
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Automated multistep synthesis of 2-pyrazolines in continuous flow†‡
Ricardo
Labes§
 a,
Julio C.
Pastre
*ab,
Richard J.
Ingham¶
a,
Claudio
Battilocchio||
a,
Henrique M.
Marçon
b,
Mariana C. F. C. B.
Damião
b,
Duc N.
Tran**
a and
Steven V.
Ley
*a
a,
Julio C.
Pastre
*ab,
Richard J.
Ingham¶
a,
Claudio
Battilocchio||
a,
Henrique M.
Marçon
b,
Mariana C. F. C. B.
Damião
b,
Duc N.
Tran**
a and
Steven V.
Ley
*a
aYusuf Hamied Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: svl1000@cam.ac.uk
bInstitute of Chemistry, University of Campinas (UNICAMP), 13083-970, Campinas, SP, Brazil. E-mail: jpastre@unicamp.br
First published on 9th November 2023
Abstract
The rapid multistep synthesis of 2-pyrazolines from aldehydes via [3 + 2] cycloaddition of unstabilised diazo species and mono and di-substituted alkenes is reported. The development encompasses semi-continuous and fully continuous methods, including a fully automated compound library generation. A graphical interface was developed for the automation control, making it more accessible for chemists without coding experience.
Introduction
Modern society relies heavily on the preparation and application of novel functional molecules. The exploration and discovery of new chemical reactivity patterns and new tools for molecular assembly are essential drivers in achieving these goals. Enabling technologies can serve in expediting this process and facilitating collaborations across disciplines. For instance, enabling tools such as continuous flow methods can provide precise control over specific reaction parameters.1–4 Flow chemistry concepts can also offer fast optimisation of synthesis protocols, that would otherwise demand laborious manual experimentation. We, as well as many others, have firmly established the power of using these methods to create opportunities for chemical processing in research laboratory settings and to assist in reaction scale-up.3–10In particular, we have been stressing the importance of machine assistance, to shift the workload in a research laboratory, to relieve staff from routine and repetitive tasks. Notably, the use of open-source technologies allows us to go beyond a ‘single-machine’ and enable iterative development of complex flow systems. Notwithstanding the improved levels of repeatability inherent in a semi-automated system.11–14 Multi-step continuous flow synthesis still poses a significant challenge.15–18 However, the ability to incorporate multiple pieces of equipment in a coordinated network of laboratory devices means it is possible to devise telescoped synthesis platforms and downstream processing operations that might otherwise be especially difficult during multi-step batch mode operation.
A distinct advantage when performing transformations in continuous flow is the ability to handle reactive or short-lived intermediates and safely determine the destiny of these species without isolation or exposure to these hazardous materials. This ability to control reactive intermediates affords greater flexibility in processing, by defining product outcomes or aiding scale-up.
We have previously reported a machine assisted approach to generate and utilise unstabilised diazo compounds safely, which greatly enhanced the applicability of such hazardous and reactive molecules. This method has been applied to cross couplings,19,20 including an iterative implementation,21 cyclopropanations,12 C–H functionalisation of aldehydes22 and synthesis of allenes23,24 (Scheme 1). We wished to extend the use of unstabilised diazo compounds to prepare 2-pyrazolines and increase the level of automation using a multistep platform, capable of compound library generation and scale-up (Scheme 1).
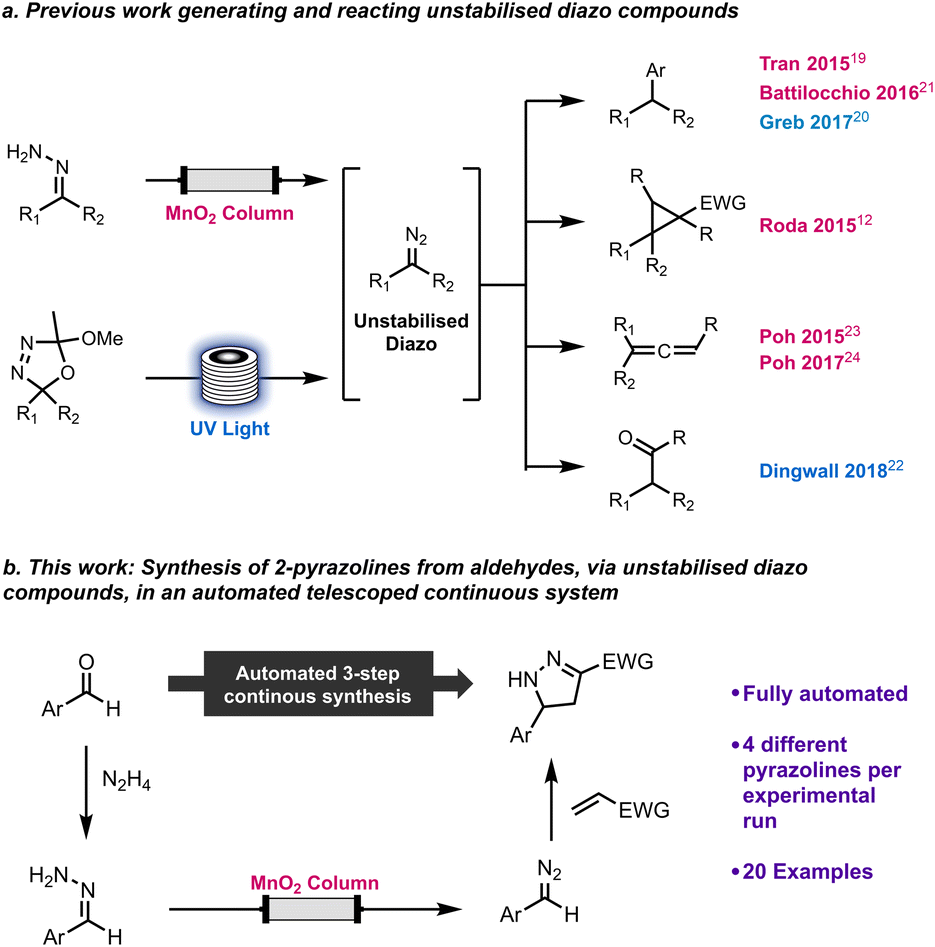 | ||
| Scheme 1 Previous work handling unstabilised diazo compounds (a) and this work on the multi-step synthesis of 2-pyrazolines from aldehydes (b). | ||
Pyrazolines occur widely in natural products and bio-active molecules.25–28 Notably, 2-pyrazolines are common motifs in pharmaceuticals agents25 owing to their biological profiles as anti-fungal, anti-inflammatory, anti-convulsant and anti-cancer agents.29 These species are also found in commercial agrochemicals and as important intermediates in the preparation of pyrazoles as their corresponding aromatic counterparts.30–33
The traditional method for the preparation of 2-pyrazolines involves the reaction of a hydrazine with an appropriate coupling partner.34,35 Alternatively a one-pot condensation between a hydrazine, an aldehyde and a ketone can be used for the preparation of these heterocycles.30 Other more recent methods have been developed for the construction of pyrazolines and its derivates, such as transition-metal catalysed reactions with copper, zinc and palladium36–38 or the use of sulfur ylides which may be converted to pyrazolines, as reported by Fang and co-workers via a formal [4 + 1] annulation.39 Aziridines have also been reported as starting materials for the synthesis of certain pyrazolines.40 A photochemical method was recently reported for the synthesis of spiro-Δ2-pyrazolines.41 This approach involved the cycloaddition of a diazo compound with an electron-deficient alkene, and, unlike previous studies that required refluxing of tosylhydrazones and base for longer times,42,43 achieved the desired reaction under mild conditions.
