
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePerformance of a helical insert in a commercial tubing as a passive micromixer to produce nanoparticles using an emulsification approach†‡
Lucia
Abengochea
 ab,
Santiago
Pina-Artal
ab,
Victor
Gonzalez
c and
Victor
Sebastian
*abde
ab,
Santiago
Pina-Artal
ab,
Victor
Gonzalez
c and
Victor
Sebastian
*abde
aInstituto de Nanociencia y Materiales de Aragón (INMA), CSIC-Universidad de Zaragoza, Zaragoza 50009, Spain. E-mail: victorse@unizar.es
bDepartment of Chemical and Environmental Engineering, Universidad de Zaragoza, Campus Rio Ebro, 50018, Zaragoza, Spain
cExella. Gran Via Corts Catalanes, 583, 08011, Barcelona, Spain
dLaboratorio de Microscopías Avanzadas, Universidad de Zaragoza, 50018 Zaragoza, Spain
eNetworking Research Center on Bioengineering, Biomaterials and Nanomedicine (CIBER-BBN), 28029 Madrid, Spain
First published on 21st March 2024
Abstract
Chemical reactions with very fast kinetics, such as nanomaterial production, depend on mixing conditions to control reactant conversion and product selectivity. Mixing at the molecular level (micromixing) is an important stage in the operation of chemical engineering and process technology, with a strong influence on the selectivity, yield, and quality of final products. Micromixers have demonstrated excellent mixing capabilities in many physical and chemical methods. In this study, a new type of passive micromixer based on a helical insert was microfabricated. Interestingly, the micromixer is easy to be assembled into conventional tubings widely used in microfluidics and has no moving parts. The proposed microsystem is robust, compact, and has a modular design that enables the proper combination of mixing units for the on-demand tuning of mixing requirements. Finally, the helical insert could be easily regenerated in the case of fouling. The mixing efficiency of the proposed insert was validated using an acetal cleavage method and was finally successfully tested in the production of single emulsions to form polymeric nanoparticles of high interest in biomedicine. The mixing efficiency of the proposed micromixers is as good as the ones currently used and also offers a plethora of advantages that are not feasible in current systems.
Introduction
Micromixing plays a pivotal role in the quality and product distribution of chemical reactions, where reagents must be brought into contact on a molecular level and collisions between molecules must be sufficiently energetic to surpass the activation energy. Molecular mixing is especially challenging for reactions, such as nanomaterial production, with fast kinetics that require fast mixing time.1 Considering that at the molecular level, diffusion is the ultimate transport process to control mixing2 and that diffusion in the liquid phase is not as fast as in the gas phase, fluid dynamics is key to promote molecular mixing. However, mixing is a complex phenomenon to study since it is coupled to several processes that occur simultaneously: mass transfer, fluid mechanics, and chemical reactions.2 One of the most intricate challenges is to achieve fast and uniform mixing in a laminar flow regime. In laminar flow, a fluid flows in parallel layers with no disruption between the layers and mixing occurs mainly via diffusion. Micromixers have demonstrated excellent mixing capabilities in a plethora of applications, ranging from microsystems in the laboratory to clinical and industrial uses.3,4 The broad variety of micromixers designed can be categorized into two types:3,5 1) active systems, which use external energy such as moving surfaces or electrical, magnetic, and sound fields to enhance mixing and 2) passive systems, which use flow energy and geometrical configuration to promote diffusion and mixing. Active micromixers are simple by design but difficult to integrate owing to the need to incorporate external energy sources. On the contrary, passive micromixers are much easier to integrate, but they require complex fabrication procedures.5The mixing time can be defined as the time required by two streams to reach a certain degree of mixing, given that the mixing time is at least ten times shorter that the characteristic reaction time.6 Passive micromixers promote mixing by decreasing mixing times and shortening diffusion paths either via multilamination, split, and recombination or promoting engulfment and vortices.6 Modelling is usually performed to evaluate the applicability of micromixers to carry out chemical processes, since it provides three-dimensional (3D) flow information that is not accessible by experimental procedures. However, computational difficulties can arise derived from surface effects, such as wettability and roughness, which prompts the need to use experimental methodologies for mixing characterization and application validation;7 and some of these experimental approaches can be summarized as: 1) visualizing the flow of a dye, 2) visualizing coloured species or colour changes of the mixed fluids that are generated by chemical reactions, and 3) chemical testing methods based on competitive reaction schemes. Among those methodologies, chemical tests based on competitive reactions are quite convenient since they do not require optical access to a microsystem (not all micromixers enable optical access) and provide very robust and reproducible data.8 The chemical reactions considered in the chemical tests are based on a set of two competing reactions occurring simultaneously and whose product selectivity depends on the competition for one reactant. The fastest reaction is favoured when the mixing time is small. On the contrary, if the mixing time is high, the slowest reaction can compete due to the local excess of the limiting reactant. Then, the reaction selectivity will be directly related to the mixing effects and the conversion of the slower reaction will indicate the mixing time. The match between the CFD calculations and the experimentally obtained mixing values by the methodology of considering competitive reactions can be good enough to consider this approach as suitable for mixing studies.9
Emulsification techniques are commonly used to produce nanoparticles with a high control of the particle size. Basically, this control is achieved by the formation of micelles,10 which can be defined as non-equilibrium systems and cannot be formed spontaneously.11 A micelle is formed when two immiscible fluids are mixed with the aid of energy and a surfactant to decrease the surface tension between the immiscible phases. Consequently, highly energetic homogenization techniques, such as probe sonication, high shear mixers, and high pressure homogenizers, have been used to effectively supply energy in the shortest time and produce monodisperse nanoparticles (NPs).11 Those techniques require a balance between maintaining the ability to control the desired size and achieving high throughput, while the reproducibility is preserved. Microchannel emulsification is a very promising procedure to fulfil the required balance between size control, throughput, and reproducibility.12 Micelles form spontaneously due to the hydrodynamic instability of the dispersed phase while it is efficiently dispersed within the non-miscible continuous phase. Considering the relevance of mixing in microchannel emulsification, where the interfacial tension between non-miscible phases is the driving force for emulsion formation,10 a variety of microsystems have been designed. Some of those microsystems require sophisticated microfabrication procedures,3 while the lack of robustness at high flow rates makes these systems unsuitable for long-term use and large-scale production. On the other hand, some designs, such as 3D flow focusing, require skilled technicians to verify the proper alignment of the microsystems.10 Even worse, microstructured devices are prone to unwanted deposition on surfaces,13 leading to local constrictions that can alter the velocity profiles and dramatically increase the pressure drop. These effects have an obvious negative repercussion on micromixing and in the worst scenario can result in the micromixer blockage13 and the need to use a new system. Consequently, micromixers targeted for use in nanoparticles production should be designed with the consideration that fouling may occur and that in such an event, complete microsystem regeneration would be needed to make the production process sustainable and competitive in the long term.
This work describes the use of a metallic insert as a passive mixer that has interesting features: straightforward to adapt to the conventional tubing widely used in microfluidics, easy numbering-up to increase the throughput, has no moving parts, is robust and compact, easy microfabrication and cost effectiveness, modular design, high mixing efficiency, and allows complete regeneration in the event of fouling because the insert can be easily disassembled from the tubing. Further, the insert can be easily introduced into commercial PTFE tubing, converting the system into a modular micromixer that does not require skilled users to assure efficient operation. The mixing efficiency was validated by the Bourne reaction test (acetal cleavage method) and finally tested in the production of single emulsions to form polymeric nanoparticles that are of high interest in biomedicine.
