
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Acetylacetone functionalized periodic mesoporous organosilicas: from sensing to catalysis†
Chunhui
Liu
 abc,
Mahdieh
Haghighat
a,
Himanshu Sekhar
Jena
a,
Dirk
Poelman
b,
Nathalie
De Geyter
d,
Rino
Morent
d,
Anna M.
Kaczmarek
*c and
Pascal
Van Der Voort
*a
abc,
Mahdieh
Haghighat
a,
Himanshu Sekhar
Jena
a,
Dirk
Poelman
b,
Nathalie
De Geyter
d,
Rino
Morent
d,
Anna M.
Kaczmarek
*c and
Pascal
Van Der Voort
*a
aCOMOC – Center for Ordered Materials Organometallics and Catalysis, Department of Chemistry, Ghent University, Krijgslaan 281 S3, 9000 Ghent, Belgium. E-mail: Pascal.VanDerVoort@UGent.be
bLumilab, Department of Solid State Sciences, Ghent University, Krijgslaan 281 S1, 9000 Ghent, Belgium
cNanoSensing Group, Department of Chemistry, Ghent University, Krijgslaan 281 S3, 9000 Ghent, Belgium. E-mail: Anna.Kaczmarek@UGent.be
dRUPT – Research Unit Plasma Technology, Department of Applied Physics, Ghent University, St-Pietersnieuwstraat 41 B4, 9000 Ghent, Belgium
First published on 2nd July 2024
Abstract
25 years after the first papers on the development of Periodic Mesoporous Organosilicas, and in this special issue in honor of prof. Ozin who stood at the cradle of PMOs, we here report for the first time an “acac-PMO” with the acac-functionality intact and embedded in the organic environment of the linker. Periodic mesoporous organosilicas (PMOs) have emerged as versatile sensors due to their stability, low toxicity, and scalability. This study reports for the first time on embedding the acetylacetone (acac) group into the PMO backbone. This integration of acac into the PMO framework opens new applications in both sensing and catalysis. The Eu3+ grafted acac-PMOs demonstrate enhanced sensitivity, with a detection limit of 108 nM, and high selectivity in aqueous copper (Cu2+) ion sensing. Additionally, these novel PMOs show high catalytic efficiency in Mannich reactions.
1 Introduction
Periodic mesoporous organosilicas (PMOs) have emerged as a promising and appealing sensor system due to their stable physicochemical properties, low toxicity, high biocompatibility, and facile scalability for large-scale production.1 PMOs were initially reported in 1999 by Ozin,2 Stein,3 and Inagaki.4 Unlike other porous materials such as MCM (Mobil Composition of Matter)5 and SBA (Santa Barbara Amorphous materials),6 PMOs offer the unique advantage of incorporating desired organic functionalities directly into the silsesquioxane framework. This is achieved by utilizing organically bridged silica precursors, where hydrolysable groups (OR) in the bis-silane (RO)3-Si-Rf-Si-(RO)3 are connected to the desired organic functionalities (Rf). The incorporation of these organic groups within the PMO structure enhances the material's hydrophobicity and hydrothermal stability, while also minimizing leaching issues commonly associated with externally grafted functionalities.7,8 Additionally, PMOs possess ordered mesopores and high surface areas (up to 2370 m2 g−1),9 making them suitable for a wide range of applications such as catalysis, adsorption, chromatography, low-k materials, drug release, and sensors.10–13 The tailored organic functionalities within the PMO framework enable the design of materials with specific properties and functionalities, allowing for enhanced performance in various applications. The ability to finely tune the pore size, surface chemistry, and hydrophobicity of PMOs provides versatility and control over their performance in different environments.Acetylacetone (acac) based homogeneous transition metal complexes have demonstrated exceptional catalytic efficacy across a variety of organic transformations.14–16 Significantly, a limited number of acetylacetone-functionalized heterogeneous systems, such as silicas and covalent triazine frameworks (CTFs), have demonstrated high reactivity and recyclability after grafting transition metals (post-synthetic functionalization) in various chemical processes. These processes include cross-coupling reactions, hydroxylation, click chemistry, Glaser-type coupling, and the oxidation of alcohols.17–22 The immobilization of acac ligands within the framework not only enhances catalytic activity but also provides a stable and recyclable catalytic system, thereby addressing the challenges associated with traditional acac complexes.17 So far, a PMO with acac functionalities embedded in the backbone has not been reported. Zhao et al. grafted the VO(acac)2 onto PMOs to create a catalyst for selective hydroxylation of benzene to phenol.20 The organic precursor was synthesized through the Schiff base condensation between the amino group of 3-aminopropyl-triethoxysilane (APTES) and the carbonyl unit of VO(acac)2. We aimed to functionalize acac group into a PMO backbone in order to design a robust acac-PMO supported transition metal catalyst for sustainable catalysis.
Next to catalysis where the acac groups are typically tethering transition metal ions, also lanthanides anchor firmly to the acac ligands, creating possibly luminescent sensors. Embedded organic groups within the PMO framework act as efficient energy transfer antennas, sensitizing trivalent lanthanide ions (Ln3+) and promoting luminescence.13,23,24 The potential applications of such sensors span environmental monitoring, biological analysis, and other areas demanding accurate metal ion detection.25 Despite this potential, research on lanthanide-based PMOs (LnPMOs) as chemical sensors remains limited, with only a few studies published in the late 2010s.24,26–30 Among these, Eu-based materials have attracted significant attention for their potential in detecting copper ions (Cu2+).31–34 Eu-based materials exhibit strong initial fluorescence that undergoes significant quenching upon interaction with Cu ions, offering a highly sensitive and selective detection method for Cu in aqueous solutions.31,35
This paper is the first paper that develops an acac-based PMO with embedded acac groups. PMOs are typically made with short rigid bridges for stiffness and extra functionalities on these organic bridges renders the synthesis much more difficult, due to additional interactions with the silane and the surfactant. So, the synthesis of PMOs with such a bulky organic bridge and the acac functionality has produced a material that has high potential.
Acac complexes are generally appreciated because the acac group can tether (hard) transition metals very effectively, this makes them interesting for metal-ion sequestration, water purification, but also for heterogeneous transition metal based catalysis as the metals do not leach from acac groups. But acac groups are also often used in lanthanide (luminescence) chemistry because of the strong interaction and a strong antenna effect. In the past, acac groups have been grafted on oxide materials, such as silica, but the hydrolytic unstable Si–O–Si (siloxane) bond between the silica and the acac containing silane renders these materials less useful in aqueous environments and leaching occurs. This acac-PMO is completely different, the acac groups are covalently bonded to the phenyl rings of the organic bridge, rendering also the local environment more hydrophobic.
Out of a multitude of possible applications for these materials, we have chosen a sensing application and a heterogeneous catalysis application, showcasing that these materials can be used in these applications.
2 Experimental
2.1 Materials and instrumentation
All chemicals were purchased from Sigma Aldrich, Fluorochem, Alfa Aesar, or TCI Europe and used without further purification.Nitrogen adsorption experiments were conducted at −196 °C using a TriStar II gas analyzer. Prior to analysis, the samples underwent vacuum degassing at 120 °C for 24 h. Powder X-ray diffraction (PXRD) patterns were obtained using a Bruker D8 Advance diffractometer equipped with an autochanger and LynxEye XE-T Silicon Strip Line detector, operating at 40 kV and 30 mA with Cu-Kα radiation (λ = 1.5406 Å) in Bragg–Brentano geometry. Fourier transform infrared spectroscopy (FT-IR) measurements were performed using a Thermo Nicolet 6700 FT-IR spectrometer equipped with a nitrogen-cooled MCT-A (mercury–cadmium–tellurium) detector and a KBr beam splitter. X-ray fluorescence (XRF) was measured by XRF Supermini200 Rigaku to analyze the relative metal contents. Transmission electron microscopy (TEM) images were acquired using a JEOL JEM-2200FS transmission electron microscope with a post-sample Cs corrector and an accelerating voltage of 200 kV. Using V2O5 as a catalyst, elemental analysis (CHNS) was finished using the Thermo Flash 2000 elemental analyzer. X-ray photoelectron spectroscopy (XPS) measurements were carried out on a PHI 5000 VersaProbe II spectrometer equipped with a monochromatic Al-Kα X-ray source (hν = 1486.6 eV). Photoluminescence measurements were conducted using an Edinburgh Instruments FLSP920 UV-vis-NIR spectrometer setup. The emission signals were detected using a Hamamatsu R928P photomultiplier tube, and a monochromated 450 W xenon lamp was employed as the steady-state excitation source.