Continuous flow methods have also been reported, one study utilised tert-butyl nitrite and fluorinated amines to generate diazoalkanes, followed by a [3 + 2] cycloaddition,44 while another report generated various diazo compounds from their respective hydrazones with a silver(I) column and reacted further with various carboxylic acids and alkenes to give different functionalities, including pyrazolines.45
We report below the use of a modular system to generate unstabilised diazo compounds on demand then coupled these with electron deficient alkenes to obtain a small library of 2-pyrazolines via [3 + 2] cycloaddition. The system was further developed into a fully automated platform capable of preparing and collecting 4 different products in one experiment. The method was also scaled-up to afford 7.71 g of one of these products (8) in 2.5 hours.
Results and discussion
This study began by evaluating the preparation of 2-pyrazolines from hydrazones in flow, building on a previously-reported protocol19 in which a hydrazone solution was passed through a column containing MnO2 powder to generate the respective diazo intermediate, which was then combined with an electron-deficient olefin to afford the desired 2-pyrazoline.Our original protocol19 included a conditioning phase of the packed-bed column using a solution of organic base. We believe that this step neutralises residual acidic areas on the surface of the manganese dioxide that would otherwise hinder reactivity. Previously, this was performed using a solution of the hydrazone and diisopropylethylamine (DIPEA) over 20 minutes (10 mL of a 0.1 M solution with 2 equivalents of DIPEA to a column with 0.9 g of MnO2). We were able to improve upon this method, by substituting these reagents for a solution of triethylamine (TEA) in methanol, which afforded an equivalent activation in just 3 minutes. In addition, this activation could be performed prior to, rather than along with, injection of the hydrazone, meaning that the hydrazone intermediate could be fully utilised. By comparison to DIPEA, the methanolic solution of triethylamine is a lower-cost reagent, and is more volatile, so the residual base can be removed via rotary evaporation.
The choice of solvent (THF/MeOH 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was informed by preliminary experiments for preparing the hydrazone in continuous flow (see ESI,‡ S3). In our initial experiments, the hydrazone solutions were introduced into the flow stream using an injection loop, so as to make a discrete and precise amount of the reactive intermediate (Scheme 2). The presence of the high-energy diazo in solution was indicated by a characteristically bright colour, varying between orange, pink and red depending on the particular diazo compound. The diazo intermediate was directed into a round bottom flask that was then charged with a solution of a reactive olefin; a [3 + 2] reaction of the diazo intermediate with the olefin within the collection vessel afforded the desired 2-pyrazoline. For safety, the collection vessel was maintained under inert atmosphere (N2), at 0 °C and behind a safety screen.
:1) was informed by preliminary experiments for preparing the hydrazone in continuous flow (see ESI,‡ S3). In our initial experiments, the hydrazone solutions were introduced into the flow stream using an injection loop, so as to make a discrete and precise amount of the reactive intermediate (Scheme 2). The presence of the high-energy diazo in solution was indicated by a characteristically bright colour, varying between orange, pink and red depending on the particular diazo compound. The diazo intermediate was directed into a round bottom flask that was then charged with a solution of a reactive olefin; a [3 + 2] reaction of the diazo intermediate with the olefin within the collection vessel afforded the desired 2-pyrazoline. For safety, the collection vessel was maintained under inert atmosphere (N2), at 0 °C and behind a safety screen.
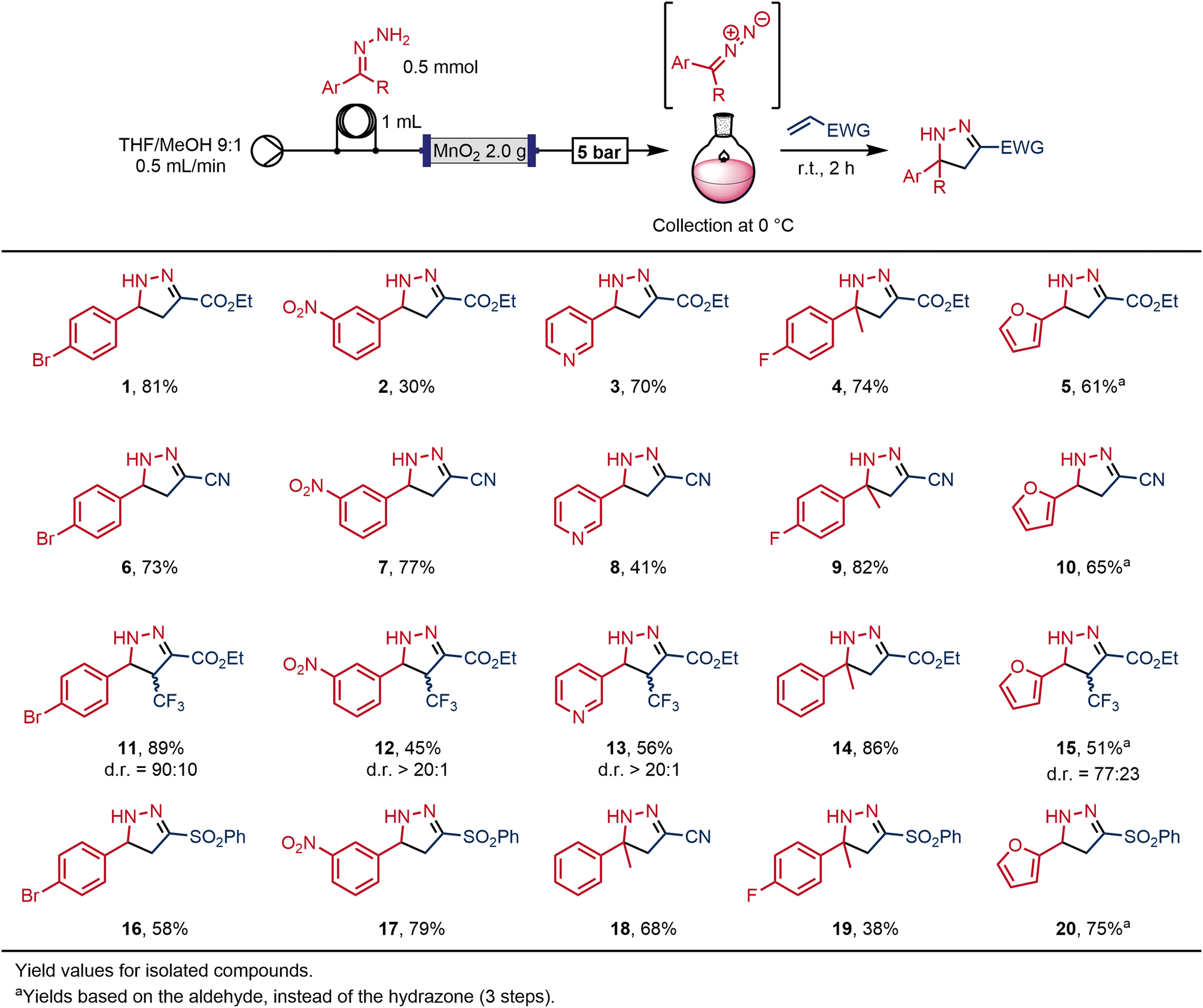 | ||
| Scheme 2 Synthesis of 2-pyrazolines from hydrazones. The specific reactor configuration comprised of a pump, a six-port injection valve with a sample loop, a packed-bed column, and a back-pressure regulator. | ||