Experimental section
Chemical reagents
All the reagents involved in the study of the mixing characterization were purchased from Sigma-Aldrich (St. Louis, MO, USA) and used without further treatment, and included: NaOH, HCl (37%), 2,2-dimethoxypropane (DMP) (98%), and ethanol (99%). Regarding the emulsification process, ethyl acetate was purchased from Sigma-Aldrich (St. Louis, MO, USA) and used without further treatment. The polymers, poly(D,L-lactide-co-glycolide) 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 (PLGA; molecular weight 38–54 kDa), under the commercial names Resomer® RG 504 and Eudragit® RS100 were supplied by Evonik Industries (Darmstadt, Germany) and the surfactant Pluronic® F-68 was purchased from PanReac AppliChem ITW Reagents. All references to water relate to the use of distilled water.
:50 (PLGA; molecular weight 38–54 kDa), under the commercial names Resomer® RG 504 and Eudragit® RS100 were supplied by Evonik Industries (Darmstadt, Germany) and the surfactant Pluronic® F-68 was purchased from PanReac AppliChem ITW Reagents. All references to water relate to the use of distilled water.
Insert and micromixer assembly
Inserts with a helical morphology were made in stainless steel AISI-304 using a microfabrication and wet etching approach, whereby the length and external diameter could be tuned on demand. The inserts were microfabricated using the well-known chemical wet etching approach. Here, a stainless-steel plate AISI-304 with a thickness of 300 μm was first cleaned with a mixture of water and ethanol using ultrasound. The plate was then covered by a UV-photosensitive film, and a predefined mask with the desire design was aligned to the plate. The plate was exposed in UV light to harden the clear sections. Afterwards, the plate was stripped using a NaOH solution to wash away the unexposed areas to UV light and unprotect those areas to be etched. The etching was performed using an aqueous solution of ferric chloride. After the etching process, the resulting inserts were rinsed with water and adapted to the tubing diameter. Three different inserts were microfabricated to be held in PTFE tubings of 0.04, 0.03, and 0.02 inch inside diameter (ID) and 1/16 inch outside diameter (OD), respectively. Fig. 1A–C presents the optical and electron microscopy images of the designed inserts and the details of each mixing unit. The micromixers were fully three-dimensional, being an advantage since, although 2D planar micromixers seem to be simple to fabricate their mixing performance is not as good as that of 3D micromixers.14 The lengths of the mixing units were 4, 2.4, and 1.6 mm for the inserts housed in the 0.04′′, 0.03′′, and 0.02′′ ID tubings, respectively. The length of the inserts was approximately 18 cm, implying that the number of mixing units was 45, 75, and 112 for the inserts housed in 0.04′′, 0.03′′, and 0.02′′ ID tubings, respectively. These inserts were based on the approach of flow division, where the liquid to be mixed is divided into smaller streams, and the streams are divided according to the microchannel dimension of each insert and are interwoven using a helical pathway to reduce the diffusion distance and the mixing time. The microchannels etched in the inserts designed for the 0.04′′ tubing were larger. Consequently, oval holes (∼250 μm × 1000 μm) were microfabricated to create supplementary inlets and outlets for the passage of fluid and to increase the streams interweaving. The volume of the insert was required to determine the proper mean residence time for each volume flow rate condition considered. To calculate the volume, the mass of the insert was divided by the stainless-steel density. The mean residence time was calculated by dividing the total volume flow rate by the calculated volume in each insert. Finally, the insert could be manually assembled or disassembled in the tubing with the assistance of a tweezer.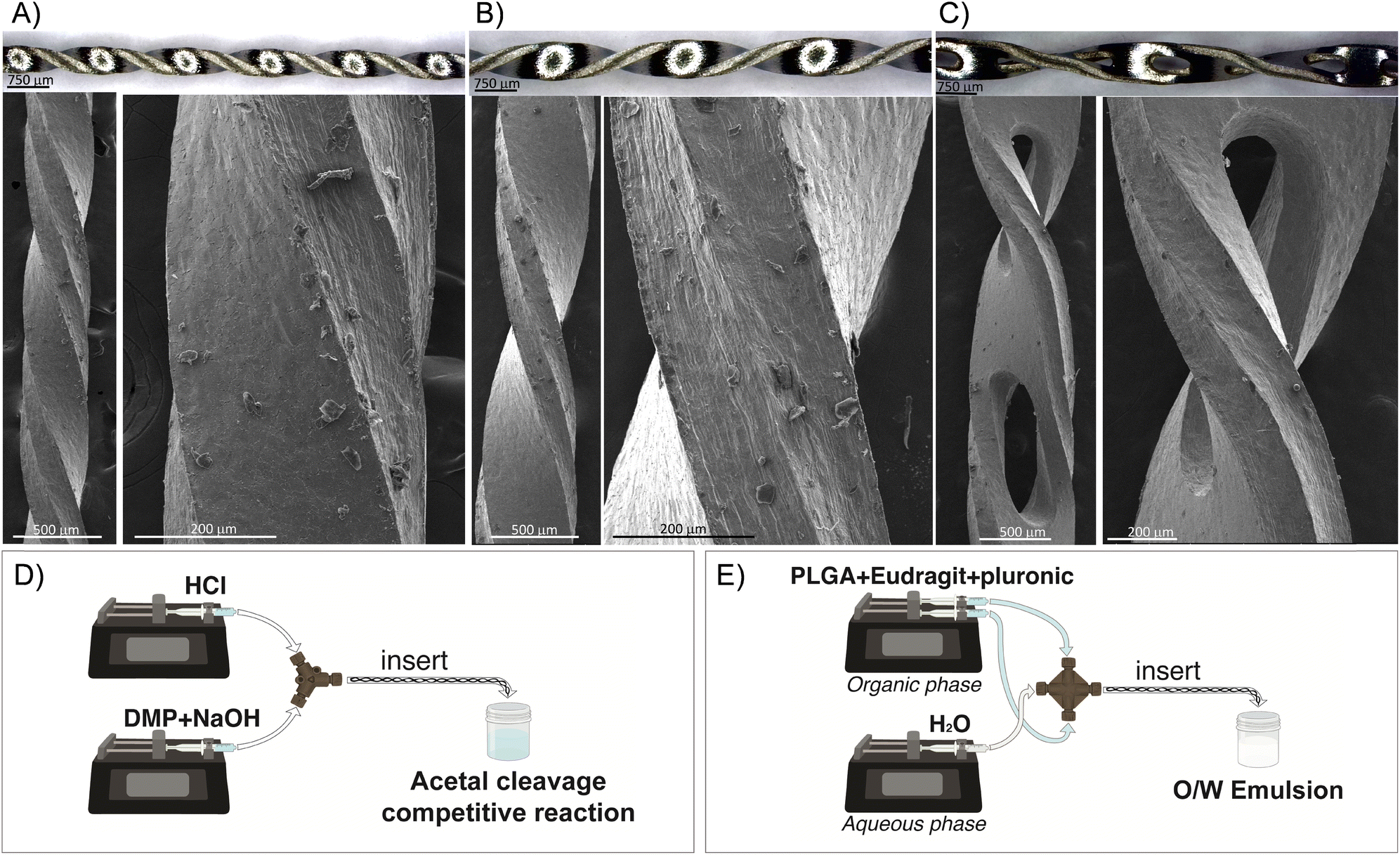 | ||
| Fig. 1 Representative optical and electronic images of the helical inserts designed in this work to be housed in PTFE tubings with an internal diameter of 0.02′′ (A), 0.03′′ (B), and 0.04′′ (C). D and E present schematics of the setup for the study of the acetal cleavage competitive reaction (D) and the O/W simple emulsification process (E). In both cases, the insert was placed at the outlet tubing. | ||
Acetal cleavage method involving the parallel reaction of neutralization of HCl and NaOH and the acid-catalyzed cleavage of 2,2-dimethoxypropane (DMP)
We selected the parallel-competitive reaction acetal cleavage method for mixing characterization. This reaction is based on the competition of the acid-catalyzed cleavage of 2,2-dimethoxpropane (DMP) with the neutralization of HCl with NaOH.15 This competitive reaction has well defined kinetics and it has been widely used to determine micromixing efficiency and mixing time. The fast reaction is the neutralization of a strong acid (HCl) and a strong base (NaOH) (eqn (1)), which is essentially instantaneous compared to any achievable mixing process, independent of the temperature and reagent concentration (k1 = 1.4 × 108 m3 mol−1 s−1).15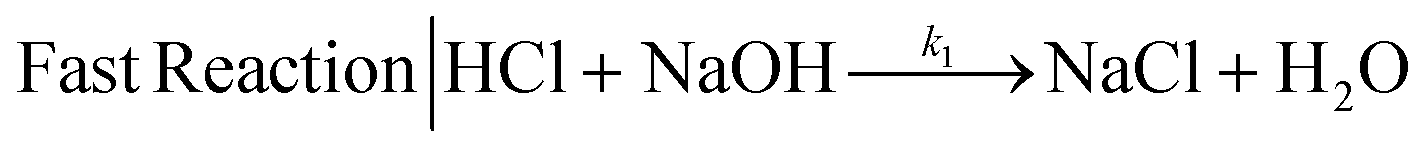 | (1) |
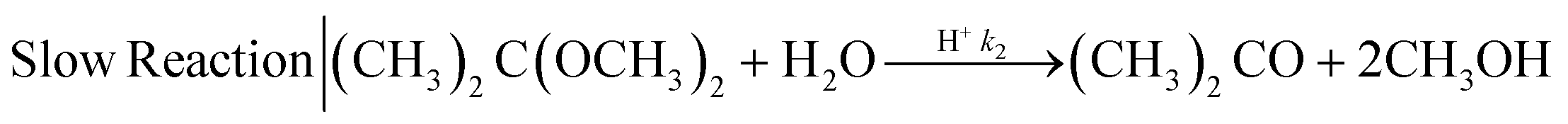 | (2) |
