
Mechanism and selectivity of MOF-supported Cu single-atom catalysts for preferential CO oxidation†
Sarawoot
Impeng
a,
Evaristo
Salaya-Gerónimo
b,
Benny
Kunkel
b,
Stephan
Bartling
 b,
Kajornsak
Faungnawakij
a,
Bunyarat
Rungtaweevoranit
a and
Ali M.
Abdel-Mageed
*b
b,
Kajornsak
Faungnawakij
a,
Bunyarat
Rungtaweevoranit
a and
Ali M.
Abdel-Mageed
*b
aNational Nanotechnology Center (NANOTEC), National Science and Technology Development Agency (NSTDA), Pathum Thani 12120, Thailand
bLeibniz-Institut für Katalyse (LIKAT), D-18059, Rostock, Germany. E-mail: ali.abdelmageed@catalysis.de
First published on 24th October 2023
Abstract
Zr-based UiO-66 metal–organic frameworks are ideal platforms for the design and development of heterogeneous single-atom catalysts (SACs) because of their thermal and chemical stability with the presence of structural defects, enabling the introduction of isolated metal atoms. Elucidating the structure–reactivity relationships and understanding reaction mechanisms for these catalysts are crucial for their industrial applications. We focus here on these aspects for a technically important reaction, preferential CO oxidation (PROX) on a UiO-66-supported Cu SAC by following temperature perturbations in catalytic performance in correlation with changes in the electronic and adsorption properties, which are validated by comprehensive DFT computations. In situ DR-UV-VIS, XANES and NAP-XPS measurements indicated an increase of Cu1+-like states and partial reduction of ZrOx nodes with the increase in reaction temperature, which correlated with a decrease in PROX selectivity. Under similar conditions, DRIFTS measurements revealed a decay of COad adsorption on Cu (i.e., COad@Cu1+ species) and a corresponding red-shift under PROX conditions compared to CO oxidation, suggesting reduction-mediated charge transfer at the Cu–ZrOx interface. In contrast to the CO oxidation cycle which commences by CO adsorption on Cu1+-like sites, DFT computations revealed that the H2 oxidation cycle starts with the reaction of H2 with a pre-adsorbed O2 molecule on Cu1+-like sites, resulting in the generation of a H2O molecule and Cu2+-like sites, which are subsequently reduced to Cu1+-like sites through a successive reaction with a second H2 (or CO) molecule. Adsorption configurations and energies of CO and H2O co-adsorption indicated a competitive adsorption phenomenon on Cu species, which depends on the oxidation state of the Cu ion with a preference for CO adsorption on Cu1+-like sites, while H2O exhibits a stronger affinity for Cu2+-like sites. These results are discussed in terms of the reaction mechanism and PROX selectivity in Cu SAC catalysts and present a model for understanding the catalytic phenomena on MOF-supported SACs.
1. Introduction
Metal–organic frameworks (MOFs) have recently emerged as an efficient platform for catalysis. This is due to not only their large specific surface area and microporosity,1 but also their hybrid molecular structure made of metal oxide clusters/nodes connected by organic linkers.2 These unique properties allow for control of the electronic properties of active metal sites, making them ideal candidates for catalysis.3 Specifically the use of MOFs is more attractive for the architecture of heterogeneous single-atom catalysts (SACs) both for thermal catalysis,4–7 and also in photocatalytic applications.8–10 With their well-defined molecular structure, MOF-based SACs can be considered as a model system for fundamental understanding of heterogeneous SACs, which is hardly accessible for oxide-supported catalysts.11,12 Additionally, the use of MOFs as supports allows for loading higher density of isolated metal single sites compared to what can be achieved with typical catalyst supports, where limited loading in the latter case is essential to prevent sintering during reactions.12–15 As an example, between 1.6 and 2 wt% of atomically dispersed Cu can be loaded into the UiO-66 framework,16,17 compared to ≤0.05 wt% on CeO2–TiO2 supports needed to ensure atomic dispersion of the loaded metal.11,12,16 This enhances the gravimetric and volumetric density of these catalysts which is necessary for their practical applications, where size and mass of the reactor are decisive. The tradeoff between the total catalyst-mass based activity and gravimetric efficiency in both cases should be optimized for possible technical applications.Heterogeneous SACs have been widely investigated for CO oxidation, both in exhaust catalysis and in preferential CO oxidation (PROX).11–13,15,18 The PROX process is important for the removal of CO from hydrogen-rich feed gases (produced from hydrocarbon reforming) that are used in low-temperature fuel cells or in ammonia power plants.19,20 Considering the sustainable production of hydrogen through the reforming of renewable hydrocarbon sources (e.g., biomass or synthetic natural gas), the PROX process would also play a crucial role in the processing of solar fuels derived from renewable resources.21
We reported earlier that Cu ions can be covalently bonded to the structural defects of Zr-based UiO-66 frameworks,22,23 resulting in stable and highly active Cu-SAC catalysts.24 These catalysts showed high CO oxidation activity in the temperature range from 80 to 350 °C and high stability during reaction on stream. Under PROX conditions at 120 °C, these catalysts exhibited high selectivity for CO oxidation to CO2 (CO + ½O2 → CO2) in hydrogen rich fuel (80% H2, 1% CO, 1% O2), essentially around 100% at temperatures up to 120 °C.
Recently, we studied the CO oxidation reaction mechanism employing an array of experimental methods coupled with DFT calculations.17 Based on this study we mapped out the reaction cycle and identified different reaction intermediates and transition states. In total, we inferred that CO oxidation proceeds via a dissociative activation of O2 which results in the continuous formation of Oad bridging Zr4+ ions in the MOF oxide node to the isolated Cu single ions. The reaction starts with the adsorption of CO on a Cu1+-like state and involves the oxidation of two CO molecules by one O2 molecule per one reaction cycle. Understanding the origin of PROX selectivity of these Cu/UiO-66 catalysts is more complicated in the presence of hydrogen and water molecules, generated during the reaction.
In this contribution, we made use of the well-defined molecular structure of the Cu-SACs supported by a UiO-66 MOF to better understand the origin of PROX selectivity on these catalysts in the PROX reaction, and thus mapped out the reaction mechanism including the determination of different molecular intermediates and transition states involved. In our approach we investigated the activity and selectivity of the single-atom Cu/UiO-66 catalyst by systematic variation of reaction temperature between 120 and 250 °C, and thus monitored the resulting perturbations in electronic and adsorption properties of the catalysts using a combination of spectroscopic methods, including in situ diffuse reflectance UV-vis/FTIR, X-ray photoelectron (XPS), and X-ray absorption spectroscopy. On the basis of earlier findings on the CO oxidation reaction and experimental results, DFT calculations were employed to study the PROX reaction mechanism based on the computations of different intermediates and transition states, facilitating the identification of the rate limiting step in the reaction and the origin of PROX selectivity. This also involved the energy minimization of the co-adsorption configuration of reactants and products in competition for active sites of Cu/UiO-66 catalysts.
2. Experimental section
2.1 Synthesis of UiO-66 and Cu/UiO-66
UiO-66 was synthesized following the procedure described in ref. 1 and 2. Next, UiO-66 (600 mg) powder was dispersed in a solution containing CuCl2·2H2O (Aldrich, 99.99%, 540 mg) dissolved in N,N-dimethylformamide (DMF, 9 mL) in a 20 mL scintillation vial. To ensure proper sealing, the vial's thread was wrapped with PTFE tape. The vial was capped, and placed in an isothermal oven preheated to 85 °C and maintained at that temperature for 24 h. The product was collected by centrifugation (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm), and washed with DMF four times (30 mL × 4) over 24 h, and with acetone four times (30 mL × 4). Finally, the catalyst was dried under dynamic vacuum overnight at room temperature.
000 rpm), and washed with DMF four times (30 mL × 4) over 24 h, and with acetone four times (30 mL × 4). Finally, the catalyst was dried under dynamic vacuum overnight at room temperature.