This approach allowed a rapid generation of different products while avoiding any requirement to collect or isolate the diazo compound, and did not require any hardware changes between experiments using different reagents. In each case, once the collection vessel had been removed, the system was washed for a further 5 minutes with the solvent mixture before injecting the following hydrazone into the sample loop and beginning the next experiment.
The method was reliable and showed high levels of robustness: we were able to quickly and safely prepare a library of twenty 2-pyrazolines with moderate to excellent yields (Scheme 2). Interestingly, the dipolarophile could be added before, after or during the diazo collection, yielding the same results.
The reaction outcome could be expected to follow the matching molecular orbitals energies driving the regioselectivity of the cycloaddition reaction46 and, in general, this was found to be the case. The best conversions for all diazo compounds were achieved with acrylonitrile and ethyl acrylate as the dipolarophile partner. By contrast, phenyl vinyl sulfone led to lower yields of the corresponding pyrazolines. With respect to the diazo compounds, the presence of electron-donating groups, such as furane, resulted in lower yields. However, improved yields with the electron-rich diazo partner with the least electron-poor alkenes when realised in contrast to diazo compounds with more electron withdrawing substituents.
Telescoped 3-step continuous synthesis
With a view to extending this flow process to give access to a wider variety of 2-pyrazolines from commercially available starting materials, we studied the formation of the initial hydrazones from the respective aldehydes and a commercial solution of hydrazine (1 mol L−1 in THF).We immediately identified a material incompatibility between the hydrazine reagent and the flow system: when a solution of hydrazine was infused continuously through the reciprocating piston pump (in this case on the Vapourtec R-series), the performance of the pump would become compromised after only a short period of operation. Swelling and degradation of its polymeric secondary seals in the presence of hydrazine solution gave rise to particulates that caused the check valves to malfunction.
This problem was mitigated by removing the secondary seals from the hydrazine pump and including 10% methanol in the solvent system. With this modification in place no further issues were encountered. Fortunately, the operation of the pumps was not affected. In order to keep the primary seal clean, the back of the pump was flushed with solvent periodically using the flushing ports to remove the hydrazine reagent collecting in this area (for more information see ESI‡).
We equipped the system with an in-line infra-red spectrometer (Mettler Toledo FlowIR®), and this meant that the progress of the reaction could be monitored in real-time by observation of the carbonyl stretching region around 1700 cm−1. Thus, the FlowIR enabled rapid optimisation of the reaction conditions until full conversion of the aldehyde was achieved. Using 4-bromobenzaldehyde as a model substrate, optimised conditions were developed whereby the aldehyde was combined with 1.2 equivalents of the hydrazine. A subsequent residence time of 10 minutes in a coil reactor held at 70 °C and 100 psi led to full conversion to the corresponding hydrazone (Scheme 3).
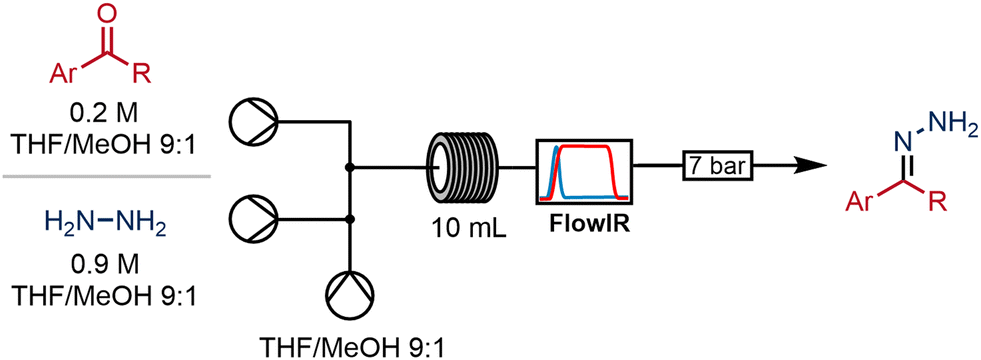 | ||
| Scheme 3 Continuous synthesis of hydrazones from aldehydes. | ||
With a process for generating the hydrazone reagents in situ from an aldehyde starting material in hand, we turned our attention to integrating this step with the diazo formation reaction. With both steps proceeding in the same solvent system and at comparable concentrations and flow rates, we had hoped that this would prove to be a trivial modification. However, matching the concentration profile of the diazo species with the sequential [3+2] cycloaddition reaction presented more of a challenge. The combined effects of the flow profile broadening resulting from the passage of the hydrazone through a packed column, and the lifetime of the activity of the MnO2 itself, meant that the concentration of the diazo compound followed an angled plateau-like profile. Under the specific flow conditions, the system took approximately 20 minutes to reach a steady state. If we were to wait for the steady-state condition to be reached before introducing the dipolarophile coupling partner, this could lead to significant material wastage – a particular issue if we wished to apply this system to a precious starting material.
