
Manganese oxide-based mesoporous thin-film electrodes: manganese disproportionation reaction in alkaline media†‡
Irmak Karakaya
Durukan
a,
Işıl
Ulu
 a and
Ömer
Dag
*ab
a and
Ömer
Dag
*ab
aDepartment of Chemistry, Bilkent University, 06800, Ankara, Turkey. E-mail: dag@fen.bilkent.edu.tr
bUNAM—National Nanotechnology Research Center, Institute of Materials Science and Nanotechnology, Bilkent University, 06800, Ankara, Turkey
First published on 1st February 2024
Abstract
In this study, we explore the disproportionation reaction mechanism in alkaline media during the oxygen evolution reaction (OER) utilizing mesoporous electrodes, namely LiMn2O4 (m-LMO), Mn3O4 (m-Mn3O4), and Mn2P2O7 (m-MnPP). The electrodes are crafted through the molten salt-assisted self-assembly (MASA) process. The procedure commences with the application of a metal salt(s)–surfactant and metal salt–pyrophosphoric acid–surfactant (where the surfactant is P123) lyotropic liquid crystalline mesophase coating over a FTO surface by employing a clear ethanol or aqueous solution of the ingredients, followed by a subsequent calcination step at 300 °C. The electrodes are characterized by spectroscopic, diffraction, imaging, and electrochemical techniques. At low electrochemical potentials, Mn(III), and at more positive potentials, Mn(VI) disproportionation reactions make these materials highly unstable in alkaline media. The aforementioned degradation processes have been investigated by examination of the electrodes both prior to and after subsequent use in electrochemical measurements in various electrolytes. We found that the degradation process is relatively slow in m-LMO, but elevated in m-Mn3O4 and m-MnPP electrodes. m-LMO is fully converted into the λ-MnO2 phase upon its oxidation and more robust to decomposition; making it ultra-thin further improves its robustness. However, the m-Mn3O4 and m-MnPP electrodes behave similarly to each other and degrade more quickly (more pronounced in the latter), by releasing purple-colored permanganate ions into the electrolyte media. A Mn(VI) disproportionation reaction mechanism is suggested using the experimentally gathered spectroscopic, diffraction, and electrochemical data. The formation of Mn(VI) surface species and their electronegativity play vital roles in the disproportionation reaction.
1. Introduction
Clean and renewable energy production and storage have become one of the most important issues of the modern world and need to be addressed in the near future. One of these energy sources is hydrogen and its production from water splitting by electrochemical or photochemical methods are feasible paths. The electrochemical path is limited by the oxygen evaluation reaction (OER) and needs to be explored towards the OER mechanism and stable electrodes. RuO2 and IrO2 are considered to be two highly efficient and stable electrocatalysts for the water oxidation reaction. However, these metals are less abundant in the earth-crust, costly, and not promising electrocatalysts in global scale applications.1 Therefore, first-row transition metal oxides and alloys are considered alternatives to overcome the high cost and low abundance drawbacks of noble metals in OER catalysis.2–4 Among them, manganese is significantly distinguished due to its high earth-abundancy, high thermal stability, low cost, and less toxicity, bearing several oxidation states from (II) to (VII) in various forms of oxides, and provides unique catalytic sites for electrochemical processes.5,6Manganese oxides are commonly used in various types of applications such as batteries, supercapacitors, and catalytic oxidation and reduction reactions.7–10 In these applications, degradation issues of MnxOy electrodes arise due to the disproportionation reaction of Mn(III) and Mn(VI) species. The manganese-based electrodes suffer from the Mn(III) disproportionation reaction in battery and energy storage applications. Two Mn(III) sites undergo a disproportionation reaction to produce Mn(II) and Mn(IV) species. The Mn(II) sites dissolve into electrolyte media, while the Mn(IV) species re-deposit on the electrode surface as MnO2 in different polymorphs.11–13 Therefore, the Mn(II) formation reduces the active manganese amount on the cathode surface and causes electrode etching. In an efficient OER, manganese must be in a high oxidation state, such as Mn(VI) or Mn(VII) to activate the manganese oxo (Mn![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) bonds. The oxo-oxygen becomes a good electrophile when the manganese is in a such high oxidation state.14–17 However, a similar disproportionation reaction occurs among three Mn(VI) sites to produce two Mn(VII) (as permanganate sites or ions) and one Mn(IV) (as manganese dioxide) species.17,18 The Mn(VII) species are mainly responsible for the degradation of the active electrode material by producing ionic permanganate (MnO4−) ions. There are many studies demonstrating such manganese loss due to the dispersion of purple-colored permanganate ions into the electrolyte solution during OER electrocatalysis.19,20 Lithium manganese oxides might stand as an efficient and sustainable OER electrocatalyst (due to its unique structure) upon the de-lithiation process. Notice that spinel LiMn2O4 (LMO) is commonly employed in lithium-ion batteries and many studies showed that the lithium ions occupy the tetrahedral sites and its electrochemical removal yields a stable spinel λ-MnO2 at high potentials.21,22 In Li-ion batteries, the Mn(III) disproportionation causes a capacity decay in the active material due to soluble Mn(II) sites.23–25 Lithiation and de-lithiation reactions in spinel LMO have also been shown in aqueous lithium salt electrolyte solutions for energy storage applications such as supercapacitors and hybrid capacitors. Note also that the aqueous electrolytes reduce the fading rates of LMOs in charge–discharge measurements.26–28
O) bonds. The oxo-oxygen becomes a good electrophile when the manganese is in a such high oxidation state.14–17 However, a similar disproportionation reaction occurs among three Mn(VI) sites to produce two Mn(VII) (as permanganate sites or ions) and one Mn(IV) (as manganese dioxide) species.17,18 The Mn(VII) species are mainly responsible for the degradation of the active electrode material by producing ionic permanganate (MnO4−) ions. There are many studies demonstrating such manganese loss due to the dispersion of purple-colored permanganate ions into the electrolyte solution during OER electrocatalysis.19,20 Lithium manganese oxides might stand as an efficient and sustainable OER electrocatalyst (due to its unique structure) upon the de-lithiation process. Notice that spinel LiMn2O4 (LMO) is commonly employed in lithium-ion batteries and many studies showed that the lithium ions occupy the tetrahedral sites and its electrochemical removal yields a stable spinel λ-MnO2 at high potentials.21,22 In Li-ion batteries, the Mn(III) disproportionation causes a capacity decay in the active material due to soluble Mn(II) sites.23–25 Lithiation and de-lithiation reactions in spinel LMO have also been shown in aqueous lithium salt electrolyte solutions for energy storage applications such as supercapacitors and hybrid capacitors. Note also that the aqueous electrolytes reduce the fading rates of LMOs in charge–discharge measurements.26–28
λ-MnO2 and a couple of manganese oxide polymorphs have been shown as efficient OER catalysts.29 There are also a few studies that employ LMO as a water oxidation catalyst.30–32 However, the stability of λ-MnO2 electrodes has not been investigated and compared for stable OER electrocatalysis; there is no comprehensive investigation to demonstrate the correlation between the stabilities and Mn(VI) disproportionation rates in this polymorph. This correlation might be investigated by fabricating mesoporous LMO (m-LMO) electrodes with a large surface area, providing an extensive interface between the material surface and electrolyte and therefore ideal materials to investigate the above issues. Because of great interest in LiMn2O4 as a potential battery material, the mesoporous LMO has been synthesized and investigated by many groups using different synthetic approaches, including hard- and soft-templating.32–36 The hard templating method produces ordered structures in powder forms that need to be further processed to an electrode configuration. However, the soft-templating approach is very practical and uses polymers37 or surfactants in liquid crystalline phases (specifically, molten salt-assisted self-assembly, MASA) to fabricate the electrodes over a desired substrate.16,32,38,39
Here, we have fabricated mesoporous LiMn2O4 thin films by employing the MASA method to control the film- and pore wall-thickness by spin-coating and dip-coating methods. Coating of the thin films was done at various spin rates in the former method and at various dilution factors for dip-coating to adjust the thicknesses of the films for a stable OER electrode. Further annealing is proceeded on these films to comprehend the pore-wall thickness, surface area, pore size, and crystallinity effect on OER catalysis. The m-LMO electrode, resulting in the best performance and long-term stability, was also compared with the electrochemical behaviors of mesoporous Mn3O4 (m-Mn3O4) and Mn2P2O7 (m-MnPP) thin film electrodes, fabricated using the same MASA method. The Mn(VI) disproportionation mechanisms at the electrode interfaces and in electrolytes have been proposed and attributed to the instabilities of the thin films in OER electrocatalysis.