2.2 Structural characterization
2.3 Activity measurements
The catalytic performance of the catalyst (activity and selectivity) was investigated in a quartz tube flow-reactor at atmospheric pressure, using high purity gases (99.999%, Air Liquide). The composition of the influent and effluent reaction gases was analyzed by using a gas chromatograph (Agilent GC 7890A, and GC 7890B) equipped with a thermal conductivity detector (TCD) and a flame ionization detector (FID), and coupled with a mass spectrometric detector (Agilent 5977B). Helium was employed as the gas carrier in all measurements. The Cu mass-normalized reaction rates were calculated from the CO conversion (XCO) under differential reaction conditions (XCO < 10%) using the molar flow rate of CO into the reactor (ṅCO,in) and the Cu metal mass (mCu) (see eqn (1)). The selectivity for CO oxidation (S) during PROX was calculated as the ratio of the formation of CO2 (XCO2) to the sum of the formation of CO2 (XCO2) and water (XH2O) during the reaction (see eqn (2)). TOF values were obtained according to eqn (3) considering the atomic weight of Cu (MCu) and essentially ≈100% dispersion of Cu species in the sample.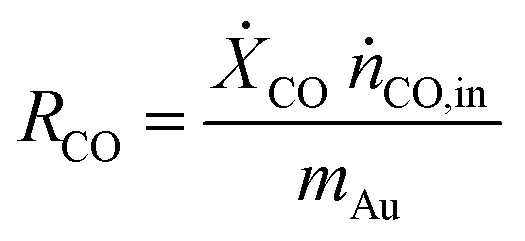 | (1) |
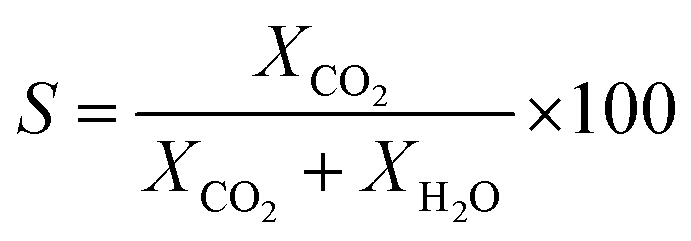 | (2) |
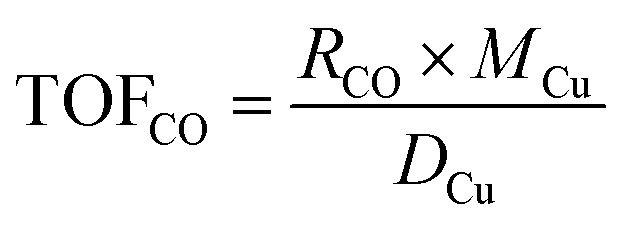 | (3) |
Activity measurements were performed in the temperature range from 250 °C and 120 °C. Studies at lower temperatures were excluded due to the non-measurable changes in the redox properties of Cu species, which correlates with very limited catalytic activity, and thus would not lead to useful insights relevant to the reaction mechanism.17
2.4 Computational details
The DFT method and the model of a single Cu atom anchored on a zirconium oxide cluster of the UiO-66 MOF used in this work follow our earlier work.24 Briefly, the model of the zirconium oxide cluster of UiO-66 was truncated from its crystal structure.29 To model a Cu1+ site in the UiO-66 MOF, one 1,4-benzenedicarboxylate (DBC) linker was removed and OH− and a single Cu atom were placed at the missing linker defect. The proton of μ3-(OH) in the zirconium oxide cluster was then removed to preserve the neutral charge of the system. The remaining BDC linkers were replaced by acetate groups to reduce computational cost. The optimized structure of Cu1+ anchored on the zirconium oxide cluster of the UiO-66 MOF is shown in Fig. 1. The effect of confinement on the reaction barrier is examined and provided in the ESI.† DFT calculations were carried out using the M06-L functional,30 and D3 dispersion correction.31 The 6-31G(d, p) basis set was used for C, O and H atoms while the Stuttgart–Dresden effective core potential (SDD) was adopted for Cu and Zr atoms.32 Frequency calculations were performed to obtain the Gibbs free energy and verify the stationary points on the potential energy surface that are either the transition state (one imaginary frequency) or local minima (all real frequencies). Further discussion on Gibbs free energy at various temperatures can be found in the ESI.† Both singlet and triplet spin states are considered during the reaction; see the ESI for more information.† The Gaussian 16 code was used in all calculations.33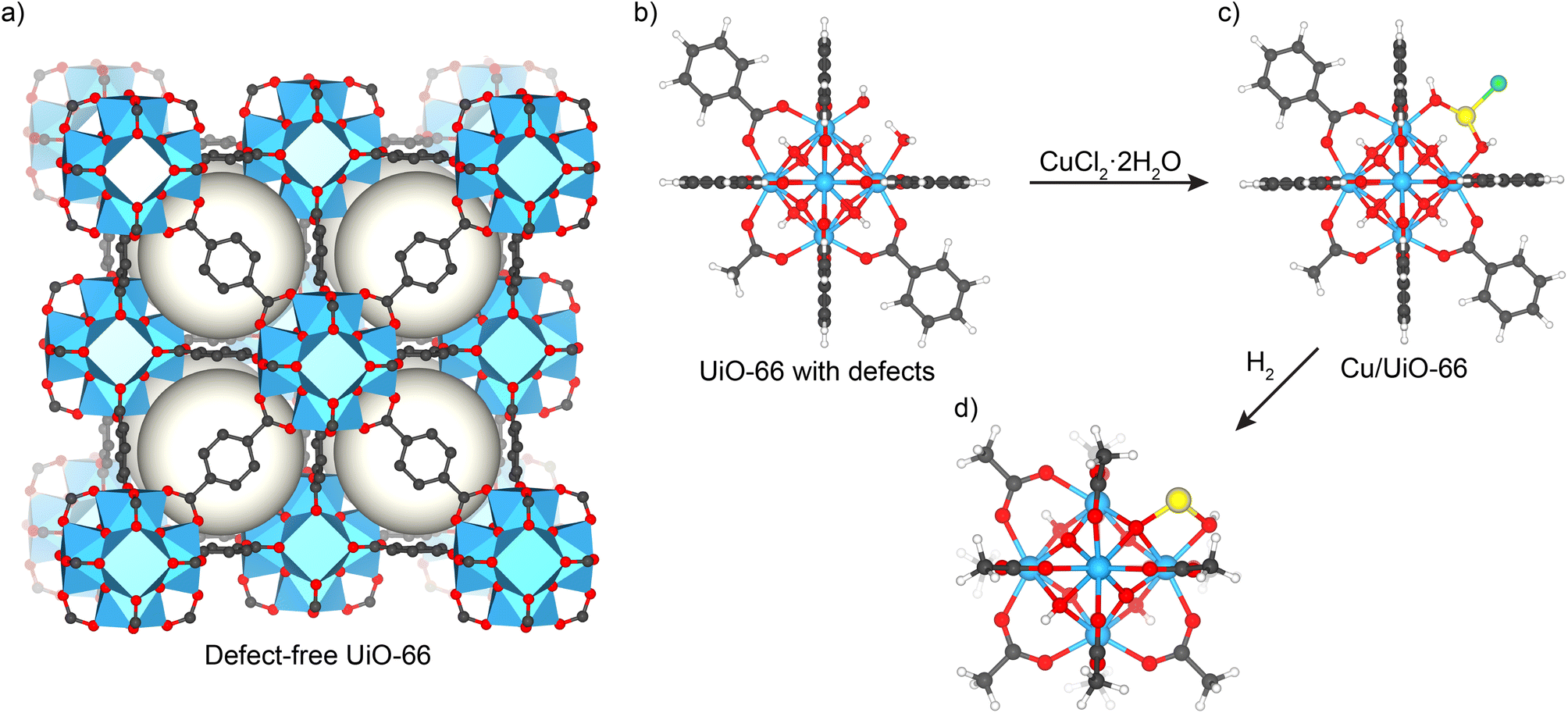 | ||
| Fig. 1 (a) Structure of defect-free UiO-66. (b) Defective UiO-66 with missing terephthalate and (c) the resulting Cu-single-atom catalyst from the reaction of CuCl2 with defective UiO-66. (d) DFT optimized structures of Cu1+ anchored on a zirconium oxide cluster of UiO-66 MOF. [Colors: Cu (dark orange), Zr (cyan), O (red), C (grey) and H (white)]. | ||
3. Results and discussion
3.1 Cu/UiO-66 catalyst
The single-atom Cu/UiO-66 catalyst was prepared in two steps. First, UiO-66 was synthesized using a solvothermal reaction between terephthalic acid and ZrCl4 in DMF.24,34 Based on the detailed characterization studies we reported previously,24 this procedure yields a MOF with a chemical formula of Zr6O4(OH)4(C8H4O4)5(CH3COO)0.7(H2O)1.3(OH)1.3. Different from the perfect UiO-66 structure (Fig. 1a),29,35 there is approximately one missing terephthalate linker per Zr6 cluster which is capped by acetate and –OH/–H2O groups (Fig. 1b) (see structural features summarized in Table S1†). The batch of the catalyst used in PROX studies is similar in chemical composition to that reported before,17 and was additionally characterized by HAADF STEM measurements and EXAFS after reduction at 250 °C to ensure synthesis reproducibility. Basic characterization including N2 adsorption isotherms and XRD measurements confirmed that the structure is retained (see details in ESI, Fig. S1 and S2†).STEM micrographs and corresponding EDS maps of the fresh reduced catalyst at 250 °C indicated a homogeneous distribution of Cu sites inside the MOF, similar to earlier findings (see Fig. S3 and S4†). In addition, EXAFS data on the catalyst used in the studies after being reduced in hydrogen at 250 °C are in good agreement with earlier results (see data discussed later, Fig. S8 and Table S2†). These –OH/–H2O groups capping molecular defects, which are on average 1.3 sites per Zr6 cluster, were then utilized to anchor Cu atoms by heating the MOF in a solution of CuCl2·2H2O in DMF at 85 °C overnight to produce Cu/UiO-66 catalysts (see Fig. 1c). Further details on the elucidation of the catalyst structure are explained elsewhere (see details in ref. 17 and 24).