We have approached this kind of problem previously in two different ways: either by using an automated pump that matched the concentration distribution of two reagents, or by using a reservoir to collect a stock of an intermediate, allowing any temporal differences in concentration to be averaged out before subsequent reactions.11,47 Whilst the high-energy diazo intermediate is not an obvious candidate for storage, we reasoned that a suitable reservoir that maintained the solution at low temperatures and under inert atmosphere could protect the unstable diazo compound over its lifetime. The presence of a reservoir would add modularity to the system such that we could use the same batch of diazo material in a diverse set of downstream reactions.
Thus, a custom-built glass attachment (see ESI‡) was mounted onto the pumping device. This then allowed the diazo intermediate to be collected at −5 °C and under a nitrogen atmosphere. We were pleased to find that the solution could be stored and pumped with no degradation being observed (see ESI‡). Due to the potential risk handling diazo compounds,48 only small amounts of the solution were kept in the double jacketed reservoir at −5 °C and nitrogen purged. The solution was prepared and immediately used in the following step and at no stage was it handled by the operator or concentrated.
In an initial test, 1-bromo-4-(diazomethyl)benzene was generated from the respective hydrazone, and subsequently pumped from the reservoir (at −5 ° C) and combined with ethyl acrylate in a tee-mixer through a 10 mL tubular reactor at 60 ° C and 1 mL min−1 (0.5 mL min−1 each pump). After removal of the solvent the product was obtained in 98% yield (using the olefin as the limiting reagent and 1.2 equivalents of diazo; yield value obtained using 1H NMR and internal standard). By repositioning the FlowIR unit we were able to follow the IR bands associated with the diazo before and after the storage in the reservoir; pleasingly, this showed no sign of decomposition.
With the addition of a multi-way selection valves, the system was then ready to prepare and collect the different 2-pyrazolines.
Automation
The automation system used for this work included a graphical user interface (GUI) developed by our group for the octopus automation project previously reported,11,49 which provides an abstraction layer allowing several different instruments to interact within a single experimental protocol. Previously, these protocols had to be defined using the Python programming language, but with the addition of the new graphical layer, this functionality can be accessible to researchers without significant programming experience. The GUI (referred to as Blocktopus) is built upon the Blockly project,50 which was created as a tool for learning software development, wherein programs can be assembled from jigsaw-puzzle-like ‘blocks’.This paradigm translated very appropriately into flow automation because, by application of only a few basic operations, programs based on sequences of steps can be prototyped rapidly. Each block corresponds to a control operation for a particular instrument, or control logic for the experiment. Stacks of blocks are executed in series, and the GUI provides real-time feedback to indicate which block or blocks are currently executing. Crucially, the blocks' parameters and even the control logic can be modified even during an experiment, creating a much closer experience to a hands-on experiment and an experience which is augmented by the automation software rather than controlled by it.
In this work, the flow equipment was connected using RS-232 via an ethernet-to-serial interface box, which were found to be much more reliable for long-term use than a direct USB-to-serial adapter. In the software, each connection is then defined by the IP address and relevant port number of the interface box. The software is pre-configured with the various components and parameters of each type of machine, and blocks corresponding to these parameters are made available allowing their values to be read and modified as part of the procedure.
For example, Fig. 1 shows an instrument definition and a stack of blocks that will be executed in sequence from top to bottom. Changes to the instrument set-points are take effect within a few milliseconds, being limited only by the latency of the RS-232 connection and the response time of the instrument. Where an operation may take some time to complete, such as a delay (‘wait’) block. In this case, the set of blocks will wait for 5 minutes, and then for the thermocouple attached to the first heater element of an R2+/R4 instrument to report a particular temperature, before switching the inlet valves from solvent to reagent.
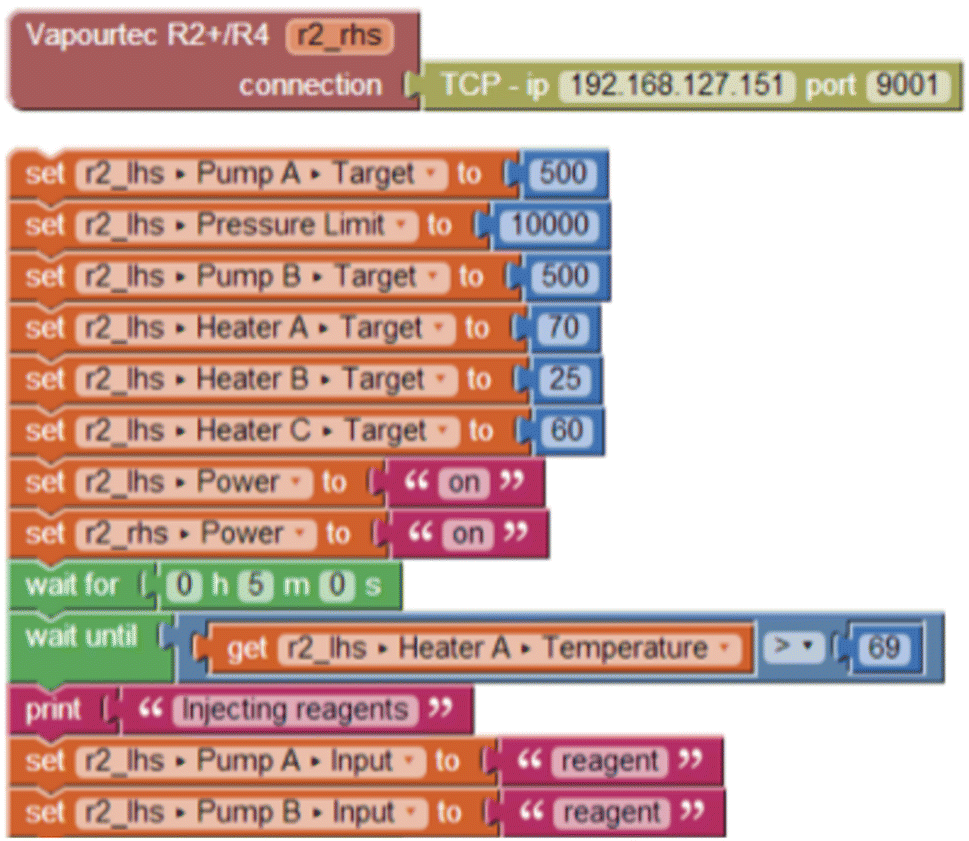 | ||
| Fig. 1 Example of automated experiment using Blocktopus user interface. | ||
During an experimental run, all sensor readings from each machine are stored to the hard disk, and can be monitored remotely, and downloaded as a spreadsheet afterwards. Further aspects of this control software are available in the ESI.‡
Automated telescoped system
An automated platform was constructed to generate a library of pyrazolines following the protocols described earlier in this report. Two Vapourtec R2+/R4 systems were used: the first provided two pumps, one coil heater, two low-pressure reagent switching valves and one low-pressure collection valve. The second provided a further two pumps and a second coil heater. A Mettler-Toledo FlowIR sensor monitored the level of diazo and aldehyde stretching absorbance in the output stream, and a VICI column-switching valve was used for the selection of the second-stage reagents and the collection output.An aldehyde solution (0.2 mol L−1) in 10% MeOH/THF with TEA (1 equivalent) was mixed with a solution of hydrazine (0.24 mol L−1 10% in MeOH/THF) through a 10 mL reactor coil at 70 °C. The coil output was connected directly to a packed column of MnO2 (3 g) and the reaction course followed using the FlowIR device. When the diazo was detected by a threshold amount, a valve placed after the in line analytical device was switched from waste to the collection reservoir. The diazo was temporarily stored at −5 °C in a closed system under inert atmosphere. Collection was stopped upon depletion of the oxidizing power of the MnO2 – defined as the level of diazo signal detected by the FlowIR dropping below a pre-set threshold (Scheme 4).