Continuous production of polymeric nanoparticles by simple emulsification
Polymeric NPs were synthesized by a simple oil-in-water emulsification (O/W) and solvent evaporation method. The organic phase consisted of PLGA (50:50) (0.5 % w/v), Eudragit RS100 (0.5% w/v), the surfactant Pluronic F-68 (3% w/v), and ethyl acetate as the organic solvent. To foster the formation of a stable emulsion, the organic phase was loaded into a plastic syringe and injected as a single stream into the central inlet of a cross-shaped polyether ether ketone (PEEK) junction, while the aqueous phase (water) was simultaneously fed into the lateral inlets of the cross junction via two syringes. Different flow ratios between the organic phase (QO) and aqueous phase (QA) were evaluated for the emulsification (QO/QA: 1/3, 1/4, 1/6, 1/8, 1/10, 1/12). After selecting the optimal QO/QA ratio, a range of total flow rates (QT) was tested, varying from 40 to 80 ml min−1. Two syringe pumps (Harvard Apparatus) were used to feed both solutions into the cross-shaped junction (Fig. 1-E). A PTFE tubing with an internal diameter of 0.04′′ and an external diameter of 1/16′′ was set at the outlet of the cross junction. The outlet tubing was set with and without the insert to evaluate its effect on the emulsification process. A schematic of the reactor assembly is shown in Fig. 1-E. Once formed, the resulting emulsion was collected in an open flask, where the organic solvent was evaporated for 3 h under magnetic stirring (600 rpm). Finally, nonreacted reagents were removed by a series of centrifugation and water resuspensions. Experiments were performed 3 times to study their reproducibility. The physicochemical characterization of the polymeric NPs was performed by scanning electron microscopy (SEM, Quanta Inspect F50; FEI Company, The Netherlands) to determine the morphology and size distribution of the NPs. Samples were prepared by deposition of the NPs onto a glass slide and later using a palladium sputter coating. SEM images were acquired at an accelerating voltage of 10 kV, with a spot size of 3.0, and the size measurements were conducted using the image analysis tool ImageJ (n ≈ 300–350 counts). In all cases, three replicates (each one of a different experiment) were subjected to SEM analysis.
The NPs size and size distribution were additionally determined by dynamic light scattering (DLS) using a Brookhaven 90 Plus system (Brookhaven Instruments Corporation, NY, USA) after diluting the samples approximately 1:200. Finally, and considering the importance of microfluidics to translate conventional bench procedures to actual practice, the productivity of the polymeric NPs (mg of polymeric NPs/min) synthesis was calculated considering the total flow rate and the concentration of NPs produced in each synthesis condition.
Results and discussion
Determination of the mixing performance by the acetal cleavage competitive reaction
The mixing performances of the three helical inserts depicted in Fig. 1A–C were studied using the acetal cleavage method. In this method, the hydrolysis of DMP is catalyzed by protons and does not take place in an alkaline environment. If the mixing is fast, all the protons will be consumed by the neutralization reaction (eqn (1)) and a DMP conversion of 0% will be achieved. On the other hand, if the mixing is not fast enough, the local excess of protons can promote DMP hydrolysis and consequently the production of acetone and methanol (0 < DMP conversion ≤ 100%). Then, it can be considered that a fast mixing process results when the DMP conversion is less than 20%,16 being the fastest mixing process in the scenario where the DMP conversion is the smallest. Considering that the DMP hydrolysis reaction is irreversible (eqn (2)), the mixing process can be determined offline by measuring the reaction progress either by the DMP reacted or by the acetone/methanol produced.8 In the present study, the DMP conversion was tracked by the measurement of the acetone concentration at the outlet.Several different flow rates ranging from 10 to 80 mL min−1 were studied in tubings with IDs of 0.02′′, 0.03′′, and 0.04′′. The pressure drop did not enable achieving a stable flow rate over 20 mL min−1 in the case of using the insert with the smallest ID tubing of 0.02′′. In this 0.02′′ tubing, the mean value of DMP conversion monotonically decreased from 71% at 10 mL min−1 to 6% at 80 mL min−1. The progression from 10 to 40 mL min−1 was sharp, while it was smooth from 40 to 80 mL min−1 (Fig. 2-A), which could infer that the micromixing was just promoted by the turbulence at flow rates larger than 50 mL min−1 since under that flow condition the critical Reynolds number was achieved.17 On the other hand, the micromixing was fully promoted when the insert was introduced in the same ID 0.02′′ tubing, and it was observed that the mean DMP conversion monotonically fell from 25% at 10 mL min−1 to 12% at 20 mL min−1. Without using the insert, the mean value of DMP conversion of 12% was achieved at a flow rate of 50 mL min−1. This micromixing improvement could be rationalized by the conversion of the fluid kinetic energy into a turbulent-like motion due to the redirection and collision of the flow promoted by the insert.