3.2 Catalytic performance and PROX selectivity
We tested the oxidation of CO and hydrogen in the PROX gas mixture at different temperatures in the range from 120 to 250 °C directly after a reductive activation step. The results indicated a continuous increase in the CO oxidation activity with the temperature between 120 °C and 220 °C with the TOF value increasing by a factor of 4 from 0.15 to 0.59 × 10−3 s−1. Further increase in temperature to 250 °C resulted in a decrease in the TOF value for CO oxidation to 0.42 × 10−3 s−1 (see Fig. 2a – see time-on-stream data in Fig. S5, ESI†). On the other hand, hydrogen oxidation under these conditions was only observed at 150 °C and increased further upon reaching 220 °C by 24 times from 0.26 × 10−3 s−1 at 150 °C to 6.3 × 10−3 s−1 at 220 °C. With a further increase in the temperature to 250 °C the rate of hydrogen oxidation to water remained almost unchanged at a value of 6.3 × 10−3 s−1. These differences in activity correlated with a change in the PROX selectivity from 100% at 120 °C to 7% at 250 °C (see Fig. 2a – right y-axis). Here it is clear that the impact of temperatures higher than 120 °C on hydrogen oxidation is much more pronounced than on the oxidation of CO, and is more decisive for the catalyst selectivity.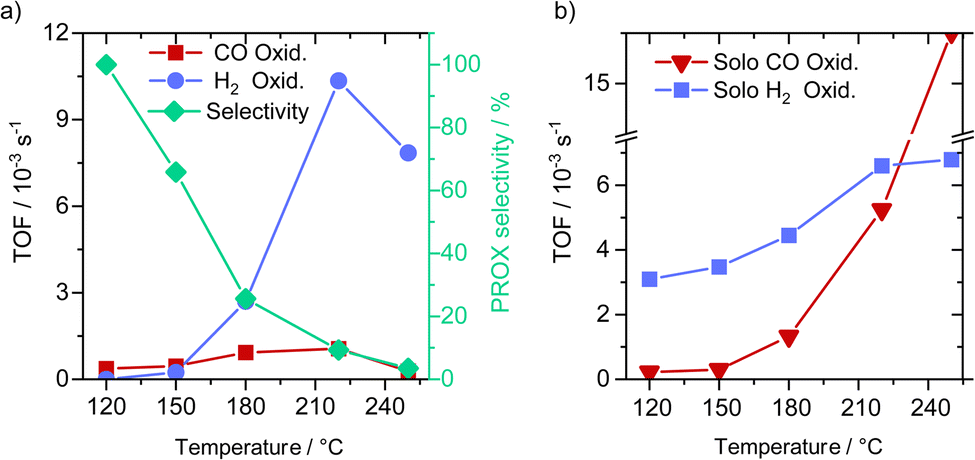 | ||
| Fig. 2 (a) CO and H2 oxidation reactions during the PROX reaction (1% CO, 1% O2, 80% H2 and N2) at different temperatures, after the H250 pretreatment (1 h in 10% H2/N2 at 250 °C) and (left y-axis) corresponding selectivity for CO oxidation derived from rates of CO2 and H2O formation (right y-axis). (b) Solo CO oxidation (1% CO, 1% O2, and N2) and hydrogen oxidation (1% O2, 80% H2 and N2) reactions after H250 treatments. | ||
To test catalyst stability and also the impact of higher CO conversion on the PROX selectivity, we increased the size of the catalyst bed by 3 times, resulting in a decrease in GHSV by 3 times (from 10975 to 3658 h−1) compared to in activity measurements reported in Fig. 3a. After this, we run the catalyst at 120 °C reaching an average CO conversion of about 10%, with essentially no trend for any deactivation with time on stream. At this conversion we observed no hydrogen oxidation, hinting at 100% selectivity for CO (see Fig. S6†). This result also agrees with earlier findings reported on similar Cu/UiO-66 catalysts.
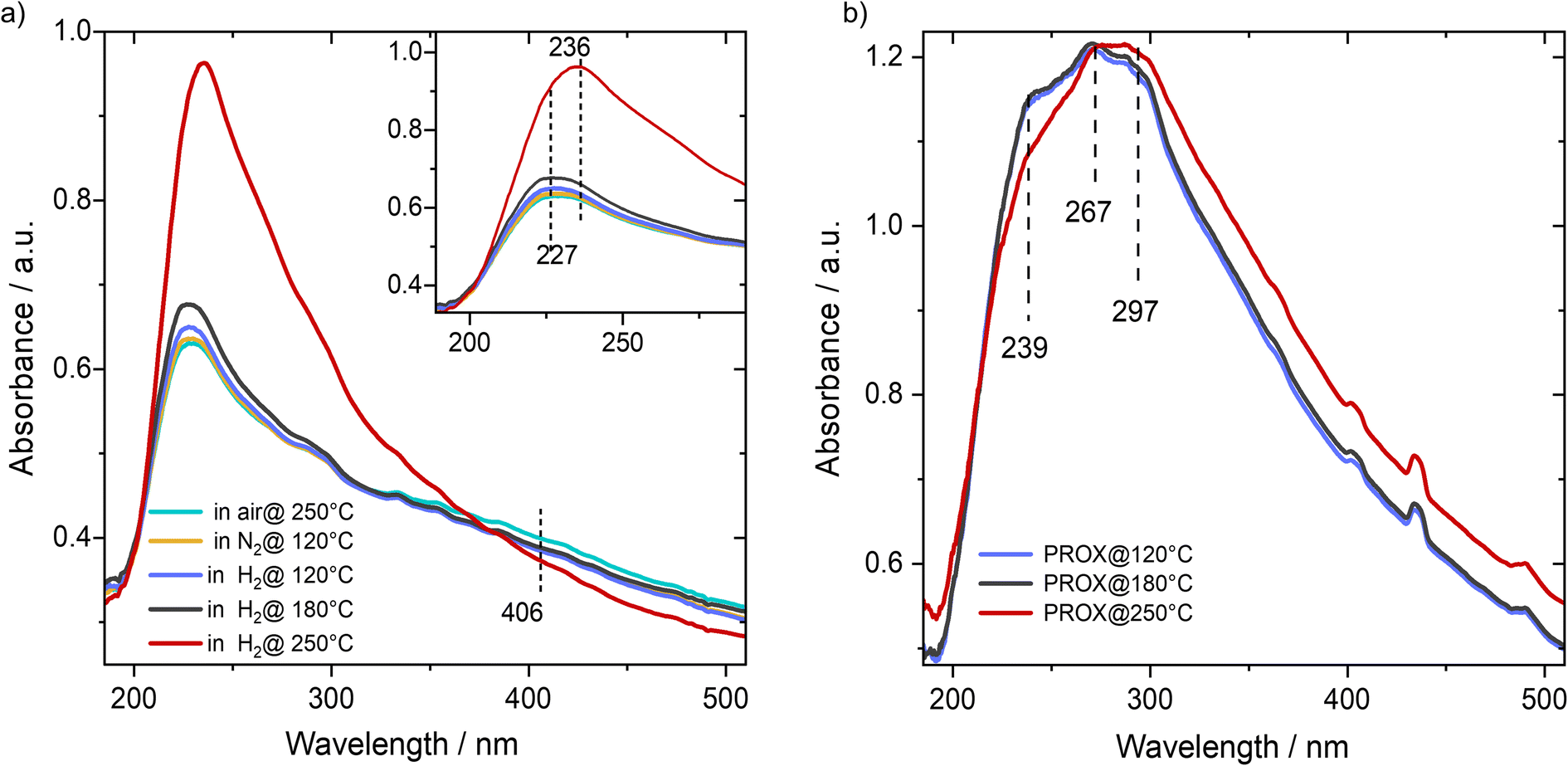 | ||
| Fig. 3 (a) DR-UV-vis spectra recorded on the Cu/UiO-66 catalyst during the activation step at 250 °C in different gas atmospheres. (b) DR-UV-vis spectra recorded during the subsequent PROX reaction (after activation step) at different reaction temperatures (at: 120 °C; 180 °C; 250 °C). | ||
As a benchmark of the above discussed experiments, we examined metal-free UiO-66 and it was found to be not active for CO oxidation at temperatures up to 250 °C. Also, we did not observe CO2 formation, which is indicative of the stability of carbon-content of the UiO-66 framework under the present reaction conditions. Additionally, we mixed CuCl2 salt with UiO-66 (1 wt% equivalent Cu2+ ions) and tested the catalyst at 120 °C after reductive treatment at 250 °C. Based on this non-wet loading of Cu ions, we observed a CO conversion of around 7.5% (see Fig. S7†). This means that active Cu species may be localized to molecular defects and become active upon reduction. The type of Cu species formed is, however, not clearly known in this case and further investigations are needed to elucidate this. Note that, based on previous studies, the intrinsic activity of isolated Cu single sites is significantly higher than that of Cu clusters supported on CeO2–TiO2 oxides.36
To gain further insight into the intrinsic activity of the catalyst toward H2 and CO oxidation, we compared CO oxidation in the absence of H2 and the oxidation of H2 fuel in the absence of CO in the feed gas. CO oxidation in the absence of H2 is much more pronounced at high temperatures (>180–250 °C) compared to the lower temperature range (120 °C to ≤180 °C). Notably, the CO oxidation TOF rates at 120 and 150 °C are not much different from that in the presence of hydrogen, while at 180, 220 and 250 °C we observed much higher CO oxidation TOF rates in the presence of H2 (i.e., during PROX) (see Fig. 2a). For the oxidation of H2 in the absence of CO (Fig. 2b) we observed a continuous increase in hydrogen oxidation between 120 and 220 °C which slows down upon further increase in temperature up to 250 °C. This indicates that the absence of CO allows for more H2 oxidation, and in total, we can infer the following two conclusions: (i) H2 and CO oxidation proceed on similar active sites, and (ii) considering the weaker adsorption of H2 on Cu compared to CO adsorption, CO oxidation is suppressed either by H2O adsorption (byproduct) or due to the strong reduction of the available Cu2+-like sites required for the activation of molecular oxygen.17 These two scenarios will be further clarified later by other experimental results and DFT simulations.
