
Construction of a graphitic carbon nitride-based photocatalyst with a strong built-in electric field via π–π stacking interactions boosting photocatalytic CO2 reduction†
Yanrui
Li
 *a,
Linda
Wang
a,
Xiang
Gao
a,
Yingying
Xue
a,
Bozhan
Li
a and
Xiaolin
Zhu
*b
*a,
Linda
Wang
a,
Xiang
Gao
a,
Yingying
Xue
a,
Bozhan
Li
a and
Xiaolin
Zhu
*b
aCollege of Materials Science and Engineering, Xi'an University of Science and Technology, Xi'an, 710054, China. E-mail: liyanrui91@xust.edu.cn
bKey Laboratory of Applied Surface and Colloid Chemistry (Ministry of Education), School of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi'an 710119, P. R. China. E-mail: xiaolinchem@snnu.edu.cn
First published on 16th February 2024
Abstract
Solar-driven photocatalytic reduction of carbon dioxide (CO2) into fuels, particularly over a graphitic carbon nitride (GCN)-based photocatalyst, presents a promising solution to address energy and CO2 emission issues. However, the limited charge-carrier dynamics significantly hampers the photocatalytic performance of CO2 reduction. Enhancing the built-in electric field at the interface has been revealed as an effective strategy to improve the activity of photocatalytic reactions. In this work, a high-efficiency photocatalyst TPNCNm was fabricated by anchoring sodium 2,5,8-tri(4′-pyridyl)-1,3,4,6,7,9-hexaazaphenalenate (TPHAP), with a similar tri-s-triazine unit to GCN, on the surface of crystalline nitride carbon (NCN) via π–π stacking interactions. The fabricated photocatalyst TPNCNm demonstrates efficient solar-driven CO2 reduction into CO and CH4 through the gas–solid mode, with evolution rates of CO and CH4 reaching 10.43 and 4.88 μmol g−1 h−1, respectively. Furthermore, the investigation of the band structure and charge carrier dynamics revealed the formation of a strong built-in electric field between TPHAP and NCN. The directional built-in electric field from NCN to TPHAP effectively accelerated the charge carrier separation and migration, thereby boosting CO2 photoreduction performance. In addition, theoretical calculations have verified that TPHAP molecules not only serve as active sites, but also effectively lower the Gibbs free energy of crucial intermediate COOH*, which is the rate-determining step for CO2-to-CO photoreduction. This work highlights the importance of utilizing coplanar materials to construct efficient hybrid graphitic nitride carbon-based photocatalysts with a strong built-in electric field through π–π interactions for enhanced photocatalytic CO2 reduction.
Introduction
Artificial photocatalytic carbon dioxide (CO2) reduction into valuable fuels offers a promising and sustainable approach to simultaneously alleviate increasing energy demands and climate change.1,2 Recently, there has been a rising interest in exploring organic semiconductors with earth-abundant elements and lightweight for photocatalytic CO2 reduction.3,4 Among the various organic semiconductors, graphite carbon nitride (GCN) with a two-dimensional (2D) layered structure has emerged as a particularly noteworthy candidate for CO2 photocatalytic reduction, due to its high chemical stability, ease of preparation, and suitable band structure for CO2 photoreduction.5,6 However, the conversion efficiency of CO2 photoreduction is significantly hindered owing to the limitations of dull charge carrier dynamics, including poor electron–hole separation ability, slow charge transfer rate, and indecisive catalytic reaction at the active sites of catalysts.7 To overcome these limitations, employing hybrid photocatalytic systems has been found as a favorable approach for developing highly efficient photocatalysts with enhanced CO2 photoreduction performance.8Developing new hybrid photocatalysts via matching another metal or semiconductor nanoparticle with GCN, such as polymers,9 metal–organic frameworks,10 metal oxides,11 or metals,12 has been widely employed to inhibit electron–hole pair recombination and facilitate charge carrier migration. Notably, the construction of a built-in electric field at the photocatalyst interface provides a driving force for charge separation and migration. Then, enhancing the built-in electric field has been revealed as an effective strategy to improve the activity of photocatalytic reactions.13,14 However, weak interfacial contacts between GCN and semiconductors would result in the formation of interfacial defects, acting as the photogenerated electron–hole recombination sites, which hardly inhibit charge carrier separation and migration with low effective photocatalytic activity. Fortunately, utilization of π–π stacking interactions between planar molecules and 2D semiconductors has been evidenced to improve the transfer efficiency of charge carriers in the interface of photocatalysts, consequently enhancing the photocatalytic performance. Ouyang et al. modified the photosensitizers and catalyst with pyrenyl groups to construct π–π stacking interactions for high-performance CO2 photoreduction.15 It is noted that the π–π stacking interaction significantly promotes the charge carrier migration from the excited-state photosensitizers to catalysts. Jing et al. synthesized a dimension-matched perylene diimide/g-C3N4 (PDI/GCN), in which the interfacial π–π interactions accelerated the dispersion of PDI on the surface of GCN as well as improving the interfacial charge migration, boosting the CO2 photoreduction performance.16 It is reported that the efficiency of electron transport through π–π stacking interactions relies on the degree of matching in the conjugated macrocycle structure.17 However, pyrenyl groups or perylene diimide possesses the conjugate macrocycle with a low matching degree with the tri-s-triazine unit, which makes it difficult to maximize the interfacial π–π interactions to drive the charge migration. Hence, organic molecules with a similar conjugated macrocycle structure to GCN are well suitable for constructing strong-contact GCN-based hybrid photocatalysts via the π–π stacking interaction, providing an interlayer charge-transfer channel along the π-stacking direction.
Sodium 2,5,8-tri(4′-pyridyl)-1,3,4,6,7,9-hexaazaphenalenate (TPHAP), featuring three pyridyl groups linked with a hexaazaphenalenate π-conjugated macrocycle (Scheme 1), has been explored in self-assembled molecule networks through π–π stacking interactions, ascribed to its D3h symmetrical aromatic plane.18–20 Interestingly, the molecular structure of the hexaazaphenalenate unit is very similar to the tri-s-triazine unit, in that a carbon atom replacing the central tertiary nitrogen of the tri-s-triazine unit forms the hexaazaphenalenate unit. Satisfactorily, the highly matched conjugate macrocycle structure between the hexaazaphenalenate unit and tri-s-triazine unit makes TPHAP an excellent candidate to construct a strong-contact GCN-based hybrid photocatalyst via the π–π stacking interaction. In addition, the coordinating groups, three pyridyl groups, provide the linkage to promote the migration of the photogenerated charge carriers from the hexaazaphenalenate conjugated rings to pyridyl groups.21
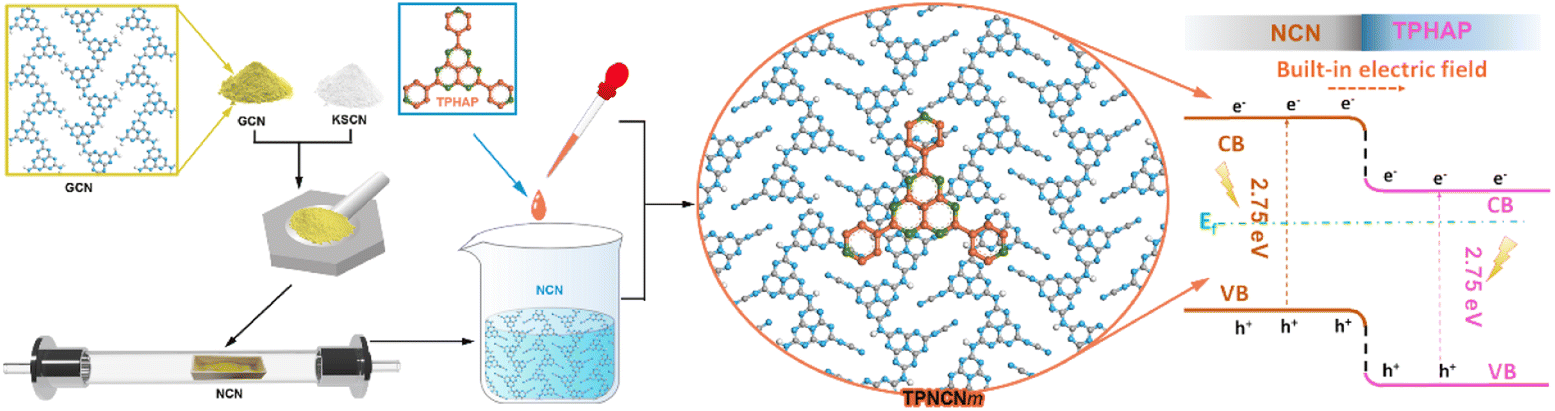 | ||
| Scheme 1 Schemed fabrication process of the TPNCNm photocatalyst. | ||
However, the bent plane forming in amorphous GCN through tri-s-triazine conjugate rings connected by sp3 imide N inhibits the effective interface matching between GCN and TPHAP. At present, the molten-salt method has been widely reported as an effective strategy to improve the crystallinity of GCN, along with an extended planar plane and stabilized π electron system,21–23 which may benefit interface matching between the crystalline carbon nitride (NCN) and TPHAP to form a strong-contact hybrid photocatalyst via strong π–π stacking interactions, named as crystalline NCN/TPHAP (TPNCN). Thus, it is expected that photogenerated charges, driven by the strong built-in electric field of TPNCN, could be induced and transferred from NCN into TPHAP in the π-stacking direction. Subsequently, these charges are expected to be captured by the three pyridyl groups, serving as active sites for the photocatalytic CO2 reduction reaction.
