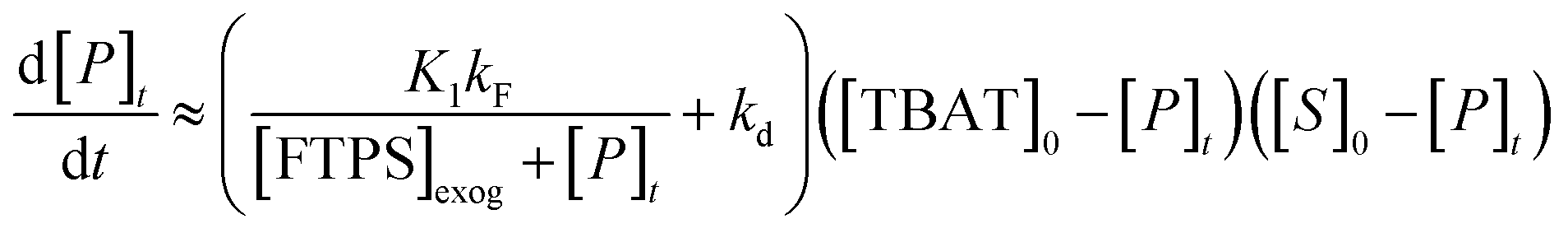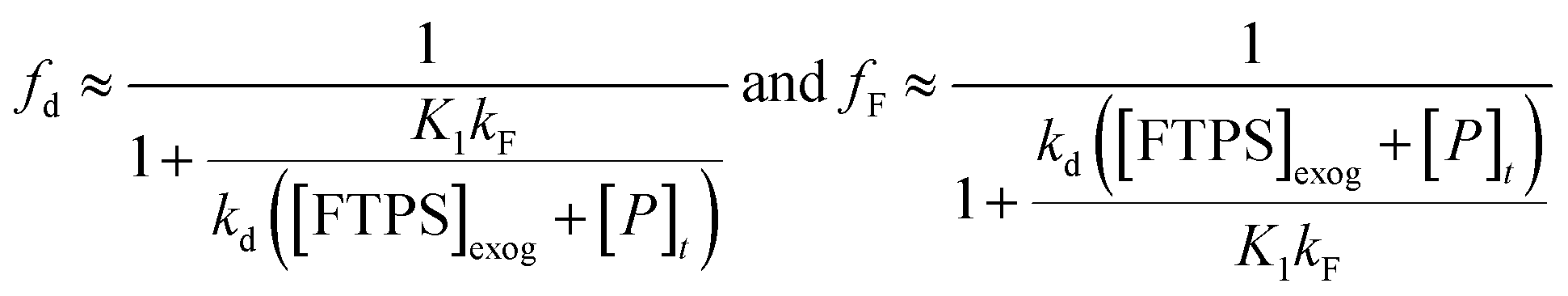DOI:
10.1039/D3SC05776C
(Edge Article)
Chem. Sci., 2024,
15, 4331-4340
Speciation and kinetics of fluoride transfer from tetra-n-butylammonium difluorotriphenylsilicate (‘TBAT’)†
Received
29th October 2023
, Accepted 6th December 2023
First published on 7th December 2023
Abstract
Tetra-n-butylammonium difluorotriphenylsilicate (TBAT) is a conveniently handled anhydrous fluoride source, commonly used as a surrogate for tetra-n-butylammonium fluoride (TBAF). While prior studies indicate that TBAT reacts rapidly with fluoride acceptors, little is known about the mechanism(s) of fluoride transfer. We report on the interrogation of the kinetics of three processes in which fluoride is transferred from TBAT, in THF and in MeCN, using a variety of NMR methods, including chemical exchange saturation transfer, magnetisation transfer, diffusion analysis, and 1D NOESY. These studies reveal ion-pairing between the tetra-n-butylammonium and difluorotriphenylsilicate moieties, and a very low but detectable degree of fluoride dissociation, which then undergoes further equilibria and/or induces decomposition, depending on the conditions. Degenerate exchange between TBAT and fluorotriphenylsilane (FTPS) is very rapid in THF, inherently increases in rate over time, and is profoundly sensitive to the presence of water. Addition of 2,6-di-tert-butylpyridine and 3 Å molecular sieves stabilises the exchange rate, and both dissociative and direct fluoride transfer are shown to proceed in parallel under these conditions. Degenerate exchange between TBAT and 2-naphthalenyl fluorosulfate (ARSF) is not detected at the NMR timescale in THF, and is slow in MeCN. For the latter, the exchange is near-fully inhibited by exogenous FTPS, indicating a predominantly dissociative character to this exchange process. Fluorination of benzyl bromide (BzBr) with TBAT in MeCN-d3 exhibits moderate progressive autoinhibition, and the initial rate of the reaction is supressed by the presence of exogenous FTPS. Overall, TBAT can act as a genuine surrogate for TBAF, as well as a reservoir for rapidly-reversible release of traces of it, with the relative contribution of the pathways depending, inter alia, on the identity of the fluoride acceptor, the solvent, and the concentration of endogenous or exogenous FTPS.
Introduction
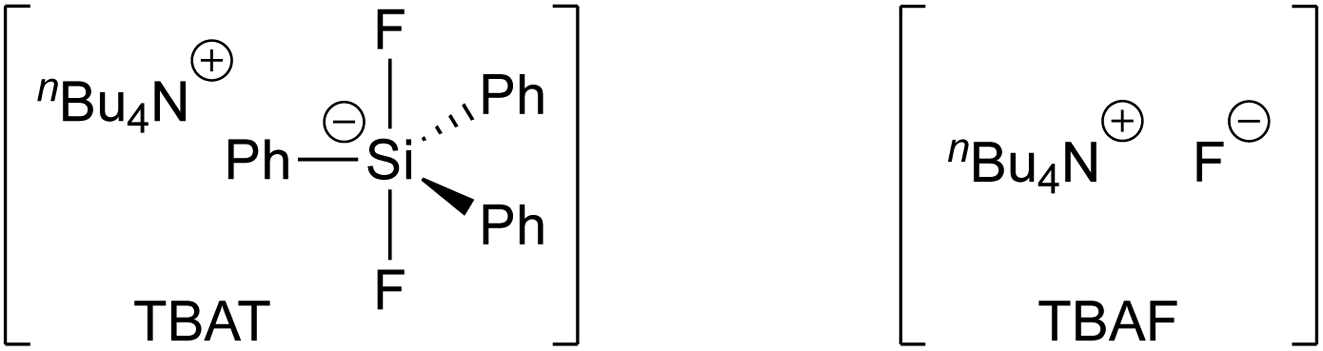
Tetra-n-butylammonium difluorotriphenylsilicate (TBAT, Fig. 1) was introduced by DeShong1 in 1995 as a convenient alternative to tetra-n-butylammonium fluoride (TBAF), and is widely employed, inter alia, for C–F generation,1a,2 deprotection,3 benzyne generation,4 and anion generation by Si–X cleavage (X = C,5 N,6 O,7 S;7d).8,9
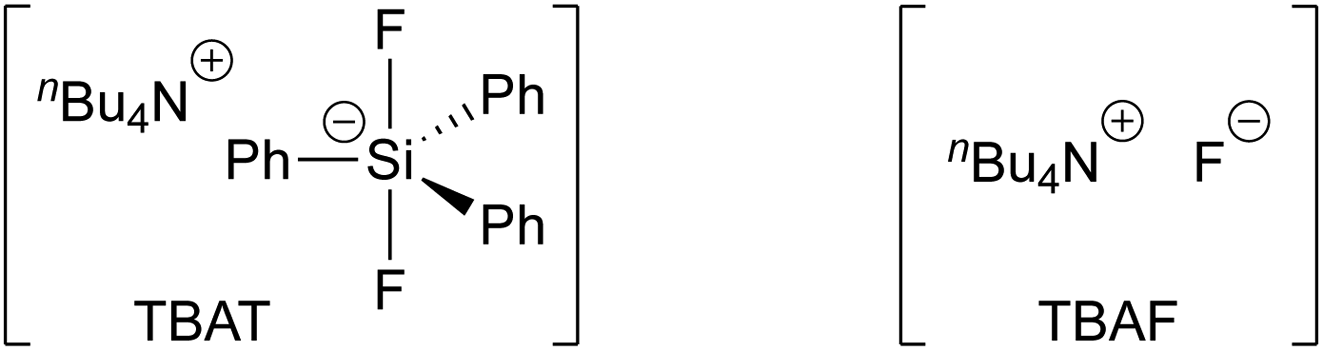 |
| | Fig. 1 Tetra-n-butylammonium difluorotriphenylsilicate (TBAT) and tetra-n-butylammonium fluoride (TBAF). | |
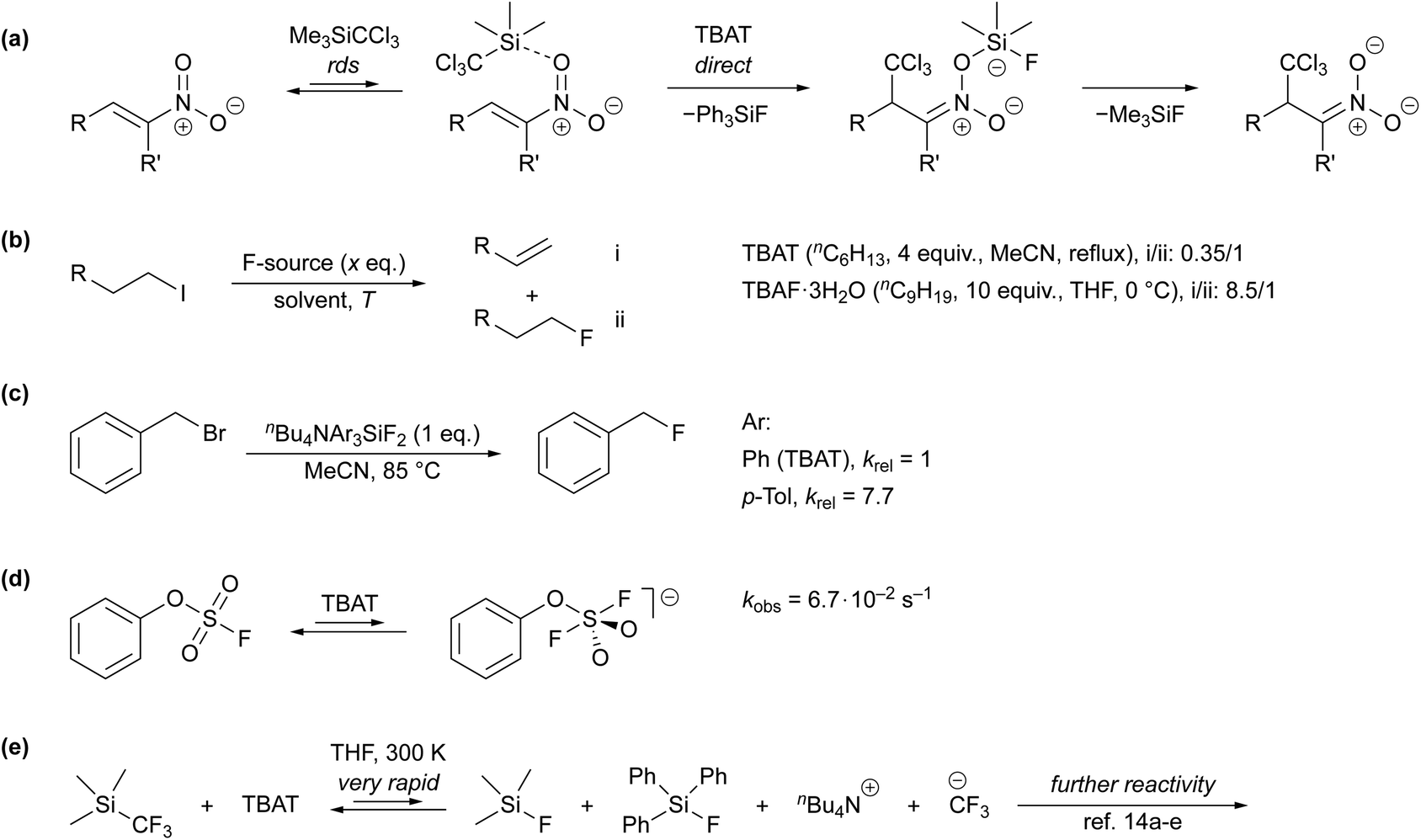
Despite its extensive application, very little has been reported about the kinetics and mechanism by which TBAT transfers fluoride.7b,10–16 Direct fluoride transfer from TBAT to silicon was proposed for the anion-initiated 1,4-addition of TMSCCl3 to nitroalkenes (Scheme 1a).10 This conclusion was primarily based on the lack of signals attributable to free fluoride ions in solution-phase 19F NMR spectra of the reaction mixture. In the conversion of 1-iodoalkanes to 1-fluoroalkanes, TBAT results in significantly less competing β-elimination than TBAF·3H2O (Scheme 1b), albeit under markedly different conditions,1a,11 again leading to the conclusion that the fluoride transferred from TBAT is much less “naked” than that in TBAF. Mąkosza and Bujok found that the tris-p-tolyl analogue of TBAT reacts with benzyl bromide (BzBr) more than seven-fold faster than TBAT itself, Scheme 1c.12 However, whether the transfer occurs directly from the silicate was not established. Finally, a recent study by Zheng et al. reported a pseudo-first-order rate constant, kobs = 6.7 × 10−2 s−1, for the nominally direct transfer of fluoride from TBAT (180 mM) to the sulfur in phenyl fluorosulfate (PhOSO2F, 18 mM) at 298 K in MeCN-d3 (Scheme 1d). The rate was estimated by time-dependent saturation-transfer NMR spectroscopy.13
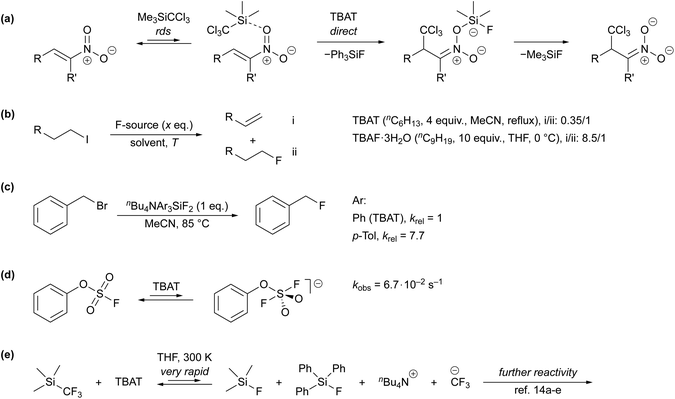 |
| | Scheme 1 (a) Mechanistic proposal in a study on the addition of TMSCCl3 to nitroalkenes,10 (b) ratios of β-elimination-to-substitution products in reactions of 1-iodoalkanes with TBAT and TBAF·3H2O,1a,11 (c) relative rate constants of fluorination of benzyl bromide using nBu4NAr3SiF2, where Ar = Ph or p-Tol,12 (d) rapid fluoride exchange between TBAT and phenyl fluorosulfate studied via time-dependent saturation transfer NMR spectroscopy,13 (e) prior mechanistic work on TBAT-initiated reactivity of TMSCF3, where the fluoride transfer from TBAT is very rapid with respect to NMR spectroscopy. In the presence of excess TMSCF3, the CF3− anion(oid) is transient, undergoing rapidly-reversible exergonic equilibrium with [TMS(CF3)2]−.14 | |
We have recently studied chain reactions initiated by liberation of a CF3− anion(oid) from [TMS(CF3)F]−. The latter is generated in situ by very rapid transfer of fluoride from TBAT to TMSCF3, concomitantly producing fluorotriphenylsilane (FTPS), Scheme 1e.14 Herein we report on a detailed investigation into the kinetics and mechanism of fluoride transfer from TBAT. We have studied this under three sets of conditions, two involving degenerate fluoride exchange (with FTPS and with 2-naphthalenyl fluorosulfate, ARSF) and one involving fluoride transfer to benzyl bromide (BzBr). The primary focus of the work has been the distinction of whether fluoride is transferred directly between TBAT and the fluoride acceptor, or whether there are pre-dissociation step(s) to liberate TBAF as the transient agent for fluoride delivery.