3.3 In situ spectroscopy measurements – electronic properties
To get insight into the electronic properties of the catalyst we combined results from different spectroscopic methods including DR-UV-vis, Cu K-edge XANES, and NAP-XPS. With DR-UV-vis we first examined the changes in the oxidation state of Cu species as a function of temperature during reduction in hydrogen. Before reduction the catalysts were oxidized at 250 °C in synthetic air to start from the full oxidized state as a reference for subsequent experiments. For a fully oxidized catalyst we observed an absorption band centered at 227 nm beside a weaker and broader shoulder centered at 406 nm (Fig. 3a). Based on the reported UV-vis spectra on Cu-exchanged zeolite the first peak would be attributed to ionic Cu species, basically referring to Cu1+-like species, while there is no clear absorption peak which would fit to Cu(II) ions,25,26 except a weak feature was observed at 406 nm, which may correlate with a slight concentration of pure Cu2+ coordinated by two oxygens.25 Analogous with the DFT calculated structure,17,22,23 which is based on a multitude of experimental methods, and considering the exposure of the catalyst to oxygen-rich gas (synthetic air), the 227 nm feature would fit to a Cu single-ion coordinated by two oxygen atoms and a bridging OH between a Cu ion and Zr ion in the UiO-66 structure which bears a charge density of +1.46 (Int. I, Fig. 5; see DFT computations later). This oxidation state is the average of pure Cu(I) and pure Cu(II) states.After this oxidative treatment we cooled the catalyst in N2 to 120 °C and started stepwise reduction in a flow of hydrogen from 120 °C up to 250 °C. Upon switching to H2 flow at 120° we observed a slight increase in the intensity of the peak at 227 nm, which agrees with our earlier XANES and EPR results showing that reduction of the Cu2+-like state to the Cu1+-like state at this temperature is not measurable when exposed to a reducing feed gas.14 Increasing the temperature in H2 to 180 °C did not result in a big change in the peak at 227 nm except for a broadening at the right shoulder of the main band, which was observed at ∼236 nm. This may agree with DFT calculations ensuring that H2 oxidation is essentially activated by a preceding adsorption of O2 molecule on a Cu1+-like state (see DFT computations later). Upon increasing the temperature to 250 °C we observed a substantial increase in the intensity of this peak (236 nm) and complete disappearance of the 406 nm shoulder. Note that the peak position at 227 nm may be contributing to the new peak which appeared at 236 nm and is assignable to the Cu1+-like state. This peak fits to another DFT optimized structure where the Cu site is bound to Zr atoms via one oxygen and a hydroxyl group (Int. I, Fig. 6). This observation hints at the significant temperature dependence of the oxidation properties of Cu species and change in the coordinating environment upon switching from an oxidizing (O2) to reducing feed gas (H2 or CO).
Next, we examined the catalyst using DR-UV-vis spectroscopy during the PROX reaction at different temperatures after the H250 treatment. Running the PROX reaction resulted in more complex spectra than those obtained in H2. At 120 °C we can observe a peak at 239 nm, which is close to the peak observed at 236 nm (Cu1+-like state coordinated by –O and –OH groups). At the same time, we observed two maxima at 267 and 297 nm which can be assigned to other intermediates (e.g. CO and OH2 adsorbed on the Cu1+-like state., i.e., on Int. 2). Increasing the temperature to 180 °C during PROX resulted in a slight decrease in the peak intensity at 239 nm, while the other two peaks at 267 and 297 nm did not change much. Further increase in reaction temperature to 250 °C resulted in a further decrease in the intensity of the 3 features. However, we cannot assign the two features at 267 and 297 to changes in the oxidation sate. The peak at 239 nm could only be observed for a fully reduced catalyst (see Fig. 3b). Finally, we would like to note that based on the comparison of the measured spectra of CuO, Cu2O, Cu2Cl2 and CuCl2 references to the collected spectra of the Cu/UiO-66 catalyst, we can exclude the presence of Cu in the form of bulk oxides or chloride, which is in good agreement with our earlier findings on these catalysts (see Fig. S8†). In total, these in situ DR-UV-vis measurements indicated the strong dependence of the reducibility of Cu/UiO-66 on temperature both in H2 and under PROX reaction conditions. Also, it should be noted that we would refer to Cu ions as the Cu1+-like state and to oxidized Cu ions as the Cu2+-like state since neither the pure Cu1+ nor the Cu2+ states would exist in this coordination environment.
