
Plasma-based post-processing of colloidal nanocrystals for applications in heterogeneous catalysis
Julia J.
Chang
a,
Xinchun
Tian
a and
Ludovico
Cademartiri
 *b
*b
aDepartment of Materials Science & Engineering, Iowa State University of Science and Technology, 2220 Hoover Hall, Ames, IA 50011, USA
bDepartment of Chemistry, Life Sciences and Environmental Sustainability, University of Parma, Parco Area delle Scienze 17/A, 43012, Parma, Italy. E-mail: ludovico.cademartiri@unipr.it
First published on 3rd June 2024
Abstract
This review summarizes the work on the use of plasmas to post-process nanostructures, in particular colloidal nanocrystals, as promising candidates for applications of heterogeneous catalysis. Using plasma to clean or modify the surface of nanostructures is a more precisely controlled method compared to other conventional methods, which is preferable when strict requirements for nanostructure morphology or chemical composition are necessary. The ability of plasma post-processing to create mesoporous materials with high surface areas and controlled microstructure, surfaces, and interfaces has transformational potential in catalysis and other applications that leverage surface/interface processes.
Introduction
The use of plasmas in nanoscience ranges from nanoparticle synthesis to surface modification, from anisotropic etching to doping, and for applications ranging from electronic materials to optical materials, and from magnetic materials to energy materials. Excellent reviews have been written on one or more of these areas in the past few years and we encourage the reader to explore those resources.1–7This review aims to summarize the recent developments in the use of plasmas to control the structure and chemistry of the surface of nanostructures, especially assemblies of colloidal nanocrystals, with a focus towards a demonstrated or plausible application in catalysis. As we discuss in the next pages, there is significant potential in the use of colloidal nanocrystals and their post-processing with plasmas to produce heterogeneous catalysts for both fundamental studies and commercial applications.
Catalysis is a technologically important application of materials and molecules whereby an active site on a molecule (in the case of homogeneous catalysts), or a surface (in the case of heterogeneous catalysts) activates an adsorbed “substrate” (i.e., a molecule) by lowering its activation energy towards the production of a specific product.8 The effectiveness of catalysis makes it ubiquitous in industry (its market is poised to reach 40 billion dollars by 20209 or 34.3 billion dollars by 2024 depending on estimates10), in research laboratories, and in biochemical networks.11,12
In the case of heterogeneous catalysts, catalysis is considered mostly a surface process. Almost every aspect of a surface can affect the activity (quantified, for example, by a “turnover number”, i.e., the maximum number of chemical conversions per second) and selectivity (i.e., the ability to produce one out of several possible products) of a catalytic process: its chemistry (e.g., stoichiometry, composition13,14), its structure15 (e.g., crystallographic orientation, defects), and the presence of contaminants or adsorbates.16,17 Furthermore, since heterogeneous catalysts are often obtained from rare and costly elements, they are usually supported on other inert materials (“supports”). Interfaces between materials can have their own peculiar catalytic properties and affect the catalytic activity and selectivity of both components.18–20
Colloidal nanocrystals are nanoscale single crystals of (usually) inorganic compounds that are stabilized in solution as colloids by either steric or charge repulsion. They are usually synthesized in solution by precipitation, decomposition, or reduction reactions in the presence of (usually) organic molecules (“ligands”).21 By coordinating the surface atoms of the nanocrystals, these ligands play multiple roles: beside providing colloidal stabilization (i.e., preventing agglomeration), they control the surface energy of the nanocrystals (often in a crystallographically specific manner), thereby controlling the rate of nucleation and growth of the particles during their synthesis. In some cases, the ligands are used to govern the assembly of the particles into ordered superlattices on surfaces.22–24
Colloidal nanocrystals are a potentially ideal building block for heterogeneous catalysts. The exquisite level of control over the rate of nucleation and growth provided by ligands, colloidal nanocrystals makes colloidal synthesis unparalleled in materials chemistry. Colloidal nanocrystals can be produced with controlled size (usually from 1.5 to 20 nm, cf. Fig. 1a),25 shape (from sphered, to rods, to platelets, to wires – cf. Fig. 1b and c),26 internal structure (from core–shell particles, cf. Fig. 1d, to Janus particles, cf. Fig. 1e, to supraparticles, cf. Fig. 1f), surface chemistry (controlled surface stoichiometry),27 crystallographic structure (controlled surface termination, cf. Fig. 1c), and composition (from metals, to semimetals, to oxides, to chalcogenides, to pnictides). Due to their small size, colloidal nanocrystals offer the potential for very large surface areas, therefore maximizing the activity of the catalyst per unit mass. The small size also implies large convex curvatures as well as local exposure of surface defects (e.g., terraces, high-index facets), which increase in the activity of the surface28 What's more, colloidal stability of the particles allows for dispersions containing more than one type of particle. These dispersions can be used to create multicomponent heterogeneous catalysts that can catalyze more than one step in a reaction.29
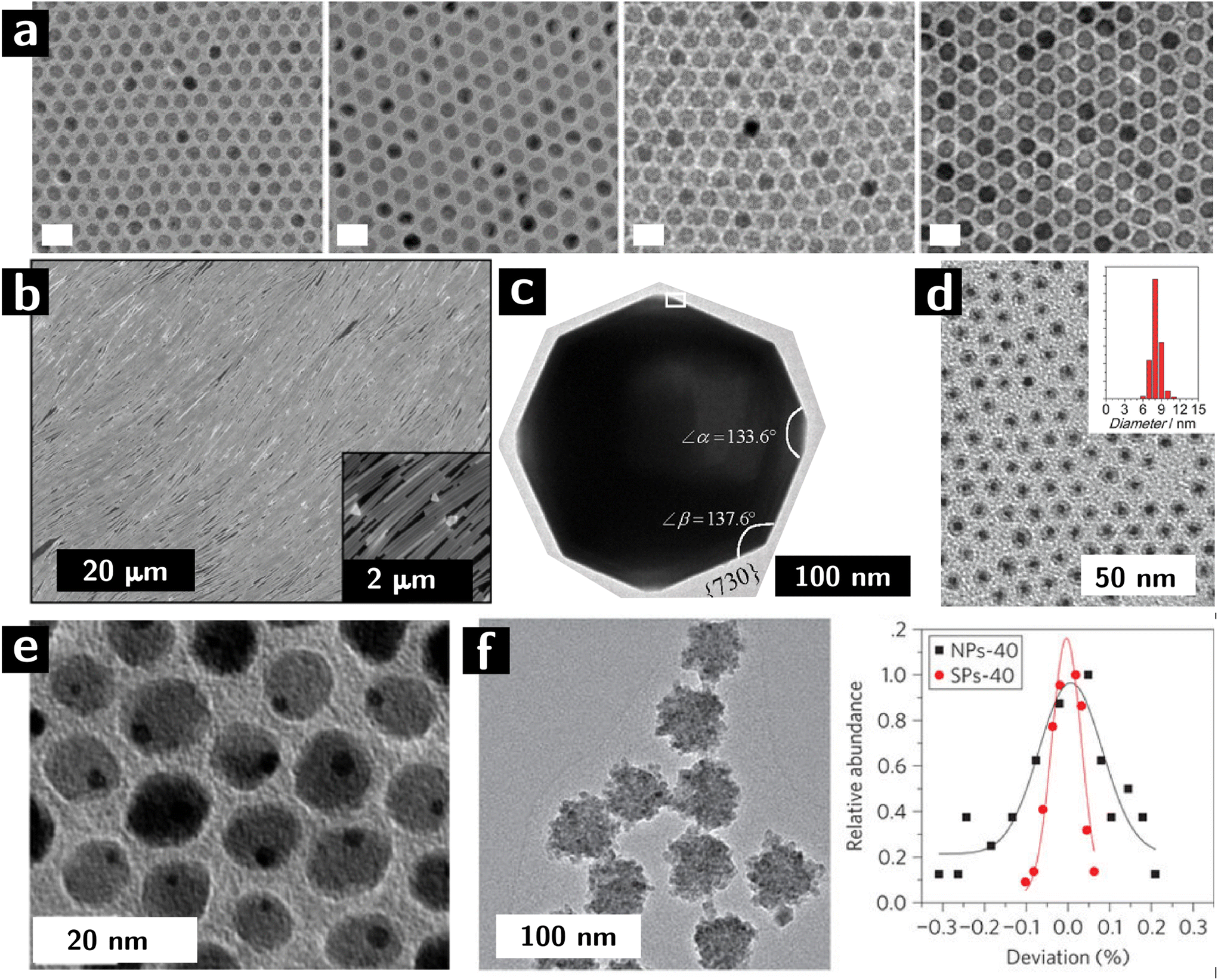 | ||
| Fig. 1 Control of microstructural characteristics of colloidal nanocrystals. (a, left to right) 10, 11, 12, and 13 nm iron oxide nanocrystals synthesized by colloidal route.30 (b) Self—assembled Ag nanowires adopting a liquid crystalline, nematic structure.31 (c) High-index facets in Pt nanocrystals.32 (d) Core shell nanocrystals (Au cores, Co3O4 shells).33 (e) Janus nanoparticles (dimers of Au and Fe3O4 nanocrystals).34 (f) Supraparticles (colloidal particles resulting from the assembly of smaller particles) of CdSe: left panel shows a TEM image, while right panel shows size distribution variance of building blocks (black) and supraparticles (red).35 | ||
In spite of these highly attractive assets, colloidal nanocrystals face challenges in their application as catalysts. The ligands that are crucial for controlling morphology and surface chemistry are usually detrimental to the catalytic activity and need to be removed (cf.Fig. 2),16,17 leaving alone that the surface obtained through chemical synthesis might not be ideal for catalytic application and can require tuning of composition and structure.36,37 The large area and chemical potential of the surface of the nanocrystals (upon removal of the ligands) could also serve as a counteractive effect: it make the surfaces highly susceptible to reconstruction, adsorption of contaminants (adventitious organics and water), non-specific reaction with other molecules, and coarsening.38 It is worth noting that the capping ligands are sometimes used to tune the catalytic selectivity of the nanocrystals.39,40 For this type of applications, the ligands should remain attached to the nanocrystals.
 | ||
| Fig. 2 Diagram comparing traditional material manufacturing with a nanoparticle-based approach that leverages the unique assets of colloidal nanocrystals. | ||
Removing ligands from monolayers of nanocrystals on exposed surfaces is relatively easy. It is instead challenging when – as in the case of many commercial heterogeneous catalysts – the particles are embedded in a high surface area, porous template with small pore sizes. In these conditions, mass transport slows down the removal of organics.41,42 Furthermore, if the fraction of organics is substantial, their removal is accompanied by a large volume loss. The resulting strain can (and usually does) compromise the structure of the material.