2.2 Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100% and 20:80% ratios, respectively, along with 1 mL of ethanol. The precursor solution was then injected promptly into the stirred aqueous surfactant solution, resulting in the formation of a yellow suspension. The mixture was stirred for an additional 24 h at 80 °C. After cooling down and filtration, the solid material was rinsed repeatedly with deionized water and ethanol, followed by drying at 120 °C. To remove the surfactant, a solvent-extraction method was employed with the following procedure: approximately 1 g of the synthesized material was stirred in a solution of 200 mL of ethanol and 3 mL of concentrated hydrochloric acid at 80 °C for 4 hours. This extraction process was repeated twice to ensure the complete removal of all surfactants from the material. Finally, the sample was dried overnight in an oven at 120 °C. The obtained products were denoted as acac(20)-PMO and acac(100)-PMO, for which the molar ratio of acac-Si to BTESE were 20:80 and 0:100, respectively.
:100% and 20:80% ratios, respectively, along with 1 mL of ethanol. The precursor solution was then injected promptly into the stirred aqueous surfactant solution, resulting in the formation of a yellow suspension. The mixture was stirred for an additional 24 h at 80 °C. After cooling down and filtration, the solid material was rinsed repeatedly with deionized water and ethanol, followed by drying at 120 °C. To remove the surfactant, a solvent-extraction method was employed with the following procedure: approximately 1 g of the synthesized material was stirred in a solution of 200 mL of ethanol and 3 mL of concentrated hydrochloric acid at 80 °C for 4 hours. This extraction process was repeated twice to ensure the complete removal of all surfactants from the material. Finally, the sample was dried overnight in an oven at 120 °C. The obtained products were denoted as acac(20)-PMO and acac(100)-PMO, for which the molar ratio of acac-Si to BTESE were 20:80 and 0:100, respectively.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch).
O stretch).
3 Results and discussion
3.1 Synthesis and characterization of acac-PMOs
The designed approach for producing specific acac-PMO materials is shown in Scheme 1. This method encompasses an initial base-catalysed condensation between 4′-bromoacetophenone and methyl 4-bromobenzoate.38 Subsequently, a coupling reaction is performed, involving vinyl-triethoxysilane and using Heck reaction conditions. Through the silylation of acac-Br, the resultant structure of acac-Si is achieved. To create a series of acac-functionalized PMO materials with varying precursor ratios, a combination of acac-Si and bis(triethoxysilyl)ethane (BTESE) is prepared in two distinct ratios: 20% acac-Si to 80% BTESE, and 100% acac-Si to 0% BTESE. The generated products are designated as acac(20)-PMO and acac(100)-PMO, wherein the molar ratios of acac-Si to BTESE are 20:80 and 100:0, respectively. In the design of acac(20)-PMO@Eu_acac for copper ion sensing, a 20% acetylacetonate (acac) functionalization is strategically chosen to address aggregation-caused quenching (ACQ). This lower acac content ensures an optimal dispersion of Eu3+ complexes within the mesoporous framework, maintaining their luminescent efficiency vital for sensitive and accurate copper detection. The tailored PMO structure facilitates effective interaction with copper ions while preserving the unique luminescent properties of Eu, overcoming the common challenge of luminescence quenching in aggregated lanthanide materials.39
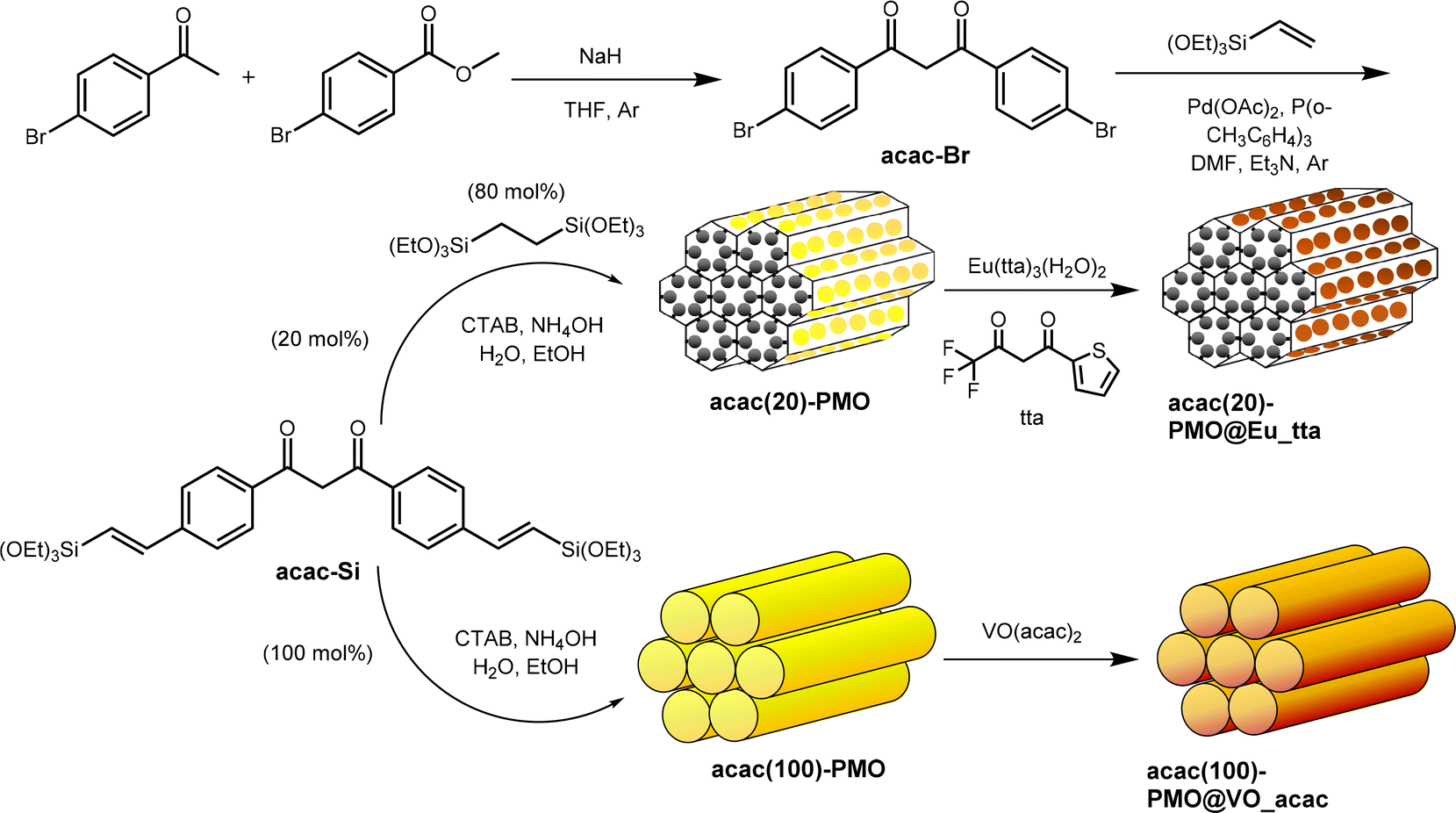 | ||
| Scheme 1 Schematic illustration of the preparation of the acac(20)-PMO@Eu_tta, acac(100)-PMO@VO_acac. | ||
The successful coupling was validated through FT-IR measurements. In Fig. 1a and d, we present a spectral analysis comparing the spectrum of pure acac(100)-PMO with that of acac(20)-PMO, as well as their respective metal-grafted counterparts. In the spectra of acac(20)-PMO and acac(100)-PMO, in addition to observing the characteristic C–H and Si–O–Si stretching vibrations at 2950–2800 cm−1, 1200–1000 cm−1, and 800 cm−1, a distinct and sharp band at 1700 cm−1 (–CO) was detected in both acac(20)-PMO and acac(100)-PMO.17,40 This observation provides clear evidence of the persistence of the acac functional group even after the synthesis of PMO.
