
The critical role of water molecules in the development of aqueous electrolytes for rechargeable metal-ion batteries
Tong
Xu
ab,
Jiaojiao
Yu
a,
Junchao
Ma
a,
Wei
Ren
a,
Mingliang
Hu
*a and
Xifei
Li
 *b
*b
aSchool of Science, Xi'an University of Posts and Telecommunications, Xi'an 710121, Shaanxi, PR China. E-mail: mlhu0301@163.com
bInstitute of Advanced Electrochemical Energy, School of Materials Science and Engineering, Xi'an University of Technology, Xi'an 710048, PR China. E-mail: xfli2011@hotmail.com
First published on 30th April 2024
Abstract
Aqueous rechargeable metal ion batteries (AMIBs) have emerged as a promising option for large-scale electrical energy storage due to their environmental friendliness, low cost, and high safety. This review examines the latest advanced technology orientation for AMIB electrolytes from these perspectives: (1) dilute to high concentrations, (2) room temperature to extreme temperatures, and (3) liquid to quasi-solid states. Typically, the significant impact of water molecule content on the solvation sheath, SEI formation, and enhancement of electrochemical performance in high concentration electrolytes was thoroughly reviewed and discussed. Furthermore, the utilization of salt concentration, organic additives, and hydrogel electrolytes was explored in breaking and re-forming hydrogen bonds between water molecules, with the aim of improving the low temperature performance of water electrolytes. Additionally, we also focus on the storage of water molecules within hydrophilic matrices, leading to advancements in the transition of water electrolytes from a liquid state to a quasi-solid state. This review provides insights into current research directions for aqueous electrolytes and an emphasis on the crucial role played by water molecules in this process.
1. Introduction
Lithium-ion batteries are used in a wide range of important applications such as consumer electronics, transport and large power grids. However, their large-scale development has been hindered by several challenges, including the safety issues arising from electrolyte flammability and toxicity, high cost of electrode and electrolyte raw materials and complicated cell manufacturing procedures.1 Aqueous metal-ion batteries (AMIBs) have been proposed and designed as a candidate to solve the concerns of current commercial organic electrolytes,2,3 and show considerable potential to become a key technology in the field of energy storage. Aqueous electrolytes are environmentally friendly, safe, cost effective, and allow cell assembly under air conditions, which mitigate the safety hazards of AMIBs and reduce their management costs.4 Due to the reduced risk of thermal runaway, aqueous electrolytes are inherently less flammable and pose a lower safety risk in case of battery failure or mishandling. AMIBs usually exhibit higher rate performance and power density compared to traditional nonaqueous LIBs due to the high electrolyte dielectric constant and ionic conductivity, and low viscosity.The utilization and advancement of metal ion carriers beyond aqueous lithium-ion batteries have diversified the development trajectory of AMIBs. Alternative aqueous metal ion batteries, including those with Na+, K+, Zn2+, Ca2+, Mg2+, and Al3+ metal ions, present various advantages over lithium-based systems. These metal ions are more abundant and readily accessible than lithium, leading to lower production expenses and alleviating concerns regarding resource availability.5–7 More diversified metal ionophores greatly facilitate the selection of electrolyte and electrode materials. Aqueous battery electrode materials determine the energy density of the battery. In recent years, a number of excellent reviews have provided a comprehensive summary and shed light on the prospect of future development of electrode materials for aqueous rechargeable ion batteries in different directions and aspects.8–11 Nevertheless, the electrolyte also plays a crucial role in the overall improvement of battery performance, which acts as an ion bridge connecting the electrode materials and provides the ion transport current medium support for the cell reaction.12 Hence, comprehensive and in-depth understanding of aqueous electrolytes, as well as research on their optimization and modification, are particularly essential for tailoring the electrolyte of AMIBs to the electrochemical behavior and properties of the electrode material.
Water molecules, as the core component of aqueous electrolytes, play an important role in influencing the properties of aqueous metal-ion batteries. For example, the relative content of water molecules versus salt results in an unusual solvated structure. The chemical environment around the water molecules is affected by the hydrogen bonding between water molecules and interactions of water molecules with ions and organic additives,13 which contribute to the different behaviours and morphologies of the aqueous electrolyte, such as changes in ionic conductivity, widening of the electrochemical window, and lowering of the freezing point of the electrolyte, as well as endowing the aqueous electrolyte with high mechanical strength and deformability. This review provides an in-depth exploration of the effects of water molecules in aqueous electrolytes, highlighting their role in creating diverse application attributes for aqueous metal-ion batteries. The discussion encompasses a wide range of conditions, including (1) from dilute to high concentrations, (2) room temperature to extreme temperatures and (3) liquid to quasi-solid states. Detailed descriptions are provided to elucidate the influence of water molecules in each of these contexts.
2. Issues and challenges of AMIB electrolytes
Aqueous electrolytes contain a significant amount of water molecules, which can affect the safety and thermal behavior of the battery system. Water content also plays a role in the ionic conductivity, viscosity, and solubility of ions in the electrolyte. More importantly, the electrolysis voltage of water defines the voltage window range of the electrolyte. The Gibbs free energy of the water electrolysis reaction can be expressed as follows:| ΔG = −nEF |
| Eop = 1.23 V + ηa + ηc + ηother |
However, there are also some challenges associated with aqueous rechargeable ion batteries. Due to the inherent thermodynamic instability, pure water has a narrow voltage window. Exceeding the operating voltage range will continue to involve water decomposition, namely two half reactions of oxygen evolution reaction (OER) and hydrogen evolution reaction (HER). The OER and HER processes can be divided into three steps: aggregation of reactants on the surface of the electrode, formation of intermediate products, and production of oxygen and hydrogen gas.18 The hydrolysis of aqueous electrolytes can result in the degradation of the electrolyte and the formation of gas bubbles during cycling, leading to a decrease in the capacity and stability of the battery. Another main challenge of AMIBs is their limited voltage window, which is mainly due to the narrow electrochemical stability window of water-based electrolytes, limiting the energy density and power output of AMIBs. The limited voltage window can restrict the choice and compatibility of electrode materials. Many electrode materials are incompatible with each other in traditional dilute aqueous electrolytes, which further restricts the range of materials that can be used for AMIBs. Additionally, at decreasing temperatures, a significant population of free water molecules in the aqueous electrolyte readily engage in hydrogen bonding with one another, leading to the formation of crystalline structures. The performance of AMIBs can be affected by temperature, which can limit their usefulness in some applications. In addition, the use of free-flowing liquid state aqueous electrolytes, which primarily consist of a large number of free water molecules, poses challenges due to potential leakage and poor mechanical strength. These limitations make it difficult to adapt such electrolytes to meet specific environmental and usage requirements.
Overall, the challenges of aqueous electrolytes include the following points: (1) limited voltage window and low energy density: aqueous electrolytes typically have a narrower electrochemical stability window compared to non-aqueous electrolytes, due to the hydrogen and oxygen evolution side reactions. The voltage window restricts the selection of electrode materials. This limitation poses challenges in choosing electrode materials that can operate within the desired voltage range while achieving higher energy density for improved battery performance. (2) Limited cycle life: water molecules with high chemical activity can react with certain electrode materials, potentially leading to side reactions, passivation, or degradation of the electrode/electrolyte interface. These interactions can negatively impact the performance and stability of the battery, highlighting the importance of understanding the role of water molecules in aqueous electrolytes for optimizing battery design and function. (3) Limited operating temperature range: the narrower operating temperature range of aqueous electrolytes can restrict the performance and stability of AMIBs, particularly in cold environments where hydrogen bonds can easily form between water molecules, leading to the destruction of battery structure and impeding ion transfer. This highlights the challenges in designing aqueous electrolytes that can maintain stable performance over a wider temperature range. (5) Low mechanical strength and flexibility: liquid aqueous electrolytes may not withstand mechanical stresses or deformations, leading to leakage or loss of electrolyte integrity, which can indeed limit their application in certain scenarios.
Addressing these challenges will require a combination of innovative electrolyte design and deepen our understanding of the fundamental electrochemical processes involved in AMIBs. As research in the area of aqueous electrolyte continues, there have been numerous advances in their design and performance. The recent advancements in the development directions and technological path of aqueous battery electrolytes are summarized in this review. As shown in Fig. 1, in light of the crucial role that water molecules play in the properties and performance of aqueous electrolytes, our discussion will center around three key developments: (1) widening the voltage window and improving stability by increasing the electrolyte salt concentration; (2) broadening the operating temperature range to make AMIBs suitable for extreme environments; (3) from liquid to quasi-solid electrolytes to allow aqueous batteries to adopt versatile forms.
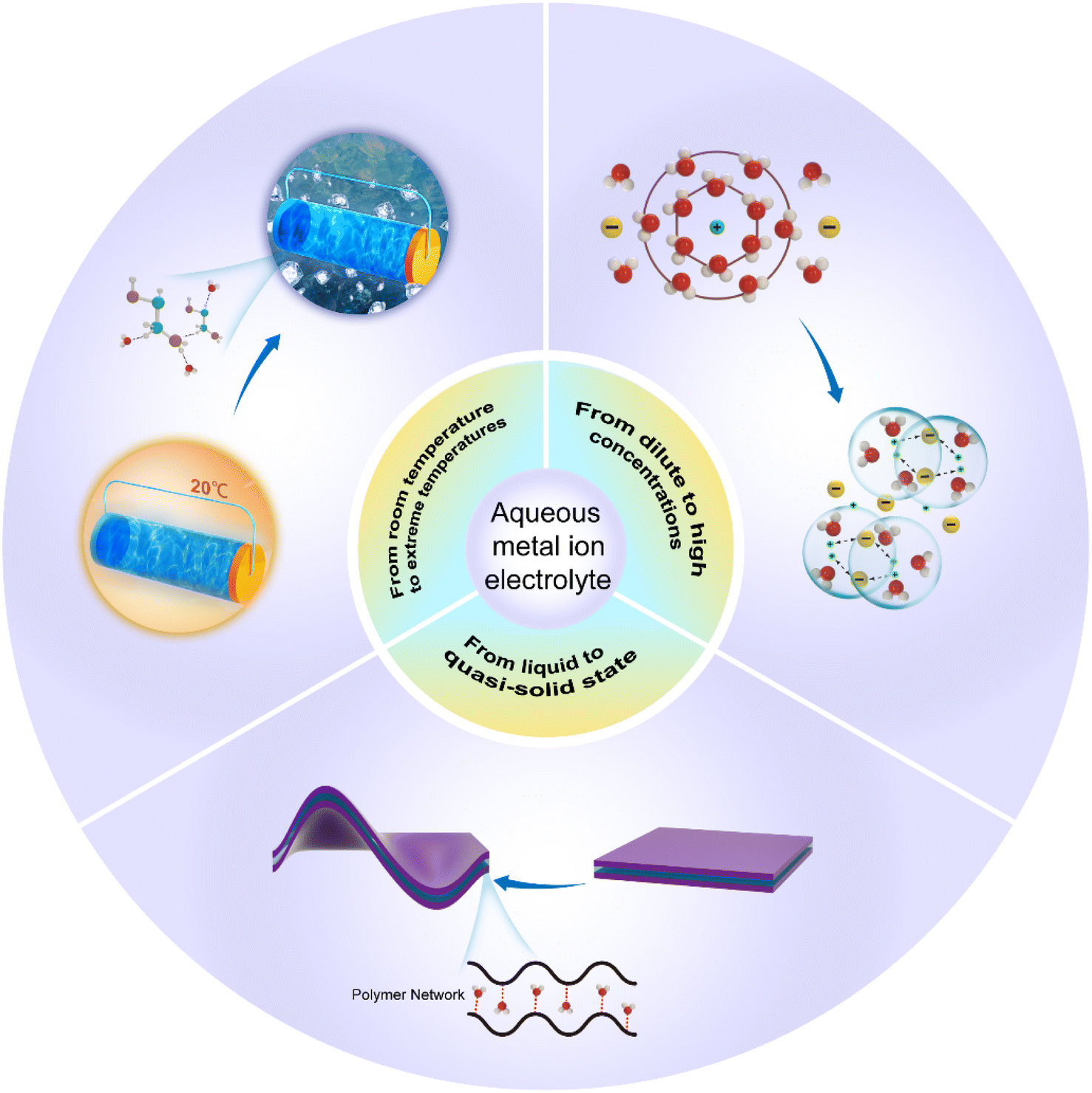 | ||
| Fig. 1 Schematic diagram of the development directions of aqueous metal ion batteries. | ||
3. Development strategy for AMIB electrolytes
3.1 From dilute to high concentrations
The first aqueous lithium-ion battery was designed and developed by Dahn et al. using a 5 M NaNO3 dilute aqueous electrolyte with an average voltage of approximately 1.5 V.19 As discussed previously, HER and OER potentials determine the electrochemical voltage window, both of which are functions of the pH value, allowing for the categorization of electrode materials that can be used in aqueous dilute electrolytes. The thermodynamic stability of the Li+ intercalation potential in aqueous media was discussed based on the following equation:20| V(x) = 3.885 − 0.118x (vs. Li/Li+) (x = pH) | (1) |
V(y) = 2.23 − 2kT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(y) (vs. Li/Li+) (y = Li+ concentration) ln(y) (vs. Li/Li+) (y = Li+ concentration) | (2) |
In diluted aqueous solutions, the coordination of ions by water molecules leads to the formation of anion/cation pairs surrounded by solvent molecules, with free water molecules also present as the dominant species. For LiTFSI and similar fluorinated electrolytes (Fig. 2a), when the molar concentration is below 5 M, they can be classified as dilute salt electrolytes. In this scenario, the weight and volume of the salt are lower than those of the solvent (water), and the average number of water molecules associated with each ion in the electrolyte satisfies the criteria for the formation of solvent-separated ion pairs (SSIPs).22,23 During the transport of ions in dilute aqueous electrolytes, they remain surrounded by water molecules, forming a solvation sheath. Fig. 2b illustrates the structure of the salt solvation sheath, highlighting how water molecules encapsulate the ions to maintain their solubility and facilitate their movement. This hydration shell plays a crucial role in the ion migration mechanism and influences various properties of the electrolyte, such as conductivity and diffusivity. In the context of dilute aqueous electrolytes, this migration mode, wherein ions are transported while being hydrated by water molecules, can be referred to as the “vehicle transport mechanism”.
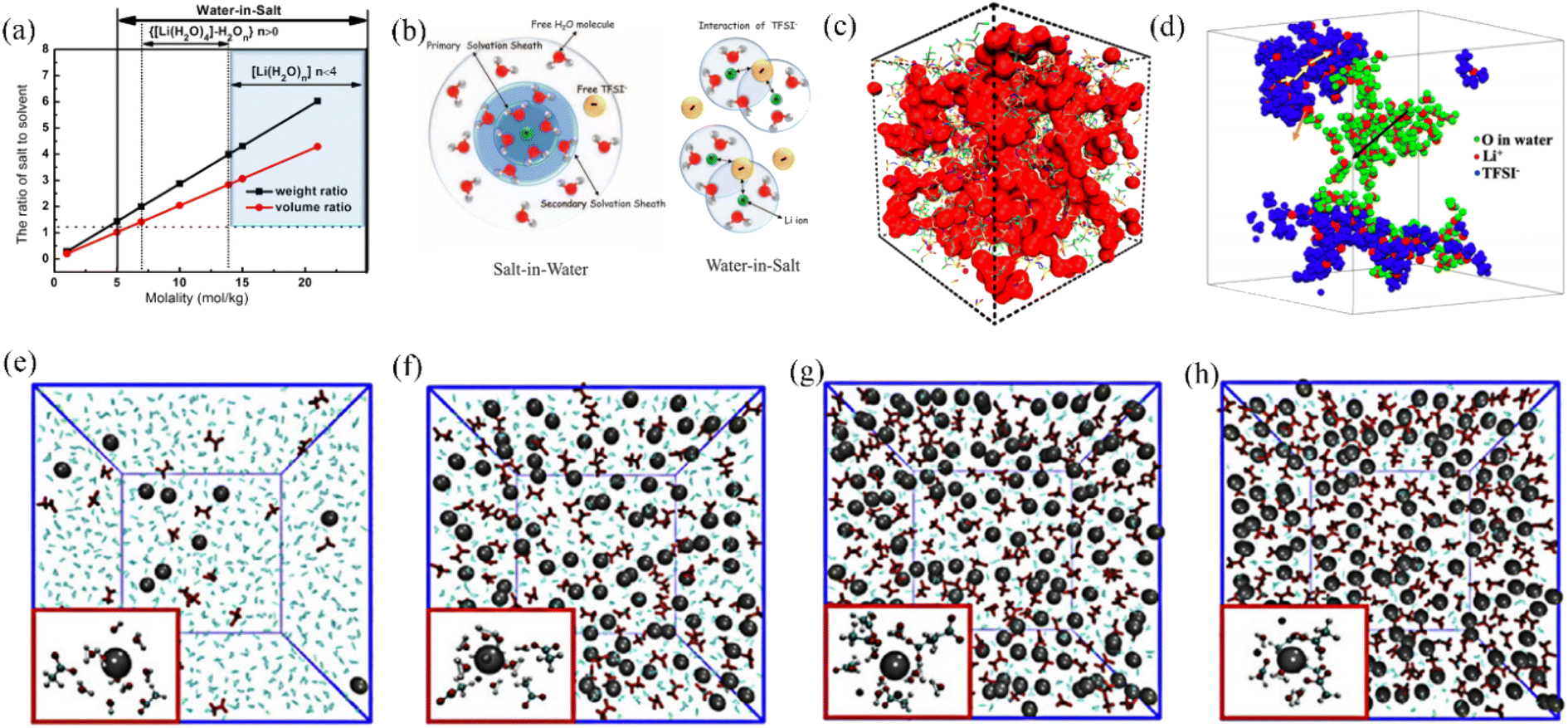 | ||
| Fig. 2 (a) The relationship between the molarity of LiTFSI in H2O and the corresponding changes in the weight and volume ratio of LiTFSI to H2O. (b) Structure of the solvation sheath of Li+ in aqueous dilute and high concentration LiTFSI electrolytes.22 Copyright © 2015 American Association for the Advancement of Science. (c) 21 M LiTFSI water-based electrolyte MD simulation.35 Copyright © 2017 American Chemical Society. (d) 20 M LiTFSI water and ionic aggregates simulations with Li+ diffusion pathways in the corresponding regions.36 Copyright © 2020 American Chemical Society. Simulation of equilibrium electrolyte visualization for (e) 1 m KAc, (f) 10 m KAc, (g) 20 m KAc, and (h) 30 m KAc, with insets showing the solvation environment around potassium ions with a radius of 0.4 nm.37 Copyright © 2020 Elsevier. | ||
The low voltage range for dilute electrolyte limits the choice of a wide range of materials, especially for anode materials at lower potential (approximately −1.08 to 0.25 V vs. SCE at different pH),21 which face severe hydrogen evolution side reactions. Increasing the pH of the aqueous electrolyte can reduce the reduction potential of hydrogen ions in water, allowing lower potential anode materials to be applied steadily.20,24 However, the overall voltage window of the aqueous electrolyte is not broadened, and the alkaline electrolyte obtained by adjusting the pH is not suitable for most electrode materials.25 So far the mild electrolytes are the dominant choice for dilute AMIBs.26 Nevertheless, some materials face serious dissolution problems in aqueous dilute salt electrolytes, resulting in severe degradation of capacity as the cycle progresses.8,27–31 In addition, the lower voltage window prevents the dilute AMIBs from achieving high energy density and power density.