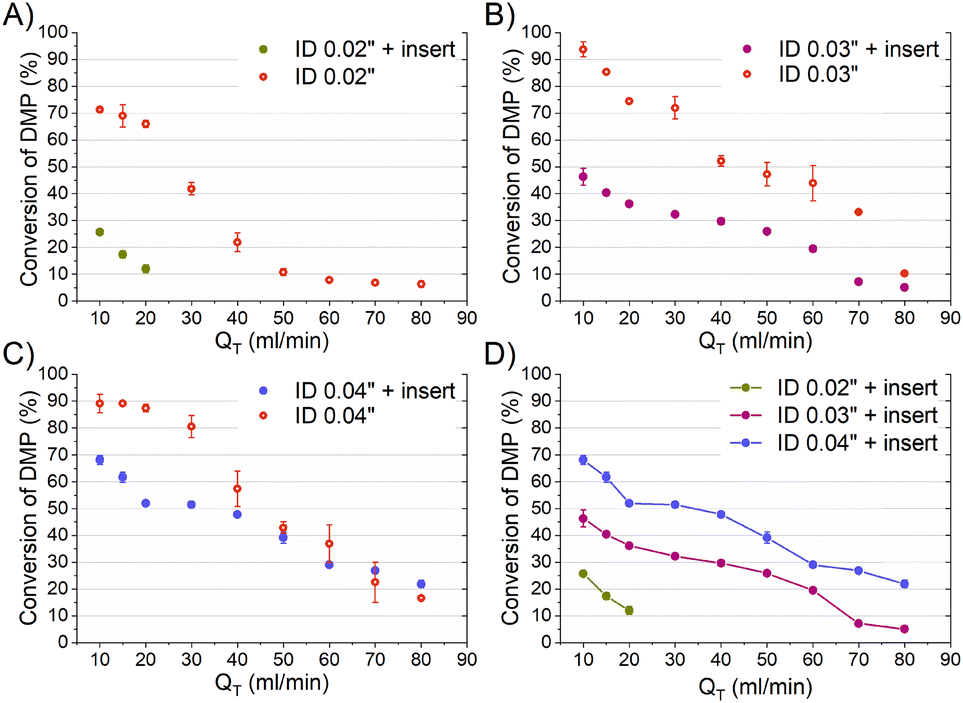 | ||
| Fig. 2 Conversion of DMP over the total volume flow rate (mL min−1) with and without an insert for tubings of ID: A) 0.02′′, B) 0.03′′, and C) 0.04′′. D) Comparison of DMP conversion for different inserts versus the total volume flow rate (mL min−1). | ||
To compare properly the experiments with and without the insert, since the mean velocity was different for the same reagent volume flow rate, the residence time was calculated considering the effective volume inside the tubing (Fig. 3-A). The curve of the DMP conversion with an insert was generally under that of the case without an insert, observing that a short residence time was required to get the same micromixing grade when the reaction occurred with an insert. The mean DMP conversions achieved for a residence time of 60 ms (the shortest achieved with an insert) were 12% and 22% with and without the insert, respectively. According to the mixing model developed by Rave et al.8 to determine the mixing time in the acetal cleavage competitive reaction, DMP conversions of 12% and 22% implied mixing times as short as 11 ms and 30 ms, respectively. Consequently, the helical insert designed for the 0.02 tubing clearly improved the micromixing.
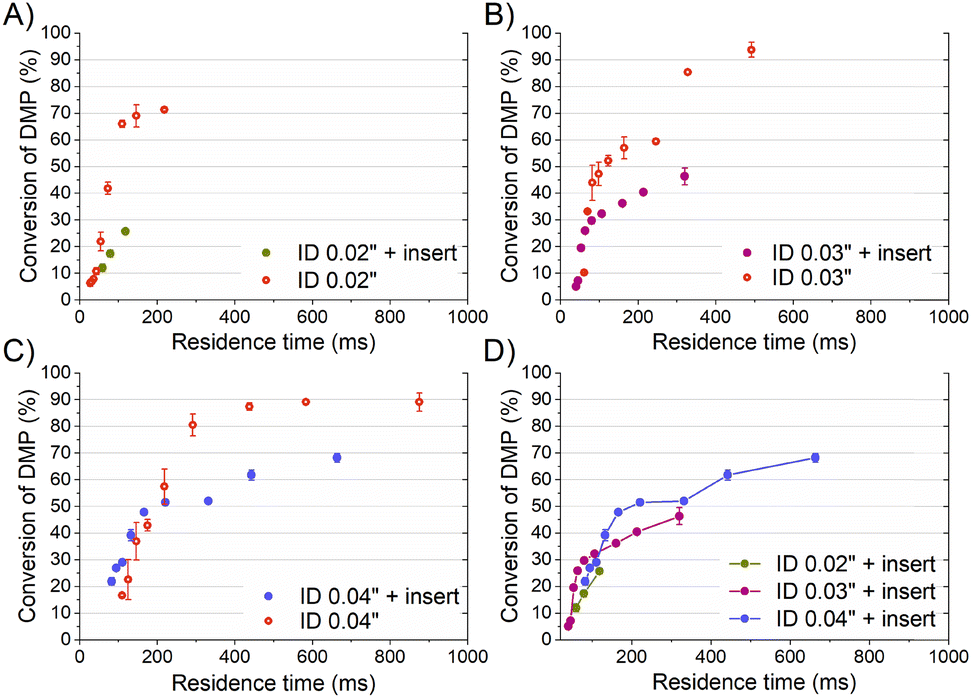 | ||
| Fig. 3 Conversion of DMP over the residence time (ms) with and without an insert for tubings of ID: A) 0.02′′, B) 0.03′′, and C) 0.04′′. D) Comparison of DMP conversion for the different inserts versus the residence time. | ||
The inserts designed to be enclosed inside 0.03′′ and 0.04′′ ID tubings performed similarly to that of the 0.02′′ insert in the acetal cleavage competitive reaction (Fig. 2B and C). The results for the 0.03′′ tubing and the 0.03′′ tubing with the insert are shown in Fig. 2-B. In this case, the pressure drop was not a limitation and the effect of the insert was studied in the range from 10 to 80 mL min−1. The mean DMP conversion monotonically decreased from 46% at 10 mL min−1 to 5% at 70 mL min−1 when the insert was assembled. Similarly, the presence of the insert enabled decreasing the DMP conversion as the flow rate increased. However, the DMP conversion reached a plateau of 5% DMP conversion at flow rates larger than 70 mL min−1, equally. This was also due to the turbulence conditions achieved at Re number > 2000 (flow regime achieved at the experimental conditions with Q > 70 mL min−1). When comparing the progression of the DMP conversion with the residence time in both scenarios, with and without the insert, it could be clearly observed that conversion curve with the insert was under that of the curve without the insert up to a residence time of 75 ms (Fig. 3-B). When comparing the DMP conversion at residence times smaller than 75 ms, the mixing performance was similar since the flow regime was not laminar (Fig. 3-B). It can be inferred from this observation that the mixing was clearly improved by the use of the insert under laminar flow conditions, which was the main aim of this work.
Like the 0.02′′ and 0.03′′ inserts, the 0.04′′ insert overperformed the mixing achieved in the tubing without an insert in the range from 10 to 60 mL min−1 (Fig. 2-C). The mixing efficiency was higher as the residence time diminished than in the case of not using an insert up to a residence time of approximately 140 ms (Fig. 3-C). Concluding similarly to the studies with the 0.02′′ and 0.03′′ inserts, the 0.04′′ insert could improve the mixing efficiency in the laminar flow regime. Fig. 2-D and S1† present a comparison of the progression of the DMP conversion as the volume flow rate was increased when the different inserts and tubings were used. It can be seen that the larger the tubing ID and the insert diameter, the worse the mixing achieved as the volume flow rate decreased. As a result, to achieve a DMP conversion of 20%, volume flow rates of approximately 12, 60, and 80 mL min−1 are required for the 0.02′′, 0.03′′, and 0.04′′ inserts, respectively (Fig. 2-D). When comparing the mixing performance of the inserts in terms of residence time, it could be observed that the 0.03′′ insert slightly outperformed the 0.04′′ at residence times larger than 150 ms; whereas below the threshold of 150 ms (approx. 30% DMP conversion), both inserts improved the mixing equally. On the other hand, the 0.02′′ insert was the best insert when a residence time smaller than 60 ms was required. All in all, the inserts designed here could all improve the mixing process of conventional microfluidics tubings due to the high energetic efficiency of mixing. Considering that the throughput is a key variable to consider in the rational design of chemical engineering processes and that chemical reactors should combine fast mixing with low pressure drops and high flow rates, the 0.04′′ insert is considered the best candidate. However, the 0.02′′ and 0.03′′ inserts could also be considered as proper micromixers in chemical processes where high throughput is not demanded, but fast mixing at low flow rates is required.