2. Results and discussion
2.1. Fabrication and characterization of m-LMO thin and ultra-thin films
The stock solution, described in the Experimental section, was coated on glass substrates at various spin rates such as 2000, 3000, 5000, 7000, and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm. This ensures a rapid evaporation of the volatile solvent (namely ethanol), leaving salts and surfactant in an ordered lyotropic liquid crystalline (LLC) mesophase. The coated sample diffracts at small angles (1–5°, 2θ), indicating the formation of an orientated thin film of a gel-like mesophase, see Fig. S1.‡ This is an important step to obtain mesoporous materials with uniform pores after the calcination of the gel films.32,38 The LLC thin films on a glass substrate and FTO were calcined at 300 °C for 2 and 1 h, respectively. Then the coated microscope glass slides were scraped to collect the powder sample (denoted as m-LMO-300) and used to record its XRD pattern, see Fig. S2.‡ The diffraction lines are indexed to a cubic-spinel LMO (ICDD card no. 00-054-0252). The most intense line at 18.7°, 2θ, due to the (111) plane provides a unit cell parameter a of 8.21 Å. Fig. S2‡ also displays the XRD pattern of the bared FTO and FTO coated samples (namely m-LMO-300-F2, where F2 stands for FTO coated at 2000 rpm and 300 is the calcination temperature in Celsius). The diffraction pattern of the m-LMO-300-F2 film was directly recorded over the FTO surface and displays additional sharp diffraction lines due to the FTO substrate, see Fig. S2.‡ Further analysis has been carried out using the most intense diffraction line (at 18.7°, 2θ) of the m-LMO-300-F2 film to determine the spin rate-dependent film thickness. Fig. 1a displays the XRD patterns of the m-LMO films, fabricated by spin coating between 2000 and 10000 rpm. The intensity of the diffraction line decreases with the increasing spin-rate and it is attributed to the film thickness. Furthermore, scanning electron microscope (SEM) cross-sectional images of the films were also collected (see Fig. S3‡) to check the film thickness. The film thickness was obtained by averaging the thickness at different parts in the SEM images of the m-LMO-300-F# films, and then the mean thickness values were plotted with respect to the spin-rates, see Fig. 1b. It is evident from both plots that the film thickness almost linearly follows the spin-rate. The plot of X-ray intensity versus spin-rate also shows that the X-ray diffraction is collected from the whole LMO film without any thickness problem. The diffraction line was further analyzed using Scherrer's equation to evaluate the LMO crystallite particle size on FTO surfaces and found that the crystalline domains are almost 18.5 nm and independent of the spin-rate. The films display similar particle-like morphology, building into a mesoporous film at all spin-rates, see Fig. S4.‡
000 rpm. This ensures a rapid evaporation of the volatile solvent (namely ethanol), leaving salts and surfactant in an ordered lyotropic liquid crystalline (LLC) mesophase. The coated sample diffracts at small angles (1–5°, 2θ), indicating the formation of an orientated thin film of a gel-like mesophase, see Fig. S1.‡ This is an important step to obtain mesoporous materials with uniform pores after the calcination of the gel films.32,38 The LLC thin films on a glass substrate and FTO were calcined at 300 °C for 2 and 1 h, respectively. Then the coated microscope glass slides were scraped to collect the powder sample (denoted as m-LMO-300) and used to record its XRD pattern, see Fig. S2.‡ The diffraction lines are indexed to a cubic-spinel LMO (ICDD card no. 00-054-0252). The most intense line at 18.7°, 2θ, due to the (111) plane provides a unit cell parameter a of 8.21 Å. Fig. S2‡ also displays the XRD pattern of the bared FTO and FTO coated samples (namely m-LMO-300-F2, where F2 stands for FTO coated at 2000 rpm and 300 is the calcination temperature in Celsius). The diffraction pattern of the m-LMO-300-F2 film was directly recorded over the FTO surface and displays additional sharp diffraction lines due to the FTO substrate, see Fig. S2.‡ Further analysis has been carried out using the most intense diffraction line (at 18.7°, 2θ) of the m-LMO-300-F2 film to determine the spin rate-dependent film thickness. Fig. 1a displays the XRD patterns of the m-LMO films, fabricated by spin coating between 2000 and 10000 rpm. The intensity of the diffraction line decreases with the increasing spin-rate and it is attributed to the film thickness. Furthermore, scanning electron microscope (SEM) cross-sectional images of the films were also collected (see Fig. S3‡) to check the film thickness. The film thickness was obtained by averaging the thickness at different parts in the SEM images of the m-LMO-300-F# films, and then the mean thickness values were plotted with respect to the spin-rates, see Fig. 1b. It is evident from both plots that the film thickness almost linearly follows the spin-rate. The plot of X-ray intensity versus spin-rate also shows that the X-ray diffraction is collected from the whole LMO film without any thickness problem. The diffraction line was further analyzed using Scherrer's equation to evaluate the LMO crystallite particle size on FTO surfaces and found that the crystalline domains are almost 18.5 nm and independent of the spin-rate. The films display similar particle-like morphology, building into a mesoporous film at all spin-rates, see Fig. S4.‡
 | ||
| Fig. 1 (a) The XRD patterns of m-LMO-300-F# films between 17.5 and 20.0°, 2θ, coated at (I) 2000, (II) 3000, (III) 5000, (IV) 7000 and (V) 10000 rpm, (b) the plots of the film thickness and X-ray intensity of the m-LMO-300-F# films versus spin-rate (black dots from SEM images and red squares are (111) diffraction line intensity values at 18.7°, 2θ), and (c) the SEM images (top view) of the thin films at indicated spin-rates. | ||
Fig. 1c shows the top view of the SEM images of all the m-LMO-300-F# films. The m-LMO-300-F# films, fabricated at lower spin-rates such as 2000 and 3000 rpm, display crack like features over the films, and also be correlated with a rougher surface, observed in the cross-sectional images in Fig. S3a and b.‡ Further increase in the spin-rate produces a smoother film morphology up to 6000 rpm, see Fig. 1c. From 7000 to 10000 rpm, the films start to mimic bare FTO morphology rather than smoother film surfaces. A TEM image of a typical m-LMO-300 sample and more SEM images of the m-LMO-300-F# films are shown in Fig. S4.‡ The TEM image displays highly crystalline nanoparticles even at 300 °C, see Fig. S4a.‡ The morphologies at all spin-rates are very similar and particle-like LMOs are accumulated over each other to form a film morphology, inducing mesoporosity.
Thicker films were also synthesized by the drop-casting method followed by calcination at 300 °C and annealing at 350, 400, 450, and 500 °C for 1 hour to analyze the thermal properties of the m-LMOs. Fig. S5‡ displays the temperature-dependent XRD patterns of these samples. The pattern can be indexed using the ICDD card no. 00-054-0252 to spinel LiMn2O4. Notice that the diffraction lines become slightly sharper with increasing the annealing temperature and attributed to the growth of crystalline domains (pore-walls) from 14.6 to 18.4 nm (calculated using the FWHM of the (111) diffraction line and Scherrer equation) by increasing annealing temperatures from 300 to 500 °C. The Butler–Emmett–Teller (BET) surface area and Barrett–Joyner–Halenda (BJH) pore size distribution (from N2 adsorption–desorption isotherms) of the thick m-LMO films have been previously reported as 98, 90, 69, and 33 m2 g−1 (surface area) and 11.1, 10.8, 12.9, and 22.5 nm (average pore size) at annealing temperatures of 300, 400, 500 and 600 °C, respectively, see Fig. S6.‡32
Thin m-LMO-F2 and m-LMO-F5 films (annealed at different temperatures) were also characterized by recording their XRD patterns between 15 and 30°, 2θ, directly from the FTO surface, see Fig. S7a and b,‡ respectively. Notice that the FWHM of the diffraction lines of m-LMO-F2 almost remains the same with increasing calcination temperatures. Similarly, the thinner m-LMO-F5 sample also shows a very small alteration in the FWHM, indicating that there is almost no effect of annealing temperature between 300 and 500 °C on the crystallite size and diffraction lines that is consistent with the thermal stability of manganese-based materials. The SEM images of the thinner m-LMO-X-F5 films are also given in Fig. S8‡ at annealing temperatures from 300 to 600 °C. All images display a similar particle-like morphology with a similar particle-size and accord well with the XRD results.
Ultra-thin m-LMO-300-G films (G stands for a graphite rod, a graphite rod coated with m-LMO) were prepared by dip-coating of diluted stock solution and calcination at 300 °C. Then, the m-LMO-300-G#, where # is 5, 10, 20, 50, and 100 (number of dilutions of the stock solution) films were only characterized by SEM, see Fig. S9.‡ Notice that m-LMO-300-G5 and m-LMO-300-G10 have film formations on graphite surfaces with some large spherical particles. The large particles disappear on the graphite rod with 20 times dilution; instead much smaller particles are observed on the surface of the entire graphite rod. It is more challenging to identify the film and its thickness over the graphite substrates. Therefore, further characterization has been carried out using the electrochemical methods, see latter.
2.2. Electrochemical characterization of the m-LMO-X-F# and m-LMO-X-G# electrodes and inspection of the Mn(III) and Mn(VI) disproportionation reactions
The m-LMO thin films were electrochemically investigated in several electrolytes at various pH values in order to investigate both the Mn(III) and Mn(VI) disproportionation reactions. First, cyclic voltammograms (CVs) of the m-LMO-300-F2 electrode were recorded in neutral electrolytes (pH of ∼6.6), such as in 1.0 M LiNO3 and 1.0 M KNO3 aqueous solutions in a −0.1 to 1.9 V vs. RHE potential window with a 5 mV s−1 scan-rate. Fig. 2a and b display the 5 CV curves of the m-LMO-300-F2 electrode in each electrolyte solution. Notice that the voltammogram of the film in the LiNO3 solution displays reversible distinct Mn(III)/Mn(VI) redox peaks in a 1.1 and 1.5 V potential window, because the lithium ion in the LiNO3 electrolyte supports the lithium-ion intercalation/de-intercalation processes, see Fig. 2a. This is a typical behavior of a LiMn2O4 electrode and has been investigated by many groups under the topics of batteries and energy storage materials.22 The reduction peak between 0.2 and 0.8 V is due to an electrochemical lithium insertion into LiMn2O4 by reducing the other Mn(IV) to Mn(III), leading to the formation of Li2Mn2O4, see later. Fig. 2b displays the CV curves of the m-LMO-300-F2 electrode in 1.0 M KNO3 electrolyte solution. Two oxidation peaks due to the de-lithiation process (between 1.1 and 1.5 V) in the 1st cycle gradually converged into a single oxidation peak at 1.4 V by further cycling. This new redox couple between 0.8 and 1.7 V might be attributed to the Mn(III)/Mn(IV) redox couple due to partial de-intercalation and intercalation of K+ ions. This assumption has been supported by X-ray photoelectron spectroscopy (XPS) survey analysis. Fig. S10‡ displays the XPS survey spectra of the m-LMO-300-F2 electrode before and after 3 CV cycles. Potentials were reported as (rev)x V when an electrode is swept in the reverse direction, from 1.9 V to x V, in the 3rd cycle and the spectrum at (rev)0.8 V was compared with the spectrum of a fresh m-LMO-300-F2 electrode. In the XPS survey spectrum, the peak at 293 eV is attributed to potassium ions and proves that the K+ ions are kept in the structure after a thorough washing procedure. m-LMO-300-F2 has 3.9% potassium and 96.1% manganese content at (rev)0.8 V, which correlates with the broad reduction peak at 1.2 V and is responsible for the K+ insertion into the MnO2 structure, likely close to surface sites. Also, a sharp reduction peak at 0.35 V might be attributed to the oxygen reduction reaction (ORR).40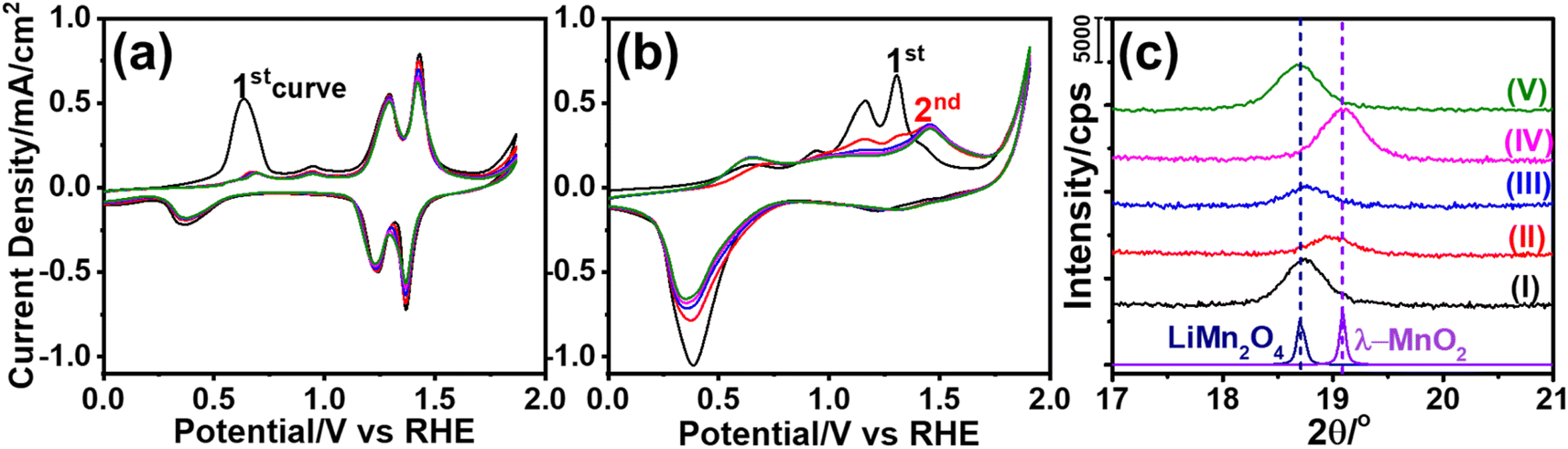 | ||
| Fig. 2 The CV curves of the m-LMO-300-F2 electrode with a 5 mV s−1 sweep rate in neutral electrolytes of (a) 1.0 M LiNO3 and (b) 1.0 M KNO3. (c) The XRD patterns of the m-LMO-300-F2 electrode before and after electrochemical measurements: (I) before cycling, (II) after sweeping from −0.1 to 1.7 V in the 3rd cycle in 1.0 M KNO3, (III) after reverse sweeping from 1.9 to 0.8 V in the 3rd cycle in 1.0 M KNO3, (IV) after sweeping from −0.1 and 1.7 V of the 1st cycle in 1.0 M LiNO3, and (V) after reverse sweeping from 1.9 to 0.8 V of the 1st cycle in 1.0 M LiNO3 aqueous solution (bottom patterns are from the LiMn2O4 and λ-MnO2 references in ICDD cards, 00-054-0252 and 00-042-1169, respectively). | ||
Another m-LMO-300-F2 electrode was used for CV cycles in a 1.0 M KNO3 solution and the CV measurement was stopped at 1.7 V in the forward direction in the 3rd cycle, removed and washed several times and then its XRD pattern was recorded, see Fig. 2c. The diffraction line at 18.7° shifts to 19.0°, 2θ, upon oxidation, see Fig. 2c. It is slightly shifted to a lower angle than the λ-MnO2 (111) line, which is observed at 19.1°, 2θ. This behavior might be attributed to a small number of inserted potassium ions in the reverse cycles or not a full de-intercalation of the Li+ ions in the 1st and 2nd cycles. The XPS spectrum in Fig. S10‡ confirms the above prediction that the potassium and manganese percentiles are 3.9 and 96.1%, respectively, in the used electrode. However, when the electrode is swept back to 0.8 V from 1.9 V in the 3rd cycle, the (111) diffraction line shifts back to 18.7°, 2θ, corresponding to the (111) line of LiMn2O4 crystallites with a reduced intensity, see Fig. 2c. Note also that the same diffraction line appears at 19.1°, 2θ, with a similar intensity upon sweeping to 1.7 V in the 1.0 M LiNO3 electrolyte solution, indicating that the electrode is almost fully converted into λ-MnO2 in the forward cycle, see Fig. 2c. When the electrode is reversed and swept from 1.9 to 0.8 V, the Li+ ions are intercalated back into the electrode to form the LiMn2O4 phase causing the diffraction line to shift back to 18.7°, 2θ, with its original intensity.