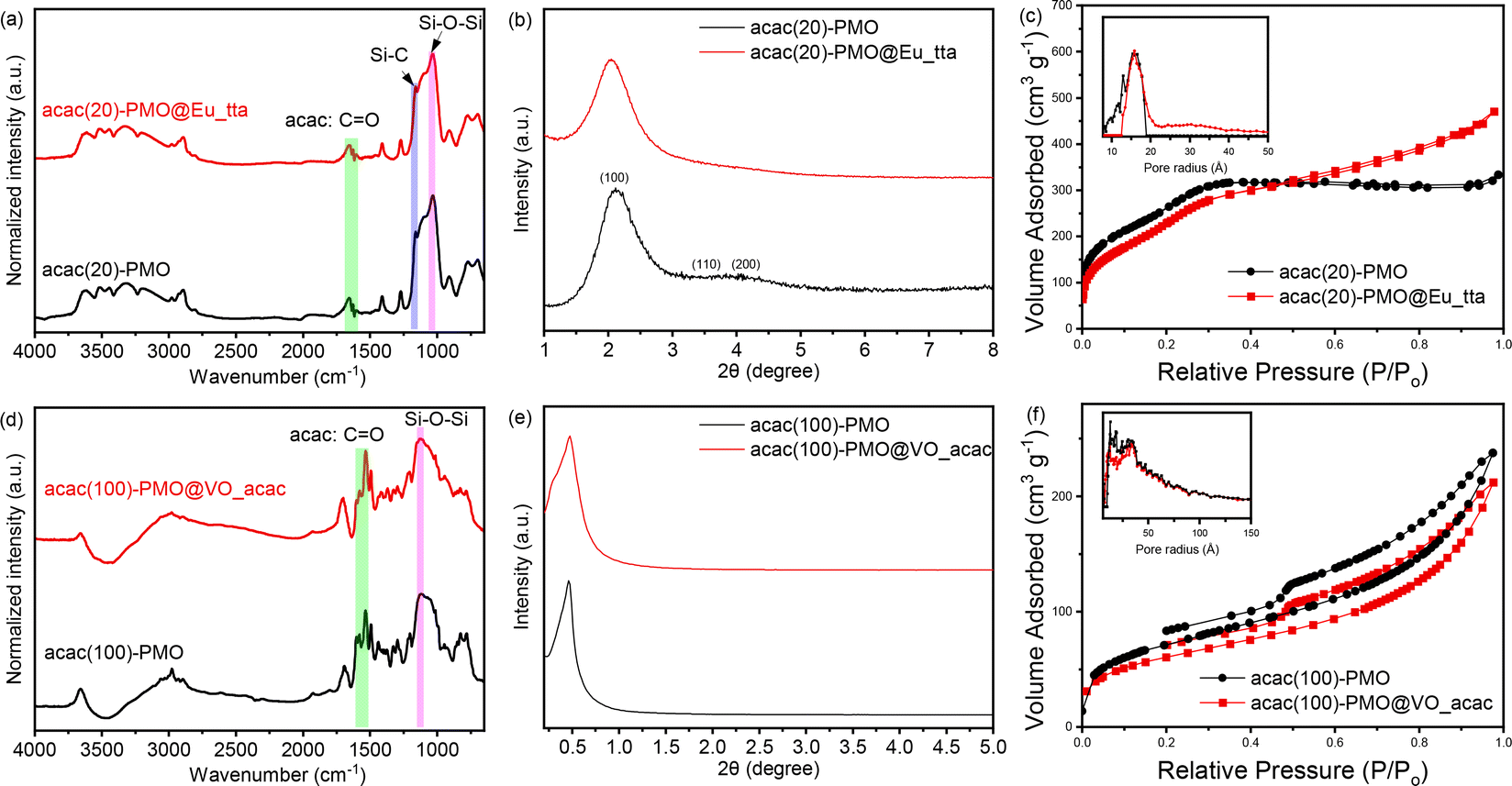 | ||
| Fig. 1 FT-IR spectra of (a) acac(20)-PMO and (d) acac(100)-PMO; XRD patterns of (b) acac(20)-PMO and (e) acac(100)-PMO; N2 adsorption–desorption isotherms and pore size distributions of (c) acac(20)-PMO and (f) acac(100)-PMO. | ||
To investigate the impact of material functionalization on structural characteristics, we conducted powder X-ray diffraction (PXRD) and N2-sorption analyses. Fig. 1b displays the X-ray diffraction (XRD) pattern of the acac(20)-PMO within the 2θ range of 1–8°. An intense peak at around 2° with a small shoulder was observed in Fig. 1b, signifying the presence of hexagonally (or cubic) arranged mesoporous structures.41,42 The XRD pattern of acac(100)-PMO reveals a distorted structure. The intense XRD peak, assigned to the (100) plane, affirms a substantial reduction in long-range ordering when compared to the co-condensation sample.43 The formation of a wormlike pore structure in acac(100)-PMO nanoparticles may be attributed to the reduced solubility of the acetylacetone-bridged silane, primarily due to the highly hydrophobic benzene moiety, in comparison to the other organosilica source (BTESE) in aqueous media.43 In Fig. 1c and f, the N2-sorption isotherms of acac-PMOs are presented. The acac(20)-PMO displays a Type IV isotherm with an H1 hysteresis loop, indicative of a uniform mesoporous structure.31,44 Conversely, the acac(100)-PMO shows a Type IV isotherm with an H2(b)-shaped hysteresis loop, suggestive of non-uniform mesopores that are blocked for the desorption (cavitation).45,46Table 1 summarizes the Brunauer–Emmett–Teller surface area (SBET), total pore volume at P/P0 = 0.95 (V), and non-local density functional theory most prominent pore diameter (dNLDFT). It is clear from the XR diffractograms, the Table 1 and the isotherms that the acac(20)-PMO, formed from 80% BTESE and only 20% acac-precursor forms the typical (expected) hexagonal structure (P6mm) whereas the acac(100)-PMO forms a distorted structure with uniform but unordered worm-like pores.
| Sample | S BET (m2 g−1) | V (cm3 g−1) | d NLDFT (nm) |
|---|---|---|---|
| a Calculated from NLDFT, N2 at 77 K, using the kernel of silica cylindrical pore, adsorption branch. | |||
| acac(20)-PMO | 1040 | 0.47 | 3.2 |
| acac(20)-PMO@Eu_tta | 984 | 0.46 | 3.2 |
| acac(100)-PMO | 264 | 0.36 | 2.6 |
| acac(100)-PMO@VO_acac | 220 | 0.32 | 2.6 |
3.2 Luminescence properties
In the context of luminescence applications, we departed from using Eu3+ salts and instead employed Eu3+ complexes for grafting into acac(20)-PMO. Therefore, we created a composite material denoted as acac(20)-PMO@Eu_tta. The tta (thenoyltrifluoroacetonate) ligand acts as an ancillary ligand that can serve as a secondary antenna ligand. In addition, this ligand can act as a shielding agent, safeguarding the lanthanide ions from quenching by water molecules.47Fig. 2a presents the excitation–emission spectra of the acac(20)-PMO under ambient conditions. The excitation spectrum displays a broad band spanning from 250 to 400 nm, with the maximum excitation wavelength at 360 nm. The emission spectrum extends from 390 to 600 nm, with the emission peak maximum at 420 nm. The luminescent properties of this hybrid material were further investigated through the grafting of Eu(tta)3 complexes onto the PMO material. The combined excitation-emission spectra for acac(20)-PMO@Eu_tta are illustrated in Fig. 2b. The excitation spectrum displays a broad band spanning from 250 to 400 nm with a maximum at λmax = 352 nm. This spectral feature can be attributed to transitions from the ground state (π)S0 to the excited state (π*)S1 of the organic ligands. Conversely, upon excitation at the maximum of the broad band, the emission spectrum exhibits characteristics indicative of intra-4f6 transitions of Eu3+ ions.48 A comprehensive assignment of the emission peaks observed is provided in Table 2. When the sample is exposed to UV light (302 nm), it emits a purplish-red color, as seen in the Fig. 2b inset and the CIE diagram of Fig. S2a.† It consists of contributions from both acac(20)-PMO and Eu3+. The luminescence decay time of the sample was assessed by excitation at 352 nm and detection at 611 nm. A satisfactory fit was achieved utilizing a mono-exponential decay curve (Fig. 2c).