Considering that for an exact comparison between mixers consideration of the hydrodynamic parameters, such as the Reynolds number, pressure drop, or even energy dissipation, is required, we just qualitatively compared the performance of the here designed inserts with the state of the art8,16 and at a total mass flow rate of 5 kg h−1 (the maximum achieved in this work) (Table 1). According to Table 1, the inserts designed in this work had qualitative mixing performances as good as the ones reported in literature, but with the following advantages: straightforward to adapt to conventional tubings that are widely used in microfluidics, modular design to tune the number of mixing stages to the required mixing efficiency and throughput, robust, compact, easy microfabrication and cost effectiveness, high mixing efficiency, and do not require skilled users to assure an efficient operation.
| Type of mixer | % Conversion of DMP | Main size | Ref. |
|---|---|---|---|
| Temperable mixer | 45 | Channels 0.2 mm × 0.1 mm | 16, 18 |
| LTF mixer | 15 | Channels 4 mm ID | 16, 18 |
| Cyclone type mixer | 3 | Swirl chamber 0.5 mm × 5 mm | 16, 18 |
| ART plate reactor | 30 | Channel 0.8 mm × 1 mm | 8 |
| 0.04 in insert | 20 | Helical insert: thickness of 0.3 mm for 1 mm ID tubing | This work |
| 0.03 in insert | 5 | Helical insert: thickness 0.3 mm for 0.7 mm ID tubing | This work |
Application of the insert in the emulsification process to produce polymeric nanoparticles for biomedical use
In a previous publication, our group studied the importance of achieving a high shear stress at the interphase of two immiscible streams to control the emulsification process of polymers.12 If the reduction of diffusion distance achieved by a fast diffusive mixing between the immiscible fluids is short enough, the shear stress can endow enough energy to the system to promote the formation of polymeric micelles without the need for external mechanical energy.The mixing time required to form the emulsion drops depends on the high shear applied, ranging from milliseconds to seconds.19 For instance, the residence time in the dispersing zone of the emulsification in rotor–stator systems (i.e. mixers, colloid mills) is in the order of 100 ms to 1 s, whereas in high pressure systems (radial diffusers, jet dispersers), it is in the order of 3 ms to 100 ms.19 Considering that the inserts studied in the previous section were promoting a very efficient mixing in the range of 50–250 ms, where the shear stress should be high enough20 to promote the emulsification process, the use of the insert was considered to try to achieve molecular mixing and high shear stress to generate nanoemulsions of a mixture of polymers widely considered in the biomedical field. The insert modifies the cylindrical geometry of the tubing, boosting a reduction in the diffusion length and maximizing the interfacial area between the immiscible fluids to create chaotic advection.21 On the other hand, it is important to highlight that chaotic advection is usually the proper option to improve mixing in a laminar regime when using viscose solutions, since using a turbulent regime is not a viable solution due to pressure drop generated. According to our previous studies,12 the high lamination of the aqueous (QA) and organic (QO) streams is key to promote local mixing and consequently the emulsification process with a small polydispersity.
Nanoparticles targeted for biomedicine use need to be produced with a controlled size because it can affect their pharmacodynamic and pharmacokinetic profiles, as well as their release profiles and cellular uptake. Generally, the emulsification process is influenced by the hydrodynamic conditions and the presence of surfactants that decrease the interfacial tension between the immiscible phases. In this case, we just considered the effect of hydrodynamics and performed a pre-formulation study where the more relevant variables were tested: 1) the flow ratio of aqueous (continuous phase) and organic (disperse phase) streams and 2) the residence time. The emulsification was performed using the same microfluidic system as reported in the previous section, but instead of using a Y junction, a cross junction was selected to better confine the disperse phase by the continuous phase in the O/W single emulsification process (Fig. 1-E). Pre-formulation experiments were carried out at different QO/QA flow ratios and a constant total flow rate (QA+ QO = QT = 48 ml min−1). The study was performed in the presence and absence of a 0.04′′ insert. Fig. 4 and S2† allow a comparison of the representative SEM images of the emulsions synthesized using just the 0.04′′ PTFE tubing and assembling the 0.04′′ insert in the same tubing. Emulsions produced with QO/QA flow ratios of 1/3, 1/4, and 1/6 showed a high heterogeneity, since large irregularly-shaped particles with widely varying sizes were observed. These results are indicative of the existence of an improper emulsification process. As the aqueous content was increased in the emulsification process for the QO/QA flow ratio of 1/8, the nanoparticles exhibited an enhanced homogeneity in size and shape, particularly when the insert was added. This result is in agreement with the literature12,22 and can be rationalized because the increase in water content diminishes the emulsion viscosity, resulting in a low viscous resistance. The viscous force becomes less important against the shear force during the emulsification process and then, a high shear stress is generated. Table 2 shows the statistics of the particle size analysis determined from the SEM images, and it can be seen that the mean particle size decreased as QO/QA decreased.
 | ||
| Fig. 4 Representative SEM images of polymeric NPs synthesized via O/W emulsification, with a 0.04′′ (id) insert in the system (A, B, and C) and without an insert (D, E, and F) at different flow ratios: A and D) QO/QA = 1/4, QO = 9.6 ml min−1, QA = 38.4 ml min−1. B and E) QO/QA = 1/8, QO = 5.33 ml min−1, QA = 42.67 ml min−1. C and F) QO/QA = 1/12, QO = 3.69 ml min−1, QA = 44.30 ml min−1. QT was maintained at 48 ml min−1 across all conditions. | ||
| Q O/QA|QT = 48 mL min−1 | Mean size ± SD, nm | PDI | |
|---|---|---|---|
| 1/3 | With insert | 308 ± 89 | 0.08 |
| Without insert | 302 ± 102 | 0.11 | |
| 1/4 | With insert | 223 ± 104 | 0.22 |
| Without insert | 321 ± 153 | 0.23 | |
| 1/6 | With insert | 193 ± 62 | 0.11 |
| Without insert | 259 ± 107 | 0.17 | |
| 1/8 | With insert | 159 ± 48 | 0.09 |
| Without insert | 151 ± 45 | 0.09 | |
| 1/10 | With insert | 167 ± 45 | 0.07 |
| Without insert | 164 ± 49 | 0.09 | |
| 1/12 | With insert | 149 ± 32 | 0.05 |
| Without insert | 164 ± 41 | 0.06 | |
The presence of the insert improved the mixing process between the disperse and continuous phases and generally resulted in a lower PDI than the case where no insert was used. Finally, the best flow QO/QA ratio was 1/12, since it could be observed in Fig. 4(C and F) and Table 2 that these conditions promoted the production of the most homogenous and smallest mean-sized nanoparticles among all the performed conditions.
Once the QO/QA flow ratio was fixed to 1/12, the effect of the residence time (or total volume flow rate (QT)) variation in the production of NPs with and without an insert was assessed. Data on the nanoparticle size and its distribution were obtained by the analysis of the SEM Images depicted in Fig. 5 and are summarized in Table 3. A two-way ANOVA analysis of the data in Table 3, followed by Tukey's post hoc test, revealed that the presence of an insert in the mixing process was a significant factor influencing both the mean diameter of the nanoparticles (F(1,20) = 23.8, p < 0.05) and the PDI (F(1,20) = 85.9, p < 0.05).