:1, it tends to create a water-in-salt (WIS) system with a high salt concentration.34,38–41 Elimination of free water molecules in the WIS system also mitigates the water oxidation, which improves the electrolyte oxidation stability. As a consequence, the high-concentration aqueous electrolytes can operate at voltages higher than 3 V,42 which are much higher than the typical voltage limit for low-concentration electrolytes. This can enable much higher energy density and power output in AMIBs, making them more suitable for applications such as electric vehicles, renewable energy storage, and portable electronics.
Theoretical calculations and simulations provide insight into the interaction patterns between the high concentration of ions and water molecules in the WIS based aqueous electrolyte.43 Molecular dynamics (MD) simulations reveal the existence of a nanoscale heterogeneous ion–water solvated shell structure in a highly concentrated aqueous electrolyte system, which controls the migration of ion carriers.34 As shown in the red region in Fig. 2c, in the 21 M LiTFSI electrolyte, water molecules are present in the form of inhomogeneous interconnected nanoclusters. This heterogeneous structure enables cations to escape from the anion during the charging and discharging processes and forms a three-dimensional network structure with water molecules to facilitate nearly half of the rapid cation transport.35 The migration mode of ions in the WIS system differs from the “vehicle transport mechanism” observed in aqueous dilute electrolytes. In the WIS system, ions move through a cross-linked network created by anions and bound water molecules, a process known as the “hopping transport mechanism”. For example, in the aqueous electrolyte formed by LiTFSI, at dilute salt concentrations, sufficient free water molecules and ionized lithium ions are available, thus forming a solution with ions in a uniformly dispersed state.36 As the concentration increases, solvent molecules are gradually decrease and a nanometric heterogeneous shell distribution was formed.36,44 As shown in Fig. 2d, a 20 M LiTFSI aqueous electrolyte simulation diagram contains water-rich (red-green region) and anion-domains (red-blue region), with three potential diffusion modes for lithium ions: (i) migration along the black arrow in the water-rich separated region; (ii) diffusion along the yellow arrow in the anion-rich aggregation region; and (iii) lithium ions can exchange between TFSI− aggregates and water domains.36 Femtosecond infrared spectroscopy, combined with MD simulations, indicated that the anions and cations in the aqueous WIS system spontaneously agglomerate and hydrogen bond with water molecules to form a nanometric three-dimensional network bulk electrolyte. This porous electrolyte transport structure, formed by the ionic electrostatic interactions of water molecules and TFSI− ions, along with the lubricity of interfacial water, plays important roles in the diffusion of lithium ions.45 Normally high salt concentration leads to a decrease in the ionic conductivity of the electrolyte, but thanks to this nanometric water porous channel solvent structure, the ionic conductivity of the LiTFSI-based aqueous electrolyte is about 10 mS cm−1 even at a concentration of 21 M, which is still slightly higher than that of the conventional organic electrolyte system.26
In addition to the TFSI− anion, Han et al. combined Raman spectroscopy and MD simulations to analyze the liquid structure of a potassium acetate WIS electrolyte on a molecular scale (Fig. 2e–h). In a 1 M KAc aqueous electrolyte, the K+ ions are solvated by water molecules and distributed evenly. As the concentration of KAc increases, the number of free water molecules decreases significantly, leading to a more pronounced WIS effect. Unlike the hydrophobicity of fluoride-containing anions, where water molecules can only be coordinated with cations, at high concentrations of acetate-based aqueous electrolytes, water molecules are more prone to be solvated by acetate anions, which leads to a lower inhibition of water activity.37 Based on this observation, it can be inferred that anions in the aqueous electrolyte are crucial for the optimal performance of the WIS system. Anions demonstrate a dual functionality in this regard – they have the ability to disrupt hydrogen bonding, thereby increasing the electrochemical window, whilst also modulating the solvation structure of the electrolyte, leading to optimized electrochemical activity of free water. Certainly, the influence of cations on expanding the electrolyte window should not be overlooked. Cations with larger radii and greater masses have a tendency to strengthen polymer chains and simultaneously weaken hydrogen bonds. In Fig. 3a, it is evident that the WIS system consisting of K+ and Cs+ in an acetate-based aqueous electrolyte with a concentration of 7 M showcases a significantly broader electrochemical window of 2.5 V, surpassing that of Li+ and Na+. This highlights the importance of cation selection in optimizing the performance of the WIS system.46
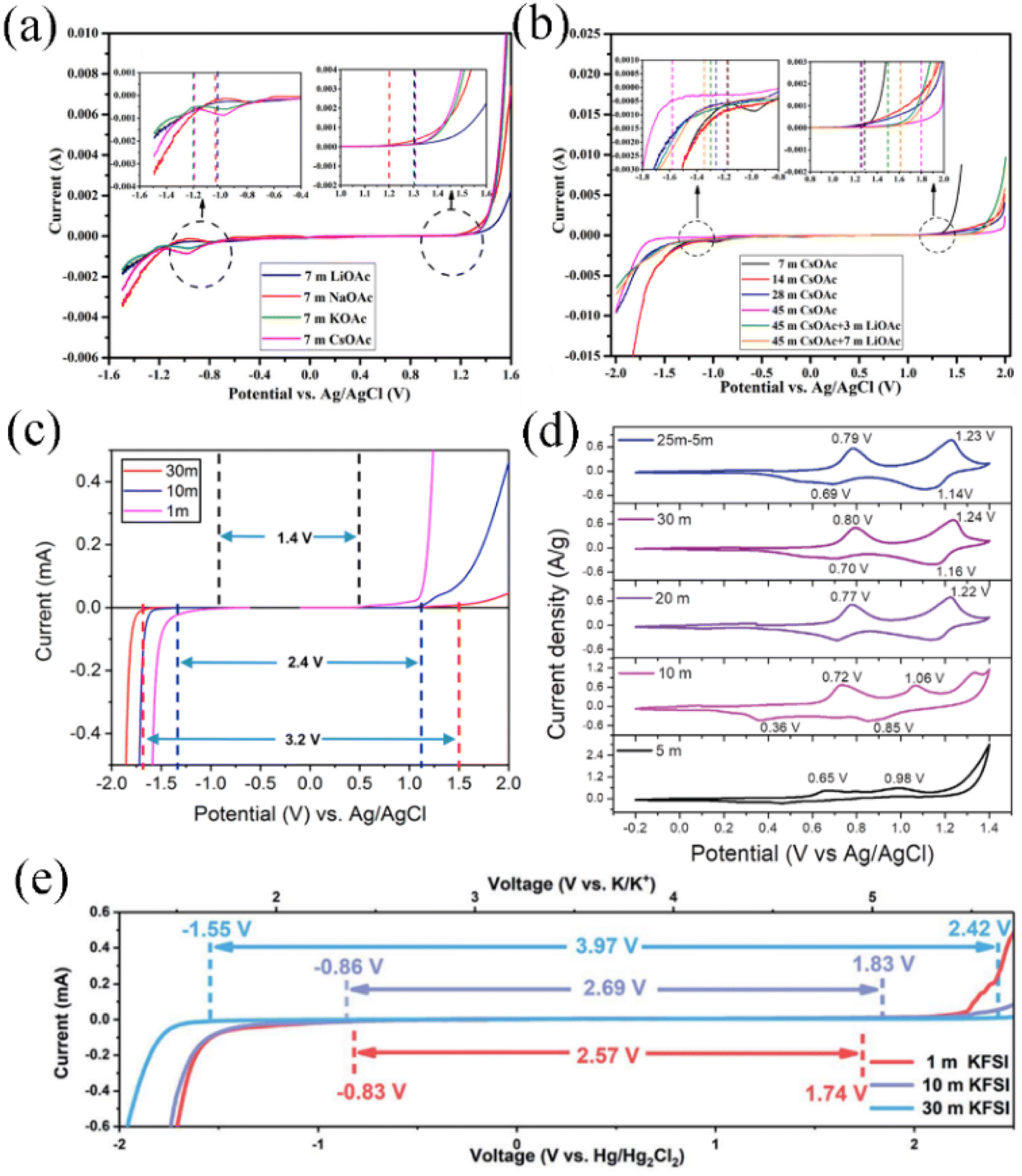 | ||
| Fig. 3 (a) Electrochemical window on 7 M acetate-based alkali metal ion aqueous electrolytes; (b) stabilized voltage windows for aqueous electrolytes with different concentrations of CsAc and LiAc + CsAc.46 Copyright © 2020 Elsevier. (c) Electrochemical stabilization windows recorded at 1 mV s−1 for 1, 10 and 30 M KAc electrolytes.47 Copyright © 2018 American Chemical Society. (d) CV curves of NVPOF electrodes at different concentrations of ZnCl2 and NH4Cl aqueous electrolytes.48 Copyright © 2020 Wiley-VCH. (e) Electrochemical stabilization windows recorded at 10 mV s−1 for 1, 10 and 30 M KFSI aqueous electrolytes.49 Copyright © 2020 The Royal Society of Chemistry. | ||
| Electrolyte | Voltage range | Ion conductivity | Anode//cathode | Cycle stability (rate) (capacity retention) | Ref. | |
|---|---|---|---|---|---|---|
| ALIBs | 21 M LiTFSI | 3 V (1.9–4.9) vs. Li/Li+ | 10 mS cm−1 | Mo6S8//LiMn2O4 | 1000 (4.5C) (68%) | 22 |
| 42 M LiTFSI + 21 M Me3EtN TFSI | 3.25 V (1.75–5.0) vs. Li+/Li | 0.91 mS cm−1 | Li4Ti5O12//LiMn2O4 | 100 (1C) (88%) | 85 | |
| 42 M LiTFSI + 21 M Pyr14 TFSI | — | 0.86 mS cm−1 | Li4Ti5O12//LiMn2O4 | 100 (1C) (90%) | 85 | |
| 35 M LiFSI | 2.5 V (−0.9 to 1.6) vs. Ag/AgCl | >30 mS cm−1 | — | — | 86 | |
| 14 M LiCl + 4 M CsCl | 2.7 V (−1.4 to 1.3) vs. Ag/AgCl | 31 mS cm−1 | TiO2//LMO | 1500 (5C) (87%) | 87 | |
| AZIBs | 1 M Zn(OTf)2 + 19 M LiTFSI | — | — | Zn//1,4-bis(diphenylamino)benzene | 1000 (6C) (75%) | 88 |
| 1 M Zn(OAc)2 + 31 M KOAc | 3.4 V (−1.45 to 1.95 V) vs. Ag/AgCL | 2.96 × 10−2 S cm−1 | Zn//α-MnO2-TiN/TiO2 | 600 (100 mA g−1) (79.7%) | 55 | |
| 30 M ZnCl2 | — | Zn//Ca0.20V2O5·0.80H2O | 1000 (1.6 A g−1) (70%) | 89 | ||
| ASIBs | 9.2 M NaOTF | — | — | Na2VTi(PO4)3/C//Na2VTi(PO4)3/C | 1000 (20C) (100%) | 90 |
| 17 M NaClO4 | 2.7 V (1.7–4.4) vs. Na/Na+ | 108 mS cm−1 | Na2VTi(PO4)3//Na4Fe3(PO4)2(P2O7) | 200 (1C) (75%) | 58 | |
| 9.26 M NaOTF | 2.5 V (1.7–4.2) vs. Na | 50 mS cm−1 | NaTi2(PO4)3//Na0.66[Mn0.66Ti0.34]O2 | 1500 (1C) (91.4%) | 91 | |
| 35 M NaFSI | 2.6 V (−1.15 to 1.45) vs. Ag/AgCl | 8 mS cm−1 | NaTi2(PO4)3//Na3(VOPO4)2F | — | 59 | |
| 25 M NaFSI + 10 M NaTFSI | — | 11.8 mS cm−1 (30 °C) | NaTi2(PO4)3//Na3(VOPO4)2F | 500 (0.2C) (77%) | 92 | |
| 9 M NaOTF + 22 M TEAOTF | 3.3 V (−1.7 to 1.6) vs. Ag/AgCl | 11.2 mS cm−1 | NaMnHCF//NaTiOPO | 800 (1C) (76%) | 93 | |
| AKIBs | 61.7 M K(FSI)0.55(OTf)0.45·0.9H2O | 2.7 V (2.05–4.75) vs. K+/K | 12 mS cm−1 (30 °C) | Pt//KVPO4F and ATi2(PO4)3 (A = Li, Na, and K) | — | 94 |
| 22 M KCF3SO3 | 3 V (−1.3 to 1.7) vs. Ag/AgCl | 76 mS cm−1 (25 °C) | Organic 3,4,9,10-perylenetetracarboxylic diimide//KxFeyMn1−y[Fe(CN)6]w·zH2O | 1000 (20C) (85%) | 56 | |
| 10 mS cm−1 (20 °C) | ||||||
| 30 M KAc | 3.2 V (−1.7 to 1.5) vs. Ag/AgCl | 28.1 mS cm−1 | KTi2(PO4)3//— | 11000 (1 A g−1) (69%) |
95 | |
| 30 M KFSI | 3.97 V (1.55–2.42) vs. Hg/Hg2Cl2 | — | β-PTCDA//KFHCF | 1000 (12.5C) (89%) | 49 | |
| AMgIBs | 4 M Mg(TFSI)2 | 2.0 V (1.7–3.7) vs. Mg/Mg2+ | — | Poly pyromellitic dianhydride//Li3V2(PO4)3 | 6000 (20C) (92%) | 80 |
| 3.5 M Mg(ClO4)2 + 1 M Zn(ClO4)2 | — | 1.41 mS cm−1 (−70 °C) | Zn//PTO or PNZ | 100 (0.5C) (100%) | 96 | |
| AAIBs | 5 M Al(OTF)3 | 3.6 V (−0.3 to 3.3) vs. Al/Al3+ | — | Al foil//AlxMnO2·nH2O | 65 (30 mA g−1) (80%) | 82 |
| 5 M Al(CF3SO3)3 | 2.65 V (−1.0 to 1.65) vs. Ag/AgCl | 26 mS cm−1 | —//Fe[Fe-(CN)6]0.79·2.1H2O | — | 97 | |
| AlCl3·6H2O:H2O = 16:1 |
4 V (0–4) vs. Ag/AgCl | — | Graphite//Al foil | 1000 (3C) (95%) | 84 | |
| 1 M Al(OTf)3 + 17 M LiTFSI + 0.02 M HCl | 3.2 V (−0.3 to 2.9) vs. Al3+/Al | — | Aluminum//S/C | 30 (200 mA g−1) (29%) | 98 |
Due to the limited lithium resources resulting in increased battery costs, the WIS strategy has also been applied in other AMIB systems with ion carriers like Na+, K+, Zn2+ and Al3+.37,47,48,51–56 Suo et al. first applied a WIS system in aqueous sodium ion batteries using 9.26 M sodium trifluoromethane sulfonate (NaOTF).57 The HER and HOR are effectively suppressed in aqueous sodium ion batteries and the voltage window is widened to 2.5 V. The Na0.66[Mn0.66Ti0.34]O2//NaTi2(PO4)3 full cell in the WIS system exhibits high cycle reversibility. NaTFSI salts, capable of concentrations up to 8 M, are also an option for the formation of WIS systems for aqueous sodium ion batteries.32,58 However, compared to the WIS system in aqueous Li-ion batteries (LiTFSI can reach 21 M), the proportion of free water in saturated NaTFSI aqueous electrolytes is higher. The electrochemical activity of water is not sufficiently suppressed. By means of preheating, Kühnel et al. obtained a 35 M sodium bis(fluorosulfonyl)imide (NaFSI) super-concentrated aqueous electrolyte at room temperature.59 The voltage window for aqueous sodium ion batteries is further broadened to 2.6 V. The preliminary electrochemical properties of the NaTi2(PO4)3 anode and Na3(VOPO4)2F cathode in 35 M NaFSI electrolyte were studied. Nevertheless, the chemical bond between S and F in the FSI anion is not very stable and prone to hydrolysis reactions in aqueous electrolytes.59 Saturated NaClO4 can reach 17 M in an aqueous electrolyte with a voltage window of 2.8 V, allowing more electrode materials to be selected in aqueous sodium ion batteries.60 An Na2MnFe(CN)6 cathode material shows a stable voltage plateau and good cyclability in 17 M NaClO4 aqueous electrolyte, while facing severe degradation when cycled in dilute aqueous electrolytes. An Na2Mn[Fe(CN)6] anode and KMn[Cr(CN)6] cathode were investigated in highly concentrated NaClO4 aqueous electrolyte systems. The Na2Mn[Fe(CN)6]//KMn[Cr(CN)6] full battery exhibited over 2 V operating voltage. The high concentration of NaClO4 can maintain a high ionic conductivity, so this aqueous sodium ion full cell also exhibits superior rate performance. However, NaClO4 is an explosive chemical reagent, whose hazardous properties limit large-scale production and application under high concentration conditions. On the basis of safety, in order to further improve the stable voltage window of the aqueous electrolyte, WIBS systems, such as hydrate melts asymmetric Na(SO2CF3)(SO2C2F5)−(PTFSI−),61 Na(PTFSI)0.65(TFSI)0.14(OTf)0.213H2O62 and 25 M NaFSI coupled with 10 M NaFTFSI,63 were also introduced in aqueous sodium ion batteries. Jiang et al. introduced tetraethylammonium triflate (TEAOTF) into an aqueous electrolyte for sodium ion batteries.64 As an inert cation, TEA+ possesses a large ionic radius which will not insert/extract into the electrode like sodium ions and is able to increase the concentration up to 31 M (22 M TEAOTF + 9 M NaOTF). The electrolyte potential window can ultimately reach 3.3 V. The NaMnHCF//NaTiOPO4 aqueous sodium ion full battery exhibits an operating voltage of 1.74 V and achieves excellent cycling stability with a capacity retention of 76% at 1C over 800 cycles. This unique system provides a new insight for increasing the concentration of AMIB electrolytes.