The dominant oxidation peak at 0.6 V in the 1st cycle in LiNO3 electrolyte is due to de-intercalation of the first lithium ion from Li2Mn2O4 that is formed by Li+ ion intercalation into LiMn2O4 at starting potentials, see Fig. 2a.41 However, this faradaic peak disappears in the 2nd and further cycles. In its place, two weak oxidation peaks are observed in further cycles. The m-LMO-300-F2 electrode was removed from the 1.0 M LiNO3 electrolyte solution at certain potentials during the 1st CV cycle and its XPS spectra were recorded. Fig. S11a‡ displays a set of O 1s XPS spectra of the electrode that was removed at various potentials from the electrolyte solution during forward and reverse cycles. The main O 1s peak at 529.8 eV originates from the lattice oxygen. A small shoulder is visible at 531.1 eV at 0.1, 0.8 (forward cycle), and 0.1 V (reverse cycle from 1.9 to 0.1 V) due to surface hydroxides and water, see Fig. S10a.‡ Notice also that the high-energy shoulder disappears at higher potentials (above 1.7 V). Fig. S11b‡ displays the Mn 2p spectra of the same samples. The Mn(III) and Mn(IV) peaks appear at 642.2 and 643.3 eV, respectively. Small amounts of surface Mn(II) species are also detected and observed as a shoulder at 641.1 eV. Also notice that the Mn(II) 2p region amplifies at 0.1 and 0.8 V, providing strong evidence for the formation of Mn(OH)2 on the electrode surface. This was also correlated with an enhancement of the shoulder at 531.1 eV (assigned to hydroxide oxygens) in the O 1s spectra. Fig. S11c‡ displays the XPS spectra in the Mn 3s region. The Mn 3s region displays two distinct peaks due to coupling of the 3s electron with the valence electrons, causing a multiplet splitting.6,42 The splitting energy is sensitive to the oxidation state of the Mn species.6,42 This region also confirms that the manganese oxidation state decreases at 0.8 V with a splitting energy of 5.2 eV, and it becomes 4.7 eV upon oxidation of Mn3+ to Mn4+. The formation of the Mn(OH)2 species could be due to the Mn(III) disproportionation reaction at these potentials. Two Mn(III) species disproportionate into Mn(II) and Mn(IV) species. The Mn(II) spontaneously redeposits as Mn(OH)2 in alkaline media on the electrode surface. Furthermore, when the electrode is swept to 1.7 V (vs. RHE), the Mn 3s spectrum displayed the Mn 3s peaks with a splitting energy of 4.4 eV. This is also a strong indication for the formation of Mn(IV) species upon complete de-lithiation at 1.7 V, see Fig. S11c.‡42 Then the electrode was reversed from 1.9 V and swept to 0.1 V. The Mn(II) feature re-appears at (rev)0.1 V in the XPS Mn 2p and 3s regions and indicates that the Mn(III) disproportionation occurs at this potential.
The m-LMO-300-F2 electrodes were further investigated to shine some light on the Mn(III) disproportionation reaction by cycling the electrodes in a non-aqueous electrolyte. Multiple CVs were recorded using a 1.0 M LiClO4 acetonitrile solution as the electrolyte to follow the lithiation and de-lithiation processes that can be used to monitor manganese degradation by a Mn(III) disproportionation reaction. Fig. 3a displays a set of CV curves, recorded using an m-LMO-300-F2 electrode between −0.5 and 1.5 V vs. Ag/Ag+ with a 10 mV s−1 scan-rate in 1.0 M LiClO4 acetonitrile solution. The current density quickly vanishes (in 15 CV cycles) because of the Mn(III) disproportionation. The Mn(II) disproportionation product dissociates into the electrolyte solution during CV cycling. When, the electrode is cycled between 0.75 and 1.5 V vs. Ag/Ag+ (see Fig. S12‡), the charge densities at redox peaks of Mn(III)/Mn(IV) are almost identical and show that there is no Mn(III) disproportionation in this potential window. However, the Mn(III) disproportionation takes place below −0.1 V vs. Ag/Ag+ in the acetonitrile solution. This behavior was investigated by cycling the electrode between x and 0.3 V, where x is −0.6, −0.5, −0.4, and −0.3 V, see Fig. S13a–d,‡ respectively. The charge density of the oxidation peak at 0 V was plotted against time spent in the cycling process and the plots were fitted to an exponential function. Notice that the decay of charge density follows almost a linear behavior when the electrode is cycled between −0.6 and 0.3 V and becomes more exponential by reducing the potential window to between −0.3 and 0.3 V. This could be due to spending more time below −0.1 V and leading to a more Mn(III) disproportionation reaction.
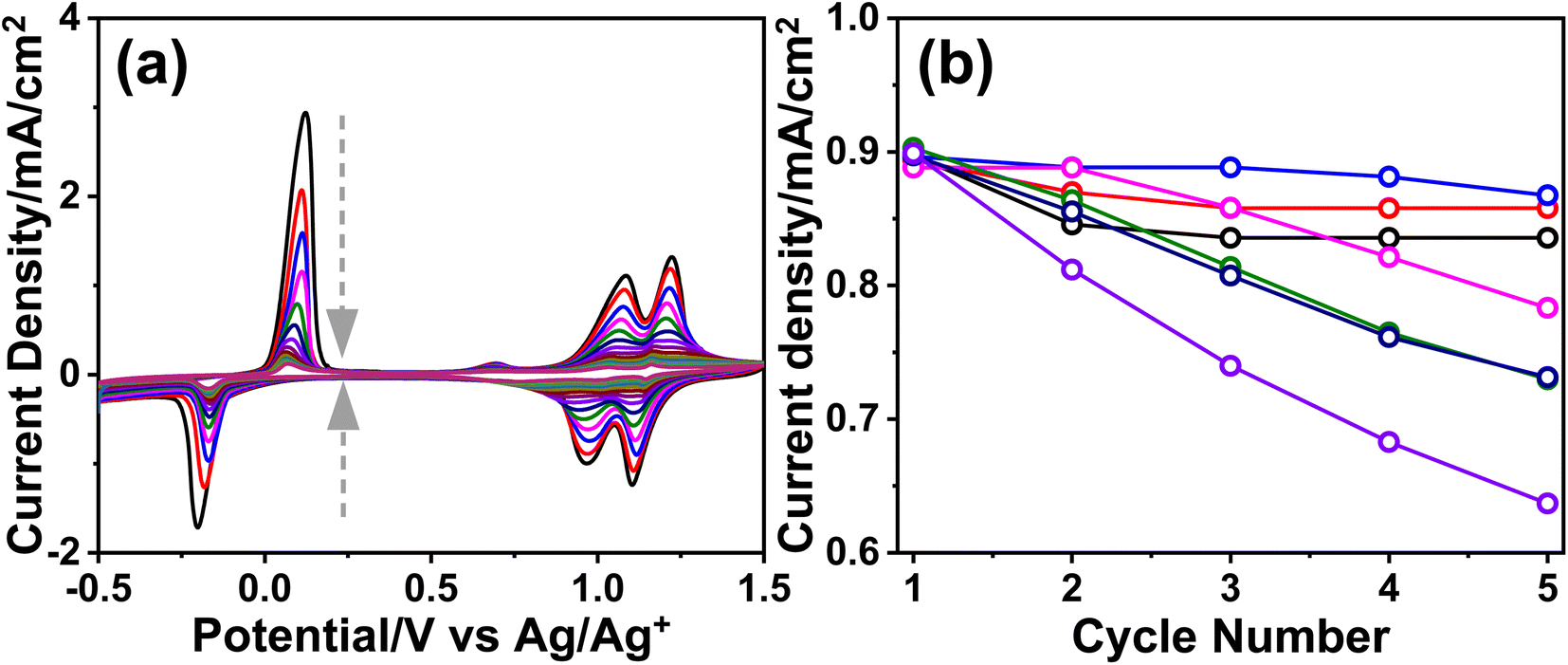 | ||
| Fig. 3 (a) The CV curves of the m-LMO-300-F2 electrode in 1.0 M LiClO4 acetonitrile solution (arrows represent the cycle order) and (b) the plot of the current density at 1.4 V of the m-LMO-300-F2 electrode versus cycle numbers in 1.0 M LiNO3 aqueous solution with respect to the starting potential of a forward sweep in the CV curves, where black: 0.75 V, red: 0.45 V, blue: 0.15 V, pink: 0.05 V, green: −0.05 V, dark blue: −0.15 V, and purple: −0.2 V. | ||
The Mn(III) disproportionation reaction was also investigated electrochemically in a 1.0 M LiNO3 aqueous electrolyte solution. Here, the starting potential of the CV curves was varied systematically from 0.75 to −0.2 V vs. RHE to understand at which potential the electrode undergoes Mn(III) disproportionation and degradation, see all the CV curves in various potential windows in Fig. S14.‡ Then, the current density of the oxidation peak at 1.4 V is plotted to show how the active manganese species decrease by pushing the CV starting potential to more negative values. Fig. 3b shows the current density variation in 5 cycles with respect to CV starting potentials. Notice that when the CV curve was recorded between 0.75 and 1.9 V and 0.15 and 1.9 V (vs. RHE), the degradation of the manganese species was slow. The ICP-MS analysis of the electrolytes was carried out to quantify the degradation of the electrodes.