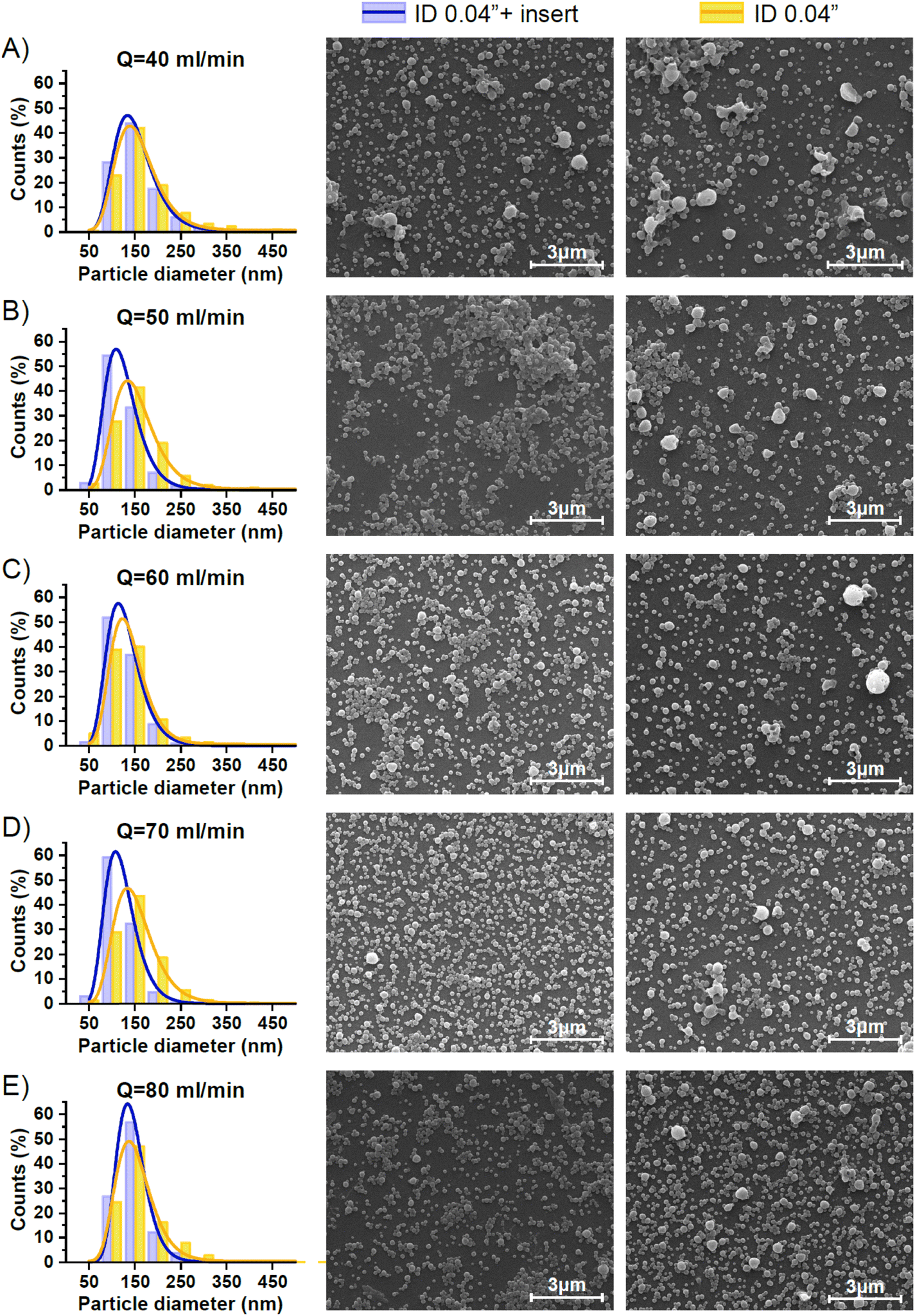 | ||
| Fig. 5 SEM particle size distribution histograms of PLGA/Eudragit NPs synthesized via O/W emulsification with and without an insert in the system and under different flow rates: 40 (A), 50 (B), 60 (C), 70 (D), and 80 ml min−1 (E). In all conditions, the QO/QA flow ratio was 1/12. Histograms were fitted to a lognormal distribution curve. SEM images corresponding to emulsions synthesized with and without insert (ID 0.04′′ + insert and ID 0.04′′, respectively) are displayed at the right of the histograms. In the latter, the emulsification QT corresponds to that of the histogram in the same row. | ||
| Q T (mL min−1)|QO/QA = 1/12 | Mean size ± SD, nm | PDI | |
|---|---|---|---|
| 40 | With insert | 157 ± 52 | 0.11 |
| Without insert | 164 ± 68 | 0.17 | |
| 50 | With insert | 130 ± 35 | 0.07 |
| Without insert | 151 ± 60 | 0.16 | |
| 60 | With insert | 129 ± 31 | 0.06 |
| Without insert | 140 ± 49 | 0.12 | |
| 70 | With insert | 127 ± 33 | 0.07 |
| Without insert | 148 ± 51 | 0.12 | |
| 80 | With insert | 133 ± 33 | 0.06 |
| Without insert | 150 ± 51 | 0.12 | |
As it was confirmed in the previous section by the acetal cleavage competitive reaction, the increase in the total flow rate and correspondingly decrease in the residence time, promoted a more homogenous emulsification process for both case studies, i.e. both with and without the insert. However, the presence of the insert notably improved the sample size homogeneity (Table 3).
Specifically, the mean size of polymeric nanoparticles decreased from 164 ± 68 nm to 140 ± 49 nm when no insert was used at the flow rates of 40 and 60 mL min−1, respectively (Fig. 6-A and Table 3). On the other hand, the presence of the insert resulted in a mean size reduction from 157 ± 52 to 129 ± 31 at 40 and 60 mL min−1, respectively (Fig. 6-A and Table 3). When comparing the effect of the residence time on the particle size, the size obtained at residence times < 200 ms was smaller in the case of using the insert. This evidence confirmed that the emulsion droplet size achieved by using the insert was due to the higher shear stress boosted by a high mixing efficiency. The advantage of using the insert can be rationalized because at similar viscous forces, the higher the shear stress is, the smaller is the resulting emulsion droplet size,19 and therefore the nanoparticle size.
