. Zhang, W. Tian, X. Duan, H. Sun, S. Liu and S. Wang, Adv. Mater., 2020, 32, 1904037 CrossRef CAS.
- N. Han, X. Zhang, C. Zhang, S. Feng, W. Zhang, W. Guo, R. Zheng, R. Zheng, P. Liu, Y. Li, J. Fransaer and B. Su, Matter, 2024, 7, 1–14 CrossRef.
- W. Guo, W. Monnens, W. Zhang, S. Xie, N. Han, Z. Zhou, N. Chanut, K. Vanstreels, R. Ameloot, X. Zhang and J. Fransaer, Microporous Mesoporous Mater., 2023, 350, 112443 CrossRef CAS.
- T. Luo, L. Gilmanova and S. Kaskel, Coord. Chem. Rev., 2023, 490, 215210 CrossRef CAS.
- W. Guo, W. Zhang, N. Han, S. Xie, Z. Zhou, W. Monnens, O. M. Mora, Z. Xue, X. Zhang, X. Zhang and J. Fransaer, Chem.–Eur. J., 2023, 8, e202302338 CrossRef PubMed.
- C. Zhang, Y. Yan, H. Huang, X. Peng, H. Song, J. Ye and L. Shi, ACS Catal., 2023, 13, 15351–15359 CrossRef CAS.
- W. Wang, C. Deng, S. Xie, Y. Li, W. Zhang, H. Sheng, C. Chen and J. Zhao, J. Am. Chem. Soc., 2021, 143, 2984–2993 CrossRef CAS.
- Z. Jiang, X. Xu, Y. Ma, H. S. Cho, D. Ding, C. Wang, J. Wu, P. Oleynikov, M. Jia, J. Cheng, Y. Zhou, O. Terasaki, T. Peng, L. Zan and H. Deng, Nature, 2021, 590, E16 CrossRef CAS PubMed.
- Z. Meng, J. Luo, W. Li and K. A. Mirica, J. Am. Chem. Soc., 2020, 142, 21656–21669 CrossRef CAS PubMed.
- M. Yoshizawa, M. Tamura and M. Fujita, Science, 2006, 312, 251–254 CrossRef CAS PubMed.
- T. Murase, S. Horiuchi and M. Fujita, J. Am. Chem. Soc., 2010, 132, 2866 CrossRef CAS PubMed.
- S. Qiu and G. Zhu, Coord. Chem. Rev., 2009, 253, 2891–2911 CrossRef CAS.
- J. Li, X. Jing, Q. Li, S. Li, X. Gao, X. Feng and B. Wang, Chem. Soc. Rev., 2020, 49, 3565–3604 RSC.
- Z. Li, T. Deng, S. Ma, Z. Zhang, G. Wu, J. Wang, Q. Li, H. Xia, S. W. Yang and X. Liu, J. Am. Chem. Soc., 2023, 145, 8364–8374 CAS.
- P. Bhanja, S. Mishra, K. Manna, A. Mallick, K. D. Saha and A. Bhaumik, ACS Appl. Mater. Interfaces, 2017, 9, 31411–31423 CrossRef CAS PubMed.
- D. K. Singha, R. I. Mohanty, P. Bhanja and B. K. Jena, Mater. Adv., 2023, 4, 4679–4706 RSC.
- Y. Wang, A. Vogel, M. Sachs, R. S. Sprick, L. Wilbraham, S. J. A. Moniz, R. Godin, M. A. Zwijnenburg, J. R. Durrant, A. I. Cooper and J. Tang, Nat. Energy, 2019, 4, 746–760 CrossRef CAS.
- R. I. Mohanty, L. Pradhan, S. Chongdar, S. Basu, P. Bhanja and B. K. Jena, Catal. Today, 2023, 424, 113789 CrossRef CAS.
- P. Bhanja, K. Bhunia, S. K. Das, D. Pradhan, R. Kimura, Y. Hijikata, S. Irle and A. Bhaumik, ChemSusChem, 2017, 10, 921–929 CrossRef CAS PubMed.