Several methods have been used and characterized for the removal of organics from colloidal nanocrystals, e.g., calcination, UV-ozone and ozone treatments, solution processing, and plasma processing. Calcination uses high temperatures in an oxidizing atmosphere to remove the ligands. Its major limitation is its tendency to coarsen the nanocrystals and oxidize their surface.43 Depending on the composition of the nanocrystals, it is possible to avoid coarsening by using relatively low temperatures for a long time (185 °C for 5 h on oleylamine-capped Pt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 44) or very high temperature for a much shorter time (500–700 °C for 1 min on oleylamine-capped Pd45). UV-ozone treatment applies ozone and ultraviolet light to oxidize and decompose the ligands. However, studies have shown UV-ozone treatment either can not remove the ligands completely when compared to calcination46 or it is just effective in removing the aliphatic part of the ligands.47 Solution processing uses different principles (e.g., solvent extraction,48 acid washing,49 or ligand replacement50) to remove the ligands from the nanocrystals, but it induces capillary strain that can compromise the integrity of the film, undesired surface reactions, and the adsorption of solvent (or trace amounts of impurities). After the removal of the ligands, the nanocrystal surfaces can be further modified by calcination in O2 or H2 or other gases to achieve oxidation, reduction, or doping of the surfaces.
44) or very high temperature for a much shorter time (500–700 °C for 1 min on oleylamine-capped Pd45). UV-ozone treatment applies ozone and ultraviolet light to oxidize and decompose the ligands. However, studies have shown UV-ozone treatment either can not remove the ligands completely when compared to calcination46 or it is just effective in removing the aliphatic part of the ligands.47 Solution processing uses different principles (e.g., solvent extraction,48 acid washing,49 or ligand replacement50) to remove the ligands from the nanocrystals, but it induces capillary strain that can compromise the integrity of the film, undesired surface reactions, and the adsorption of solvent (or trace amounts of impurities). After the removal of the ligands, the nanocrystal surfaces can be further modified by calcination in O2 or H2 or other gases to achieve oxidation, reduction, or doping of the surfaces.
In the next section we will discuss the advantages and challenges of plasma processing of colloidal nanocrystals for the removal of ligands and the control of surface structure and chemistry.
Plasma processing of colloidal nanocrystals
Plasma processing offers significant advantages over other approaches for the control of surfaces of colloidal nanocrystals. These advantages are especially attractive for applications in catalysis.1. It is a near-room-temperature process. Vacuum plasmas have average temperatures in the order of 40–60 °C. This low temperature avoids sintering of the nanocrystals, as well as carbonization of the organic matrix or the formation of carbides seen in the high temperature decomposition processes of many hybrid systems.51
2. It can remove all accessible carbon from the system. The avoidance of high temperatures, and therefore of carbonization, circumvents the “binder burnout problem”.
3. It does not cause capillary strain. It is a gas-phase process.
4. It can selectively modify the surfaces. Plasmas are far-from-equilibrium systems where minority components have much greater kinetic energies than what expected from the average temperature. This means that chemical reactions that would be too costly to conduct at room temperature can easily occur in a plasma. A side effect of high energy collisions in a plasma is the formation of long-lived radicals which can perform high energy chemical reactions without the side effect of ablation which is associated with the accelerated charged species.
5. Ablation in a plasma, while not completely avoidable, can be greatly ameliorated by a careful choice of the carrier gas.
Of course, plasma processing poses challenges which will depend on the type of plasma that is used. While plasma processing in dilute plasmas does not expose films to high temperatures, high power plasmas with feed gases such as Ar will cause ablation, which can compromise the structure of colloidal nanocrystal assemblies or contaminate the films with elements sputtered from the chamber walls. Furthermore, scalability can be challenging for low pressure plasmas due to the vacuum requirements.
Lastly, to apply plasmas for removing the ligands from colloidal nanocrystal assemblies and modifying their surface requires the initial film to be porous (i.e., essentially devoid of free ligands), to allow for radicals and other excited species to diffuse into the material and etch the organic ligands, as well as the escape of the etching byproducts.
Plasma processing to remove organic ligands from colloidal nanocrystals
Early work by the groups of J. Spatz, H. G. Boyen, and J. Buriak showed that O2, H2, and Ar plasmas can be used to remove the organic fraction from block copolymer micelles infiltrated with metal-containing compounds (especially Au).52–56 The Au compounds segregated to the domains of one of the blocks and the subsequent removal of the organic fraction by plasma left behind Au nanostructures on surfaces that were templated by the self-assembled block copolymer. Later work expanded significantly on these ideas by using DNA-templated assembly and other strategies.54,57–60Work by the Ozin group showed that low pressure, low power inductively-coupled plasmas of air could significantly remove the ligands from hundreds of layers of colloidal nanocrystals of PbS61 and Bi2S3.62 Importantly, the work showed that the removal of ligands deep into the films was accompanied by the oxidation of the surfaces to oxide, sulfate, and sulfite species. The process was demonstrated to work also within templates that had been infiltrated with nanocrystals, leading to hierarchical assemblies of nanocrystals (e.g., microrods, inverse opals) after template removal.63
Recent work in this area has focused on using plasma processing to improve the properties of nanocrystal assemblies, using a variety of starting compositions (Table 1). In most cases the emphasis lies in using plasmas as tools, rather than pursuing a fundamental understanding of the mechanism by which plasmas can remove ligands from within these colloidal nanocrystal assemblies. Our laboratory has made some advances in this fundamental understanding by using ZrO2 nanocrystals as a model system due to their small size (3–3.5 nm), high reproducibility, relatively high yield (gram scale), exceptional colloidal stability and highly stable phase, and no strongly absorbing in the UV range. These assets endowed us the opportunity to test the mass transport limitations within the porous films with controlled thickness (typically between 300 and 450 nm) spin-coated from thoroughly cleaned ZrO2 nanocrystal solution.
| Phase | Ligand | Film thickness | Power | Feed gas | Pressure | Catalyzed reaction | Ref. |
|---|---|---|---|---|---|---|---|
| ZrO2 | TOPO/OLA | 200–400 nm | 7–30 W | O2 | 500 mTorr | 42, 64–66 | |
| PbS | OA, EDT | 90 nm | 75 W | O2 | — | 67 | |
| Au@Ag | Thiolated polystyrene | Monolayer | — | O2 | 0.5 L min−1 flow rate | 68 | |
| Au | Dodecanethiol or thiol-terminated polystyrene | Monolayer | 20 W | O2 | 0.5 mbar | 69 | |
| H2 | 0.15 mbar | ||||||
| Au nanowires | Oleylamine | 10 nm | 100 W | 5% H2/Ar | 0.3 mbar | 70 | |
| O2 | |||||||
| ZrO2 | TOPO/OLA | 200–400 nm | 7–30 W | He | 100–500 mTorr | 71 | |
| HfO2 | TOPO | 200–400 nm | 7–30 W | O2 | 500 mTorr | Benzaldehyde acetalization | 72 |
| Si | ODE | 50 nm | 7–30 W | He | 100 mTorr | 73 | |
| Au | Hexadecyltrimethylammonium-bromide | Monolayer | 160 W | O2 | 600 mTorr | 74 | |
| Fe2O3 | Monolayer | — | Ar/O2 (5:1) |
10 Pa | 75 | ||
| CoPt3 | 1-Adamantane-carboxylic-acid and hexadecylamine | 20–30 W | H2 | 3.0 mbar | 76 | ||
| N2 | 3.5 mbar | ||||||
| O2 | 3.5 mbar | ||||||
| Co | Oleic acid and oleylamine | Monolayer | 100 W | O2 | 7 Pa | 77 | |
| H2 | 5 Pa | ||||||
| FePt | Oleic acid and oleylamine | Monolayer | 50 W | H2 | 5 Pa | 78, 79 | |
| Pd on SiO2 | PVP | 7.8 W | He/2% O2 | 1 atm | Furfural hydrogenation | 80 | |
| Au | Triphenylphosphine | On zeolite | ? | O2 | 0.5 mbar | Propylene epoxidation | 81 |
| Au | Triphenylphosphine | On zeolite | ? | O2 | 0.5 mbar | Epoxidation of propylene | 82 |
| Au | APTES | On SiO2 | 130 W | O2 | 400 mTorr | Hydrogenation of acetylene | 83 |
The major challenge in the characterization of the effect of plasma exposure on these films was the quantification of the carbon concentration. The films were too thick to be characterized by X-ray photoelectron spectroscopy (XPS) in one scan and too thin to be characterized by thermogravimetric analysis (TGA). FTIR and Raman spectroscopy can encounter challenges in quantifying small amounts of carbon residue, especially if amorphous.84 Electron energy loss spectroscopy (EELS) could characterize film cross-sections but at a high cost. We resolved this issue by using Ion Beam Analysis (IBA) techniques such as Elastic Backscattering Spectroscopy (EBS), which provide quantitative characterization of the areal concentration of all elements (typically in thin film units, TFU, i.e., 1015 atoms per cm2) in films as thick as 1 μm, in one scan, without requiring manipulation of the film (e.g., sectioning). The data can be processed and modeled to extract the concentration of all elements as a function of depth. By using the EBS resonance for carbon at 4265 keV, the sensitivity to carbon was below 1 at%.
The highly quantitative nature of this technique, and the ability to extract information for all elements allowed, with few clear assumptions, to extract the volume fraction of each phase in the film with the knowledge of the thickness of the film. These main assumptions were that the particles were spherical, phases were stoichiometric (e.g., ZrO2) and having the same density of the bulk state. We attributed any O atom that could not be to the ZrO2 or to the ligands to physisorbed H2O and, to the degree that it was possible, to hydroxyls on the surface of the particles. The ability to extract the volume fractions of the different phases was essential to infer the porosity of the film and therefore gather information on the structural evolution of the colloidal nanocrystal assemblies.