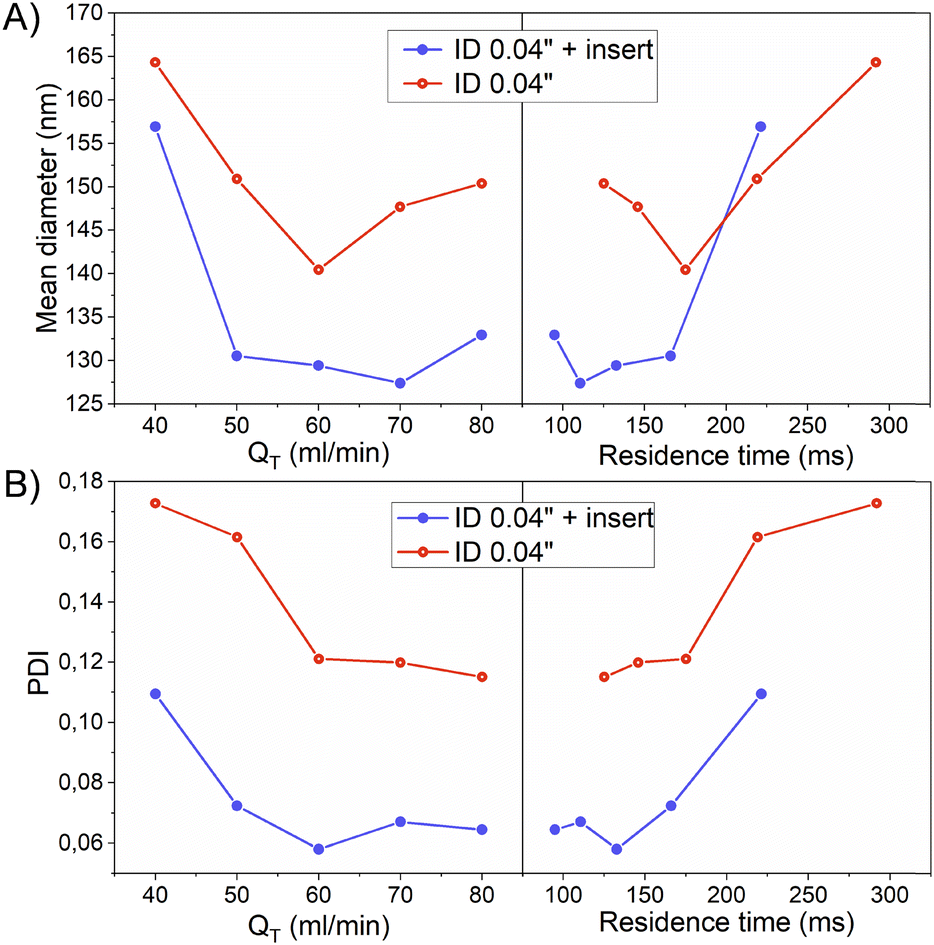 | ||
| Fig. 6 Effect of QT (mL min−1) and residence time (ms) on polymeric NP mean diameter (A) and polydispersity index (B) considering the presence/absence of an insert. All the emulsions were produced with a constant QO/QA flow ratio of 1/12 and the following composition: QO: PLGA and Eudragit (0.5% w/v QO) each, Pluronic (3% w/v QO) in ethyl acetate; QA: water. The data represent the mean values derived from 3 replicated experiments. | ||
The presence of the insert not only promoted a reduction of the emulsion droplet size, but also decreased the heterogeneity of the emulsion at all the flow rates compared (Fig. 5 and 6-A). The PDI is a parameter that is generally used to estimate the average uniformity and broadness of a particle population. The value of PDI ranges from 1 when a sample is highly polydisperse and with multiple particle size populations to ideally a value of 0 for a perfectly uniform sample with respect to the particle size. It is well considered that values of 0.2 and below are acceptable in practice for polymer-based nanoparticle materials.23 The PDI curve obtained when the insert was used was under the PDI curve of the case without the insert, decreasing the PDI from 0.17 to 0.11, from 0.16 to 0.07, and from 0.12 to 0.05, at 40, 50, and 60 mL min−1, respectively (Fig. 6-B and Table 3). The same observation could be noted when comparing the effect of the residence time on the PDI while using the insert (Fig. 6-B and Table 3). It was concluded that a residence time smaller than 130 ms was enough to decrease the PDI to 0.05 and achieve a mean size of 130 nm.
Previous results were based on SEM imaging, an analysis technique with a high accuracy to determine the nanoparticle size, but with some limitations since the number of particles analysed is in the local range and cannot provide any information about aggregation or agglomeration phenomena; beside the measurements were provided for dry samples. Thus, we also considered DLS as a complementary technique, as it is able to provide information about a sample in bulk range and in a wet environment. Although DLS provides the particle size as a hydrodynamic radius (considering the solvation shell), it is subjected to the presence of measurement artefacts due to the overestimation of larger particles.24 Then, as became evident from above, the combination of several techniques gave the most realistic information about the sample quality. Fig. S2† depicts the particle size histograms provided by DLS after the analysis of the polymeric nanoparticles obtained at different volume flow rates. In general, there was a good agreement between the DLS and SEM results in terms of the size distribution, obtaining that the narrowest size distribution was when the insert was used. According to DLS histograms, the presence of the insert during the emulsification process also confirmed the reduction of the emulsion droplet size since the size of nanoparticles was smaller. Finally, the quality of the nanoparticles produced in terms of their mean size and homogeneity was similar when comparing the outcome of the experiments at 70 and 80 mL min−1, but the pressure drop at these fluid dynamic conditions was too high and some instabilities were observed.
The interaction of the emulsion components with the microfluidic system interfaces can result in fouling, creating additional mass flow resistance and increasing the pressure drop. This phenomenon can alter the fluid dynamics and even block microfluidic systems.
Also, regeneration of the microfluidic components is key to preserve the proper operation. However, not all microfluidic systems are easy to regenerate by surface cleaning due to the compactness of these microstructured systems, whereby the cleaning agent (generally organic solvents and acids) cannot get access to the blockage. In this particular case, the insert could be easily dissembled from the PTFE tubing, regenerated in a beaker, and then re-assembled again without affecting the mixing performance, as we demonstrated to study the reproducibility of each experimental condition.
Finally, the production throughput required for clinical translation of nanoparticles targeted for biomedical use is still a challenge, being highly desired the design of high-throughput procedures.12 The insert enabled a robust operation at flow rates as large as 60–80 mL min−1, while the nanoparticles quality could be preserved. The estimated productivity values were 11.1 and 14.8 g h−1 at total volume flow rates of 60 and 80 mL min−1, respectively. Considering that the production rates typically required for biomedical applications in clinical studies and the industrial-scale production of nanoparticles are in the order of 4 and 40 g h−1, respectively,25 it is remarkable that a single insert could fulfil the production for clinical studies and, by numbering up the inserts, the industrial scale could also be feasible.
Conclusions
We reported a metallic insert as a passive mixer that has interesting features as a passive micromixing platform: straightforward to adapt to the conventional tubing widely used in microfluidics, easy numbering-up to increase the throughput, has no moving parts, robust, compact, easy microfabrication and cost effectiveness, modular design, high mixing efficiency, complete regeneration in case of fouling, and does not require skilled users to assure an efficient operation. The mixing efficiency was validated by the acetal cleavage method and the emulsification of a mixture of biomedical polymers to yield monodisperse nanoparticles. The presence of the insert enhanced the degree of mixing by passively inducing a high shear stress and diffusive advection. The insert could promote an efficient mixing of immiscible streams at residence times smaller than 130 ms, obtaining nanoparticles with a mean size of 130 nm and PDI as a small as 0.05. Finally, the throughput is suitable for in vivo studies and could be feasible for industrial scale by numbering up.Author contributions
L. A. performed and planned the experiments of emulsification, did the data curation, contributed to the interpretation of the results and wrote the manuscript. S. P. performed and planned the experiments of acetal cleavage reaction did the data curation, and contributed to the interpretation of the results. V. G. performed the insert microfabrication. V. S. conceived the present idea, designed the experiments, work out in the insert manufacturing, supervised the experiments and the project, was in charge of overall direction and planning, contributed to the interpretation and wrote the manuscript in consultation with L. A., S. P. and V. G.Conflicts of interest
There are no conflicts to declare.Acknowledgements
VS acknowledge funding from project PID2021-127847OB-I00 MCIN/AEI/10.13039/501100011033 and PDC2022-133866-I00 MCIN/AEI/10.13039/501100011033 (Unión Europea Next GenerationEU/PRTR). We also thank CIBER-BBN, an initiative funded by the VI National R&D&i Plan 2008–2011 financed by the Instituto de Salud Carlos III and by Fondo Europeo de Desarrollo Regional (Feder) ‘Una manera de hacer Europa’, with the assistance of the European Regional Development Fund. LMA-ELECMI and NANBIOSIS ICTs are gratefully acknowledged.Notes and references
- V. Sebastian, Toward Continuous Production of High-Quality Nanomaterials Using Microfluidics: Nanoengineering the Shape, Structure and Chemical Composition, Nanoscale, 2022, 14(12), 4411–4447, 10.1039/D1NR06342A.