- S. T. Emmerling, R. Schuldt, S. Bette, L. Yao, R. E. Dinnebier, J. Kaestner and B. V. Lotsch, J. Am. Chem. Soc., 2021, 143, 15711–15722 CrossRef CAS PubMed.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef PubMed.
- C. Li, L. Liu, J. Kang, Y. Xiao, Y. Feng, F.-F. Cao and H. Zhang, Energy Storage Mater., 2020, 31, 115–134 CrossRef.
- S. Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- J. Tang, C. Su and Z. Shao, Small Methods, 2021, 5, 2100945 CrossRef CAS PubMed.
- S. Huang, G. Chen and G. Ouyang, Chem. Soc. Rev., 2022, 51, 6824–6863 RSC.
- Z. Fan, Y. Zou, C. Liu, S. Xiang and Z. Zhang, Chem.–Eur. J., 2022, 28, e202200422 CrossRef CAS PubMed.
- X. Song, Y. Wang, C. Wang, D. Wang, G. Zhuang, K. O. Kirlikovali, P. Li and O. K. Farha, J. Am. Chem. Soc., 2022, 144, 10663–10687 CrossRef CAS.
- W. K. Qin, C. H. Tung and L. Z. Wu, J. Mater. Chem. A, 2023, 11, 12521–12538 RSC.
- L. Barreto, A. Makihira and K. Riahi, Int. J. Hydrogen Energy, 2003, 28, 267–284 CrossRef CAS.
- G. Zhang, Z. A. Lan, L. Lin, S. Lin and X. Wang, Chem. Science, 2016, 7, 3062–3066 RSC.
- S. L. Li and Q. Xu, Energy Environ. Sci., 2013, 6, 1656–1683 RSC.
- S. Huang, G. Chen and G. Ouyang, Chem. Soc. Rev., 2022, 51, 6824–6863 RSC.
- R.-B. Lin, Y. He, P. Li, H. Wang, W. Zhou and B. Chen, Chem. Soc. Rev., 2019, 48, 1362–1389 RSC.
- A. Wu, Y. Xie, H. Ma, C. Tian, Y. Gu, H. Yan, X. Zhang, G. Yang and H. Fu, Nano Energy, 2018, 44, 353–363 CrossRef CAS.
- Z. Yang, C. Zhang, G. Zeng, X. Tan, D. Huang, J. Zhou, Q. Fang, K. Yang, H. Wang, J. Wei and K. Nie, Coord. Chem. Rev., 2021, 446, 214103 CrossRef CAS.
- K. Meyer, M. Ranocchiari and J. A. van Bokhoven, Energy Environ. Sci., 2015, 8, 1923–1937 RSC.
- Y. Liu, D. Huang, M. Cheng, Z. Liu, C. Lai, C. Zhang, C. Zhou, W. Xiong, L. Qin, B. Shao and Q. Liang, Coord. Chem. Rev., 2020, 409, 213220 CrossRef CAS.
- H. Luo, Z. Zeng, G. Zeng, C. Zhang, R. Xiao, D. Huang, C. Lai, M. Cheng, W. Wang, W. Xiong, Y. Yang, L. Qin, C. Zhou, H. Wang, Y. Zhou and S. Tian, Chem. Eng. J., 2020, 383, 123196 CrossRef CAS.
- F. Leng, H. Liu, M. Ding, Q.-P. Lin and H.-L. Jiang, ACS Catal., 2018, 8, 4583–4590 CrossRef CAS.
- T. Song, P. Zhang, J. Zeng, T. Wang, A. Ali and H. Zeng, Int. J. Hydrogen Energy, 2017, 42, 26605–26616 CrossRef CAS.
- D. Sun, W. Liu, M. Qiu, Y. Zhang and Z. Li, Chem. Commun., 2015, 51, 2056–2059 RSC.
- L. Liu, S. Du, X. Guo, Y. Xiao, Z. Yin, N. Yang, Y. Bao, X. Zhu, S. Jin, Z. Feng and F. Zhang, J. Am. Chem. Soc., 2022, 144, 2747–2754 CrossRef CAS PubMed.