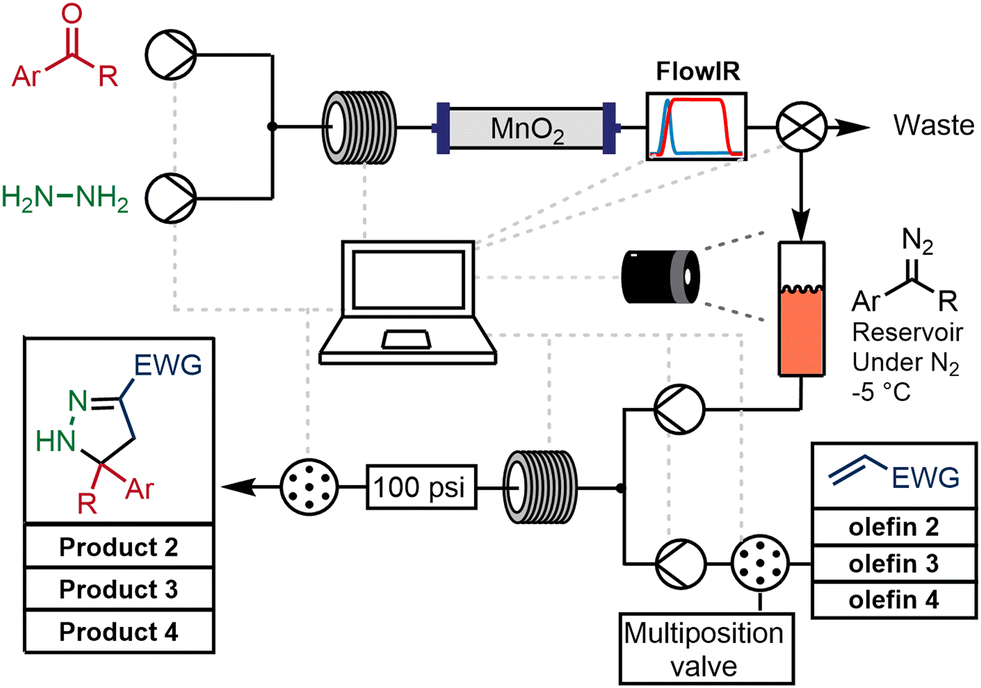 | ||
| Scheme 4 Automated continuous flow chemistry approach for the assembly of a library of 2-pyrazolines. | ||
It is important to note that the sampling protocol reliably generated a library for each diazo compound with a varying alkene input. The sampling method could also be initiated prior to completion of the first step thereby reducing the amount of stored diazo intermediate. Once a small amount of the diazo (approximately 5 mL) was present in the intermediate reservoir the cycloaddition step could begin. A proof-of-concept experiment was performed using 4-bromobenzaldehyde, and 4 different olefins by simply selecting different alkenes and collecting the different products through selection valve transfer (Scheme 4). The results from these fully automated experiments were in agreement with previous observations using batch techniques.
Scale-up experiment
An advantage of continuous-flow synthesis is the potential to scale up reactions simply by running the reactor system for longer. However, this is sometimes not possible where solid reagents are involved, as they are present in a finite quantity within the system. This is also the case for this 2-pyrazoline synthesis process. Increasing the size of the column would at some point become impractical, and also considering the dispersive effect of the packed-bed column, we decided instead to evaluate a two-column system.For this, a switching valve was introduced such that during the process, while one column was being used to generate the diazo compound, the other was being conditioned with a solution of base in 10% MeOH/THF. In this experiment DIPEA was used, but both DIPEA and TEA provided similar outcomes. After depletion of the first column (observed by the FlowIR) valves can be switched, such that the diazo compound was being generated from the freshly conditioned second MnO2 column. Subsequently, the depleted reactor column was removed for re-conditioning off-system. We can envisage a different valve setup such that the re-conditioning could be performed automatically by an additional set of pumps.
This system was used to scale up the diazo generation, reducing time for necessary pre-conditioning of the oxidizer column. More specifically, the hydrazone from 3-pyridinecarboxaldehyde was oxidised then coupled with acrylonitrile as an illustrative example. The corresponding diazo intermediate was collected in a reservoir in line, then transferred and mixed in a tee-piece with a solution of acrylonitrile in THF and reacted in a coil reactor at 60 °C for 23 minutes (Scheme 5). This experiment consumed nine reloaded 7.3 g MnO2 columns and generated 7.71 g of the pyrazoline 8 in 73% yield for the 2 steps and with an overall productivity of 3.1 g h−1. The reaction time in the last reactor was not optimized, but afforded essentially the same yield obtained in the semi-continuous approach.