For aqueous potassium ion batteries, potassium acetate (KAc),47 KCFSO3 (ref. 56) and KFSI49,65 possess high solubility levels that enable the formation of WIS systems. Leonard et al. reported a WIS aqueous potassium ion battery based on KAc, which provides the advantages of low price and non-toxicity compared to fluorine-based salts, and is capable of forming a 30 M ultra-high concentration aqueous electrolyte with a voltage window of approximately 3.2 V (Fig. 3c).47 A KTP anode was first applied in the aqueous potassium ion battery system and showed stable electrochemical performance. Jiang et al. investigated the electrochemical behaviour and performance of Fe substituted for an Mn-based Prussian blue cathode and 3,4,9,10-perylenetetracarboxylic diimide (PTCDI) anode in 22 M KCF3SO3-based aqueous electrolyte.56 The WIS system achieved a wide voltage window of 3 V, effectively suppressing the dissolution of both electrode materials in active free water, and a KFeMn[Fe(CN)6]·H2O cathode material at 100C high rate capable of 10000 cycles with 70% capacity retention. Due to the wider ionic radius of potassium ions compared to lithium ions, KFSI-based aqueous electrolytes can be used up to a concentration of 30 M (Fig. 3e), enabling a stable electrochemical window of 3.97 V with cathodic and anodic potential limits of −1.55 V to 2.42 V vs. Hg/Hg2Cl2.49 The hydrated potassium salt melt, K(FSI)0.55(OTf)0.45·0.9H2O, formed through the combination of KFSI and KOTf, can achieve a molar concentration of 61.7 M, with a voltage window extending up to 2.7 V. Although its salt concentration is the highest degree attainable in known alkali metal ion aqueous electrolytes, it still has a high ionic conductivity of about 12 mS cm−1 at 30 °C.65
Aqueous zinc ion batteries, a representative of aqueous multivalent ion batteries, have received a great deal of research interest in recent years. In dilute aqueous electrolytes, the presence of a large number of free water molecules can lead to corrosion of the zinc anode surface. This corrosion process generates by-products such as H2, Zn(OH)2, or ZnO. As a consequence, the coulombic efficiency of the system is reduced.66–69 ZnCl2 exhibits high solubility of up to 30 M in aqueous electrolytes. As the concentration of ZnCl2 increases, the structure of the solvated sheath surrounding Zn2+ ions changes from octahedral [Zn(OH2)2Cl4]2 to tetrahedral [ZnCl4]2−. This structural change suppresses the formation of electrochemically non-active species such as Zn(OH)2 and ZnO. Additionally, the incomplete hydration shells of Zn2+ ions tightly bind water molecules, limiting their reactivity with the zinc anode. Consequently, a higher coulombic efficiency can be achieved in the 30 M ZnCl2 WiS electrolyte.70 In addition, the ultra-high salt concentration greatly reduces the free water content, avoiding the formation of side products in the Zn anode and widening the electrochemical working voltage window to 2.3 V.70 Ni et al. investigated the electrochemical behavior of an Na3V2(PO4)2O1.6F1.4 (NVOPF) cathode material in ZnCl2 and NH4Cl WIBS aqueous electrolytes. The two pairs of redox potentials of NVOPF in an AZIB were gradually revealed and they shifted to higher potentials with the increase of the ZnCl2 concentration (Fig. 3d). Compared to 30 M ZnCl2, a 25 M ZnCl2 + 5 M NH4Cl WIBS electrolyte with neutral pH was able to inhibit the dissolution of vanadium in NVOPF, resulting in a stable cycle life over 7000 cycles at 2 A g−1.48 In WIS (water-in-salt) aqueous zinc ion battery systems, the selection of the anion in the electrolyte is pivotal as it significantly influences the battery's performance and characteristics. Wang et al. demonstrated that the six coordination oxygen atoms in the TFSI− anion completely occupied the solvated sheath of Zn2+.71 This solvation sheath structure could inhibit the formation of Zn dendrites during the plating/stripping of Zn2+. However, Zn(TFSI)2 is not as highly soluble as ZnCl2 in aqueous electrolyte. To realise the WIS system, aqueous zinc ion electrolytes are commonly utilized in a dual-salt hybrid configuration. A novel hybrid WIS aqueous electrolyte (1 M Zn(TFSI)2 + 20 M LiTFSI) was designed to achieve the concentration required for the anion to form the WIS system, widening the stable voltage platform to 3.2 V.72,73
AZIBs can utilize a range of electrolytes, including both inorganic zinc salts like Zn(ClO4)2, Zn(NO3)2, ZnSO4, and ZnCl2, as well as organic zinc salts such as Zn(CH3COO)2, Zn(CF3SO3)2, and Zn(TFSI)2.66 The options of ultra-high concentration electrolytes in AZIBs are restricted by the physical properties of solubility of the solute. ZnCl2 displays exceptional solubility, enabling it to reach ultra-high concentrations of up to 30 M in an aqueous solution at 25 °C. In contrast, other zinc salts, such as ZnSO4, Zn(NO3)2, and Zn(CH3COO)2, have limited solubility and cannot be used to prepare similar ultra-high concentration electrolytes. For instance, ZnSO4 has a solubility of 3.4 M, for Zn(NO3)2 it is 0.67 M, and for Zn(CH3COO)2 it is 2.3 M, and they are all confined by their limited solubility. Zn(ClO4)2 exhibits a stable structure and lower reactivity due to the tetrahedral coordination of four O atoms around the central Cl atom. The electrolyte Zn(ClO4)2 demonstrates stability; however, the presence of by-products like ZnO and Zn(OH)2 on the zinc anode significantly impacts reaction kinetics.74,75 SO42− shows a stable structure attributed to the presence of four similar S–O bonds within the SO42− groups. ZnSO4 is known for its stable electrochemical performance in aqueous solutions, thanks to the inclusion of SO42−. However, ZnSO4 is often hindered by the formation of Zn4(OH)6SO4·nH2O, which can adversely affect the cycling stability of the system.76,77 The system utilizing a Zn(CF3SO3)2 electrolyte demonstrates remarkable reversibility and fast zinc deposition/dissolution kinetics, which can be attributed to the relatively large size of the CF3SO3− anion. The anion's large size facilitates a reduction in water molecules surrounding Zn2+ ions. Furthermore, the higher conductivity of CF3SO3− compared to SO42− promotes the migration and charge transfer of Zn2+.78 The large volume of TFSI− in the electrolyte also helps to reduce the amount of water around Zn2+, minimizing the solvation effect.79 However, as discussed above, different zinc salts exhibit varying levels of solubility in aqueous solutions. The ionization degree of a salt is influenced by the binding strength between its anions and cations; Zn(TFSI)2, due to the strong binding affinity between TFSI− anions and zinc ions, exhibits low ionization and limited solubility. In contrast, zinc chloride, with weaker binding strength, can easily ionize, making it suitable for high-concentration aqueous electrolytes.
Aqueous high concentration systems also have relevant applications and studies with other multivalent ions, such as Mg2+, Ca2+ and Al3+. Similarly, water in these aqueous multivalent ion electrolytes also plays a crucial role in the electrochemical behavior of the cell and the ion migration process. In an aqueous magnesium ion battery, 4 M concentration of Mg(TFSI)2 can form a stable voltage window of 2 V, which can effectively suppress hydrogen and oxygen evolution side reactions, and enable an Li3V2(PO4)3 cathode and polypyromellitic dianhydride (PPMDA) anode to carry out a stable charging and discharging process.80 In aqueous calcium ion batteries with α-V2O5 as the host material, the co-intercalation of H+ and Ca2+ is correlated with the Ca(TFSI)2 electrolyte concentration. However, as the salt concentration increases, lower water content will lead to an increase in electrochemical overpotential.81 For the aqueous aluminium-ion battery, the standard reaction potential of aluminium metal is −1.68 V, which is lower than the hydrogen evolution reaction potential of water in dilute salt electrolyte. Although metallic aluminium has a high theoretical capacity (2980 mA h g−1), it cannot be deposited and stripped stably in an aqueous electrolyte. Therefore, it is promising and valuable to study the utilization of WIS electrolytes to improve the aqueous aluminium ion battery.82–84 The concentration of Al(OTF)3 in aqueous solution can reach 6 M, forming a WIS system and a stable voltage window up to 3.6 V. The WIS aqueous electrolyte formed by the low-cost aluminum salt AlCl3·6H2O can increase the electrochemical stability window to 4 V, allowing metallic aluminium to be used directly as the anode electrode to assemble aqueous aluminium ion full cells. The capacity is 165 mA h g−1 at 500 mA g−1 current density and can be stable for 200 cycles.84
For the oxygen reduction reaction (ORR), intermediate products like H2O2 and OH− on the electrode surface in a dilute aqueous electrolyte will diffuse rapidly and quickly dissolve into the dilute aqueous electrolyte, which will accelerate the ORR kinetics (Fig. 4a), while in a high salt concentration electrolyte (21 M LiTFSI + 8 M LiOTf), the oxygen content and free water molecules will be reduced, thus inhibiting the process of ORR. Although a minimal amount of H2O2 is detected in the 28 M WIS electrolyte, its concentration is significantly lower compared to that in dilute electrolytes. Furthermore, the slow solubility and diffusion rates of H2O2 at high concentrations greatly restrict the progress of the ORR in the WIS system. In addition, the final reaction product on the electrode surface beyond the potential in the WIS electrolyte will further generate Li2O2. This insulating Li2O2 nano-passivation layer can slow the diffusion of oxygen and reduce the ORR (Fig. 4b).99
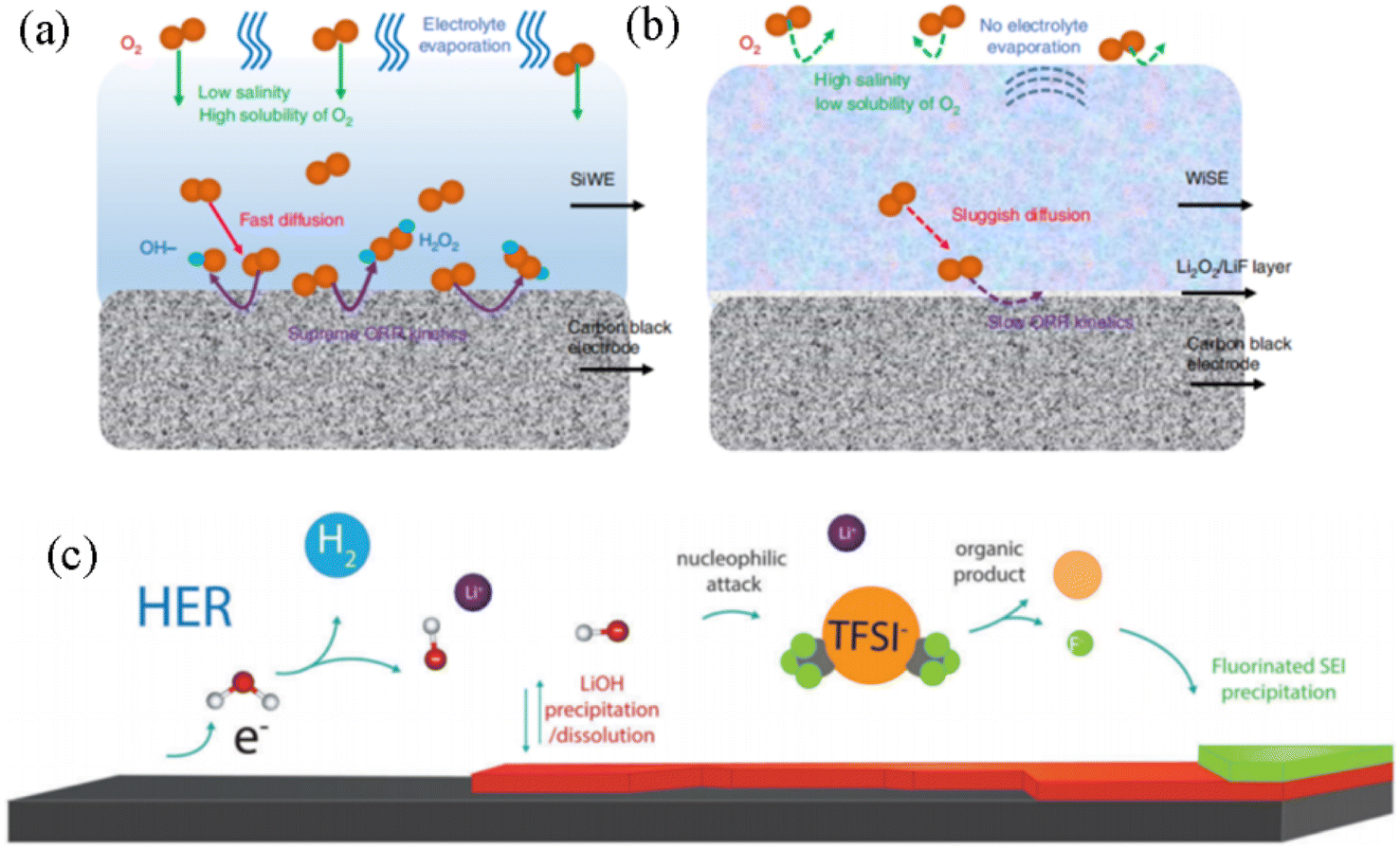 | ||
| Fig. 4 Schematic diagram of the ORR process on the electrode surface in (a) 1 M dilute aqueous electrolyte and (b) 28 M high concentration aqueous electrolyte.99 Copyright © 2020 Springer. (c) Schematic diagram of the SEI formation mechanism and process in 20 M LiTFSI WIS electrolyte.100 Copyright © 2018 The Royal Society of Chemistry. | ||
For EER side effects, the bound water and free water in the WIS electrolyte correspond to different reaction potentials. The decomposition of a small amount of water molecules on the electrode surface triggers dynamic precipitation and dissolution of high salt concentrations, and decomposition and deposition on the electrode surface to form SEI films.101 The chemical properties of the anions play a key role in expanding the electrochemical voltage window. Incorporating the anion into the first solvation shell of the cation facilitates anion decomposition, enabling the formation of less soluble compounds that create an electrode/electrolyte interphase on the anode surface. This process kinetically suppresses electrolyte decomposition and enhances overall stability. For example, based on the process and mechanism of SEI film formation on the electrode surface in 21 M LiTFSI aqueous electrolyte, the TFSI− anion will be preferentially reduced. Firstly, at excessively low potentials, a certain amount of water is reduced to generate H2 and OH−. Partial LiOH will also be formed on the anode electrode surface. Under reducing conditions, the highly reactive OH− and LiOH will nucleophilically attack and interact with the TFSI− anion to degrade and generate CF3 groups, and catalyze the formation of fluorine-containing products (LiF, CFx) on the surface of the electrode to form an SEI passivation layer (Fig. 4c).100 An inhomogeneous and highly fluorinated electrolyte additive is added directly to the anode electrode surface (graphite or Li-metal) of the aqueous battery,102 which will form an inner-Helmholtz layer on the electrode surface after cyclic reduction, excluding free water molecules from direct contact with the electrode surface. Characterization by XPS and TEM reveals that the SEI film formed on the electrode surface exists in amorphous form and consists of fluorinated hydrocarbons and inorganic fluorides. The assembled series of AMIB full cells are capable of achieving an output voltage of 4 V.102
The formation of a stable SEI film is an important consideration in the development of high-performance AMIBs, as it can help to improve the stability and cycle life of the AMIBs. However, the formation of the film can also be a complex process, and further research is needed to optimize the formation and performance of SEI films in AMIBs.
To minimize the use of salt, electrolyte additives like acetonitrile (AN),103 dimethyl sulfoxide (DMSO),104 methanol,105 and polymer poly(ethylene glycol) (PEG)106 were introduced, leading to the emergence of new concepts and patterns in electrolyte design. Using acetonitrile (AN) as a water co-solvent helps reduce the presence of water at the interface by creating a uniform and thin interphase on the negatively charged electrode surface. This interphase is composed of a sulfamide and nitrile-based organic outer layer along with a LiF-rich inner layer. This structure significantly enhances the electrochemical stability window up to 4.5 V by inhibiting water molecule decomposition. Cao et al. demonstrated that the incorporation of DMSO as a co-solvent into the ZnCl2–H2O electrolyte could significantly alter the coordination environment of the electrolyte. The presence of DMSO as a co-solvent leads to the effective replacement of H2O molecules in the Zn2+ solvation sheath with DMSO molecules. The residual water molecules then undergo strong interaction with DMSO. This adjustment results in the effective customization of the solvation structure of water, even at 1.3 M ZnCl2 dilute aqueous electrolyte. The inclusion of DMSO in the ZnCl2–H2O electrolyte effectively mitigates the side reactions triggered by water molecules on the surface of the zinc negative electrode. This ZnCl2–H2O–DMSO electrolyte enables zinc anodes in a half-cell configuration to achieve a notably high average coulombic efficiency of 99.5%. Hao et al. introduced the concept of an antisolvent, a solvent that can be mixed with the original solvent but cannot dissolve the solutes in the solution. In their study, methanol was added to the ZnSO4-based aqueous electrolyte. The investigation of ZnSO4 in various ratios of antisolvent electrolytes revealed a gradual disappearance of the Zn2+–nH2O solvation structure as the amount of antisolvent increased. This disappearance was attributed to the strong interaction between methanol and H2O, which disrupted the interaction between H2O and Zn. Interestingly, when the concentration of methanol surpassed a certain level, some Zn2+ ions were observed to recombine with SO42−. These alterations in solvation structures, caused by the antisolvent effect, effectively reduced water activity, leading to an expanded voltage window within the electrolyte system. What's more, the concept of molecular crowding electrolytes was utilized to efficiently modify the bonding network of water by confining water molecules within macromolecules. Xie et al. used PEG as the crowding agent and constructed a series of molecular crowding electrolytes based on 2 M LiTFSI, with PEG ratios ranging from 71% to 100%. The results showed that the ESW of the electrolyte increased as the PEG concentration increased, reaching 3.2 V when the PEG content was 94 wt%. The negatively charged ethereal oxygen in PEG forms strong H–O bonds with water molecules, which are stronger than the hydrogen bonding between water molecules. This hydrogen bonding between PEG and water disrupts the hydrogen bonding between water molecules, leading to a reduction in water activity.
3.2 From room temperature to extreme temperatures
The discharge capacity, cycling stability and rate performance are all important electrochemical indicators of AMIBs which are very sensitive to the test temperature. It is common to keep the environmental temperature variable of the battery test room strictly constant (usually 20 °C) when studying the electrochemical properties and conducting data analysis of AMIBs. However, many storage applications require electrolytes that can function efficiently under a wide range of conditions. The practical application of AMIBs will involve harsh and variable external factors. Especially at subzero temperatures, normal AMIBs will be frozen, which will cause sluggish kinetics and deteriorated electrochemical performance. The adaptation of AMIBs under extreme conditions, especially at excessively high or low temperatures, is an essential research area that needs to be investigated.107,108The AMIB electrolyte may face the following challenges under low temperature conditions: (a) aqueous electrolyte solidification leads to retarded reaction kinetics. (b) The electrolyte resistance and the charge transfer resistance of AMIBs increase significantly. (c) Precipitation or crystallisation of solutes and freezing of the aqueous electrolyte reduce cycle stability. (d) Low ionic conductivity leads to poor rate performance. (e) The infiltration angle will increase as the temperature drops, resulting in insufficient wettability between the aqueous electrolyte and the electrode material.109,110 Currently, lowering the freezing point of aqueous electrolytes is the mainstream and core view to solve these mentioned issues.
| ΔT = Tsolution − Twater = K·m |
The hydrogen bonding between water molecules significantly influences the freezing point of the electrolyte. Various salts dissolved in aqueous solutions display different interaction strengths with water molecules. As per the Hofmeister series,113 anions tend to exhibit stronger interactions with water compared to cations. Furthermore, the hydration of ions is directly related to the surface charge density. Ions with higher surface charge density tend to enter the hydrated shell layer and form bonds with water molecules, thereby disrupting the hydrogen bonding network among water molecules.114 Consequently, this disruption enables the attainment of lower freezing point temperatures. Yushin et al. investigated the low-temperature performance of saturated aqueous electrolytes with low-cost inorganic salts for aqueous lithium-ion battery single-salt systems including LiNO3, Li2SO4, and LiCl, respectively.112 Among them, the saturated LiCl (16 M) aqueous solution remained ice-free even at −50 °C, where the intercalated Prussian blue LiCoO2 is able to maintain 72% of its room temperature capacity. Interestingly, compared to conventional organic electrolytes, all three saturated aqueous electrolytes exhibit better rate performance and less charge transfer impedance at a low temperature of−10 °C. Zhang et al. investigated the effect of different anion types on the low-temperature performance of aqueous zinc ion batteries.115 Five anions (SO42−, NO3−, Cl−, I−, and CF3SO3−) were studied in depth experimentally and theoretically. The electrostatic potential value of CF3SO3− was the highest, calculated using density functional theory (DFT), indicating that it is easier to form hydrogen bonds with the hydrogen atoms of water molecules. From the FTIR spectra, the Zn(CF3SO3)2 aqueous electrolyte has weaker strength compared to the other zinc salt solution, accounting for its lower freezing temperature. The relationship between the macroscopic low temperature parameters of anions corresponding to aqueous electrolytes is shown in Fig. 5a. At the same concentration of zinc salts (2 M), Zn(CF3SO3)2 exhibits an impressive low-temperature performance, with a freezing point of −34 °C and an electronic conductivity of 4.47 mS cm−1 at −30 °C.