The removal of ligands from the assemblies was complete and selective (cf.Fig. 3a), albeit slow (complete removal took between 2 and 7 days) in inductively coupled plasmas (7 W, 500 mTorr, O2 feed).64 No element other than C and H were affected. Most significantly the removal was found to be homogeneous throughout the thickness of the films, even for partial etching: the etching process proceeds with a uniform rate throughout the thickness of the film, indicating that the etching is not diffusion limited within the film in spite of the nanoscale porosity and the low pressure (Knudsen regime).42 Limitations to etch rates still arises due to diffusion at the surface of the film (boundary layers still exist in the vacuum).
 | ||
| Fig. 3 Characterization of structural and chemical evolution of ZrO2 colloidal nanocrystal films exposed to O2 and He inductively coupled plasmas. (a) Depth profile of the carbon concentration (in at%) before (black) and after calcination (blue and green) and plasma processing (red). (b) Graph of the total volume loss of ZrO2 film (black open circles) and pore volume (gray open squares) as a function of the processing time. (c) Scanning electron microscopy (SEM) images of the surface and cross-section of a ZrO2 nanocrystal film after ligand removal showing the absence of cracks. The lack of cracks was due to the disordered arrangement of the particles in the film. (d) XPS characterization of the P 2p3/2 orbital in TOPO samples before and after plasma processing showing oxidation of phosphine oxides to likely phosphate groups. (e) Carbon concentration in ZrO2 nanocrystal assemblies during He (black squares) and O2 (red circles) plasma processing. (f) Histogram of the relative contributions to etching of different plasma species in He (black) and O2 (red) plasma. | ||
Modeling the concentration of the elements in the film as a function of processing time gave information about the structural evolution of the films during etching. As shown in Fig. 3b, the porosity of the films (calculated from the EBS data) grows significantly in the first hours of etching from ∼26% to over 50% after 2 days, only to then drop back to 38% after 7 days. The initial and final porosity of the films were both consistent with a random close packed arrays of spheres with interdigitating ligand shells and no ligand shells, respectively. The highly porous intermediate stages suggested the formation of an open structure: the carbon concentration between 2 days and 7 days of processing differed by only 4%, but accounted for ∼17% of pore volume. On the basis of these data and of prior observations on the plasma processing of polystyrene colloids,85 we postulated that the etching caused the formation of highly crosslinked necks between the particles which prevent a complete collapse of the films, thereby increasing the porosity at the intermediate stages of ligand removal. The hypothesis was supported by a characterization of the mechanical properties of these films which showed that intermediate etching led to materials with significantly larger moduli than expected from a granular system of equal filling fraction.64,86
More recently, our group has compared the results from plasma processing with calcination in oxygen at high temperatures (400–800 °C) as a function of time (up to 12 h). The work showed that, while Raman spectroscopy indicated the disappearance of C–H bonds in the films, the EBS characterization (cf.Fig. 3a) revealed that between 20 and 40% of the original carbon atoms in the as-prepared assemblies were left behind after calcination (depending on calcination temperature).42
Two other contributions looked at the structural and chemical modifications of the colloidal nanocrystal assemblies during plasmas and how these could be manipulated by controlling the structure and chemistry of the as-deposited assembly. The first study showed that, differently from all other ligand removal approaches, the removal of the ligands by the plasma can occur without cracking of the assemblies due to volume loss (cf.Fig. 3c).64 We found that the ability of these assemblies to resist cracking depended very strongly on the arrangement of the nanocrystals in the assembly: ordered assemblies cracked while disordered assemblies resisted cracking for thickness below 450 nm.66 The second study showed that the composition of the surfaces of the nanocrystals after etching could be controlled by choosing appropriate ligands in the starting colloidal building blocks. Inorganic elements associated with the ligands (e.g., P in trioctylphosphine oxide) can be left behind on the surface of the nanocrystals. As an example, we compared two systems, ZrO2 nanoparticles capped with trioctylphosphine oxide or with oleic acid (OA). After O2 plasma treatment, the phosphine oxide was converted to phosphate (according to XPS result, cf. Fig. 3d), while the oleic acid was completely removed. This change in the surface chemistry of the nanocrystals had profound effects on the thermal evolution of the nanocrystal assemblies, their phase transition temperatures, and their mechanical properties.65 Importantly, this strategy could provide an avenue for the molecular control of interfacial chemistry in nanocrystalline materials, and therefore a tool to understand the effect of interfaces on the macroscopic properties of extended polycrystalline solids, like catalysis.65
Lastly, we showed how He plasmas compare favorably to O2 plasmas for removing ligands from oxygen-sensitive materials (e.g., chalcogenides).87,88 One of the main limitations of calcination and O2 plasmas is that they both oxidize the surface or the entirety of the nanocrystals, which can drastically affect physical properties of the system (e.g., charge transport). We found that He plasma successfully removes ligands from our ZrO2 model system, albeit slightly more slowly than O2 plasma (cf.Fig. 3e). A control experiment with hard masks89 determined the relative influence of different components of the plasma (e.g., charged species, neutral species, UV photons, VUV photons) on the etching rate (cf.Fig. 3f). Surprisingly, we found that even in the case of He plasma, most of the etching could be attributed to excited neutral species (e.g., metastable He species) that can cleave C–C bonds to form carbon radicals that can easily recombine with radicals from the cleavage of C–H bonds leading to the progressive fragmentation and crosslinking of the organic fraction.90 Our current work is investigating quantitatively the kinetics of etching to determine what is the rate limiting step in the process, in an attempt to speed up the etching rate. Current optimized conditions allow us to remove all ligands from 300–400 nm films within 6 h, which compares favorably to calcination.
Other groups over the past few years have made significant contributions by using plasmas on colloidal nanocrystals to remove ligands with the goal of improving optical and electrical properties for applications in Surface-Enhanced Raman Scattering (SERS), solar cells, and catalysis.
Nam et al. used O2 plasma to remove the ligands from the surface of PbS quantum dots (QD) to achieve more efficient charge transfer in solar cell. The heterojunction solar cell used in this study was deposited in sequential layers of FTO (150 nm)/ZnO (40 nm)/PbS QD film (90 nm)/polymer (15–20 nm)/Au (100 nm), in which the PbS film was treated by O2 plasma at 75 W for 1 min to remove the ligands. To evaluate the effect of O2 plasma on PbS QDs, FTIR and XPS characterizations were applied to 3 types of PbS QD films, namely (i) unprocessed OA-capped PbS QDs, (ii) unprocessed 1,2-Ethanedithiol (EDT)-capped PbS QDs and (iii) O2 plasma processed EDT-capped PbS QDs. FTIR results (cf.Fig. 4a) showed an incomplete ligand exchange of OA by EDT in type (ii) film. The 100% transmittance of type (iii) film indicated instead the complete removal of ligands. However, presence of PbSO4, PbSO3 and PbO in XPS O 1s and S 2p spectra of type (iii) films indicated surface oxidation of PbS QDs. The J–V curve of solar cells composed of type (ii) and (iii) films in Fig. 4b showed that O2 plasma induced an increase of open-circuit voltage while a decrease of short-circuit current due to the formation of surface oxide layer.67