Results and discussion
Prior to our NMR-investigation of the kinetics and mechanism of fluoride transfer, we analysed the NMR-spectroscopic features of TBAT, in THF and in MeCN, including its speciation. The latter comprises two aspects: the extent of interaction between the difluorotriphenylsilicate and tetra-n-butylammonium ions, Ph3SiF2− and nBu4N+, and the extent of fluoride dissociation from the anion, Ph3SiF2−.
Solution-phase 1H,19F NMR-spectroscopic parameters of TBAT
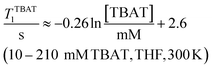
The 1H and 19F nuclei in TBAT were selected for study based on their high abundance and receptivity, 1/2-spin character, and relatively short longitudinal relaxation times. The difluorotriphenylsilicate anion is pseudo-trigonal bipyramidal in both the solid state,1a and in solution,1,17 with the fluorine atoms axial and the phenyl groups equatorial. 1H NMR (400 MHz) spectra of TBAT in THF-d8 (see Section S2.1 of ESI†) comprise two sets of signals in the aromatic region (p-, m- and o-protons in Ph) and three sets of signals in the aliphatic region (C(1), C(2,3) methylenes and C(4) methyl in nBu). In MeCN-d3 the three methylene units are all resolved in the aliphatic region, the aromatic region is similar to that in THF. The 19F NMR (377 MHz) spectra display a singlet corresponding to the two chemically and magnetically equivalent fluorine atoms in the Ph3SiF2− anion, with satellites (1JSiF = (253.8 ± 0.4) Hz and 2JFC = (41.4 ± 0.3) Hz) that are consistent with the 29Si and 13C{1H} NMR data (both in CDCl3) reported by DeShong.1b The concentration dependencies of the chemical shift, δTBAT, and the longitudinal relaxation time constant, TTBAT1, of the fluorine atoms were correlated empirically, eqn (1)–(4). The chemical shift of TBAT is concentration dependent in THF (varying between −96.8 and −95.3 ppm, in the range studied, eqn (1)), whereas it is essentially invariant in MeCN in the concentration range studied (−95.3 ppm; eqn (3)). This phenomenon may arise from the much lower dielectric constant of THF, compared to MeCN, and thus greater impact of changes in the ionic strength of the medium as the TBAT concentration in THF is raised. The longitudinal relaxation of the 1H and 19F nuclei in TBAT takes longer in MeCN than in THF, at all concentrations studied, see Sections S3.2 and S3.3 of ESI.†| | 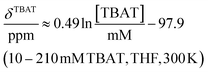 | (1) |
| |  | (2) |
| |  | (3) |
| | 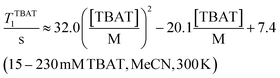 | (4) |
Ion pairing.
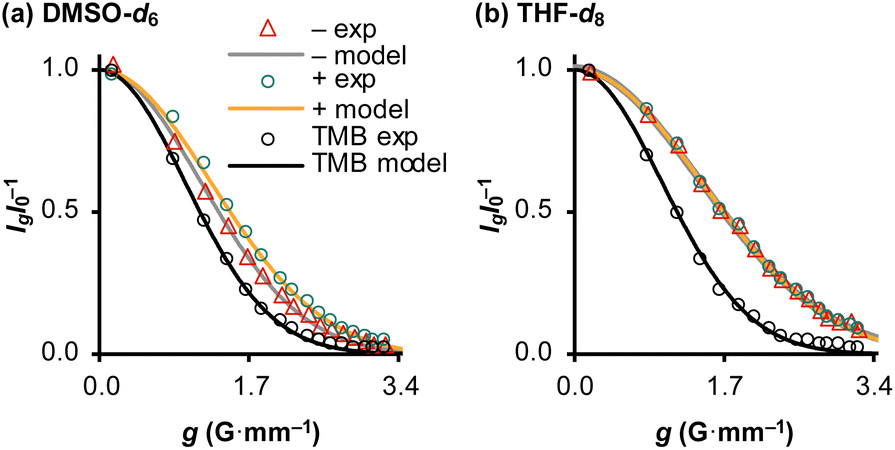
The translational diffusion (D) and mutual proximity (NOESY) of the Ph3SiF2− and nBu4N+ ions were used to probe the extent of ion pairing in TBAT, see Section S4.1 of ESI† for details. The relative translational self-diffusion coefficients, D−/D+ were determined via1H pulsed field gradient NMR experiments at three concentrations, in both THF-d8 and MeCN-d3, and at three temperatures, Table 1, entries 1–18. The average relative translational self-diffusion coefficient, D−/D+, is close to unity in both THF-d8 (0.987 ± 0.008) and MeCN-d3, (1.02 ± 0.02). These values indicate either that the ions are strongly paired, or that the separated ions have coincidental diffusive properties. To distinguish these possibilities, we determined translational diffusion coefficients relative to 1,3,5-trimethoxybenzene, TMB, at low concentration in DMSO-d6, and at high concentration in THF-d8, Fig. 2a and b. The TMB-normalised coefficients in DMSO-d6 are not only different to each other, but also significantly larger than in THF-d8, Table 1, entries 19 and 20. Moreover, the TMB-normalised diffusion coefficient of a reference nBu4N+ ion liberated from [nBu4N+][B(3,5-(CF3)2–C6H3)4] in THF-d8 is very similar to the D+/DTMB value of the TBAT-derived nBu4N+ ion in DMSO-d6, Table 1, entries 19 and 21.
Table 1 (a) Ratios of translational diffusion coefficients of Ph3SiF2− and nBu4N+, D−/D+, in solutions of TBAT in THF-d8 and MeCN-d3 (entries 1–18). (b) Translational diffusion coefficients of Ph3SiF2− and nBu4N+, relative to the internal diffusion standard, TMB, in solutions of TBAT in DMSO-d6 and THF-d8 (entries 19 and 20). (c) Translational diffusion coefficient of nBu4N+, relative to the internal diffusion standard, TMB, in a solution of [nBu4N+][B(3,5-(CF3)2–C6H3)4] in THF-d8 (entry 21)
| Entry |
Solvent |
[TBAT] (mM) |
T (K) |
D
−/D+ |
|
[nBu4N+][B(3,5-(CF3)2–C6H3)4].
|
| 1 |
THF-d8 |
220 |
300 |
0.994 |
| 2 |
THF-d8 |
220 |
310 |
0.984 |
| 3 |
THF-d8 |
220 |
320 |
0.981 |
| 4 |
THF-d8 |
110 |
300 |
0.980 |
| 5 |
THF-d8 |
110 |
310 |
0.973 |
| 6 |
THF-d8 |
110 |
320 |
1.01 |
| 7 |
THF-d8 |
30 |
300 |
0.981 |
| 8 |
THF-d8 |
30 |
310 |
0.980 |
| 9 |
THF-d8 |
30 |
320 |
1.00 |
| 10 |
MeCN-d3 |
220 |
300 |
1.04 |
| 11 |
MeCN-d3 |
220 |
320 |
0.986 |
| 12 |
MeCN-d3 |
220 |
335 |
0.971 |
| 13 |
MeCN-d3 |
120 |
300 |
1.04 |
| 14 |
MeCN-d3 |
120 |
320 |
1.01 |
| 15 |
MeCN-d3 |
120 |
335 |
0.994 |
| 16 |
MeCN-d3 |
40 |
300 |
1.03 |
| 17 |
MeCN-d3 |
40 |
320 |
1.05 |
| 18 |
MeCN-d3 |
40 |
335 |
1.01 |
| 19 |
DMSO-d6 |
1.5 |
300 |
1.21 |
|
D
−/DTMB = 0.710 |
|
D
+/DTMB = 0.586 |
| 20 |
THF-d8 |
207 |
300 |
1.00 |
|
D
−/DTMB = 0.467 |
|
D
+/DTMB = 0.466 |
| 21 |
THF-d8 |
201a |
300 |
0.931 |
|
D
−/DTMB = 0.536 |
|
D
+/DTMB = 0.576 |
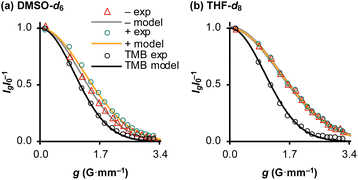 |
| | Fig. 2 Diffusion profiles of Ph3SiF2− (“−”), nBu4N+ (“+”) and the internal diffusion standard (“TMB”) in: (a) 1.5 mM TBAT and 263 mM TMB in DMSO-d6, and (b) 207 mM TBAT and 252 mM TMB in THF-d8; both at 300 K. | |
1H 1D NOESY analysis of 210 mM solutions of TBAT confirmed extensive close-contacts between the ions, in both THF-d8 and in MeCN-d3 (see Section S4.2 of ESI† for details). For example, perturbation of selected aromatic protons resulted in extensive inter-ion NOEs at the aliphatic protons, as well as the expected intra-ion NOEs at the residual aromatic protons. The combined analysis of ion-diffusion and ion-proximity in TBAT indicate that Ph3SiF2− and nBu4N+ are extensively paired in THF-d8 and in MeCN-d3, even at moderately low concentration, and predominantly dissociated in dilute solutions in DMSO-d6.
Exchange of fluoride with TBAT.
To the best of our knowledge, the reversible liberation of TBAF from TBAT has not been reported, and TBAF is not detected in standard NMR spectra of solutions of purified TBAT.1 However, in such solutions we do detect exchange processes between TBAT and a range of species, including TBAF vide infra, using 19F chemical exchange saturation transfer (19F-CEST) NMR spectroscopy (see Sections S4.3 and S7.1.1 in the ESI†). In one 19F-CEST regime, the chemical shift corresponding to a low-concentration undetected spin is selectively pre-saturated to probe for its chemical exchange with a detectable spin present at higher concentration. If the exchange occurs at a rate that is sufficiently large relative to the concentration and longitudinal relaxation time, T1, of the higher concentration spin, then there is a detectable attenuation in its intensity in the subsequent pulse-acquire 1D NMR spectrum.
The experiments were conducted using concentrated solutions18 of TBAT that were prepared and sealed, in J Young valve NMR tubes, in the glove-box. 19F-CEST to TBAT was detected when pre-saturating at −170 ppm (FTPS) and at −149/147 ppm in THF/MeCN. The latter species was assigned as tetra-n-butylammonium bifluoride, TBABF (δTBABF = −147 ppm, THF and MeCN-d3).19,20 The 19F-CEST was greater in MeCN than in THF. When the solutions were stored over 3 Å molecular sieves for 2 months, 19F-CEST was supressed to below the detection limit in THF. In MeCN the 19F-CEST profile showed exchange with species at −77 ppm, −115 ppm, −128 ppm, −147 ppm (TBABF) and −169 ppm (FTPS). The species at −77 ppm is tentatively assigned as partially-hydrous TBAF (δTBAF = −72 ppm, MeCN-d3).19 The additional signals (at −115 ppm and −128 ppm) possibly arise from co-products of Hofmann elimination, e.g.nBu3N(HF)x. Lastly, solutions of TBAT containing exogenous FTPS (∼5 mol%)21 were studied. The 19F-CEST profiles exhibited saturation over a broader frequency range, indicative of rapid exchange between TBAT and FTPS. All other 19F-CEST effects were supressed to below the detection limit, indicative that exogenous FTPS drives dissociated fluoride equilibria towards TBAT.