The LiNO3 electrolyte solution (used in the CV experiment) contains 76 ppb manganese, corresponding to 1.74 μg Mn in 20 mL electrolyte solution, after 3 cycles between 0.75 and 1.9 V. However, if the electrode is used by sweeping from 0.05 to 1.9 V, a vigorous Mn(III) disproportionation reaction occurs, as evidenced by a fast current drop at 1.4 V, see Fig. 3b and S14d.‡ Shifting the starting potentials to a more negative potential further enhances the Mn(III) disproportionation reaction and the manganese amount after the 3rd CV cycle between −0.15 and 1.9 V is 562 ppb (corresponding to 12.85 μg Mn in 20 mL electrolyte solution).
The Mn(VI) disproportionation reaction was also investigated in neutral electrolyte solution by employing a linear sweep voltammetry (LSV) experiment and surface analysis of the electrode using the XPS technique. The LSV was recorded between −0.05 and 3.4 V, see Fig. 4a. The XPS spectra in the O 1s, Mn 2p and 3s have been collected upon exposing the electrode to various potentials over the lithium de-intercalation process. Notice that the MnO2 features were observed in the O 1s, Mn 2p, and 3s spectra at 1.6 V, see Fig. 4b–d, respectively. The color of the m-LMO electrode is dark green and gets lighter upon sweeping the electrode at low potentials from 0.5 to −0.05 V. This is attributed to the formation of the Li2Mn2O4 phase. Over the first de-lithiation peak at 0.7 V, the electrode's color changes to its original dark-green. The color of the electrode becomes orange, when it is swept to 1.6 V, corresponding to an almost complete conversion of LiMn2O4 to λ-MnO2, see Fig. 4a. Further increase in the potential to 1.8 V enhances the shoulder at 532 eV in the O 1s XPS spectrum, which might be attributed to surface manganese–oxo (MnO, oxo-bond) bond formation that is responsible for the OER catalysis.15,16 Then, this shoulder decreased by a further increase in the potential to 2.0 V, at which the OER takes place on the active MnO sites and simultaneously consumes these species, see later. The MnO2 features are preserved at these potentials as evidenced by the Mn 2p and 3s spectra (see Fig. 4c and d) and the color of the electrode (still orange, see insets in Fig. 4a). Fig. 4e displays the changes in the ATR-FTIR spectra during the above electrochemical processes. A weak peak at 906 cm−1, due to the MnO double bond,14 appears in the spectra by increasing the oxidation potential, see Fig. 4e, indicating the formation of surface MnO species. Further oxidation of the manganese to a 6+ and/or 7+ oxidation state on the MnO sites makes the oxo-oxygen highly electrophilic and becomes accessible to a nucleophilic attack of a hydroxide ion to form Mn–O–O–H bonds. This step is also the slow and rate-determining step of the OER15,16,39 and is highly important to detect.14
 | ||
| Fig. 4 (a) The LSV curve of the m-LMO-300-F2 electrode between −0.05 and 3.4 V in 1.0 M LiNO3 with a 5 mV s−1 scan rate (insets: color of the electrodes at indicated potentials) and the XPS spectra of m-LMO-300-F2 at various potentials in the (b) O 1s, (c) Mn 2p, and (d) Mn 3s regions, where black: 1.6 V, red: 1.7 V, blue: 1.8 V, pink: 1.9 V, and green: 2.0 V. (e) The FTIR spectra of m-LMO-300-F2 at various potentials. | ||
The O 1s and Mn 2p and 3s XPS spectra of the m-LMO-300-F2 electrode show typical MnO2 features, when the final potential is pushed up to 3.4 V, see Fig. S15a–c,‡ respectively. However, the color over the electrode surface is dark purple over 2.5 V, indicating the formation of permanganate species over the electrode surface. Combining this observation with the spectra provides strong evidence for a fast consumption of active manganese species during the OER through a Mn(VI) disproportionation reaction into stable MnO2 on the FTO surface and a release of permanganate ions into the electrolyte solution. Notice that a dominant purple color of the permanganate (MnO4−) ion is observed on the FTO surface with oxygen bubbles at 2.5 V, see photos embedded in Fig. 4a and Video S1.‡ The oxidation peak, which is irreversible at 2.2 V, might be responsible for the MnO4− formation through the electrochemical oxidation of manganese species and/or disproportionation reaction of the surface manganates (MnO42−) into MnO2 and MnO4− ions. The appearance of an irreversible peak at 2.2 V also proves that the oxidation product is consumed likely either by the Mn(VI) disproportionation reaction and/or OER. Notice that the Mn(VI) disproportionation reaction still occurs at more positive potentials, including OER potentials. Here, the CV cycling has been initiated at 0.75 V to prevent the Mn(III) disproportionation reaction. Decay of the charge density in the redox peaks (between 1.0 and 1.6 V) is enhanced by further cycling, when the electrode is swept to a more positive potential, see Fig. S16.‡ Moreover, the m-LMO-300-F2 electrode was cycled 3 times in 1.0 M LiNO3 solution in two potential ranges (namely between 0.75 and 1.7 V and 0.75 and 2.8 V) to quantify the manganese amount, dispersed into the electrolyte solution, by ICP-MS. The manganese, in the electrolyte, was detected to be 76 ppb (1.74 μg Mn) when the electrode was cycled between 0.75 and 1.7 V, see Fig. S17a‡ and 1163 ppb (26.58 μg Mn) upon cycling between 0.75 and 2.8 V in 20 mL of 1.0 M LiNO3 electrolyte solution, see Fig. S17b.‡
Notice that the reverse sweeping from 2.8 to 0.75 V gives a reduction peak at 1.4 V (see inset CVs in Fig. S17b‡) that might be attributed to the reduction of the MnO2 surface sites to MnOOH. The m-LMO-300-F2 electrode was also used in CV cycling in 1.0 M KNO3 electrolyte in a wide potential window, see Fig. S17c.‡ According to the ICP-MS data, the manganese species amount, which is dispersed into the aqueous electrolyte solution, is 2738 ppb (62.58 μg of Mn) upon 3 CV cycles between 0.75 and 2.8 V in 20 mL 1.0 M KNO3 solution. It is 2.35 times more in KNO3 solution compared to the LiNO3 solution, indicating that the Mn(VI) disproportionation reaction is further enhanced in the KNO3 electrolyte, as evident from the enhanced purple color at high potentials, see Video S1.‡
Furthermore, a new electrode was prepared using a similar stock solution of the ingredients without LiNO3 to obtain a mesoporous Mn3O4 electrode over FTO, labeled m-Mn3O4-300-F2. Like the m-LMO-300-F2 electrode, the new electrode was also tested by an m-CV cycling experiment in 1.0 M KNO3 aqueous solution between 0.75 and 2.8 V, see Fig. S17d.‡ After 3 CV cycles, the manganese amount in the electrolyte was detected to be 4635 ppb (105.94 μg of Mn) in the 20 mL 1.0 M KNO3 electrolyte solution and consistent with the observed enhanced purple color during CV cycling, compare the videos in Video S1.‡ However, it is also important to mention that the Mn amount (detected by ICP-MS) seems more than the coated LMO or Mn3O4 over the FTO surface by the spin coating technique. The KO+ and ArNH+ (with a mass to charge ratio of 55 as of Mn+ ion)43 interfere with manganese ICP-MS signal and therefore it is not reliable to quantify the manganese release and/or catalytic load by ICP-MS sıgnal at 55 (see later), but it can be qualitatively used to say, the manganese release is enhanced in the m-Mn3O4-300-F2 electrode and also during oxidation of the electrodes at more positive potentials in the KNO3 solutions. Moreover, the oxidation peak, which has been assigned to the Mn(VI) formation over the m-LMO-300-F2 electrode surface has a higher current density in the m-Mn3O4-300-F2 electrode. Here, the Mn3O4 surface species are oxidized to another MnO2 phase (no orange color due to λ-MnO2) at lower than OER potentials.
The Mn(IV) formation (likely as MnO2) was also detected in the Mn 2p and 3s XPS spectra upon sweeping the m-Mn3O4-300-F2 electrode up to 1.7 V in 1.0 M LiNO3 electrolyte solution, see Fig. S18a.‡ Notice also that the CV curves of the m-Mn3O4-300-F2 electrode are very similar to that of the m-LMO-300-F2 electrode in 1.0 M KNO3 aqueous solution, see Fig. 2b. The XPS spectra (Fig. S18b and c‡) display the characteristic line shape of MnO2 in the 2p and 3s regions (with a 4.6 eV splitting).6 Unlike in the m-LMO-300-F2 electrode, the release of the purple-colored species from the m-Mn3O4-300-F2 electrode surface continues at the OER potentials, see Video S1.‡ This observation is another piece of strong evidence for the fact that the λ-MnO2 phase is more robust to a Mn(VI) disproportionation reaction, compared to the MnO2 phase, obtained from the m-Mn3O4-300-F2 electrode. Furthermore, this electrode was also tested in the LiNO3 electrolyte solution and found that there is no Li+ ion insertion into this MnO2 phase (obtained from the m-Mn3O4 electrode) in the reverse cycles, also indicating how special the λ-MnO2 phase for both the OER and Li+ ion intercalation processes.
2.3. Thickness and temperature-dependent properties of the mesoporous LiMn2O4 films
To investigate the role of the electrode thickness in the above behaviors, the ultra-thin LMO electrodes were also tested in electrochemical experiments. Quantitative analysis of these electrodes has also been carried out by evaluating the charge capacities, obtained from the CV cycling (between 0.7 and 1.7 V, where the m-LMO-X-F2 electrodes are stable and undergo reversible de-lithiation/lithiation) experiments in 1.0 M LiNO3 aqueous solution. m-LMO-300-F2 was swept to 1.7 V to de-lithiate the electrode fully. Then the electrode was annealed at 600 °C for 1 hour. The aim of this experiment is to quantify the lithium content left in the structure by converting the de-lithiated product, namely LixMn2O4. It decomposes into (1 − x) mol of Mn2O3 and x mol of LiMn2O4 at 600 °C, see eqn (1). Fig. S19‡ displays the diffraction patterns of the m-LMO-600-F2 (pattern I) and m-LMO-300-F2 (pattern II) electrodes and m-LMO-300-F2 after sweeping from 0.7 to 1.7 V (pattern III) and the same electrode in pattern III annealed at 600 °C (pattern IV). Note also that the used electrodes were washed before the thermal treatment. The (111) line of the λ-MnO2 phase appears after LSV measurement (see pattern III in Fig. S19‡). The m-LMO-600-F2 diffraction pattern only displays sharper LiMn2O4 diffraction lines (no decomposition). However, the λ-MnO2 phase is converted to the Mn2O3 phase at 600 °C, see eqn (1). Intense Mn2O3 diffract lines appear in the pattern with the thermal treatment at 600 °C. Additionally, a weak line at 18.7°, 2θ, corresponding to the LiMn2O4 phase, also appears as a decomposition product of the LixMn2O4 phase (the used electrode).