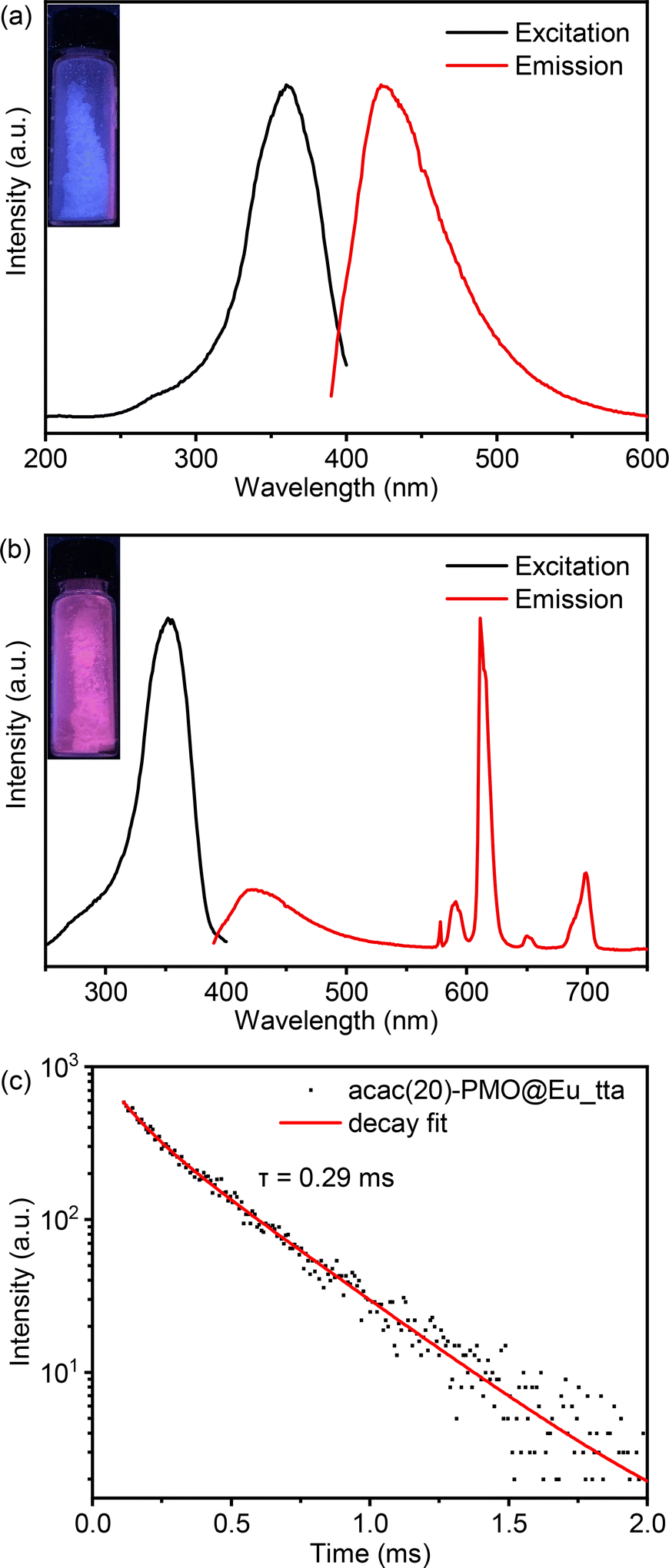 | ||
| Fig. 2 (a) Combined RT excitation-emission spectrum of the acac(20)-PMO (excited at 360 nm observed at 420 nm); (b) combined RT excitation-emission spectrum of the acac(20)-PMO@Eu_tta, ex = 352 nm, em = 611 nm; (c) luminescence decay profile of as prepared acac(20)-PMO@Eu_tta, ex = 352 nm, em = 611 nm. Measurements refer to the sample in solid form. | ||
| Wavelength (nm) | Wavenumber (cm−1) | f–f transition |
|---|---|---|
| 578 | 17301 |
5D0 → 7F0 |
| 591 | 16920 |
5D0 → 7F1 |
| 611 | 16367 |
5D0 → 7F2 |
| 650 | 15385 |
5D0 → 7F3 |
| 699 | 14306 |
5D0 → 7F4 |
The photophysical characteristics of acac(20)-PMO@Eu_tta are primarily ascribed to the f–f transitions of Ln3+ ions. The luminescence properties and the porosity of PMOs give them the ability to discern variations in their surrounding environment. This attribute allows applications in turn-off chemical sensing, achieved by modulating guest motifs through host–guest interactions.49 To investigate the metal ion selectivity exhibited by Acac(20)-PMO@Eu_tta materials, fluorescence experiments were systematically conducted, as depicted in Fig. 3. The ion sensing performance of this material was assessed through the use of a suspension with a concentration of 1 mg mL−1. In Fig. 3a, we have compiled and compared the photoluminescence spectra of interest. Most suspensions containing various metal ions (Pb2+, Na+, Al3+, K+, Mg2+, Ca2+, Sr2+, Fe3+, Hg2+, Cr3+, Y3+, Zn2+, and Cu2+) exhibit the characteristic purplish-red luminescence attributed to acac(20)-pmo and Eu3+ transitions. However, only the presence of Cu2+ results in a substantial reduction in luminescence intensity, indicating a pronounced quenching effect. This observation underscores the high selectivity of acac(20)-PMO@Eu_tta for the detection and specific identification of Cu2+ in aqueous solutions. Notably, under UV light excitation at 302 nm, this quenching effect is visible to the naked eye (Fig. 3a inset), where the color transition from purplish-red to blue is observed. It shows the color primarily originates from acac(20)-PMO after the addition of Cu2+ (Fig. S2b†). It can be, in part, elucidated by the Cu(II) static quenching mechanism, which arises from the formation of non-fluorescent ground-state complexes.50 In the PL decay time analysis shown in Fig. 3b, we also observed a decrease in the PL lifetime upon the addition of Cu2+. The initial PL lifetime, recorded in the absence of Cu2+, is established at 201 μs. A titration of Cu2+ into the system results in a measurable decrease in the PL lifetime, reaching a minimum of 175 μs. This trend confirms the quenching effect, emphasizing the selective interaction between Cu2+ ions and the acac(20)-PMO@Eu_tta framework. The change in decay time suggests an underlying mechanism involving energy or electron transfer, likely indicative of the formation of a charge transfer complex with Cu2+, which may be attributed to both dynamic quenching and the paramagnetic nature of the Cu2+ ions.51,52 Luminescence titration experiments were conducted to investigate the luminescent response of acac(20)-PMO@Eu_tta in the presence of Cu2+ ions. With an increasing concentration of Cu2+ ions, there was a marked reduction in the emission intensity of acac(20)-PMO@Eu_tta, as depicted in Fig. 3c. This effect resulted in the complete quenching of fluorescence emission in the Eu3+ hybrid suspension when the Cu2+ concentration reached 30 μM (Fig. S3†). To quantitatively assess this luminescence quenching effect, we employed the Stern–Volmer (S–V) equation. The Stern–Volmer quenching constant (KSV) was calculated as a measure of the quenching efficiency and enabling a precise characterization of the interaction between acac(20)-PMO@Eu_tta and Cu2+ ions:
