
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSequential proton coupled electron transfer events from a tetraruthenium polyoxometalate in photochemical water oxidation†
Elena
Rossin
 a,
Marcella
Bonchio
a,
Mirco
Natali
*b and
Andrea
Sartorel
*a
a,
Marcella
Bonchio
a,
Mirco
Natali
*b and
Andrea
Sartorel
*a
aDepartment of Chemical Sciences, University of Padova, Via Marzolo 1, 35131 Padova, Italy. E-mail: Andrea.sartorel@unipd.it
bDepartment of Chemical Pharmaceutical and Agricultural Sciences (DOCPAS), University of Ferrara, Ferrara, Italy. E-mail: Mirco.natali@unife.it
First published on 18th March 2024
Abstract
The tetraruthenium polyoxometalate {RuIV(H2O)4(μ-OH)2(μ-O)4[SiW10O36]2}10− (Ru4POM) shows multiple oxidative proton coupled electron transfer (PCET) events in a [Ru(bpy)3]2+/S2O82− photochemical cycle for catalytic water oxidation, with electrons conveyed to the photogenerated [Ru(bpy)3]3+ oxidant and protons transferred to aqueous bases. As shown by laser flash photolysis, in aqueous phosphate buffer the consumption of the [Ru(bpy)3]3+ oxidant by Ru4POM shows bi-exponential kinetics with a fast component and a slow component that feed the Ru4POM catalyst with up to 6 oxidative equivalents through PCET in ca. 50 ms. The apparent rates of both the fast and slow components depend linearly on HPO42− and on the pH of the aqueous medium, suggesting the involvement of the buffer base, of water and of OH− in assisting removal of the protons from Ru4POM. In particular, the beneficial role of HPO42− is reflected in a proportional improvement in the oxygen evolution activity, reaching quantum efficiency approaching 14%, although an excessive increase of buffer concentration is detrimental to the [Ru(bpy)3]3+ stability and leads to the abatement of the O2 evolution.
Introduction
Proton coupled electron transfer (PCET) is a pervasive process in many biological and artificial chemical transformations.1–4 PCET events are often associated with photoinduced charge separation,5–10 which is a primary step in chemical processes associated with the photosynthetic conversion of small molecules, while recently, its interest has broadened to synthetic organic chemistry.11–15In the field of water oxidation, investigation of PCET was pioneered by T. J. Meyer, who recognized that for the [RuII(H2O)(py)(bpy)2]2+ coordination complex (where py = pyridine; bpy = 2,2′-bipyridine) two stepwise oxidative PCET events occurring in a narrow potential window of 110 mV lead to the formation of a RuIV–oxo species;16 these findings were pivotal in the design of the blue dimer [(bpy)2RuIII(H2O)(μ-O)RuIII(H2O)(bpy)2]4+ as the first molecular water oxidation catalyst.17 The importance of PCET was then recognised in many ruthenium based catalytic manifolds,18–22 including the case of the tetraruthenium polyoxometalate {RuIV(H2O)4(μ-OH)2(μ-O)4[SiW10O36]2}10− (Ru4POM) investigated in this work (Scheme 1). Ru4POM is the first structurally characterized polyoxometalate based water oxidation catalyst,23,24 and has been extensively investigated in electrochemical,25,26 photochemical27,28 and photoelectrochemical systems;29–32 its tetraruthenium active core [RuIV(H2O)4(μ-OH)2(μ-O)4]6+ can indeed undergo stepwise oxidation processes associated with the formation of high-valent intermediates characterised spectroscopically or with computational tools, up to the formation of Ru–oxo moieties active towards oxygen evolution at low overpotential.
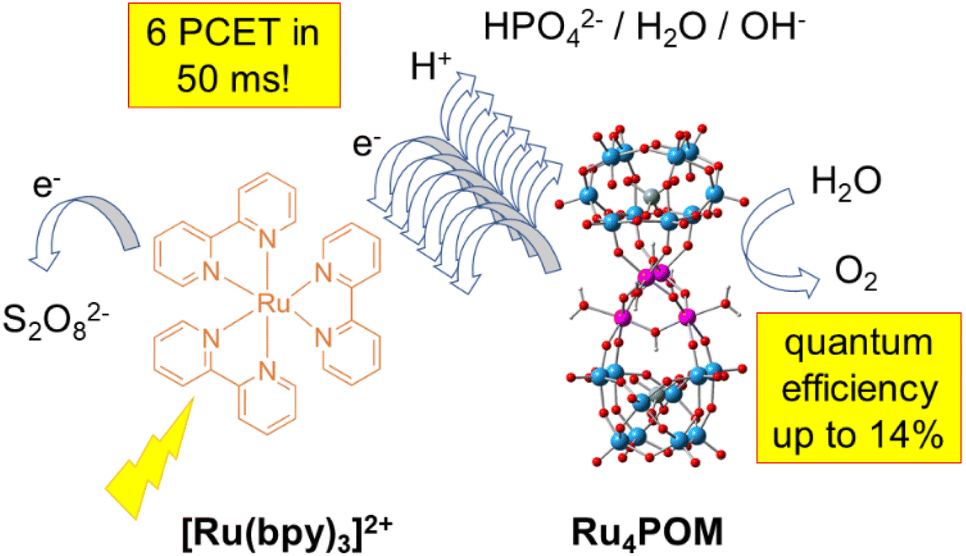 | ||
| Scheme 1 Multiple and sequential proton coupled electron transfer events from a tetraruthenium polyoxometalate Ru4POM to photogenerated [Ru(bpy)3]3+ investigated in this work through laser flash photolysis, with HPO42−, H2O and OH− being responsible for proton transfer. | ||
Despite ruthenium catalysts being reported to show excellent performance in oxygenic photosynthetic systems,18,33–35 the managing of the necessary PCET events under photochemical conditions has been poorly investigated. Moreover, most of the PCET studies in photosynthetic systems for water oxidation refer to a single event,33 and thus represent only the primary step in the water oxidation process.
In this study, we report multiple, sequential oxidative PCET events from Ru4POM to a photogenerated [Ru(bpy)3]3+ oxidant (electron acceptor) highlighting the role of bases as the proton acceptors (Scheme 1). We will show by laser flash photolysis studies that the nature and concentration of the buffer impact the dynamics and the number of oxidation processes at the Ru4POM catalyst, showing a consistent effect in the O2 evolving rate.