As inspired by the consideration above, we successfully designed and synthesized crystalline TPNCN via molten-salt and solution-assembly process, which exhibited enhanced CO2 photoreduction performance with CO and CH4 evolution rates reaching up to 10.43 and 4.88 μmol g−1 h−1, respectively. The improved crystallinity for NCN was proved to accelerate the charge separation and transfer efficiency. Based on the enhanced charge dynamics of NCN, the introduction of TPHAP firmly anchored on the surface of NCN was demonstrated further to enhance the charge separation from NCN to TPHAP, and promote the charge carrier migration, as well as act as the active site for photocatalytic reactions. Furthermore, in situ spectral characterization and density functional theory (DFT) calculations and other various characterization tests were carried out to further investigate the band structure, charge dynamics and reaction mechanisms for excellent CO2 photoreduction performance.
Results and discussion
Constructing the TPNCN photocatalyst
The preparation procedure of TPNCN is illustrated in Scheme 1. Starting from thermal-polymerization of GCN as the precursor, highly crystalline nitride carbon (denoted as NCN) is obtained through the molten-salt method with high-temperature calcination treatment of the GCN and KSCN molten-salt mixture in the tube furnace under argon. With a similar π-conjugated macrocycle structure to NCN, TPHAP, 2,5,8-tri(4′-pyridyl)-1,3,4,6,7,9-hexaazaphenalene, could be easily synthesized via a one-step condensation reaction between tricyanomethanide and 4-pyridylamidine hydrochloride (Scheme S1†). Depending on the strong π–π interaction between TPHAP and NCN, the TPHAP solution was dropwise injected into the NCN suspension and then TPNCNm was formed, in which m represents the mass percentage (m = 1 (1 w%), 5 (5 w%) and 10 (10 w%)) of TPHAP used for deposition on the NCN surface.The crystal structures of NCN and TPNCNm were investigated by X-ray diffraction (XRD). For GCN, we observed two consistent peaks at 2θ = 13.0° (100) assigned to the in-plane periodic tri-s-triazine units (d100 = 6.595 Å) and 2θ = 27.4° (002) ascribed to the periodic stacking of the nanosheets layers (d002 = 3.249 Å) (Fig. 1a).24 Compared with GCN, the (002) peak of NCN displayed a shift to 28.2° with a reduced interlayer distance (d002 = 3.163 Å), suggesting the strongly compressed interlayer stacking after the molten-salt treatment.25 After molten-salt treatment, the in-plane periodicity (100) peak of NCN is weakened and two additional peaks appear at 2θ = 7.9° and 2θ = 10.1° (Fig. 1a), attributed to the in-plane periodicity (110) (d110 = 10.740 Å) and (020) planes (d020 = 8.525 Å),26,27 respectively. Given sp3 imide N resulting in the non-coplanar tri-s-triazine conjugate rings and then bending the conjugated area in amorphous GCN, the size of the periodic tri-s-triazine unit is increased from 6.595 Å for GCN to 8.525 or 10.740 Å for NCN, due to the improved crystallization extending the planar plane of NCN,25 which benefits the further interface matching for TPHAP with NCN in TPNCNm. The improved crystallization could be further confirmed by the reduced full width at half maximum (FWHM) from 1.424° for GCN to 1.107° for NCN (Table S1†). Similar to the NCN, the characteristic peaks of NCN could be observed in all TPNCNm clearly (Fig. 1b), suggesting that the crystalline structure of NCN remains unchanged after TPHAN being introduced. Further comparison reveals that only low-angel reflection peaks of TPHAP with slight displacement are observed in TPNCNm, which could be owing to the different stacking structure of TPHAP in TPNCNm to TPHAP powder as well as the relatively low mass percentage for TPHAP in TPNCNm.
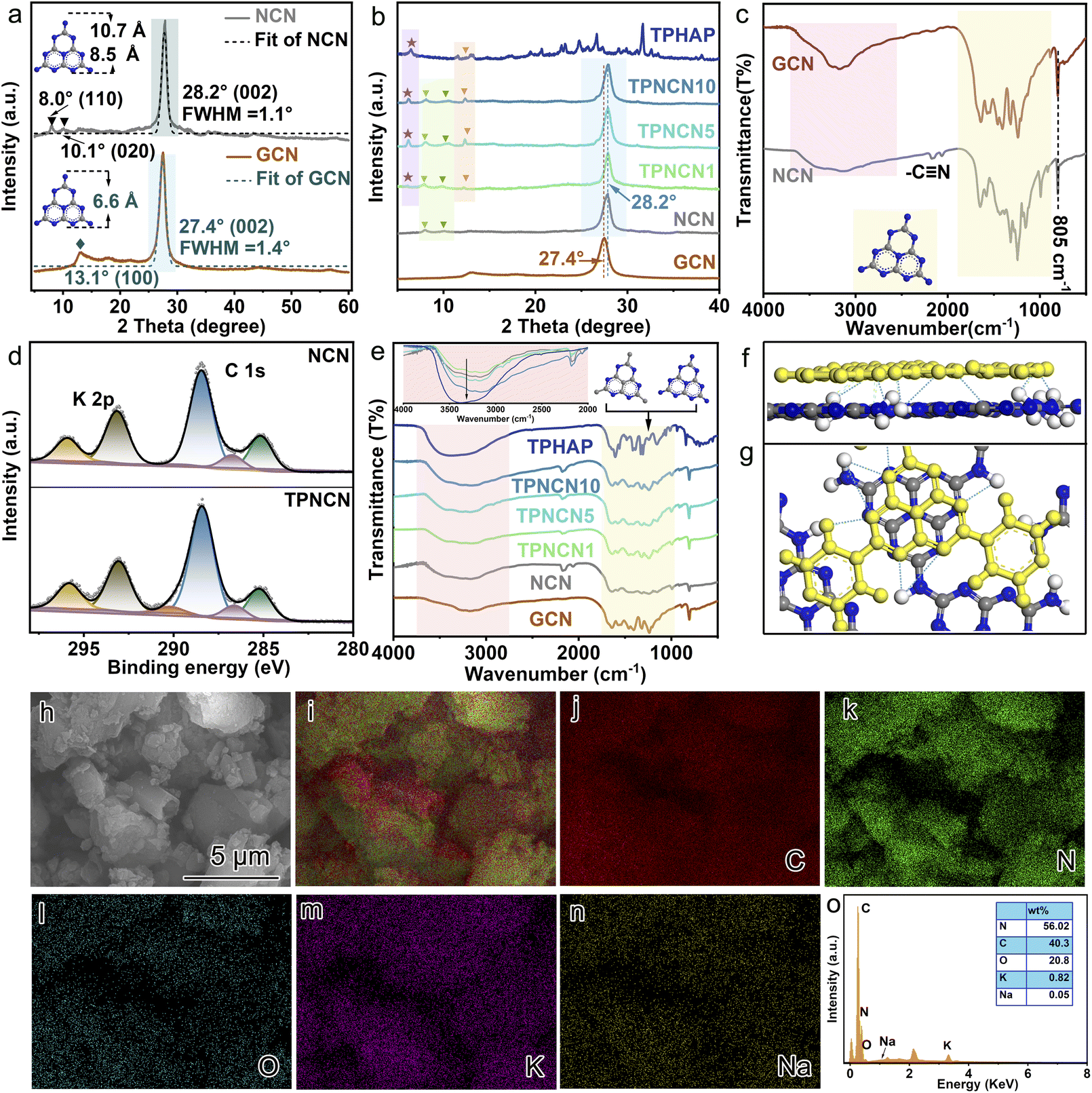 | ||
| Fig. 1 X-ray diffraction (XRD) patterns of (a) GCN, NCN and (b) TPNCNm. (c) Fourier transform infrared spectroscopy (FTIR) spectra of GCN and NCN. (d) C 1s and K 2p X-ray photoelectron spectroscopy (XPS) spectra of NCN and TPNCN. (e) FTIR spectra of TPNCNm. Schematic illustration for TPNCNm in (f) horizontal and (g) vertical directions. (h) SEM image and SEM-mapping scan images of (j) C, (k) N, (i) O, (m) K, and (n) Na of TPNCN. (o) The mass percentage of all elements. | ||