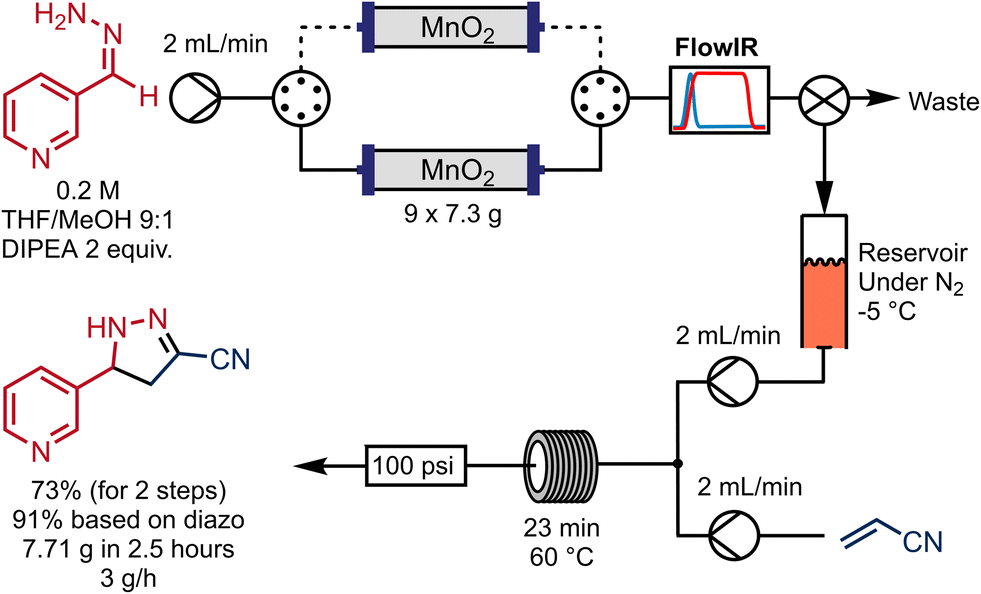 | ||
| Scheme 5 Multi-column setup for scale out of the pyrazoline synthesis using a pre-conditioned MnO2 columns. | ||
Implementation in an industrial setting
We also became interested in transferring these flow chemistry technologies to wider audiences and, in particular, situations where larger scale processing is of interest and where safety becomes a major issue. The instability and toxicity of diazo compounds such as those reported in this work presents a particular challenge within an industrial setting.With a goal of implementing this reaction process in the laboratories of Pfizer R&D, we further modified the above procedure to remove the intermediate-storage aspect. Whilst this removes the advantages of this element that are described above for small scale operations, within an industrial setting it could present an undesirable safety hazard when the process is scaled up. Instead, we implemented an alternative method to solve the variable reagent concentration profile of the generated diazo intermediates by introducing an additional pump for the olefin reagent, and matching the stoichiometry of the reagents by controlling the pumping rate based on the measured concentration of the diazo intermediate.
We encountered a challenge during the detection of the diazo intermediate by infrared spectrometry using equipment available in Pfizer's laboratory at the time. The standard diamond-windowed IR sensor has a strong intrinsic absorption between 2000 and 2250 cm−1, which can mask the typical absorption band of the diazo compounds at around 2070 cm−1. We therefore chose to infer the degree of conversion from the diazo compound by observing the disappearance of the aldehyde stretching frequency at 1713 cm−1.
We began, therefore, by measuring the response from a diamond-windowed infrared spectrometer when an aldehyde reagent had progressed through the reactor, recording the signal response to different concentrations of input. We identified a characteristic peak for p-bromobenzaldehyde at 1713 cm−1. Next, we constructed the first half of the reactor platform to measure the dispersion effect of a reactor coil and the column of heterogeneous manganese dioxide. By switching between solvent only or the hydrazine reagent on the second stream, we could observe the aldehyde being fully consumed when the hydrazine was present (Fig. 2). Hydrazine was generally used in excess and the manganese dioxide column served to safely decompose any residual hydrazine into nitrogen gas as well as generating the diazo species.
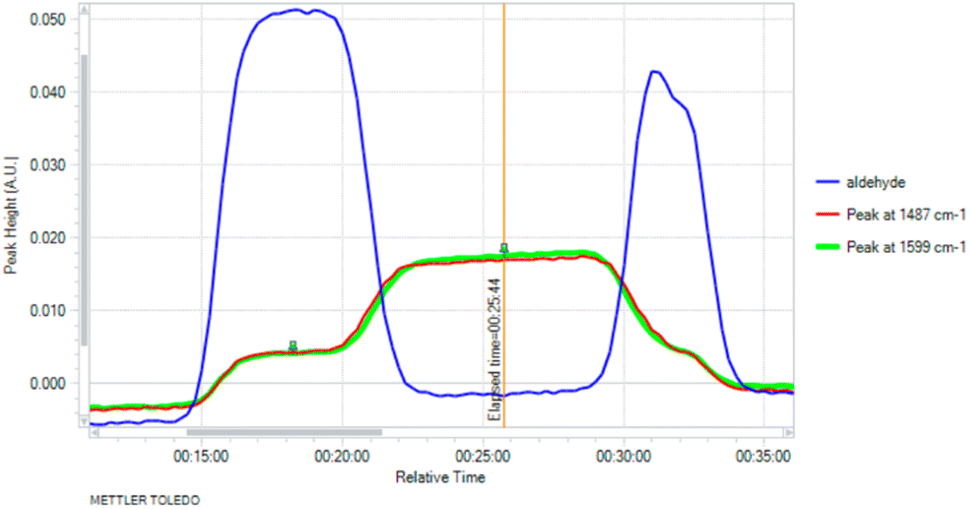 | ||
| Fig. 2 Consumption of aldehyde starting material when hydrazine reagent is injected, and identification of peaks at 1487 cm−1 and 1599 cm−1 indicative of the presence of the intermediate. | ||
With these data in hand, we then moved on to a full reaction configuration using two Vapourtec R2+/R4 systems. Reagent switching valves were used to introduce the reagent plugs, and the resulting mixture was passed through a reactor coil held at 70 °C, followed by a MnO2 column, and lastly the IR spectrometer. One of the high-pressure injection valves directed this output stream of the diazo intermediate either to waste or into the second stage. In the second stage, the diazo intermediate was combined with an olefin reagent, and the mixture incubated at 60 °C in a residence coil. The output was collected using a low-pressure collection valve.
We chose to use a pair of pumps for delivery of the olefin reagent into the second stage, one with solvent and the other with the reagent. Their flow rates were coupled by the control software such that a constant total flow rate of 0.1 mL min−1 was maintained. This meant that the amount of reagent could be adjusted without affecting the resulting downstream flow rate. The residence time in the second column was then independent of the reaction stoichiometry. The initial control protocol was controlled entirely based on precalculated residence times (see ESI‡). We introduced control of the initiation of the second phase based on the infrared sensor data.