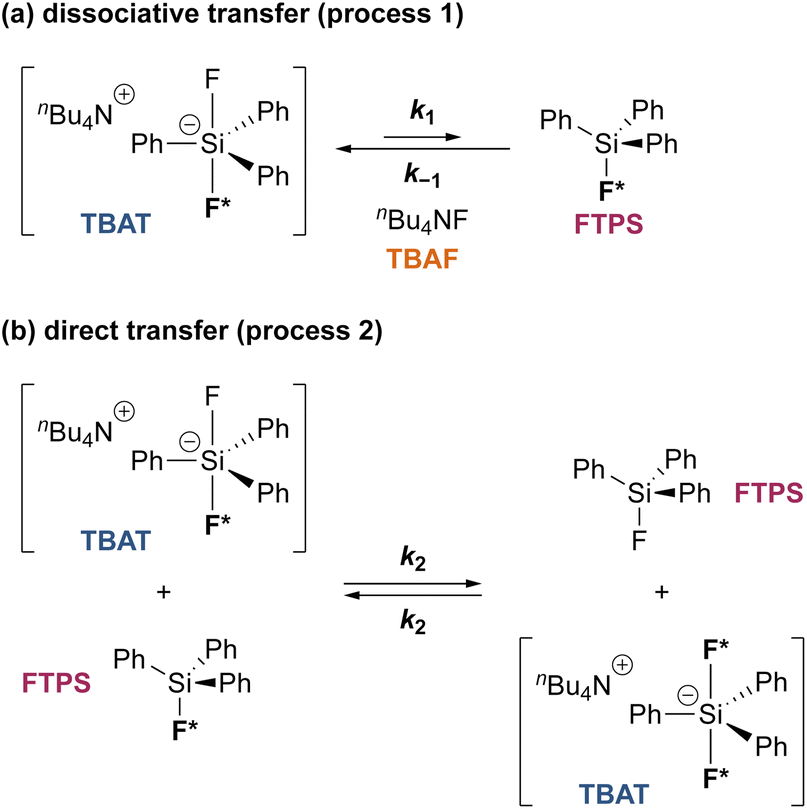
Analytical model for the kinetics of TBAT exchange with FTPS.
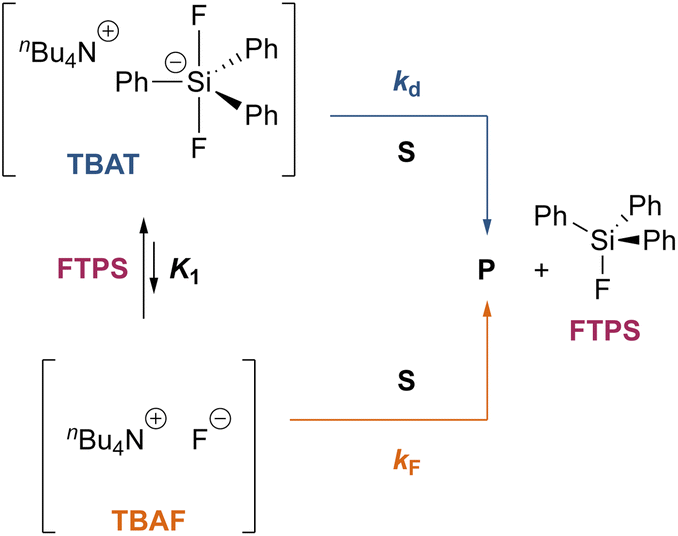
To gain quantitative insight into the exchange processes detected by 19F-CEST, we conducted magnetisation transfer NMR-spectroscopic analysis of TBAT solutions, using FTPS as the fluoride acceptor22 (see Section S7.1.2 of ESI,† for further discussion). The two limiting pathways for fluoride transfer are outlined in Schemes 2a and 2b. In the dissociative transfer pathway (process 1), endogenous TBAF is the reactive intermediate. The net rate coefficients are k1 for dissociation of TBAT into FTPS + TBAF, and k−1 for their recombination to generate TBAT; thus K1 = k1/k−1 ≪ 1. In the direct fluoride transfer pathway (process 2), TBAT undergoes a bimolecular elementary reaction with FTPS. The net rate coefficient for this process is k2, in both directions (ΔG° = 0).23
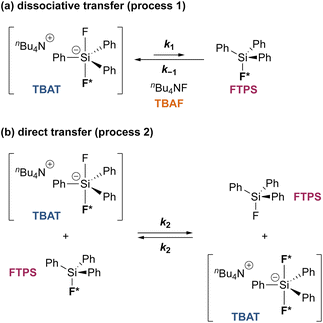 |
| | Scheme 2 Two limiting pathways of fluoride transfer from TBAT to FTPS. (a) Dissociative transfer via TBAF, where K1 = k1/k−1 ≪ 1. (b) Direct bimolecular transfer (the rate constants in each direction, k2, are identical, as ΔG° = 0). In each transfer pathway, examples of exchanging fluorine atoms are marked in bold with an asterisk. | |
The kinetic models below are derived using a discrete spin formalism,24 in which the spin-half nuclei (19F) within a large ensemble are opposed (19F∇) or aligned (19FΔ) to the +z-axis. In this formalism a species ‘S’ with n equivalent 19F nuclei has (n + 1) magnetic states: thus, FTPS and TBAF both have two states, while TBAT has three. The populations (N) of 19F∇ and 19FΔ in each species (N∇ and NΔ) dictate its fractional magnetisation, mS, eqn (5), and this is readily correlated with the integral of the measured NMR signal (MzS, eqn (6)).
| | 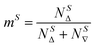 | (5) |
At equilibrium, the fractional magnetisation is unity (meqS = 1) and when fully inverted, it is zero (mS = 0). Rate laws that describe the change in fractional magnetisation (dm/dτ) can be derived using the spin formalism, with a steady-state approximation applied to the magnetisation of TBAF. The rate laws for mTBAT and mFTPS can then be solved analytically to give their temporal magnetisation when undergoing dissociative and/or direct transfer (and longitudinal relaxation), eqn (7) and (8), see Section S5.2.1 of ESI† for full derivation.
| | | mTBAT = c1e(β+γ)τ + c2e(β−γ)τ + 1 | (7) |
| | | mFTPS = c3e(β+γ)τ + c4e(β−γ)τ + 1 | (8) |
where:
| | 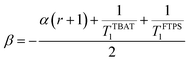 | (9) |
| | 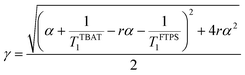 | (10) |
| | 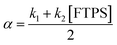 | (11) |
| | 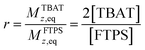 | (12) |
The coefficients for these equations are defined below, in which mTBAT0 and mFTPS0 are fractional magnetisations of the spins at τ = 0, the starting point of the relaxation occurring under the spin-formalism.
| | 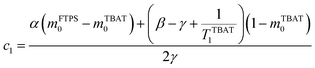 | (13) |
| | 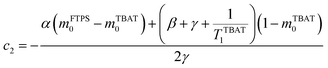 | (14) |
| | 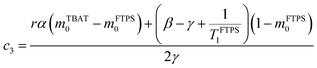 | (15) |
| |  | (16) |
Magnetisation transfer in THF and condition-sensitivity.
At 300 K in THF the 19F longitudinal relaxation of FTPS, TFTPS1 = 12.0 s, is concentration independent and significantly longer than that of TBAT. When the two species were mixed ([TBAT] = 52.5 mM, [FTPS] = 83.9 mM), both 19F spins relaxed with equal rates (Tobs1 = (2.45 ± 0.02) s), after non-selective inversion. This value is very close to the weighted combination, Tcalc1 = 2.54 s, of the separated spins (see eqn (S5.51) in Section S5.2.1 of ESI†) and indicative of rapid exchange of 19F at the longitudinal relaxation timescale.25 This allows use of a simplified kinetic model, eqn (17), to analyse the temporal fractional magnetisation of TBAT after selective inversion of FTPS, and thus extract α(r + 1).| | 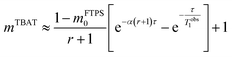 | (17) |
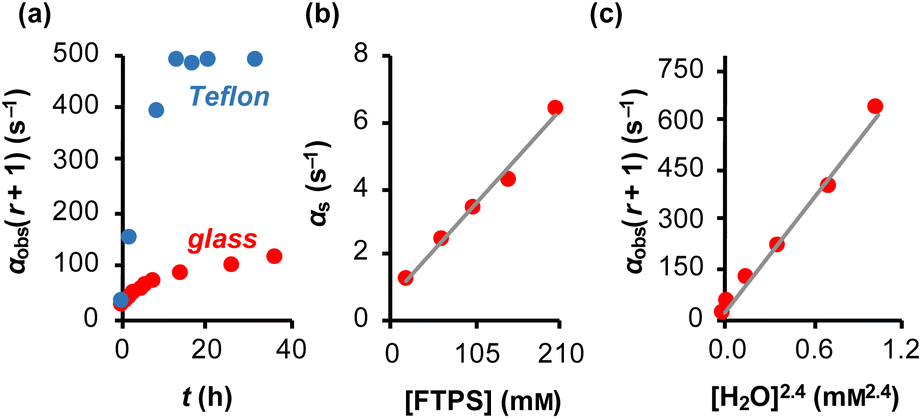
The magnitude of α(r + 1) depends on the rate coefficients of exchange, k1 and k2, and the TBAT and FTPS concentrations. However, the experimental values are sensitive to the conditions of sample preparation,26 and found to inherently increase with time.27 Conducting the reaction inside a dry Teflon insert located within a sealed NMR tube substantially exacerbated the problem, Fig. 3a. A range of tests were conducted to find additives which would afford temporal stability by sequestration of the unidentified acids/ions that were accelerating fluoride exchange between TBAT and FTPS. This eventually led to the use of a combination of 2,6-di-tert-butylpyridine (DTBP), as a hindered base, with 3 Å molecular sieves (3 Å-MS), as a passive dehydrating agent. DTBP and 3 Å-MS were not effective in isolation, see Section S5.2.4 of ESI† for further discussion. This allowed us to systematically explore the kinetics of the fluoride transfer from TBAT to FTPS under stabilised, anhydrous and non-acidic conditions,28 in THF.
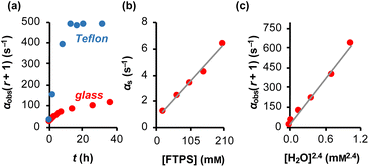 |
| | Fig. 3 Magnetisation transfer between TBAT and FTPS in THF (at 300 K). (a) An inherent increase of the magnetisation transfer rate with time in reactions conducted inside a dry and sealed glass NMR tube, and inside a dry Teflon insert (located within the NMR tube). (b) Under anhydrous, stabilised, non-acidic conditions, TBAT transfers fluoride to FTPS via parallel direct and dissociative mechanisms, with 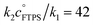 . αs/s−1 = 0.0273[FTPS/mM] + 0.656, R2 = 0.989. (c) Exchange between the species is catalysed by water, with the order in water estimated as approximately 2. αobs(r + 1)/s−1 = 577[H2O/mM]2.4 + 17.2, R2 = 0.989. . αs/s−1 = 0.0273[FTPS/mM] + 0.656, R2 = 0.989. (c) Exchange between the species is catalysed by water, with the order in water estimated as approximately 2. αobs(r + 1)/s−1 = 577[H2O/mM]2.4 + 17.2, R2 = 0.989. | |
Exchange pathways in the temporally-stabilised system.
Solutions comprising TBAT (101 mM), FTPS (22.6, 62.9, 101, 144, and 204 mM), DTBP (20 mM) and 1-fluoronaphthalene (internal standard) in THF were sealed in NMR tubes preloaded with 3 Å-MS beads. The exchange rates were then measured periodically by magnetisation transfer over a period of two weeks. The kinetic model, eqn (17), gave excellent fits to all 50 experimental datasets (the average RMSE/MTBATz,eq = 1%). The resulting plots of αobs against t were fitted to an empirical exponential decay, eqn (18), to allow evaluation of the underlying temporally-stabilised exchange rate, αs, see Table S5.10 in Section S5.2.5 of ESI† for fitted parameters and estimated errors.| | | αobs = (α0 − αs)e−kst + αs | (18) |
Evaluation of αs as a linear function of [FTPS], eqn (11), allows the rate coefficients for the two pathways to be determined as k1 = 1.3 s−1 and k2 = 55 M−1 s−1, Fig. 3b. Thus, under anhydrous, stabilised, non-acidic conditions in THF, TBAT transfers fluoride to FTPS via parallel direct and dissociative mechanisms, with 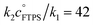 . Analysis of the non-stabilised anhydrous system indicated that the dissociative pathway is again a significant contributor to fluoride transfer from TBAT at low acceptor concentrations, see Sections S5.2.6 and S7.2.3 of ESI† for experimental details and mathematical considerations. The rate of 19F transfer from TBAT to FTPS in THF is markedly accelerated by exogenous water which may assist fluoride dissociation from TBAT via H-bonding or autoionisation.29 Analysis of αobs(r + 1) against [H2O], Fig. 3c, indicates that this predominantly involves interaction with a water dimer, or sequential reaction with two water molecules (see Section S5.2.4 of ESI†).