Results and discussion
Primary electron transfer from Ru4POM to [Ru(bpy)3]3+
In the laser flash photolysis set-up, [Ru(bpy)3]3+ is photogenerated in a few ns upon laser excitation (λexc = 355 nm) of a solution containing the [Ru(bpy)3]2+ sensitizer and the S2O82− sacrificial acceptor (a quantum yield of 2 is reported for Ru(III) formation, since two equivalents of Ru(III) are generated upon one photon absorption according to the reaction steps in Scheme S1 in ESI†). Formation of Ru(III) is confirmed by the decrease of the absorbance at 450 nm (usually referred to as “bleach”, Fig. 1).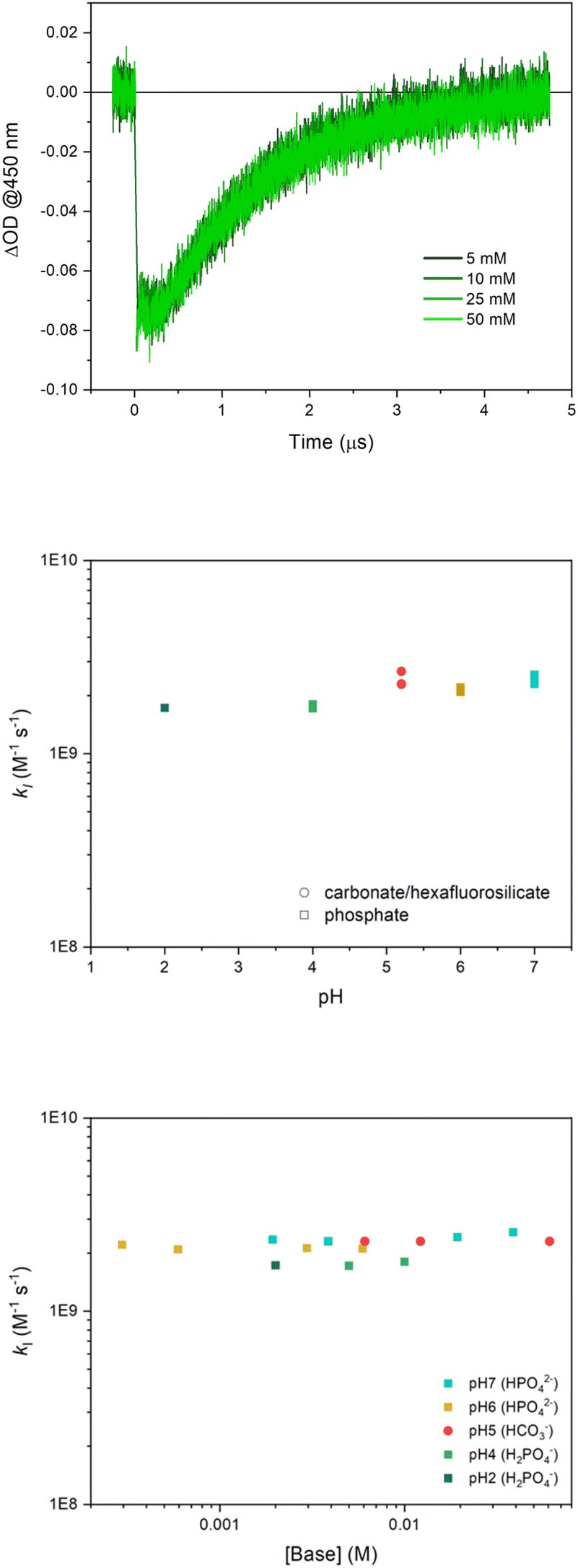 | ||
| Fig. 1 (Top panel) Kinetic traces at 450 nm obtained by laser flash photolysis of 50 μM Ru(bpy)3Cl2, 5 mM Na2S2O8, 30 μM Na10Ru4POM, 0.1 M Na2SO4 in 5–50 mM phosphate buffer at pH 7.0. (Medium panel) Bimolecular rate constant kI (in logarithmic scale) for the primary oxidation of Ru4POM by [Ru(bpy)3]3+vs. pH (the different data at a specific pH refer to the different concentrations of the buffer employed). (Bottom panel) kI (in logarithmic scale) vs. buffer base concentration for all experimental conditions tested (the different colours refer to the conditions employed, with the base specified in brackets in the legend panel). | ||
In the presence of Ru4POM, the Ru(II) initial absorption is restored in a few μs (“bleach recovery”, Fig. 1), indicating consumption of the [Ru(bpy)3]3+ oxidant by Ru4POM, thus leading to the formation of the singly oxidized form of the Ru4POM catalyst, Ru4POMox.‡
Under pseudo first order conditions with [Ru4POM] ≫ [[Ru(bpy)3]3+], fitting of the bleach recovery traces leads to the determination of the bimolecular rate constant kI for the primary oxidative process in Ru4POM (eqn (1)), involving oxidation of one RuIV–OH2 moiety into RuV–OH.
| [Ru(bpy)3]3+ + Ru4POM → [Ru(bpy)3]2+ + Ru4POMox, kI | (1) |
In order to map the possible involvement of a PCET event in this primary step, we performed the laser flash photolysis investigation in aqueous solutions at different pH values (in the range 2–7, conditions associated with the stability of Ru4POM in aqueous solution) employing different buffer and buffer concentrations. In Fig. 1, traces correspond to phosphate buffer at pH 7, while traces under other reaction conditions are reported in ESI (Fig. S1–S4).†
The main outcome of this analysis is that the primary oxidation of Ru4POM by [Ru(bpy)3]3+ occurs under diffusion control under all the conditions explored (kI in the range 1.7 ÷ 2.5 × 109 M−1 s−1).27 The classical model for bimolecular ET reactions foresees the formation of the {[Ru(bpy)3]3+·Ru4POM} encounter complex, eqn (2), followed by ET to form the successor complex {[Ru(bpy)3]2+·Ru4POMox}, eqn (3), and product diffusion, eqn (4); assuming a steady state condition for the encounter and successor complexes the equation of the rate constant kI can be expressed by eqn (5).36
| [Ru(bpy)3]3+ + Ru4POM ⇌ {[Ru(bpy)3]3+·Ru4POM}, k1, k−1 | (2) |
| {[Ru(bpy)3]3+·Ru4POM} ⇌ {[Ru(bpy)3]2+·Ru4POMox}, k2, k−2 | (3) |
| {[Ru(bpy)3]2+·Ru4POMox} → [Ru(bpy)3]2+ + Ru4POMox, k3 | (4) |
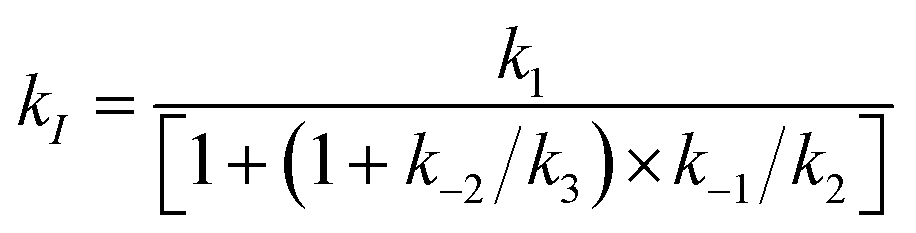 | (5) |
A diffusion limited kI is expected when both k3 ≫ k−2 and k2 ≫ k−1, in which case kI ≈ k1; this occurs under all the conditions explored for the process in eqn (1). This is beneficial towards the accumulation of the first oxidation equivalent in Ru4POMox (i.e. backward processes do not significantly compete with forward processes in eqn (2)–(4)), but limits a mechanistic comprehension of the Ru4POM to Ru4POMox conversion, according to the expected PCET event from electrochemistry data.37
We then focused our analysis on multiple accumulation of oxidation equivalents in Ru4POM by photogenerated [Ru(bpy)3]3+.
Multiple electron transfer events from Ru4POM to [Ru(bpy)3]3+: effect of pH, buffer, and buffer concentration
The fast accumulation of oxidation equivalents in Ru4POM by photogenerated [Ru(bpy)3]3+ can be investigated by performing flash photolysis experiments using a low concentration of Ru4POM to guarantee [Ru4POM] ≪ [[Ru(bpy)3]3+] conditions.27 With this setup, the amount of bleach recovery is associated with the number n of ET events from Ru4POM to [Ru(bpy)3]3+ occurring in the timeframe of the experiment (50 ms), and generating a multiply oxidized form of the catalyst (eqn (6)), while the single steps can be represented as in eqn (7).| n[Ru(bpy)3]3+ + Ru4POM → n[Ru(bpy)3]2+ + Ru4POMnox | (6) |
| [Ru(bpy)3]3+ + Ru4POMjox → [Ru(bpy)3]2+ + Ru4POM(j+1)ox | (7) |
Experiments aimed at tracing multiple electron transfer events were conducted under aqueous conditions at different pH, buffer, and buffer concentrations. A representative example is shown in Fig. 2 (pH 7, 5–100 mM phosphate buffer), see ESI† for other traces (Fig. S5–S7†). The initial concentration of photochemically generated [Ru(bpy)3]3+ in Fig. 2 (top panel) is ca. 2 × 10−5 M and is obtained from the Δ(OD)450 abatement;§ considering the concentration of 2.5 μM of Ru4POM employed in the experiment, the traces indicate that the amount of [Ru(bpy)3]3+ being reduced by Ru4POM actually exceeds the amount of Ru4POM and shows a dependence on the buffer concentration. The role of Ru4POM in the reactivity of [Ru(bpy)3]3+ was confirmed by conducting experiments by varying the concentration of Ru4POM (in the range 0.5–2.5 μM) and keeping the buffer concentration constant at 100 mM: the amount of Δ(OD) recovery of [Ru(bpy)3]3+ depends linearly on Ru4POM concentration (Fig. S8 in ESI† and further discussion).