- A. Aziz, A. R. Ruiz-Salvador, N. C. Hernández, S. Calero, S. Hamad and R. Grau-Crespo, J. Mater. Chem. A, 2017, 5, 11894–11904 RSC.
- C. Wang, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2012, 134, 7211–7214 CrossRef CAS PubMed.
- Q. Mo, L. Zhang, S. Li, H. Song, Y. Fan and C.-Y. Su, J. Am. Chem. Soc., 2022, 144, 22747–22758 CrossRef CAS PubMed.
- J. D. Xiao, Q. Shang, Y. Xiong, Q. Zhang, Y. Luo, S. H. Yu and H. L. Jiang, Angew. Chem., Int. Ed., 2016, 55, 9389–9393 CrossRef CAS PubMed.
- M. Wang, Y. Xu, C.-K. Peng, S. Y. Chen, Y. G. Lin, Z. Hu, L. Sun, S. Ding, C. W. Pao, Q. Shao and X. Huang, J. Am. Chem. Soc., 2021, 143, 16512–16518 CrossRef CAS PubMed.
- R. Chen, Y. Wang, Y. Ma, A. Mal, X. Y. Gao, L. Gao, L. Qiao, X.-B. Li, L. Z. Wu and C. Wang, Nat. Commun., 2021, 12, 1354 CrossRef CAS PubMed.
- H. Zhang, Z. Lin, P. Kidkhunthod and J. Guo, Angew. Chem., Int. Ed., 2023, 62, e202217527 CrossRef CAS PubMed.
- W. K. Han, Y. Liu, X. Yan, Y. Jiang, J. Zhang and Z. G. Gu, Angew. Chem., Int. Ed., 2022, 61, e202208791 CrossRef CAS.
- M. Lu, S. B. Zhang, M. Y. Yang, Y. F. Liu, J. P. Liao, P. Huang, M. Zhang, S. L. Li, Z. M. Su and Y. Q. Lan, Angew. Chem., Int. Ed., 2023, 62, e202307632 CrossRef PubMed.
- W. Zhou, Q. W. Deng, H. J. He, L. Yang, T. Y. Liu, X. Wang, D. Y. Zheng, Z. B. Dai, L. Sun, C. Liu, H. Wu, Z. Li and W. Q. Deng, Angew. Chem., Int. Ed., 2022, 62, e202214143 CrossRef PubMed.
- Y. Zang, R. Wang, P. P. Shao, X. Feng, S. Wang, S. Q. Zang and T. C. W. Mak, J. Mater. Chem. A, 2020, 8, 25094–25100 RSC.
- P. Dong, Y. Wang, A. Zhang, T. Cheng, X. Xi and J. Zhang, ACS Catal., 2021, 11, 13266–13279 CrossRef CAS.
- J. Ming, A. Liu, J. Zhao, P. Zhang, H. Huang, H. Lin, Z. Xu, X. Zhang, X. Wang, J. Hofkens, M. B. J. Roeffaers and J. Long, Angew. Chem., Int. Ed., 2019, 58, 18290–18294 CrossRef CAS PubMed.
- N. Zhang, Q. Yin, S. Guo, K. K. Chen, T. F. Liu, P. Wang, Z. M. Zhang and T. B. Lu, Appl. Catal., B, 2021, 296, 120337 CrossRef CAS.
- G. Wang, Q. Sun, Y. Liu, B. Huang, Y. Dai, X. Zhang and X. Qin, Chem.–Eur. J., 2014, 21, 2364–2367 CrossRef PubMed.
- L. Li, Z. B. Fang, W. Deng, J. D. Yi, R. Wang and T. F. Liu, CCS Chem., 2022, 4, 2782–2792 CrossRef CAS.
- J. Chen, X. Tao, C. Li, Y. Ma, L. Tao, D. Zheng, J. Zhu, H. Li, R. Li and Q. Yang, Appl. Catal., B, 2020, 262, 118271 CrossRef CAS.