. Analysis of the non-stabilised anhydrous system indicated that the dissociative pathway is again a significant contributor to fluoride transfer from TBAT at low acceptor concentrations, see Sections S5.2.6 and S7.2.3 of ESI† for experimental details and mathematical considerations. The rate of 19F transfer from TBAT to FTPS in THF is markedly accelerated by exogenous water which may assist fluoride dissociation from TBAT via H-bonding or autoionisation.29 Analysis of αobs(r + 1) against [H2O], Fig. 3c, indicates that this predominantly involves interaction with a water dimer, or sequential reaction with two water molecules (see Section S5.2.4 of ESI†).
The exchange between non-stabilised TBAT and FTPS at 300 K in MeCN was found to be significantly more rapid than in THF. Indeed, the initial fractional magnetisation of TBAT (mTBAT0) was less than unity due to non-negligible exchange with FTPS during the 1.3 ms selective inversion pulse. In freshly prepared samples, a statistical distribution of 19F spins was achieved within 5 ms, and the exchange rate again increased with time. Use of DTBP + 3 Å-MS afforded a four-fold decrease in the initial magnetisation transfer rate in freshly prepared samples, accompanied by reduced line broadening, e.g., the 29Si satellites could be detected (see Fig S5.22c and S5.24a in Section S5.2.7 of ESI† for the appearance of the signals in the absence and in the presence of DTBP + 3 Å-MS, respectively). However, the stabilisation was short-lived and unsuitable for detailed kinetic interrogation. We thus switched to the use of less-reactive fluoride acceptors to facilitate study of the kinetics of transfer from TBAT in MeCN, vide infra.
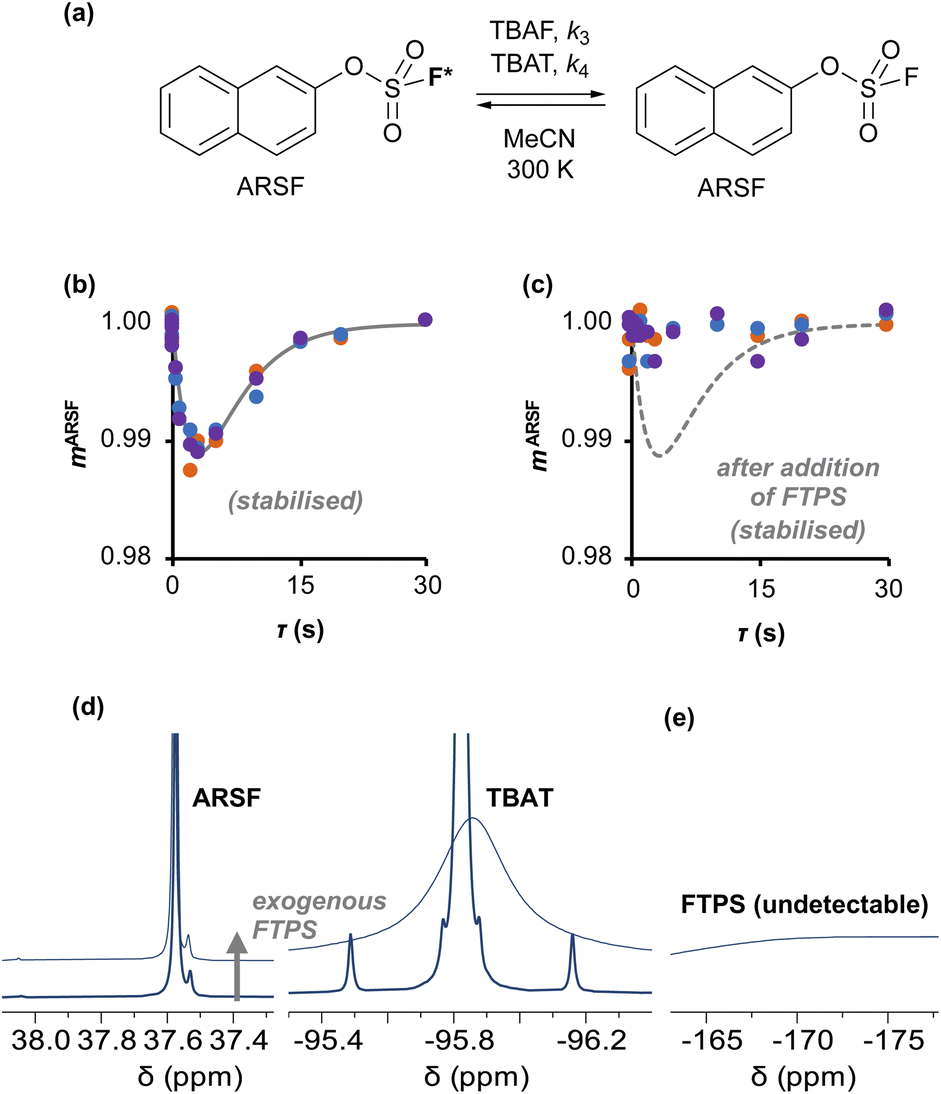
Aryl fluorosulfate 19F-exchange with TBAT in MeCN and inhibition by exogenous FTPS
Zheng et al. reported on exchange between aryl fluorosulfates and several fluoride sources, including TBAT in MeCN.13 They proposed that the ‘SuFEx’ process proceeds via an endergonic equilibrium association of fluoride at sulfur. To test for dissociative versus direct transfer pathways (processes 3 and 4, respectively; Fig. 4a), we selected 2-naphthalenyl fluorosulfate (ARSF) and derived a kinetic model for its magnetisation after selective inversion of TBAT, eqn (19); see Section S5.3.1 of ESI† for the derivation.| | 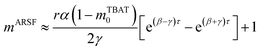 | (19) |
where:| | 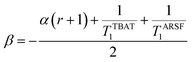 | (20) |
| | 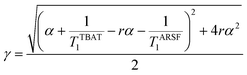 | (21) |
| | 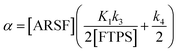 | (22) |
| | 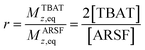 | (23) |
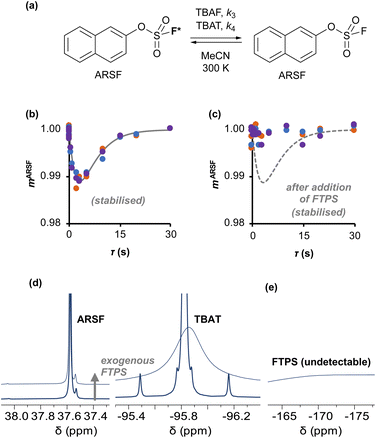 |
| | Fig. 4 (a) An approximation for concerted fluoride transfer between TBAT and ARSF in MeCN (at 300 K; exchanging fluorine atoms are marked with an asterisk for clarity). (b) Magnetisation transfer between the spins in MeCN; first measurement, prior to stabilisation, not shown. Solid line show fitted model. (c) Exchange inhibited by addition of a small amount of exogenous FTPS to the solution; dashed line shows fitted model from (b) for reference. (d) The 19F NMR signal of ARSF does not exhibit line broadening upon addition of exogenous FTPS, unlike that of TBAT. (e) The FTPS signal is too broad to detect. | |
Exchange was detected at 300 K and four magnetisation transfer measurements were performed on the same sample over a period of 3 hours immediately after its preparation (188 mM TBAT; 52 mM ARSF, MeCN; Section S5.3.3 of ESI†), Fig. 4b. The rate of exchange initially decreased then became stable and the kinetic model, eqn (19), correlated well with the experimental datasets, affording an average (stabilised) exchange rate constant αs = 1.4 × 10−3 s−1.
In the above experiment the endogenous FTPS is at very low concentration (K1 ≪ 1). However, as evident from eqn (22), while FTPS does not affect the direct transfer pathway (k4; TBAT), α is a decreasing function of [FTPS] for the dissociative pathway (k3; TBAF). Magnetisation transfer measurements were thus repeated on the sample after addition of FTPS, ∼4 mM. The rate of 19F-transfer between TBAT and ARSF was inhibited to below the qualitative detection limit, Fig. 4c; individual mARSF profiles before and after FTPS addition are presented in Fig. S5.28 in Section S5.3.3 of ESI.† Fitting the data to eqn (19) gave an average value of αs = 8.1 × 10−5 s−1 across all four runs, i.e., 94% inhibition of the rate of fluoride transfer. Furthermore, comparison of the 19F NMR spectra before and after addition of FTPS (Fig. 4d) showed that while the ARSF signal was unaffected, the TBAT signal exhibited very significant line broadening, and the FTPS signal itself was broadened to an extent which rendered it undetectable, Fig. 4e. These features indicate that fluoride exchange between TBAT and aryl fluorosulfates is predominantly via the dissociative pathway, with an estimated standard state partitioning 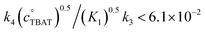 for 2-naphthalenyl fluorosulfate (ARSF).
for 2-naphthalenyl fluorosulfate (ARSF).
TBAT-mediated aryl fluorosulfate decomposition.
Rapid defluorosulfation of phenyl fluorosulfate by nominally anhydrous TBAF was reported by Zheng et al.13 We detected analogous decomposition of ARSF by TBAT in MeCN at 335 K, as indicated by the growth of two low intensity signals identified as [FSO2O−][nBu4N+] and SO2F2.30 The third by-product of the decomposition is nominally FTPS, but this is not observed in the spectra, even on cooling to 233 K, possibly due to the rapid exchange line-broadening.31 However, a series of magnetisation transfer measurements (see Section S5.3.4 of ESI† for details) showed a progressive decrease in the rate of 19F exchange between TBAT and ARSF, arising from the impact of the growing FTPS concentration on the dissociative pathway, eqn (24) and Fig. 5a.| | 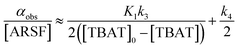 | (24) |
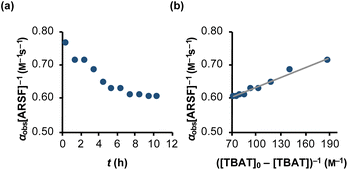 |
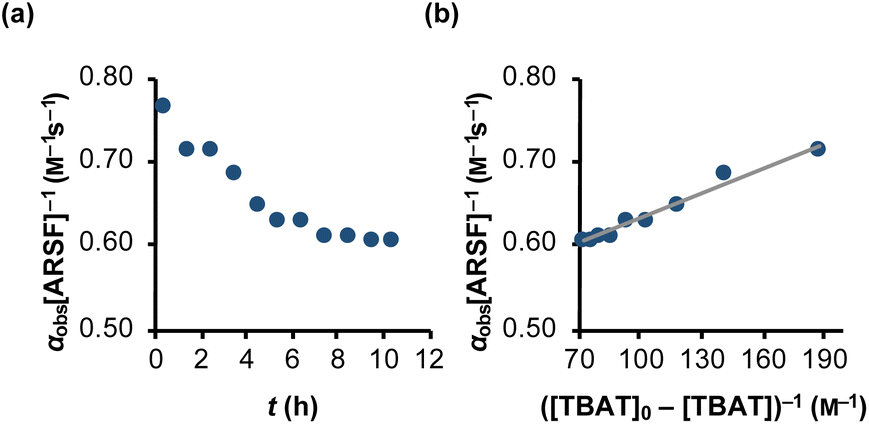
| | Fig. 5 Temporal evolution of the rate of magnetisation transfer between TBAT and ARSF in MeCN (at 335 K). (a) Exchange between the spins is progressively inhibited due to accumulating FTPS, formed from decomposing TBAT, indicating at least partially dissociative character to the exchange. (b) Fitting of the model of eqn (24) showed that fluoride is transferred to ARSF via both dissociative and direct transfer pathways, with k3/k4 = 43. αobs[ARSF]−1/M−1 s−1 = 1 × 10−3 ([TBAT]0-[TBAT])−1 /M−1 + 0.53; R2 = 0.974. | |
The experimental data corresponding to αobs, [ARSF], and [TBAT] was fitted against eqn (24) and Fig. 5b, to estimate k3/k4 = 43, and thus that TBAF is a significantly more potent direct fluoride donor than TBAT.
FTPS inhibition of the reaction of benzyl bromide with TBAT
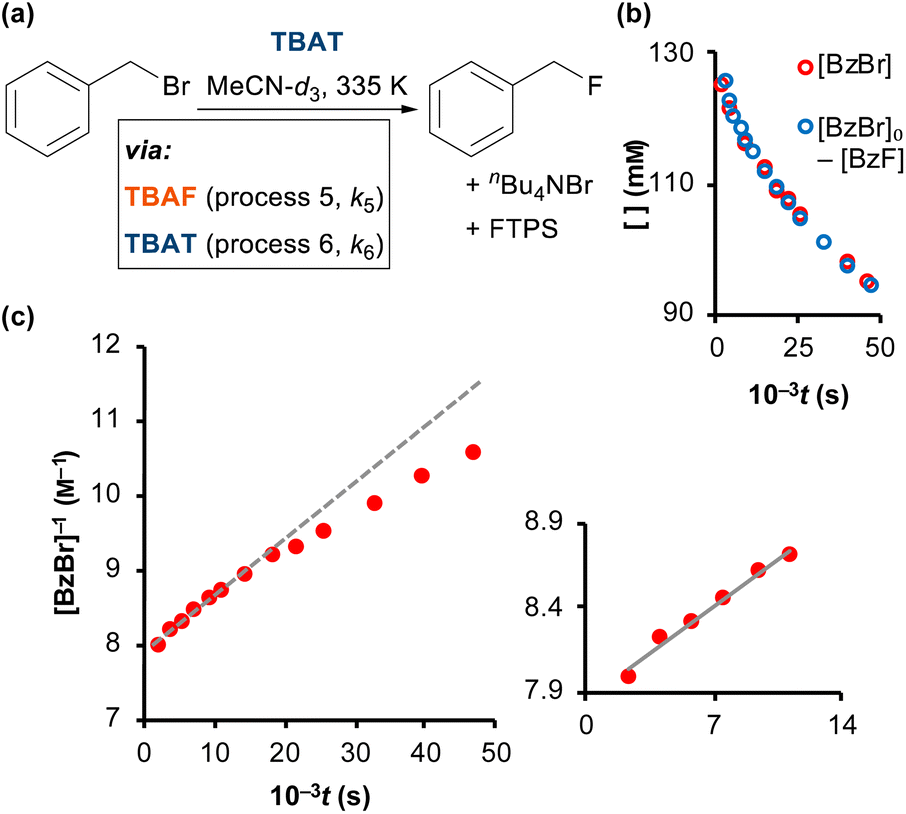
As reported by Mąkosza and Bujok, TBAT reacts with benzyl bromide (BzBr) at elevated temperatures in MeCN, Fig. 6a.12 However, unlike the degenerate exchange processes explored thus far, vide supra, the process stoichiometrically co-generates FTPS. Thus, fluoride transfer to BzBr that proceeds via a dissociative pathway (process 5) will undergo progressive inhibition by FTPS. Conversely, if the fluoride transfer proceeds predominantly, or exclusively, via direct transfer from the difluorotriphenylsilicate anion (process 6), then there should be no significant inhibition by accumulating, or exogenous, FTPS. The reaction of [TBAT]0 = 129 mM with [BzBr]0 = 131 mM was readily analysed in situ by 1H/19F NMR spectroscopy at 335 K in MeCN-d3, under N2; with the decay in BzBr mirroring the growth of BzF, Fig. 6b; see Section S6.1 of ESI† for details.