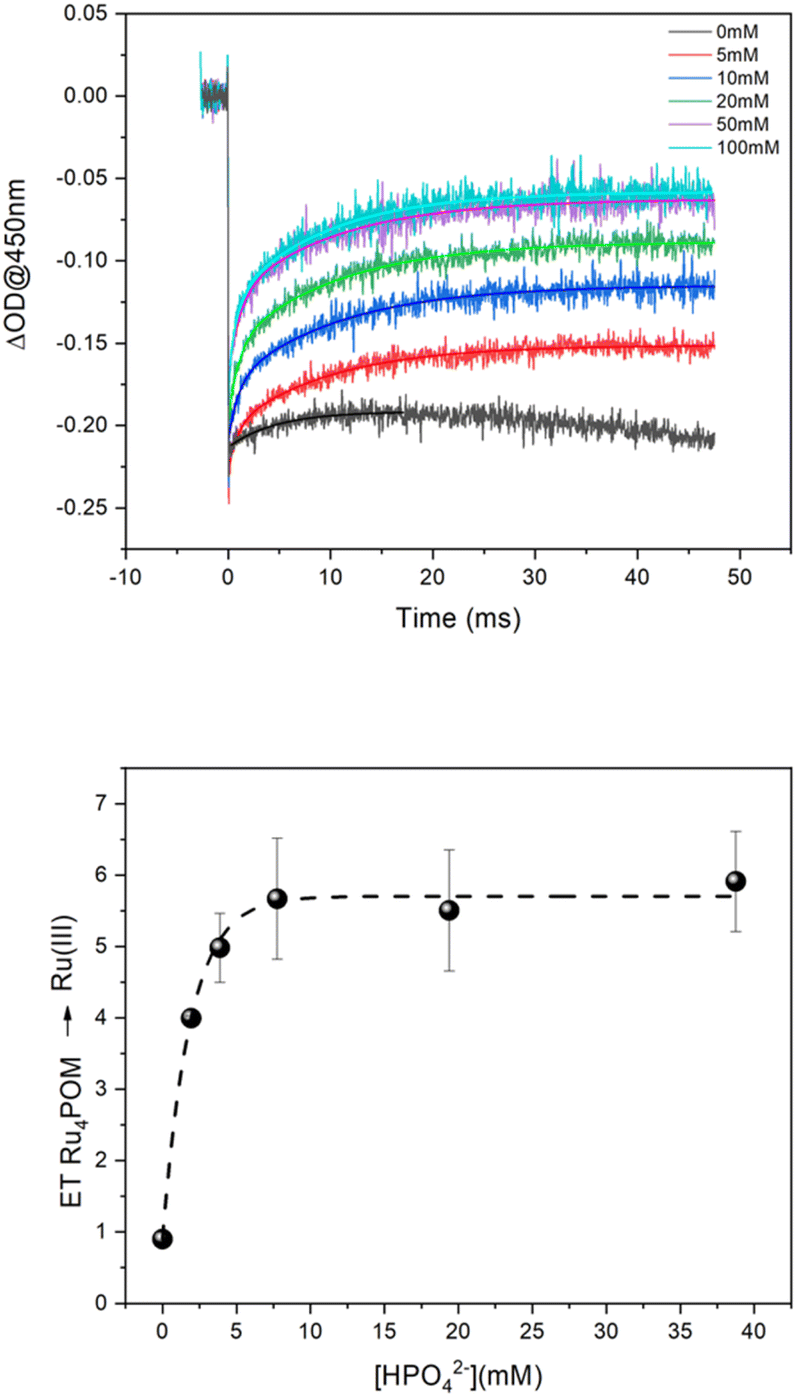 | ||
| Fig. 2 (Top panel) Kinetic traces at 450 nm obtained by laser flash photolysis of 100 μM Ru(bpy)3Cl2, 5 mM Na2S2O8, 2.5 μM Na10Ru4POM, 0.1 M Na2SO4 in unbuffered water and in 5–100 mM phosphate buffer at pH 7.0. The solid lines indicate the biexponential fittings of the traces (monoexponential fitting in the trace in water). (Bottom panel) Plot of the number of Ru4POM → [Ru(bpy)3]3+ ET events in 50 ms vs. the concentration of HPO42− (base of the buffer). The number of ET events is estimated from the experimental recovery of the bleach in the kinetic traces in the top panel. The error bars are given as semidispersion of two separate experiments. | ||
These results speak in favour of the occurrence of multiple PCET events from Ru4POM, with the HPO42− base of the buffer being responsible for the transfer of the protons, while electrons are conveyed to [Ru(bpy)3]3+. The number of ET events increases up to a HPO42− base concentration of ca. 15 mM, above which a plateau value of ca. 6 events is reached (Fig. 2, bottom panel). This concentration threshold is likely due to the availability of the HPO42− base in proximity of the Ru4POM site reacting through PCET. Indeed, in the case of Ru4POM, accumulation of 6 oxidizing equivalents is associated with the generation of RuVIoxo states through RuIV(H2O)/RuV(OH)/RuVI(O) manifolds, subjected to a water nucleophilic attack as the first step finally releasing dioxygen in the catalytic cycle (Scheme 2).38
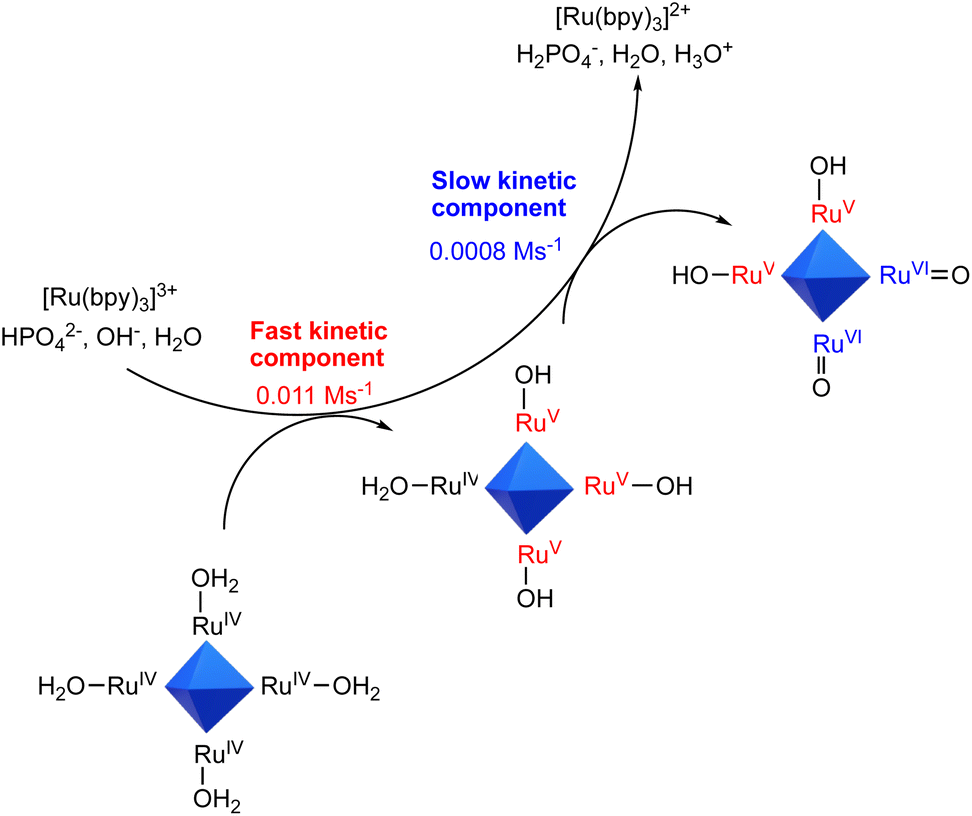 | ||
| Scheme 2 Schematic representation of the accumulation of oxidation equivalents on Ru4POM through sequential PCET involving [Ru(bpy)3]3+ and aqueous bases within two kinetic regimes. | ||
In order to extract kinetic parameters from the experimental traces in Fig. 2 we focused on the profile of consumption of [Ru(bpy)3]3+ and used bi-exponential functions to describe the recovery of the Δ(OD) along the experiments, (eqn (8)):¶
| Δ(OD) = −AF × exp(−t/τF) − AS × exp(−t/τS) + Δ(OD)@50ms | (8) |
| Different pH and phosphate buffer concentrationsa | ||||
|---|---|---|---|---|
| pH ([buffer], mM) | A F (τF, ms) | A S (τS, ms) | App. RateFast × 103, M s−1 | App. RateSlow × 103, M s−1 |
| a 100 μM Ru(bpy)3Cl2, 5 mM Na2S2O8, 2.5 μM Na10Ru4POM, 0.1 M Na2SO4 in 5–100 mM phosphate buffer at pH 7.0, 100 mM phosphate buffer pH 6.0 or water. b 100 μM Ru(bpy)3Cl2, 5 mM Na2S2O8, 0.5–2.5 μM Na10Ru4POM, 0.1 M Na2SO4 in 100 mM phosphate buffer at pH 7.0. In all cases the photogenerated [Ru(bpy)3]3+ estimated from the initial Δ(OD) is ca. 20 μM. | ||||
| 7 (5) | 0.025 ± 0.001 (0.85 ± 0.04) | 0.051 ± 0.004 (9.7 ± 0.1) | 3.12 ± 0.19 | 04.54 ± 0.04 |
| 7 (10) | 0.034 ± 0.001 (0.99 ± 0.06) | 0.060 ± 0.001 (10.7 ± 0.2) | 3.56 ± 0.24 | 0.58 ± 0.01 |
| 7 (20) | 0.045 ± 0.001 (1.05 ± 0.04) | 0.063 ± 0.004 (10.7 ± 0.2) | 4.43 ± 0.19 | 0.61 ± 0.04 |
| 7 (50) | 0.051 ± 0.001 (0.92 ± 0.05) | 0.062 ± 0.001 (10.1 ± 0.2) | 5.75 ± 0.33 | 0.64 ± 0.02 |