For further investigation of the chemical structure of TPHAP, NCN and TPNCNm, Fourier transform infrared spectroscopy (FTIR) and X-ray photoelectron spectroscopy (XPS) were carried out. Similar to GCN, two characteristic absorption bands, broad bands at 1700–900 cm−1 originating from the tri-s-triazine derivatives and the band at 805 cm−1 indexed to the out-of-plane bending of the tri-s-triazine rings, could be detected for NCN (Fig. 1c),28 indicating that the molecular structure of NCN remains unchanged after the molten-salt treatment,29 with tri-s-triazine rings well maintained in NCN, which could be evidenced by the N 1s XPS (Fig. S1†) of NCN similar to that of GCN. Meanwhile, all the characteristic FTIR bands of NCN are sharper than those of GCN, revealing an improved interlayer stacking and in-plane periodicity order for NCN, originating from the molten-salt treatment.30 On careful comparison, one should note that an additional band at 2164 cm−1 corresponding to –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N is detected over NCN, related to partial replacement of amino groups by cyanamide groups (–CN) during the KSCN molten-salt treatment, which could be confirmed by the existence of C 1s XPS at 286.2 eV (Fig. 1d).31 In addition, this replacement of groups results in weakening of the peaks ranging from 3000 to 3500 cm−1 ascribed to amino groups and reduction of the XPS ratio of amino groups over NCN in contrast to GCN (Table S2†).32 It is observable that the FTIR patterns of TPNCNm featuring all the characteristic bands of NCN indicate the molecular structure of NCN remaining in TPNCNm (Fig. 1e) via π–π stacking interactions. However, the characteristic bands of TPHAP in the range of 2500–800 cm−1 are undistinguishable in TPNCNm, due to the relatively low amount of TPHAP in TPNCNm. However, the FTIR peaks of TPNCNm centered at 3450 cm−1 assigned to hydrogen bonds gradually increased with the increasing ratio of TPHAP, revealing that, in addition to the π–π interaction, hydrogen bonds are another interaction mode for TPHAP connecting with NCN (Fig. 1f). The π–π stacking interaction between NCN and TPHAP could be investigated by C 1s XPS (Fig. 1d). In addition to the C 1s peak of the –CN group, another two C 1s peaks at 284.6 and 288.4 eV, ascribed to the graphitic carbon and sp2-bonded carbon in the aromatic ring (N
N is detected over NCN, related to partial replacement of amino groups by cyanamide groups (–CN) during the KSCN molten-salt treatment, which could be confirmed by the existence of C 1s XPS at 286.2 eV (Fig. 1d).31 In addition, this replacement of groups results in weakening of the peaks ranging from 3000 to 3500 cm−1 ascribed to amino groups and reduction of the XPS ratio of amino groups over NCN in contrast to GCN (Table S2†).32 It is observable that the FTIR patterns of TPNCNm featuring all the characteristic bands of NCN indicate the molecular structure of NCN remaining in TPNCNm (Fig. 1e) via π–π stacking interactions. However, the characteristic bands of TPHAP in the range of 2500–800 cm−1 are undistinguishable in TPNCNm, due to the relatively low amount of TPHAP in TPNCNm. However, the FTIR peaks of TPNCNm centered at 3450 cm−1 assigned to hydrogen bonds gradually increased with the increasing ratio of TPHAP, revealing that, in addition to the π–π interaction, hydrogen bonds are another interaction mode for TPHAP connecting with NCN (Fig. 1f). The π–π stacking interaction between NCN and TPHAP could be investigated by C 1s XPS (Fig. 1d). In addition to the C 1s peak of the –CN group, another two C 1s peaks at 284.6 and 288.4 eV, ascribed to the graphitic carbon and sp2-bonded carbon in the aromatic ring (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–N), are observed in NCN and TPNCN.32 Significantly, a new C 1 s peak at 290.2 eV, corresponding to π–π satellite bonds, appears in TPNCN, which confirms the π–π stacking interaction between TPHAP and NCN.33–35 In addition, the Na 1s peak at 1071.3 eV reveals the existence of Na+ ions originating from TPHAP,36,37 which proves the successful anchoring of TPHAP on the surface of NCN (Fig. S2†).
C–N), are observed in NCN and TPNCN.32 Significantly, a new C 1 s peak at 290.2 eV, corresponding to π–π satellite bonds, appears in TPNCN, which confirms the π–π stacking interaction between TPHAP and NCN.33–35 In addition, the Na 1s peak at 1071.3 eV reveals the existence of Na+ ions originating from TPHAP,36,37 which proves the successful anchoring of TPHAP on the surface of NCN (Fig. S2†).