The bright yellow/orange colour of the intermediate diazo material meant that we were able to improve the control over this section of the flow system with the application of a camera-based observation platform.51 A simple colorimeter was constructed from clear PFA tubing wound around a 3D printed part, which was observed by a USB webcam (see ESI‡). Pleasingly, this setup afforded comparable information about the diazo concentration in the flow stream. We were then able to move the FlowIR spectrometer to the end of the flow system to monitor the generated product, the level of which could then be used to control the output collection valve.
Since the colorimeter reported the presence of the diazo intermediate (rather than the absence of the aldehyde) the intermediate waste valve could then be controlled using the original thresholding logic, with adjusted threshold parameters for the signal being produced by the colorimetry algorithm.
The solvent and olefin pumps for the second stage were also controlled based on the diazo colorimetry data. Once the diazo had been detected over a certain threshold, the diazo was directed through to the second phase of the process by the injection valve, so long as the level was maintained over this threshold. If there were temporary drops in the amount of diazo intermediate detected, the output from the first stage of the process was directed to waste. The flow rate of the olefin reagent pump was set to deliver a calibrated amount based on the colorimeter detection level. The flow rate of the paired solvent pump was linked such that it would make the total flow rate of the delivered olefin reagent up to 0.1 mL min−1 (or 1.1 mL min−1 if the diazo intermediate stream was being directed to waste). The flow rate in the second phase of the process was then maintained at a constant 1.1 mL min−1.
Once the diazo detection level had fallen low consistently for 10 minutes, the control system assumed that stage 1 of the protocol was complete. This aspect of control was stopped and the THF pump was set at a constant 1.1 mL min−1 for the remainder of the process. The collection of the final product was controlled in a similar way, by switching the collection valve to the collection position when the colour intensity detected by the second colorimeter was over some threshold level. Further details of the automation protocol are provided in the ESI.‡
This implementation, in an industrial setting, further showcases the robustness of the system. In addition, the removal of the reservoir, although suitable for small scale operations, increased safety and the replacement of the FlowIR with a home-made webcam colorimeter reduced the automation cost.
Conclusion
In summary we have developed a platform for generation of unstabilised diazo compounds and synthesis of 2-pyrazolines from aldehydes. This synthesis was performed by forming and oxidizing a hydrazone with MnO2, followed by a [3 + 2] cycloaddition reaction with an electron-poor alkene under continuous flow conditions. A small library of 20 pyrazolines were successfully isolated and characterized in 30–89% yield. The process could also be scaled out using a multi-column approach, yielding pyrazoline 8 in 73% with a productivity of 3.1 g h−1.Moreover, this study presents an automation solution for generating a library of 2-pyrazolines. Automation and PAT tools were utilized through a new GUI that allowed easy reprogramming of the platform by chemists without programming experience. The analytical tools also played a major role enabling the scale out, determining the MnO2 depletion and switching reagent inputs. This approach successfully demonstrates the enabling aspect of automation and should encourage similar approaches towards handling unstable intermediates for library synthesis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Robert Maguire and Peter H. Dorff for useful discussions. We thank Pfizer inc. for a brief study visit (RJI). The authors gratefully acknowledge financial support from the São Paulo Research Foundation – FAPESP (JCP, award No. 2014/26378-2, 2014/25770-6 and 2020/11578-7; HMM, award No. 2018/25233-1; MCFCB, award No. 2015/18572-6), National Council for Scientific and Technological Development – CNPq (HMM, award No. 130504/2019-0) and CAPES (RL, No. 9865/13-6).Notes and references
- S. V. Ley, D. E. Fitzpatrick, R. J. Ingham and R. M. Myers, Angew. Chem., Int. Ed., 2015, 54, 3449–3464 CrossRef CAS PubMed.
- S. V. Ley, D. E. Fitzpatrick, R. M. Myers, C. Battilocchio and R. J. Ingham, Angew. Chem., Int. Ed., 2015, 54, 10122–10136 CrossRef CAS PubMed.
- K. F. Jensen, AIChE J., 2017, 63, 858–869 CrossRef CAS.
- C. P. Breen, A. M. K. Nambiar, T. F. Jamison and K. F. Jensen, Trends Chem., 2021, 3, 373–386 CrossRef.
- I. R. Baxendale, J. Deeley, C. M. Griffiths-Jones, S. V. Ley, S. Saaby and G. K. Tranmer, Chem. Commun., 2006, 2566–2568 RSC.
- C. A. Hone and C. O. Kappe, Chemistry–Methods, 2021, 1, 454–467 CrossRef CAS.
- T. Razzaq and C. O. Kappe, Chem. – Asian J., 2010, 5, 1274–1289 CrossRef CAS PubMed.
- S. Newton, C. F. Carter, C. M. Pearson, L. De, C. Alves, H. Lange, P. Thansandote and S. V. Ley, Angew. Chem., Int. Ed., 2014, 53, 4915–4920 CrossRef CAS PubMed.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed.
- M. Guidi, P. H. Seeberger and K. Gilmore, Chem. Soc. Rev., 2020, 49, 8910–8932 RSC.
- R. J. Ingham, C. Battilocchio, J. M. Hawkins and S. V. Ley, Beilstein J. Org. Chem., 2014, 10(56), 641–652 CrossRef PubMed.
- N. M. Roda, D. N. Tran, C. Battilocchio, R. Labes, R. J. Ingham, J. M. Hawkins and S. V. Ley, Org. Biomol. Chem., 2015, 13, 2550–2554 RSC.
- D. E. Fitzpatrick, C. Battilocchio and S. V. Ley, Org. Process Res. Dev., 2016, 20, 386–394 CrossRef CAS.
- D. E. Fitzpatrick, T. Maujean, A. C. Evans and S. V. Ley, Angew. Chem., Int. Ed., 2018, 57, 15128–15132 CrossRef CAS PubMed.
- J. Jiao, W. Nie, T. Yu, F. Yang, Q. Zhang, F. Aihemaiti, T. Yang, X. Liu, J. Wang and P. Li, Chem. – Eur. J., 2021, 27, 4817–4838 CrossRef CAS PubMed.
- C. A. Shukla and A. A. Kulkarni, Beilstein J. Org. Chem., 2017, 13(97), 960–987 CrossRef CAS PubMed.
- C. Raston, J. Britton, S. J. Britton and C. L. Raston, Chem. Soc. Rev., 2017, 46, 1250–1271 RSC.