| (1) |
Fig. 5a compares the XRD patterns of the unused m-LMO-600-F2 electrode with its used/annealed counterpart. Since both electrodes were prepared in a similar way, the ratio of the line intensity can be used to quantify the de-lithiated part of the electrode during LSV (from 0.75 to 1.7 V). The diffraction lines were fitted using a Lorentzian function to compare the areas under the (111) lines of each sample. The area under the line at 18.7°, 2θ, of the used electrode is ca. 16.3% that of the unused electrode, indicating that 83.7% of the m-LMO-300-F2 electrode is de-lithiated during LSV from 0.75 to 1.7 V. Thus, the de-lithiation efficiency could be used in the calculations of the catalytic load of the electrodes. Suppose the de-lithiation efficiency of the electrodes is precisely determined, then the mass of the LMO electrodes, which cannot be accurately measured using even a 4-digit balance (mass of the electrodes is usually less than 100 μg), can be calculated using the CVs and the charge capacities under the oxidation (de-lithiation) peaks.
 | ||
| Fig. 5 (a) The XRD patterns of m-LMO-600-F2 and m-LMO-300-F2 after LSV in 1.0 M LiNO3 aqueous solution followed by annealing at 600 °C (relative area under diffraction lines (I) 100 and (II) 16.3) and (b) the XRD patterns of m-LMO-300-F2 after LSV from 1.0 to 1.6 V and in reverse cycles from 1.6 to 1.0 V (repeated 3 times), and (c) 3 CVs of the m-LMO-300-F2 electrode (insets show the color of the electrodes on each side of the potential window). | ||
Further CV cycling has been carried out to investigate the reversibility of the Li+ de-intercalation and intercalation process using XRD. The XRD patterns between 17 and 20°, 2θ, have been sequentially collected after CV measurements from 1.0 to 1.6 V and 1.6 to 1.0 V, see Fig. 5b. The (111) diffraction line reversibly shifts upon oxidation and reduction cycles to the λ-MnO2 and LiMn2O4 line positions at 1.6 and 1.0 V, respectively, also indicating the reversibility of the de-lithiation and lithiation processes and the stability of the LiMn2O4 and λ-MnO2 phases during these measurements in the 1.0 to 1.6 V potential window. Reversible color-change (from dark green to orange and orange to dark green) of the electrode during electrochemical CV cycling has also been demonstrated, see Video S2.‡ Notice also that the color of the electrode remains orange when the electrode is cycled in 1.0 M KNO3 solution in the 1.0 and 1.6 V potential window (see Video 2‡), indicating not much K+ insertion (determined to be only 3.1% from K and Mn XPS analysis) in the reverse cycles. It might also be concluded that the lithiation process does not take place in the KNO3 solution as expected and the interior (bulk) of the electrode remains λ-MnO2 (even though the surface manganese species are reduced over the electrode surface, not visible to the naked eye) and keeps its orange color throughout the CV cycling. The above observation has been visualized for the first time in this investigation, see Video S2,‡ and is important and contributes to understanding of the stability of the manganese-based electrodes. The m-Mn3O4-300-F2 electrode was also cycled between −0.1 and 1.9 V in 1.0 M LiNO3 (see Fig. S19‡) and its XRD pattern at 1.9 V was recorded. Diffraction lines are indexed to the reference XRD pattern of the Mn3O4 phase (ICDD card no. 00-024-0734) and found that the Mn3O4 structure is still preserved at 1.9 V. The electrochemical oxidation is limited to the oxidation of the surface manganese. Moreover, the electrode is swept back to −0.1 V, and still the Mn3O4 diffraction pattern remains unaltered (Fig. S20‡) and accords well with the above conclusion that the redox reaction occurs on the electrode surface unless if there is a lithium intercalation/de-intercalation reaction.
Galvanostatic charge–discharge (GCD) measurements, using the m-LMO-300-F2 electrode, were also conducted at various charge–discharge current densities to investigate the de-lithiation efficiencies at low and high current densities, see Fig. S21a.‡ Fig. S21b‡ shows the charge capacity versus current density plot of a m-LMO-300-F2 electrode in 1.0 M LiNO3 electrolyte solution. There are no significant changes in the charge capacity of the electrode between 0.05 and 0.2 mA cm−2 current densities, indicating that slow or fast de-intercalation has no effect on the charging/discharging performance of the m-LMO-F electrodes.
The charge capacities of the ultra-thin m-LMO-X-F electrodes were evaluated from their CV curves (collected at a 20 mV s−1 scan rate in 1.0 M LiNO3 electrolyte solution), see Fig. S22a.‡ The total charge capacity under the oxidation peaks was calculated by extracting the mathematical area under the oxidation peaks and dividing it by the scan rate (∫idV/ν, where ∫idV is the mathematical area and ν is the scan rate). Fig. S22b‡ shows a plot of the charge density versus the spin-rate of the electrodes. An increase in the spin-rate from 2000 to 7000 rpm results in a gradual decrease in the charge capacity that accords well with the electrode thickness. However, further increasing the spin-rate does not alter the electrode thickness and charge capacity; both m-LMO-300-F10 and m-LMO-300-F7 electrodes have similar charge capacities, due to the FTO surface roughness as discussed in the XRD and SEM characterization section of these electrodes, see Fig. 1c. The catalytic loads on these electrodes have been calculated using the mathematical equation given below (eqn S(1)‡), by considering that the de-lithiation performance of the m-LMO-300-F2 electrodes is 83.7%, see Table S1.‡ The catalytic load varies from 61.1 to 38.1 μg cm−2 by increasing the spin-rate of coating from 2000 to 7000 rpm. To correlate these values, we also prepared a new set of standard solutions (similar in composition to electrolyte solutions used in CV measurements) and recorded their ICP-MS spectra to obtain new calibration curves for the KNO3 and LiNO3 solutions, see Fig. S29‡ (details are given in the ESI‡). The calibration curve gave us about 2.46 times deviation in the manganese concentration in KNO3 and 1.21 times in LiNO3 solutions; the corrected ICP-MS data of the above measurements are also tabulated in Table S4.‡ The corrected manganese release is close to or lower than the estimated catalytic load as expected and accords well with the CV measurements.
The m-LMO-X-F2 and m-LMO-X-F5 (where X is 300, 350, 400, 450, and 500) electrodes have also been fabricated and used to investigate the effect of annealing temperature on the charge capacity by CV measurements, see Fig. S23a and b,‡ respectively. We found that the annealing temperature has no effect on the catalytic load as expected (approximately ca. 60 μg cm−2 and 40 μg cm−2 in the m-LMO-F2 and m-LMO-F5 electrodes, respectively), see Table S2 and Fig. S23c, d.‡ Ultra-thin m-LMO-300-G# electrodes were also quantitatively analyzed by cycling in the 1.0 M LiNO3 electrolyte, see Fig. S24a.‡ Notice that the current density of the Mn(III)/Mn(IV) redox peaks (between 1.0 and 1.6 V) decreases by increasing the dilution of the stock solution and attributed to the film thickness; the higher the dilution, the thinner the electrodes as a result the lower the charge density, see Fig. S24b.‡ Table S3‡ lists the calculated catalytic load on the graphite surface. The charge capacity and sample amount exponentially decay by increasing the dilution factor and become almost constant over 50 times dilution similar to the spin-coated electrodes over 7000 rpm. Therefore, the dip-coating method by dilution is also limited by the surface morphology (roughness) of the graphite rods as observed in the FTO-coated electrodes.
2.4. Degradation of the electrodes during the OER in alkaline media
The m-LMO-300-F2 and m-Mn3O4-300-F2 electrodes were further used in the CV cycling experiment in 1.0 M LiOH and 1.0 M KOH electrolyte solutions to investigate the Mn(VI) disproportionation reaction during the OER, see Fig. 6a–c. Each electrode was cycled 300 times between 0.8 and 1.8 V and their current density values at 1.8 V were plotted against the cycle number to investigate the degradation behavior of the electrodes, see Fig. 6d. Notice that the OER performance is better in the KOH solution as evidenced by a higher current density at 1.8 V in all cycles. However, the degradation is also faster in the KOH solution, evidenced by a continuous decay of the current density with a higher slope. The electrode degradation, in the LiOH electrolyte, is slow in early cycling and almost stops in the further cycles, see Fig. 6d. Also, the reduction peaks (due to lithium intercalation) appear at around 1.6 V in the reverse cycles and might be attributed to the stability of the λ-MnO2 phase in the LiOH solution. The m-LMO-300-F2 electrode was also compared with the m-Mn3O4-300-F2 electrode. The Mn(VI) disproportionation reaction and degradation of the electrode are much faster in the m-Mn3O4-300-F2 electrode, which only survives up to 150 CV cycles, see Fig. 6d. The permanganate formation is low over the m-LMO-300-F2 electrode–electrolyte interface at higher potentials, see Video S3,‡ compared to the m-Mn3O4-300-F2 electrode, indicating that the λ-MnO2 phase (or m-LMO electrode) is more robust to a Mn(VI) disproportionation reaction.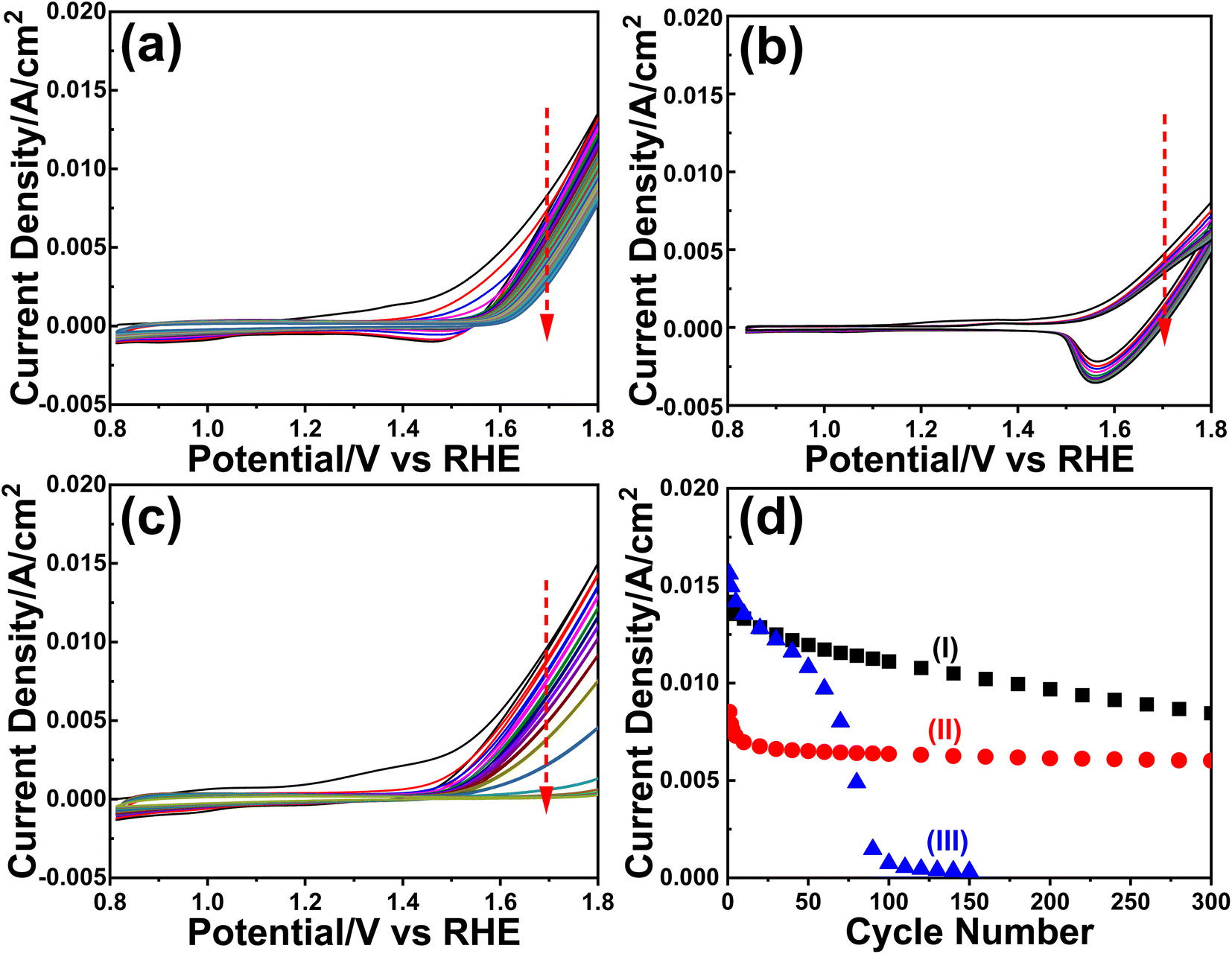 | ||
| Fig. 6 The CV curves (50 mV s−1 scan rate) of the m-LMO-300-F2 electrode in (a) 1.0 M KOH and (b) 1.0 M LiOH and (c) the CV curves (50 mV s−1 scan rate) of the m-Mn3O4-300-F2 electrode in 1.0 M KOH (red arrows indicate the cycling order). (d) Plot of current density values at 1.8 V versus CV cycle numbers of the electrodes, (I) m-LMO-300-F2 in 1.0 M KOH, (II) m-LMO-300-F2 in 1.0 M LiOH, and (III) m-Mn3O4-300-F2 in 1.0 M KOH. | ||
m-LMO-300-F2 was also cycled between 0.8 and 2.3 V in 1.0 M KOH and 1.0 M LiOH solutions to enhance the Mn(VI) disproportionation reaction, see Fig. S25a and b,‡ respectively. Notice that the degradation rate is still slow in the LiOH electrolyte likely due to regeneration of some stable LiMn2O4 phase in reverse cycles. However, in the KOH electrolyte, the degradation is much faster and the electrode survives only up to 250 CV cycles, see Fig. S25c.‡ Notice that the surface of the electrode is also reduced at similar potentials in the reverse cycle, likely to MnOOH and KMn2O4 (as detected by XPS). Therefore, the bulk spinel KMn2O4 does not form to stabilize the entire electrode (bulk of the electrode remains λ-MnO2) and only the surface Mn(IV) species are reduced Mn(III) species (similarly in the Mn3O4 phase) in reverse cycles and makes the electrodes undergo a faster decay.