In support of these results, Cu K-edge XANES data collected after H250 treatment and the subsequent PROX reaction at 120, 180 and 250 °C indicated a mixed valence between Cu2+ and Cu1+ (Fig. S9†), which is in good agreement with earlier findings.37 Upon increasing the temperature from 120 °C to 180 °C, and further to 250 °C the spectra show features more characteristic of the Cu1+-like species (Fig. S7, ESI†), in agreement with DR-UV-vis spectra. It can generally be inferred that the increasing temperature under the PROX reaction results in an increase in the reduced state of the catalyst. Next we analyzed the EXAFS data collected at 250 °C after catalyst reduction in hydrogen at 250 °C. EXAFS data analysis (see Fig. S10† and fitting parameters in Table S1†) indicates that Cu ions bind entirely to O atoms/OH with a small coordination number of 2.3 ± 0.5 and a Cu–O distance of 1.96 ± 0.02 Å. Note that the Cu–O bond distance is rather longer than typical distances for CuO (1.88 ± 0.02 Å) and Cu2O (1.91 ± 0.02 Å).38,39 The results of the present Cu/UiO-66 catalyst are in good agreement with the results reported on similar catalysts after an identical treatment.17,24
To get further insights into the electronic nature of Cu species and ZrOx oxide nodes of the UiO-66 framework during reductive activation and the subsequent PROX reaction, we performed in situ NAP-XPS measurements. Measurements were carried out at 250 °C using a total pressure of 2 mbar of the following gas atmospheres: (i) He; (ii) 10% H2/He; (iii) 1% CO, 1% O2, 80% H2, He balance; and (iv) back to He. Note that due to massive charging effects at lower temperatures we could not reliably collect XP spectra at temperatures <250 °C. The energy correction was carried out with respect to the C–C/C–H bonds in the C1s signal of the native carbon species (see Fig. S11†) which did not change under all study conditions. The fresh sample was first evacuated at room temperature and subsequently heated up to 250 °C at 2 mbar of He. At 250 °C the Cu 2p NAP-XP spectra are shown in Fig. 4a and indicate a binding energy of the Cu 2p3/2 peak of 933.0 eV. Together with the absent 2p satellite, this feature is indicative of the presence of Cu species as Cu1+ and/or Cu0 sites.40 Unfortunately, both oxidation states cannot be distinguished unambiguously due to the same binding energies and peak shapes. Based on the XANES/UV-vis spectra as well as DRIFT spectra discussed later, we can exclude any contribution from Cu0 species. Furthermore, Cu2+ species might be partially reduced by the X-ray beam.40 Based on the DR-UV-vis spectra we exclude, however, any measurable contribution from pure Cu2+-like states.
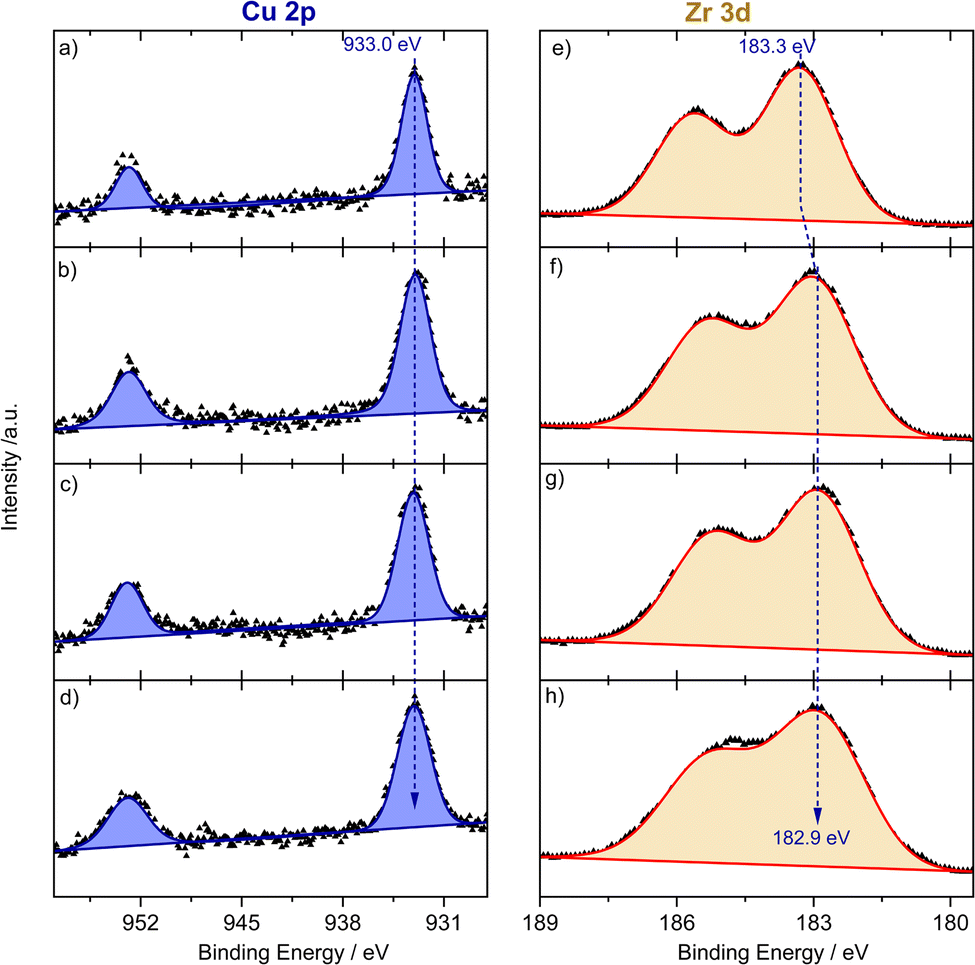 | ||
| Fig. 4 Operando NAP-XP spectra for the Cu 2p region (left panels: (a)–(d)) and Zr 3d region (right panels: (e) and (f)) at 250 °C and a pressure of 2 mbar and with different feed gases ((a) and (e): in He after heating from RT; (b) and (f): in 10% H2/He; (c) and (g): in PROX gas (1% CO, 1% O2, 80% H2, He balance); (d) and (h): back again in He). | ||
For ZrOx oxide nodes, we detected a 3d core level energy of 183.3 eV (see Fig. 4e), which is slightly higher than typically reported binding energies of Zr4+ species.41 The exposure to 10% H2/He for 1 h did not change the oxidation state of Cu species (see Fig. 4b), while for Zr we observed an energy shift to a lower binding energy of 182.9 eV (see Fig. 4f), which indicates partial reduction of ZrOx species under flow of a reductive atmosphere. During the PROX step both Cu 2p and Zr 3d are rather unchanged, which is expected under these conditions (see Fig. 4c and g). Finally, upon returning to a He atmosphere at 250 °C the oxidation state of both species remained constant under these inert conditions (Fig. 4d and h). These results agree in total with findings from DR-UVis/XANES and are additionally indicative of an interface mediated charge transfer from ZrOx oxide nodes to the bonding Cu sites, both in H2 and under PROX reaction conditions.