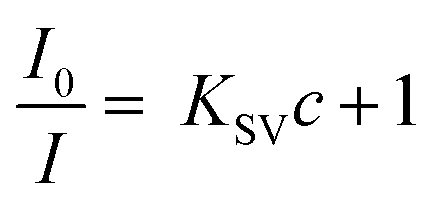 | (1) |
198 M−1. The limit of detection (LOD) was calculated to be 108 nM (26 ppb) according to the method used by Armbruster et al., assuming that| LOD = 3σ/KSV | (2) |
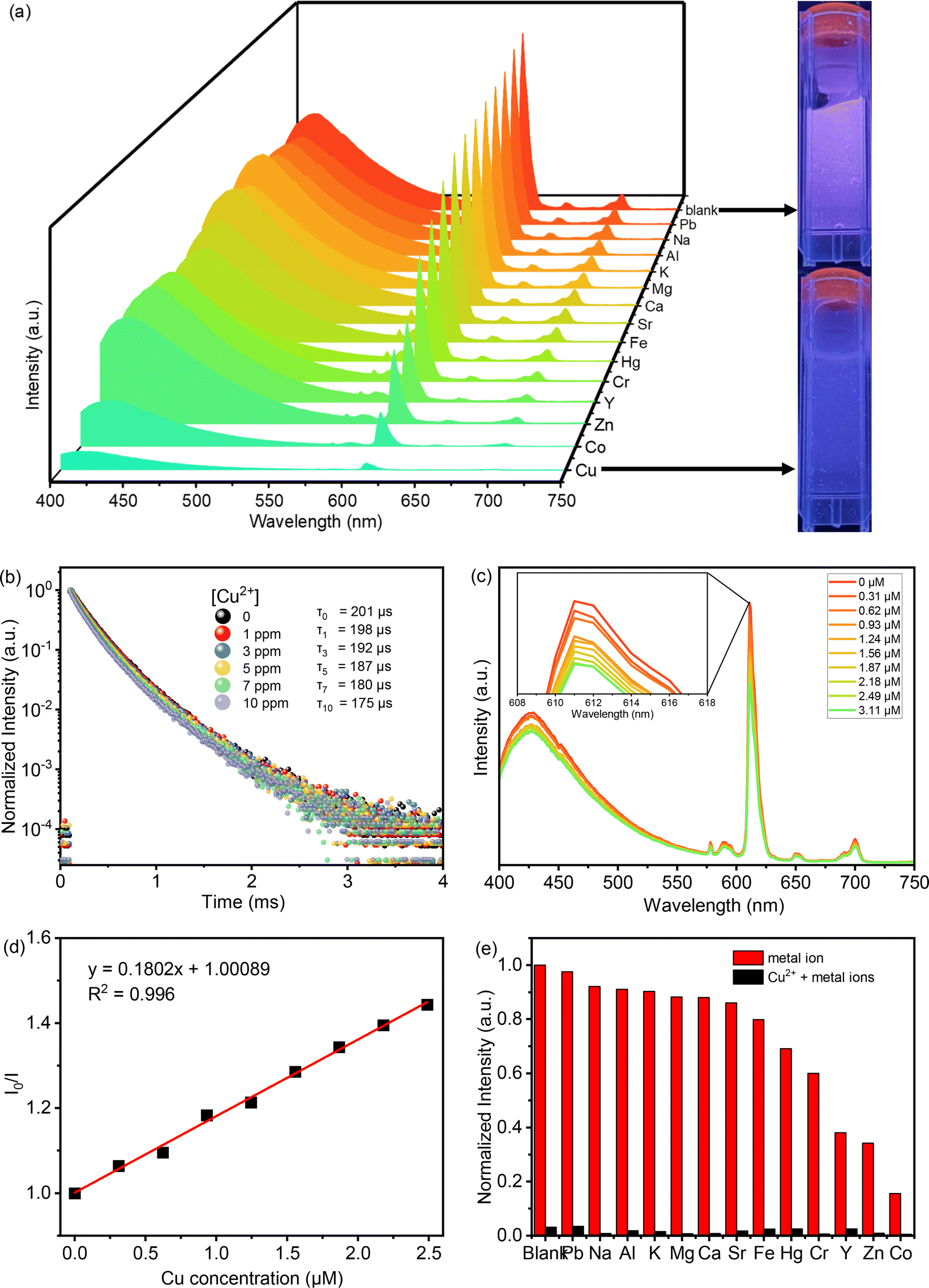 | ||
| Fig. 3 (a) Emission spectra of the colloidal suspensions of acac(20)-PMO@Eu_tta in the presence of different metal ions (1000 ppm) (Pb2+, Na+, Al3+, K+, Mg2+, Ca2+, Sr2+, Fe3+, Hg2+, Cr3+, Y3+, Zn2+, and Cu2+) when excited at 343 nm; (b) luminescence decay profile of colloidal suspensions of acac(20)-PMO@Eu_tta before and after the addition of Cu2+ ions. (c) Luminescence spectra of acac(20)-PMO@Eu_tta under different concentrations of Cu2+ aqueous solutions; (d) plot of the Stern–Volmer curve between the luminescence intensity and Cu2+ concentration in an aqueous solution; (e) comparison of the luminescence intensities (611 nm) of acac(20)-PMO@Eu_tta with varied ions (1000 ppm) (Pb2+, Na+, Al3+, K+, Mg2+, Ca2+, Sr2+, Fe3+, Hg2+, Cr3+, Y3+, and Zn2+) before and after treated by Cu2+. | ||
3.3Catalytic properties
The acac(100)-PMO was utilized as a heterogeneous catalysis by anchoring VO(acac)2 onto the material, resulting in a catalyst containing 0.59 mmol V g−1 (cat). The surface elemental composition of acac(100)-PMO and acac(100)-PMO@VO_acac was analyzed using XPS. Fig. 4a and d illustrate the presence of elements such as V, C, O, and Si on the surfaces of these nanocomposites. In Fig. 4b and e, the C 1s region reveals the presence of multiple chemical states of carbon. Four distinct peaks were discerned and attributed to C–Si(282.6 eV), C–C and C–H (284.8 eV), C–O (286.9 eV), and CO species (289.8 eV).61 The increasing peak of CO can be attributed to the grafting of VO(acac)2. In the XPS O 1s spectra, two distinct peaks were observed at 531.9 and 532.9 eV. These peaks can be attributed to the presence of CO and –C–OH groups, likely arising from the tautomerization of the acac group in both its keto and enol forms, as illustrated in Fig. 4c and f. The high-resolution XPS spectrum of V 2p can be deconvoluted into two primary doublet peaks, as depicted in Fig. S5.† In this detailed spectrum, the doublet peaks at 515.3 eV and 522.2 eV correspond to V 2p3/2 and V 2p1/2, respectively.62 This observation suggests the presence of V(IV) in the acac(100)-PMO@VO_acac.
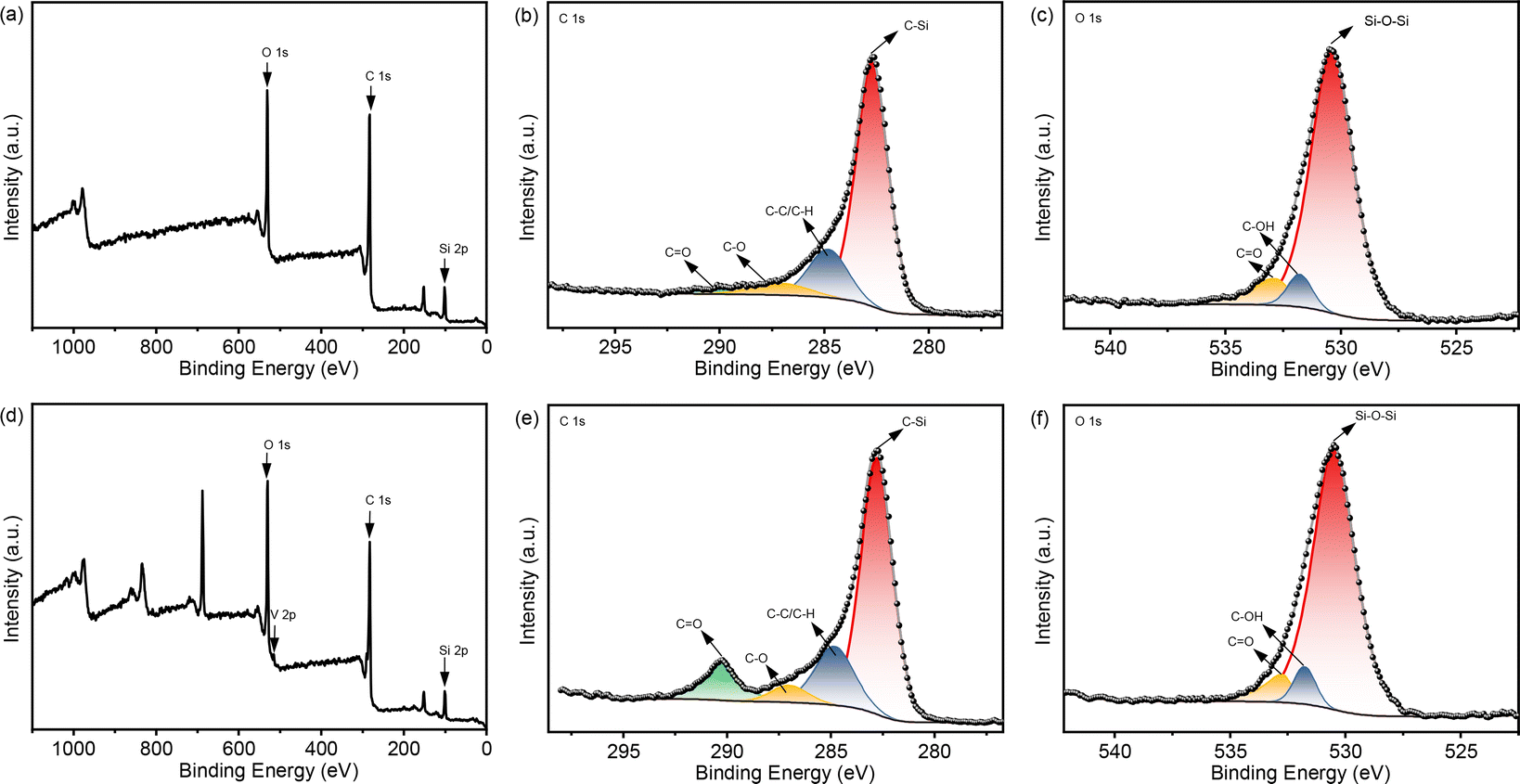 | ||
| Fig. 4 XPS spectra of (a) acac(100)-PMO, in the regions of (b) C 1s and (c) O 1s; and (d) acac(100)-PMO@VO_acac, in the regions of (e) C 1s and (f) O 1s. | ||
Upon completing the material characterization, the catalytic performance of the acac(100)-PMO@VO_acac material was tested for the model Mannich-type reaction, as shown in Table 3. During a typical catalytic test, predetermined quantities of 2-naphthol and N-methylmorpholine N-oxide (NMO), along with the catalyst were combined. The mixture was then refluxed in anhydrous DCM within a 10 mL Schlenk tube under an inert atmosphere. At specific time intervals, the catalyst was separated, and the filtrate was evaporated until dryness, resulting in the formation of the pure Mannich base with a quantitative yield.17 The confirmation of product formation was achieved through 1H NMR spectroscopy.