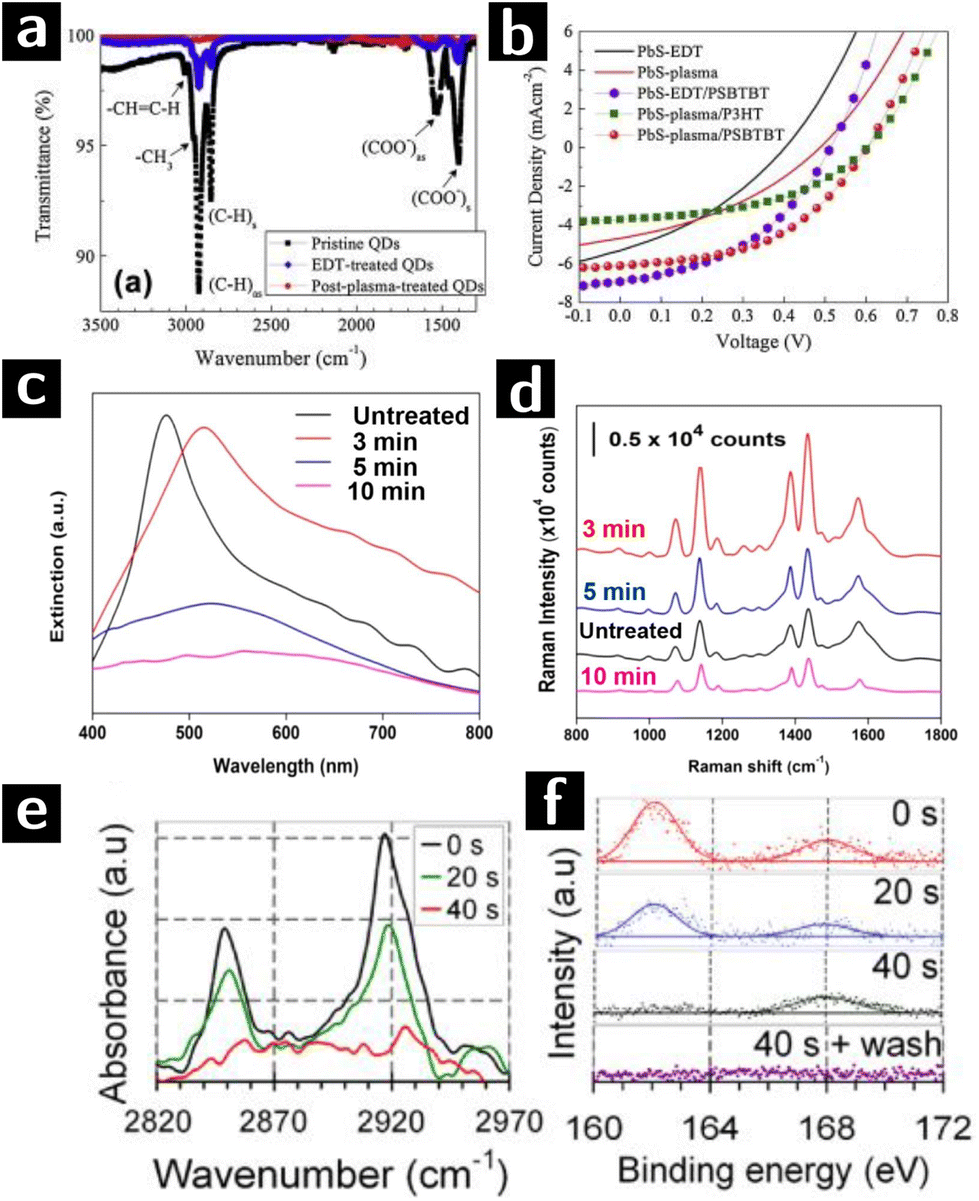 | ||
| Fig. 4 Plasma processing of PbS and Au colloidal nanocrystals to remove ligands. (a) FTIR results of unprocessed OA-capped PbS QD film (sample I), unprocessed EDT-capped PbS QD film (sample II), and O2 plasma processed EDT-capped PbS QD film (sample III). (b) J–V curves of solar cells composed of sample II (black line) and III (red line) PbS QD films.67 (c) Spectral evolution of plasmene sheets treated by 0 min (black), 3 min (red), 5 min (blue) and 10 min (purple) plasma processing. (d) 4-Aminothiophenol SERS spectra at 1 μM concentration of plasmene sheets with corresponding plasma processing times.68 (e) FTIR and (f) S 2p XPS data of dodecanethiol-capped Au nanocrystals after 0 s, 20 s or 40 s H2 plasma treatment.69 | ||
SERS is a common method for detecting extremely small amounts of molecules at metal surfaces. Si and co-workers developed a new type of SERS substrate composed of Au and Ag nanocube (35 nm) plasmene sheet. In this study, thiolated polystyrene capped Au@Ag nanocubes monolayer was used as the starting materials. These sheets were then treated in a UV-ozone chamber with O2 plasma at a flow rate of 0.5 L min−1 for 3, 5, and 10 min. Plasma treatment caused a red shift of the plasmonic resonance (cf.Fig. 4c). However, optimized treating time (3 min) was crucial to decrease the nanocube inter-particle spacing to provide strong plasmonic coupling as well as avoid the aggregation of particles induced by van der Waals attraction. As proved by the SERS spectra (cf.Fig. 4d), the plasmene sheet treated by O2 plasma for 3 min showed an enhanced response to 4-aminothiophenol compared to untreated one, indicating a successful removal of ligand. Longer treatments (≥5 min) decreased the performance due to the agglomeration of the nanocubes and oxidation of Ag nanocubes.68
Sivaraman and Santhanam successfully obtained chemically and thermally stable 2D Au nanoparticle arrays with different inter-particle spacing (2–13 nm) by using plasma processing. Dodecanethiol-capped Au nanocrystals were treated by H2 plasma of 20 W, 0.5 mbar, while polystyrene-capped nanocrystals were treated by O2 plasma of 20 W, 0.15 mbar to prevent polystyrene cross linking. In dodecanethiol-capped Au nanocrystals, 40 s of H2 plasma treatment eliminated the FTIR signal of methylene moieties (cf.Fig. 4e) and XPS S 2p signal of thiols, leaving just the signal from sulfonate groups. A following water rinsing step removed the physisorbed sulfonate and formed ligand-free Au nanoparticle arrays (cf.Fig. 4f). In the case of polystyrene-capped Au nanocrystals, signal from methylene stretch in FTIR spectra showed that polystyrene could not be completely removed by O2 plasma before nanoparticle aggregation (cf.Fig. 5a). The SERS enhancement factor of ligand-free 2D nanoparticle arrays was compared to that of unprocessed arrays showing a difference in enhancement of 5 orders of magnitude.69
 | ||
| Fig. 5 Plasma processing of Au colloidal nanowires to remove ligands. (a) FTIR and (b) S 2p XPS data of polystyrene-capped Au nanocrystals after 0 min or 9 min O2 plasma treatment.69 (c) Sheet resistances of oleylamine-capped Au nanowire processed by H2 plasma (red triangle), O2 plasma (blue circle) or ligand exchange (green square) at various treatment time. (d) SEM and GISAXS (left corner inset) patterns of Au nanowire sheets treated by H2 or O2 plasma for 1 or 5 min.70 | ||
Au nanoparticle systems are of great interest for applications beyond SERS. For example Au nanoparticles are very active catalysts for a range of reactions.91 Au nanowires could also serve as the active component in electrocatalytic reactions, and the catalytic efficiency of Au nanowires is better than Au nanoparticle under certain circumstances.92 Plasmas have been applied in removal ligands from various structures composed of Au nanowires, e.g. honeycomb macroporous pattern films.93 Maurer and coworkers showed that, when compared with O2 plasma, H2 plasma treatment is more effective at removing the ligands while retaining the microstructure of Au nanowires (cf.Fig. 5c and d). Oleylamine-capped, 1.6 nm-thick Au nanowires were dip-coated onto glass substrate and formed 10 nm-thick films. The films were then treated by either H2 plasma (0.3 mbar, 100 W, 5% H2/Ar2) or O2 plasma (0.3 mbar, 100 W, 100% O2). After just 1 min of exposure, H2 plasma treatment showed significant reduction of sheet resistivity (from >40 MΩ to 200 Ω sq−1). Longer processing times decreased the resistance further (50 Ω sq−1) but sintered the nanowires, as indicated by the disappearance of grazing-incidence small-angle X-ray scattering (GISAXS) single wire scattering pattern. However, 1 min O2 plasma treatment could only reduce the resistance to the MΩ range and caused drastic changes in the morphology of the nanowire arrays, possibly due to surface oxidation.70
Plasma processing to control the structure and chemistry of the surface of colloidal nanocrystals
Plasma processing of polymers is an industrially used process to change the chemical composition of polymer surfaces to improve adhesion,94,95 change wettability,96etc. Similarly, plasma processing can change the composition of the surface of inorganic nanocrystals by chemical reactions with the plasma species, depending on the material and feed gas.Boyen et al. studied the oxidation and reduction effects of different plasma types on monolayers of ligand-free metal/metal alloy nanocrystals, e.g. Au,97 Co98 and FePt.99 They proved that O2 plasma is capable of removing ligands as well as oxidize the metal nanocrystals, while H2 plasma could reduce the oxidized nanoparticle back to the original metallic phase. For example, a 5 min O2 plasma processing (50 W, 0.005 mbar) oxidized Co nanoparticle into oxides, while 5 min H2 plasma processing (50 W, 0.005 mbar) could reduce most of the oxides back to metallic state. These results obtained in monolayers of nanocrystals were reproduced by Ozin and colleagues in multilayers of PbS and Bi2S3 nanocrystals where oxidation of the surface by air plasmas (5 W) resulted in the formation of oxide, sulfate and sulfite groups. X-ray diffraction (XRD) characterization demonstrated that the size of the nanocrystalline chalcogenide phase was not modified, therefore indicating that the oxidation was limited to the surface atoms.61,62 Oxidation of the nanostructure surface could also be achieved indirectly by activating the surface atoms. Ar plasmas can break bonds due to bombardment therefore activating the surface towards further reaction100 (e.g., surface grafting of carbon nanotubes101,102).
More recent examples show the potential for these surface chemistry approaches for the postprocessing of colloidal nanocrystals. The encapsulation of Au nanoparticles with graphene is considered a valuable strategy to improve the catalytic efficiency of Au nanoparticles by preventing their aggregation.103 However, the low solubility of carbon in Au makes it difficult to deposit graphene on nanocrystals directly by chemical vapor deposition (CVD).104 O2 plasma processing partially oxidizes the surface of the gold nanocrystals, allowing the surface oxide to serve as catalysts for graphene growth during CVD.105 Wu et al. studied the effect of O2 plasma on gold nanocrystals with the goal of optimizing the conditions for carbon CVD.