3.4 DRIF spectroscopy – CO-adlayer
We further employed diffuse reflectance FTIR spectroscopy to gain information on adsorption properties of the active Cu species during the PROX reaction at different temperatures and for comparison during CO oxidation. Following the H250 pre-treatment, the DRIFTS spectra collected on the catalyst under steady state conditions at 120 °C after 2 h reaction showed a COad band centred at 2116 cm−1 (Fig. 5a), which is characteristic of the adsorption of CO on Cu1+-like sites, whereas we did not detect any feature related to Cu0 or Cu2+ like-species.42,43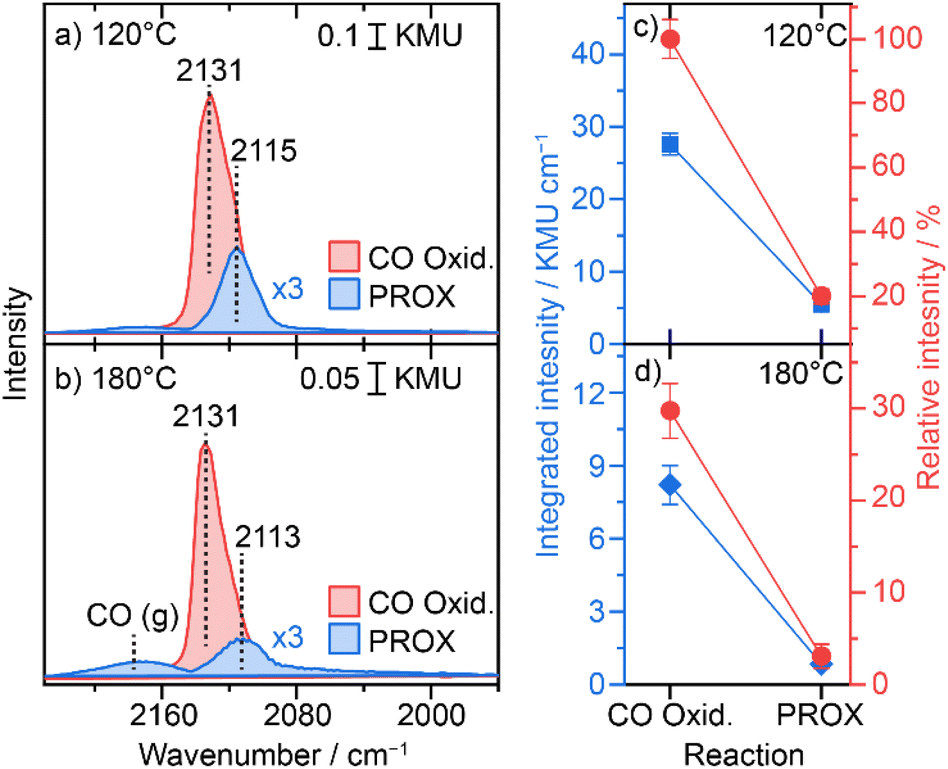 | ||
| Fig. 5 COad bands during preferential CO oxidation (1% CO, 1% O2, 80% and N2 balance) PROX (1% CO, 1% O, 80% and N2 balance) ((a): at 120 °C; (b) at 180 °C) and their relative intensity ((c): at 120 °C; (d): at 180 °C). The integration is carried out over a widow from 2160 to 2060 cm−1. Relative intensity is a normalization of all integrated intensities by that for CO oxidation at 120 °C (highest intensity). Integrated intensities are corrected for the contribution from gas phase CO. | ||
On the same layer we showed the COad spectrum during CO oxidation (i.e., in the absence of hydrogen), which appears at 2131 cm−1 but with about 5-fold higher intensity (see Fig. 5d). Interestingly, these two bands observed during PROX (i.e., in the presence of hydrogen) and during CO oxidation (i.e., in the absence of hydrogen) have about the same FWHM value (∼22 cm−1), which may hint at similar adsorption sites but otherwise with electronically different states. The presence of hydrogen promotes the reduction of UiO-66 oxide nodes as concluded from the Zr 3d NAP-XPS results. This correlates with the formation of O-vacancy defects in the ZrOx oxide nodes which leads to a rather higher electron density on the ZrOx cluster. As a result, induced electron back-donation into CO antibonding p* orbitals is expected which would translate into a CO-stretch red-shift.
At 180 °C the peak position shifted to 2113 cm−1 hinting at further electronic modification of Cu at higher temperature. Note that this wavenumber is still significantly higher than that of the Cu0-relevant CO stretch (<2080 cm−1) and the observed red-shift would only correlate with higher charge density close to the Cu1+-states. Also, the intensity of the COad peak at this temperature is 5 times lower than that observed at 120 °C. The position of the COad band during CO oxidation, however, did not change on going from 120 to 180 °C (see Fig. 4b and d), which confirms that the shift observed at 180 °C during PROX is mainly due to the presence of hydrogen and subsequent reduction of UiO-66 oxide nodes rather than resulting from increasing the temperature. Note that the peak intensity of COad species during CO oxidation (2131 cm−1) is also more than 6-times higher than that during PROX at 180 °C (see Fig. 4 c and d). With further increase in reaction temperature to 250 °C the COad peak shifted to an even lower wavenumber of 2106 cm−1 during the PROX reaction with almost ∼5 times lower intensity compared to that detected at 180 °C (see Fig. S12†). In the meantime, during CO oxidation at 250 °C the band position did not shift (2132 cm−1) but its intensity is ∼31 times lower than that at 180 °C. The much lower intensity in both cases can be explained by the higher conversion of CO at higher temperature which is apparently more pronounced for CO oxidation at 250 °C than the PROX reaction, as observed in the kinetic results discussed above. Note that due to a strong change in the background at 250 °C we refrained from the integration of the COad peaks (see spectra in Fig. S12†).
In total these results indicate that (i) charge density on the Cu single sites increases in the presence of hydrogen, (ii) although the concentration of Cu1+ sites is higher under the PROX reaction than under the CO oxidation reaction, the COad coverage on Cu1+ sites is much lower during PROX, which can be explained by competitive adsorption between CO and H2 or its by-products (i.e., H2O), and (iii) increased reduction with increasing temperature leads to a more pronounced loss in COad intensity during PROX which goes along with the observed decrease in CO oxidation selectivity (i.e., increase in H2 oxidation).
3.5 Mechanistic investigation by DFT calculations
To better understand the PROX reaction, we performed DFT calculations of H2 oxidation which follow the same procedure reported before for CO oxidation on similar Cu/UiO-66 catalysts.17 In brief, the mechanisms of both H2 and CO oxidation reactions were studied on a Cu/UiO-66 catalyst where isolated Cu single sites exist in the Cu1+-like state, as shown in Fig. 6. For H2 oxidation, the reaction begins by the adsorption of O2 on oxygen vacancy sites between the Cu and Zr atom with a calculated adsorption energy of −122 kJ mol−1 (I → II), which is different from CO oxidation where the reaction cycle starts by CO adsorption as reported before.17 The H2 molecule then adsorbs on the Cu site on intermediate II with a relative adsorption energy of −154 kJ mol−1. The adsorbed H2 subsequently reacts with the adsorbed O2via a transition state (TS I), which involves an activation energy barrier of 94 kJ mol−1. The collapse of TS I gives intermediate IV with a relative energy of −159 kJ mol−1. To yield the first H2O product, H2 oxidation then proceeds via TS II with an activation energy barrier of 124 kJ mol−1, which is much greater than that of the first transition state (TS I). The subsequent collapse of TS II leads to intermediate V with H2O adsorbed on the Cu active site and the remaining O atom at the oxygen vacancy site. We note here that this remaining O atom is bound to the Zr and Cu atoms. Finally, the removal of the H2O product from intermediate V is an activated step and requires a desorption energy of 98 kJ mol−1. Overall, the first H2 oxidation reaction is exothermic with a relative energy of −313 kJ mol−1 and the oxidation state of the Cu active site changes from +1 to +2 after the first H2 oxidation.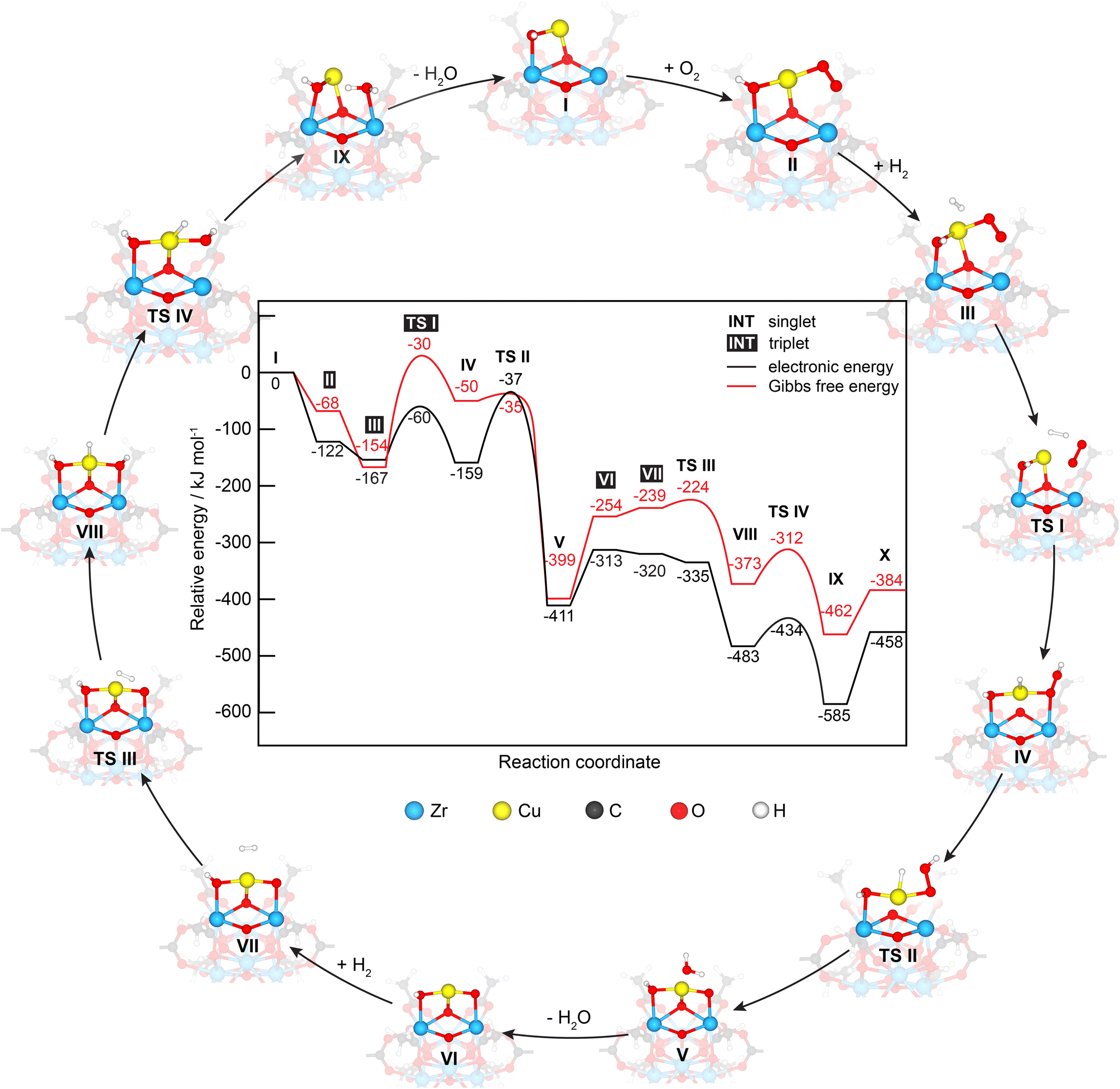 | ||
| Fig. 6 Energy profile and corresponding optimized structures of adsorption, intermediate and transition states for H2 oxidation by O2 on the Cu/UiO-66 model catalyst. | ||
The second H2 oxidation reaction begins with a very weak adsorption of the second H2 molecule on the newly generated Cu2+ site with an adsorption energy of −7 kJ mol−1 (VII). The adsorbed H2 molecule then reacts with the remaining O atom, forming an intermediate VIIIvia TS III, which has an energy barrier of 78 kJ mol−1. The second water product is then produced via TS IV with an energy barrier of 49 kJ mol−1, leading to the formation of intermediate IX with a relative energy of −585 kJ mol−1. Finally, the desorption of this water molecule from the Cu1+ site requires an energy barrier of 127 kJ mol−1. Despite the H2O adsorption configuration on Zrcus site is energetically less stable than the H2O adsorption configuration on Cu1+ site by 6 kJ mol−1 (see Fig. S13†), its optimized structure (i.e. H2Oad@Zrcus) fits more to the TS IV. Overall, the H2 oxidation reaction is exothermic by −458 kJ mol−1 and is less exothermic compared to CO oxidation (−658 kJ mol−1).