The morphological structures of the as-prepared photocatalysts were monitored by scanning electron microscopy (SEM). The SEM images with no apparent change in Fig. S3† reveal that the introduced TPHAP could not affect the morphological structure of NCN in the TPNCNm, due to TPHAP just anchored on the surface of NCN via π–π stacking interactions. For further studying the distribution of TPHAP on the surface of CCN, the SEM-mapping scan was performed (Fig. 1h–o). The elements of C, N, O, and K are uniformly dispersed throughout the surface of NCN. Specifically, the uniform dispersion of the Na element originating from TPHAP confirms the successful anchoring of TPHAP on the surface of NCN in TPNCNm without any accumulation, which is favorable for the fast catalytic reaction of CO2 photoreduction. In addition, TPNCNm reveals a high thermal stability below 500 °C according to thermogravimetry analysis (TG) (Fig. S4†)
Efficient charge separation and migration driven by the strong built-in electric field
After successfully anchoring TPHAP on the surface of NCN, the band structure of TPHAP and NCN was evaluated by UV-visible diffuse reflectance (DRS) spectroscopy and Mott–Schottky measurements. As illustrated in Fig. 2a, all the samples feature distinct optical absorption in the range of 240–420 nm, corresponding to the π → π* transition from the valence band to the conduction band in the conjugated aroma system of NCN.38 Distinct from the optical absorption of GCN, NCN exhibited a slightly wider optical absorption, related to the n → π* electronic transition in the cyanamide group translated from the amino groups after the molten-salt (KSCN) method. This phenomenon is consistent with the smaller band gap values (Eg) calculated from the Kubelka–Munk equation over NCN (2.75 eV) than that of GCN (2.77 eV) (Fig. S5 and Table S3†). Furthermore, the spectrum of TPHAP displays an enhanced absorption intensity in the range of 450–600 nm, which is not very obvious in TPNCNm, due to the small mass percentage of TPHAP in TPNCNm. As further determined from the Mott–Schottky plots (Fig. S6†), flat band potentials (Efb) of GCN, NCN and TPHAP are −0.71 V, −0.65 V and −0.35 V vs. NHE (PH = 7), respectively. In generation, the conduction band potential (ECB) minimum was about 0.2 V more negative than the Efb.24 Thus, the ECB values of GCN, NCN and TPHAP are calculated to be −0.91 V, −0.85 V, and −0.55 V vs. NHE (PH = 7), respectively. Given the calculations of Eg and ECB, the corresponding band positions of GCN, NCN and TPHAP are outlined in Fig. S7a.† Notably, all the samples possessed more negative ECB values than the redox potential of CO2–CO (−0.53 V) and CO2–CH4 (−0.24 V), demonstrating enough thermodynamics reduction ability for photocatalytic CO2 reduction. It is worth pointing out that NCN exhibited a more negative CB position than that of TPHAP, suggesting that a built-in electric field would be formed between them, driving photogenerated charge carrier migration from NCN to TPHAP and inhibiting charge recombination.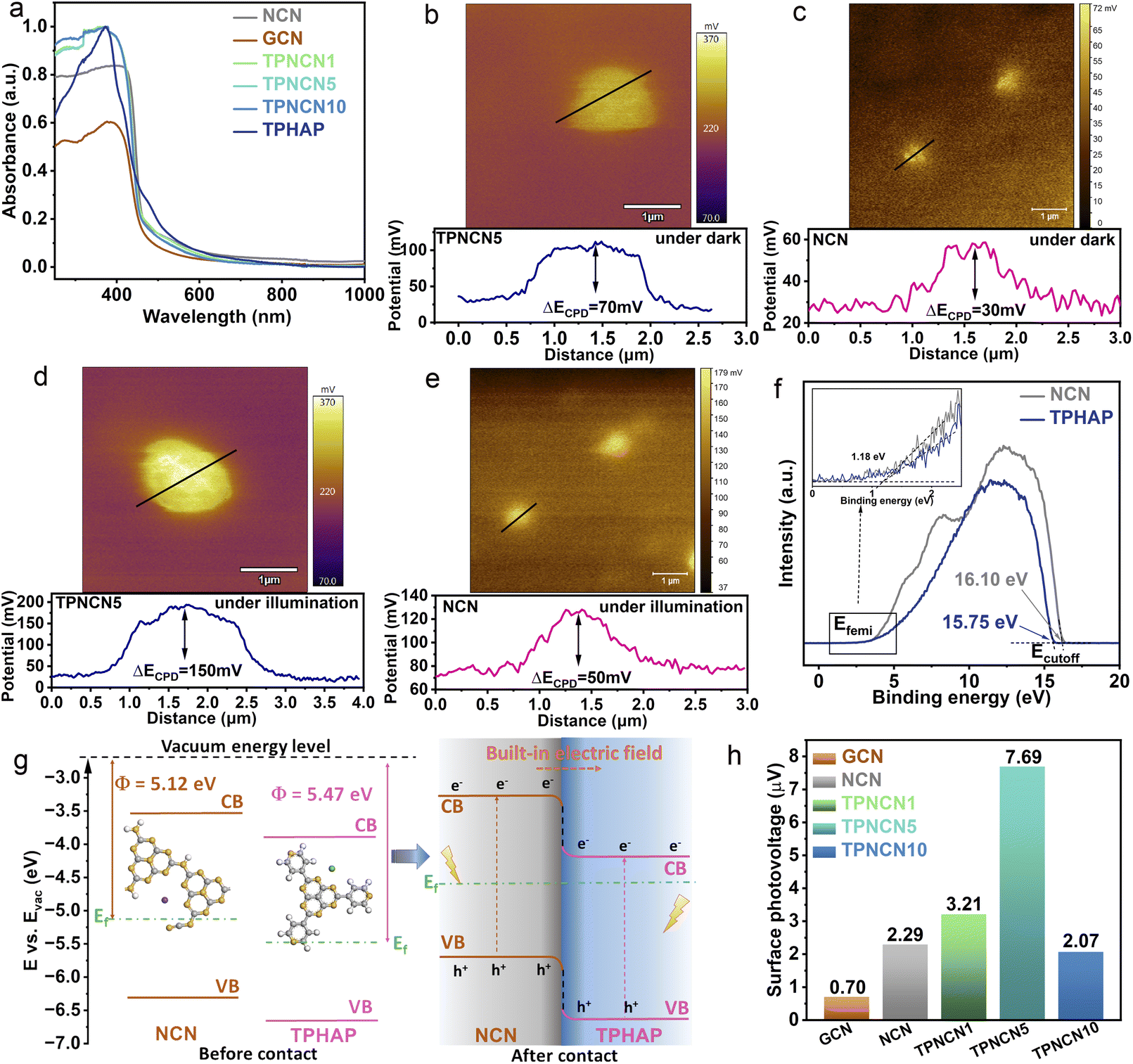 | ||
| Fig. 2 (a) UV-visible diffuse reflectance spectra of GCN, NCN and TPNCNm. Kelvin probe force microscopy images (top) and surface potential difference (bottom) according to the cross-section in (b), (c) in the dark and (d), (e) under irradiation. (f) Ultraviolet photoelectron spectroscopy (UPS) of NCN and TPHAP. (g) Schematic diagram of forming a built-in electric field between TPHAP and NCN. (h) The surface voltage intensity of GCN, NCN and TPNCNm. | ||
To directly confirm the built-in electric field between NCN and TPHAP and investigate its direction, Kelvin probe force microscopy (KPFM), ultraviolet photoelectron spectroscopy (UPS) and density functional theory calculation (DFT) were further carried out. After anchoring TPHAP on the surface of NCN, the contact potential difference (ΔECPD) detected from KPFM of TPNCN5 (ΔECPD = 70 mV, Fig. 2b) increases in contrast to NCN (ΔECPD = 30 mV, Fig. 2c), which provides direct evidence for the formation of the built-in electric field between TPHAP and NCN.39 Driven by this built-in electric field, TPNCN5 possesses a larger difference between before and after illumination of ΔECPD = 150 mV than that of NCN (ΔECPD = 50 mV) due to the accumulation of electrons and holes on TPHAP and NCN (Fig. 2d and e), respectively, which further demonstrates the accelerated photoexcited charge carrier separation by the built-in electric field.36,40,41
Based on the secondary electron cutoff energy (Fig. 2f), the work function (Φ) of NCN and TPHAP could be calculated to be −5.12 and 5.47 eV (Fig. 2g, S7b and Table S4†). Generally speaking, the Φ could be used to judge the direction of the built-in electric field, as the electrons tend to migrate from one sample with a low Φ to another sample with high Φ.42,43 Hence, it is pertinent to determine that, when TPHAP is anchored on the surface of NCN, electrons of NCN with low Φ would migrate to TPHAP with high Φ until equilibrium of the Fermi energy levels of NCN and TPHAP, forming a stable built-in electric field with the direction from NCN to TPHAP. Additionally, DFT calculation further confirms this phenomenon as NCN (Φ = 5.055 eV) possesses a more lower Φ than TPHAP (Φ = 5.319 eV) (Fig. S8†) and this difference in Φ could drive the electron migration from NCN to TPHAP.