Hexadecyltrimethylammonium-bromide-capped Au nanoparticles were drop-casted on Si substrates and then treated with O2 plasma (160 W, 600 mTorr) for different exposure durations. When the nanoparticles were processed for short (<30 min) or long (>45 min) durations, nanoparticle aggregation would happen due to the formation of new particles from unreacted precursors or coalescence, respectively. Intermediate processing times produced non-aggregated, surface-oxidized Au nanoparticles without surfactants and reactants, as suggested by the SEM results (cf.Fig. 6a). The XPS results (cf.Fig. 6b) showed that surface AuOx was composed of Au2O and Au2O3. A kinetic study revealed that the thickness of the AuOx shell was a function of processing time and was independent of oxide stoichiometry. The oxidized Au nanocrystals were then used as electron acceptors in the xylene CVD process to facilitate graphene encapsulation. TEM result showed the formation of a 1.6 nm-thick graphene shell surrounding the nanoparticles.74
 | ||
| Fig. 6 Using plasmas to modify the surfaces of colloidal nanostructures. (a) SEM images of Au NPs treated with O2 plasma for 0 min, 15 min, 45 min and 60 min from left to right. (b) Au 4f XPS result of Au NPs treated with O2 plasma for 45 min.74 (c) O 1s XPS spectra of unprocessed and Ar plasma processed α-Fe2O3. (d) SEM images of Au NPs grafted on α-Fe2O3 with (above) or without (below) Ar plasma treatment.75 (e) XPS spectra of the C 1s signal before and after plasma treatment with H2, N2, O2, and O2 followed by a brief H2 plasma. (f) Reduced (metallic states indicated by solid lines) Pt and Co after a sequential O2–H2 plasma treatment (O2: 120 min at 20 W, H2: 45 min at 20 W).76 (g) XA spectra at the Co–L3,2, O–K, and C–K edges of Co/CoO nanoparticles: (a) as prepared and after (b) 6, (c) 26, and (d) 46 min hydrogen-plasma etching, and (e) after 46 min hydrogen-plus 20 min oxygen-plus 30 min hydrogen-plasma exposure.77 (h) XANES at the carbon K edge and (i) Fe L3,2 absorption edges of 6 nm FePt nanoparticles before (dashed line) and after plasma processing.78 | ||
A new type of SERS substrate was created by grafting Au nanocrystals on dendritic α-Fe2O3. Tang et al. used Ar/O2 plasma to treat α-Fe2O3 nanostructure to increase the concentration of –OH groups on the surface and promote the grafting of Au nanocrystals. Dendritic α-Fe2O3 was synthesized by autoclaving an aqueous solution of K3Fe(CN)6. The obtained material was then treated by Ar/O2 (5:1) plasma (10 Pa) for 30 min before grafting with Au nanoparticles. O 1s XPS spectra show that the intensity of the peak at 531.2 eV (attributed to Fe–OH) is stronger after plasma processing (cf.Fig. 6c), indicating the successful activation of the surface. The SEM characterization (cf.Fig. 6d) showed a higher Au nanoparticle coverage on the plasma processed dendritic α-Fe2O3 structure. The Au nanoparticle grafted α-Fe2O3 showed good performance when used as SERS substrates.75
Gehl et al. studied the effect of plasma processing on the surface modification of CoPt3 nanocrystals capped with 1-adamantane-carboxylic-acid and hexadecylamine. The nanoparticle solution was spin-coated on Si or Al2O3 substrates and formed layers of nanoparticles. The films were then treated by different plasmas (H2: 30 sccm, 3.0 mbar; N2: 30 sccm, 3.5 mbar; O2: 10 sccm, 3.5 mbar; 20–30 W power). The C 1s XPS spectrum showed that O2, O2 followed by H2 (O2/H2), H2, and N2 plasmas could all remove the surface ligands (cf.Fig. 6e). However, N2 plasma (30 W, 120 min) and H2 plasma treatment (20 W, 140 min) sputtered or interconnected the nanocrystals, respectively. O2 plasma processing (20 W, 120 min) preserved the morphology of nanocrystals but caused surface oxidation. A subsequent H2 plasma treatment (20 W, 45 min) could reduce all the platinum oxide and most of cobalt oxide back into metallic state (cf.Fig. 6f) as well as maintain the mesoscopic order. The nanoparticles processed by O2/H2 plasma showed a cobalt-rich surface due to the surface-energy-driven surface segregation of the cobalt oxide produced during O2 plasma.76
Wiedwald et al. showed that H2 plasma is capable of reducing both ligand-free Co3O4 nanocrystals (that had been previously processed by O2 plasma to remove the ligands), and ligand capped Co/CoO nanocrystals. Oleic acid and oleylamine-capped Co nanocrystals were deposited on a Si substrate as a monolayer, where they oxidized and formed a passivating layer (Co/CoO). Some of the samples were first treated by O2 plasma (100 W, 7 Pa) to remove ligands, and then processed by H2 plasma (100 W, 5 Pa), while the other samples were directly processed by H2 plasma. SEM characterization showed that the nanocrystals could preserve their morphology and dispersion on the substrate. X-ray absorption spectroscopy (XAS) results (cf.Fig. 6g) showed that, as H2 plasma processing went on, the intensity of the C–K edges gradually decreased, until 46 min processing when there was only 40% of the original intensity. The H2/O2/H2 plasma processing could remove all the ligands thus no C–K absorption could be detected, while the Co–L3,2 spectra showed the reduction of the surface oxide into metal.77
Antoniak et al. studied the effect of H2 plasma on oxidized FePt particles by using a submonolayer of oleic acid and oleylamine capped FePt nanoparticles on Si substrates. The films were treated with H2 plasma (50 W, 5 Pa, 30 min) to remove ligands. From the X-ray absorption near-edge structure (XANES) results (Fig. 6h and i), the intensity of the carbon K absorption in H2-plasma-treated films reduced, indicating a removal of organic ligands. At the same time, multiplet features of the Fe L3,2 edge vanished indicating the reduction to the metallic state.78,79 While the above materials were chosen for their magnetic properties, they also are useful for heterogeneous catalysis.106,107
Doping of TiO2 with N2 plasma was recently reported.108,109 Titanium dioxide is a widely used photocatalyst. However the wide bandgap of the anatase limit its use to visible light. Ever since Morikawa et al. reported the band gap narrowing effect of doping N into TiO2 lattice,110 there has been a significant effort devoted to understanding the N-doping of TiO2. Islam et al. used microwave-assisted N2/Ar plasma to process titanium dioxide films deposited from TiO2 sols. The result showed that N2/Ar plasma successfully introduced nitrogen atom into TiO2, as well as introduced catalytically active surface defects on the particles. Preliminary results from our work showed that similar effects can be obtained from colloidal nanocrystals, thereby avoiding the high-temperature calcination step.
Application of plasma-processed colloidal nanocrystals in catalysis
In the last part of this review we discuss most significant examples of how the capabilities of plasmas in cleaning and modifying the surfaces of colloidal nanocrystals were leveraged for use in catalysis.113Banerjee et al. synthesized octadecanethiol-capped colloidal nanocrystals consisting of a Au core coated by a Pt shell (Au@Pt). Different batches were created with different Pt loading and tested for catalytic efficiency towards methanol oxidation. The nanoparticle solution was deposited on gold-coated substrates to form very thin films (bilayers), which were then treated by Ar plasma (100 W, 0.5 mbar, 2 min) to remove the ligands. The removal of the ligands was demonstrated by the absence of signal from sulfur species in XPS characterization (cf.Fig. 7a and b). Furthermore, the original ligand-capped nanoparticle bilayer did not show any electrocatalytic activity indicating the blockage of the redox species by the organic ligands. While samples showed electrocatalytic activity only when they had been treated with plasma.114
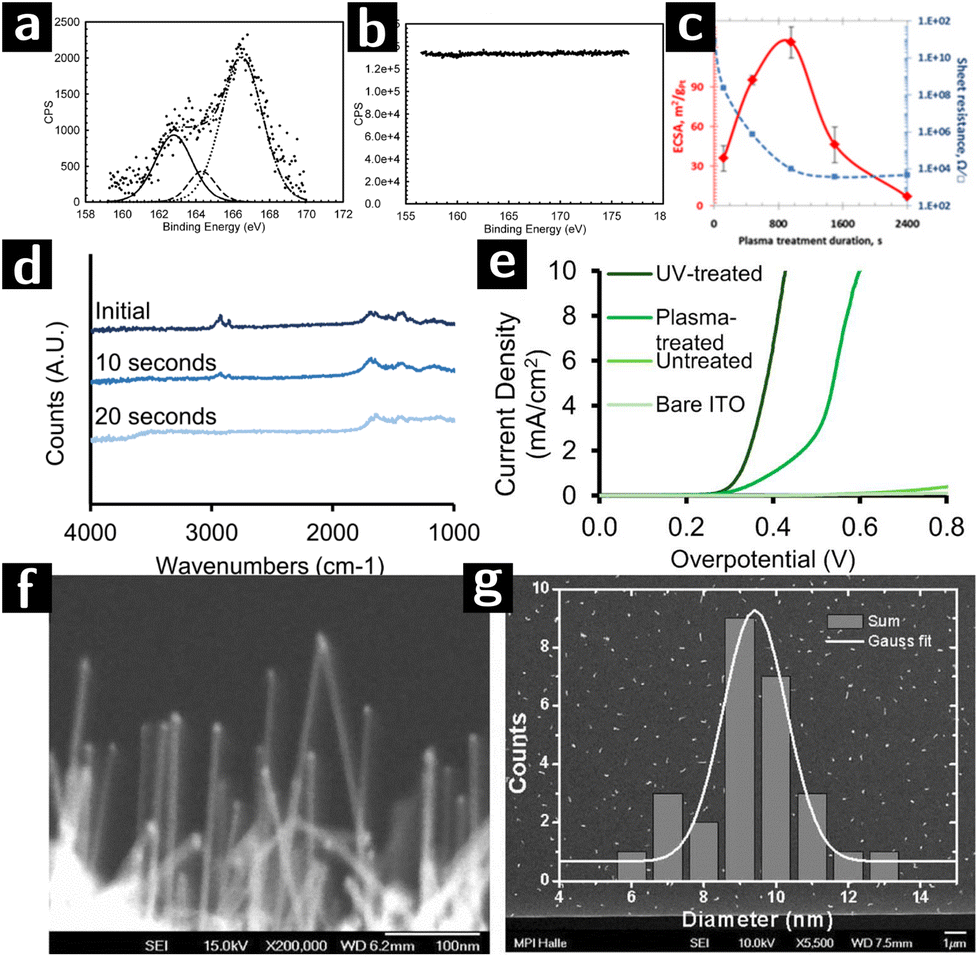 | ||
| Fig. 7 Research on using plasmas to create heterogeneous catalysts from colloidal nanocrystals. (a) S 2s XPS spectra of Au@Pt nanoparticles (Pt/Au – 0.39) before and (b) after argon plasma treatment for 2 minutes. (c) ECSA values (red) and lateral electrical sheet resistance values (blue) at various RF argon plasma treatment durations of of Au@Pt nanoparticles (Pt/Au – 0.39).111 (d) FTIR spectra of a [Ni,Fe]O (Ni:Fe 51:49) nanoparticle film spin-coated on high resistivity Si (∼10 Ω·cm) after 0 s, 10 s or 20 s of air plasma treatment, at 0.2 torr. (e) Voltammograms of UV-treated, plasma-treated, untreated electrodes (functionalized with 51:49 [Ni,Fe]O nanoparticles), and a nanoparticle-free ITO control, in 0.1 M KOH.112 (f) SEM image of SiNWs catalyzed by Au-apo, immobilized on a Si (111) substrate after O2-plasma treatment. (g) SEM image and the size distribution of 25 randomly selected SiNWs with unspecific growth directions. | ||
In a following work, the same authors investigated the effect of plasma exposure time on the electronic conductivity and electrocatalytic activity of the Au@Pt nanocrystals with different Pt loadings. The other plasma processing parameters were kept constant (100 W, 0.5 mbar Ar plasma). The highest electrocatalytically active surface area (ECSA) as well as the lowest sheet resistance could be obtained at optimum plasma processing time, resulting in an enhanced electrocatalytic efficiency (cf.Fig. 7c). For example, the magnitude of the resistance change for closed packed Au@Pt samples with Pt/Au ratio greater than 0.19 before and after plasma exposure is of the order of 107. Optimum plasma processing time prevents the migration of Au atoms from the core to the surface which was observed after extensive plasma processing, resulting in a strong electronic interaction between surface Pt atoms and underlying gold cores.111
Nickel/iron oxide ([Ni,Fe]O) is an important candidate for the catalysis of the oxygen evolution reaction (OER) of water splitting.115 Bau and co-workers compared the effectiveness of air plasma and UV irradiation for removing the surface ligands from the nanocrystals. The oleate-capped [Ni,Fe]O nanoparticle solution was spin-coated on an ITO substrate and formed a monolayer, which was then treated with either air plasma (18 W, 0.2 Torr, 20 s) or a Hg lamp (15 mW, 254 nm, 1 h). The FTIR results (cf.Fig. 7d) showed that air plasma could achieve complete removal of oleate. However, the current density-overpotential results (cf.Fig. 7e) showed that the overpotential for UV-irradiated sample was smaller than the plasma processed one. The performance and morphological stability of the nanocrystal films during electrocatalysis was better for the UV-treated films than for the plasma treated films, possibly due to the crosslinking effect of UV-radiation on residual ligands.112
Zhang and coworkers synthesized Au nanocrystals inside the protein cavity of apoferritin (Au-apo) on a Si substrate. The goal was to control the size distribution of the Au nanocrystals and to use them as catalysts for the CVD synthesis of highly uniform, ultrathin Si nanowires with superior monodispersity of diameter. The Au-apo was synthesized by reducing a mixture of apoferritin and HAuCl4 with NaBH4. The aqueous solution of Au-apo was then deposited on the Si substrate and treated with O2 plasma (100 W, 0.9 Torr, 2 min) to remove the apo matrix. After plasma treatment, UHV-CVD was applied to the sample to form Si nanowires on the bare surface of Au nanoparticle. SEM results showed that the Si nanowire obtained by this method produced nanowires with a size distributions consistent with the size distribution of the catalyst particles (cf Fig. 7f and g).116
Recent work by Vlachos et al.80 showed convincingly the use of non-thermal atmospheric-pressure plasmas (a He/O2 mixture, at atmospheric pressure can produce a non-thermal plasma by simple dielectric barrier discharge, basically a corona) to remove polyvinylpyrrolidone (PVP) ligands from SiO2-supported Pd-colloidal nanoparticles and test their catalytic activity in furfural conversion. Comparison with calcination was done to assess the benefits of plasma processing. Consistently with previous work at reduced pressures, the authors did not see thermal effects (sintering, shape changes) in the plasma treated samples. Importantly they observed much improved activity of the nanoparticle catalysts when the ligands were removed by plasma treatment rather than calcination (cf.Fig. 8).