| 7 (100) | 0.060 ± 0.002 (0.57 ± 0.03) | 0.069 ± 0.001 (8.9 ± 0.1) | 10.87 ± 0.67 | 0.80 ± 0.02 |
| 6 (100) | 0.036 ± 0.001 (2.15 ± 0.12) | 0.035 ± 0.001 (11.6 ± 0.5) | 1.73 ± 0.11 | 0.31 ± 0.01 |
| Water | 0.022 ± 0.001 (4.0 ± 0.1) | — | 0.56 ± 0.02 | — |
| Different concentrations of Ru4POM (Fig. S8)b | ||||
|---|---|---|---|---|
| [Ru4POM] μM | A F (τF, ms) | A S (τS, ms) | App. RateFast × 103, M s−1 | App. RateSlow × 103, M s−1 |
| 2.5 | 0.060 ± 0.002 (0.57 ± 0.02) | 0.069 ± 0.001 (8.9 ± 0.1) | 10.87 ± 0.67 | 0.80 ± 0.20 |
| 1 | 0.043 ± 0.001 (1.29 ± 0.05) | 0.058 ± 0.001 (9.7 ± 0.2) | 3.44 ± 0.13 | 0.62 ± 0.17 |
| 0.5 | 0.038 ± 0.002 (3.05 ± 0.16) | 0.039 ± 0.002 (13.7 ± 0.6) | 1.30 ± 0.10 | 0.30 ± 0.20 |
First, it is interesting to note that the relative amplitude contributions of the fast and slow components to the reactivity of [Ru(bpy)3]3+ depend on the concentration of the buffer base (Fig. 3 top panel). The relative contributions of the fast and slow components – calculated as AF × 100/(AF + AS) and AS × 100/(AF + AS), respectively – at low HPO42− concentrations are indeed 33% and 67%, respectively, while at high HPO42− concentrations the relative contributions reach plateau values of 45% and 55% for the fast and slow components, respectively.
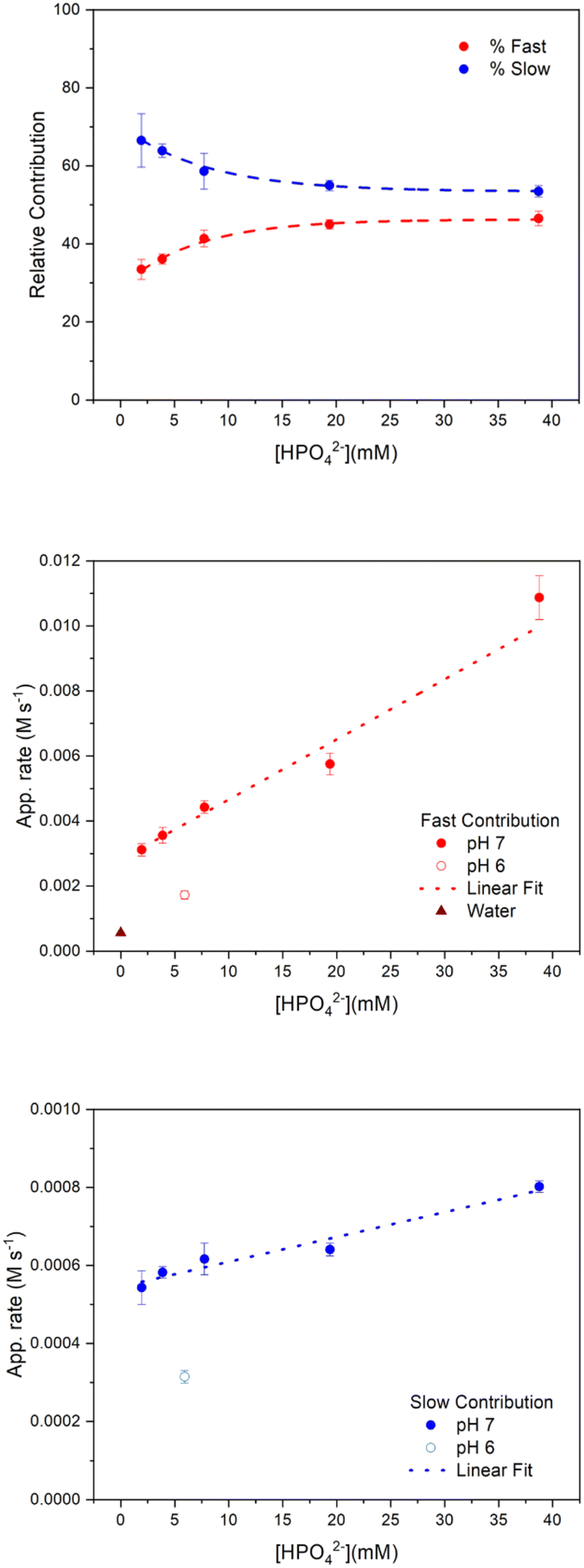 | ||
| Fig. 3 (Top panel) Relative contribution of the two components to the recovery of the traces, calculated as AF × 100/(AF + AS) and AS × 100/(AF + AS), respectively. (Medium panel) Plot of apparent rates for [Ru(bpy)3]3+ consumption vs. the concentration of base of the buffer for the fast contribution at different pH. (Bottom panel) Plot of apparent rates for [Ru(bpy)3]3+ consumption vs. the concentration of base of the buffer for the slow contribution at different pH. | ||
From the fitting parameters we then derived two apparent rates for the reactivity of [Ru(bpy)3]3+ with Ru4POM due to the fast and slow components (eqn (9) and (10); the 1.04 × 10−4 factor converts the Δ(OD) units of AF and AS into the corresponding concentration of [Ru(bpy)3]3+):||
| App. RateFast (M s−1) = AF/τF × 1.04 × 10−4 | (9) |
| App. RateSlow (M s−1) = AS/τS × 1.04 × 10−4 | (10) |
As shown in Fig. 3 (bottom panel and inset), the apparent rates determined above depend linearly on the concentration of the HPO42− base of the phosphate buffer (the log–log plot analysis supports a first order in [HPO42−] in both apparent rates, see ESI Fig. S10†).
| App. RateFast (M s−1) = 2.62 × 10−3 + 2.04 × 10−4 [HPO42−] | (11) |
| App. RateSlow (M s−1) = 5.45 × 10−4 + 6.38 × 10−6 [HPO42−] | (12) |
The linear dependence of the rates on [HPO42−] confirms the role of the base in assisting the removal of the protons from Ru4POM along its conversion to highly oxidized states through reactivity with [Ru(bpy)3]3+ (both apparent rates are indeed linearly dependent on Ru4POM concentration, see Fig. S11†).