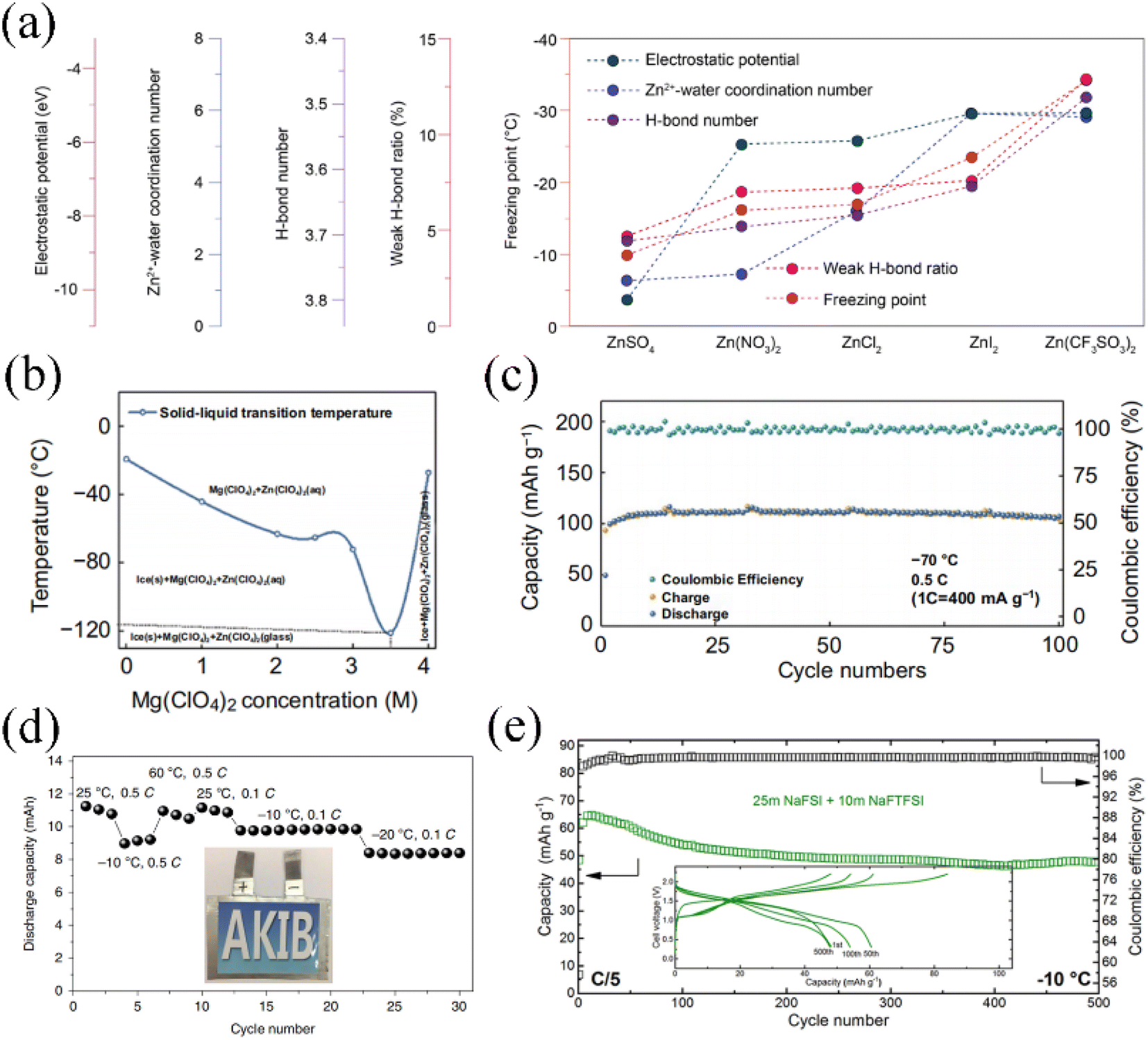 | ||
| Fig. 5 (a) Summary of the trends in properties' values for different aqueous electrolytes for ZnSO4, Zn(NO3)2, ZNCl2, ZnI2 and Zn(CF3SO3)2.115 Copyright © 2021 American Chemical Society. (b) Profiles of Mg(ClO4)2 concentration versus freezing point for Mg(ClO4)2 + 1 M Zn(ClO4)2 aqueous electrolyte. (c) Cycling stability of the Zn//PTO full battery in 3.5 M Mg(ClO4)2 + 1 M Zn(ClO4)2 aqueous electrolyte at−70 °C.96 Copyright © 2021 Springer. (d) The 22 M KCF3SO3 AKIB cycling performance at different temperatures.56 Copyright © 2019 Springer. (e) The 25 M NaFSI + 10 M NaFTSI ASIB cycling performance at −10 °C.63 Copyright © 2019 American Chemical Society. | ||
The freezing point of the electrolyte is highly correlated with the salt concentration. Sun et al. applied 3.5 M Mg(ClO4)2 and 1 M Zn(ClO4)2 dilute salt concentration aqueous electrolytes to an AZIB,96 which achieves an extremely low freezing point and ensures high ionic conductivity and a low viscosity coefficient. Fig. 5b shows the relationship between Mg(ClO4)2 concentration and freezing point for this aqueous electrolyte. The solid–liquid transition temperature reaches a minimum of −121 °C at a Mg(ClO4)2 concentration of 3.5 M. In addition, the synergistic effect of cations and anions in the aqueous electrolyte can effectively reduce the water–water hydrogen bonding. In this case, Mg2+ can capture the oxygen atoms of water molecules to form hydrogen bonds, while the hydrogen atoms in water molecules are attracted by the strong electrostatic force of ClO4−. The discharge capacity of the assembled Zn/Pyrene-4,5,9,10-tetraone (PTO) full cell is 101.5 mA h g−1 at a current density of 400 mA g−1, with almost no capacity decay for 100 cycles (Fig. 5c). When the concentration reaches the WIS level, as described in the previous section, the amount of salt far exceeds that of water molecules, which proves to be effective in decreasing the freezing point of the aqueous electrolyte. Wang et al. investigated the low-temperature performance of LiTi2(PO4)3/Li3V2(PO4)3 aqueous lithium-ion full cells in 21 M LiTFSI electrolyte. Thanks to the very low content of free water molecules in the WIS system and the high ionic conductivity of NASICON-type electrode materials, the capacity of the full cell at −20 °C is 91% of the capacity at room temperature.116 Aqueous potassium ion batteries based on a 22 M KCF3SO3 WIS system are also capable of achieving stable performance at temperatures ranging from −20 to 60 °C (Fig. 5d).56 Reber et al. employed the symmetric anionic salt sodium bis(fluorosulfonyl)imide (NaFSI) mixed with the asymmetric anionic salt sodium (fluorosulfonyl)(trifluoromethylsulfonyl)imide (NaFTFSI) to form the WIS system for an ASIB.63 By incorporating FTFSI− asymmetric anions, the WIS electrolyte structure and long-range ordering were modified, which stabilizes the crystallization kinetics for the salt at high concentrations and allows for a wider low-temperature window. In this type of asymmetric mixed WIS aqueous electrolyte, the Na3(VOPO4)2F/NaTi2(PO4)3 full cell exhibited excellent low temperature performance, with a capacity retention of 74% at −10 °C for 500 cycles at 0.2C (Fig. 5e). Similarly, upon adding the asymmetric salt lithium (pentafluoroethanesulfonyl)(trifluoromethanesulfonyl)imide (LiPTFSI), the ALIB in this WIS system remains thermodynamically stable at −10 °C.117 This asymmetric anion-based method successfully further broadens the temperature range of the WIS aqueous electrolyte in the liquid state over 30 °C and maintains high ionic conductivity and stable electrochemical properties. For WIS electrolytes with suitable anions, the low content of free water molecules thermodynamically contributes to a reduced freezing point. To a certain extent, an acceptable electronic conductivity of the aqueous electrolyte is preserved.
Nevertheless, the WIS system with ultra-high salt concentration is near the verge of saturation, which is very sensitive to the ambient temperature. During cycling at low temperatures, precipitation or crystallization of the salt will occur, which can influence the stability of the electrochemical performance. Besides, most WIS electrolytes working at low temperatures will suffer from low ionic conductivity and high viscosity coefficients. The strong interactions between ions and water molecules lead to high viscosity and poor mobility.
Aqueous electrolytes contain water–water hydrogen bonds and water–solvent hydrogen bonds. When solvent components have both acceptors and donors, another type of solvent–solvent hydrogen bond can occur. If the binding energy of the hydrogen bond between water and the solvent is greater than that of the water–water hydrogen bond, water molecules will preferentially form hydrogen bonds with the solvent.120
Introducing organic solvent additives into aqueous electrolytes to form a co-solvent with water is an effective method for widening the liquid low-temperature range. This approach requires the solvent molecule to contain functional groups that bind more strongly with water molecules. The organic solvents replace a certain proportion of water molecules in the aqueous electrolytes. The added solvent molecules are more likely to form hydrogen bonds with water molecules, disrupting the bonding network between water molecules, making it challenging for the organic/aqueous hybrid electrolyte to form a solid crystalline structure at low temperatures. The solvent additives need to possess the following traits: (a) an inherently low melting point; (b) solubility or compatibility with aqueous solutions; (c) higher hydrogen bonding energy with water than that of water–water. The organic additives that have been reported to contribute effectively to the low-temperature performance of aqueous electrolytes include ethylene glycol,121,122 acetonitrile103,123 and dimethyl sulfoxide.124 However, it should be noted that the dielectric constants of organic solvents are usually lower than those of aqueous solutions. The co-solvent method achieves excellent low-temperature performance at the expense of its ionic conductivity to some extent.
Ethylene glycol (EG) is a common anti-freeze additive for aqueous solutions, which can effectively widen the low temperature range. Varying the content of the EG (0–40 wt%) organic additive affects the low-temperature performance of aqueous electrolytes. Although 40 wt% EG corresponds to the lowest freezing point, excessive introduction of EG will reduce the ionic conductivity of the electrolyte. When the co-solvent electrolyte is composed of 10 wt% EG and 1 M Li2SO4 aqueous solution, the LiFePO4 cathode ALIB exhibited improved low-temperature electrochemical performance compared to other EG contents. The ALIB composed of 20 wt% EG exhibits better properties at −20 °C.121 EG can form strong hydrogen bonds with water, which inhibits the freezing of electrolytes at low temperatures. Experimental and theoretical calculations have also demonstrated that the addition of EG weakens the solvation of Li+ and Zn2+ ions with water, facilitating rapid and reversible ion conduction. In Fig. 6a, it is shown that the coordination number of Zn2+ and H2O decreases with increasing EG content, indicating that the addition of EG significantly disrupts the solvated shell of Zn2+ with H2O. Importantly, the diffusion coefficient of Zn2+ initially increases and then decreases with the increase in EG content, suggesting that the solvation structure formed by an appropriate amount of EG molecules with Zn+ and water can promote the rapid transport of Zn2+. The Zn//V2O5 full cells in an EG-containing co-solvent electrolyte provide stable cycling performance (250 cycles with almost no degradation shown in Fig. 6b) and high energy density (121 W h kg−1).122 Chen et al. introduced acetonitrile (AN) with 15.3 M aqueous LiTFSI to form a co-solvent electrolyte,103 which substantially suppresses water content and achieves an extremely wide electrochemical stability window of 4.5 V. Compared to dimethyl carbonate as a nonaqueous solvent, AN exhibited higher ionic conductivity in the temperature range of −20 to 60 °C (Fig. 6c).The assembled LiMn2O4/Li4Ti5O12 full cell at 0 °C can exhibit a capacity of 110 mA h g−1 after 120 cycles with a capacity retention of 95% (Fig. 6d). The freezing point of the AN organic additive is −48 °C and its dielectric constant is nearly half that of water, which allows the AN–water co-solvent electrolyte to inherit the advantages of high conductivity and low viscosity coefficient of aqueous electrolytes. By changing the ratio of AN and water, it is feasible to obtain a hybrid solvent WIS electrolyte with 21 M LiTFSI, which has a wider temperature range (−30 °C to 50 °C).123 Interactively, the WIS system also achieved an AN breakthrough of its inherent instability at low voltage. This co-solvent electrolyte system is compatible with the lithium ion intercalation reactions into graphite.38 DMSO is an antifreeze additive that contains a highly hydrophilic sulfinyl group. In a hybrid DMSO–water solution, it exhibits a core–shell solvent structure where DMSO molecules encapsulate the water molecules, preventing the tendency of water to form an ordered crystal structure as it cools down. This behavior helps inhibit the freezing of the solution at low temperatures. Adjusting the optimal ratio of DMSO and water molecules can yield an apparent freezing temperature of −130 °C (Fig. 6e). Nian et al. employed DMSO–water in a ratio of 0.3 as the co-solvent electrolyte. The intermolecular hydrogen bonding between DMSO and water enables the system to exhibit good low-temperature electrochemical performance at −50 °C when applied to aqueous lithium/sodium/potassium ion batteries (Fig. 6f).124
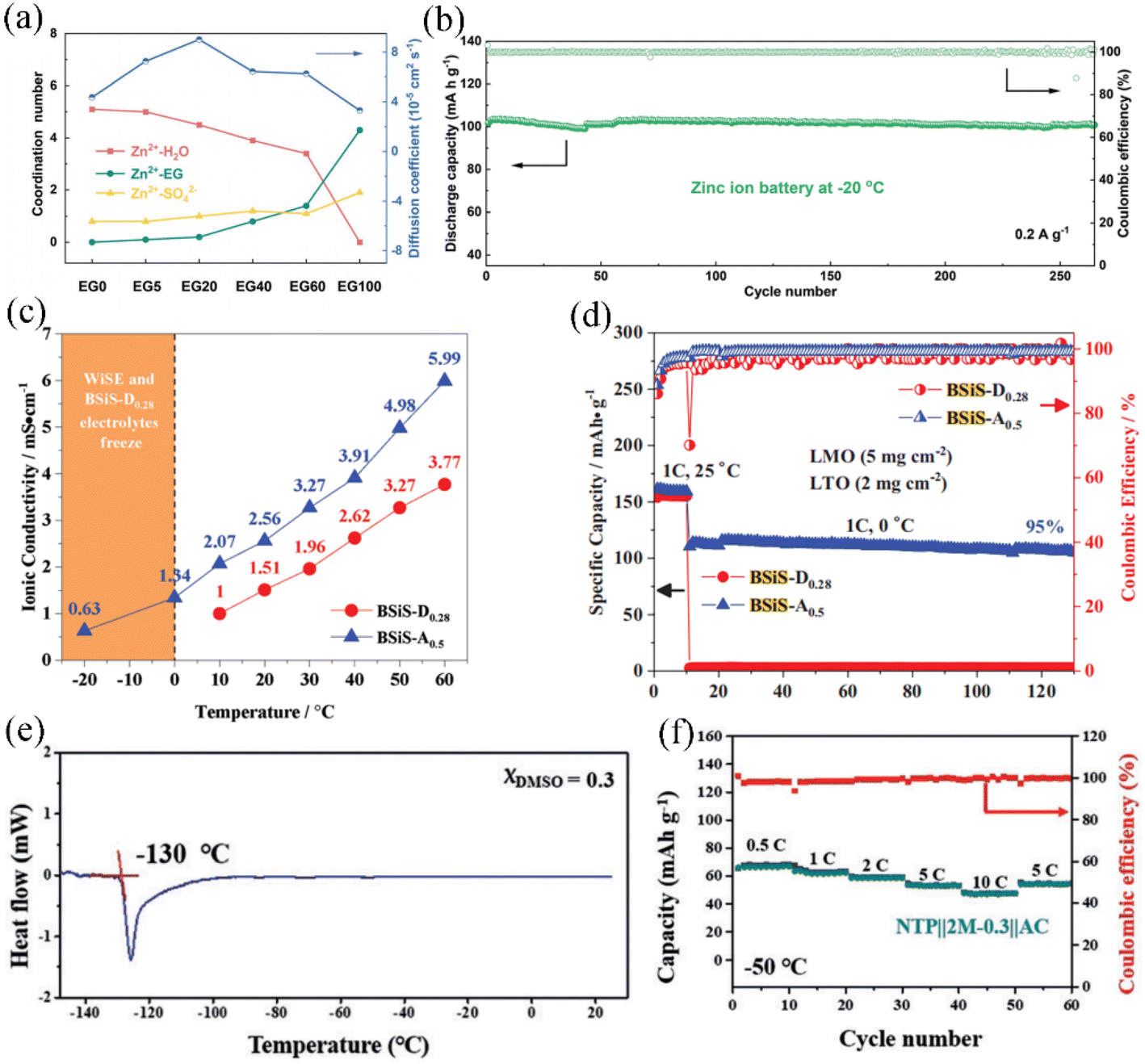 | ||
| Fig. 6 (a) Variation of coordination number and Zn2+ diffusion coefficient in aqueous electrolytes with different contents of EG.122 Copyright © 2020 The Royal Society of Chemistry. (b) Cycling performance of a Zn//V2O5 full cell in an aqueous EG-containing co-solvent electrolyte at −20 °C. (c) Comparison of ionic conductivity of hybrid aqueous electrolytes containing DMC and AN in the temperature range of −20 to 60 °C.103 Copyright © 2019 Wiley-VCH. (d) The cycling performance of LMO//LTO aqueous cells containing DMC and AN at 25 °C and 0 °C. (e) Differential scanning calorimetry (DSC) of the aqueous electrolyte with a DMSO molar fraction of 0.3. (f) Rate performance for NTP//AC ASIBs at −50 °C.124 Copyright © 2019 Wiley-VCH. | ||
Organic additives not only significantly lower the freezing point, but also replace water molecules in the solvation shell of cations. This interaction between organic molecules and cations promotes the rapid ion transport of AMIBs. Co-solvent electrolytes inherit the advantages of aqueous electrolytes and organic solvents. The premise is that the organic additives must be selected with high safety to preserve the vital safety properties of the aqueous electrolyte.