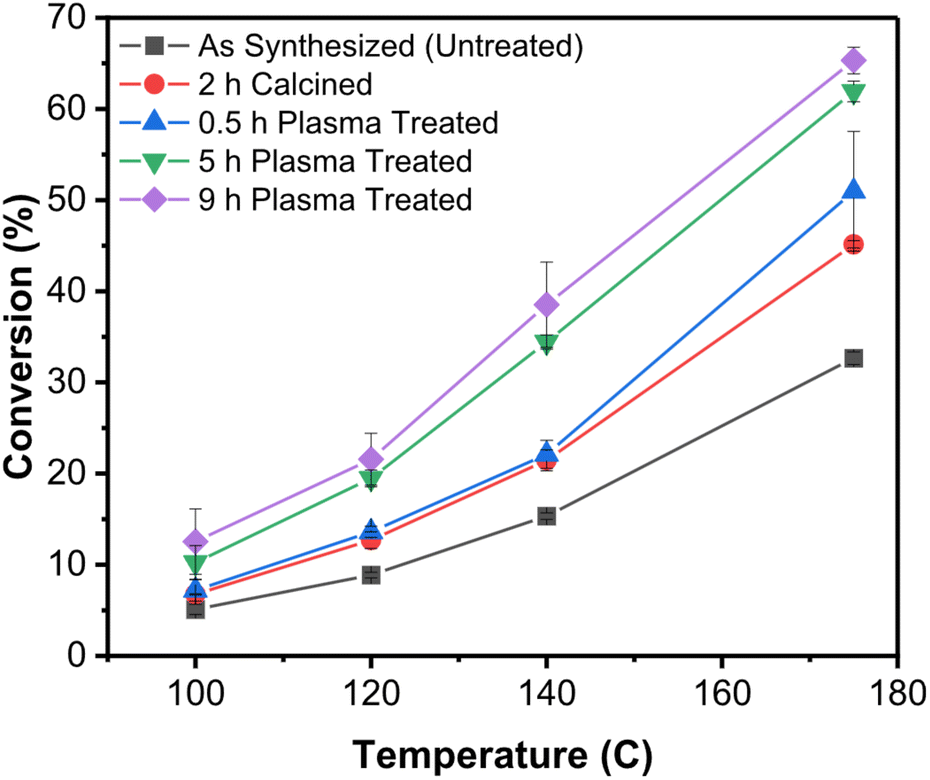 | ||
| Fig. 8 Improved catalytic activity of SiO2-supported Pd nanocrystals upon removal of the ligands by plasma treatment.80 This graph shows the conversion (furfural hydrogenation) as a function of temperature for SiO2-supported Pd nanocrystals capped with PVP and subjected to different postprocessing treatments. In black is shown the untreated control. In red is shown the sample calcined for 2 h. The remaining data show samples that were plasma-treated in He/O2 for different lengths of time. | ||
Summary of known effects of plasmas on colloidal nanocrystal surfaces
For the purpose of clarity we here summarize the current know-how of how plasmas affect nanocrystal surfaces, especially directed at the Colleagues exploring this approach to the post-processing of hybrid nanomaterials.1. Low-pressure plasmas have near-room-temperature average temperatures.117 In such plasmas, thermal effects are not observed. Non-thermal atmospheric pressure plasmas can be used to similar effect.
2. The most well-known components of plasmas are ions resulting from ionizing collisions. Those species are “heavy” and charged, hence carrying large kinetic energies upon collision with the surface. They can cause ablation of the topmost surface of the material.117 They will not cause ablation inside a porous material, especially if mesoporous.72,118 Ablation is moderated/eliminated by using light gases (e.g., He rather than Ar71,73), reducing plasma power and (sometimes) increasing pressure.119 Ablation is followed by redeposition of the ablated material all over the plasma chamber. Care must be taken not to use easily ablatable material for the chamber walls or the sample holder.
3. The second-most well-known component of plasmas are free electrons. They interact only with the topmost surface of the samples, either rupturing single bonds or implanting into the material.119 They do not cause ablation.
4. The least well-known component of plasmas are radicals and other excited species. These are produced by a variety of processes in the gas phase. They are usually neutral, so they are not accelerated in the plasma. Given the low pressure of the gas, these species are much longer lived than the charged species: “remote” plasma processing places the sample downstream from a plasma, therefore exposing the sample purely to a radical-rich atmosphere.120 Radicals are highly reactive and usually dominate the reactivity of plasmas towards nanocrystals.119 In the case of nanocrystal superlattices, the vast majority of the removal of organics is due to radicals produced in the plasma, diffusing into the pores between the particles.119 The chemical nature of the radicals depend mostly on the composition of the feed gas.
5. Another important consequence of plasma formation is the generation of UV radiation that can affect organic matter in the plasma.119
6. To some extent, the material being removed by the plasma (i.e., by ablation or reaction) becomes part of the plasma itself. The feed gas only determines the majority component. Water adsorbed on the walls of the chamber will become part of the plasma, at least until it is flushed out (flushing of a plasma is very inefficient due to the low-pressure and consequent high velocity at which feed gas moves through the chamber).117 Hydrogen removed with the organic moieties will become part of the plasma and can induce some unintended reduction on the nanocrystal surfaces.
7. The composition of the plasma is not homogeneous. Plasma chambers have a gradient in composition usually oriented between the feed gas inlet and outlet. Feed gas often creates a high-velocity stream in the line-of-sight path between the feed gas inlet and outlet.73,119 The position of a sample in the chamber is an important parameter. These inhomogeneities can be reduced by structuring the gas flow in the chamber through obstacles.
8. The removal of organics from the nanocrystal superlattices, and the chemical modifications caused to the surface will tend to a steady state that will persist as long as the plasma is present. Turning off the plasma, and removing the sample from the plasma chamber creates a different environment that inevitably leads to a modification of the surfaces: water and adventitious carbon from the atmosphere will very rapidly return to the exposed surfaces to reduce their chemical potential. In our studies, the mesopores between ZrO2 nanoparticles that had been plasma treated became saturated with water which remained bound even in ultra-high vacuum.118
9. With “inert” feed gases (e.g., N2, He), organic moieties are removed while inorganic components to the ligands (e.g., P in TOPO) seems to be left behind.72 With “reactive” feed gases (e.g., O2), organic moieties are removed and the surface of the nanocrystals is oxidized.
10. So far, most of the work in plasma processing of nanocrystal superlattices has concentrated on understanding the influence of the plasma parameters (e.g., feed gases, pressure, power, temperature) on the removal of the ligands. Much work still needs to be done on how to functionalize/protect the exposed surfaces so that the extraction of the sample from the plasma does not impair the function of the films.
Conclusion
The design and control of nanostructures and surfaces allowed by the combination of colloidal chemistry and plasma processing is especially attractive for applications in catalysis. The colloidal synthesis of nanocrystals provides exceptional control on the size, shape, habit, interface chemistry, and interactions of nanocrystals thereby providing the means to form assemblies of nanocrystals with controlled mesostructure or their infiltration into porous supports. The use of dilute plasmas allows for the removal of the surface-coordinating organic ligands to expose bare surfaces, without incurring in the limitations of sintering (coarsening, surface reconstruction, diffusion, incomplete removal of organics, etc.…). The use of reactive gases as feeds allows for the control over the surface chemistry in post-processing. Recently it was found that Cu nanostructures after plasma processed with certain conditions showed enhanced efficiency when served as catalyst for carbon dioxide reduction.121 Gao et al. used Cu nanocubes as the starting materials and found that O2 plasma processing would enhance the O content of Cu nanocubes greatly after CO2 electroreduction reaction, which has strong effect on the catalytic efficiency and selectivity.122 The study of plasma interaction with nanostructures is a promising topic, and extensive use of colloidal nanoparticles as catalyst is just around the corner.While these assets bode well for the use of plasmas for the creation of heterogeneous catalysts with designed structure and composition, several challenges and opportunities await.