For both the fast and slow apparent rates, the non-null intercept in eqn (11) and (12) indicates that HPO42− is not the only base assisting the reactivity of [Ru(bpy)3]3+ towards Ru4POM. When conducting the analysis at lower pH (100 mM phosphate buffer pH 6) a significant abatement of both components of apparent rates was observed,**Table 1 and Fig. 3, medium and bottom panels. Registration and fitting of a trace in water in the absence of any buffer (in this case through a monoexponential fitting) lead to the obtainment of a single component of the apparent rate, which was significantly abated with respect to the values previously determined. These results indicate that both OH− and H2O are involved in the reactivity of [Ru(bpy)3]3+ towards Ru4POM (Scheme 2).36,39†† The possibility for a base to assist PCET events is associated with the libido rule40,41 and with the strength of the base, expressed in terms of the pKa of the conjugate acid/base couples (in this case: pKa = 7.2 for H2PO4−/HPO42−, pKa = 0 for H3O+/H2O, pKa = 14 for H2O/OH−). Although H2O is the least basic, the possibility of preorganisation of water channels at the hydrophilic surface of polyoxometalates may favour its role in assisting the transfer of protons during oxidation of Ru4POM.36 Finally, superimposable kinetic traces and unchanged fitting parameters were obtained when registering the experiment in deuterated medium (Fig. S12 in ESI,† obtained by laser flash photolysis of 100 μM Ru(bpy)3Cl2, 5 mM Na2S2O8, 2.5 μM Na10Ru4POM, 0.1 M Na2SO4 in 100 mM deuterated phosphate buffer at pD 7.0), indicating a negligible H/D isotopic effect in the multiple ET dynamics. In single PCET events, small H/D isotope effects are indicative of a low modification of the overlap integrals of the donor–acceptor states along the proton transfer coordinate, when replacing H with D.42
The whole mechanistic scenario is represented in Scheme 2, where [Ru(bpy)3]3+ feeds Ru4POM with 6 oxidizing equivalents up to RuVI![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O states, with the assistance of aqueous bases in two distinct kinetic regimes. We finally attempted to estimate rate constants for the stepwise PCET processes by employing a kinetic model that considers 6 consecutive bimolecular events involving [Ru(bpy)3]3+ and Ru4POM (previous eqn (7)), each one associated with a bimolecular rate constant kj (j = 1–6). Under the optimal conditions investigated (100 mM phosphate buffer, pH 7, purple trace in Fig. 2 top panel), the fitting provides values in the range 1.3 × 108 ÷ 2 × 109 M−1 s−1 for k1–k3 (associated with the fast component of the reactivity of [Ru(bpy)3]3+ and with the generation of the primary oxidized intermediates of Ru4POM) and 1.2 × 107 ÷ 4.5 × 107 M−1 s−1 for k4–k6 (associated with the slow component of the reactivity of [Ru(bpy)3]3+ and with the generation of the highly oxidized intermediates of Ru4POM). The slowing down of the rate for reaching highly oxidized Ru4POM intermediates is expected based on less favorable thermodynamics, according to the higher potentials associated with such species.37,38‡‡
O states, with the assistance of aqueous bases in two distinct kinetic regimes. We finally attempted to estimate rate constants for the stepwise PCET processes by employing a kinetic model that considers 6 consecutive bimolecular events involving [Ru(bpy)3]3+ and Ru4POM (previous eqn (7)), each one associated with a bimolecular rate constant kj (j = 1–6). Under the optimal conditions investigated (100 mM phosphate buffer, pH 7, purple trace in Fig. 2 top panel), the fitting provides values in the range 1.3 × 108 ÷ 2 × 109 M−1 s−1 for k1–k3 (associated with the fast component of the reactivity of [Ru(bpy)3]3+ and with the generation of the primary oxidized intermediates of Ru4POM) and 1.2 × 107 ÷ 4.5 × 107 M−1 s−1 for k4–k6 (associated with the slow component of the reactivity of [Ru(bpy)3]3+ and with the generation of the highly oxidized intermediates of Ru4POM). The slowing down of the rate for reaching highly oxidized Ru4POM intermediates is expected based on less favorable thermodynamics, according to the higher potentials associated with such species.37,38‡‡
Impact of the PCET mechanism in light driven O2 evolution
We finally investigated the implication of the PCET mechanism and the effect of the buffer concentration on the O2 evolution of the system, by irradiating 15 mL of an aqueous buffered solution containing 0.1 M Na2SO4, 1 mM Ru(bpy)3Cl2, 5 mM Na2S2O8 and 5 μM of Na10Ru4POM with a blue LED (λem = 450 nm, FWHM 10 nm). The O2 evolution traces are reported in Fig. 4, and show an initial linear production of O2, before reaching a plateau after 1–2 h irradiation. The key performance indicators are collected in Table 2; in this case, we considered the maximum O2 evolving rate, Rate(O2)MAX, as the most significant one to evaluate the efficiency of the system and the effect of the buffer concentration. The main outcome can be summarised as follows: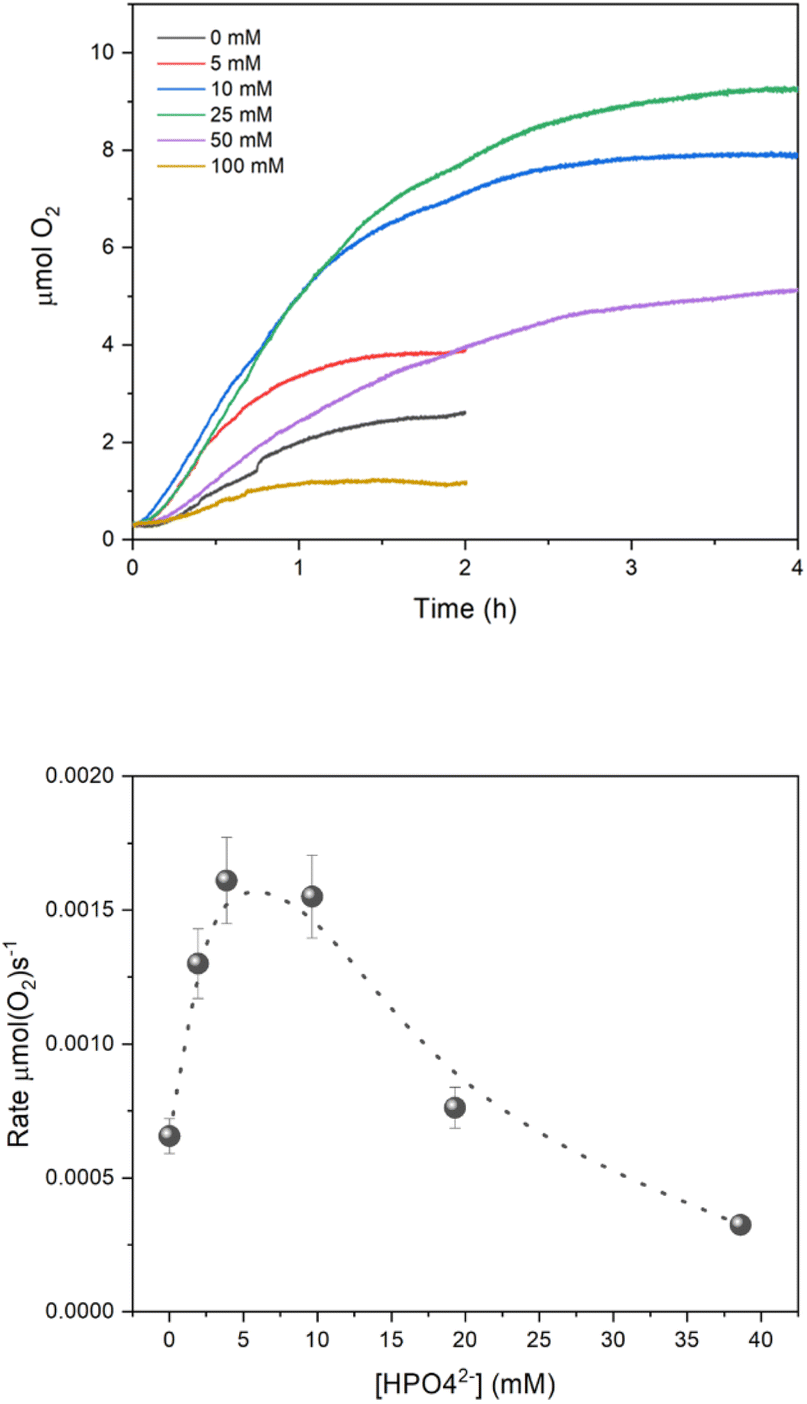 | ||
| Fig. 4 (Top panel) O2 evolution kinetics (see Table 2 for conditions). (Bottom panel) Plot of Rate(O2)MAXvs. the concentration of HPO42− buffer base. Error bars result from repetitive runs. | ||
| [Buffer] mM | Rate(O2)MAX μmol(O2) s−1 | TOF × 102 s−1 | μmol(O2) | TON | QE% |
|---|---|---|---|---|---|
| a Photon flux = 3.33 × 10−8 einstein s−1. b Photon flux = 0.87 × 10−8 einstein s−1. | |||||
| — | 6.56 × 10−4 | 0.87 ± 0.1 | 2.8 ± 0.3 | 37 ± 3 | 1.6 ± 0.2 |
| 5 | 1.30 × 10−3 | 1.73 ± 0.1 | 4.3 ± 0.4 | 57 ± 6 | 3.2 ± 0.2 |
| 10 | 1.61 × 10−3 | 2.15 ± 0.2 | 8.0 ± 0.8 | 107 ± 9 | 3.8 ± 0.4 |
| 25 | 1.55 × 10−3 | 2.07 ± 0.2 | 9.5 ± 0.9 | 127 ± 12 | 3.8 ± 0.4 |