- E. Zhou, X. Zhang, L. Zhu, E. Chai, J. Chen, J. Li, D. Yuan, L. Kang, Q. Sun and Y. Wang, Sci. Adv., 2024, 10, eadk8564 CrossRef CAS PubMed.
- Y. Yang, X. Chu, H. Y. Zhang, R. Zhang, Y. H. Liu, F. M. Zhang, M. Lu, Z.-D. Yang and Y. Q. Lan, Nat. Commun., 2023, 14, 593 CrossRef CAS PubMed.
- Y. Wan, L. Wang, H. Xu, X. Wu and J. Yang, J. Am. Chem. Soc., 2020, 142, 4508–4516 CrossRef CAS PubMed.
- T. Devic and C. Serre, Chem. Soc. Rev., 2014, 43, 6097–6115 RSC.
- A. de Oliveira, G. F. de Lima and H. A. De Abreu, Chem. Phys. Lett., 2018, 691, 283–290 CrossRef CAS.
- Z. Zhan, X. Liang, X. Zhang, Y. Jia and M. Hu, Dalton, 2019, 48, 1786–1794 RSC.
- A. Torrisi, R. G. Bell and C. Mellot-Draznieks, Cryst. Growth Des., 2010, 10, 2839–2841 CrossRef CAS.
- L. Z. Dong, L. Zhang, J. Liu, Q. Huang, M. Lu, W. X. Ji and Y. Q. Lan, Angew. Chem., Int. Ed., 2020, 59, 2659–2663 CrossRef CAS PubMed.
- Q. Huang, Q. Niu, X. F. Li, J. Liu, S. N. Sun, L. Z. Dong, S. L. Li, Y.-P. Cai and Y. Q. Lan, Sci. Adv., 2022, 8, eadd559 Search PubMed.
- M. Dan-Hardi, C. Serre, T. Frot, L. Rozes, G. Maurin, C. Sanchez and G. Ferey, J. Am. Chem. Soc., 2009, 131, 10857 CrossRef CAS PubMed.
- Y. Fu, D. Sun, Y. Chen, R. Huang, Z. Ding, X. Fu and Z. Li, Angew. Chem., Int. Ed., 2012, 51, 3364–3367 CrossRef CAS.
- D. Gao, R. M. Aran-Ais, H. S. Jeon and B. Roldan Cuenya, Nat. Catal., 2019, 2, 198–210 CrossRef CAS.
- S. Xie, C. Deng, Q. Huang, C. Zhang, C. Chen, J. Zhao and H. Sheng, Angew. Chem., Int. Ed., 2023, 62, e20221671 Search PubMed.
- J. Y. Zeng, X. S. Wang, B. R. Xie, Q. R. Li and X. Z. Zhang, J. Am. Chem. Soc., 2022, 144, 1218–1231 CrossRef CAS PubMed.
- J. W. Wang, D. C. Zhong and T. B. Lu, Coord. Chem. Rev., 2018, 377, 225–236 CrossRef CAS.
- J. Li, H. Huang, W. Xue, K. Sun, X. Song, C. Wu, L. Nie, Y. Li, C. Liu, Y. Pan, H. L. Jiang, D. Mei and C. Zhong, Nat. Catal., 2021, 4, 719–729 CrossRef CAS.
- W. Liu, X. Li, C. Wang, H. Pan, W. Liu, K. Wang, Q. Zeng, R. Wang and J. Jiang, J. Am. Chem. Soc., 2019, 141, 17431–17440 CrossRef CAS PubMed.
- Y. N. Gong, W. Zhong, Y. Li, Y. Qiu, L. Zheng, J. Jiang and H. L. Jiang, J. Am. Chem. Soc., 2020, 142, 16723–16731 CrossRef CAS PubMed.
- M. Lu, J. Liu, Q. Li, M. Zhang, M. Liu, J. L. Wang, D. Q. Yuan and Y. Q. Lan, Angew. Chem., Int. Ed., 2019, 58, 12392–12397 CrossRef CAS PubMed.