The organic hydrogels formed by the addition of cryoprotectants (glycerol,129 EG,130,131 acetonitrile and oil132) with the hydrophilic polymer network are effective in preventing water from freezing. Introduction of the EG organic additive into a polyvinyl alcohol (PVA)-based aqueous electrolyte resulted in a binary system electrolyte with high flexibility, strong mechanical strength and self-healing properties, while also being able to operate in a wide temperature range between −55.0 and 44.6 °C.130 Another similar binary system, polydopamine (PDA)-modified CNT, forms a conductive hydrogel that crosslinks catechol groups on the network adhering to water molecules to achieve the antifreeze effect.129 Mo et al. designed a novel polymer matrix, EG-waterborne polyurethane acrylates, with polyacrylamide (EG-waPUA/PAM) hydrogel electrolyte. Compared with the traditional single PAM-based hydrogel electrolyte, the EG-waPUA/PAM system could cooperatively anchor water molecules, enabling better freeze-resistant and superior low-temperature electrochemical performance.133 A Zn//EG-waPUA/PAM//MnO2 zinc ion full cell can deliver a reversible capacity of 226 mA h g−1 at −20 °C, which is 82.18% the capacity at room temperature. Chen et al. incorporated borax and glycerol into a PVA-based gel electrolyte (PVA-B-G) which formed a cross-linked structure with a glycerol–water biosolvent.125 This unique design of the borax-crosslinked PVA/glycerol electrolyte presents good flexibility, strong mechanical properties, high ionic conductivity (10.1 mS cm−1) and wide temperature range. Glycerol further suppresses the crystallization of water in the PVA-based gel, allowing this electrolyte to remain in the liquid state at ultra-low temperatures. The PVA-B-G electrolyte can remain very soft and retain high flexibility even at −35 °C (Fig. 7a). PVA-B-G based aqueous batteries show a slight increase in charge transfer impedance during the ambient temperature drop from 25 to −35 °C, indicating that faster reaction kinetics and ion transport rates are still ensured at low temperatures (Fig. 7b). The MnO2//Zn AZIB applied to this quasi-solid electrolyte showed excellent cycling stability in the temperature range of −35 to 25 °C, with 90% capacity retention after 2000 cycles (Fig. 7c). In addition to incorporating an organic antifreeze agent in combination with the hydrogel electrolyte, adding a suitable amount of ionic compounds is another effective strategy. The interaction between ions and hydrogels and the synergistic hydration between ions can both achieve ultra-low temperature performance by disrupting the hydrogen bonds between water molecules. For example, when polyacrylamide–alginate gels are immersed in aqueous calcium chloride solutions, the quasi-solid gels are able to resist freezing at −57 °C.134 Wei et al. reported that in a PAM-based functional gel electrolyte incorporating ZnSO4, glycerol and acetonitrile (ZS/GL/AN gel), the cation Zn2+ and water molecules would interact with the oxygen-containing functional groups, allowing the zinc anode to remain highly reversible at a wide temperature range.126 The Zn//V2O5 aqueous full battery in the ZS/GL/AN electrolyte showed a high reversible capacity of 138 mA h g−1 at −20 °C and 262 mA h g−1 at 60 °C and was able to cycle stably for 500 cycles at both extreme temperatures (Fig. 7d and e). Compared with the aqueous solution ZnSO4 electrolyte conditions, the battery low-temperature performance is significantly improved. Chen et al. reported the introduction of 2 mol L−1 ZnSO4 and 4 mol L−1 LiCl into a polyacrylamide hydrogel.127 The mixed hydrated cations formed by lithium and zinc ions synergistically dissociated the hydrogen bonds between water molecules and significantly reduced the freezing point of the aqueous electrolyte. From the CV test of the Zn//LiFePO4 full AMIBs, it retains highly reversible and symmetrical redox peaks, although the charge transfer and conversion kinetics are impacted at −20 °C, which indicates a good low-temperature electrochemical performance (Fig. 7f). This aqueous full cell exhibited good electrochemical stability in the rate performance study at −20 °C, even outperforming itself at room temperature with low current densities (Fig. 7g). According to the Hofmeister effect, anions such as ClO4− can weaken the hydrogen bonds between water molecules.128 Huang et al. designed a dual network hydrogel which combines with the polysaccharides carboxymethyl chitosan (CMCS) and PAM with ClO4− inserted. This design aims to break the hydrogen bonds within the polymer chains and water molecules, effectively lowering the freezing point of aqueous electrolytes. Upon incorporating ClO4− ions into the structure, they disrupt the hydrogen bonding network between the polymer chains and water molecules, thereby improving the low-temperature performance of the electrolyte in applications such as energy storage systems (Fig. 7h). The Zn//PANI aqueous batteries exhibit stable cycling performance in this deformable hydrogel electrolyte, with 2500 cycles at a high current density of 5 A g−1 at −30 °C (Fig. 7i).
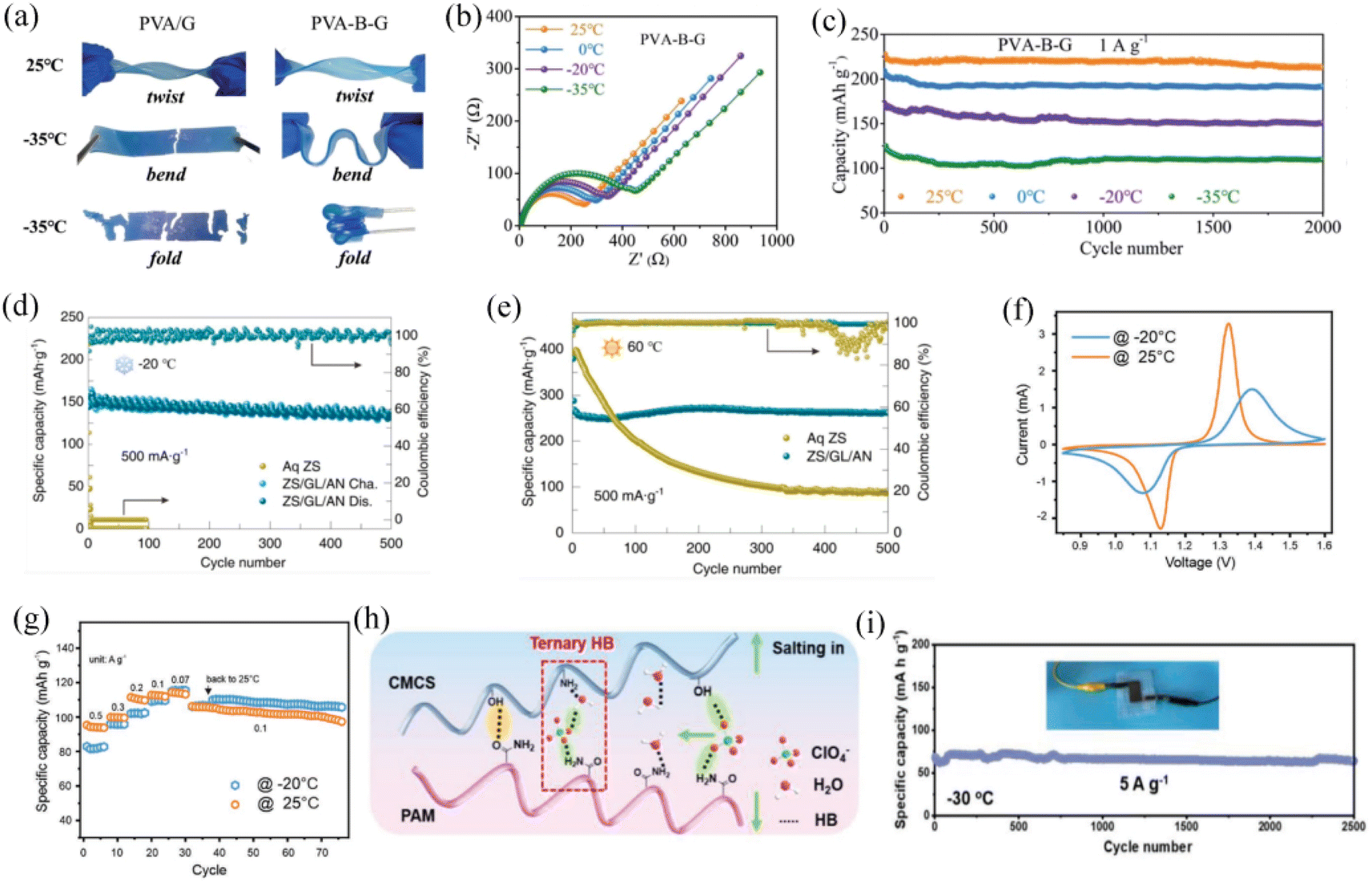 | ||
| Fig. 7 (a) Flexibility comparison between PVA-G and PVA-B-G gel electrolytes at 25 and −35 °C. (b) Electrochemical impedance spectroscopy of a PVA-B-G based aqueous battery from 20 to −35 °C. (c) Cycling performance of a PVA-B-G based AZIB from 20 to −35 °C.125 Copyright © 2020 The Royal Society of Chemistry. (d and e) The cycling performance of a Zn//V2O5 aqueous battery in ZnSO4 aqueous electrolyte and ZS/GL/AN gel electrolyte at 60 and −20 °C.126 Copyright © 2022 Elsevier. (f and g) Comparison of CV measurement and rate performance for a Zn//LiFePO4 hybrid aqueous battery at −20 and 25 °C.127 Copyright © 2020 Wiley-VCH. (h) Structural simulations of ClO4− anion insertion in CMCS and PAM organic chains. (i) Cycling curve of the Zn//PANI flexible battery at 5 A g−1 at −30 °C.128 Copyright © 2022 Wiley-VCH. | ||
Table 2 shows the operating temperature range for different aqueous electrolyte compositions. Although the temperature range is widely extended, operating AMIBs in sub-zero temperatures is still a significant challenge. Aqueous electrolytes used in cold environments need to maintain their liquid state and ionic conductivity. The essence of several approaches to improve the low-temperature performance of aqueous electrolytes is to break and reduce the hydrogen bonding between water molecules. In recent years, the influence of anions and cations, organic additives, and hydrogel polymers on the hydrogen bonding of water molecules has been more thoroughly studied with encouraging progress achieved.
| Electrolyte composition | Temperature range | Ref. |
|---|---|---|
| 22 M KCF3SO3 | −20 to 60 °C | 56 |
| 8 M NaTFSI | −10 °C | 63 |
| 9 M NaOTf | −10 °C | 63 |
| 16 mol kg−1 LiCl | −45 to −50 °C | 112 |
| 9 mol kg−1 LiNO3 | −20 to −30 °C | 112 |
| 3.5 mol kg−1 Li2SO4 | −30 to −35 °C | 112 |
| 2 M ZnSO4 | −9.8 °C | 115 |
| 2 M Zn(NO3)2 | −16.1 °C | 115 |
| 2 M ZnI2 | −23.4 °C | 115 |
| 2 M Zn(CF3SO3)2 | −34.1 °C | 115 |
| 3.5 M (mol L−1) Mg(ClO4)2 + 1 M Zn(ClO4)2 | −121 °C | 96 |
| 21 M LiTFSI | −20 °C | 116 |
| LiOTf:LiPTFSI = 10:10 |
−10 °C | 117 |
| 1 M Li2SO4 + 20 wt% EtG | −20 °C | 121 |
| 2 M ZnSO4 (EG:H2O = 6:10) |
−40 °C | 122 |
| 5 M LiTFSI (H2O:acetonitrile = 1:2.6) |
−30 °C | 123 |
| 0.3 M DMSO + 2 M NaClO4 | −50 °C | 124 |
| GW-hydrogel | −20 to 60 °C | 129 |
| 66.6 wt% EG + 33.3 wt% H2O + 9 wt% PVA + 0.2wt% PEDOT:PSS |
−55.0 to 44.6 °C | 130 |
| AF-SH-CPAM | −20 °C | 131 |
| EG-waPUA/PAM | −20 °C | 133 |
| Borax-crosslinked polyvinyl alcohol (PVA)/glycerol | −60 °C | 125 |
| Polyacrylamide–alginate double network hydrogels + 30 wt% CaCl2 | −57 °C | 134 |
| ZS/GL/AN | −20 to 60 °C | 126 |
| 2 mol L−1 ZnSO4 + 4 mol L−1 LiCl + polyacrylamide hydrogel | −20 °C | 127 |
3.3 From liquid to quasi-solid states
Traditionally, aqueous electrolytes have been used in liquid form and widely studied in various energy storage applications due to their high ionic conductivity, low cost, and low toxicity. The lack of flexibility in liquid electrolytes restricts their compatibility with certain form factors or designs. Devices that require unconventional shapes or conformal designs may not be easily accommodated by liquid electrolytes due to their inability to conform to complex geometries. Aqueous quasi-solid-state batteries have thus been proposed and widely studied.The concept of quasi-solid electrolyte changes the contact surface between the electrode material and electrolyte from solid–liquid to solid–solid. It may alleviate the side reactions of hydrogen and oxygen evolution, surface passivation, and dissolution of electrode materials compared with the liquid aqueous electrolyte. Besides, the quasi-solid electrolyte possesses additional physical merits including flexibility,136–140 stretchability,135,141 compressibility,142 self-healing131,143,144 and shape memory.135,145 Adjusting and modifying the gel polymer can also change its chemical properties.146 Quasi-solid electrolytes can avoid electrolyte leakage due to their deformation capability compared to liquid electrolytes. They play the dual role of electrolyte and separator in aqueous battery systems, which is the cornerstone for obtaining high-performance aqueous batteries under complex mechanical deformations. Based on these characteristics, quasi-solid electrolytes can be used in a wide range of applications such as wearable consumer electronics, medical electronics, artificial intelligence, aerospace and national security.18,66,146
AMIBs with quasi-solid-state electrolytes are typically assembled into two types of shape, planar and fibrous structures. The planar structure requires high contact and adhesion between the quasi-solid electrolyte and the surface of the electrode material, as it could be prone to slip or even detachment during deformation, resulting in localized failure of the battery. Integrating quasi-solid electrolyte and electrode materials to form a tightly and seamlessly connected full cell can alleviate the negative effects of deformation. Reducing the thickness of the quasi-solid electrolyte also improves the overall deformation capability of the AMIBs. Fiber-structured quasi-solid electrolyte batteries have higher volume specific capacity and adjustable structure compared to planar structures.147,148 The fibrous structure is usually paired with a tubular or linear electrode material, wrapped around a quasi-solid electrolyte, and packaged in a fiber-like material. The fiber structure requires a note that the design and assembly process should try to avoid direct contact between the cathode and anode electrodes.149 Fibrous coaxial structure and spring structure can increase the effective contact surface of the electrodes while avoiding a short circuit.150
Quasi-solid aqueous electrolytes are required to retain high ionic conductivity, high stability, low cost, and good adhesion to the electrode surface. The use of quasi-solid-state aqueous electrolytes is still a relatively new area of research. To optimize their performance, there are several challenges waiting to be addressed: (i) achieving high ionic conductivity while maintaining mechanical stability; (ii) the solid–solid contact between the quasi-solid electrolyte and the electrode material is prone to slip or disconnect during its deformation, affecting the performance of the aqueous battery; (iii) the quasi-solid electrolyte in the deformation state is more likely to cause electrode active material detachment during charge and discharge. Overall, the fundamental electrochemical and chemical processes within quasi-solid-state aqueous electrolytes still demand thorough investigations.
To avoid repeated and long-term deformation of AMIBs during operation and the consequential battery degradation or failure, quasi-solid electrolytes are developed with other advanced functions, such as self-healing and shape memory. The quasi-solid self-healing ability derives from hydrogen bonding, metal–ligand coordination, and reversible cross-linking reconstruction between polymers. The shape memory function provides the quasi-solid electrolyte with the ability to return to its original shape after the withdrawal of an external force and to maintain the original electrochemical properties.
The flexibility and electrochemical properties of quasi-solid electrolytes are directly related to the internal polymer matrix, which is the core component for the quasi-solid electrolyte. The design of the physical structure and modification of the chemical properties mainly focused on their polymer matrix. The topics of interest for polymer matrixes are preparation methods, safety, chemical stability, mechanical strength, hydrophilicity and compatibility with metal salts. The ionic conductivity, cycling performance, tensile strength and self-healing properties of aqueous quasi-solid electrolytes based on different polymer matrices are compared in Fig. 8. The detailed electrochemical performance of the quasi-solid-state electrolyte AMIBs is summarized below in terms of the representative polymer matrix.