| 50 | 7.62 × 10−4 | 1.02 ± 0.1 | 5.1 ± 0.5 | 68 ± 6 | 1.8 ± 0.2 |
| 100 | 3.24 × 10−4 | 0.43 ± 0.04 | 1.2 ± 0.1 | 16 ± 1 | 0.8 ± 0.1 |
| 10a | 1.16 × 10−3 | 1.55 ± 0.1 | 8.4 ± 0.8 | 112 ± 11 | 6.8 ± 0.6 |
| 10b | 5.93 × 10−4 | 0.79 ± 0.1 | 6.4 ± 0.6 | 85 ± 8 | 13.6 ± 1.2 |
(i) The system shows O2 evolution activity also in the absence of buffer, with a Rate(O2)MAX of 6.56 × 10−4 μmol(O2) s−1; upon addition of the phosphate buffer (5–10 mM), an increase in Rate(O2)MAX is observed up to 1.61 × 10−3 μmol(O2) s−1 (Fig. 4 bottom panel; for consistency with previous data, we reported the HPO42− base concentration in the abscissa). In oxygen evolution, the effect of the buffer can be related to multiple factors, including the aid in generating oxidized intermediates of the catalyst (vide supra), assisting the water nucleophilic attack and the oxygen–oxygen bond formation,38 managing the protons released in the water oxidation process.
(ii) A further increase of buffer concentration in the reaction conditions leads to a progressive abatement of Rate(O2)MAX (Fig. 4). This effect is ascribed to a rapid deactivation of the [Ru(bpy)3]2+ photosensitizer,43–45 confirmed by UV-vis analysis of the reaction solution, which visibly turns from brilliant orange to brownish along the first minutes of irradiation (Fig. S13†). Recently, two decomposition pathways were elucidated for [Ru(bpy)3]2+ when combined with light and persulfate: a dark one, occurring at pH > 6, in which photogenerated [Ru(bpy)3]3+ reacts with OH− to form OH˙ radicals which then attack the bpy ligands, and a light-induced one, starting from excitation of the [Ru(bpy)3]3+ oxidised state, promoting its reactivity with S2O82−.45 Given the specific conditions employed in our study (pH 7, irradiation at 450 nm corresponding to the MLCT band of [Ru(bpy)3]2+) it is plausible that the dark decomposition pathway prevails, with HPO42− being also involved in the radical reactivity.46 Detrimental effects of high buffer concentration in water oxidation catalysis were previously documented under both light driven39 and electrochemical conditions.47
(iii) Finally, under the optimized buffer conditions (10 mM), we explored the effect of light intensity on O2 generation. When reducing the photon flux from 8.28 × 10−8 to 3.33 × 10−8 and to 0.87 × 10−8 einstein s−1, a progressive decrease of Rate(O2)MAX was observed from 1.61 × 10−3 to 1.16 × 10−3 and to 5.93 × 10−4 μmol(O2) s−1, indicative of light being a limiting reagent (Fig. S14†). Lowering the light intensity leads to an optimization of photon exploitation, as demonstrated by the quantum yield ϕ(O2) of the process reaching 6.8; this corresponds to a quantum efficiency for oxygen evolution of 13.6%, given that a theoretical maximum quantum yield ϕ(O2) = 0.50 is expected with the [Ru(bpy)3]2+/S2O82− cycle, where the production of one oxygen molecule theoretically requires the absorption of two photons (see Scheme S1 in ESI†).48 A value of 9% for quantum efficiency was previously reported in the literature under similar reaction conditions but with 420–520 nm irradiation.28
Conclusions and perspectives
We investigated the accumulation of oxidizing equivalents on the Ru4POM water oxidation catalyst through PCET events in a [Ru(bpy)3]2+/S2O82− photochemical cycle. Laser flash photolysis experiments indicate the occurrence of 6 oxidative PCET events on Ru4POM in ca. 50 ms, leading to the generation of competent RuVI–oxo intermediates. While electrons are conveyed from Ru4POM to the [Ru(bpy)3]3+ oxidant, protons are transferred to aqueous bases. The flash photolysis traces show indeed two components of the rate of [Ru(bpy)3]3+ reactivity, both linearly dependent on the HPO42− buffer base concentration; a contribution of H2O and OH− in managing protons was also highlighted. The effect of HPO42− buffer base was evident also in O2 evolving kinetics, inducing a progressive increase of O2 rate up to 5–10 mM, above which the system loses activity due to [Ru(bpy)3]2+ photosensitizer fast decomposition. Under optimal buffer composition, managing light intensity led to reaching a quantum efficiency up to 13.6%.This work highlights the importance of considering the molecular nature of PCET events under photochemical conditions, since this can significantly impact the efficiency of the overall process. The investigation of this aspect is expected to be general and broad, given the fact that PCET events are pervasive in many chemical transformations.
Experimental part
Instrumentation and procedures
Na10{RuIV(H2O)4(μ-OH)2(μ-O)4[SiW10O36]2} (Na10Ru4POM) was synthesised as previously reported.29–31Nanosecond transient absorption measurements were performed with a custom laser spectrometer consisting of a Continuum Surelite II Nd:YAG laser (FWHM 6–8 ns) with an option to double (532 nm) or triple (355 nm) the frequency, an Applied Photo-physics xenon light source including a mod. 720![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 150 W lamp housing, a mod. 620 power-controlled lamp supply and a mod. 03-102 arc lamp pulser. Laser excitation was provided at 90° with respect to the white light probe beam. Light transmitted by the sample was focused onto the entrance slit of a 300 mm focal length Acton SpectraPro 2300i triple grating, flat field, double exit monochromator equipped with a photomultiplier detector (Hamamatsu R3896) and a Princeton Instruments PIMAX II gated intensified CCD camera, using an RB Gen II intensifier, an ST133 controller and a PTG pulser. Signals from the photomultiplier (Hamamatsu R928) were processed by means of a Teledyne LeCroy 604Zi digital oscilloscope (400 MHz, 20 GS s−1).