To clarify the effect of the electrode thickness on the stability of the electrodes, the m-LMO-300-F2 and m-LMO-300-F5 electrodes were also used to collect 300 CVs in the 1.0 M KOH solution and the current density values at 1.8 V versus CV cycle number are plotted, see Fig. S26a.‡ Notice that the degradation is slightly faster in the m-LMO-300-F2 electrode, compared to the m-LMO-300-F5 electrode. This may be attributed to the film quality and partial mechanical degradation of the thicker electrodes. The ultra-thin m-LMO-300-G# electrodes were also tested by recording 300 CVs (at a 50 mV s−1 scan rate) in the 1.0 M KOH electrolyte to clarify the role of electrode thickness in degradation. Fig. S26b‡ displays a plot of current density values at 1.8 V versus CV cycle numbers. Notice that the m-LMO-300-G5, m-LMO-300-G10, m-LMO-300-G20 and m-LMO-300-G50 electrodes also undergo gradual degradation by CV cycling with a similar rate, but the m-LMO-300-G100 electrode behaves similarly to bare graphite, indicating that the 100-times diluted stock solution is not suitable for an effective coating.
Note also that the m-LMO-300-F5 and m-LMO-300-G5 electrodes have similar LMO loadings as quantified using the two characteristic Mn(III)/Mn(IV) oxidation peaks in the CV curves in the 1.0 M LiNO3 electrolyte solution. Then, both electrodes were used to collect 300 CV curves in 1.0 M KOH electrolyte solution. Fig. S26c‡ shows the 1st and 300th CV curves of the electrodes. The m-LMO-300-G5 electrode displays a much higher current density compared to the m-LMO-300-F5 electrode and can be attributed to a better conductivity (lower IR losses) of the graphite substrate. However, the drop in the current densities at 1.8 V, in the 1st to 300th CV curves on both electrodes, is very similar and it is 37.4% in the m-LMO-300-G5 and 32.5% in the m-LMO-300-F5 electrodes. We previously showed that the graphite rod is a better substrate for the fabrication of mesoporous CaFe2O4 and M2P2O7 (fabricated using the MASA method); the mesoporous CaFe2O4 and M2P2O7 electrodes undergo mechanical degradation in an alkaline media over the FTO surface, but the same electrodes that are fabricated over the graphite surface are robust and unaffected during the electrochemical test.39,44,45 However, this is not the case in the m-LMO electrodes, which undergo disproportionation reactions and display a similar degradation over both substrates. The only advantage of using a graphite substrate might be a smaller IR-drop in the OER potentials and better overpotential values. Therefore, the m-LMO-300-F# electrodes were also used for further investigation by multi-step chronopotentiometry (CP) experiments from 1 mA cm−2 to 50 mA cm−2 in 1.0 M KOH solution, see Fig. S27a.‡ A sudden increase of the overpotentials in Fig. S27‡ at higher current densities is attributed to a complete degradation of the m-LMO film over the FTO surface; the high overpotential is due to the bare FTO.
The m-LMO-300-F2 and m-LMO-300-F3 (thicker) electrodes display higher overpotential values and degrade at lower current densities, compared to the m-LMO-300-F5 electrode. This behavior might be due to poor film quality in the thicker electrodes, resulting in also some mechanical material losses under the harsh OER conditions. Moreover, the m-LMO-300-F7 and m-LMO -300-F10 (thinner) electrodes also have higher overpotentials than the m-LMO-300-F5 electrode and might be attributed to the surface roughness as previously discussed. Therefore, the most stable electrode is the m-LMO-300-F5 electrode and the overpotentials at high current densities might be correlated with the film quality. The effect of annealing temperature was also tested using the m-LMO-300-F5 electrode by annealing at various temperatures to obtain the m-LMO-X-F5 electrodes (where X is 350, 400, 450, 500) and used to collect their multi-step chronopotentiometry data, see Fig. S27b.‡ Notice that the m-LMO-X-F5 electrodes degrade at a lower current density with increasing annealing temperature; the m-LMO-300-F5 electrode performs the best among all tested electrodes in the OER.
2.5. The Mn(VI) disproportionation reaction mechanism
Considering the above experimental data and observed electrochemical behaviors, we suggest the following Mn(VI) disproportionation reaction mechanism during the OER. As shown in Scheme 1, the electrochemical formation of the Mn(VI) species over the LiMn2O4 surface (or λ-MnO2) triggers the disproportionation reaction at the OER potentials. We believe that the Mn(VI) disproportionation reaction requires adjacent three Mn(VI) sites to produce one Mn(IV) and two Mn(VII) species as suggested by the charge-balance of this reaction. Notice that all three Mn(VI) species are not equivalent in the proposed structure; the one in the middle connected by bridging oxygens to the other two Mn(VI) sites is more electronegative than the other two Mn(VI) members. Because the end Mn(VI) sites are connected by bridging oxygen to the lattice Mn(IV) sites and they are relatively less electronegative. Simply, by calculating Sanderson's electronegativity (χ) using the geometric mean of the electronegativity46 of the atomic constituents (χMnxOy = (χMnxχOy)1/(x+y)), the middle Mn(VI) site has a local geometry with a Mn3O8.66 atomic composition and the terminal Mn(VI) site with Mn3O5.83 (see Scheme 1) and their mean electronegativities are ca. 6.28 and 5.92 eV, respectively, with a 0.36 eV difference (calculated using the atomic Mullikan electronegativity of 3.72 eV for Mn and 7.54 eV for O atoms). This electronegativity unbalance triggers an electron transfer from each less electronegative terminal Mn(VI) site to the more electronegative middle Mn(VI) to reduce it to Mn(IV) and remains stable as MnO2 on the electrode surface (see Scheme 1). However, the oxidized two terminal Mn(VI) to Mn(VII) species, which are unstable on the electrode surface, will leach into the electrolyte solution as permanganate ions. A typical purple color of the MnO4− ion appears on the electrode surface during the OER at high oxidation potentials, see Video S1.‡ Therefore, the reaction in the second step occurs to release permanganate ions into the electrolyte solution, see Scheme 1. This leaching is enhanced in the KOH solutions, compared to the LiOH solution. | ||
| Scheme 1 Electrode degradation mechanism of LiMn2O4 electrodes during the OER. | ||
The same process likely makes the thinner electrodes relatively more robust. The driving force for permanganate release into the electrolyte solution is the concentration of the MnO4− surface sites on the electrode surface, which is simply higher over the thicker electrodes. Thus, the permanganate species may stay stable (see the product in the first step), used for water oxidation, and be more extensively converted back into the MnO2 species over the thinner electrode surface by a similar mechanism, given in Scheme 2.