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst |
1a:2a |
Time (h) | Yieldb (%) |
| a Reaction conditions: 2-naphthol (1 mmol), NMO, DCM (2 mL) and catalyst (15 mg). b Yield was calculated from 1H NMR analysis using mesitylene as internal standard. c 10 mol%. | ||||
| 1 | — | 1:2 |
12 | — |
| 2 | VO(acac)2c | 1:3 |
12 | 92 |
| 3 | acac(100)-PMO | 1:3 |
12 | — |
| 4 | acac(100)-PMO@VO_acac | 1:3 |
12 | 98 |
| 5 | acac(100)-PMO@VO_acac | 1:2 |
12 | 98 |
| 6 | acac(100)-PMO@VO_acac | 1:1.5 |
12 | 96 |
| 7 | acac(100)-PMO@VO_acac | 1:1 |
12 | 70 |
| 8 | acac(100)-PMO@VO_acac | 1:2 |
1 | 16 |
| 9 | acac(100)-PMO@VO_acac | 1:2 |
6 | 40 |
| 10 | acac(100)-PMO@VO_acac | 1:2 |
24 | 98 |
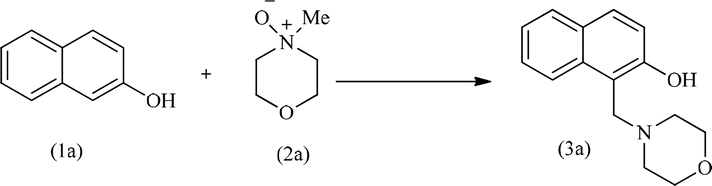
Initially, the Mannich type reaction between 2-naphthol (1a) and 4-methylmorpholine N-oxide (NMO) (2a) was conducted in the absence of a catalyst at 40 °C. After 12 hours of reaction, no substrate conversion occurred, highlighting the necessity of the presence of a catalyst (Table 3, entry 2). Subsequently, we investigated the catalytic activity of VO(acac)2 for this model reaction. This complex exhibited a yield of 92% after 12 hours of reaction. However, it is hard to recycle this homogeneous catalyst.17,18,20 For the heterogeneous acac(100)-PMO@VO_acac catalyst, a 16% yield of the major product, 1-(morpholinomethyl)naphthalen-2-ol (3a), was obtained within 60 minutes.17,63 An increase in the yield from 40% to 98% was obtained upon extending the reaction time from 6 to 12 hours. Moreover, we conducted the reaction multiple times with varying equivalents of NMO. When employing 1, 1.5, 2, or 3 equivalents of NMO under the optimized reaction conditions, we observed yields of 70%, 96% and 98% of 3a, respectively. The highest yield of 92% for 3a was achieved using 3 equivalents of NMO.
The recyclability and structural stability of acac(100)-PMO@VO_acac material were studied during the preparation of 1(morpholinomethyl)naphthalen-2-ol under optimized reaction conditions. After completing the reaction, the recyclability test was conducted using the recovered catalyst. The catalyst was thoroughly washed with dichloromethane and dried under vacuum at 120 °C before being utilized in the subsequent catalytic reaction. The catalyst demonstrated consistent activity, with no significant decrease up to the four tested cycles (Fig. 5).
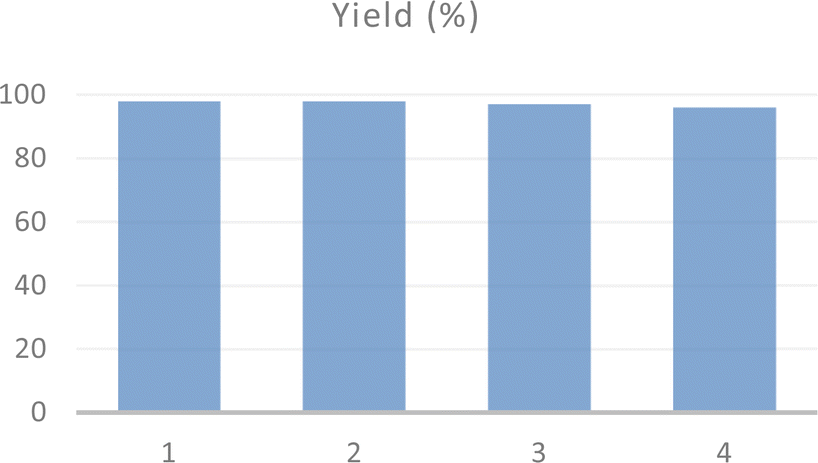 | ||
| Fig. 5 Recycling test of acac(100)-PMO@VO_acac catalyst for Mannich-type reaction. | ||
Furthermore, a hot filtration test was performed to assess the potential leaching of vanadium ions from the support. The catalyst was removed from the reaction mixture after 6 hours, and the reaction was allowed to continue for an additional 18 hours (Fig. 6). No further progress was observed in the reaction, indicating the absence of leaching.
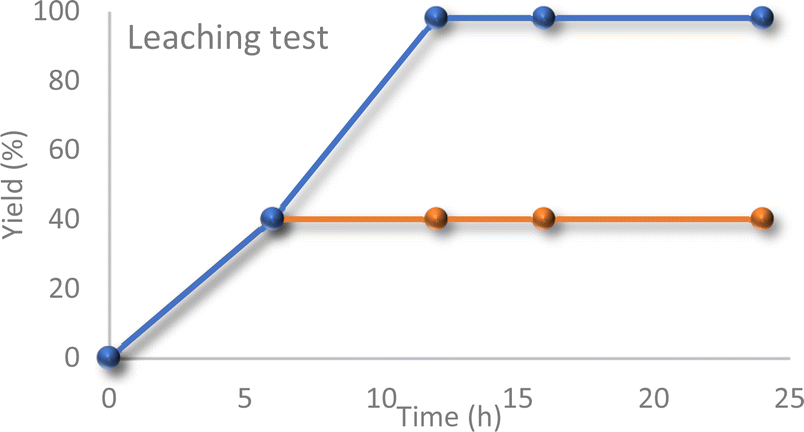 | ||
| Fig. 6 Reaction profile with or without the catalyst (after separating the catalyst from the reaction mixture via hot-filtration technique). | ||
To examine the physio-chemical properties of the catalyst, FT-IR and XPS analyses were conducted, as depicted in Fig. 7. The recovered catalyst exhibited all the characteristic peaks, similar to the fresh catalyst. Additionally, the XPS analysis of the recycled catalyst (Fig. 7b–d) displayed the same specific peaks as the fresh catalyst. These results confirm that the chemical structure of the catalyst was preserved during the recycling process.
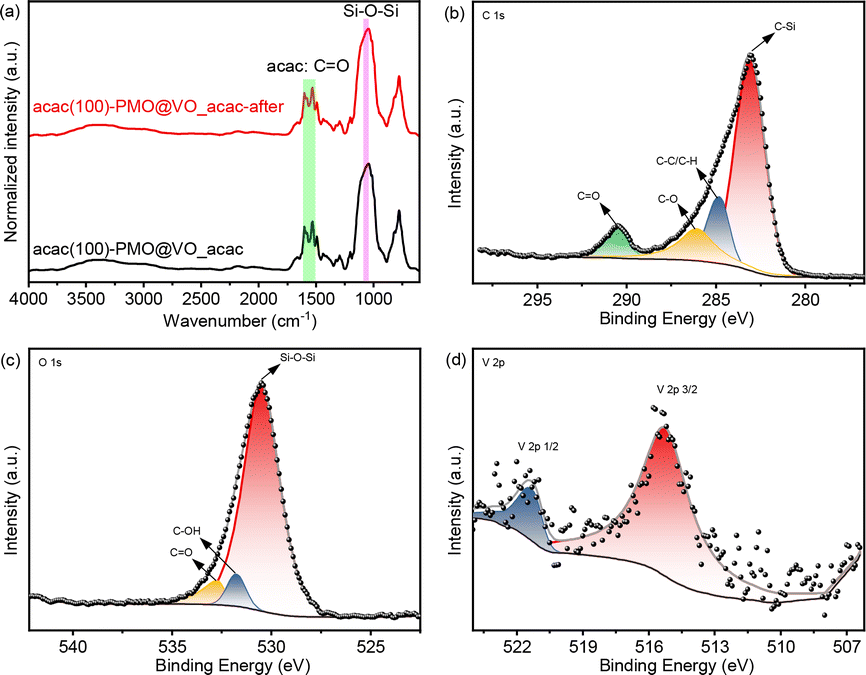 | ||
| Fig. 7 (a) DRIFT spectra of acac(100)-PMO@VO_acac before and after Mannich-type reaction; (b) deconvoluted XPS C 1s spectra, (c) O 1s spectra and (d) vanadium in the V 2p region of the recycled acac(100)-PMO@VO_acac. | ||
The catalytic reactivity of the acac(100)-PMO@VO_acac catalyst was evaluated using a diverse range of substrates, including 2-naphthol, 1-naphthol, and phenol derivatives, under the optimized reaction conditions (Table 4). Substantial catalytic conversion was achieved for nearly all the substrates, regardless of whether they had electron-donating or electron-withdrawing substituents on the aromatic ring.