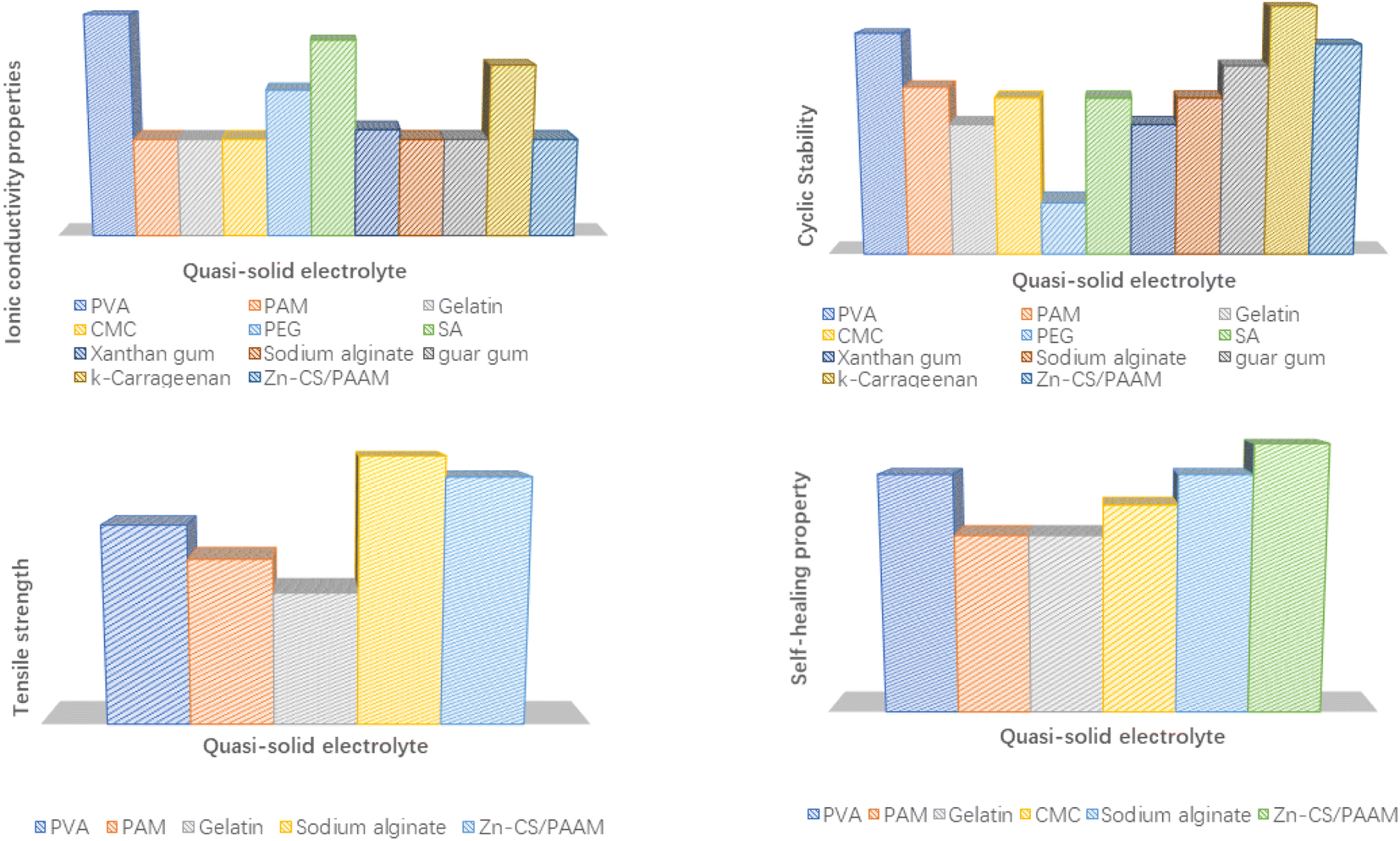 | ||
| Fig. 8 Comparison of the ionic conductivity, cycling performance, tensile strength and self-healing properties of aqueous quasi-solid electrolytes with different polymer matrixes. | ||
Polyvinyl alcohol (PVA) contains an abundance of functional groups that have the ability to anchor water molecules. This property has made PVA a popular choice for effectively imparting quasi-solid electrolyte properties to AMIBs. The PVA homopolymer can improve the electrochemical window of an aqueous electrolyte by minimizing water content; for example, in Zn–Li hybrid electrolytes, the addition of PVA reduces the electrochemical activity of the LiClO4 aqueous electrolyte, which in turn improves the energy density and cycling stability of the full cell.151 The addition of PVA enables the wearability and flexibility of AMIBs. For example, the introduction of PVA into the 21 M LiTFSI WIS system applied to the aqueous electrolyte of the LiVPO4F symmetric AMIBs not only dramatically improves the electrochemical stability, but also greatly enhances its flexibility.152 A chemically cross-linked PVA hydrogel electrolyte was reported and used for Co3O4/Zn AMIBs. Unlike physical cross-linking, chemically cross-linked PVA-based quasi-solid electrolytes exhibit enhanced mechanical strength and variability, and can deliver significant rate performance (116C).153 PVA as a polymer matrix also exhibits good compressing and stretching properties.141,154 The PVA-Zn(CF3SO3)2 quasi-solid electrolyte in aqueous zinc ion batteries survived 1000 cycles of a 60% pressing/releasing test without losing its initial electrochemical properties.154 Wang et al. developed a cable-type AZIB based on a ZnTFS-PVA quasi-solid electrolyte. The Zn–MnO2 full cell achieved high flexibility without sacrificing electrochemical performance, demonstrating high discharge capacity (188 mA h g−1) and durable capacity retention (95% for 100 cycles).140 Niu et al. prepared a Zn(CF3SO3)2-PVA hydrogel electrolyte with recovery and healing properties through hydrogen bonding by the freeze–thaw method.144 The PVA polymer matrix forms a cross-linked porous network structure in which Zn2+ and CF3SO3− were uniformly dispersed. This transparent quasi-solid electrolyte achieved superior ionic transport rates. Mechanically split Zn(CF3SO3)2-PVA hydrogel electrolytes can self-recover and bind to become like the original state after half an hour under the effect of hydrogen bonding without external stimulation (Fig. 9a).The polyacrylic acid (PAA) polymer additive, similar to PVA, possesses a physically cross-linked form based on hydrogen bonding, which results in an excellent mechanical ductility and self-healing ability. Zhi et al. observed almost no cracks under SEM in the prepared PAA electrolyte after stretching it to 37 times of its original size.155
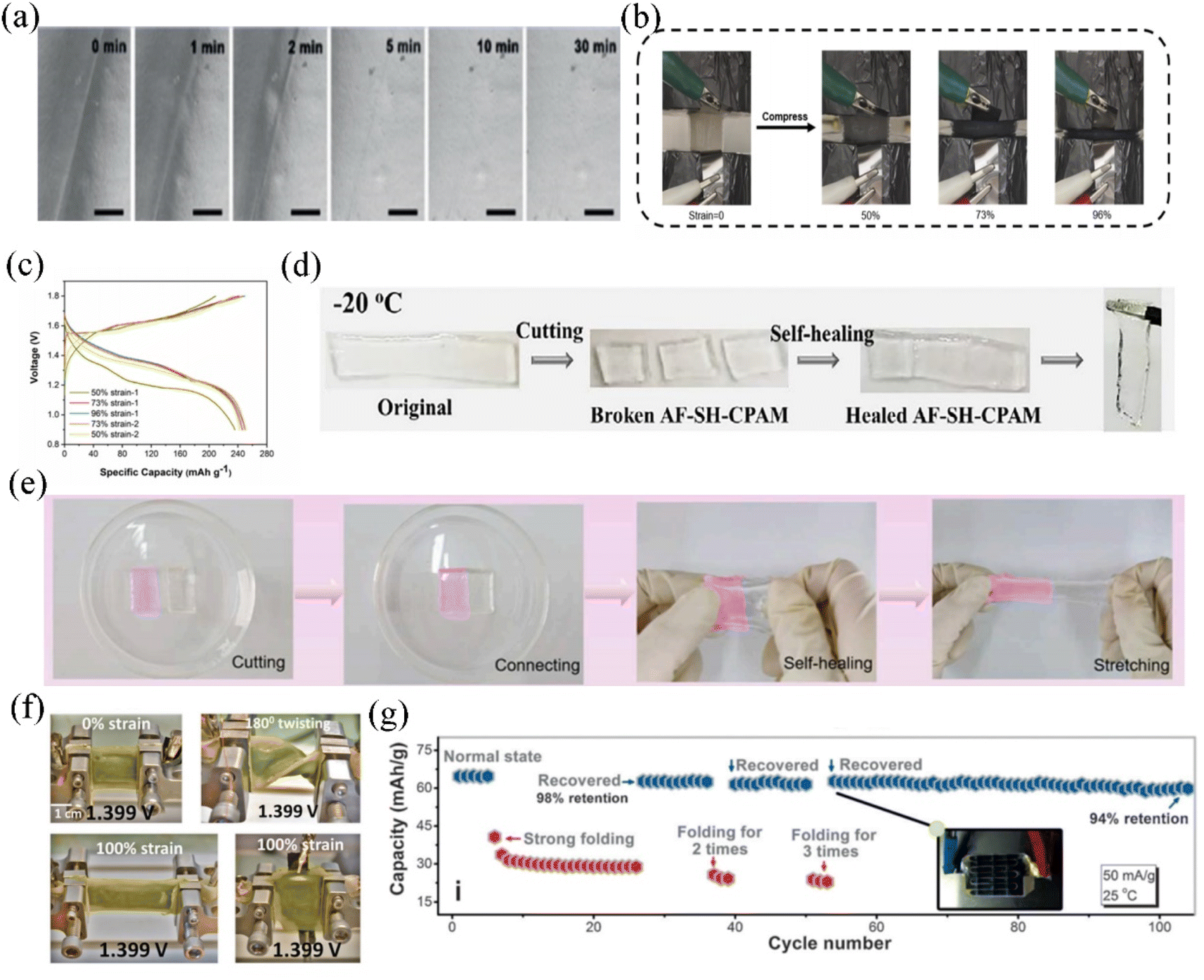 | ||
| Fig. 9 (a) SEM image of the self-recovery process of the quasi-solid state electrolyte.144 Copyright © 2019 Wiley-VCH. (b) The Zn–MnO2 aqueous quasi-solid state batteries under different compressive stresses of 0, 50%, 73% and 96%. (c) Charge–discharge curves under different compressive stresses.142 Copyright © 2021 Elsevier. (d) Self-recovery processes of the quasi-solid state electrolyte at −20 °C.131 Copyright © 2021 Elsevier. (e) Self-healing processes of the sheared hydrogel electrolytes.143 Copyright © 2022 Elsevier. (f) The final cell tested under twisted and stretched 100% deformation for the open circuit voltage shot image.160 Copyright © 2021 Wiley-VCH. (g) Charge/discharge curves of an AMIB with strong folding/recovery three times during the charge/discharge process.161 Copyright © 2017 Wiley-VCH. | ||
Zhi et al. introduced another chemically cross-linked PAM polymer combining ZnSO4 and MnSO4 as a hydrogel electrolyte applied in an MnO2/Zn AZIB.156 Thanks to the three-dimensional porous structure of the gel electrolyte and the large number of hydrophilic functional groups on the PAM polymer, it exhibits a high ionic conductivity of 1.65 × 10−2 S cm−1 and good suitability of salt solutes and gel polymers. This novel AZIB was assembled into a rope-like shape with assistance from CNT in both the anode and cathode. The capacity retentions of such quasi-solid-state full cells were nearly 100% under various forms (straight, bent, knotted, twisted, and straight again). In addition, PAM-based solid-state electrolytes enable excellent mechanical stretching and compression properties.142,157 A quasi-solid-state Zn–MnO2 cell with high compression reversibility was designed by Jiang et al.142 The polymer matrix consists of negatively charged soybean protein isolated aggregated nanoparticles in a PAM polymer network structure by reversible electrostatic adsorption. The assembled quasi-solid Zn–MnO2 full cells were able to maintain the original charge/discharge curves without significant changes under 96% compression pressure (Fig. 9b). The battery charging and discharging curves remain basically unchanged with applied compression strain between 50% and 96% (Fig. 9c). By in situ polymerization of the acrylamide monomer in water/glycol, Jin et al. developed a PAM based solid-state electrolyte which simultaneously realized stable electrochemical performance and excellent self-healing properties at a low temperature of −20 °C (Fig. 9d).131 Liu et al. fabricated a double crosslinked hydrogel electrolyte by combining chitosan and PAM, introducing zinc ions as a cross-linking agent and ion carrier, which enabled long-lasting cycling stability of the Zn//PANI AZIB.143 The interactions among the coordinated zinc ions and amino groups, and hydrogen bonding provide this quasi-solid electrolyte with excellent auto-repair performance. As shown in Fig. 9e, two pieces of cut hydrogel electrolyte can be dynamically linked together through supramolecular interaction. The connected quasi-solid electrolyte still exhibits good stretched mechanical properties.
Gelatin, as a large hydrophilic gel, consists of three polypeptide chains intertwined to form a helix structure, which features high mechanical strength (7.76 MPa) and recovery memory.158 The novel structure of gelatine can combine and accommodate a large number of water molecules, which results in a high ionic conductivity of 1.76 × 10−2 S cm−1.138 Wang et al. designed a wire-shaped AZIB using gelatin and borax as the polymer matrix for the quasi-solid-state electrolyte with nitinol wire as the substrate material.145 This quasi-solid aqueous battery not only exhibits flexible characteristics and maintains electrochemical performance, but also has a shape memory function. After bending the battery by 90°, the initial shape can be restored quickly by immersing it in water at 45 °C.
Another commonly used polymer host material is carboxymethyl cellulose (CMC). The 20 M KAc aqueous electrolyte can be made gelatinous by adding 2 wt% CMC.46 In addition, this CMC-based quasi-solid aqueous electrolyte can effectively suppress the dissolution of electrode materials, such as Mn and Fe in the Prussian blue analogue (KMHCF) cathode material. Gel polymer additives have the capability to establish a densely packed molecular environment within the aqueous electrolyte, disrupting the hydrogen-bonded pattern and diminishing the electrochemical activity of water molecules. Lu et al. introduced 6 wt%. PEG to 2 M LiTFSI and widened the electrochemical window to 3.2 V. The assembled Li4Ti5O12//LiMn2O4 full cell showed 110 W h kg−1 energy density.106 Pan et al. developed a water in gel (WIG) hybrid electrolyte consisting of NaCl/ZnCl2/sodium alginate (SA) for application in a Zn–Na hybrid aqueous battery.159 The polar functional groups, such as OH and COO−, present on SA exhibit a strong affinity for water molecules. This characteristic allows for ample storage of water molecules within the WIG (water-in-glycerol) electrolyte, ensuring a high ionic conductivity of 62.2 mS cm−1. The internal structure of this novel WIG electrolyte was investigated by Fourier-transform infrared spectroscopy (FTIR) tests. On the one hand, the coordination of carboxylate groups to cations provides a fast transport channel for Na+ and Zn2+ hybrid ions, and on the other hand, the three-dimensional porous fibrous structure of the sodium alginate-based polymer network promotes fast ion transport. In WIG AMIBs, the electrode material comes into contact with the SA gel interface instead of with the free water molecules, which achieved a 2.72 V voltage window compatible with the copper hexacyanoferrate (CuHCF)-CNT/Zn aqueous full battery. Poly(styrene-isobutylene-styrene) (SIBS) as a polymer matrix is capable of encapsulating the entire Zn–MnO2 cell. The assembled full flexible battery is able to maintain stable operating voltage and electrochemical properties even when twisted 180° and stretched to double length (Fig. 9f).160 Zhao et al. reported a thermal responsive polymer matrix, poly(ethylene oxide)–poly(propylene oxide)–poly(ethylene oxide) (PEO–PPO–PEO), as a recoverable functional quasi-solid electrolyte. This novel electrolyte matrix exists as a quasi-solid at room temperature and changes to a liquid state at low temperatures. The assembled flexible aqueous battery was tested for strong folding and cooling recovery performance during cell operation. After three cycles of strong folding–cooling recovery tests at different battery locations, it still maintains a stable cycling performance and 98% initial capacity retention rate (Fig. 9g).161
Quasi-solid electrolytes in AMIBs alter the characteristics of the solvents, adjust the bonding interactions among water molecules, and decrease the activity of water molecules. This enables the storage of water molecules within the polymer matrix in a quasi-solid state, providing flexibility, a wide voltage range, and stable electrochemical properties to the AMIBs. The high mechanical strength of the hydrogel polymer electrolyte allows AMIBs to exhibit outstanding compatibility with harsh environments. However, the poor contact between the quasi-solid electrolyte and the electrode material, and the inherent poor ionic conductivity of organic polymers limit the extensive application of flexible AMIB devices. There are still some technical issues and theoretical mechanisms that deserve further consideration. The transition from traditional liquid electrolytes towards quasi-solid-state electrolytes is stimulated by the need for safer, more stable and flexible high-performance energy storage systems. Overall, the development of quasi-solid-state aqueous electrolytes is a meaningful research direction that holds great promise for the development of high-performance and safe energy storage devices.
4. Conclusions
Water molecules play a crucial role in the development and modification of aqueous metal-ion electrolytes. As the concentration of water molecules varies from dilute to WIS electrolyte, it directly impacts the solvated sheath structure surrounding carrier cations. This influence leads to the emergence of a novel SEI formation mechanism on the electrode surface. The voltage limit of aqueous electrolytes is surpassed, enabling the realization of a stable and wide electrochemical window. The development from a room-temperature to a low-temperature electrolyte is characterized by the disruption and re-formation of hydrogen bonds between water molecules, driven by the interactions among water, ions, and additives. This dynamic process ultimately leads to outstanding electrochemical performance at low temperatures. The transition from a liquid state to a quasi-solid state is centred around the capture and storage of water molecules within the hydro-polymer matrix. This integration allows for the retention of a significant amount of water, thereby leveraging the numerous advantages of aqueous electrolytes. The solid-like properties and exceptional mechanical characteristics simultaneously enhance the adaptability of AMIBs to a wider range of complex application scenarios.4.1. Prospects of the WIS electrolyte in AMIBs
The high concentration electrolyte system restrains the activity of water molecules, and the novel solvation sheath structure significantly improves the electrochemical stability of aqueous batteries, enabling many high-voltage electrode materials to be compatible in aqueous battery systems. However, there are still some problems with the WIS electrolyte system that need to be solved. One of the challenges is the decrease of ionic conductivity compared to dilute aqueous electrolytes. Ionic conductivity refers to the ability of ions to move through the electrolyte solution, and it is an important parameter that affects the performance and efficiency of AMIBs. For instance, LiTFSI reaches its maximum ionic conductivity at a concentration of 3.5 M. In addition, the increasing electrostatic interactions and coulombic friction between ions will result in high viscosity coefficients for WIS, which can reduce its flowability and make it difficult to transport through the electrolyte. Ionic conductivity and viscosity play critical roles in determining the electrochemical properties of aqueous electrolytes. Although the voltage window and the energy density are effectively increased, the use of large amounts of salt in the WIS system makes it more expensive than dilute electrolytes. Therefore, a balance between these properties of aqueous electrolytes is necessary to optimize the performance of AMIBs.4.2. Future advancements for low-temperature performance of aqueous electrolytes
The inherently high freezing point of water limits the electrochemical performance of aqueous electrolytes at low temperatures. In order to solve a series of problems caused by the freezing of aqueous electrolytes under cold conditions, various strategies, including the formulation of optimal salt concentration, the introduction of effective anions, the addition of antifreeze and organic co-solvents, and incorporation of hydrogel matrixes, have been reported. The essence of these strategies is to disrupt the network of hydrogen bonds between water molecules by stronger interactions. Challenges for improvement measures for the low-temperature performance of aqueous electrolytes coexist, for example, the high cost of electrolytes at high salt concentrations and the problem of low ionic conductivity. Although organic additives have improved the freezing point of aqueous electrolytes, the overall impact on the battery due to the mixing of organic systems and aqueous solutions still requires systematic and in-depth research.4.3. Quasi-solid aqueous electrolytes: balancing ionic conductivity, interfacial compatibility, and performance in AMIBs
Quasi-solid aqueous electrolytes have no leakage, high mechanical strength, deformability, and high electrochemical and thermodynamic stability. Meanwhile, the quasi-solidification of aqueous electrolytes involves basic requirements, e.g. sufficiently high ionic conductivity and interfacial compatibility with electrode materials. Ionic conductivity is an essential performance indicator for assessment of aqueous quasi-solid electrolytes, representing their ability to transport metal ions. However, the quasi-solid state of the electrolyte is usually achieved with the sacrifice of ionic conductivity. Designing polymer matrices capable of achieving high ionic conductivity in aqueous quasi-solid electrolytes requires continuous research. The balance between flexibility and ion conductivity of aqueous quasi-solid electrolytes requires a deliberate compromise. The interface between the electrode material and the quasi-solid electrolyte is an important issue. The physical contact and chemical stability of the interface play an important role in the performance of aqueous batteries. An insightful contact surface mechanism study is an important support for structural optimization and performance enhancement of aqueous batteries.4.4. Combining theoretical calculations and advanced characterization techniques to elucidate the crucial role of water molecules in aqueous electrolytes
In addition, theoretical calculations and advanced characterization are indispensable for a profound insight into the role of aqueous electrolytes and the influence of different systems on the aqueous battery mechanism. Theoretical simulations can assist in realistically revealing the solvation sheath structure of aqueous electrolytes, which is difficult to view by experiment. Density functional theory (DFT) and molecular dynamics (MD) simulations allow researchers to study the behavior of aqueous electrolytes at the atomic and molecular levels. They provide valuable information about the solvated shell layer structure, dynamics of metal ion diffusion at the electrode/electrolyte interface, and thermodynamic stability of electrolyte solutions. These calculations can elucidate the solvation properties of ions, ion–ion interactions, and ion transport mechanisms, helping to design electrolytes with improved performance. Ab initio molecular dynamics (AIMD) as a computational method that combines ab initio quantum mechanics calculations with classical molecular dynamics simulations could allow researchers to investigate the microscopic interactions and dynamics of ions and solvent molecules at an atomistic level. AIMD can elucidate how ions interact with water molecules and the surrounding solvent structure. It provides details on ion hydration shells and local solvation structures around ions. It enables the investigation of metal ion dynamics, hydrogen bonding networks, and the role of solvent molecules in facilitating metal ion transfer reactions. For example, in the WIS system, combined with first principles, the stable electrochemical window of the aqueous electrolyte is predicted by calculating the lowest unoccupied molecular orbital (LUMO) and highest occupied molecular orbital (HOMO) of the electrolyte. Density functional theory calculations can involve atomic-scale electronic properties such as energy band structures, density of states and charge distributions, leading to a wide range of applications in aqueous electrolyte systems. By calculating the migration energies and diffusion coefficients of ions, DFT can predict the rates and mechanisms of ion migration in solution. By calculating the formation energies and electronic properties of electrolyte components, DFT can assess the stability of the electrolyte and identify potential degradation mechanisms.Traditional electrochemical test methods such as cyclic voltammetry, galvanostatic cycling and electrochemical impedance spectroscopy are widely used to evaluate the electrochemical behavior of aqueous batteries. Electrochemical measurement and analysis techniques, including differential voltage, incremental capacity, and GITT, facilitate an in-depth understanding of electrode/electrolyte interfacial reactions, capacity decay mechanisms in aqueous electrolytes, and the determination of metal ion diffusion coefficients and transport kinetics. Furthermore, Raman spectroscopy, Fourier transform infrared spectroscopy and nuclear magnetic resonance instrumental characterization techniques have been widely used to study the solvation structure of aqueous electrolytes, the interaction between anions and water molecules, and to analyze the effect of electrode material dissolution composition and salt concentration on the transport properties of metal ions. In situ characterization experimental techniques for real-time monitoring and characterization of electrochemical processes occurring within aqueous electrolytes provide valuable insights into the dynamic behavior and mechanisms of electrochemical reactions. In situ characterization techniques are increasingly being applied to the study of aqueous electrolytes, such as in situ electrochemical quartz crystal microbalance,162,163in situ atomic force microscopy,163in situ neutron powder diffraction164 and in situ nuclear magnetic resonance imaging spectroscopy,165 which provide a profound understanding of electrochemical reaction mechanisms, solid–liquid reaction interfaces, and kinetic processes for the transport of metal ions in aqueous electrolytes.