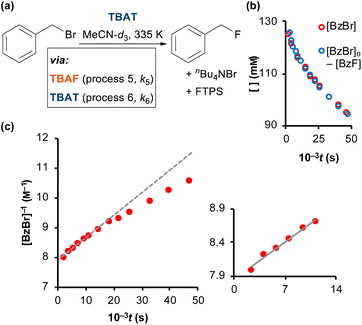 |
| | Fig. 6 (a) Fluorination of benzyl bromide with TBAT in MeCN-d3 (at 335 K). (b) The reaction was followed by 1H and 19F NMR spectroscopy, and the decay of BzBr mirrored the growth of BzF. (c) The reaction exhibits progressive inhibition, see dashed line, due to accumulating FTPS. Inset shows linear second-order correlation over initial 30% conversion: 1/[BzBr]/M−1 = 9 × 10−5t/s + 1/[BzBr]0/M−1; R2 = 0.985. | |
Standard graphical analysis, Fig. 6c, afforded an initial pseudo second-order rate coefficient of 9 × 10−5M−1 s−1. The curvature in the reciprocal plot is consistent with progressive inhibition of the reaction. Graphical analysis of an identical reaction conducted in the presence of one equivalent of exogenous FTPS (140 mM), afforded a pseudo second-order rate coefficient of 4 × 10−5M−1 s−1, and without any evident progressive inhibition (see Section S6.2 of ESI†). These results indicate that the reaction of TBAT with BzBr at 335 K in MeCN initially proceeds with significant flux via both the dissociative and direct transfer pathways.
Conclusions
The speciation of tetra-n-butylammonium difluorotriphenylsilicate ([Ph3SiF2−][nBu4N+], TBAT) and the mechanism of its reaction with three fluoride acceptors has been studied in detail by a range of 1H/19F NMR-spectroscopic and kinetic methods. A combination of 1H 1D NOESY and 1H diffusion analysis showed the Ph3SiF2− and nBu4N+ ions to be strongly paired in THF-d8, and in MeCN-d3, but separated in DMSO-d6. A series of 19F CEST NMR experiments identified that the ion-pairs undergo endergonic interconversion with fluorotriphenylsilane (FTPS) and tetra-n-butylammonium fluoride (TBAF), both of which are below the detection limit in standard 19F pulse-acquire NMR spectra. TBAF undergoes further equilibria and decomposition leading, inter alia, to the formation of tetra-n-butylammonium bifluoride (TBABF).
The kinetics of degenerate fluoride transfer from TBAT to FTPS, and to 2-naphthalenyl fluorosulfate (ARSF), were then studied by 19F magnetisation transfer. The rate of exchange between TBAT and FTPS in THF is much more rapid than the longitudinal relaxation timescale of the spins, increases with time, and is profoundly accelerated by traces of water. Addition of a combination of 2,6-di-tert-butylpyridine (DTBP) and 3 Å molecular sieves (3 Å-MS) to the samples prepared in gas-tight sealed NMR tubes under N2 afforded a stabilised, anhydrous, non-acidic medium suitable for detailed and systematic kinetic analysis. The exchange rates were analysed by magnetisation transfer at various concentrations of FTPS, and the kinetics characterised using a model that includes direct (k2) transfer from [Ph3SiF2−] and indirect transfer (k1) via TBAF. The standard state partitioning was estimated to be 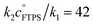 , and thus at 1 M FTPS the transfer is near-exclusively direct (>97%), while at 1 mM FTPS it is near-exclusively dissociative (>95%). The analogous system in MeCN undergoes very rapid fluoride exchange, approaching the limit of practicality of the measurement method, and is only transiently stabilised by 2,6-di-tert-butylpyridine and 3 Å molecular sieves.
, and thus at 1 M FTPS the transfer is near-exclusively direct (>97%), while at 1 mM FTPS it is near-exclusively dissociative (>95%). The analogous system in MeCN undergoes very rapid fluoride exchange, approaching the limit of practicality of the measurement method, and is only transiently stabilised by 2,6-di-tert-butylpyridine and 3 Å molecular sieves.
Exchange of fluoride between TBAT and ARSF was analysed by magnetisation transfer in MeCN at 300 K, again using a model that includes direct (k4) transfer from [Ph3SiF2−] and indirect transfer (k3) via TBAF. The exchange rate is very much slower than between TBAT and FTPS, and approaches the longitudinal relaxation timescale of the spins. Moreover, the addition of FTPS (4 mM) results in the rate of transfer between TBAT and ARSF being attenuated to the limits of detection. The standard state partitioning was estimated to be 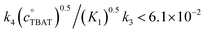 , and thus the direct exchange pathway would only become favoured over the dissociative pathway at unachievable TBAT concentrations (>67 M). On heating to 335 K the system undergoes slow decomposition, resulting in co-generation of FTPS, [FSO2O−][nBu4N+] and SO2F2, and gradual inhibition of the rate of fluoride exchange between TBAT and ARSF. The reaction of benzyl bromide (BzBr) with TBAT in MeCN-d3 at 335 K proceeds with non-degenerate fluoride transfer and is progressively and exogenously inhibited by FTPS, again due to competing dissociative and direct transfer mechanisms. The initial standard state partitioning in this case is estimated to be
, and thus the direct exchange pathway would only become favoured over the dissociative pathway at unachievable TBAT concentrations (>67 M). On heating to 335 K the system undergoes slow decomposition, resulting in co-generation of FTPS, [FSO2O−][nBu4N+] and SO2F2, and gradual inhibition of the rate of fluoride exchange between TBAT and ARSF. The reaction of benzyl bromide (BzBr) with TBAT in MeCN-d3 at 335 K proceeds with non-degenerate fluoride transfer and is progressively and exogenously inhibited by FTPS, again due to competing dissociative and direct transfer mechanisms. The initial standard state partitioning in this case is estimated to be 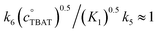 .
.
Overall, the investigation shows that both dissociative and direct pathways contribute to fluoride transfer from TBAT to fluoride acceptors in THF, and in MeCN. The rate of transfer, and the pathway partitioning, are strongly dependent on the solvent, the presence of water, the affinity of the substrate to fluoride, and the concentrations of TBAT, the substrate and FTPS. The most common application of TBAT is for stoichiometric non-degenerate fluoride transfer. Under these conditions, the reaction efficiency (rate) and selectivity (e.g., addition versus elimination) will be dependent on the pathways and their partitioning. The situation can be generalised for a substrate, ‘S’, undergoing stoichiometric reaction with TBAT via direct (kd) or dissociative (kF) pathways, to generate product, ‘P’, plus FTPS, Scheme 3 (see Section S7.2.4 of ESI†). Since K1 is small, soon after the start of reaction the rate and pathway fractionations (fd and fF) can be approximated by eqn (25) and (26), respectively.
| | 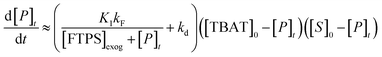 | (25) |
| | 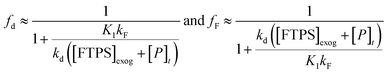 | (26) |
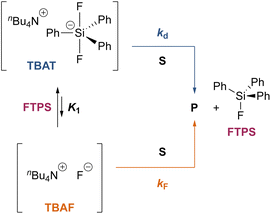 |
| | Scheme 3 Generic processes involving a substrate ‘S’ undergoing stoichiometric reaction with TBAT to generate product ‘P’ via direct (kd) and dissociative (kF) pathways. | |
For systems where the direct pathway is desired (fd ≫ fF), then high substrate concentrations, together with the use of exogenous FTPS, will be beneficial. Conversely, where the dissociative pathway is desired (fF ≫ fd), then low concentrations of both substrate and TBAT will be beneficial, albeit at the cost of significantly attenuated rate.
Data availability
Experimental data, NMR spectra, derivation of kinetic models, and further discussion and analysis can be found in the ESI.†
Author contributions
MMK conducted the experimental work. MMK and GCL-J analysed the data. All authors contributed to the preparation of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank CRITICAT-EPSRC-CDT (scholarship to MMK) and the EPSRC Programme Grant “Boron: Beyond the Reagent” (EP/W007517) for support.
Notes and references
-
(a) A. S. Pilcher, H. L. Ammon and P. DeShong, J. Am. Chem. Soc., 1995, 117, 5166–5167 CrossRef CAS;
(b) C. J. Handy, Y. F. Lam and P. DeShong, J. Org. Chem., 2000, 65, 3542–3543 CrossRef CAS PubMed.
-
(a) V. Gracias, G. L. Milligan and J. Aubé, J. Org. Chem., 1996, 61, 10–11 CrossRef CAS;
(b) V. Gurvich, H. Y. Kim, R. P. Hodge, C. M. Harris and T. M. Harris, Nucleosides Nucleotides, 1999, 18, 2327–2333 CrossRef CAS PubMed;
(c) Y. Kasai, N. Matsumori, H. Ueno, K. Nonomura, S. Yano, M. Michio and T. Oishi, Org. Biomol. Chem., 2011, 9, 1437–1442 RSC;
(d) B. B. Liau, B. C. Milgram and M. D. Shair, J. Am. Chem. Soc., 2012, 134, 16765–16772 CrossRef CAS PubMed;
(e) E. Lee, J. M. Hooker and T. Ritter, J. Am. Chem. Soc., 2012, 134, 17456–17458 CrossRef CAS;
(f) J. Naidoo, H. De Jesus-Cortes, P. Huntington, S. Estill, L. K. Morlock, R. Starwalt, T. J. Mangano, N. S. Williams, A. A. Pieper and J. M. Ready, J. Med. Chem., 2014, 57, 3746–3754 CrossRef CAS PubMed;
(g) J. Y. Yang, X. H. Xu and F. L. Qing, J. Fluorine Chem., 2016, 186, 45–51 CrossRef CAS;
(h) R. F. Gamache, C. Waldmann and J. M. Murphy, Org. Lett., 2016, 18, 4522–4525 CrossRef CAS PubMed;
(i) P. Adler, C. J. Teskey, D. Kaiser, M. Holy, H. H. Sitte and N. Maulide, Nat. Chem., 2019, 11, 329–334 CrossRef CAS PubMed;
(j) C. D. Vogt, A. G. Bart, R. Yadav, E. E. Scott and J. Aubé, Org. Biomol. Chem., 2021, 19, 7664–7669 RSC.
-
(a) O. Tamura, A. Toyao and H. Ishibashi, Synlett, 2002, 1344–1346 CrossRef CAS;
(b) A. E. Mattson, A. M. Zuhl, T. E. Reynolds and K. A. Scheldt, J. Am. Chem. Soc., 2006, 128, 4932–4933 CrossRef CAS;
(c) M. J. Campbell, P. D. Pohlhaus, G. Min, K. Ohmatsu and J. S. Johnson, J. Am. Chem. Soc., 2008, 130, 9180–9181 CrossRef CAS PubMed;
(d) J. C. Killen, L. C. Axford, S. E. Newberry, T. J. Simpson and C. L. Willis, Org. Lett., 2012, 14, 4194–4197 CrossRef CAS PubMed;
(e) X. Frogneux, N. von Wolff, P. Thuéry, G. Lefèvre and T. Cantat, Chem. - Eur. J., 2016, 22, 2930–2934 CrossRef CAS PubMed;
(f) M. E. McCallum, G. M. Smith, T. Matsumaru, K. Kong, J. A. Enquist and J. L. Wood, J. Antibiot., 2016, 69, 331–336 CrossRef CAS;
(g) N. von Wolff, J. Char, X. Frogneux and T. Cantat, Angew. Chem., Int. Ed., 2017, 56, 5616–5619 CrossRef CAS.