(i) While the breadth of compositions accessible by colloidal chemistry is significant, many important phases (carbides, nitrides, most transition metals) remain either challenging to obtain or inaccessible. For those phases, plasma synthesis offer a tremendous alternative, as discussed in other reviews.5,6
(ii) While the use of low pressure plasmas allows for the reduction of the thermal input into the nanocrystals and their protection from coarsening, it also limits the scalability of these approaches. Can atmospheric pressure plasmas be adopted to yield similar or superior results in terms of etching selectivity and efficiency, and surface modification?
(iii) While there is significant literature and understanding concerning the effect of plasmas on surfaces directly exposed to them, much less work deals with the effect of plasmas on the interior volumes of porous materials. Recent work shows that even mesoporous nanocrystal assemblies can be effectively processed by plasmas through the diffusion of radicals and exposure to UV and VUV radiation generated by the plasmas. Nonetheless the fundamental limitations of this type of processing are less clear: how thick a film can be homogeneously processed before diffusion limitations within the film become limiting? How fast can the removal of the ligands be? How homogeneous is the processing across the plasma chamber?
(iv) Looking forward, can these approaches be integrated into additive manufacturing processes to create nanostructured materials, catalysts, and devices from the bottom-up?
We believe that answering these questions will require the combined expertise of plasma scientists and engineers, materials chemists, computational scientists, characterization experts. Powerful techniques of characterization, such as EBS, could prove invaluable in providing firm quantitative basis on which to compare experimental results with theoretical models and simulations.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the Member-Specific-Research-Intel Program of Semiconductor Research Corporation under Award No. 2015-IN-2582. X. C. T. is grateful to the Chinese Scholarship Committee for a scholarship.References
- Q. Chen, J. Li and Y. Li, J. Phys. D: Appl. Phys., 2015, 48, 424005 CrossRef.
- S. Misra, L. Yu, W. Chen, M. Foldyna and P. R. i. Cabarrocas, J. Phys. D: Appl. Phys., 2014, 47, 393001 CrossRef.
- K. Ostrikov, J. Phys. D: Appl. Phys., 2014, 47, 224009 CrossRef.
- K. Ostrikov, E. C. Neyts and M. Meyyappan, Adv. Phys., 2013, 62, 113–224 CrossRef CAS.
- U. Kortshagen, Plasma Chem. Plasma Process., 2016, 36, 73–84 CrossRef CAS.
- U. R. Kortshagen, R. M. Sankaran, R. N. Pereira, S. L. Girshick, J. J. Wu and E. S. Aydil, Chem. Rev., 2016, 116, 11061–11127 CrossRef CAS PubMed.
- Z. Wang, Y. Zhang, E. C. Neyts, X. Cao, X. Zhang, B. W. L. Jang and C.-j. Liu, ACS Catal., 2018, 8, 2093–2110 CrossRef CAS.
- G. Ertl, Angew. Chem., Int. Ed., 2008, 47, 3524–3535 CrossRef CAS PubMed.
- S. Dwivedi Dheeraj and Y. Singh, Catalyst market by Type (Zeolites, Chemical Compounds), Application (Petroleum Refining, Chemical Synthesis, Polymer Catalysis and Environmental) – Global Opportunity Analysis and Industry Forecast, 2014–2022, Allied Market Research, 2014 Search PubMed.
- Catalyst Market Analysis By Raw Material, By Product, Application And Segment Forecasts To 2024, Grand View Research, 2016 Search PubMed.
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493 RSC.
- D. J. Hayes, Catal. Today, 2009, 145, 138–151 CrossRef CAS.
- W. H. Chiang and R. M. Sankaran, Nat. Mater., 2009, 8, 882–886 CrossRef CAS PubMed.
- G. A. Camara, R. B. de Lima and T. Iwasita, Electrochem. Commun., 2004, 6, 812–815 CrossRef CAS.
- W. Huang, Acc. Chem. Res., 2016, 49, 520–527 CrossRef CAS PubMed.
- Z. Niu and Y. Li, Chem. Mater., 2013, 26, 72–83 CrossRef.
- W. Huang, Q. Hua and T. Cao, Catal. Lett., 2014, 144, 1355–1369 CrossRef CAS.
- A. Bruix, J. A. Rodriguez, P. J. Ramirez, S. D. Senanayake, J. Evans, J. B. Park, D. Stacchiola, P. Liu, J. Hrbek and F. Illas, J. Am. Chem. Soc., 2012, 134, 8968–8974 CrossRef CAS PubMed.
- C. T. Campbell, Nat. Chem., 2012, 4, 597–598 CrossRef CAS PubMed.
- Y. Lykhach, S. M. Kozlov, T. Skala, A. Tovt, V. Stetsovych, N. Tsud, F. Dvorak, V. Johanek, A. Neitzel, J. Myslivecek, S. Fabris, V. Matolin, K. M. Neyman and J. Libuda, Nat. Mater., 2016, 15, 284–288 CrossRef CAS PubMed.
- R. A. Sperling and W. J. Parak, Philos. Trans. R. Soc., A, 2010, 368, 1333–1383 CrossRef CAS PubMed.
- R. L. Whetten, J. T. Khoury, M. M. Alvarez, S. Murthy, I. Vezmar, Z. L. Wang, P. W. Stephens, C. L. Cleveland, W. D. Luedtke and U. Landman, Adv. Mater., 1996, 8, 428–433 CrossRef CAS.
- J. S. Yin and Z. L. Wang, J. Phys. Chem. B, 1997, 101, 8979–8983 CrossRef CAS.
- Z. L. Wang, S. A. Harfenist, R. L. Whetten, J. Bentley and N. D. Evans, J. Phys. Chem. B, 1998, 102, 3068–3072 CrossRef CAS.
- J. Xian, Q. Hua, Z. Jiang, Y. Ma and W. Huang, Langmuir, 2012, 28, 6736–6741 CrossRef CAS PubMed.
- K. Zhou and Y. Li, Angew. Chem., Int. Ed., 2012, 51, 602–613 CrossRef CAS PubMed.
- Z. Wu, A. K. P. Mann, M. Li and S. H. Overbury, J. Phys. Chem. C, 2015, 119, 7340–7350 CrossRef CAS.
- T. Fujita, P. Guan, K. McKenna, X. Lang, A. Hirata, L. Zhang, T. Tokunaga, S. Arai, Y. Yamamoto, N. Tanaka, Y. Ishikawa, N. Asao, Y. Yamamoto, J. Erlebacher and M. Chen, Nat. Mater., 2012, 11, 775–780 CrossRef CAS PubMed.
- Y. Yamada, C. K. Tsung, W. Huang, Z. Y. Huo, S. E. Habas, T. Soejima, C. E. Aliaga, G. A. Somorjai and P. D. Yang, Nat. Chem., 2011, 3, 372–376 CrossRef CAS PubMed.
- J. Park, E. Lee, N. M. Hwang, M. Kang, S. C. Kim, Y. Hwang, J. G. Park, H. J. Noh, J. Y. Kim and J. H. Park, Angew. Chem., 2005, 117, 2932–2937 CrossRef.
- A. R. Tao, S. Habas and P. Yang, Small, 2008, 4, 310–325 CrossRef CAS.
- N. Tian, Z.-Y. Zhou, S.-G. Sun, Y. Ding and Z. L. Wang, Science, 2007, 316, 732–735 CrossRef CAS PubMed.
- Z. Zhongbin, S. Wenchao and Y. Yushan, Adv. Mater., 2014, 26, 3950–3955 CrossRef PubMed.
- H. Yu, M. Chen, P. M. Rice, S. X. Wang, R. L. White and S. Sun, Nano Lett., 2005, 5, 379–382 CrossRef CAS PubMed.
- Y. S. Xia, T. D. Nguyen, M. Yang, B. Lee, A. Santos, P. Podsiadlo, Z. Y. Tang, S. C. Glotzer and N. A. Kotov, Nat. Nanotechnol., 2011, 6, 580–587 CrossRef CAS PubMed.
- J. Lee, J. C. Park and H. Song, Adv. Mater., 2008, 20, 1523–1528 CrossRef CAS.
- J. Y. Park, Y. Zhang, M. Grass, T. Zhang and G. A. Somorjai, Nano Lett., 2008, 8, 673–677 CrossRef CAS PubMed.
- L. Xu, H.-W. Liang, Y. Yang and S.-H. Yu, Chem. Rev., 2018, 118(7), 3209–3250 CrossRef CAS PubMed.
- S. H. Pang, C. A. Schoenbaum, D. K. Schwartz and J. W. Medlin, Nat. Commun., 2013, 4, 2448 CrossRef PubMed.
- S. M. Rogers, C. R. A. Catlow, C. E. Chan-Thaw, A. Chutia, N. Jian, R. E. Palmer, M. Perdjon, A. Thetford, N. Dimitratos, A. Villa and P. P. Wells, ACS Catal., 2017, 7, 2266–2274 CrossRef CAS.
- P. Calvert and M. Cima, J. Am. Ceram. Soc., 1990, 73, 575–579 CrossRef CAS.
- P. Mohapatra, S. Shaw, D. Mendivelso-Perez, J. M. Bobbitt, T. F. Silva, F. Naab, B. Yuan, X. Tian, E. A. Smith and L. Cademartiri, Nat. Commun., 2017, 8, 2038 CrossRef PubMed.
- L. Menard, F. Xu, R. Nuzzo and J. Yang, J. Catal., 2006, 243, 64–73 CrossRef CAS.
- D. Li, C. Wang, D. Tripkovic, S. Sun, N. M. Markovic and V. R. Stamenkovic, ACS Catal., 2012, 2, 1358–1362 CrossRef CAS.
- M. Cargnello, C. Chen, B. T. Diroll, V. V. Doan-Nguyen, R. J. Gorte and C. B. Murray, J. Am. Chem. Soc., 2015, 137, 6906–6911 CrossRef CAS PubMed.
- Z. Liu, M. Shamsuzzoha, E. T. Ada, W. M. Reichert and D. E. Nikles, J. Power Sources, 2007, 164, 472–480 CrossRef CAS.
- C. Aliaga, J. Y. Park, Y. Yamada, H. S. Lee, C.-K. Tsung, P. Yang and G. A. Somorjai, J. Phys. Chem. C, 2009, 113, 6150–6155 CrossRef CAS.