| a Yields are calculated from 1H NMR using mesitylene as an internal standard. |
|---|
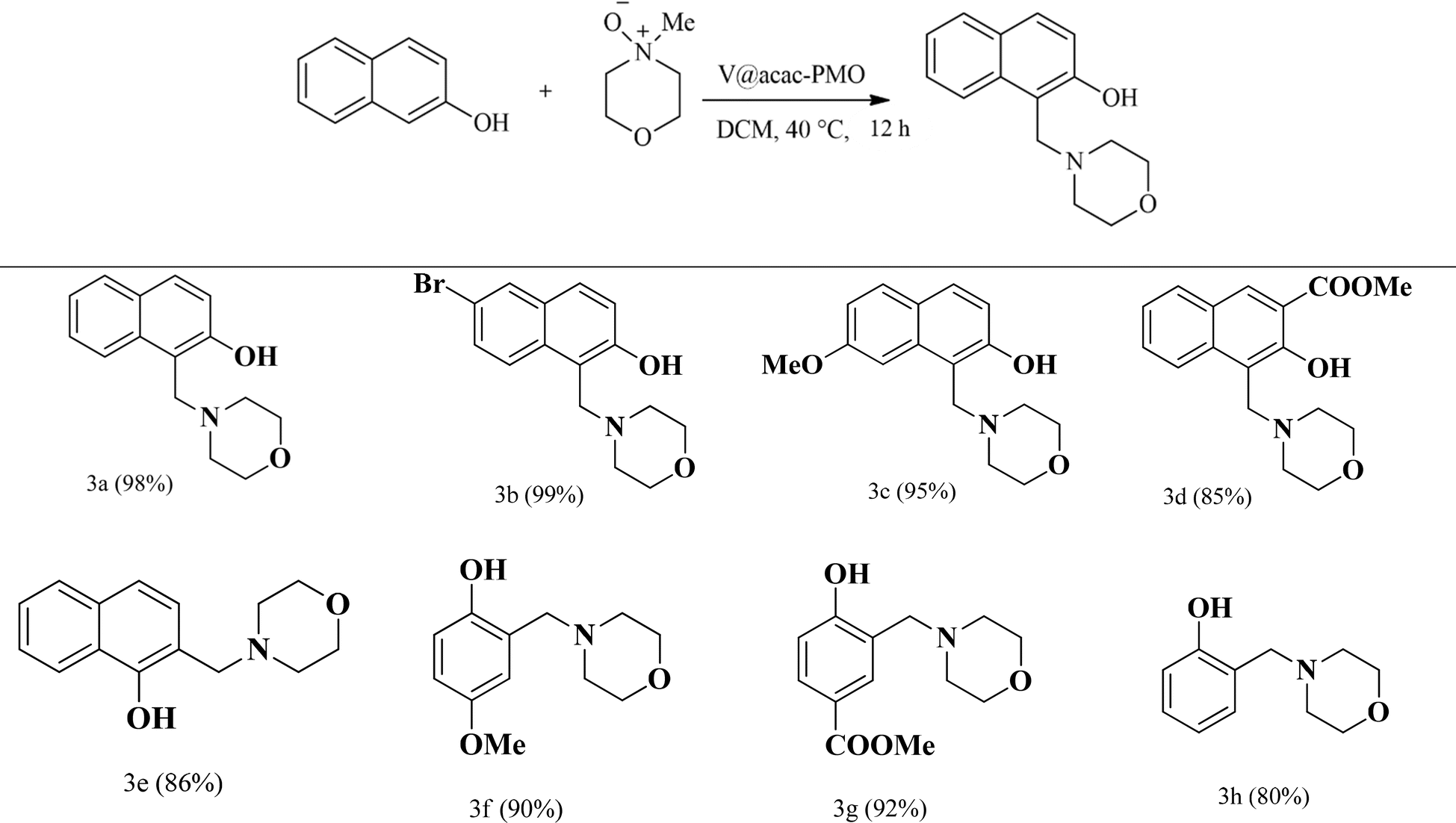
|
In Scheme S1,† a plausible pathway for the preparation of the Mannich base was proposed, following similar steps as previously reported by Uang et al.17,63–65 Initially, the active VO groups of the catalyst formed complexes with 4-methylmorpholine N-oxide, leading to the formation of a six-membered structure (A). An intramolecular elimination process via this transition structure resulted in the formation of an iminium ion B structure. Second, the [V(OH)(O−)] species played a crucial role in abstracting an acidic proton from 2-naphthol, generating a negatively charged oxygen species. This species further creates an acidic carbon center. A target Mannich base formed through a reaction of this acidic carbon center with iminium ions. Finally, the elimination of water molecules from V(OH)2 regenerated the catalyst, completing the catalytic cycle.
The catalytic performance of acac(100)-PMO@VO_acac was compared to its homogeneous counterpart, VO(acac)2, as well as acac-based covalent triazine frameworks (CTFs) and covalent organic frameworks (COFs) in Table S2.†17,63,64,66 It is evident from Table S2† that the acac(100)-PMO@VO_acac catalyst resulted in the targeted product with a high yield and conversion rate. When compared with VO(acac)2, it exhibited a higher yield (98%) and turnover number (TON) of 111, which is 12 times greater than the TON achieved by the homogeneous catalyst. Moreover, we evaluated the catalytic activity of acac(100)-PMO@VO_acac in comparison to acac-based COFs, such as vanadium-docked COFs (VO-TAPT-2,3-DHTA COF, VO-PyTTA-2,3-DHTA COF). In these cases, the catalysts resulted in the targeted product with yield of 98% and 96%, along with TON values of 29 and 36, respectively. This combination of high conversion and TON suggests potential for acac(100)-PMO@VO_acac as a heterogeneous catalyst. These findings substantiate the promising future of acac-PMO materials as versatile supports for the development of advanced heterogeneous catalysts.
4 Conclusions
This study introduces a novel PMOs with embedded acetylacetone (acac) groups, showcasing its dual utility in catalysis and sensing. The acac(20)-PMO grafted with Eu(tta)3 demonstrates effectiveness as a luminescent sensor towards aqueous Cu2+ ions, achieving a LOD of 108 nM. In addition, a heterogeneous catalyst (acac(100)-PMO@VO_acac) was prepared by anchoring VO(acac)2 on pristine acac-PMO that showed high reactivity in a modified Mannich reaction with a turnover number (TON) of 111 and a wide substrate scope. Furthermore, the embedded acac functionality's capacity to tether “hard” metal ions suggests potential applications in sensing, catalysis, and water purification. These findings underscore the versatility of acac-PMOs and open new avenues for their application in environmental and industrial contexts.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. H. L. gratefully acknowledges the financial support from the China Scholarship Council (201908110280). P. V. D. V. acknowledges Hercules Project AUGE/17/07 for the UV VIS DRS spectrometer and UGent BASBOF BOF20/BAS/015 for the powder X-Ray Diffractometer. The authors thank Prof. Rik Van Deun for access to the spectrofluorometer in his lab. The authors thank Katrien Haustraete for STEM-EDX measurements, within the UGent TEM core facility. The authors thank Dr Karen Leus for the XPS measurements.References
- S. S. Park, M. S. Moorthy and C. S. Ha, NPG Asia Mater., 2014, 6, e96 CrossRef CAS.
- T. Asefa, M. J. MacLachlan, N. Coombs and G. A. Ozin, Nature, 1999, 402, 867–871 CrossRef CAS.
- B. J. Melde, B. T. Holland, C. F. Blanford and A. Stein, Chem. Mater., 1999, 11, 3302–3308 CrossRef CAS.
- S. Inagaki, S. Guan, Y. Fukushima, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 1999, 121, 9611–9614 CrossRef CAS.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS PubMed.
- M. Ferré, R. Pleixats, M. Wong Chi Man and X. Cattoën, Green Chem., 2016, 18, 881–922 RSC.
- P. Van der Voort, D. Esquivel, E. De Canck, F. Goethals, I. Van Driessche and F. J. Romero-Salguero, Chem. Soc. Rev., 2013, 42, 3913–3955 RSC.
- Y. Wei, X. Li, R. Zhang, Y. Liu, W. Wang, Y. Ling, A. M. El-Toni and D. Zhao, Sci. Rep., 2016, 6, 20769 CrossRef CAS PubMed.
- A. M. Kaczmarek, S. Abednatanzi, D. Esquivel, C. Krishnaraj, H. S. Jena, G. B. Wang, K. Leus, R. Van Deun, F. J. Romero-Salguero and P. Van der Voort, Microporous Mesoporous Mater., 2020, 291, 109687 CrossRef CAS.
- A. A. Gismatulin, V. A. Gritsenko, D. S. Seregin, K. A. Vorotilov and M. R. Baklanov, Appl. Phys. Lett., 2019, 115, 082904 CrossRef.
- X. Feng, F. Li, L. Zhang, W. Liu, X. Wang, R. Zhu, Z. A. Qiao, B. Yu and X. Yu, Acta Biomater., 2022, 143, 392–405 CrossRef CAS PubMed.
- A. M. Kaczmarek and P. Van der Voort, J. Mater. Chem. C, 2019, 7, 8109–8119 RSC.
- M. A. Bennett, M. J. Byrnes and I. Kovacik, J. Organomet. Chem., 2004, 689, 4463–4474 CrossRef CAS.
- P. Muraoka, D. Byun and J. I. Zink, Coord. Chem. Rev., 2000, 208, 193–211 CrossRef CAS.
- W. H. Wang, T. L. Yung, S. S. Cheng, F. Chen, J. B. Liu, C. H. Leung and D. L. Ma, Sens. Actuators, B, 2020, 321, 128486 CrossRef CAS.
- H. S. Jena, C. Krishnaraj, G. B. Wang, K. Leus, J. Schmidt, N. Chaoui and P. Van der Voort, Chem. Mater., 2018, 30, 4102–4111 CrossRef CAS.
- A. R. Hajipour and G. Azizi, Green Chem., 2013, 15, 1030–1034 RSC.
- A. Hajipour and G. Azizi, Appl. Organomet. Chem., 2015, 29, 247–253 CrossRef CAS.