- For selected recent examples of the use of TBAT in in situ generation of reactive benzyne intermediates, see:
(a) S. M. Bronner, D. Lee, V. Bacauanu and P. Cyr, Synlett, 2017, 28, 799–804 CrossRef CAS;
(b) T. Ikawa, S. Masuda, H. Nakajima and S. Akai, J. Org. Chem., 2017, 82, 4242–4253 CrossRef CAS PubMed;
(c) B. Cheng, B. Bao, B. Zu, X. Duan, S. Duan, Y. Li and H. Zhai, RSC Adv., 2017, 7, 54087–54090 RSC;
(d) Y. Okugawa, Y. Hayashi, S. Kawauchi, K. Hirano and M. Miura, Org. Lett., 2018, 20, 3670–3673 CrossRef CAS;
(e) A. Shamsabadi and V. Chudasama, Chem. Commun., 2018, 54, 11180–11183 RSC;
(f) H. Ryu, J. Seo and H. M. Ko, J. Org. Chem., 2018, 83, 14102–14109 CrossRef CAS;
(g) B. Cheng, Y. Li, B. Zu, T. Wang, R. Wang, Y. Li and H. Zhai, Tetrahedron, 2019, 75, 1–6 CrossRef;
(h) J. Seo, D. Kim and H. M. Ko, Adv. Synth. Catal., 2020, 362, 2739–2743 CrossRef CAS;
(i) Q. Wang, Y. An, G. Du, Z. H. Cai, B. Dai and L. He, J. Org. Chem., 2020, 85, 14210–14218 CrossRef CAS;
(j) K. Donnelly and M. Baumann, J. Org. Chem., 2022, 87, 8279–8288 CrossRef CAS PubMed.
- For selected recent examples of the use of TBAT as an initiator involving Si–C cleavage, see:
(a) M. Fuertes, C. Masdeu, E. Martin-Encinas, A. Selas, G. Rubiales, F. Palacios and C. Alonso, Molecules, 2020, 25, 3252 CrossRef CAS PubMed;
(b) A. Ben Jamaa, M. Latrache, E. Riguet and F. Grellepois, J. Org. Chem., 2020, 85, 9585–9598 CrossRef CAS;
(c) K. Kikushima, H. Koyama, K. Kodama and T. Dohi, Molecules, 2021, 26, 1365–1378 CrossRef CAS PubMed;
(d) R. Shimazumi, K. Morita, T. Yoshida, K. Yasui and M. Tobisu, Synthesis, 2021, 53, 3037–3044 CrossRef CAS;
(e) Y. Cai, W. Zhu, S. Zhao, C. Dong, Z. Xu and Y. Zhao, Org. Lett., 2021, 23, 3546–3551 CrossRef CAS;
(f) N. Hara, N. Uemura and Y. Nakao, Chem. Commun., 2021, 57, 5957–5960 RSC;
(g) S. Keereewan, C. Kuhakarn, P. Leowanawat, S. Saithong, V. Reutrakul and D. Soorukram, J. Org. Chem., 2022, 87, 15963–15985 CrossRef CAS PubMed . For selected early examples of the use of TBAT as an initiator involving Si–C cleavage, see: ;
(h) A. S. Pilcher and P. DeShong, J. Org. Chem., 1996, 61, 6901–6905 CrossRef CAS PubMed;
(i) E. DiMauro and A. J. Fry, Tetrahedron Lett., 1999, 40, 7945–7949 CrossRef CAS;
(j) G. K. S. Prakash, M. Mandal and G. A. Olah, Angew. Chem., Int. Ed., 2001, 40, 589–590 CrossRef CAS;
(k) G. K. S. Prakash, M. Mandal and G. A. Olah, Org. Lett., 2001, 3, 2847–2850 CrossRef CAS;
(l) G. K. Friestad and H. Ding, Angew. Chem., Int. Ed., 2001, 40, 4491–4493 CrossRef CAS;
(m) G. K. S. Prakash, M. Mandal and G. A. Olah, Synlett, 2001, 77–78 CrossRef CAS.
-
(a) E. D. Soli and P. DeShong, J. Org. Chem., 1999, 64, 9724–9726 CrossRef CAS;
(b) S. Large, N. Roques and B. R. Langlois, J. Org. Chem., 2000, 65, 8848–8856 CrossRef CAS;
(c) H. Fujioka, T. Okitsu, T. Ohnaka, R. Li, O. Kubo, K. Okamoto, Y. Sawama and Y. Kita, J. Org. Chem., 2007, 72, 7898–7902 CrossRef CAS;
(d) L. Finck and M. Oestreich, Chem. - Eur. J., 2021, 27, 11061–11064 CrossRef CAS.
- For selected examples of the use of TBAT as an initiator involving Si–O cleavage, see:
(a) D. C. Behenna, J. T. Mohr, N. H. Sherden, S. C. Marinescu, A. M. Harned, K. Tani, M. Seto, S. Ma, Z. Novák, M. R. Krout, R. M. McFadden, J. L. Roizen, J. A. Enquist, D. E. White, S. R. Levine, K. V. Petrova, A. Iwashita, S. C. Virgil and B. M. Stoltz, Chem. - Eur. J., 2011, 17, 14199–14223 CrossRef CAS;
(b) B. Lecachey, L. Palais, B. de Courcy, S. Bouauli, M. Durandetti, H. Oulyadi, A. Harisson-Marchand, J. Maddaluno, H. Gérard, E. Vrancken and J. M. Campagne, Chem.–Eur. J., 2021, 27, 7942–7950 CrossRef CAS PubMed;
(c) T. Kawatsu, S. Kataoka, N. Fukaya, J. C. Choi, K. Sato and K. Matsumoto, ACS Omega, 2021, 6, 12853–12857 CrossRef CAS;
(d) Z. Nelson, N. A. Romero, J. Tiepelt, M. Baldo and T. M. Swager, Macromolecules, 2021, 54, 6698–6704 CrossRef CAS;
(e) J. Takino, A. Kotani, T. Ozaki, W. Peng, J. Yu, Y. Guo, S. Mochizuki, K. Akimitsu, M. Hashimoto, T. Ye, A. Minami and H. Oikawa, Angew. Chem., Int. Ed., 2021, 60, 23403–23411 CrossRef CAS.
-
N. S. Simpkins, F. E. Nytko, P. DeShong, M. Vayer and N. Maulide, in Encyclopedia of Reagents for Organic Synthesis, Wiley, 2020, DOI:10.1002/047084289X.rn00469.pub3.
- TBAT has also been used as a source of the phenyl synthon in cross-coupling reactions, which is beyond the scope of this study, see:
(a) M.-R. Brescia and P. DeShong, J. Org. Chem., 1998, 63, 3156–3157 CrossRef CAS;
(b) M. E. Mowery and P. DeShong, J. Org. Chem., 1999, 64, 3266–3270 CrossRef CAS PubMed;
(c) M. E. Hoke, M. Brescia, S. Bogaczyk, P. DeShong, B. W. King and M. T. Crimmins, J. Org. Chem., 2002, 67, 327–335 CrossRef CAS.
- N. Wu, B. Wahl, S. Woodward and W. Lewis, Chem.–Eur. J., 2014, 20, 7718–7724 CrossRef CAS PubMed.
- R. Schwesinger, R. Link, P. Wenzl and S. Kossek, Chem.–Eur. J., 2005, 12, 438–445 CrossRef CAS PubMed.
- M. Mąkosza and R. Bujok, J. Fluorine Chem., 2005, 126, 209–216 CrossRef.
- Q. Zheng, H. Xu, H. Wang, W. G. H. Du, N. Wang, H. Xiong, Y. Gu, L. Noodleman, K. B. Sharpless, G. Yang and P. Wu, J. Am. Chem. Soc., 2021, 143, 3753–3763 CrossRef CAS.
-
(a) C. P. Johnston, T. H. West, R. E. Dooley, M. Reid, A. B. Jones, E. J. King, A. G. Leach and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2018, 140, 11112–11124 CrossRef CAS;
(b) A. García-Domínguez, T. H. West, J. J. Primozic, K. M. Grant, C. P. Johnston, G. G. Cumming, A. G. Leach and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2020, 142, 14649–14663 CrossRef;
(c) A. García-Domínguez, P. H. H. De Oliveira, G. T. Thomas, A. R. Sugranyes and G. C. Lloyd-Jones, ACS Catal., 2021, 11, 3017–3025 CrossRef;
(d) A. García-Domínguez, A. G. Leach and G. C. Lloyd-Jones, Acc. Chem. Res., 2022, 55, 1324–1336 CrossRef PubMed;
(e) H. B. Minshull and G. C. Lloyd-Jones, J. Org. Chem., 2023, 88, DOI:10.1021/acs.joc.3c02219.
- O. Planas, V. Peciukenas, M. Leutzsch, N. Nöthling, D. A. Pantazis and J. Cornella, J. Am. Chem. Soc., 2022, 144, 14489–14504 CrossRef CAS.
- Although a different difluorotriorganosilicate, computational calculations were provided for direct fluoride transfer from difluorotrimethylsilicate (Me3SiF2−) to aryldimethylvinylsilane (ArMe2SiCH
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2) in THF. The transition states were located at ΔG‡ = +92 kJ mol−1 for phenyl and 2-pyridyl aryl substituents; see ref. 3e.
CH2) in THF. The transition states were located at ΔG‡ = +92 kJ mol−1 for phenyl and 2-pyridyl aryl substituents; see ref. 3e.
- R. Damrauer and S. E. Danahey, Organometallics, 1986, 5, 1490–1494 CrossRef CAS.
- Increasing the concentration of TBAT maximises the chance of observing any low-concentration spins in exchange with it in CEST. Additionally, the longitudinal relaxation time constant of TBAT decreases with concentration in both solvents hence allowing for more scans in a given time period. T1TBAT in the saturated solutions were determined, immediately prior to the corresponding CEST experiments, as 1.10 s in THF and 3.60 s in MeCN; see Section S3.3.1 of ESI.†.
- The 19F chemical shift of anhydrous TBAF is −86 ppm in THF and −72 ppm in MeCN-d3, and the signal moves upfield as the water content in the solution is increased; see: H. Sun and S. G. DiMagno, J. Am. Chem. Soc., 2005, 127, 2050–2051 CrossRef CAS . The upfield extreme of the chemical shift of TBAF in these two (or any other) solvents is not known..
- R. K. Sharma and J. L. Fry, J. Org. Chem., 1983, 48, 2112–2114 CrossRef CAS.
- FTPS was detected in a 19F pulse-acquire NMR spectrum of the solution in THF (∼5 mol% FTPS by integration against TBAT). In MeCN, the FTPS signal was just above qualitative detection limit in the 19F pulse-acquire NMR spectrum, due to very significant line broadening. Application of baseline correction renders the signal undetectable.
- For selected examples of tetravalent silanes acting as fluoride acceptors, see ref. 5c–f, 5i, 5l, 10, and 14c.
- The values of k2 and k1 correspond to the net rate coefficients; the appropriate coefficients per (chemically equivalent) exchangeable fluoride in TBAT are k2/2 and k1/2.
- F. W. Dahlquist, K. J. Longmuir and R. B. D. Vernet, J. Magn. Reson., 1975, 17, 406–410 CAS.
- J. B. Lambert and J. W. Keepers, J. Magn. Reson., 1980, 38, 233–244 CAS.
- This observation is supported by previous findings in NMR spectroscopic studies of the dynamic properties of fluoroorganosilicates; see:
(a) F. Klanberg and E. L. Muetterties, Inorg. Chem., 1968, 7, 155–160 CrossRef CAS;
(b) R. K. Marat and A. F. Janzen, Can. J. Chem., 1977, 55, 1167–1174 CrossRef CAS.
- Control experiments confirmed that this did not arise from ingress of moisture, nor was it caused by exposure of the NMR tube to light, or through any detectable change in the TBAT or FTPS concentrations, although both signals exhibited progressive line broadening; see Fig. S5.8a in Section S5.2.4 of ESI† for the appearance of the FTPS signal. The inherent temporal rate increase in the system was negligible compared to the rate increase resulting from added water; see Fig. S5.14 in Section S5.2.4 of ESI† for details.
- The decomposition of TBAT has been shown by DeShong to be accelerated by trace amounts of acid, see ref. 1b.
- H. Gunaydin and K. N. Houk, J. Am. Chem. Soc., 2008, 130, 15232–15233 CrossRef CAS PubMed.
- Fluorosulfates with electron-poor aryl moieties are susceptible to nucleophilic aromatic substitution by fluoride; see: S. D. Schimler, R. D. J. Froese, D. C. Bland and M. S. Sanford, J. Org. Chem., 2018, 83, 11178–11190 CrossRef CAS PubMed . Under the conditions explored herein, no fluoronaphthalenes were detected, see Section S5.3.4 of ESI† for details..
- FTPS was not observed in the 19F NMR spectrum of the solution of TBAT and ARSF in MeCN incubated at 335 K for 15 h, even at 233 K. Similarly, in the 19F NMR spectra at 233 K, calculations of the mass balance of decomposition of TBAT and ARSF with respect to Si and F gave vastly different expected concentrations of FTPS; see Section S5.3.4 of ESI† for details..
|
| This journal is © The Royal Society of Chemistry 2024 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Allan J. B.
Watson
a,
Allan J. B.