 | ||
| Scheme 2 Water oxidation by permanganate ions. Green species are the products; red species are consumed reactants at each step. | ||
Schema 2 shows a likely mechanism of water oxidation in the presence of permanganate ions in the alkaline aqueous electrolyte solution and can be used to evaluate the chemical water oxidation process by permanganate species on the electrode surface. It is the same process that converts the permanganate ion into Mn(IV) and to precipitate as solid MnO2 from the electrolyte solution (as evidenced by observing brown particles at the bottom of the electrolyte solution in cycling the electrodes). Therefore, the manganese concentration, detected by ICP-MS, originates from the above disproportionation reaction. It is the same reaction that makes the LiMn2O4 or manganese oxide species unstable during the charge–discharge process in batteries and the water oxidation process that requires manganese to be at a high oxidation state, such as 6+ or 7+. Otherwise, the oxygen in the oxo bond (MnO) is not electrophilic enough for a hydroxide attack to form an oxygen–oxygen (Mn–O–O–H) bond.15,16,39 This step is also the key and rate-determining step in the water oxidation process.15,16,39
Remember that the disproportionation reaction is enhanced in the m-Mn3O4-300-F2 electrodes, see Videos S1 and S3.‡ To further enhance the disproportionation reaction and collect some spectroscopic evidence, we also tested mesoporous Mn2P2O7 in 3.0 M KOH. Recently, we have shown that the mesoporous M2P2O7 materials (see Fig. S28‡) undergo a chemical transformation into layered β-M(OH)2 in alkaline media.44,45 The characterization and transformation processes of Mn2P2O7 have been extensively investigated in our previous publication.45 Therefore, we will focus on the disproportionation reaction on the graphite rod-coated mesoporous Mn2P2O7 electrode, which is already transformed into β-Mn(OH)2 before any electrochemical tests upon dipping the electrode into a 3.0 M KOH electrolyte solution. Since β-Mn(OH)2 is a layered material, the Mn(VI) disproportionation reaction is significantly enhanced and also supports our mechanism in Scheme 1. One can easily observe that a large number of MnO4− ions are dispersed into the electrolyte solution in every CV cycle (between −0.4 and 1.6 V, vs. NHE) at positive potentials even in such a basic solution, see Video S4.‡ The MnO4− dispersion from the electrode surface is strong enough to observe a spray of a purple-colored species around the electrode surface towards the electrolyte solution. The purple color turns into a greenish color over time. Both purple and green-colored solutions, in different time intervals, were collected and their visible absorption spectra were recorded to confirm the formation of permanganate and manganate ions, respectively, see Fig. 7. To collect the spectrum of the permanganate ion, a 1.0 mL electrolyte solution (from a close proximity to the sprayed permanganate region) was immediately taken upon release of the permanganate ions into a cuvette and acidified to stabilize the permanganate ion for a duration of recording its visible absorption spectrum. Otherwise, the permanganate ions quickly convert into manganate ions and the solution becomes green over time. The permanganate ion leaching continues until the 10th cycle during CV measurement around the OER potentials. After the 10th cycle, either decomposition stops due to transformation or a minimal number of permanganate ions are released out so it cannot be observed by the naked eye. The spectrum in Fig. 7a corresponds to a solution, collected immediately after permanganate release and acidification. The peak with vibronic features corresponds to oxygen to manganese charge transfer transition and is very characteristic of the MnO4− ion.47 The spectrum shown in Fig. 7b corresponds to an aged electrolyte solution after 20 CV cycles and is characteristic for the MnO42− ions in an aqueous solution. Note also that the peaks due to the MnO4− ion peak decay over time even in acidic solution but the peaks at 430 and 606 nm due to MnO42− ions do not appear immediately, even though these two manganese species have similar extinction coefficients. Therefore, it is reasonable to conclude that the MnO42− species also undergo a reduction in the electrolyte solution to form MnO2 particles (likely faster in an acidic solution); one can observe an accumulation of dark brown particles at the bottom of the electrolyte solution over time and also mechanistically shown in Schemes 2 and 3.
 | ||
| Fig. 7 The visible absorption spectrum of electrolyte during (a) MnO4− release in acidic media and (b) upon aging the same solution after 20 CV cycles. | ||
 | ||
| Scheme 3 Disproportionation of manganate ions into permanganate ions and manganese dioxide in aqueous solution. | ||
We propose that the permanganate and manganate decomposition reactions in the alkaline solution follow the steps in Scheme 2. First the MnO4− ion reacts with the hydroxide ion to form Mn–O–O–H (O–O bond) in the first step, which could be the rate-determining step in the OER. It takes place as a redox process, in which the oxygens are oxidized to produce peroxides and Mn7+ is reduced to Mn5+. The manganese in the Mn5+–O–O site gets oxidized by two other MnO4− ions in the later steps to produce highly unstable Mn7+–O–O that quickly releases an O2 molecule through another redox process, in which the O–O2− peroxide species is oxidized to an O2 molecule and Mn7+ is reduced to back to Mn5+. In the final step, the fourth MnO4− and Mn5+ species quickly react with each other to form two manganate ions. Notice that Mn5+ species were never detected in the electrolyte solution or on the electrode surface during the OER. Therefore, related steps are fast and Mn5+ species never accumulate in the electrolyte or on the electrode surface. Overall, all four MnO4− are converted to four MnO42− and one O2 molecule, as shown in eqn (2) below (this makes MnO42− ions accumulate in the electrolyte solution and is also the origin of the green color in the electrolyte solution). For every four manganate ions that undergo a disproportionation reaction produce 8/3 MnO4− back and 4/3 MnO2 as shown in Scheme 1 on the electrode surface and in Scheme 3 in the electrolyte solution. The formation of permanganate ions in eqn (3) feeds the reaction in eqn (2) until all the manganese species become MnO2 and precipitate from the electrolyte solution. In an overall reaction (eqn (4)), eqn (2) and (3) repeat until all the permanganate ions are converted to a solid MnO2 precipitate by producing 3/4 mole of O2 using half a mole of water per MnO4− ion.
| 4MnO−4 + 4OH− ⇌ 4MnO2−4 + O2 + 2H2O | (2) |
| 4MnO2−4 + 8/3H2O ⇌ 8/3MnO−4 + 4/3MnO2 + 16OH− | (3) |
| Overall: MnO−4 + 1/2H2O ⇌ MnO2(s) + 3/4O2 + OH− | (4) |
The disproportionation reaction on the electrode surface likely takes place more quickly because the Mn(VI) sites are connected by oxygen bridges and in a very close vicinity, however in the solution media, the interaction between the manganate ions requires hydrogen bonding as shown in Scheme 3 or frequent collisions (less likely) for the electron transfer. We believe that similar processes occur on our electrode surface and lead to a decomposition of the electrodes from the FTO or graphite surfaces. The source for the permanganate ions in the electrolyte solution is the reactions shown in Schema 1. If one can block this path, one can produce a more stable manganese-based electrocatalyst for the OER. There are some attempts in the literature that introduce another metal ion (such as Co or Ni) to form LiMn2-xMxO4 materials to stabilize the LiMn2O4 electrodes. The surface of the m-LMO electrode was modified by incorporating Co(II) ions and calcining the electrode, which displays relatively better stability.15 We believe that it is important to avoid reaction two (eqn (2)) by replacing the middle Mn(VI) by either Co or Ni ions, which cannot be electrochemically oxidized to a 6+ oxidation state. Further studies are needed to improve the stability of LMO electrodes using the concepts highlighted in this investigation.
3. Experimental section
3.1. Preparation of the stock solution
All chemicals, used in this investigation, are Sigma-Aldrich grade and used without further purification. A solution of the salts and surfactants is prepared by dissolving LiNO3, [Mn(H2O)4](NO3)2, P123 (H(OCH2CH2)20(OCH(CH3)CH2)70(OCH2CH2)20OH) and cetyltrimethylammonium bromide (CTAB), and concentrated HNO3 (65%) in absolute ethanol (99.9%). First, 719 mg (0.125 mmol) P123 is completely dissolved in 5 mL ethanol by stirring and then 46 mg (0.125 mmol) CTAB is added to the clear solution and stirred further until all CTAB completely dissolves. Then, to this clear solution, 173 mg (2.5 mmol) LiNO3 is added at once and after 5 min, 550 mg 70% concentrated HNO3 is added dropwise and stirred to obtain a homogeneous clear solution. Finally, 1255 mg (5 mmol) [Mn(H2O)4](NO3)2 is added to the above clear solution in portions with 5 min intervals and then the solution vial is sealed and stirred for 1 day to obtain a homogeneous and clear stock solution.3.2. Synthesis of mesoporous LiMn2O4, Mn3O4 thin and thick films
The above clear stock solution is coated on microscope slides by drop-casting or spin-coating methods for various purposes. In drop-casting, 8 drops of the solution were placed with equal spaces over a glass slide and then kept for 1 hour for the evaporation of the excess solvent to obtain a thick LLC film. In the spin coating case, 8 drops of solution were placed over a slide and spun at various spin rates from 2000 to 10000 rpm for 10 seconds resulting in instantaneous evaporation of solvent to obtain thinner LLC films. The drop-cast and spin-coated LLC films were calcined at 300 °C for 3 and 2 hours, respectively, to obtain m-LMO films. The m-LMO powders were collected by scraping the films from the microscope slides and further annealed at 350, 400, 450, and 500 °C for 1 hour. Mesoporous Mn3O4 thin films were also synthesized using the same procedure and a similar stock solution was prepared using all the above ingredients without LiNO3 salt. The manganese salt/P123 mole ratio was kept at 40. The coated gel-films were calcined at 300 °C for various time periods depending on coating techniques similar to m-LMO films. The Mn2O3 films were obtained by calcination of scraped powder at 600 °C for 1 hour.
3.3. Fabrication of mesoporous LiMn2O4, Mn3O4 thin and their ultra-thin film electrodes
Fluorine-doped tin oxide (FTO) is used as a conductive substrate for electrode fabrication. Half of a 1 by 2 cm FTO surface is covered with tape and the other 1 cm2 half of the FTO surface is coated by placing 3 drops of the stock solutions and spun at various rates (2000, 4000, 6000, 8000, and 10000 rpm) for 10 seconds. Afterward, the tape was removed and the 1 cm2 LLC thin film on FTO was calcined at 300 °C for 1 h. The fabricated electrodes were further annealed by heating them from 300 °C to the desired temperature. The mesoporous LiMn2O4 thin film electrodes were denoted as m-LMO-X-F# where m, LMO, X, F, and # stand for mesoporous, LiMn2O4, calcination/annealing temperature in Celsius, FTO, and the first digit of the spin rate, respectively (for instance m-LMO-400-F6 is spin-coated on FTO at 6000 rpm and annealed at 400 °C). Ultra-thin films were also synthesized using a 3 cm graphite rod (3 mm diameter) as a conductive substrate. The stock solution was diluted by a desired dilution factor (5, 10, 20, 50, and 100 times) by using ethanol. 2 cm of the rod was covered with tape, and 1 cm side of the rod was immersed into the diluted solution for 10 seconds and pulled out at a speed of 1 cm s−1. Then, the electrodes were calcined/annealed as described above and labeled m-LMO-X-G# (X is the calcination/annealing temperature in Celsius, G is graphite, and # is the dilution factor).
3.4. Fabrication of the mesoporous Mn2P2O7 thin film electrodes
The m-MnPP electrodes were fabricated using the following solutions. First, 2.619 g of [Mn(H2O)4](NO3)2 was dissolved in 3 mL of deionized water in a vial of 20 mL. Then, 1.000 g of melted P123 (at 80 °C) and 0.929 g of PPA were dissolved in another vial in 7 mL of deionized water. The mole ratio of the surfactant, transition metal and acid was kept constant (1:60:30 of P123:[Mn(H2O)4](NO3)2:PPA). Both mixtures were stirred for 3–4 hours until homogeneous solutions were obtained. Then, these two solutions were combined and stirred for 1 day to obtain a clear and homogeneous solution. The above stock solution was diluted 10 times and used for coating the graphite rods. Before the coating process, the graphite rod was cleaned using deionized water and pure ethanol and covered with parafilm to obtain only a 1 cm2 area of uncovered bare graphite. Then, the uncovered part was dip-coated using a diluted solution by first keeping it in the stock solution for 10 seconds and then pulling out at a speed of 1.45 mm s−1 and then aging for a few minutes under ambient conditions to evaporate the excess solvent. After removing the parafilm, the dip-coated electrode was calcined directly in a pre-heated oven at a desired temperature (as low as 300 °C) for 1 h and labeled m-MnPP-X-G (X is the calcination/annealing temperature in Celsius and G is the graphite rod).