We note here that another co-adsorption configuration of O2 and H2 with a pre-adsorbed O2 on Cu is observed, which is 23 kJ mol−1 more stable in energy than intermediate II (see Fig. S14, ESI – intermediate 2†). According to this co-adsorption configuration, we infer that O2 dissociation on the Cu site is a prerequisite for further H2 oxidation reaction. The O2 dissociation step requires an activation energy of 175 kJ mol−1,17 which is much higher than the energy barrier of the TS I of 94 kJ mol−1 and that of the rate determining step TS II of 124 kJ mol−1 for H2 oxidation. In addition, O2 dissociation could result in Cu-oxo species (i.e., unbound to ZrOx clusters) which has not been observed. Accordingly, our focus was on the mechanistic study of H2 oxidation on intermediate II.
Next, we compared the energy profile for H2 oxidation herein with that for CO oxidation (see comparison in Fig. S14, ESI†). The results reveal that the activation energies of the rate-determining step of H2 and CO oxidation reactions are 124 and 141 kJ mol−1, respectively. However, H2/O2 co-adsorption is 90 kJ mol−1 less stable in energy than CO/O2 co-adsorption. Thus, CO oxidation is likely to dominate in the PROX reaction.
During the PROX reaction, H2O can be formed and competitively adsorbed on the catalytic active sites with other reactants (CO, H2 and O2) as proposed from kinetics and in situ DRIFT spectroscopy experiments. We thus inspected the adsorption of these molecules on the Cu1+ (intermediate I) and Cu2+ (intermediate VI) sites. The optimized structures of the most stable adsorption configurations are shown in Fig. S16, ESI.† The calculated adsorption energies of CO, H2O, O2 and H2 on Cu1+ and [Cu2+] are −209 [−58], −133 [−80], −157 [−33] and −93 [−7] kJ mol−1, respectively. Based on these results it can be inferred that Cu1+ and Cu2+ could be predominately covered by CO and H2O, respectively. Furthermore, the formation of H2O during the reaction would have a higher tendency than CO to block the Cu2+, while the opposite is true for Cu1+-like where CO is preferentially more adsorbed than water. We note here that the calculated O2 adsorption on the Cu1+ site is −157 kJ mol−1, which is more stable in energy than that at the oxygen vacancy site (−122 kJ mol−1) as discussed previously in the mechanistic study of H2 oxidation.17 The relative Gibbs free energy (ΔG) at 25, 120, 180 and 250 °C reveals that both steps of H2 oxidation are exergonic, which agrees well with the relative electronic energy (ΔE) results (see more detailed discussion in the ESI of data in Fig. S17 and Table S3†).
In addition, we considered singlet and triplet spin states in our calculations (see energy profiles compared in Fig. S18†). Our calculations showed that the singlet state is the ground spin state for Cu1+ (intermediate I) with the triplet state being 147 kJ mol−1 higher in energy. During the reaction, spin state changes occurred. Following O2 adsorption, the spin state changed from singlet to triplet and then returned to singlet after the first transition state TS I. During the first H2- oxidation step (intermediate VI), a spin state change from singlet to triplet occurred, suggesting the oxidation of Cu1+ to Cu2+. Then the spin state returned to singlet for the second H2 oxidation step (from TS III to X), signifying the change in the oxidation state of the Cu active site from +2 to +1.
Finally, to gain insights into the effect of water on the catalytic performance of Cu active sites during the PROX reaction, we calculated the co-adsorption energies of O2/H2O, O2/CO, and H2O/CO co-adsorption on the Cu1+ and Cu2+ sites. For Cu1+, the calculated co-adsorption energies are −218, −244 and −351 kJ mol−1, respectively (see co-adsorption configurations in Fig. S19, ESI†). The calculated co-adsorption energies of H2O–CO are much larger than that of O2–CO and O2–H2O, hinting at a competitive adsorption of H2O with CO compared to that with O2 for these Cu1+ like-species. This finding agrees well with the strongly suppressed CO coverage during the PROX reaction and the pronounced loss of PROX selectivity especially at high temperatures (>150 °C). For the Cu2+-like state, we could not find the co-adsorption configuration of the co-adsorbed O2–H2O, O2–CO, and H2O–CO pairs on the Cu2+ site probably due to the strong adsorption of H2O which obstructs the exchange of the Cu2+ ↔ Cu1+ redox shuttle and make it less facile, which affects the rate of CO and H2 oxidation.
4. Conclusions
The controlled perturbation of activity/selectivity of the PROX reaction through the variation of temperature led to changes in electronic and structural features of the Cu1+/UiO-66 catalysts, which were monitored using in situ/operando (DR-UV-vis, NAP-XPS, XANES and DRIFT) spectroscopy measurements. Selectivity was found to decrease with increasing temperatures which was correlated with measurable changes in the redox properties of Cu single sites (relative [Cu1+-like state: Cu2+-like state] ratio increases with temperature). Detailed study of the reaction mechanism by DFT computations indicated that hydrogen oxidation starts with the adsorption of H2 on a pre-adsorbed O2 molecule on a Cu1+-like site and then proceeds through two transition states to produce a H2O product. This is different from the CO oxidation pathway which starts with CO adsorption on a Cu1+-like state. The hydrogen cycle is completed by the reaction of a second hydrogen molecule with the remaining oxygen adsorbed on the Cu2+-like state. Based on energy minimized co-adsorption configurations (total adsorption energies) of CO and H2O competitive adsorption is assumed to affect the occupation of Cu species relying on the underlying oxidation state. While CO adsorbs stronger than H2O on Cu1+-like sites, the H2O product adsorbs preferentially on Cu2+-like sites over CO. This would indirectly reduce the rate of molecular oxygen activation, deaccelerating the CO oxidation pathway and affecting the selectivity. These findings agree well with the decrease in the CO oxidation rate during the PROX reaction at higher temperatures, which is not observed in CO oxidation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
B. K., and A. M. A. acknowledge the financial support from the BMWK project “MEGA- Methanol reformer with innovative gas purification for H2 supply of a LT-PEMFC (03EN5006A)”. We thank Dr S. Wohlrab for the use of DR-UV-vis setup and for discussions. We thank the beamtime access at the SAMBA (Soleil light source) and P65 (DESY, Germany) beamlines and the user support by Dr Emiliano Fonda and Dr Edmund Welter. S. I., B. R. and K. F. thank the Office of National Higher Education Science Research and Innovation Policy Council (NXPO) via the Program Management Unit for Human Resources & Institutional Development Research and Innovation (PMU-B), Thailand, and the access to data and the literature bank at the central library of Cairo University. B. Rungtaweevoranit acknowledges the research grant by National Research Council of Thailand (NRCT) grant no. N42A650179. The Nanoscale Simulation Laboratory at National Nanotechnology Center (NANOTEC) and NSTDA Supercomputer center (ThaiSC) are acknowledged for providing computational resources.Notes and references
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS PubMed.