Watson
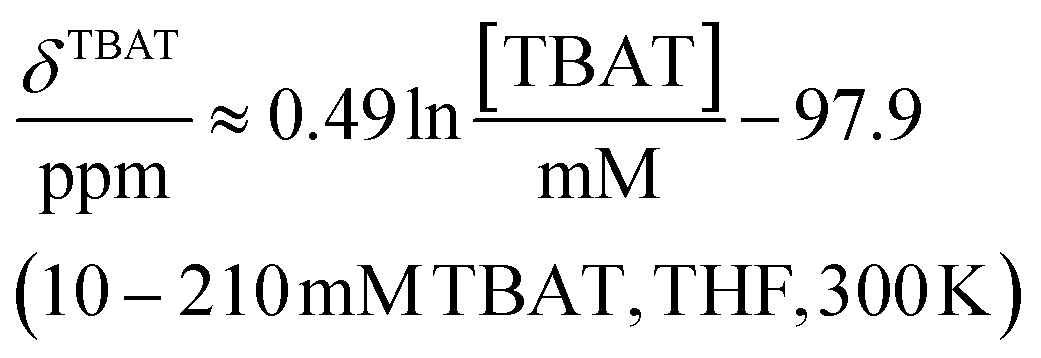
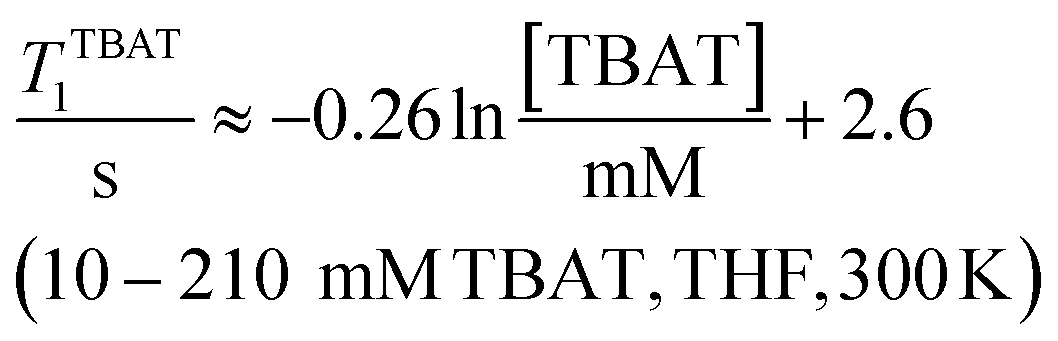
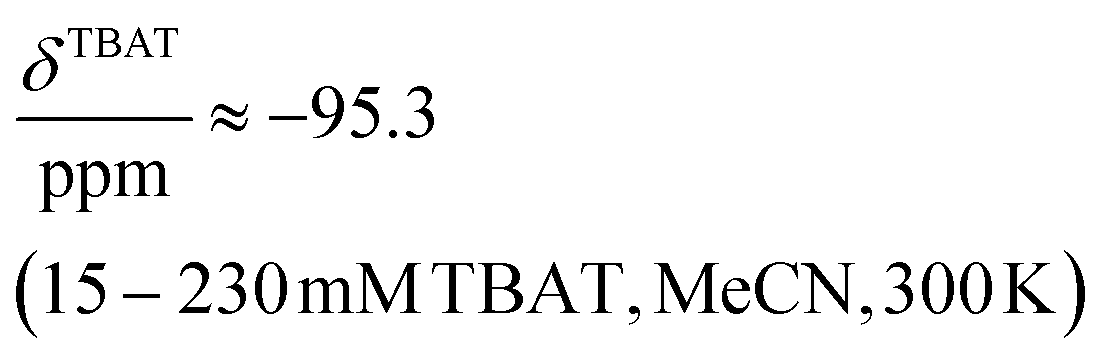
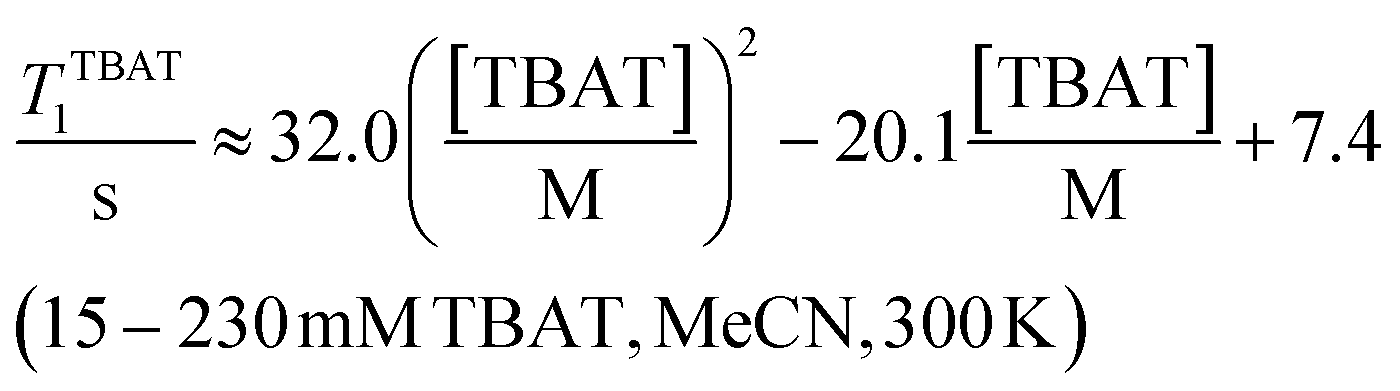
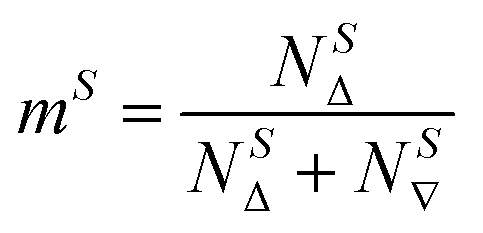
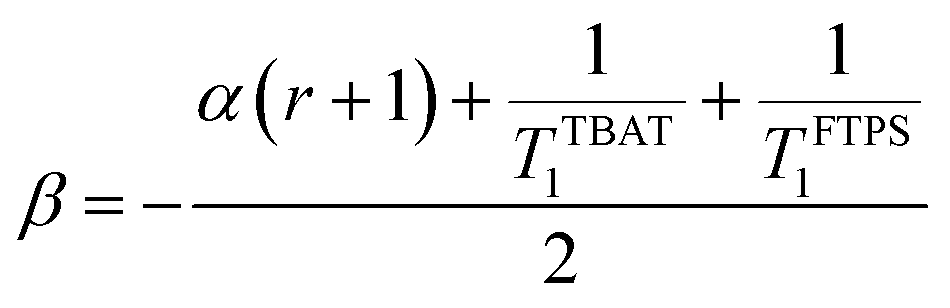
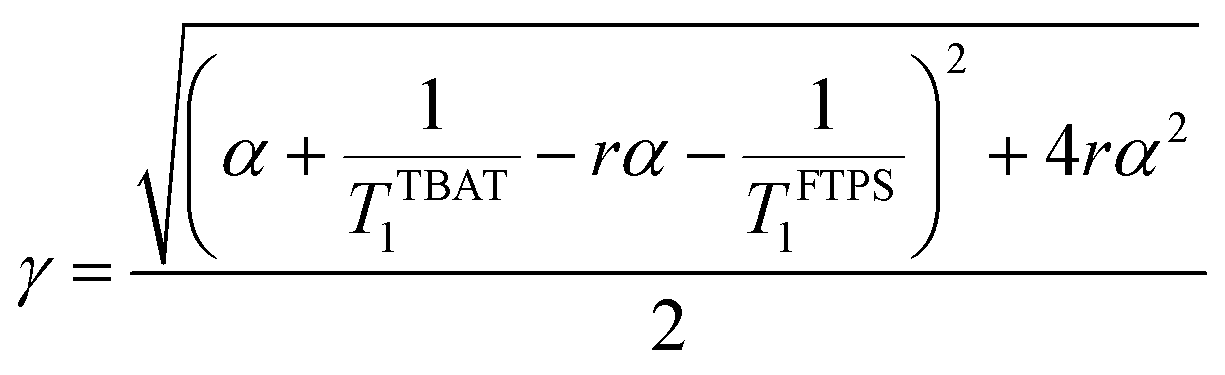
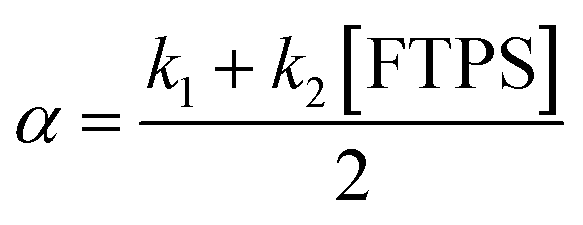
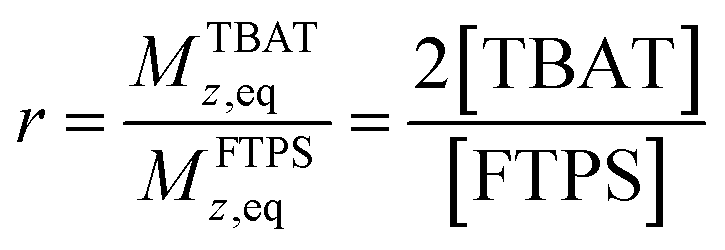
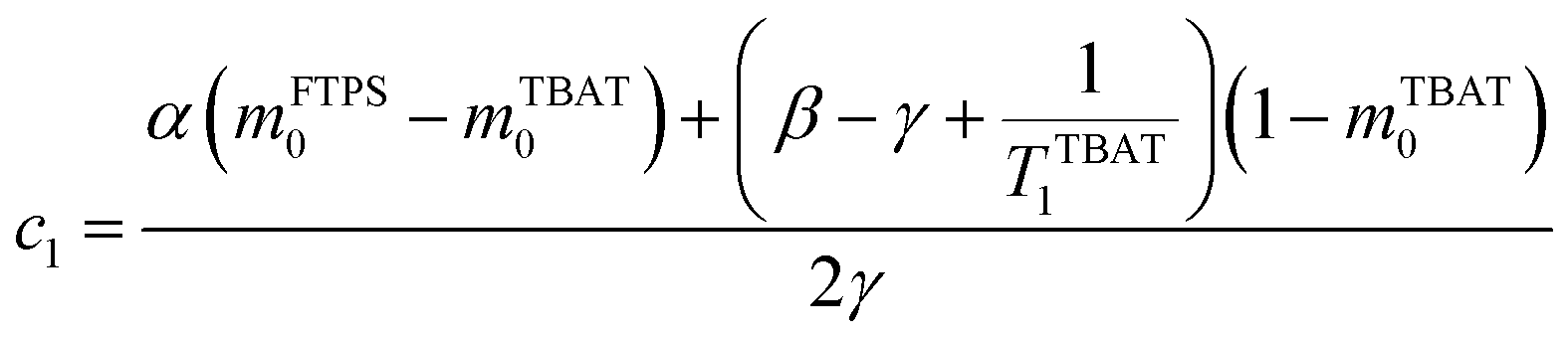
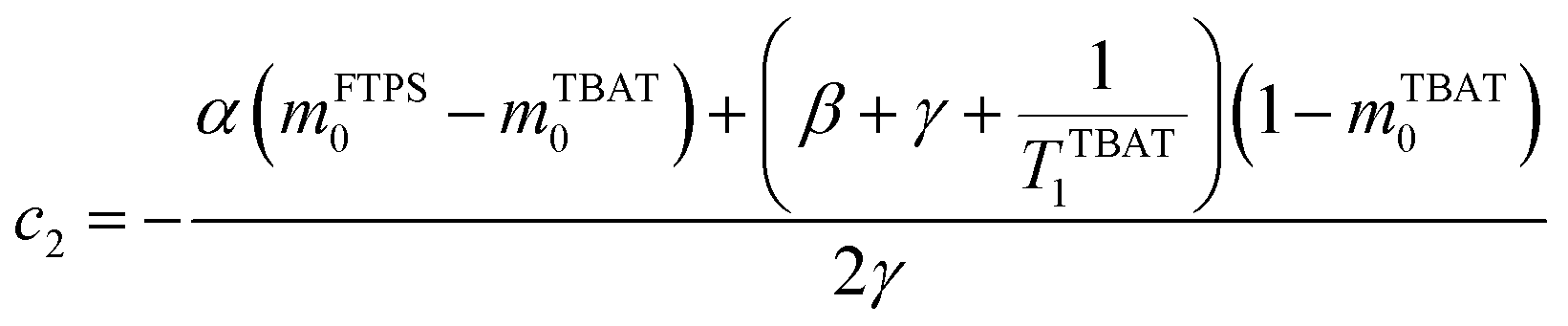
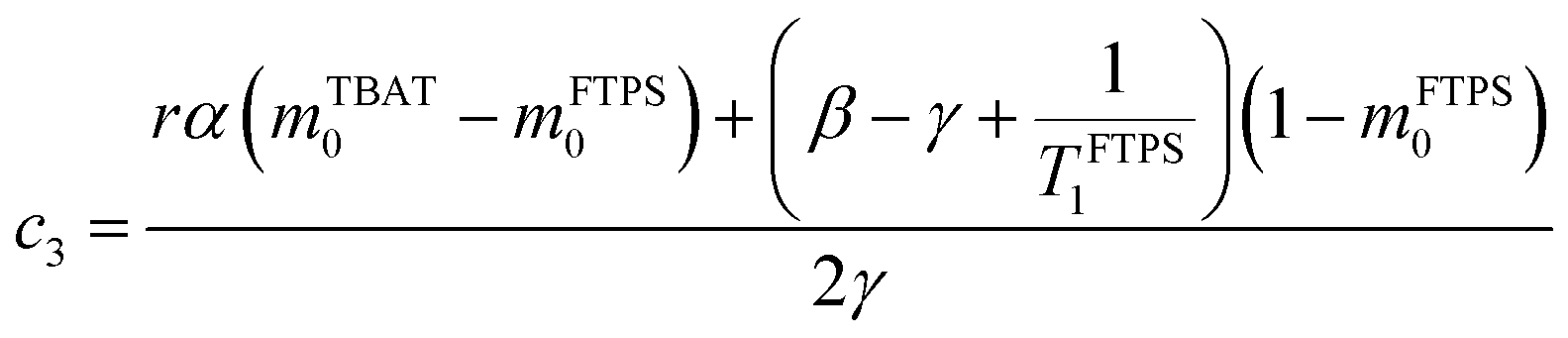
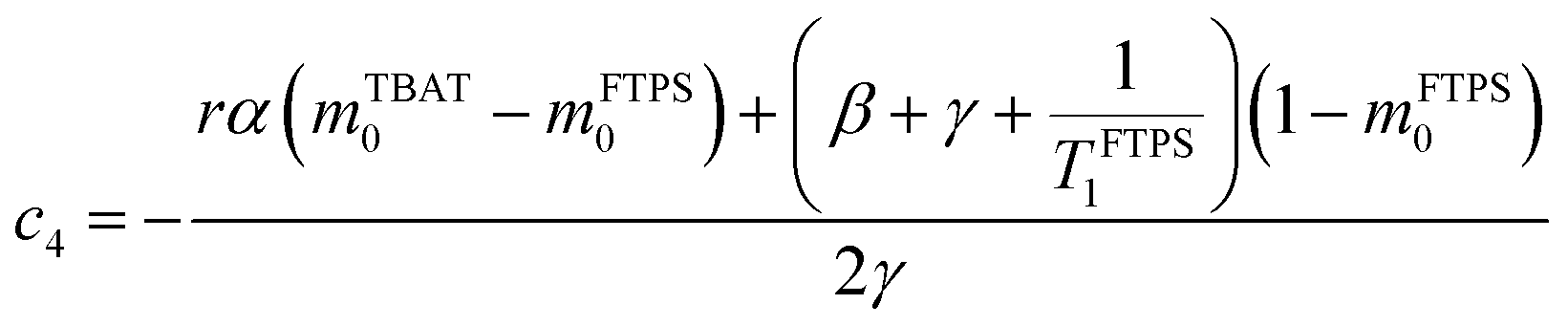
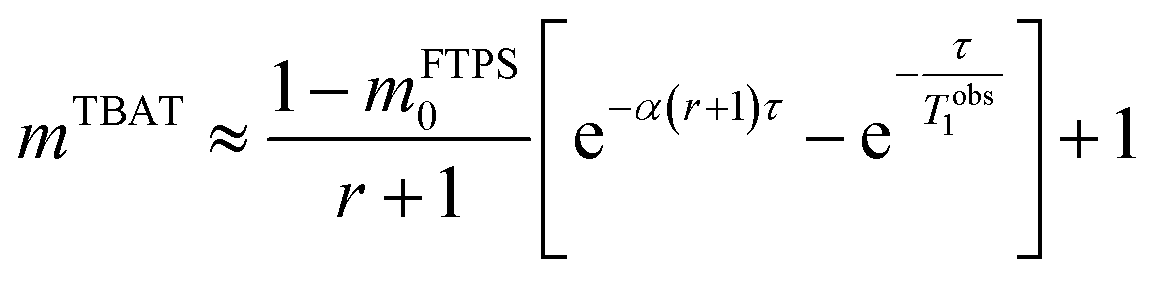
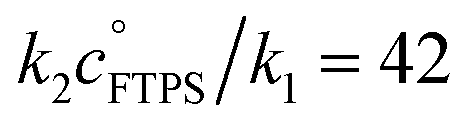 . αs/s−1 = 0.0273[FTPS/mM] + 0.656, R2 = 0.989. (c) Exchange between the species is catalysed by water, with the order in water estimated as approximately 2. αobs(r + 1)/s−1 = 577[H2O/mM]2.4 + 17.2, R2 = 0.989.