- D. Webb and T. F. Jamison, Chem. Sci., 2010, 1, 675–680 RSC.
- D. N. Tran, C. Battilocchio, S. B. Lou, J. M. Hawkins and S. V. Ley, Chem. Sci., 2015, 6, 1120–1125 RSC.
- A. Greb, J. S. Poh, S. Greed, C. Battilocchio, P. Pasau, D. C. Blakemore and S. V. Ley, Angew. Chem., Int. Ed., 2017, 56, 16602–16605 CrossRef CAS PubMed.
- C. Battilocchio, F. Feist, A. Hafner, M. Simon, D. N. Tran, D. M. Allwood, D. C. Blakemore and S. V. Ley, Nat. Chem., 2016, 8, 360–367 CrossRef CAS PubMed.
- P. Dingwall, A. Greb, L. N. S. Crespin, R. Labes, B. Musio, J. S. Poh, P. Pasau, D. C. Blakemore and S. V. Ley, Chem. Commun., 2018, 54, 11685–11688 RSC.
- J. S. Poh, D. N. Tran, C. Battilocchio, J. M. Hawkins and S. V. Ley, Am. Ethnol., 2015, 127, 8031–8034 Search PubMed.
- J. S. Poh, S. Makai, T. von Keutz, D. N. Tran, C. Battilocchio, P. Pasau and S. V. Ley, Angew. Chem., Int. Ed., 2017, 56, 1864–1868 CrossRef CAS PubMed.
- M. R. Shaaban, A. S. Mayhoub and A. M. Farag, Expert Opin. Ther. Pat., 2012, 22, 253–291 CrossRef CAS PubMed.
- Y. Katsuyama and K. Matsuda, Curr. Opin. Chem. Biol., 2020, 59, 62–68 CrossRef CAS PubMed.
- G. Le Goff and J. Ouazzani, Bioorg. Med. Chem., 2014, 22, 6529–6544 CrossRef CAS PubMed.
- D. Havrylyuk, O. Roman and R. Lesyk, Eur. J. Med. Chem., 2016, 113, 145–166 CrossRef CAS PubMed.
- Q.-S. Li, B.-N. Shen, Z. Zhang, S. Luo and B.-F. Ruan, Curr. Med. Chem., 2020, 28, 940–962 CrossRef PubMed.
- V. Lellek, C. Y. Chen, W. Yang, J. Liu, X. Ji and R. Faessler, Synlett, 2018, 29, 1071–1075 CrossRef CAS.
- J. P. Waldo, S. Mehta and R. C. Larock, J. Org. Chem., 2008, 73, 6666–6670 CrossRef CAS PubMed.
- N. Nakamichi, Y. Kawashita and M. Hayashi, Synthesis, 2004, 2004, 1015–1020 Search PubMed.
- Y. Shao, J. Tong, Y. Zhao, H. Zheng, L. Ma, M. Ma and X. Wan, Org. Biomol. Chem., 2016, 14, 8486–8492 RSC.
- A. Lévai, J. Heterocycl. Chem., 2002, 39, 1–13 CrossRef.
- A. Lévai, Chem. Heterocycl. Compd., 1997, 33(6), 647–659 CrossRef.
- K. Alex, A. Tillack, N. Schwarz and M. Beller, Org. Lett., 2008, 10, 2377–2379 CrossRef CAS PubMed.
- M. Chen, L. J. Wang, P. X. Ren, X. Y. Hou, Z. Fang, M. N. Han and W. Li, Org. Lett., 2018, 20, 510–513 CrossRef CAS PubMed.
- M. N. Yang, D. M. Yan, Q. Q. Zhao, J. R. Chen and W. J. Xiao, Org. Lett., 2017, 19, 5208–5211 CrossRef CAS PubMed.
- Z. Wang, Y. Yang, F. Gao, Q. Luo and L. Fang, Org. Lett., 2018, 20, 934–937 CrossRef CAS PubMed.
- S. L. Cui, J. Wang and Y. G. Wang, Org. Lett., 2008, 10, 13–16 CrossRef CAS PubMed.
- V. George and B. König, Chem. Commun., 2023, 59, 11835–11838 RSC.
- R. R. Merchant, D. M. Allwood, D. C. Blakemore and S. V. Ley, J. Org. Chem., 2014, 79, 8800–8811 CrossRef CAS PubMed.
- T. L. Wootton and D. M. Allwood, Org. Biomol. Chem., 2022, 20, 2255–2260 RSC.
- J. Britton and T. F. Jamison, Angew. Chem., Int. Ed., 2017, 56, 8823–8827 CrossRef CAS PubMed.
- P. Rullière, G. Benoit, E. M. D. Allouche and A. B. Charette, Angew. Chem., Int. Ed., 2018, 57, 5777–5782 CrossRef PubMed.
- R. Sustmann, Pure Appl. Chem., 1974, 40, 569–593 CrossRef CAS.
- H. Lange, C. F. Carter, M. D. Hopkin, A. Burke, J. G. Goode, I. R. Baxendale and S. V. Ley, Chem. Sci., 2011, 2, 765–769 RSC.
- S. P. Green, K. M. Wheelhouse, A. D. Payne, J. P. Hallett, P. W. Miller and J. A. Bull, Org. Process Res. Dev., 2020, 24, 67–84 CrossRef CAS PubMed.
- Octopus is available under an MIT open source license at https://www.github.com/richardingham/octopus.
- Blockly, Google Developers, https://developers.google.com/blockly, (accessed 17 September 2023).
- S. V. Ley, R. J. Ingham, M. O'Brien and D. L. Browne, Beilstein J. Org. Chem., 2013, 9(118), 1051–1072 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Klavs F. Jensen on the occasion of his 70th birthday. |
| ‡ Electronic supplementary information (ESI) available: Additional experimental details, including photographs of experimental setup, 1H and 13C NMR spectra for isolated compounds. See DOI: https://doi.org/10.1039/d3re00515a |
| § Current address: Syngenta, Jealott's Hill International Research Centre, Bracknell, UK. |
| ¶ Current address: Single Technologies AB, Sankt Eriksgatan 48F, 112 34 Stockholm, Sweden. |
| || Current address: Syngenta Crop Protection, Schaffhauserstrasse, Stein, CH-4332, Switzerland. |
| ** Current address: Janssen Pharmaceutica NV, Turnhoutseweg 30, B-2340, Beerse, Belgium. |
| This journal is © The Royal Society of Chemistry 2024 |