- J. Ding, X. Guan, J. Lv, X. Chen, Y. Zhang, H. Li, D. Zhang, S. Qiu, H. L. Jiang and Q. Fang, J. Am. Chem. Soc., 2023, 145, 3248–3254 CrossRef CAS PubMed.
- W. Liu, X. Li, C. Wang, H. Pan, W. Liu, K. Wang, Q. Zeng, R. Wang and J. Jiang, J. Am. Chem. Soc., 2019, 141, 17431–17440 CrossRef CAS PubMed.
- X. Wang, X. Ding, T. Wang, K. Wang, Y. Jin, Y. Han, P. Zhang, N. Li, H. Wang and J. Jiang, ACS Appl. Mater. Interfaces, 2022, 14, 41122–41130 CrossRef CAS.
- Y. Kuramochi, O. Ishitani and H. Ishida, Coord. Chem. Rev., 2018, 373, 333–356 CrossRef CAS.
- S. Yang, W. Hu, X. Zhang, P. He, B. Pattengale, C. Liu, M. Cendejas, I. Hermans, X. Zhang, J. Zhang and J. Huang, J. Am. Chem. Soc., 2018, 140, 14614–14618 CrossRef CAS PubMed.
- Q. Pan, M. Abdellah, Y. Cao, W. Lin, Y. Liu, J. Meng, Q. Zhou, Q. Zhao, X. Yan, Z. Li, H. Cui, H. Cao, W. Fang, D. A. Tanner, M. Abdel-Hafiez, Y. Zhou, T. Pullerits, S. E. Canton, H. Xu and K. Zheng, Nat. Commun., 2022, 13, 845 CrossRef CAS PubMed.
- Y. Z. Cheng, W. Ji, P. Y. Hao, X. H. Qi, X. Wu, X. M. Dou, X. Y. Bian, D. Jiang, F. T. Li, X. F. Liu, D. H. Yang, X. Ding and B. H. Han, Angew. Chem., Int. Ed., 2023, 62, e202308523 CrossRef CAS PubMed.
- Y. Zhang, L. Cao, G. Bai and X. Lan, Small, 2023, 19, 2300035 CrossRef CAS PubMed.
- L. Ran, Z. Li, B. Ran, J. Cao, Y. Zhao, T. Shao, Y. Song, M. K. H. Leung, L. Sun and J. Hou, J. Am. Chem. Soc., 2022, 144, 17097–17109 CrossRef CAS PubMed.
- Q. Zhang, S. Gao, Y. Guo, H. Wang, J. Wei, X. Su, H. Zhang, Z. Liu and J. Wang, Nat. Commun., 2023, 14, 1147 CrossRef CAS PubMed.
- Y. J. Li, W. R. Cui, Q. Q. Jiang, Q. Wu, R. P. Liang, Q. X. Luo and J. D. Qiu, Nat. Commun., 2021, 12, 1–11 CrossRef PubMed.
- M. Lu, M. Zhang, J. Liu, T. Y. Yu, J. N. Chang, L. J. Shang, S. L. Li and Y. Q. Lan, J. Am. Chem. Soc., 2022, 144, 1861–1871 CrossRef CAS PubMed.
- Y. Kurarnochi, Y. Fujisawa and A. Satake, J. Am. Chem. Soc., 2020, 142, 705–709 CrossRef PubMed.
- D. Song, W. Xu, J. Li, J. Zhao, Q. Shi, F. Li, X. Sun and N. Wang, Chin. J. Catal., 2022, 43, 2425–2433 CrossRef CAS.
- L. J. Gong, L. Y. Liu, S. S. Zhao, S. L. Yang, D.-H. Si, Q. J. Wu, Q. Wu, Y. B. Huang and R. Cao, Chem. Eng. J., 2023, 458, 141360 CrossRef CAS.
- L. Gong, D. Zhang, Y. Shen, X. Wang, J. Zhang, X. Han, L. Zhang and Z. Xia, J. Catal., 2020, 390, 126–134 CrossRef CAS.