- O. M. Yaghi, M. J. Kalmutzki and C. S. Diercks, Introduction to Reticular Chemistry: Metal-Organic Frameworks and Covalent Organic Frameworks, John Wiley & Sons, 2019 Search PubMed.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- H. Zhang, G. Liu, L. Shi and J. Ye, Adv. Energy Mater., 2018, 8, 1701343 CrossRef.
- B. Rungtaweevoranit, A. M. Abdel-Mageed, P. Khemthong, S. Eaimsumang, K. Chakarawet, T. Butburee, B. Kunkel, S. Wohlrab, K. Chainok and J. Phanthasri, ACS Appl. Mater. Interfaces, 2023, 15, 26700–26709 CrossRef CAS PubMed.
- K.-i. Otake, S. Ahn, J. Knapp, J. T. Hupp, J. M. Notestein and O. K. Farha, Inorg. Chem., 2021, 60, 2457–2463 CrossRef CAS PubMed.
- J. Sui, H. Liu, S. Hu, K. Sun, G. Wan, H. Zhou, X. Zheng and H. L. Jiang, Adv. Mater., 2022, 34, 2109203 CrossRef CAS PubMed.
- X. Ma, H. Liu, W. Yang, G. Mao, L. Zheng and H.-L. Jiang, J. Am. Chem. Soc., 2021, 143, 12220–12229 CrossRef CAS PubMed.
- L. Jiao and H.-L. Jiang, Chem, 2019, 5, 786–804 CAS.
- H. Abed, J. Mosrati, A. M. Abdel-Mageed, S. Cisneros, T. Huyen Vuong, N. Rockstroh, S. Bartling, S. Wohlrab, A. Brückner and J. Rabeah, ChemCatChem, 2022, e202200923 CrossRef CAS.
- J. Mosrati, A. M. Abdel-Mageed, T. H. Vuong, R. Grauke, S. Bartling, N. Rockstroh, H. Atia, U. Armbruster, S. Wohlrab and J. Rabeah, ACS Catal., 2021, 11, 10933–10949 CrossRef CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- M. Yang, S. Li, Y. Wang, J. A. Herron, Y. Xu, L. F. Allard, S. Lee, J. Huang, M. Mavrikakis and M. Flytzani-Stephanopoulos, Science, 2014, 346, 1498–1501 CrossRef CAS.
- W. Sangkhun, J. Ponchai, C. Phawa, A. Pengsawang, K. Faungnawakij and T. Butburee, ChemCatChem, 2021, e202101916 Search PubMed.
- A. M. Abdel-Mageed, S. Cisneros, J. Mosrati, H. Atia, T. H. Vuong, N. Rockstroh, S. Wohlrab, A. Brückner and J. Rabeah, ChemCatChem, 2023, 15, e202201669 CrossRef CAS.
- A. M. Abdel-Mageed, B. Rungtaweevoranit, S. Impeng, J. Bansmann, J. Rabeah, S. Chen, T. Häring, S. Namuangrak, K. Faungnawakij and A. Brückner, Angew. Chem., 2023, e202301920 CAS.
- A. Beniya and S. Higashi, Nat. Catal., 2019, 2, 590–602 CrossRef.
- A. F. Ghenciu, Curr. Opin. Solid State Mater. Sci., 2002, 6, 389–399 CrossRef.
- W. Lubitz and W. Tumas, Chem. Rev., 2007, 107, 3900–3903 CrossRef CAS PubMed.
- S. Chen and A. M. Abdel-Mageed, Int. J. Hydrogen Energy, 2023, 48, 24915–24935 CrossRef CAS.
- M. Kandiah, M. H. Nilsen, S. Usseglio, S. Jakobsen, U. Olsbye, M. Tilset, C. Larabi, E. A. Quadrelli, F. Bonino and K. P. Lillerud, Chem. Mater., 2010, 22, 6632–6640 CrossRef CAS.
- A. J. Howarth, Y. Liu, P. Li, Z. Li, T. C. Wang, J. T. Hupp and O. K. Farha, Nat. Rev. Mater., 2016, 1, 1–15 Search PubMed.
- A. M. Abdel-Mageed, B. Rungtaweevoranit, M. Parlinska-Wojtan, X. Pei, O. M. Yaghi and R. J. Behm, J. Am. Chem. Soc., 2019, 141, 5201–5210 CrossRef CAS PubMed.
- C. La Fontaine, L. Barthe, A. Rochet and V. Briois, Catal. Today, 2013, 205, 148–158 CrossRef CAS.
- N. Muddada, U. Olsbye, G. Leofanti, D. Gianolio, F. Bonino, S. Bordiga, T. Fuglerud, S. Vidotto, A. Marsella and C. Lamberti, Dalton Trans., 2010, 39, 8437–8449 RSC.
- I. M. Hamadeh and P. R. Griffiths, Appl. Spectrosc., 1987, 41, 682–688 CrossRef CAS.
- J. Sirita, S. Phanichphant and F. C. Meunier, Anal. Chem., 2007, 79, 3912–3918 CrossRef CAS PubMed.
- C. A. Trickett, K. J. Gagnon, S. Lee, F. Gandara, H. B. Bürgi and O. M. Yaghi, Angew. Chem., Int. Ed., 2015, 54, 11162–11167 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- M. Dolg, H. Stoll, H. Preuss and R. M. Pitzer, J. Phys. Chem., 1993, 97, 5852–5859 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- L. Valenzano, B. Civalleri, S. Chavan, S. Bordiga, M. H. Nilsen, S. Jakobsen, K. P. Lillerud and C. Lamberti, Chem. Mater., 2011, 23, 1700–1718 CrossRef CAS.
- A. M. A.-M. Jawaher Mosrati, T. Huyen Vuong, R. Grauke, S. Bartling, H. Atia, U. Armbruster, S. Wohlrab, J. Rabeah and A. Brückner, ACS Catal., 2021, 11, 10933–10949 CrossRef.
- A. Martini, E. Borfecchia, K. A. Lomachenko, I. A. Pankin, C. Negri, G. Berlier, P. Beato, H. Falsig, S. Bordiga and C. Lamberti, Chem. Sci., 2017, 8, 6836–6851 RSC.
- A. Caballero, J. J. Morales, A. M. Cordon, J. P. Holgado, J. P. Espinos and A. R. Gonzalez-Elipe, J. Catal., 2005, 235, 295–301 CrossRef CAS.
- Y. Okamoto, T. Kubota, H. Gotoh, Y. Ohto, H. Aritani, T. Tanaka and S. Yoshida, J. Chem. Soc., Faraday Trans., 1998, 94, 3743–3752 RSC.
- M. C. Biesinger, Surf. Interface Anal., 2017, 49, 1325–1334 CrossRef CAS.
- M. Guittet, J. Crocombette and M. Gautier-Soyer, Phys. Rev. B, 2001, 63, 125117 CrossRef.
- F. Giordanino, P. N. R. Vennestrom, L. F. Lundegaard, F. N. Stappen, S. Mossin, P. Beato, S. Bordiga and C. Lamberti, Dalton Trans., 2013, 42, 12741–12761 RSC.
- K. Hadjiivanov and H. Knözinger, Phys. Chem. Chem. Phys., 2001, 3, 1132–1137 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta05047e |
| This journal is © The Royal Society of Chemistry 2024 |