To further evaluate the magnitude of the built-in electric field between TPHAP and NCN, surface voltage and photocurrent studies were performed. After anchoring TPHAP on the surface of NCN, the built-in electric field magnitude could be evaluated based on surface charge density (ρ) and surface voltage (Vs),44 which could be estimated by the photocurrent density and surface photovoltage (SPV) measurement (Fig. 3a and S9†), respectively. As illustrated in Fig. 2h, TPNCNm exhibits an enhanced built-in electric field in contrast to NCN and GCN, which could provide a strong driving force for effective charge separation and migration at the interface. Significantly, TPNCN5 presents the maximum built-in electric field magnitude, which is 3.35 times higher than that of NCN.
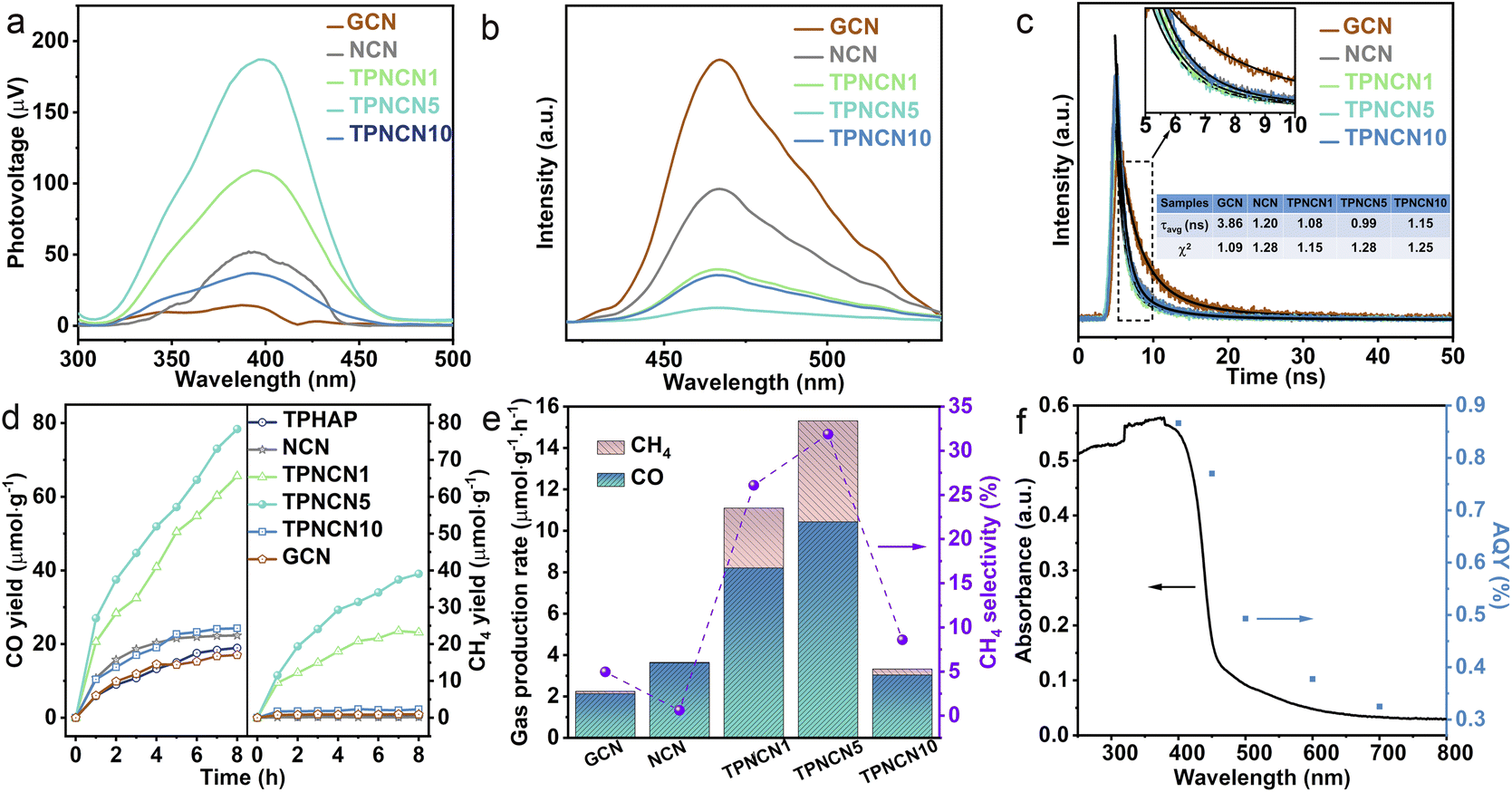 | ||
| Fig. 3 (a) Surface photovoltage spectra, (b) steady-state photoluminescence (PL) emission spectra, and (c) time-resolved PL decay spectra of GCN, NCN and TPNCNm. (d) Time dependent CO (left) and CH4 (right) yield and (e) production rates of CO and CH4 over GCN, NCN and TPNCNm under visible-light irradiation. Inset of (e) the purple curve reveals the CH4 selectivity over catalysts. (f) UV-visible DRS spectrum and wavelength-dependent AQYs of TPNCN5 for photocatalytic CO generation. | ||
It is well recognized that a built-in electric field formed in TPNCNm could accelerate the efficiency of charge transfer between NCN and TPHAP, as it is believed to enhance the photoactivity of CO2 photoreduction. For further understanding the charge-transfer dynamics of TPNCNm driven by the built-in electric field, the photophysical behaviors of the photoexcited charge carriers were investigated by photocurrent, surface photovoltage (SPV), steady-state and time-resolved photoluminescence (PL) emission spectroscopy, and electrochemical impedance spectroscopy (EIS) measurements. Given the positive SPV signals illustrated in Fig. 3a, GCN, NCN and TPNCNm feature n-type semiconductor characteristics.45 The stronger SPV signal intensity of NCN than GCN identifies that the improved crystallinity would favor effective photogenerated charge separation and then transfer to the surface. After introducing TPHAP, the SPV signals of TPNCNm increased with the maximum for TPNCN5, attributed to the built-in electric field driving photogenerated charge separation. However, the SPV signal of TPHAP10 is weakened, revealing that an excess amount of TPHAP would act as the recombination centers of e−–h+ pairs. This phenomenon could be further verified by steady-state and time-resolved PL emission spectra. It is noted that the PL emission originates from the radiative recombination of electron–hole pairs (e−–h+), meaning a lower intensity of steady-state PL emission indicates a lower density of charge carrier recombination and shortened PL average lifetimes (τavg) indicating the enhanced process of charge transfer.46,47 Given the improved crystallinity for NCN, the PL intensity of NCN is significantly reduced compared to GCN with a shortened τavg (1.20 ns) (Fig. 3b and c), due to effective charge carrier migration over NCN with a reduced interlayer distance and extended conjugate system, inhibiting charge recombination. Thanks to the built-in electric field, the steady-state PL spectra of TPNCNm are effectively quenched relative to NCN, with shortened τavg (1.08 ns for TPNCN1, 0.99 ns for TPNCN5 and 1.15 ns for TPNCN10), attributed to the efficient charge carrier separation between NCN and TPHAP, inhibiting the recombination of photoexcited e−–h+ pairs. With increasing the mass percentage of TPHAP, the PL quenching ability of TPNCNm changes among which TPNCN5 exhibits the lowest steady-state PL intensity and shortest PL lifetime, which confirms that 5% mass percentage of TPHAP is conducive to the maximum efficiency of charge-carrier separation. These observations are further solidified by the increased photocurrent response (Fig. S10†) and decreased charge transfer resistance in electrochemical impedance spectroscopy (EIS) (Fig. S11†). In detail, a high intensity of photocurrent indicates the effective separation of photoexcited e−–h+ pairs.48 Meanwhile, the small value of charge transfer resistances (Rct) detected from the Nyquist plots fitted by an equivalent circuit reveals reduced charge transfer resistance for fast charge carrier migration rate,49 synergistically improving the charge dynamics for efficient photocatalytic performance. With improved crystallinity, NCN possesses an enhanced photocurrent intensity and decreased Rct value relative to GCN. Specifically, TPNCN5 displays the largest photocurrent intensity and smallest Rct value, attributed to the driving force from the built-in electric field with a suitable mass percentage of TPHAP to facilitate charge carrier separation and migration.