3.5. Instrumentation
3.6. Electrochemical analysis
Electrochemical measurements were performed using Potentiostats PC14G750 and IFC5000-07565 from Gamry Instruments. A three electrode system was utilized for all electrochemical measurements. A platinum spring, Ag/AgCl (3.5 M KCl) and LMO (or Mn3O4 or Mn2P2O7) on the conductive substrate were used as counter, reference, and working electrodes, respectively. The electrodes were used at various sweep rates in cyclic voltammetry (CV) experiments. The reference electrode potential was mathematically converted to the normal hydrogen electrode (NHE) or reversible hydrogen electrode (RHE) in CV plots. Chronopotentiometry (CP) experiments were performed at various current densities from 1 to 50 mA cm−2 according to the desired type of analysis and voltage values were converted to overpotential by subtracting standard OER potential (0.401 V vs. NHE) at pH 14 and additional voltage resulting from the ohmic potential drop (IR-drop) from the measured voltage values.4. Conclusion
Three different manganese oxide-based (namely LiMn2O4, Mn3O4, and Mn(OH)2) electrodes were fabricated using the MASA process over FTO and graphite rods and their electrochemical stability issues have been investigated in alkaline media. The LMO electrode thickness can be controlled by the spin-rate of the initial ingredient solution in spin-coating and by dilution in the dip-coating process. The performance and stability of the m-LMO-300-F5 (coated at 5000 rpm) and m-LMO-300-G5 (diluted by 5 times) electrodes are the best among the tested at least thickness and annealing temperature dependent 15 electrodes. Oxidation of the electrodes during the OER produces MnO2 in different polymorphs. Mesoporous LMO undergoes almost a complete de-lithiation reaction, producing a λ-MnO2 phase upon oxidation. The de-lithiation process is reversible and the electrode can be fully Li+ ion intercalated back to LiMn2O4 in a reverse cycle in a lithium salt electrolyte. Insertion of a second lithium produces the Li2Mn2O4 phase, but it undergoes a Mn(III) disproportionation reaction and degradation. The λ-MnO2 phase can be further oxidized to carry out the OER at decent overpotentials, but the formation of Mn(VI) triggers another disproportionation reaction and causes electrode decomposition. The electrode of the λ-MnO2 phase is more robust and can be further improved by reducing its physical thickness. However, the MnO2 phase produced from the mesoporous Mn3O4 and Mn(OH)2 electrodes undergoes fast decomposition. The decomposition is even more pronounced in layered β-Mn(OH)2 and produces likely layered MnO2 upon electrochemical oxidation and quickly decomposes with further oxidation to Mn(VI) and Mn(VII) species through a Mn(VI) disproportionation reaction. This process can be monitored by a release of purple-colored permanganate ions that are leached out from the electrode surface and cause electrochemical etching of the electrode. The suggested Mn(VI) disproportionation reaction mechanism uses the concept of mean electronegativity, charge neutrality principle, and experimental data and explains both the experimental observations during Mn(VI) disproportionation reactions and nucleophilic addition of hydroxide to the MnO bond to form an oxygen–oxygen bond (Mn–O–O–H) in the OER mechanism.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank TÜBİTAK (under project numbers 113Z730 and 119Z877) for the financial support to this work. The authors also thank Mete Batuhan Durukan for configuring the videos. ÖD is a member of the Academy of Science, Istanbul, Turkey.References
- S. Rana, J. P. Biswas, S. Paul, A. Paik and D. Maiti, Chem. Soc. Rev., 2021, 50, 243 RSC.
- G. Liu, Y. Xu, T. Yang and L. Jiang, Nano Mater. Sci., 2023, 5, 101 CrossRef CAS.
- Z. Chen, Q. Li, H. Xiang, Y. Wang, Y. Wang, P. Yang, C. Dai, H. Zhang, W. Xiao, Z. Wu and L. Wang, Inorg. Chem. Front., 2023, 10, 1493 RSC.
- M. S. A. S. Shah, G. Y. Jang, K. Zhang and J. H. Park, EcoEnergy, 2023, 1, 344 CrossRef.
- W. Levason and C. A. McAuliffe, Coord. Chem. Rev., 1972, 7, 353 CrossRef CAS.
- E. S. Ilton, J. E. Post, P. J. Heaney, F. T. Ling and S. N. Kerisit, Appl. Surf. Sci., 2016, 366, 475 CrossRef CAS.
- Y. Meng, W. Song, H. Huang, Z. Ren, S. Y. Chen and S. L. Suib, J. Am. Chem. Soc., 2014, 136, 11452 CrossRef CAS PubMed.
- M. Kölbach, S. Fiechter, R. van de Krol and P. Bogdanoff, Catal. Today, 2017, 290, 2 CrossRef.
- K. Xiao, S. L. Zhao, M. Cao, L. Zhang, N. Li and Z. Q. Liu, J. Mater. Chem. A, 2020, 8, 23257 RSC.
- Y. G. Cho, S. H. Jung, S. H. Joo, Y. Jeon, M. Kim, K. M. Lee, S. Kim, J. M. Park, H. K. Noh, Y. S. Kim, J. E. Hong, S. I. Jeon, T. Kim, S. K. Kwak, H. Kong and H. K. Song, J. Mater. Chem. A, 2018, 6, 22483 RSC.
- S. Sun, J. Li, C. Xu, T. Zhai and H. Xia, J. Mater. Chem. A, 2022, 10, 19231 RSC.
- A. Banerjee, Y. Shilina, B. Ziv, J. M. Ziegelbaurer, S. Luski, D. Aurbach and I. C. Halalay, J. Am. Chem. Soc., 2017, 139, 1738 CrossRef CAS PubMed.
- T. Liu, A. Dai, Y. Yuan, Y. Xiao, L. Yu, M. Li, J. Gim, L. Ma, J. Liu, C. Zhan, L. Li, J. Zheng, Y. Ren, T. Wu, R. S. Yassar, J. Wen, F. Pan and K. Amine, Nat. Commun., 2019, 10, 4721 CrossRef CAS PubMed.
- B. Zhang, Q. Daniel, L. Fan, T. Liu, Q. Meng and L. Sun, iScience, 2018, 4, 144 CrossRef CAS PubMed.
- I. Karakaya, F. Karadas, B. Ulgut and Ö. Dag, Adv. Mater. Technol., 2020, 5, 2000353 CrossRef CAS.
- A. Amirzhanova, N. Akmanşen, I. Karakaya and Ö. Dag, ACS Appl. Energy Mater., 2021, 4, 2769 CrossRef CAS.
- F. R. Duke, J. Phys. Chem., 1952, 56, 882 CrossRef CAS.
- J. H. Sutter, K. Colquitt and J. R. Sutter, Inorg. Chem., 1974, 13, 1444 CrossRef CAS.
- M. Huynh, C. Shi, S. J. L. Billinge and D. G. Nocera, J. Am. Chem. Soc., 2015, 137, 14887 CrossRef CAS PubMed.
- A. Li, H. Ooka, N. Bonnet, T. Hayashi, Y. Sun, Q. Jiang, C. Li, H. Han and R. Nakamura, Angew. Chem., Int. Ed., 2019, 58, 5054 CrossRef CAS PubMed.
- M. M. Thackeray, P. J. Johnson, L. A. de Picciotto, P. G. Bruce and J. B. Goodenough, Mater. Res. Bull., 1984, 19, 179 CrossRef CAS.
- D. K. Kim, P. Muralidharan, H. W. Lee, R. Ruffo, Y. Yang, C. K. Chan, H. Peng, R. A. Huggins and Y. Cui, Nano Lett., 2008, 8, 3948 CrossRef CAS PubMed.
- Y. Xia, Y. Zhou and M. Yoshio, J. Electrochem. Soc., 1997, 144, 2593 CrossRef CAS.
- X. Hou, X. Liu, H. Wang, X. Zhang, J. Zhou and M. Wang, Energy Storage Mater., 2023, 57, 577 CrossRef.
- J. C. Hunter, J. Solid State Chem., 1981, 39, 142 CrossRef CAS.
- A. Eftekhari, Electrochim. Acta, 2001, 47, 495 CrossRef CAS.
- L. Chen, W. Zhai, L. Chen, D. Li, X. Ma, Q. Ai, X. Xu, G. Hou, L. Zhang, J. Feng, P. Si and L. Ci, J. Power Sources, 2018, 392, 116 CrossRef CAS.
- M. Jayalakshmi, M. M. Rao and F. Scholz, Langmuir, 2003, 19, 8403 CrossRef CAS.
- D. M. Robinson, Y. B. Go, M. Mui, G. Gardner, Z. Zhang, D. Mastrogiovanni, E. Garfunkel, J. Li, M. Greenblatt and G. C. Dismukes, J. Am. Chem. Soc., 2013, 135, 3494 CrossRef CAS PubMed.
- M. Baumung, L. Kollenbach, L. Xi and M. Risch, ChemPhysChem, 2019, 20, 2981 CrossRef CAS PubMed.
- C. W. Cady, G. Gardner, Z. O. Maron, M. Retuerto, Y. B. Go, S. Segan, M. Greenblatt and G. C. Dismukes, ACS Catal., 2015, 5, 3403 CrossRef CAS.
- F. M. Balci, I. Karakaya, E. P. Alsaç, M. Y. Yaman, G. Saat, F. Karadas, B. Ulgut and Ö. Dag, J. Mater. Chem. A, 2018, 6, 13925 RSC.
- S. Chen, Z. Chen and C. Cao, Electrochim. Acta, 2016, 199, 51 CrossRef CAS.
- F. Wang, J. Wang, H. Ren, H. Tang, R. Yu and D. Wang, Inorg. Chem. Front., 2016, 3, 365 RSC.
- J. Y. Luo, Y. G. Wang, H. M. Xiong and Y. Y. Xia, Chem. Mater., 2007, 19, 4791 CrossRef CAS.
- F. Jiao, J. Bao, A. H. Hill and P. G. Bruce, Angew. Chem., Int. Ed., 2008, 47, 9711 CrossRef CAS PubMed.
- Y. Ren, M. Zen and P. G. Bruce, Chem. Soc. Rev., 2012, 41, 4909 RSC.
- G. Saat, F. M. Balci, E. P. Alsaç, F. Karadas and Ö. Dag, Small, 2018, 14, 1701913 CrossRef PubMed.
- H. A. Raza, I. Karakaya and Ö. Dag, ACS Appl. Energy Mater., 2023, 6(18), 9681 CrossRef CAS.
- S. Lee, G. Nam, J. Sun, J. S. Lee, H. W. Lee, W. Chen, J. Cho and Y. Cui, Angew. Chem., Int. Ed., 2016, 55, 8599 CrossRef CAS PubMed.
- M. M. Thackeray, W. I. F. David, P. G. Bruce and J. B. Goodenough, Mater. Res. Bull., 1983, 18, 461 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717 CrossRef CAS.
- T. W. May and R. H. Wiedmeyer, At. Spectrosc., 1998, 19, 150 CAS.
- I. Ulu, B. Ulgut and Ö. Dag, J. Mater. Chem. A, 2023, 11, 22384 RSC.
- I. Ulu, B. Ulgut and Ö. Dag, Inorg. Chem., 2023, 62, 16994 CrossRef CAS PubMed.
- R. G. Pearson, Inorg. Chem., 1988, 27, 734 CrossRef CAS.
- D. G. Lee, C. R. Moylan, T. Hayashi and J. I. Brauman, J. Am. Chem. Soc., 1987, 109, 3003 CrossRef CAS.
Footnotes |
| † Dedicated to Prof. Geoffrey A. Ozin's 80th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta07973b |
| This journal is © The Royal Society of Chemistry 2024 |