150 W lamp housing, a mod. 620 power-controlled lamp supply and a mod. 03-102 arc lamp pulser. Laser excitation was provided at 90° with respect to the white light probe beam. Light transmitted by the sample was focused onto the entrance slit of a 300 mm focal length Acton SpectraPro 2300i triple grating, flat field, double exit monochromator equipped with a photomultiplier detector (Hamamatsu R3896) and a Princeton Instruments PIMAX II gated intensified CCD camera, using an RB Gen II intensifier, an ST133 controller and a PTG pulser. Signals from the photomultiplier (Hamamatsu R928) were processed by means of a Teledyne LeCroy 604Zi digital oscilloscope (400 MHz, 20 GS s−1).
Light driven catalytic tests for water oxidation were conducted in a home-made glass reactor, equipped with a TROXROB10 oxygen probe inserted in the headspace, and connected with a FirestingO2 fiber-optical oxygen meter for real time monitoring of evolved O2. 15 mL of aqueous buffer was introduced into the reactor, which was then closed and purged under a dark atmosphere with nitrogen for 20 minutes: after purging, the solution was allowed to equilibrate in the dark for 5 minutes and then illuminated with a series of six monochromatic LEDs emitting at 450 nm, photon flux = (8.28 ÷ 0.87) × 10−8 einstein s−1. The irradiation power of the LEDs was measured with an AvaSpec-2048 Fiber Optic Spectrometer from Avantes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from Ministero dell’Università e della Ricerca (PRIN 2022 PROMETEO, 2022KPK8WM), by the European Union – Next Generation EU (PRIN2022 PNRR PHOTOCORE, P2022ZSPWF) and from Fondazione Cariparo (Project “SYNERGY”, Ricerca Scientifica di Eccellenza 2018) is greatly acknowledged. We thank the National Recovery and Resilience Plan (NRRP), Mission 4 Component 2 Investment 1.3 – Call for tender No. 1561 of 11.10.2022 of MUR, funded by the European Union – NextGenerationEU, Project code PE000002 “Network 4 Energy Sustainable Transition – NEST”.Notes and references
- R. G. Agarwal, S. C. Coste, B. D. Groff, A. M. Heuer, H. Noh, G. A. Parada, C. F. Wise, E. M. Nichols, J. J. Warren and J. M. Mayer, Chem. Rev., 2022, 122, 1–49 CrossRef CAS PubMed.
- R. Tyburski, T. Liu, S. D. Glover and L. Hammarström, J. Am. Chem. Soc., 2021, 143, 560–576 CrossRef CAS PubMed.
- G. A. Parada, Z. K. Goldsmith, S. Kolmar, B. P. Rimgard, B. Q. Mercado, L. Hammarström, S. Hammes-Schiffer and J. M. Mayer, Science, 2019, 475, 471–475 CrossRef PubMed.
- M. C. Kessinger, J. Xu, K. Cui, Q. Loague, A. V. Soudackov, S. Hammes-Schiffer and G. J. Meyer, J. Am. Chem. Soc., 2024, 146, 1742–1747 CrossRef CAS PubMed.
- M. Natali, A. Amati, S. Merchiori, B. Ventura and E. Iengo, J. Phys. Chem. C, 2020, 124, 8514–8525 CrossRef CAS.
- M. Natali, A. Amati, N. Demitri and E. Iengo, Chem.–Eur. J., 2021, 27, 7872–7881 CrossRef CAS PubMed.
- E. Benazzi, J. Karlsson, Y. Ben M'Barek, P. Chabera, S. Blanchard, S. Alves, A. Proust, T. Pullerits, G. Izzet and E. A. Gibson, Inorg. Chem. Front., 2021, 8, 1610–1618 RSC.
- M. Kuss-Petermann, M. Orazietti, M. Neuburger, P. Hamm and O. S. Wenger, J. Am. Chem. Soc., 2017, 139, 5225–5232 CrossRef CAS PubMed.
- A. Pannwitz and O. S. Wenger, Chem. Commun., 2019, 55, 4004–4014 RSC.
- A. Dey, N. Ghorai, A. Das and H. N. Ghosh, J. Phys. Chem. B, 2020, 124, 11165–11174 CrossRef CAS PubMed.
- T. F. Markle, J. W. Darcy and J. M. Mayer, Sci. Adv., 2018, 4, eaat5776 CrossRef PubMed.
- P. R. D. Murray, J. H. Cox, N. D. Chiappini, C. B. Roos, E. A. McLoughlin, B. G. Hejna, S. T. Nguyen, H. H. Ripberger, J. M. Ganley, E. Tsui, N. Y. Shin, B. Koronkiewicz, G. Qiu and R. R. Knowles, Chem. Rev., 2022, 122, 2017–2291 CrossRef CAS PubMed.
- P. Franceschi, E. Rossin, G. Goti, A. Scopano, A. Vega-Penaloza, M. Natali, D. Singh, A. Sartorel and L. D. Amico, J. Org. Chem., 2023, 88, 6454–6464 CrossRef CAS PubMed.
- T. Bortolato, G. Simionato, M. Vayer, C. Rosso, L. Paoloni, E. M. Benetti, A. Sartorel, D. Lebœuf and L. Dell'Amico, J. Am. Chem. Soc., 2023, 145, 1835–1846 CrossRef CAS PubMed.
- Y. Yang, G. A. Volpato, E. Rossin, N. Peruffo, F. Tumbarello, C. Nicoletti, R. Bonetto, L. Paoloni, P. Umari, E. Colusso, L. Dell'Amico, S. Berardi, E. Collini, S. Caramori, S. Agnoli and A. Sartorel, ChemSusChem, 2023, e202201980 CrossRef CAS PubMed.
- B. P. Sullivan, D. J. Salmon and T. J. Meyer, Inorg. Chem., 1978, 17, 3334–3341 CrossRef CAS.
- S. W. Gersten, G. J. Samuels and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4029–4030 CrossRef CAS.
- M. Schulze, V. Kunz, P. D. Frischmann and F. Würthner, Nat. Chem., 2016, 8, 576–583 CrossRef CAS PubMed.
- M. A. Hoque, M. Gil-Sepulcre, A. de Aguirre, J. A. A. W. Elemans, D. Moonshiram, R. Matheu, Y. Shi, J. Benet-Buchholz, X. Sala, M. Malfois, E. Solano, J. Lim, A. Garzón-Manjón, C. Scheu, M. Lanza, F. Maseras, C. Gimbert-Suriñach and A. Llobet, Nat. Chem., 2020, 12, 1060–1066 CrossRef CAS PubMed.
- R. Matheu, P. Garrido-Barros, M. Gil-Sepulcre, M. Z. Ertem, X. Sala, C. Gimbert-Suriñach and A. Llobet, Nat. Rev. Chem, 2019, 3, 331–341 CrossRef CAS.
- L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet and L. Sun, Nat. Chem., 2012, 4, 418–423 CrossRef CAS PubMed.
- R. Matheu, M. Z. Ertem, J. Benet-Buchholz, E. Coronado, V. S. Batista, X. Sala and A. Llobet, J. Am. Chem. Soc., 2015, 137, 10786–10795 CrossRef CAS PubMed.
- A. Sartorel, M. Carraro, G. Scorrano, R. De Zorzi, S. Geremia, N. D. McDaniel, S. Bernhard and M. Bonchio, J. Am. Chem. Soc., 2008, 130, 5006–5007 CrossRef CAS PubMed.
- Y. V. Geletii, B. Botar, P. Kögerler, D. A. Hillesheim, D. G. Musaev and C. L. Hill, Angew. Chem., Int. Ed., 2008, 47, 3896–3899 CrossRef CAS PubMed.