CO2 photoreduction performance and reaction mechanism
The CO2 photoreduction performance of all the samples was assessed through the gas–solid model under simulated light irradiation. Given the improved crystallinity for NCN, the CO yield reaches 22.33 μmol g−1 with a CO product rate of 2.12 μmol g−1 h−1, 1.3 times higher in contrast to GCN. With the introduction of TPHAP, the CO product yields of TPNCNm increase first and then decrease depending on the increasing mass percentage of TPHAP (Fig. 3d). Based on the improving photocatalytic activity for NCN, TPNCN5 presents the highest CO generation rate of 10.43 μmol g−1 h−1, 3.5 times compared to NCN (Fig. 3e), which stands at a higher level than those of reported GCN-based all-organic photocatalysts and at an equivalent level to GCN coupled with compounds containing metal elements (Table S5†).No liquid products and H2 were detected (Fig. S12†). Unfortunately, instead of continuous increase, the CO generation rate of TPNCN10 begins to rapidly decrease, ascribed to the excessive TPHAP molecules acting as the centres of charge carrier recombination. Significantly, TPNCNm exhibits a fast CH4 generation rate (2.90 μmol g−1 h−1 for TPNCN1, 4.88 μmol g−1 h−1 for TPNCN5, 0.285 μmol g−1 h−1 for TPNCN10) compared to NCN with a seriously slow CH4 generation rate of 0.022 μmol g−1 h−1, indicating that introducing TPHAP could drive the multiple electron and proton reaction resulting from the built-in electric field in TPNCNm. Notably, the CH4 selectivity increased after constructing TPNCNm, reaching a maximum CH4 selectivity of 33% for TPCNCN5 and electron selectivity of 64% for CH4 (Fig. S13†). Recycling photocatalytic reactions under the same conditions reveals the stability of TPNCNm for the CO2 photoreduction reaction according to no significant reduction for the photoreduction activity of CO2–CO/CH4 (Fig. S14a†). In addition, all the samples exhibit excellent chemical stability according to the unchanged FTIR spectra after the photocatalytic CO2 reduction reaction (Fig. S14b†). The apparent quantum yield (AQY) for CO production at different wavelengths is detected under the same photocatalytic reaction conditions (Fig. 3f). It is noted for TPNCN5 that the AQY values at different wavelengths are consistent with the optical absorption edge, suggesting that the CO2 photoreduction reaction was indeed driven by the incident photons. In addition, the control experiments and 13CO2 isotopes firmly confirm that the photocatalytic products of CO and CH4 originate from the CO2 molecule reacting with H2O over the photocatalyst driven by irradiation (Fig. S15†).Based on introducing the TPHAP molecule and constructing a strong built-in electric field in TPNCNm, a further fundamental understanding of the CO2 photoreduction reaction mechanism over GCN, NCN and TPNCN (taking TPNCN5 as an example) was obtained by in situ attenuated total reflectance (ATR)-FTIR spectroscopy measurements. With prolonged absorption and illumination time, gradually strong bands at 1704 and 1262 cm−1 corresponding to the CO2* asymmetric stretching could be detected over TPNCN50 (Fig. 4a–c), which are much stronger than those of NCN and PCN, suggesting that the CO2 absorption at the surface of TPNCN is dramatically enhanced with the introduction of TPHAP molecules. Meanwhile, a band at 1652 cm−1 corresponding to the surface-adsorption of H2O on TPNCN is detected, which is more distinct than those of GCN and NCN,51 demonstrating effective H2O molecular adsorption on the surface of TPNCN, the initial step of CO2 photoreduction reaction. The intermediate species of CO2 photoreduction reduction are mainly distributed in the range of 1200–2200 cm−1 (Fig. 4a). Subsequently, some surface protons and carbon species, such as carbonate (m-CO32−), bicarbonate (HCO3−) and bidentate carbonate (b-CO32−), are generated from the reaction of surface-adsorbed H2O and CO2 molecules (CO2 + H2O → CO32− + H+, CO2 + H2O → HCO3− + H+).50–53 TPNCN presents more abundant carbon species than GCN and NCN, revealing that more surface protons would be generated and accumulated on the surface of TPNCN, which favours the subsequent protonation reaction. Depending on the increase of illumination time, new bands located at 1436 and 1352 cm−1 appear and get strengthened, which could be associated with the key intermediate of COOH* for photoreduction CO2-to-CO conversion (CO2 (g) + H+ + e− → COOH*).54 Additionally, careful observation shows the emergence of two additional bands at 2168 and 2130 cm−1, gradually increasing with prolonged illumination time, which could be attributed to the CO* intermediate accumulated on the surface of TPNCN via the reaction of COOH* + H+ + e− → CO* + H2O.55 However, this CO* intermediate band could not be detected over GCN and NCN. This comparison of the CO* intermediate band confirms that TPNCN would favour effective transfer of photogenerated electrons and surface protons, attributed to the effective reaction of CO2 and H2O molecules driven by the enhanced charge dynamics. Therefore, the above results confirm the crucial importance of two intermediates COOH* and CO* for the CO2 photoreduction reaction.
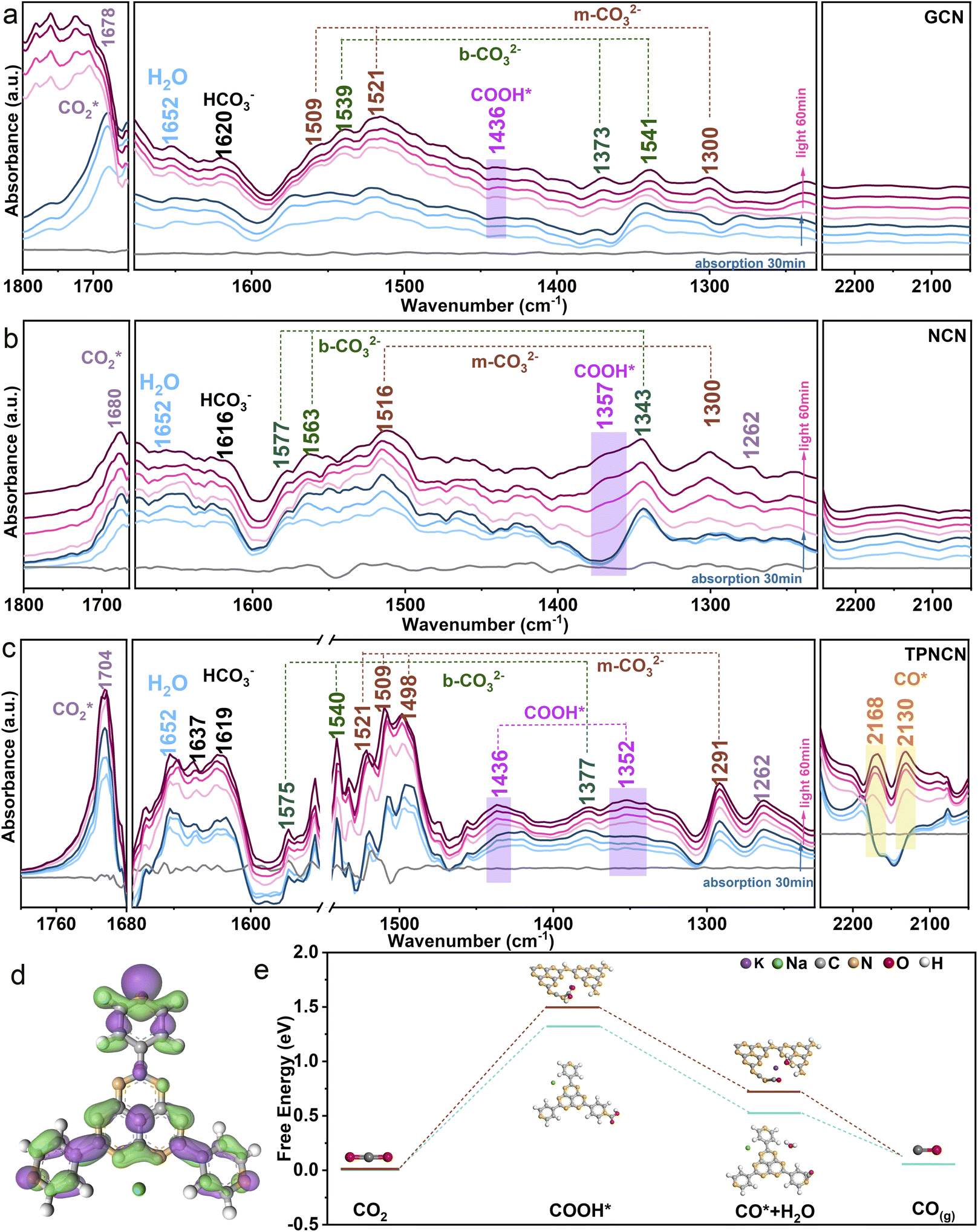 | ||
| Fig. 4 In situ ATR-FTIR spectra of (a) GCN, (b) NCN and (c) TPNCN. (d) Charge density difference of TPHAP. Green and purple regions represent electron depletion and accumulation, respectively. (e) Calculated free energy diagram for the reaction pathways of CO2 photoreduction for TPHAP and NCN. | ||