4.5. Collaborative crossover development strategies enhance the performance and broaden the application scope of AMIBs
Two or more systems can be combined to obtain a nearly complete electrochemical operating characteristic. For example, the WIS system added organic additives to form a co-solvent electrolyte, which combines the advantages of high voltage, high ionic conductivity and low temperature performance.152,166 Quasi-solid state hybrid ion aqueous electrolytes can provide a wide temperature range and high flexibility while balancing operating voltage and energy density.152,167 Combining methods such as the high salt concentration method and the introduction of hydrogel matrixes can improve the low-temperature performance of aqueous electrolytes while accompanying the attainment of a more stable wide voltage window, high coulombic efficiency and cycling stability, and even enable the battery to possess a flexible feature. Overall, aqueous electrolytes play a crucial role in numerous fields and exhibit diverse properties across different concentrations, low-temperature performance, battery states, and hybrid ion types. Ongoing research and development efforts aim to enhance their performance, expand their application range, and address specific challenges in various industries.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work is supported by the National Natural Science Foundation of China (52072298, 51802261), Scientific Research Program Funded by Shaanxi Provincial Education Department (Program No. 23JK0662), and the Youth Innovation Team of Shaanxi Universities.Notes and references
- X. H. Yuan, F. X. Ma, L. Q. Zuo, J. Wang, N. F. Yu, Y. H. Chen, Y. S. Zhu, Q. H. Huang, R. Holze, Y. P. Wu and T. van Ree, Electrochem. Energy Rev., 2021, 4, 1–34 CrossRef CAS
.
- D. L. Chao, W. H. Zhou, F. X. Xie, C. Ye, H. Li, M. Jaroniec and S. Z. Qiao, Sci. Adv., 2020, 6, 19 Search PubMed
- M. Liu, H. Ao, Y. Jin, Z. Hou, X. Zhang, Y. Zhu and Y. Qian, Mater. Today Energy, 2020, 17, 21 Search PubMed
- J. L. Liu, C. H. Xu, Z. Chen, S. B. Ni and Z. X. Shen, Green Energy Environ., 2018, 3, 20–41 CrossRef
- C. Wang, Q. H. Xie, T. L. Guo, M. Fang, W. Mao, Y. C. Zhang, H. B. Wang, X. X. Ma, Y. T. Wu, S. Li and J. Han, Nano Lett., 2023, 23, 10930–10938 CrossRef CAS PubMed
- Y. Shang, V. Kundi, I. Pal, H. N. Kim, H. Y. Zhong, P. Kumar and D. Kundu, Adv. Mater., 2024, 36, 2309212 CrossRef CAS PubMed
- F. Gao, B. Mei, X. Y. Xu, J. H. Ren, D. C. Zhao, Z. Zhang, Z. L. Wang, Y. T. Wu, X. Liu and Y. Zhang, Chem. Eng. J., 2022, 448, 137742 CrossRef CAS
- S. Liu, L. Kang, J. M. Kim, Y. T. Chun, J. Zhang and S. C. Jun, Adv. Energy Mater., 2020, 10, 71 Search PubMed
- L. N. Chen, Q. Y. An and L. Q. Mai, Adv. Mater. Interfaces, 2019, 6, 24 Search PubMed
- S. Qiu, Y. K. Xu, X. Y. Wu and X. L. Ji, Electrochem. Energy Rev., 2022, 5, 242–262 CrossRef CAS
- M. G. Wu, W. Ni, J. Hu and J. M. Ma, Nano-Micro Lett., 2019, 11, 36 CrossRef PubMed
- T. S. Zhang, Y. Tang, S. Guo, X. X. Cao, A. Q. Pan, G. Z. Fang, J. Zhou and S. Q. Liang, Energy Environ. Sci., 2020, 13, 4625–4665 RSC
- T. J. Sun, Q. S. Nian, X. D. Ren and Z. L. Tao, Joule, 2023, 7, 2700–2731 CrossRef CAS
- B. You, M. T. Tang, C. Tsai, F. Abild-Pedersen, X. L. Zheng and H. Li, Adv. Mater., 2019, 31, 1807001 CrossRef PubMed
- M. Fang, G. Dong, R. Wei and J. C. Ho, Adv. Energy Mater., 2017, 7, 1700559 CrossRef
- X. X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC
- F. Lu, M. Zhou, Y. Zhou and X. Zeng, Small, 2017, 13, 1701931 CrossRef PubMed
- M. Zhou, Z. Bo and K. K. Ostrikov, Phys. Chem. Chem. Phys., 2022, 24, 20674–20688 RSC
- W. Li, J. R. Dahn and D. S. Wainwright, Science, 1994, 264, 1115–1118 CrossRef CAS PubMed
- W. Li, W. R. McKinnon and J. R. Dahn, J. Electrochem. Soc., 1994, 141, 2310–2316 CrossRef CAS
- J. Y. Luo, W. J. Cui, P. He and Y. Y. Xia, Nat. Chem., 2010, 2, 760–765 CrossRef CAS PubMed
- L. M. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. L. Fan, C. Luo, C. S. Wang and K. Xu, Science, 2015, 350, 938–943 CrossRef CAS PubMed
- T. T. Liang, R. L. Hou, Q. Y. Dou, H. Z. Zhang and X. B. Yan, Adv. Funct. Mater., 2021, 31, 2006749 CrossRef CAS
- Y. G. Wang, J. Yi and Y. Y. Xia, Adv. Energy Mater., 2012, 2, 830–840 CrossRef CAS
- H. Kim, J. Hong, K. Y. Park, H. Kim, S. W. Kim and K. Kang, Chem. Rev., 2014, 114, 11788–11827 CrossRef CAS PubMed
- J. H. Huang, Z. W. Guo, Y. Y. Ma, D. Bin, Y. G. Wang and Y. Y. Xia, Small Methods, 2019, 3, 20 Search PubMed
- Y. C. Shi, Y. Chen, L. Shi, K. Wang, B. Wang, L. Li, Y. M. Ma, Y. H. Li, Z. H. Sun, W. Ali and S. J. Ding, Small, 2020, 16, 28 Search PubMed
- B. Y. Tang, L. T. Shan, S. Q. Liang and J. Zhou, Energy Environ. Sci., 2019, 12, 3288–3304 RSC
- C. W. Xu, Z. W. Yang, X. K. Zhang, M. T. Xia, H. H. Yan, J. Li, H. X. Yu, L. Y. Zhang and J. Shu, Nano-Micro Lett., 2021, 13, 166 CrossRef CAS PubMed
- T. Xiong, Y. X. Zhang, W. S. V. Lee and J. M. Xue, Adv. Energy Mater., 2020, 10, 2001769 CrossRef CAS
- L. Sharma and A. Manthiram, J. Mater. Chem. A, 2022, 10, 6376–6396 RSC
- H. Gao, K. Tang, J. Xiao, X. Guo, W. Chen, H. Liu and G. Wang, J. Energy Chem., 2022, 69, 84–99 CrossRef CAS
- X. M. Li, X. Y. Wang, L. T. Ma and W. Huang, Adv. Energy Mater., 2022, 12, 2202068 CrossRef CAS
- Y. H. Shen, B. Liu, X. R. Liu, J. Liu, J. Ding, C. Zhong and W. B. Hu, Energy Storage Mater., 2021, 34, 461–474 CrossRef
- O. Borodin, L. M. Suo, M. Gobet, X. M. Ren, F. Wang, A. Faraone, J. Peng, M. Olguin, M. Schroeder, M. S. Ding, E. Gobrogge, A. V. Cresce, S. Munoz, J. A. Dura, S. Greenbaum, C. S. Wang and K. Xu, ACS Nano, 2017, 11, 10462–10471 CrossRef CAS PubMed
- Z. Yu, L. A. Curtiss, R. E. Winans, Y. Zhang, T. Li and L. Cheng, J. Phys. Chem. Lett., 2020, 11, 1276–1281 CrossRef CAS PubMed
- J. Han, A. Mariani, H. Zhang, M. Zarrabeitia, X. P. Gao, D. V. Carvalho, A. Varzi and S. Passerini, Energy Storage Mater., 2020, 30, 196–205 CrossRef
- Y. Yamada, K. Furukawa, K. Sodeyama, K. Kikuchi, M. Yaegashi, Y. Tateyama and A. Yamada, J. Am. Chem. Soc., 2014, 136, 5039–5046 CrossRef CAS PubMed
- G. Horwitz, C. R. Rodriguez, P. Y. Steinberg, G. Burton and H. R. Corti, Electrochim. Acta, 2020, 359, 136915 CrossRef CAS
- S. R. Chen, J. M. Zheng, D. H. Mei, K. S. Han, M. H. Engelhard, W. G. Zhao, W. Xu, J. Liu and J. G. Zhang, Adv. Mater., 2018, 30, 1706102 CrossRef PubMed
- V. A. Azov, K. S. Egorova, M. M. Seitkalieva, A. S. Kashin and V. P. Ananikov, Chem. Soc. Rev., 2018, 47, 1250–1284 RSC
- F. Wan, J. Zhu, S. Huang and Z. Niu, Batteries Supercaps, 2020, 3, 323–330 CrossRef CAS
- P. Samanta, S. Ghosh, A. Kundu, P. Samanta, N. C. Murmu and T. Kuila, J. Energy Chem., 2023, 78, 350–373 CrossRef CAS
- K. S. Han, Z. Yu, H. Wang, P. C. Redfern, L. Ma, L. Cheng, Y. Chen, J. Z. Hu, L. A. Curtiss, K. Xu, V. Murugesan and K. T. Mueller, J. Phys. Chem. B, 2020, 124, 5284–5291 CrossRef CAS PubMed
- J. Lim, K. Park, H. Lee, J. Kim, K. Kwak and M. Cho, J. Am. Chem. Soc., 2018, 140, 15661–15667 CrossRef CAS PubMed
- S. G. Chen, R. Lan, J. Humphreys and S. W. Tao, Appl. Mater. Today, 2020, 20, 100728 CrossRef
- D. P. Leonard, Z. X. Wei, G. Chen, F. Du and X. L. Ji, ACS Energy Lett., 2018, 3, 373 CrossRef CAS
- Q. Ni, H. Jiang, S. Sandstrom, Y. Bai, H. X. Ren, X. Y. Wu, Q. B. Guo, D. X. Yu, C. Wu and X. L. Ji, Adv. Funct. Mater., 2020, 30, 8 CrossRef
- H. Chen, Z. Zhang, Z. Wei, G. Chen, X. Yang, C. Wang and F. Du, Sustainable Energy Fuels, 2020, 4, 128–131 RSC
- L. M. Suo, O. Borodin, W. Sun, X. L. Fan, C. Y. Yang, F. Wang, T. Gao, Z. H. Ma, M. Schroeder, A. von Cresce, S. M. Russell, M. Armand, A. Angell, K. Xu and C. S. Wang, Angew. Chem., Int. Ed., 2016, 55, 7136–7141 CrossRef CAS PubMed
- M. T. Wang, H. Q. Wang, H. M. Zhang and X. F. Li, J. Energy Chem., 2020, 48, 14–20 CrossRef
- V. L. Martins and R. M. Torresi, Curr. Opin. Electrochem., 2020, 21, 62–68 CrossRef CAS
- C. H. Lin, K. Sun, M. Y. Ge, L. M. Housel, A. H. McCarthy, M. N. Vila, C. H. Zhao, X. H. Xiao, W. K. Lee, K. J. Takeuchi, E. S. Takeuchi, A. C. Marschilok and Y. C. K. Chen-Wiegart, Sci. Adv., 2020, 6, 11 Search PubMed
- R. S. Kuhnel, D. Reber and C. Battaglia, ACS Energy Lett., 2020, 5, 346 CrossRef CAS
- S. G. Chen, R. Lan, J. Humphreys and S. W. Tao, Energy Storage Mater., 2020, 28, 205–215 CrossRef
- L. W. Jiang, Y. X. Lu, C. L. Zhao, L. L. Liu, J. N. Zhang, Q. Q. Zhang, X. Shen, J. M. Zhao, X. Q. Yu, H. Li, X. J. Huang, L. Q. Chen and Y. S. Hu, Nat. Energy, 2019, 4, 495–503 CrossRef CAS
- L. M. Suo, O. Borodin, Y. S. Wang, X. H. Rong, W. Sun, X. L. Fan, S. Y. Xu, M. A. Schroeder, A. V. Cresce, F. Wang, C. Y. Yang, Y. S. Hu, K. Xu and C. S. Wang, Adv. Energy Mater., 2017, 7, 1701189 CrossRef
- M. H. Lee, S. J. Kim, D. Chang, J. Kim, S. Moon, K. Oh, K. Y. Park, W. M. Seong, H. Park, G. Kwon, B. Lee and K. Kang, Mater. Today, 2019, 29, 26–36 CrossRef CAS
- R. S. Kühnel, D. Reber and C. Battaglia, ACS Energy Lett., 2017, 2, 2005–2006 CrossRef
- L. B. Hu, Z. C. Zhang and K. Amine, Electrochem. Commun., 2013, 35, 76–79 CrossRef CAS
- Q. Zheng, S. Miura, K. Miyazaki, S. Ko, E. Watanabe, M. Okoshi, C.-P. Chou, Y. Nishimura, H. Nakai, T. Kamiya, T. Honda, J. Akikusa, Y. Yamada and A. Yamada, Angew. Chem., Int. Ed., 2019, 58, 14202–14207 CrossRef CAS PubMed
- K. Liu, Y. X. Zhou, H. B. Han, S. S. Zhou, W. F. Feng, J. Nie, H. Li, X. J. Huang, M. Armand and Z. B. Zhou, Electrochim. Acta, 2010, 55, 7145–7151 CrossRef CAS
- D. Reber, R. S. Kuhnel and C. Battaglia, ACS Mater. Lett., 2019, 1, 44–51 CrossRef CAS
- L. W. Jiang, L. L. Liu, J. M. Yue, Q. Q. Zhang, A. X. Zhou, O. Borodin, L. M. Suo, H. Li, L. Q. Chen, K. Xu and Y. S. Hu, Adv. Mater., 2020, 32, 1904427 CrossRef CAS
- S. Ko, Y. Yamada and A. Yamada, Electrochem. Commun., 2020, 116, 106764 CrossRef CAS
- H. Yan, X. Zhang, Z. Yang, M. Xia, C. Xu, Y. Liu, H. Yu, L. Zhang and J. Shu, Coord. Chem. Rev., 2022, 452, 214297 CrossRef CAS
- Y. Lv, Y. Xiao, L. Ma, C. Zhi and S. Chen, Adv. Mater., 2022, 34, 2106409 CrossRef CAS PubMed
- Z. Xing, C. Huang and Z. Hu, Coord. Chem. Rev., 2022, 452, 214299 CrossRef CAS
- Z. Li and A. W. Robertson, Battery Energy, 2023, 2, 20220029 CrossRef CAS
- C. Zhang, J. Holoubek, X. Y. Wu, A. Daniyar, L. D. Zhu, C. Chen, D. P. Leonard, I. A. Rodriguez-Perez, J. X. Jiang, C. Fang and X. Ji, Chem. Commun., 2018, 54, 14097–14099 RSC
- F. Wang, O. Borodin, T. Gao, X. L. Fan, W. Sun, F. D. Han, A. Faraone, J. A. Dura, K. Xu and C. S. Wang, Nat. Mater., 2018, 17, 543 CrossRef CAS PubMed
- P. Hu, M. Y. Yan, T. Zhu, X. P. Wang, X. J. Wei, J. T. Li, L. Zhou, Z. H. Li, L. N. Chen and L. Q. Mai, ACS Appl. Mater. Interfaces, 2017, 9, 42717–42722 CrossRef CAS PubMed
- J. W. Zhao, Y. Q. Li, X. Peng, S. M. Dong, J. Ma, G. L. Cui and L. Q. Chen, Electrochem. Commun., 2016, 69, 6–10 CrossRef CAS
- N. S. V. Narayanan, B. V. Ashokraj and S. Sampath, J. Colloid Interface Sci., 2010, 342, 505–512 CrossRef CAS PubMed
- L. J. Wang, Y. Zhang, H. L. Hu, H. Y. Shi, Y. Song, D. Guo, X. X. Liu and X. Q. Sun, ACS Appl. Mater. Interfaces, 2019, 11, 42000–42005 CrossRef CAS PubMed
- B. Lee, H. R. Seo, H. R. Lee, C. S. Yoon, J. H. Kim, K. Y. Chung, B. W. Cho and S. H. Oh, ChemSusChem, 2016, 9, 2948–2956 CrossRef CAS PubMed
- V. Soundharrajan, B. Sambandam, S. Kim, M. H. Alfaruqi, D. Y. Putro, J. Jo, S. Kim, V. Mathew, Y. K. Sun and J. Kim, Nano Lett., 2018, 18, 2402–2410 CrossRef CAS PubMed
- N. Zhang, F. Y. Cheng, Y. C. Liu, Q. Zhao, K. X. Lei, C. C. Chen, X. S. Liu and J. Chen, J. Am. Chem. Soc., 2016, 138, 12894–12901 CrossRef CAS PubMed
- Z. Peng, Q. L. Wei, S. S. Tan, P. He, W. Luo, Q. Y. An and L. Q. Mai, Chem. Commun., 2018, 54, 4041–4044 RSC
- F. Wang, X. L. Fan, T. Gao, W. Sun, Z. H. Ma, C. Y. Yang, F. Han, K. Xu and C. S. Wang, ACS Cent. Sci., 2017, 3, 1121–1128 CrossRef CAS PubMed
- Y. Murata, S. Takada, T. Obata, T. Tojo, R. Inada and Y. Sakurai, Electrochim. Acta, 2019, 294, 210–216 CrossRef CAS
- C. Wu, S. C. Gu, Q. H. Zhang, Y. Bai, M. Li, Y. F. Yuan, H. L. Wang, X. Y. Liu, Y. X. Yuan, N. Zhu, F. Wu, H. Li, L. Gu and J. Lu, Nat. Commun., 2019, 10, 73 CrossRef CAS PubMed