- P. Borah, X. Ma, K. T. Nguyen and Y. Zhao, Angew. Chem., Int. Ed., 2012, 51, 7756–7761 CrossRef CAS PubMed.
- B. B. Lai, Z. P. Huang, Z. F. Jia, R. X. Bai and Y. L. Gu, Catal. Sci. Technol., 2016, 6, 1810–1820 RSC.
- C. Pereira, A. R. Silva, A. P. Carvalho, J. Pires and C. Freire, J. Mol. Catal. A: Chem., 2008, 283, 5–14 CrossRef CAS.
- S. V. Eliseeva and J. C. Bunzli, Chem. Soc. Rev., 2010, 39, 189–227 RSC.
- C. H. Liu, A. M. Kaczmarek, H. S. Jena, Z. T. Yang, D. Poelman and P. van der Voort, J. Mater. Chem. C, 2023, 11, 5634–5645 RSC.
- Y. Liu, Q.-L. Shi and J.-L. Yuan, Chin. Chem. Lett., 2015, 26, 1485–1489 CrossRef CAS.
- H. Li, Y. Li, Z. Zhang, X. Pang and X. Yu, Mater. Des., 2019, 172, 107712 CrossRef CAS.
- A. M. Kaczmarek and P. Van Der Voort, J. Mater. Chem. C, 2019, 7, 8109–8119 RSC.
- A. M. Kaczmarek, S. Abednatanzi, D. Esquivel, C. Krishnaraj, H. S. Jena, G. Wang, K. Leus, R. Van Deun, F. J. Romero-Salguero and P. Van Der Voort, Microporous Mesoporous Mater., 2020, 291, 109687–109688 CrossRef CAS.
- W. Liu, A. M. Kaczmarek, H. Rijckaert, P. Van Der Voort and R. Van Deun, Dalton Trans., 2021, 50, 11061–11070 RSC.
- W. Liu, A. M. Kaczmarek, P. Van Der Voort and R. Van Deun, Dalton Trans., 2022, 51, 11467–11475 RSC.
- H. Li, Y. J. Li, Z. Zhang, X. L. Pang and X. D. Yu, Mater. Des., 2019, 172, 107712 CrossRef CAS.
- Y. Su, D. Zhang, P. Jia, W. Gao, Y. Li, J. He, C. Wang, X. Zheng, Q. Yang and C. Yang, Eur. Polym. J., 2019, 112, 461–465 CrossRef CAS.
- R. R. Ma, Z. W. Chen, S. N. Wang, Q. X. Yao, Y. W. Li, J. Lu, D. C. Li and J. M. Dou, J. Solid State Chem., 2017, 252, 142–151 CrossRef CAS.
- N. Ding, D. L. Zhou, G. C. Pan, W. Xu, X. Chen, D. Y. Li, X. H. Zhang, J. Y. Zhu, Y. A. Ji and H. W. Song, ACS Sustainable Chem. Eng., 2019, 7, 8397–8404 CrossRef CAS.
- Z. G. Lin, F. Q. Song, H. Wang, X. Q. Song, X. X. Yu and W. S. Liu, Dalton Trans., 2021, 50, 1874–1886 RSC.
- N. Brun, B. Julian-Lopez, P. Hesemann, G. Laurent, H. Deleuze, C. Sanchez, M. F. Achard and R. Backov, Chem. Mater., 2008, 20, 7117–7129 CrossRef CAS.
- K. Binnemans, P. Lenaerts, K. Driesen and C. Görller-Walrand, J. Mater. Chem., 2004, 14, 191–195 RSC.
- L. Carlucci, G. Ciani, S. Maggini, D. M. Proserpio and M. Visconti, Chem.–Eur. J., 2010, 16, 12328–12341 CrossRef CAS PubMed.
- Z. Zhang, H. Chang, Y. F. Kang, X. P. Li, H. Jiang, B. L. Xue, Y. Y. Wang, X. Q. Lü and X. J. Zhu, Sens. Actuators, B, 2019, 282, 999–1007 CrossRef CAS.
- L. Bourda, H. S. Jena, R. Van Deun, A. M. Kaczmarek and P. Van der Voort, J. Mater. Chem. A, 2019, 7, 14060–14069 RSC.
- A. M. Kaczmarek, Y. Maegawa, A. Abalymov, A. G. Skirtach, S. Inagaki and P. Van Der Voort, ACS Appl. Mater. Interfaces, 2020, 12, 13540–13550 CrossRef CAS PubMed.
- G. Chandrasekar, K. S. You, J. W. Ahn and W. S. Ahn, Microporous Mesoporous Mater., 2008, 111, 455–462 CrossRef CAS.
- B. Guan, Y. Cui, Z. Ren, Z. A. Qiao, L. Wang, Y. Liu and Q. Huo, Nanoscale, 2012, 4, 6588–6596 RSC.
- E. Valiey and M. G. Dekamin, Nanoscale Adv., 2022, 4, 294–308 RSC.
- P. Van Der Voort, K. Leus and E. De Canck, Introduction to Porous Materials, John Wiley & Sons, 2019 Search PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- A. M. Kaczmarek, Y. Y. Liu, M. K. Kaczmarek, H. Liu, F. Artizzu, L. D. Carlos and P. Van Der Voort, Angew. Chem., 2020, 132, 1948–1956 CrossRef.
- X. Guo, X. Wang, H. Zhang, L. Fu, H. Guo, J. Yu, L. D. Carlos and K. Yang, Microporous Mesoporous Mater., 2008, 116, 28–35 CrossRef CAS.
- J. Lin, Q. Cheng, J. Zhou, X. Lin, R. C. K. Reddy, T. Yang and G. Zhang, J. Solid State Chem., 2019, 270, 339–345 CrossRef CAS.
- N. Ding, D. L. Zhou, G. C. Pan, W. Xu, X. Chen, D. Y. Li, X. H. Zhang, J. Y. Zhu, Y. A. Ji and H. W. Song, ACS Sustainable Chem. Eng., 2019, 7, 8397–8404 CrossRef CAS.
- T. Kohlmann and M. Goez, Phys. Chem. Chem. Phys., 2019, 21, 10075–10085 RSC.
- L. Li, S. Shen, R. Lin, Y. Bai and H. Liu, Chem. Commun., 2017, 53, 9986–9989 RSC.
- G. Ji, J. Liu, X. Gao, W. Sun, J. Wang, S. Zhao and Z. Liu, J. Mater. Chem. A, 2017, 5, 10200–10205 RSC.
- D. A. Armbruster and T. Pry, Clin. Biochem. Rev., 2008, 29, S49 Search PubMed.
- M. E. Zorn, R. D. Gibbons and W. C. Sonzogni, Anal. Chem., 1997, 69, 3069–3075 CrossRef CAS PubMed.
- E. Mohamed Ali, Y. Zheng, H.-h. Yu and J. Y. Ying, Anal. Chem., 2007, 79, 9452–9458 CrossRef PubMed.
- U. Latief, S. Ul Islam, Z. Khan and M. S. Khan, J. Alloys Compd., 2022, 910, 164889 CrossRef CAS.
- H. Li, Y. Li, Z. Zhang, X. Pang and X. Yu, Mater. Des., 2019, 172, 107712 CrossRef CAS.
- T. Chopra, S. Sasan, L. Devi, R. Parkesh and K. K. Kapoor, Coord. Chem. Rev., 2022, 470, 214704 CrossRef CAS.
- X. G. Liu, D. L. Huang, C. Lai, G. M. Zeng, L. Qin, H. Wang, H. Yi, B. S. Li, S. Y. Liu, M. M. Zhang, R. Deng, Y. K. Fu, L. Li, W. J. Xue and S. Chen, Chem. Soc. Rev., 2019, 48, 5266–5302 RSC.
- Y. Peng, N. Pan, D. Wang, J. Yang, Z. Guo and W. Yuan, J. Mater. Sci., 2018, 53, 12432–12440 CrossRef CAS.
- P. Borah, X. Ma, K. T. Nguyen and Y. Zhao, Angew Chem. Int. Ed. Engl., 2012, 51, 7756–7761 CrossRef CAS PubMed.
- H. Vardhan, L. X. Hou, E. Yee, A. Nafady, M. A. Al-Abdrabalnabi, A. M. Al-Enizi, Y. X. Pan, Z. Y. Yang and S. Q. Ma, ACS Sustainable Chem. Eng., 2019, 7, 4878–4888 CrossRef CAS.
- D. R. Hwang and B. J. Uang, Org. Lett., 2002, 4, 463–466 CrossRef CAS PubMed.
- X. Chen, N. Huang, J. Gao, H. Xu, F. Xu and D. Jiang, Chem. Commun., 2014, 50, 6161–6163 RSC.
- P. P. Reddy, C. Y. Chu, D. R. Hwang, S. K. Wang and B. J. Uang, Coord. Chem. Rev., 2003, 237, 257–269 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta01143k |
| This journal is © The Royal Society of Chemistry 2024 |