- Y. Zhou, L. Chen, L. Sheng, Q. Luo, W. Zhang and J. Yang, Nano Res., 2022, 15, 7994–8000 CrossRef CAS.
- B. Yu, L. Li, S. Liu, H. Wang, H. Liu, C. Lin, C. Liu, H. Wu, W. Zhou, X. Li, T. Wang, B. Chen and J. Jiang, Angew. Chem., Int. Ed., 2021, 60, 8983–8989 CrossRef CAS PubMed.
- A. A. Zhang, D. Si, H. Huang, L. Xie, Z. B. Fang, T. F. Liu and R. Cao, Angew. Chem., Int. Ed., 2022, 61, e202203955 CrossRef CAS PubMed.
- M. S. Liao and S. Scheiner, J. Chem. Phys., 2001, 114, 9780–9791 CrossRef CAS.
- Q. Yin, E. V. Alexandrov, D. H. Si, Q. Q. Huang, Z. B. Fang, Y. Zhang, A. A. Zhang, W. K. Qin, Y. L. Li, T. F. Liu and D. M. Proserpio, Angew. Chem., Int. Ed., 2021, 61, e202115854 CrossRef PubMed.
- J. M. Campos-Martin, G. Blanco-Brieva and J. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962 CrossRef CAS PubMed.
- Y. Kondo, Y. Kuwahara, K. Mori and H. Yamashita, Chem, 2022, 8, 2924–2938 CAS.
- K. Sahel, L. Elsellami, I. Mirali, F. Dappozze, M. Bouhent and C. Guillard, Appl. Catal., B, 2016, 188, 106 CrossRef CAS.
- Y. Zhang and S. J. Park, J. Mater. Chem. A, 2018, 6, 20304 RSC.
- A. M. Rice, G. A. Leith, O. A. Ejegbavwo, E. A. Dolgopolova and N. B. Shustova, ACS Energy Lett., 2019, 4, 1938–1946 CrossRef CAS.
- Y. Isaka, Y. Kawase, Y. Kuwahara, K. Mori and H. Yamashita, Angew. Chem., Int. Ed., 2019, 58, 5402–5406 CrossRef CAS PubMed.
- Y. Kawase, Y. Isaka, Y. Kuwahara, K. Mori and H. Yamashita, Chem. Commun., 2019, 55, 6743–6746 RSC.
- Y. C. Hao, L. W. Chen, J. Li, Y. Guo, X. Su, M. Shu, Q. Zhang, W. Y. Gao, S. Li, Z. L. Yu, L. Gu, X. Feng, A. X. Yin, R. Si, Y. W. Zhang, B. Wang and C. H. Yan, Nat. Commun., 2021, 12, 2682 CrossRef CAS PubMed.
- B. An, Z. Li, Z. Wang, X. Zeng, X. Han, Y. Cheng, A. M. Sheveleva, Z. Zhang, F. Tuna, E. J. L. McInnes, M. D. Frogley, A. J. Ramirez-Cuesta, L. S. Natrajan, C. Wang, W. Lin, S. Yang and M. Schröder, Nat. Mater., 2022, 21, 932–938 CrossRef CAS PubMed.
- Q. Zhi, W. Liu, R. Jiang, X. Zhan, Y. Jin, X. Chen, X. Yang, K. Wang, W. Cao, D. Qi and J. Jiang, J. Am. Chem. Soc., 2022, 144, 21328–21336 CrossRef CAS PubMed.
- Y. Liu, L. Li, H. Tan, N. Ye, Y. Gu, S. Zhao, S. Zhang, M. Luo and S. Guo, J. Am. Chem. Soc., 2023, 145, 19877–19884 CrossRef CAS PubMed.
- J. Chang, J. Shi, Q. Li, S. Li, Y. Wang, Y. Chen, F. Yu, S. Li and Y. Lan, Angew. Chem., Int. Ed., 2023, e202303606 Search PubMed.
| This journal is © The Royal Society of Chemistry 2024 |