- F. M. Toma, A. Sartorel, M. Iurlo, M. Carraro, P. Parisse, C. MacCato, S. Rapino, B. R. Gonzalez, H. Amenitsch, T. Da Ros, L. Casalis, A. Goldoni, M. Marcaccio, G. Scorrano, G. Scoles, F. Paolucci, M. Prato and M. Bonchio, Nat. Chem., 2010, 2, 826–831 CrossRef CAS PubMed.
- S. X. Guo, Y. Liu, C. Y. Lee, A. M. Bond, J. Zhang, Y. V. Geletii and C. L. Hill, Energy Environ. Sci., 2013, 6, 2654–2663 RSC.
- M. Natali, M. Orlandi, S. Berardi, S. Campagna, M. Bonchio, A. Sartorel and F. Scandola, Inorg. Chem., 2012, 51, 7324–7331 CrossRef CAS PubMed.
- Y. V. Geletii, Z. Huang, Y. Hou, D. G. Musaev, T. Lian and C. L. Hill, J. Am. Chem. Soc., 2009, 131, 7522–7523 CrossRef CAS PubMed.
- G. A. Volpato, M. Marasi, T. Gobbato, F. Valentini, F. Sabuzi, V. Gagliardi, A. Bonetto, A. Marcomini, S. Berardi, V. Conte, M. Bonchio and S. Caramori, Chem. Commun., 2020, 56, 2248–2251 RSC.
- T. Gobbato, F. Rigodanza, E. Benazzi, P. Costa, M. Garrido, A. Sartorel, M. Prato and M. Bonchio, J. Am. Chem. Soc., 2022, 144, 14021–14025 CrossRef CAS PubMed.
- M. Bonchio, Z. Syrgiannis, M. Burian, N. Marino, E. Pizzolato, K. Dirian, F. Rigodanza, G. A. Volpato, G. La Ganga, N. Demitri, S. Berardi, H. Amenitsch, D. M. Guldi, S. Caramori, C. A. Bignozzi, A. Sartorel and M. Prato, Nat. Chem., 2019, 11, 146–153 CrossRef CAS PubMed.
- J. Fielden, J. M. Sumliner, N. Han, Y. V. Geletii, X. Xiang, D. G. Musaev, T. Lian and C. L. Hill, Chem. Sci., 2015, 6, 5531–5543 RSC.
- H. Song, A. Amati, A. Pannwitz, S. Bonnet and L. Hammarström, J. Am. Chem. Soc., 2022, 144, 19353–19364 CrossRef CAS PubMed.
- Y. Xu, L. Duan, L. Tong, B. Åkermark and L. Sun, Chem. Commun., 2010, 46, 6506 RSC.
- Y. Xu, A. Fischer, L. Duan, L. Tong, E. Gabrielsson, B. Åkermark and L. Sun, Angew. Chem., Int. Ed., 2010, 49, 8934–8937 CrossRef CAS PubMed.
- F. Rigodanza, N. Marino, A. Bonetto, A. Marcomini, M. Bonchio, M. Natali and A. Sartorel, ChemPhysChem, 2021, 22, 1208–1218 CrossRef CAS PubMed.
- Y. Liu, S. Guo, A. M. Bond, J. Zhang, Y. V Geletii and C. L. Hill, Inorg. Chem., 2013, 52, 11986–11996 CrossRef CAS PubMed.
- S. Piccinin, A. Sartorel, G. Aquilanti, A. Goldoni, M. Bonchio and S. Fabris, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4917–4922 CrossRef CAS PubMed.
- G. A. Volpato, A. Bonetto, A. Marcomini, P. Mialane, M. Bonchio, M. Natali and A. Sartorel, Sustainable Energy Fuels, 2018, 2, 1951–1956 RSC.
- W. P. Jencks, J. Am. Chem. Soc., 1972, 94, 4731–4732 CrossRef CAS.
- W. Jencks, Chem. Rev., 1972, 72, 705–718 CrossRef CAS.
- S. Hammes-Schiffer, J. Am. Chem. Soc., 2015, 137, 8860–8871 CrossRef CAS PubMed.
- P. K. Ghosh, B. S. Brunschwig, M. Chou, C. Creutz and N. Sutin, J. Am. Chem. Soc., 1984, 106, 4772–4783 CrossRef CAS.
- C. T. Lin, W. Böttcher, M. Chou, C. Creutz and N. Sutin, J. Am. Chem. Soc., 1976, 98, 6536–6544 CrossRef CAS.
- U. S. Akhtar, E. L. Tae, Y. S. Chun, I. C. Hwang and K. B. Yoon, ACS Catal., 2016, 6, 8361–8369 CrossRef CAS.
- J. Lee, U. Von Gunten and J. H. Kim, Environ. Sci. Technol., 2020, 54, 3064–3081 CrossRef CAS PubMed.
- D. Wang and J. T. Groves, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15579–15584 CrossRef CAS PubMed.
- A. Volpe, C. Tubaro, M. Natali, A. Sartorel, G. W. Brudvig and M. Bonchio, Inorg. Chem., 2019, 58, 16537–16545 CrossRef CAS PubMed.
- C. Chiorboli, M. T. Indelli, M. A. R. Scandola and F. Scandola, J. Phys. Chem., 1988, 92, 156–163 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4se00146j |
| ‡ In all the experiments, 0.1 M Na2SO4 was employed to guarantee a sufficient ionic strength of the medium (I = 0.3 M), thus mitigating: (a) formation of [Ru(bpy)3]2+·Ru4POM ion pairs, where fast static quenching of the [Ru(bpy)3]2+ photosensitizer hampers the occurrence of the envisaged photosynthetic cycle,27 and (b) ET rate constant variability associated with the change of the ionic strength due to the change of buffer concentrations; the rate constant for a diffusion based ET event between charged reactants (as in the present case) is sensitive to the ionic strength according to the Debye–Eigen theory.49 |
| § The photogenerated concentration of [Ru(bpy)3]3+ is calculated from Δε450 (Ru(II)/Ru(III)) = 1.3 × 104 M−1 cm−1 and the appropriate correction for the ratio between the volume of solution probed by the analysing beam and that excited by the laser pulse (1.35, as obtained from saturation techniques). As a result, a Δ(OD) of 0.1 corresponds to a concentration of photogenerated [Ru(bpy)3]3+ of 1.04 × 10−5 M. |
| ¶ In this case, a biexponential function was necessary to provide a suitable fitting of the experimental data. The use of a biexponential function to fit the traces where six sequential oxidation processes are postulated can be explained considering the similar reaction rates of the Ru4POM oxidized intermediates with [Ru(bpy)3]3+. This is indeed not unexpected taking into account the close spacing in potential associated with the oxidation of Ru4POM according to electrochemical studies.37 The use of multiexponential functions is often exploited in the case of electron transfer involving metal oxide particles, due to the presence of surface sites with different reactivity.39 |
| || An alternative analysis considers the determination of an average time constant 〈τ〉 = (AF × τF + AS × τS)/(AF + AS), and the determination of average apparent rates as: App. RateAverage (M−1 s−1) = (AF + AS)/〈τ〉 × 1.04 × 10−4. This leads to a consistent dependence of the average App. Rate on [HPO42−], see ESI (Fig. S9 and Table S1).† |
| ** At pH 6 (phosphate buffer), investigation of other buffer concentrations leads to a marked abatement of [Ru(bpy)3]2+ recovery, thus hampering the possibility of exploring further the system. Reasonable data and fittings were obtained only for the conditions reported in the main text. |
| †† The consumption of [Ru(bpy)3]3+ by Ru4POM in the absence of buffer base could also be consistent with a stepwise mechanism ET/PT, in which electron transfer precedes proton transfer. |
| ‡‡ The kinetic model considering 6 consecutive oxidative events was applied also for the other experimental traces in the Fig. 2 top panel, but in these cases it led to overfitting of the data, see details in ESI.† |
| This journal is © The Royal Society of Chemistry 2024 |