For deeper insight into the role of the TPHAP molecule in the TPNCNm for effective CO2 photoreduction reaction, density functional theory (DFT) calculation for elementary steps was then performed depending on the above result of in situ ATR-FTIR measurement. Given charge carrier migration from NCN to TPHAP driven by the built-in electric field in TPNCNm, the conversion reaction of CO2 should be carried out on the TPHAP molecule. To verify this assumption, the contrast of Gibbs free energy of elementary steps for NCN and TPHAP was recorded by the DFT. Prior to elementary step calculation, more accurate active sites of TPHAP for CO2 photoreduction reaction are detected by the charge density difference. As shown in Fig. 4d and S9a,† pyridine N is the electron-accumulation region for TPHAP, making it the preferred active site for the conversion of CO2. As for NCN, the electrons are accumulated in the triangular edge N of the tri-s-triazine ring, acting as the active site for the reduction reaction (Fig. S16c†). For TPHAP, the free energy of formation of COOH* (Fig. 4e), the rate-determining step for the photoreduction reaction of CO2 to CO, is observably reduced from 1.50 eV for NCN to 1.31 eV, attributed to the abundant surface-protons and accumulated electrons over the TPHAP surface driven by the built-in electric field in TPNCNm. Similarly, the decomposition energy barrier for COOH* into CO* on TPHAP (0.72 eV) is also lower than that of NCN (0.52 eV), leading to fast formation of CO* and then desorption into CO gas. The above result confirmed that TPHAP acting as the active site is more conducive for the CO2 conversion reaction than GCN, which was consistent with the highly effective CO evolution rate for CO2 photoreduction on TPNCNm.
Conclusions
We successfully designed and synthesized crystalline TPNCN via moleton-salt and solution-assembly process, achieving a notable enhancement in CO2 photoreduction performance. Experimental results revealed that TPNCN5 achieved a CO and CH4 evolution rate of 10.43 and 4.88 μmol g−1 h−1, respectively, higher than the rates for both GCN and NCN. Depending on the enhanced charge dynamics of NCN with improved crystallinity, a built-in electric field was established between NCN and TPHAP via π–π stacking interactions. This built-in electric field efficiently directed the photoexcited charge carriers from NCN to TPHAP, inhibiting the recombination of electron–hole pairs. DFT calculations demonstrated that pyridine N of TPHAP could act as the active site, as well as reducing the Gibs free energy for the COOH* intermediate in the photoreduction of CO2-to-CO. This effect was attributed to the abundant surface-proton and accumulated electrons facilitated by the built-in electric field. Accordingly, this work provides a strategy in which coplanar materials are utilized to construct high-efficiency hybrid graphitic nitride carbon-based photocatalysts with a strong built-in electric field through π–π interactions to boost the photocatalytic performance.Author contributions
Y. Li and L. Wang carried out the sample synthesis and characterization. X. Gao carried out the electrochemical measurement. Y. Xue and B. Li carried out the CO2 photoreduction measurements. Y. Li and X. Zhu wrote the paper and conceived the idea and supervised the project. All the authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22005241, 22202128, and 22303064), Outstanding Youth for Xi'an University of Science and Technology (2023YQ3-07), the Natural Science Basic Research Program of Shaanxi (2018JQ2028, 2021JQ-302, and 2023-JC-YB-094), the China Postdoctoral Science Foundation (2020M673440), and the Education Department Fund in Shaanxi Province (2021JK0763).Notes and references
- W. Soontornchaiyakul, S. Yoshino, T. Kanazawa, R. Haruki, D. Fan, S. Nozawa, Y. Yamaguchi and A. Kudo, J. Am. Chem. Soc., 2023, 145, 20485 CrossRef CAS PubMed.
- S. Li, L. Hu, Z. Qian, J. Yin, J. Tang, C. Pan, G. Yu and K. A. I. Zhang, ACS Catal., 2023, 13, 12041 CrossRef CAS.
- Z. A. Lan, M. Wu, Z. Fang, X. Chi, X. Chen, Y. Zhang and X. Wang, Angew. Chem., Int. Ed., 2021, 60, 16355 CrossRef CAS PubMed.
- F. Guo, B. Hu, C. Yang, J. Zhang, Y. Hou and X. Wang, Adv. Mater., 2021, 33, 2101466 CrossRef CAS PubMed.
- Y. Li, B. Li, D. Zhang, L. Cheng and Q. Xiang, ACS Nano, 2020, 14, 10552 CrossRef CAS PubMed.
- P. Chen, B. Lei, X. Dong, H. Wang, J. Sheng, W. Cui, J. Li, Y. Sun, Z. Wang and F. Dong, ACS Nano, 2020, 14, 15841 CrossRef CAS PubMed.
- L. Zhu, F. Hu, B. Sun, S. Gu, T. Gao and G. Zhou, Adv. Sustainable Syst., 2023, 7, 2200394 CrossRef CAS.
- W. Lyu, Y. Liu, J. Zhou, D. Chen, X. Zhao, R. Fang, F. Wang and Y. Li, Angew. Chem., Int. Ed., 2023, e202310733 CAS.
- J. Ding, Q. Tang, Y. Fu, Y. Zhang, J. Hu, T. Li, Q. Zhong, M. Fan and H. H. Kung, J. Am. Chem. Soc., 2022, 144, 9576 CrossRef CAS PubMed.
- Y. Li, Y. Guo, D. Luan, X. Gu and X. W. Lou, Angew. Chem., Int. Ed., 2023, e202310847 CAS.
- K. Li, B. Peng, J. Jin, L. Zan and T. Peng, Appl. Catal., B, 2017, 203, 910 CrossRef CAS.
- B. C. He, C. Zhang, P. P. Luo, Y. Li and T. B. Lu, Green Chem., 2020, 22, 7552 RSC.
- X. Chen, J. Wang, Y. Chai, Z. Zhang and Y. Zhu, Adv. Mater., 2021, 33, 2007479 CrossRef CAS PubMed.
- J. Jing, J. Yang, W. Li, Z. i. Wu and Y. Zhu, Adv. Mater., 2022, 34, 2106807 CrossRef CAS PubMed.
- J. Wang, H. Huang, P. Wang, G. Yang, S. Kupfer, Y. Huang, Z. Li, Z. Ke and G. Ouyang, JACS Au, 2022, 2, 1359 CrossRef CAS PubMed.