- A. X. Zhou, L. W. Jiang, J. M. Yue, Y. X. Tong, Q. Q. Zhang, Z. J. Lin, B. H. Liu, C. Wu, L. M. Suo, Y. S. Hu, H. Li and L. Q. Chen, ACS Appl. Mater. Interfaces, 2019, 11, 41356–41362 CrossRef CAS PubMed
- W. D. Pan, Y. F. Wang, Y. G. Zhang, H. Y. H. Kwok, M. Y. Wu, X. L. Zhao and D. Y. C. Leung, J. Mater. Chem. A, 2019, 7, 17420–17425 RSC
- L. Chen, J. X. Zhang, Q. Li, J. Vatamanu, X. Ji, T. P. Pollard, C. Y. Cui, S. Hou, J. Chen, C. Y. Yang, L. Ma, M. S. Ding, M. Garaga, S. Greenbaum, H. S. Lee, O. Borodin, K. Xu and C. S. Wang, ACS Energy Lett., 2020, 5, 968–974 CrossRef CAS
- D. Reber, R. Figi, R. S. Kühnel and C. Battaglia, Electrochim. Acta, 2019, 321, 134644 CrossRef CAS
- M. Turgeman, V. Wineman-Fisher, F. Malchik, A. Saha, G. Bergman, B. Gavriel, T. R. Penki, A. Nimkar, V. Baranauskaite, H. Aviv, M. D. Levi, M. Noked, D. T. Major, N. Shpigel and D. Aurbach, Cell Rep. Phys. Sci., 2022, 3, 100688 CrossRef CAS
- X. L. Li, M. Li, Q. Yang, H. F. Li, H. Xu, Z. L. Chai, K. Chen, Z. X. Liu, Z. J. Tang, L. T. Ma, Z. D. Huang, B. B. Dong, X. W. Yin, Q. Huang and C. Y. Zhi, ACS Nano, 2020, 14, 541–551 CrossRef CAS PubMed
- L. Zhang, I. A. Rodriguez-Perez, H. Jiang, C. Zhang, D. P. Leonard, Q. B. Guo, W. F. Wang, S. M. Han, L. M. Wang and X. L. Ji, Adv. Funct. Mater., 2019, 29, 1902653 CrossRef
- H. Zhang, S. Jeong, B. S. Qin, D. V. Carvalho, D. Buchholz and S. Passerini, ChemSusChem, 2018, 11, 1382–1389 CrossRef CAS PubMed
- L. Suo, O. Borodin, Y. Wang, X. Rong, W. Sun, X. Fan, S. Xu, M. A. Schroeder, A. V. Cresce and F. Wang, Adv. Energy Mater., 2017, 7, 1701189 CrossRef
- D. Reber, R.-S. Kühnel and C. Battaglia, ACS Mater. Lett., 2019, 1, 44–51 CrossRef CAS
- L. Jiang, L. Liu, J. Yue, Q. Zhang, A. Zhou, O. Borodin, L. Suo, H. Li, L. Chen and K. Xu, Adv. Mater., 2020, 32, 1904427 CrossRef CAS PubMed
- S. Ko, Y. Yamada and A. Yamada, Electrochem. Commun., 2020, 116, 106764 CrossRef CAS
- D. P. Leonard, Z. Wei, G. Chen, F. Du and X. Ji, ACS Energy Lett., 2018, 3, 373–374 CrossRef CAS
- T. J. Sun, S. B. Zheng, H. H. Du and Z. L. Tao, Nano-Micro Lett., 2021, 13, 204 CrossRef CAS PubMed
- A. X. Zhou, L. W. Jiang, J. M. Yue, Y. X. Tong, Q. Q. Zhang, Z. J. Lin, B. H. Liu, C. Wu, L. M. Suo, Y. S. Hu, H. Li and L. Q. Chen, ACS Appl. Mater. Interfaces, 2019, 11, 41356–41362 CrossRef CAS PubMed
- Z. Hu, Y. Guo, H. Jin, H. Ji and L.-J. Wan, Chem. Commun., 2020, 56, 2023–2026 RSC
- L. Chen, L. S. Cao, X. Ji, S. Hou, Q. Li, J. Chen, C. Y. Yang, N. Eidson and C. S. Wang, Nat. Commun., 2020, 11, 2638 CrossRef CAS PubMed
- N. Dubouis, P. Lemaire, B. Mirvaux, E. Salager, M. Deschamps and A. Grimaud, Energy Environ. Sci., 2018, 11, 3491–3499 RSC
- R. Bouchal, Z. J. Li, C. Bongu, S. Le Vot, R. Berthelot, B. Rotenberg, F. Favier, S. A. Freunberger, M. Salanne and O. Fontaine, Angew. Chem., Int. Ed., 2020, 59, 15913–15917 CrossRef CAS PubMed
- C. Y. Yang, J. Chen, T. T. Qing, X. L. Fan, W. Sun, A. von Cresce, M. S. Ding, O. Borodin, J. Vatamanu, M. A. Schroeder, N. Eidson, C. S. Wang and K. Xu, Joule, 2017, 1, 122–132 CrossRef CAS
- J. W. Chen, J. Vatamanu, L. D. Xing, O. Borodin, H. Y. Chen, X. C. Guan, X. Liu, K. Xu and W. S. Li, Adv. Energy Mater., 2020, 10, 1902654 CrossRef CAS
- L. S. Cao, D. Li, E. Y. Hu, J. J. Xu, T. Deng, L. Ma, Y. Wang, X. Q. Yang and C. S. Wang, J. Am. Chem. Soc., 2020, 142, 21404–21409 CrossRef CAS PubMed
- J. N. Hao, L. B. Yuan, C. Ye, D. L. Chao, K. Davey, Z. P. Guo and S. Z. Qiao, Angew. Chem., Int. Ed., 2021, 60, 7366–7375 CrossRef CAS PubMed
- J. Xie, Z. J. Liang and Y. C. Lu, Nat. Mater., 2020, 19, 1006 CrossRef CAS PubMed
- Y. Wang, H. Wei, Z. Li, X. Zhang, Z. Wei, K. Sun and H. Li, Chem. Rec., 2022, 22, e202200132 CrossRef CAS PubMed
- S. Wang, W. Deng, Z. Geng, P. Li, N. Hu, L. Zhu, W. Sun and C. M. Li, Battery Energy, 2023, 2, 20220050 CrossRef CAS
- Y. W. Zhao, Z. Chen, F. N. Mo, D. H. Wang, Y. Guo, Z. X. Liu, X. L. Li, Q. Li, G. J. Liang and C. Y. Zhi, Adv. Sci., 2021, 8, 2002590 CrossRef CAS PubMed
- L. Chen, H. Wu, X. Ai, Y. Cao and Z. Chen, Battery Energy, 2022, 1, 20210006 CrossRef CAS
- F. C. Andrews, Science, 1976, 194, 567–571 CrossRef CAS PubMed
- A. Ramanujapuram and G. Yushin, Adv. Energy Mater., 2018, 8, 1802624 CrossRef
- Y. J. Zhang and P. S. Cremer, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15249–15253 CrossRef CAS PubMed
- N. Schwierz, D. Horinek and R. R. Netz, Langmuir, 2013, 29, 2602–2614 CrossRef CAS PubMed
- Q. Zhang, K. X. Xia, Y. L. Ma, Y. Lu, L. Li, J. Liang, S. L. Chou and J. Chen, ACS Energy Lett., 2021, 6, 2704–2712 CrossRef CAS
- H. Q. Wang, H. Z. Zhang, Y. Cheng, K. Feng, X. F. Li and H. M. Zhang, Electrochim. Acta, 2018, 278, 279–289 CrossRef CAS
- M. Becker, R. S. Kuhnel and C. Battaglia, Chem. Commun., 2019, 55, 12032–12035 RSC
- C. Q. Sun, X. Zhang, X. J. Fu, W. T. Zheng, J. L. Kuo, Y. C. Zhou, Z. X. Shen and J. Zhou, J. Phys. Chem. Lett., 2013, 4, 3238–3244 CrossRef CAS PubMed
- L. T. Ma, S. M. Chen, N. Li, Z. X. Liu, Z. J. Tang, J. A. Zapien, S. M. Chen, J. Fan and C. Y. Zhi, Adv. Mater., 2020, 32, 1908121 CrossRef CAS PubMed
- J. L. Dashnau, N. V. Nucci, K. A. Sharp and J. M. Vanderkooi, J. Phys. Chem. B, 2006, 110, 13670–13677 CrossRef CAS PubMed
- A. Tron, S. Jeong, Y. D. Park and J. Mun, ACS Sustain. Chem. Eng., 2019, 7, 14531–14538 CrossRef CAS
- N. N. Chang, T. Y. Li, R. Li, S. N. Wang, Y. B. Yin, H. M. Zhang and X. F. Li, Energy Environ. Sci., 2020, 13, 3527–3535 RSC
- Q. Y. Dou, S. L. Lei, D. W. Wang, Q. N. Zhang, D. W. Xiao, H. W. Guo, A. P. Wang, H. Yang, Y. L. Li, S. Q. Shi and X. B. Yan, Energy Environ. Sci., 2018, 11, 3212–3219 RSC
- Q. Nian, J. Wang, S. Liu, T. Sun, S. Zheng, Y. Zhang, Z. Tao and J. Chen, Angew. Chem., Int. Ed., 2019, 58, 16994–16999 CrossRef CAS PubMed
- M. F. Chen, W. J. Zhou, A. R. Wang, A. X. Huang, J. Z. Chen, J. L. Xu and C. P. Wong, J. Mater. Chem. A, 2020, 8, 6828–6841 RSC
- T. T. Wei, Y. K. Ren, Z. Q. Li, X. X. Zhang, D. H. Ji and L. H. Hu, Chem. Eng. J., 2022, 434, 134646 CrossRef CAS
- M. S. Zhu, X. J. Wang, H. M. Tang, J. W. Wang, Q. Hao, L. X. Liu, Y. Li, K. Zhang and O. G. Schmidt, Adv. Funct. Mater., 2020, 30, 1907218 CrossRef CAS
- S. W. Huang, L. Hou, T. Y. Li, Y. C. Jiao and P. Y. Wu, Adv. Mater., 2022, 34, 2110140 CrossRef CAS PubMed
- L. Han, K. Z. Liu, M. H. Wang, K. F. Wang, L. M. Fang, H. T. Chen, J. Zhou and X. Lu, Adv. Funct. Mater., 2018, 28, 1704195 CrossRef
- Q. F. Rong, W. W. Lei, L. Chen, Y. A. Yin, J. J. Zhou and M. J. Liu, Angew. Chem., Int. Ed., 2017, 56, 14159–14163 CrossRef CAS PubMed
- X. T. Jin, L. Song, C. L. Dai, H. Y. Ma, Y. K. Xiao, X. N. Zhang, Y. Y. Han, X. Y. Li, J. T. Zhang, Y. Zhao, Z. P. Zhang, L. Duan and L. T. Qu, Energy Storage Mater., 2022, 44, 517–526 CrossRef
- H. N. Gao, Z. G. Zhao, Y. D. Cai, J. J. Zhou, W. D. Hua, L. Chen, L. Wang, J. Q. Zhang, D. Han, M. J. Liu and L. Jiang, Nat. Commun., 2017, 8, 15911 CrossRef CAS PubMed
- F. N. Mo, G. J. Liang, Q. Q. Meng, Z. X. Liu, H. F. Li, J. Fan and C. Y. Zhi, Energy Environ. Sci., 2019, 12, 706–715 RSC
- X. P. Morelle, W. R. Illeperuma, K. Tian, R. B. Bai, Z. G. Suo and J. J. Vlassak, Adv. Mater., 2018, 30, 1801541 CrossRef PubMed
- W. J. Song, S. Lee, G. Song and S. Park, ACS Energy Lett., 2019, 4, 177–186 CrossRef CAS
- Z. X. Liu, H. F. Li, M. S. Zhu, Y. Huang, Z. J. Tang, Z. X. Pei, Z. F. Wang, Z. C. Shi, J. Liu, Y. Huang and C. Y. Zhi, Nano Energy, 2018, 44, 164–173 CrossRef CAS
- C. L. Dai, X. T. Jin, H. Y. Ma, L. Y. Hu, G. Q. Sun, H. Chen, Q. J. Yang, M. W. Xu, Q. W. Liu, Y. K. Xiao, X. Q. Zhang, H. S. Yang, Q. Guo, Z. P. Zhang and L. T. Qu, Adv. Energy Mater., 2021, 11, 2003982 CrossRef CAS
- J. Zhao, H. Ren, Q. H. Liang, D. Yuan, S. B. Xi, C. Wu, W. Manalastas, J. M. Ma, W. Fang, Y. Zheng, C. F. Du, M. Srinivasan and Q. Y. Yan, Nano Energy, 2019, 62, 94–102 CrossRef CAS
- Z. X. Liu, Q. Yang, D. H. Wang, G. J. Liang, Y. H. Zhu, F. N. Mo, Z. D. Huang, X. L. Li, L. T. Ma, T. C. Tang, Z. G. Lu and C. Y. Zhi, Adv. Energy Mater., 2019, 9, 12 Search PubMed
- K. Wang, X. H. Zhang, J. W. Hang, X. Zhang, X. Z. Sun, C. Li, W. H. Liu, Q. W. Li and Y. W. Ma, ACS Appl. Mater. Interfaces, 2018, 10, 24573–24582 CrossRef CAS PubMed
- M. J. Yao, Z. S. Yuan, S. S. Li, T. W. He, R. Wang, M. J. Yuan and Z. Q. Niu, Adv. Mater., 2021, 33, 2008140 CrossRef CAS PubMed
- D. Jiang, N. Lu, L. B. Li, H. Q. Zhang, J. S. Luan and G. B. Wang, J. Colloid Interface Sci., 2022, 608, 1619–1626 CrossRef CAS PubMed
- Y. Q. Liu, A. M. Gao, J. N. Hao, X. L. Li, J. Z. Ling, F. Y. Yi, Q. Z. Li and D. Shu, Chem. Eng. J., 2023, 452, 139605 CrossRef CAS
- S. Huang, F. Wan, S. S. Bi, J. C. Zhu, Z. Q. Niu and J. Chen, Angew. Chem., Int. Ed., 2019, 58, 4313–4317 CrossRef CAS PubMed
- Z. F. Wang, Z. H. Ruan, Z. X. Liu, Y. K. Wang, Z. J. Tang, H. F. Li, M. S. Zhu, T. F. Hung, J. Liu, Z. C. Shi and C. Y. Zhi, J. Mater. Chem. A, 2018, 6, 8549–8557 RSC
- S. Chen, M. Zhang, P. Zou, B. Sun and S. Tao, Energy Environ. Sci., 2022, 15, 1805–1839 RSC
- T. T. Gao, G. Y. Yan, X. Yang, Q. Yan, Y. K. Tian, J. W. Song, F. X. Li, X. L. Wang, J. Y. Yu, Y. J. Li and S. J. Guo, J. Energy Chem., 2022, 71, 192–200 CrossRef CAS
- F. Wan, L. L. Zhang, X. Y. Wang, S. S. Bi, Z. Q. Niu and J. Chen, Adv. Funct. Mater., 2018, 28, 1804975 CrossRef
- X. B. Zang, L. T. Li, J. X. Meng, L. J. Liu, Y. Y. Pan, Q. G. Shao and C. Ning, J. Mater. Sci. Technol., 2021, 74, 52–59 CrossRef CAS
- W. H. Wang, C. W. Li, S. Z. Liu, J. C. Zhang, D. J. Zhang, J. M. Du, Q. C. Zhang and Y. G. Yao, Adv. Energy Mater., 2023, 13, 2300250 CrossRef CAS
- G. Wang, X. Lu, Y. Ling, T. Zhai, H. Wang, Y. Tong and Y. Li, ACS Nano, 2012, 6, 10296–10302 CrossRef CAS PubMed
- S. G. Chen, R. Lan, J. Humphreys and S. W. Tao, ACS Appl. Energy Mater., 2020, 3, 2526–2536 CrossRef CAS
- Y. Huang, W. S. Ip, Y. Y. Lau, J. F. Sun, J. Zeng, N. S. S. Yeung, W. S. Ng, H. F. Li, Z. X. Pei, Q. Xue, Y. K. Wang, J. Yu, H. Hu and C. Y. Zhi, ACS Nano, 2017, 11, 8953–8961 CrossRef CAS PubMed
- D. T. Li, T. Lv, Z. L. Chen, Y. L. Yang, Y. A. Liu, J. Wan, Y. L. Qi, S. K. Cao and T. Chen, Small Struct., 2022, 3, 2200027 CrossRef CAS
- Y. Huang, M. Zhong, Y. Huang, M. S. Zhu, Z. X. Pei, Z. F. Wang, Q. Xue, X. M. Xie and C. Y. Zhi, Nat. Commun., 2015, 6, 10310 CrossRef CAS PubMed
- H. F. Li, Z. X. Liu, G. J. Liang, Y. Huang, Y. Huan, M. S. Zhu, Z. X. Pei, Q. Xue, Z. J. Tang, Y. K. Wang, B. H. Li and C. Y. Zhi, ACS Nano, 2018, 12, 3140–3148 CrossRef CAS PubMed
- T. Liu, X. Chen, E. Tervoort, T. Kraus and M. Niederberger, ACS Appl. Energy Mater., 2021, 4, 6166–6179 CrossRef CAS
- Q. Han, X. W. Chi, S. M. Zhang, Y. Z. Liu, B. A. Zhou, J. H. Yang and Y. Liu, J. Mater. Chem. A, 2018, 6, 23046–23054 RSC
- W. D. Pan, Y. F. Wang, X. L. Zhao, Y. Zhao, X. H. Liu, J. Xuan, H. Z. Wang and D. Y. C. Leung, Adv. Funct. Mater., 2021, 31, 2008783 CrossRef CAS
- T. N. Nguyen, B. Iranpour, E. Cheng and J. D. W. Madden, Adv. Energy Mater., 2022, 12, 2103148 CrossRef CAS
- J. W. Zhao, K. K. Sonigara, J. J. Li, J. Zhang, B. B. Chen, J. J. Zhang, S. S. Soni, X. H. Zhou, G. L. Cui and L. Q. Chen, Angew. Chem., Int. Ed., 2017, 56, 7871–7875 CrossRef CAS PubMed
- Z. H. Sun, Y. M. Zhang, Y. Liu, L. R. Hou and C. Z. Yuan, Chempluschem, 2021, 86, 1487–1496 CrossRef CAS PubMed
- S. Cora, S. Ahmad and N. Y. Sa, ACS Appl. Mater. Interfaces, 2021, 13, 10131–10140 CrossRef CAS PubMed
- C. Pastor-Fernández, K. Uddin, G. H. Chouchelamane, W. D. Widanage and J. Marco, J. Power Sources, 2017, 360, 301–318 CrossRef
- X. Z. Zhou, Y. Lu, Q. Zhang, L. C. Miao, K. Zhang, Z. H. Yan, F. J. Li and J. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 55476–55482 CrossRef CAS PubMed
- H. Chen, M. F. Chen, W. J. Zhou, X. Han, B. Liu, W. Q. Zhang and J. Z. Chen, ACS Appl. Mater. Interfaces, 2022, 14, 6876–6884 CrossRef CAS PubMed
- Z. X. Liu, Q. Yang, D. H. Wang, G. J. Liang, Y. H. Zhu, F. N. Mo, Z. D. Huang, X. L. Li, L. T. Ma, T. C. Tang, Z. G. Lu and C. Y. Zhi, Adv. Energy Mater., 2019, 9, 1902473 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2024 |