. αs/s−1 = 0.0273[FTPS/mM] + 0.656, R2 = 0.989. (c) Exchange between the species is catalysed by water, with the order in water estimated as approximately 2. αobs(r + 1)/s−1 = 577[H2O/mM]2.4 + 17.2, R2 = 0.989.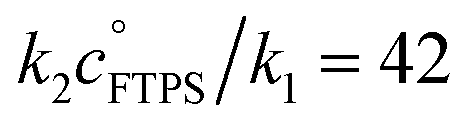 . Analysis of the non-stabilised anhydrous system indicated that the dissociative pathway is again a significant contributor to fluoride transfer from TBAT at low acceptor concentrations, see Sections S5.2.6 and S7.2.3 of ESI† for experimental details and mathematical considerations. The rate of 19F transfer from TBAT to FTPS in THF is markedly accelerated by exogenous water which may assist fluoride dissociation from TBAT via H-bonding or autoionisation.29 Analysis of αobs(r + 1) against [H2O], Fig. 3c, indicates that this predominantly involves interaction with a water dimer, or sequential reaction with two water molecules (see Section S5.2.4 of ESI†).
. Analysis of the non-stabilised anhydrous system indicated that the dissociative pathway is again a significant contributor to fluoride transfer from TBAT at low acceptor concentrations, see Sections S5.2.6 and S7.2.3 of ESI† for experimental details and mathematical considerations. The rate of 19F transfer from TBAT to FTPS in THF is markedly accelerated by exogenous water which may assist fluoride dissociation from TBAT via H-bonding or autoionisation.29 Analysis of αobs(r + 1) against [H2O], Fig. 3c, indicates that this predominantly involves interaction with a water dimer, or sequential reaction with two water molecules (see Section S5.2.4 of ESI†).
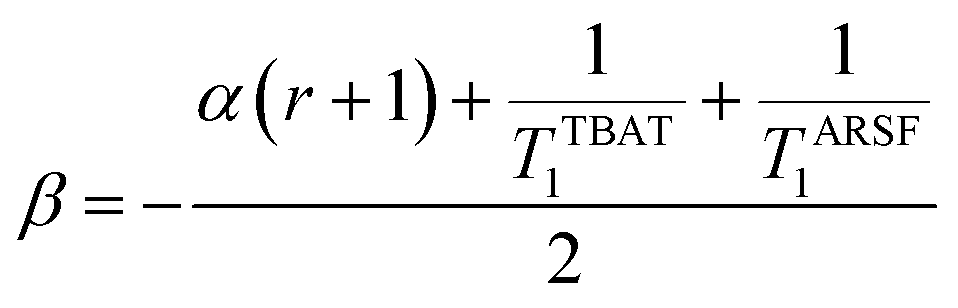
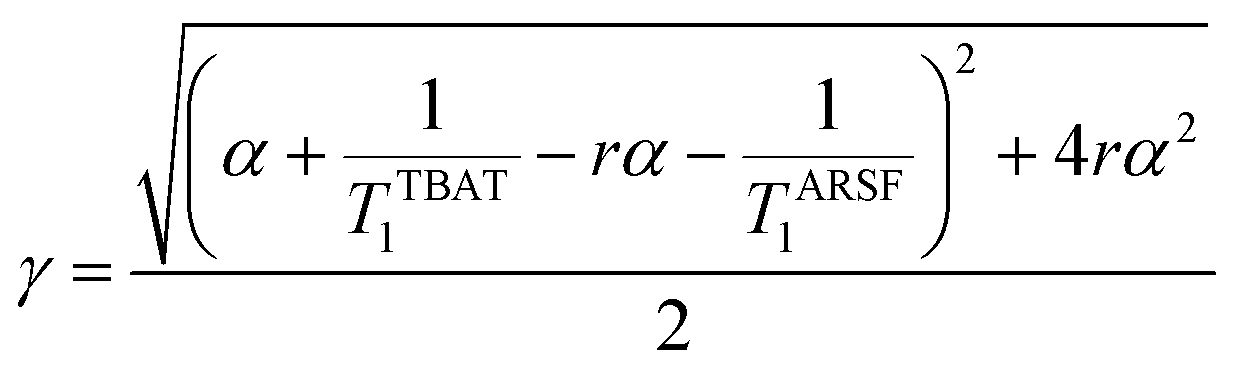
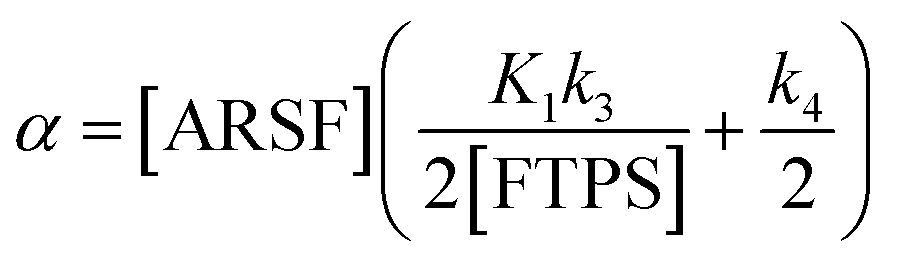
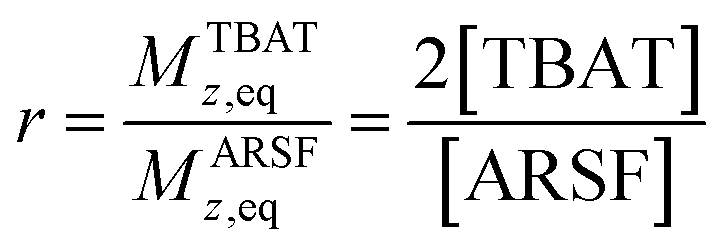
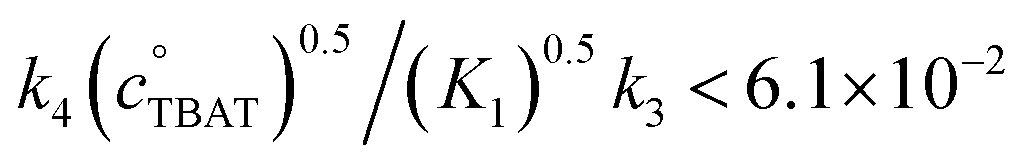 for 2-naphthalenyl fluorosulfate (ARSF).
for 2-naphthalenyl fluorosulfate (ARSF).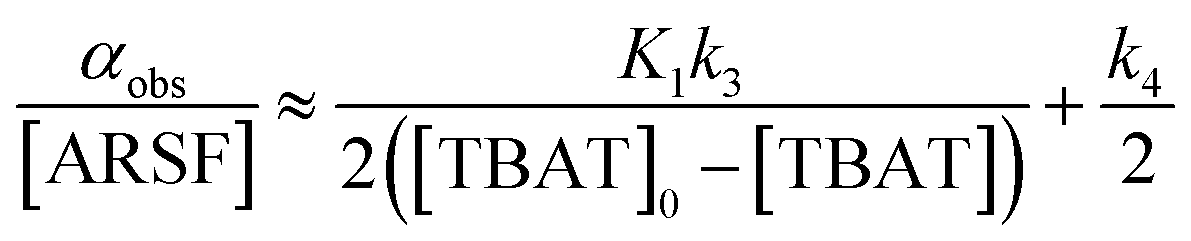
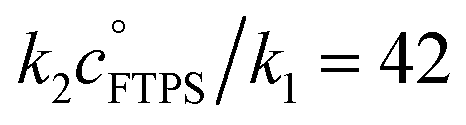 , and thus at 1 M FTPS the transfer is near-exclusively direct (>97%), while at 1 mM FTPS it is near-exclusively dissociative (>95%). The analogous system in MeCN undergoes very rapid fluoride exchange, approaching the limit of practicality of the measurement method, and is only transiently stabilised by 2,6-di-tert-butylpyridine and 3 Å molecular sieves.
, and thus at 1 M FTPS the transfer is near-exclusively direct (>97%), while at 1 mM FTPS it is near-exclusively dissociative (>95%). The analogous system in MeCN undergoes very rapid fluoride exchange, approaching the limit of practicality of the measurement method, and is only transiently stabilised by 2,6-di-tert-butylpyridine and 3 Å molecular sieves.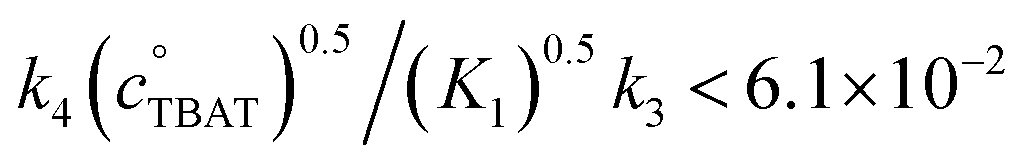 , and thus the direct exchange pathway would only become favoured over the dissociative pathway at unachievable TBAT concentrations (>67 M). On heating to 335 K the system undergoes slow decomposition, resulting in co-generation of FTPS, [FSO2O−][nBu4N+] and SO2F2, and gradual inhibition of the rate of fluoride exchange between TBAT and ARSF. The reaction of benzyl bromide (BzBr) with TBAT in MeCN-d3 at 335 K proceeds with non-degenerate fluoride transfer and is progressively and exogenously inhibited by FTPS, again due to competing dissociative and direct transfer mechanisms. The initial standard state partitioning in this case is estimated to be
, and thus the direct exchange pathway would only become favoured over the dissociative pathway at unachievable TBAT concentrations (>67 M). On heating to 335 K the system undergoes slow decomposition, resulting in co-generation of FTPS, [FSO2O−][nBu4N+] and SO2F2, and gradual inhibition of the rate of fluoride exchange between TBAT and ARSF. The reaction of benzyl bromide (BzBr) with TBAT in MeCN-d3 at 335 K proceeds with non-degenerate fluoride transfer and is progressively and exogenously inhibited by FTPS, again due to competing dissociative and direct transfer mechanisms. The initial standard state partitioning in this case is estimated to be 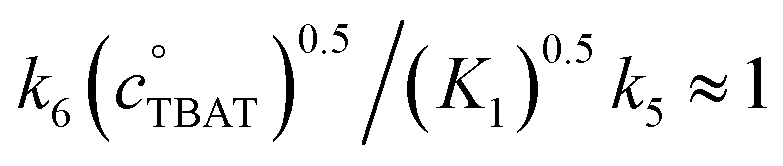 .
.