- R. Sun, H. Yin, Z. Zhang, Y. Wang, T. Liang, S. Zhang and L. Jing, J. Phys. Chem. C, 2021, 125, 23830 CrossRef CAS.
- S. A. Sharber, W. J. Mullin and S. W. Thomas, Chem. Mater., 2021, 33, 6640 CrossRef CAS.
- G. R. Lee, H. Ohtsu, J. Koo, Y. Yakiyama, M. J. Park, D. Inoue, D. Hashizume and M. Kawano, Chem. Commun., 2016, 52, 3962 RSC.
- Y. Yakiyama, A. Ueda, Y. Morita and M. Kawano, Chem. Commun., 2012, 48, 10651 RSC.
- J. Kim, H. Ohtsu, T. Den, K. Deekamwong, I. Muneta and M. Kawano, Chem. Sci., 2019, 10, 10888 RSC.
- V. W. Lau, I. Moudrakovski, T. Botari, S. Weinberger, M. B. Mesch, V. Duppel, J. Senker, V. Blum and B. V. Lotsch, Nat. Commun., 2016, 7, 12165 CrossRef CAS PubMed.
- L. Lin, H. Ou, Y. Zhang and X. Wang, ACS Catal., 2016, 6, 3921 CrossRef CAS.
- H. Ou, L. Lin, Y. Zheng, P. Yang, Y. Fang and X. Wang, Adv. Mater., 2017, 29, 1700008 CrossRef PubMed.
- Q. Liang, Z. Li, Z. H. Huang, F. Kang and Q. H. Yang, Adv. Funct. Mater., 2015, 25, 6885 CrossRef CAS.
- Z. Yu, X. Yue, J. Fan and Q. Xiang, ACS Catal., 2022, 12, 6345 CrossRef CAS.
- V. W. H. Lau, I. Moudrakovski, T. Botari, S. Weinberger, M. B. Mesch, V. Duppel, J. Senker, V. Blum and B. V. Lotsch, Nat. Commun., 2016, 7, 12165 CrossRef CAS PubMed.
- Y. Xu, X. He, H. Zhong, D. J. Singh, L. Zhangc and R. Wang, Appl. Catal., B, 2019, 246, 349 CrossRef CAS.
- Z. Liu, J. Ma, M. Hong and R. Sun, ACS Catal., 2023, 13, 2106 CrossRef CAS.
- Y. Li, Y. Wang, C. L. Dong, Y. C. Huang, J. Chen, Z. Zhang, F. Meng, Q. Zhang, Y. Huangfu, D. Zhao, L. Gu and S. Shen, Chem. Sci., 2021, 12, 3633 RSC.
- J. Yuan, X. Yi, Y. Tang, C. Liu and S. Luo, ACS Appl. Mater. Interfaces, 2020, 12, 19607 CrossRef CAS PubMed.
- J. Zhu, Y. Xu, C. Yang, C. He, P. Zhang, Y. Li, X. Ren and H. Mi, ACS Catal., 2022, 12, 4648 CrossRef.
- Y. Li, Y. Xue, X. Gao, L. Wang, X. Liu, Z. Wang and S. Shen, Adv. Funct. Mater., 2023, 2312634 CrossRef.
- X. Chen, X. Wang and D. Fang, Fullerenes, Nanotubes, Carbon Nanostruct., 2020, 28, 1048 CrossRef CAS.
- D. Yang, J. Rochette and E. Sacher, J. Phys. Chem. B, 2005, 109, 4481 CrossRef CAS PubMed.
- X. Liu, Z. Wang, S. Feng, X. Zhang, H. Xu, G. Wei, X. Gong, X. g. Wu and J. Hua, Macromolecules, 2023, 56, 8275 CrossRef CAS.
- A. Abbas, M. Nisar, Z. H. Zheng, F. Li, B. Jabar, G. Liang, P. Fan and Y. Chen, ACS Appl. Mater. Interfaces, 2022, 14, 25802 CrossRef CAS PubMed.
- Y. Yang, L. Huang and D. Pan, ACS Appl. Mater. Interfaces, 2017, 9, 23878 CrossRef CAS PubMed.
- J. Zhang, X. Chen, K. Takanabe, K. Maeda, K. Domen, J. D. Epping, X. Fu, M. Antonietti and X. Wang, Angew. Chem., Int. Ed., 2010, 49, 441 CrossRef CAS PubMed.
- Q. Tang, W. Tao, J. Hu, T. Gui, Z. Wang, Y. Xiao, R. Song, Y. Jiang and S. Guo, ACS Appl. Nano Mater., 2023, 6, 17130 CrossRef CAS.
- C. Cheng, B. He, J. Fan, B. Cheng, S. Cao and J. Yu, Adv. Mater., 2021, 2100317 CrossRef CAS PubMed.
- J. Wang, E. Kim, D. P. Kumar, A. P. Rangappa, Y. Kim, Y. Zhang and T. Kim, Angew. Chem., 2022, 134, 202113044 CrossRef.
- W. Lyu, Y. Liu, J. Zhou, D. Chen, X. Zhao, R. Fang, F. Wang and Y. Li, Angew. Chem., Int. Ed., 2023, e202310733 CAS.
- L. Zhai, X. She, L. Zhuang, Y. Li, R. Ding, X. Guo, Y. Zhang, Y. Zhu, K. Xu, H. J. Fan and S. P. Lau, Angew. Chem., 2022, 134, e202116057 CrossRef.
- P. Lefebvre, J. Allègre, B. Gil, H. Mathieu, N. Grandjean, M. Leroux, J. Massies and P. Bigenwald, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 15363 CrossRef CAS.
- G. Morello, F. Della Sala, L. Carbone, L. Manna, G. Maruccio, R. Cingolani and M. DeGiorgi, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 195313 CrossRef.
- Z. Zhang, X. Chen, H. Zhang, W. Liu, W. Zhu and Y. Zhu, Adv. Mater., 2020, 32, 1907746 CrossRef CAS PubMed.
- S. K. Das, N. K. Subbaiyan, F. D'Souza, A. S. D. Sandanayaka, T. Hasobe and O. Ito, Energy Environ. Sci., 2011, 4, 707 RSC.
- W. Xing, G. Chen, C. Li, Z. Han and Y. Hu Q. Meng, Nanoscale, 2018, 10, 5239 RSC.
- W. Che, W. Cheng, T. Yao, F. Tang, W. Liu, H. Su, Y. Huang, Q. Liu, J. Liu, F. Hu, Z. Pan, Z. Sun and S. Wei, J. Am. Chem. Soc., 2017, 139, 3021 CrossRef CAS PubMed.
- X. M. Cheng, X. Y. Dao, S. Q. Wang, J. Zhao and W. Y. Sun, ACS Catal., 2021, 11, 650 CrossRef CAS.
- Q. Zhang, S. Gao, Y. Guo, H. Wang, J. Wei, X. Su, H. Zhang, Z. Liu and J. Wang, Nat. Commun., 2023, 14, 1147 CrossRef CAS PubMed.
- W. Shangguan, Q. Liu, Y. Wang, N. Sun, Y. Liu, R. Zhao, Y. Li, C. Wang and J. Zhao, Nat. Commun., 2022, 12, 3894 CrossRef PubMed.
- J. Zhou, J. Li, L. Kan, L. Zhang, Q. Huang, Y. Yan, Y. Chen, J. Liu, S. Li and Y. Lan, Nat. Commun., 2022, 13, 4681 CrossRef CAS PubMed.
- L. Shen, Z. Xie, L. Hou, J. Yang and Q. Li, Energy Fuels, 2022, 36, 11515 CrossRef CAS.
- D. Sun, J. Li, T. Shen, S. An, B. Qi and Y. Song, ACS Appl. Mater. Interfaces, 2022, 14, 16369 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta06974e |
| This journal is © The Royal Society of Chemistry 2024 |