- L. Falk and J.-M. Commenge, Performance Comparison of Micromixers, Chem. Eng. Sci., 2010, 65(1), 405–411, DOI:10.1016/j.ces.2009.05.045.
- V. Hessel, H. Löwe and F. Schönfeld, Micromixers—a Review on Passive and Active Mixing Principles, Chem. Eng. Sci., 2005, 60(8–9), 2479–2501, DOI:10.1016/J.CES.2004.11.033.
- A. Maged, R. Abdelbaset, A. A. Mahmoud and N. A. Elkasabgy, Merits and Advances of Microfluidics in the Pharmaceutical Field: Design Technologies and Future Prospects, Drug Delivery, 2022, 29(1), 1549–1570, DOI:10.1080/10717544.2022.2069878.
- G. Cai, L. Xue, H. Zhang and J. Lin, A Review on Micromixers, Micromachines, 2017, 8(9), 274, DOI:10.3390/mi8090274.
- F. Reichmann, K. Vennemann, T. A. Frede and N. Kockmann, Mixing Time Scale Determination in Microchannels Using Reaction Calorimetry, Chem. Ing. Tech., 2019, 91(5), 622–631, DOI:10.1002/cite.201800169.
- J. Aubin, M. Ferrando and V. Jiricny, Current Methods for Characterising Mixing and Flow in Microchannels, Chem. Eng. Sci., 2010, 65(6), 2065–2093, DOI:10.1016/j.ces.2009.12.001.
- A. Rave, L. Schaare and G. Fieg, Investigation of Micromixing in the ART Plate Reactor PR37 Using the Acetal Cleavage Method and Different Mixing Models, Chem. Eng. Process.: Process Intensif., 2022, 181, 109134, DOI:10.1016/j.cep.2022.109134.
- C. Lindenberg, J. Schöll, L. Vicum, M. Mazzotti and J. Brozio, Experimental Characterization and Multi-Scale Modeling of Mixing in Static Mixers, Chem. Eng. Sci., 2008, 63(16), 4135–4149, DOI:10.1016/j.ces.2008.05.026.
- A. Larrea, A. Clemente, E. Luque-Michel and V. Sebastian, Efficient Production of Hybrid Bio-Nanomaterials by Continuous Microchannel Emulsification: Dye-Doped SiO2 and Au-PLGA Nanoparticles, Chem. Eng. J., 2017, 316, 663–672, DOI:10.1016/j.cej.2017.02.003.
- C. Solans, P. Izquierdo, J. Nolla, N. Azemar and M. J. Garcia-Celma, Nano-Emulsions, Curr. Opin. Colloid Interface Sci., 2005, 10(3), 102–110, DOI:10.1016/j.cocis.2005.06.004.
- I. O. De Solorzano, L. Uson, A. Larrea, M. Miana, V. Sebastian and M. Arruebo, Continuous Synthesis of Drug-Loaded Nanoparticles Using Microchannel Emulsification and Numerical Modeling: Effect of Passive Mixing, Int. J. Nanomed., 2016, 11, 3397–3416, DOI:10.2147/IJN.S108812.
- M. Schoenitz, L. Grundemann, W. Augustin and S. Scholl, Fouling in Microstructured Devices: A Review, Chem. Commun., 2015, 51(39), 8213–8228, 10.1039/C4CC07849G.
- M. Juraeva and D. J. Kang, Mixing Enhancement of a Passive Micromixer with Submerged Structures, Micromachines, 2022, 13, 1050, DOI:10.3390/mi13071050.
- J. Baldyga, J. R. Bourne and B. Walker, Non-Isothermal Micromixing in Turbulent Liquids: Theory and Experiment, Can. J. Chem. Eng., 1998, 76(3), 641–649, DOI:10.1002/cjce.5450760336.
- A. Kölbl and M. Kraut, Characterization of a Temperable Mixer with the Acetal Cleavage Method, Chem. Eng. J., 2013, 227, 22–33, DOI:10.1016/j.cej.2012.11.124.
- G. R. Wang, F. Yang and W. Zhao, There Can Be Turbulence in Microfluidics at Low Reynolds Number, Lab Chip, 2014, 14(8), 1452–1458, 10.1039/C3LC51403J.
- K. J. Hecht, A. Kölbl, M. Kraut and K. Schubert, Micromixer Characterization with Competitive-Consecutive Bromination of 1,3,5-Trimethoxybenzene, Chem. Eng. Technol., 2008, 31(8), 1176–1181, DOI:10.1002/ceat.200800213.
- S. M. Jafari, E. Assadpoor, Y. He and B. Bhandari, Re-Coalescence of Emulsion Droplets during High-Energy Emulsification, Food Hydrocolloids, 2008, 22(7), 1191–1202, DOI:10.1016/j.foodhyd.2007.09.006.
- H. Lu, L. Y. Koo, W. M. Wang, D. A. Lauffenburger, L. G. Griffith and K. F. Jensen, Microfluidic Shear Devices for Quantitative Analysis of Cell Adhesion, Anal. Chem., 2004, 76(18), 5257–5264, DOI:10.1021/ac049837t.
- M. Rousta, E. Shirani and Z. Habibi, A Comprehensive Study on the Characterization of Chaotic Advection Flow and Heat Transfer in a Twisted Pipe with Elliptical Cross-Section, Results Eng., 2022, 13, 100323, DOI:10.1016/j.rineng.2021.100323.
- K. Y. Hernández-Giottonini, R. J. Rodríguez-Córdova, C. A. Gutiérrez-Valenzuela, O. Peñuñuri-Miranda, P. Zavala-Rivera, P. Guerrero-Germán and A. Lucero-Acuña, PLGA Nanoparticle Preparations by Emulsification and Nanoprecipitation Techniques: Effects of Formulation Parameters, RSC Adv., 2020, 10(8), 4218–4231, 10.1039/C9RA10857B.
- M. Danaei, M. Dehghankhold, S. Ataei, F. Hasanzadeh Davarani, R. Javanmard, A. Dokhani, S. Khorasani and M. R. Mozafari, Impact of Particle Size and Polydispersity Index on the Clinical Applications of Lipidic Nanocarrier Systems, Pharmaceutics, 2018, 10(2), 57, DOI:10.3390/pharmaceutics10020057.
- H. Hinterwirth, S. K. Wiedmer, M. Moilanen, A. Lehner, G. Allmaier, T. Waitz, W. Lindner and M. Lämmerhofer, Comparative Method Evaluation for Size and Size-Distribution Analysis of Gold Nanoparticles, J. Sep. Sci., 2013, 36(17), 2952–2961, DOI:10.1002/jssc.201300460.
- J.-M. Lim, A. Swami, L. M. Gilson, S. Chopra, S. Choi, J. Wu, R. Langer, R. Karnik and O. C. Farokhzad, Ultra-High Throughput Synthesis of Nanoparticles with Homogeneous Size Distribution Using a Coaxial Turbulent Jet Mixer, ACS Nano, 2014, 8(6), 6056–6065, DOI:10.1021/nn501371n.
Footnotes |
| † Electronic supplementary information (ESI) available: Acetal cleavage method data, SEM images and DLS analysis. See DOI: https://doi.org/10.1039/d4re00033a |
| ‡ This manuscript was sent for Celebration of Klavs Jensen's 70th Birthday. Prof Klavs Jensen has been an inspiration and support for the scientific community. |
| This journal is © The Royal Society of Chemistry 2024 |