- J. A. Lopez-Sanchez, N. Dimitratos, C. Hammond, G. L. Brett, L. Kesavan, S. White, P. Miedziak, R. Tiruvalam, R. L. Jenkins, A. F. Carley, D. Knight, C. J. Kiely and G. J. Hutchings, Nat. Chem., 2011, 3, 551–556 CrossRef CAS PubMed.
- V. Mazumder and S. Sun, J. Am. Chem. Soc., 2009, 131, 4588–4589 CrossRef CAS PubMed.
- S. M. Ansar, F. S. Ameer, W. Hu, S. Zou, C. U. Pittman Jr. and D. Zhang, Nano Lett., 2013, 13, 1226–1229 CrossRef CAS PubMed.
- X. C. Tian, S. Shaw, K. R. Lind and L. Cademartiri, Adv. Mater., 2016, 19, 3677–3682 CrossRef PubMed.
- J. P. Spatz, A. Roescher and M. Moller, Adv. Mater., 1996, 8, 337–340 CrossRef CAS.
- J. P. Spatz, S. Mossmer and M. Moller, Chem. – Eur. J., 1996, 2, 1552–1555 CrossRef CAS.
- J. P. Spatz, S. Mossmer, C. Hartmann, M. Moller, T. Herzog, M. Krieger, H. G. Boyen, P. Ziemann and B. Kabius, Langmuir, 2000, 16, 407–415 CrossRef CAS.
- J. P. Spatz, T. Herzog, S. Mossmer, P. Ziemann and M. Moller, Adv. Mater., 1999, 11, 149–153 CrossRef CAS.
- J. Chai, D. Wang, X. N. Fan and J. M. Buriak, Nat. Nanotechnol., 2007, 2, 500–506 CrossRef PubMed.
- M. Haupt, S. Miller, R. Glass, M. Arnold, R. Sauer, K. Thonke, M. Möller and J. P. Spatz, Adv. Mater., 2003, 15, 829–831 CrossRef CAS.
- J. P. Zhang, Y. Liu, Y. G. Ke and H. Yan, Nano Lett., 2006, 6, 248–251 CrossRef CAS PubMed.
- J. Chai, D. Wang, X. Fan and J. M. Buriak, Nat. Nanotechnol., 2007, 2, 500–506 CrossRef PubMed.
- J. Chai and J. M. Buriak, ACS Nano, 2008, 2, 489–501 CrossRef CAS PubMed.
- L. Cademartiri, G. von Freymann, A. C. Arsenault, J. Bertolotti, D. S. Wiersma, V. Kitaev and G. A. Ozin, Small, 2005, 1, 1184–1187 CrossRef CAS PubMed.
- R. Malakooti, L. Cademartiri, Y. Akcakir, S. Petrov, A. Migliori and G. A. Ozin, Adv. Mater., 2006, 18, 2189–2194 CrossRef CAS.
- A. Ghadimi, L. Cademartiri, U. Kamp and G. A. Ozin, Nano Lett., 2007, 7, 3864–3868 CrossRef CAS.
- S. Shaw, J. L. Colaux, J. L. Hay, F. C. Peiris and L. Cademartiri, Adv. Mater., 2016, 28, 8900–8905 CrossRef CAS PubMed.
- S. Shaw, T. F. Silva, J. M. Bobbitt, F. Naab, C. L. Rodrigues, B. Yuan, J. J. Chang, X. Tian, E. A. Smith and L. Cademartiri, Chem. Mater., 2017, 29, 7888–7900 CrossRef CAS.
- S. Shaw, B. Yuan, X. Tian, K. J. Miller, B. M. Cote, J. L. Colaux, A. Migliori, M. G. Panthani and L. Cademartiri, Adv. Mater., 2016, 28, 8892–8899 CrossRef CAS PubMed.
- M. Nam, T. Lee, S. Kim, S. Kim, S.-W. Kim and K.-K. Lee, Org. Electron., 2014, 15, 391–398 CrossRef CAS.
- K. J. Si, P. Guo, Q. Shi and W. Cheng, Anal. Chem., 2015, 87, 5263–5269 CrossRef CAS PubMed.
- S. K. Sivaraman and V. Santhanam, Nanotechnology, 2012, 23, 255603 CrossRef PubMed.
- J. H. L. Maurer, L. Gonzalez-Garcia, B. Reiser, I. Kanelidis and T. Kraus, ACS Appl. Mater. Interfaces, 2015, 7, 7838–7842 CrossRef CAS PubMed.
- S. Shaw, X. Tian, T. F. Silva, J. M. Bobbitt, F. Naab, C. L. Rodrigues, E. A. Smith and L. Cademartiri, Chem. Mater., 2018, 30, 5961–5967 CrossRef CAS.
- X. Tian, T. W. Goh, O. Vandenberg, J. VanDerslice, T. F. da Silva, F. Naab, J. L. Hay, J. J. Chang, B. Yuan, F. C. Peiris, W. Huang and L. Cademartiri, J. Phys. Chem. C, 2019, 123(30), 18410–18416 CrossRef CAS.
- P. Mohapatra, D. Mendivelso-Perez, J. M. Bobbitt, S. Shaw, B. Yuan, X. Tian, E. A. Smith and L. Cademartiri, ACS Appl. Mater. Interfaces, 2018, 10, 20740–20747 CrossRef CAS PubMed.
- J. Wu, W. Shi and N. Chopra, J. Phys. Chem. C, 2012, 116, 12861–12874 CrossRef CAS.
- X. Tang, W. Cai, L. Yang and J. Liu, Nanoscale, 2013, 5, 11193–11199 RSC.
- B. Gehl, A. Frömsdorf, V. Aleksandrovic, T. Schmidt, A. Pretorius, J.-I. Flege, S. Bernstorff, A. Rosenauer, J. Falta, H. Weller and M. Bäumer, Adv. Funct. Mater., 2008, 18, 2398–2410 CrossRef CAS.
- U. Wiedwald, K. Fauth, M. Hessler, H. G. Boyen, F. Weigl, M. Hilgendorff, M. Giersig, G. Schutz, P. Ziemann and M. Farle, ChemPhysChem, 2005, 6, 2522–2526 CrossRef CAS PubMed.
- C. Antoniak, M. Spasova, A. Trunova, K. Fauth, M. Farle and H. Wende, J. Phys.: Conf. Ser., 2009, 190, 012118 CrossRef.
- C. Antoniak, M. Spasova, A. Trunova, K. Fauth, F. Wilhelm, A. Rogalev, J. Minar, H. Ebert, M. Farle and H. Wende, J. Phys.: Condens.Matter, 2009, 21, 336002 CrossRef CAS PubMed.
- D. K. Nguyen, V. Vargheese, V. Liao, P. Dimitrakellis, S. Sourav, W. Zheng and D. G. Vlachos, ACS Nano, 2023, 17, 21480–21492 CrossRef PubMed.
- N. Kapil, T. Weissenberger, F. Cardinale, P. Trogadas, T. A. Nijhuis, M. M. Nigra and M. O. Coppens, Angew. Chem., 2021, 133, 18333–18341 CrossRef.
- N. Kapil, F. Cardinale, T. Weissenberger, P. Trogadas, T. A. Nijhuis, M. M. Nigra and M.-O. Coppens, Chem. Commun., 2021, 57, 10775–10778 RSC.
- X. Liu, C.-Y. Mou, S. Lee, Y. Li, J. Secrest and B. W.-L. Jang, J. Catal., 2012, 285, 152–159 CrossRef CAS.
- Y. Lifshitz, Diamond Relat. Mater., 2003, 12, 130–140 CrossRef CAS.
- G. von Freymann, S. John, V. Kitaev and G. A. Ozin, Adv. Mater., 2005, 17, 1273–1276 CrossRef CAS.
- K. Kendall, N. M. Alford and J. D. Birchall, Nature, 1987, 325, 794–796 CrossRef CAS.
- S. Shaw, X. C. Tian, T. F. Silva, J. M. Bobbitt, F. Naab, C. L. Rodrigues, E. A. Smith and L. Cademartiri, Chem. Mater. , 2018, 30(17), 5961–5967, DOI:10.1021/acs.chemmater.8b02095.
- P. Mohapatra, D. Mendivelso-Perez, J. M. Bobbitt, S. Shaw, B. Yuan, X. Tian, E. A. Smith and L. Cademartiri, ACS Appl. Mater. Interfaces, 2018, 10(24), 20740–20747, DOI:10.1021/acsami.8b03771.
- S. Uchida, S. Takashima, M. Hori, M. Fukasawa, K. Ohshima, K. Nagahata and T. Tatsumi, J. Appl. Phys., 2008, 103, 073303 CrossRef.
- S. Shaw, T. F. Silva, P. Mohapatra, D. Mendivelso-Perez, X. Tian, F. Naab, C. L. Rodrigues, E. A. Smith and L. Cademartiri, Phys. Chem. Chem. Phys., 2019, 21, 1614–1622 RSC.
- M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed.
- A. Leelavathi, G. Madras and N. Ravishankar, J. Mater. Chem. A, 2015, 3, 17459–17468 RSC.
- Y. He, Y. Chen, Q. Xu, J. Xu and J. Weng, ACS Appl. Mater. Interfaces, 2017, 9, 7826–7833 CrossRef CAS PubMed.
- K. Chau, B. Millare, A. Lin, S. Upadhyayula, V. Nuñez, H. Xu and V. I. Vullev, Microfluid. Nanofluid., 2010, 10, 907–917 CrossRef.
- P. Rezai, P. R. Selvaganapathy and G. R. Wohl, J. Micromech. Microeng., 2011, 21, 065024 CrossRef.
- H. Hillborg, J. F. Ankner, U. W. Gedde, G. D. Smith, H. K. Yasuda and K. Wikstrom, Polymer, 2000, 41, 6851–6863 CrossRef CAS.
- H.-G. Boyen, G. Kastle, F. Weigl, B. Koslowski, C. Dietrich, P. Ziemann, J. P. Spatz, S. Riethmuller, C. Hartmann, M. Moller, G. Schmid, M. G. Garnier and P. Oelhafen, Science, 2002, 297, 1533–1536 CrossRef CAS PubMed.
- H. G. Boyen, G. Kästle, K. Zürn, T. Herzog, F. Weigl, P. Ziemann, O. Mayer, C. Jerome, M. Möller, J. P. Spatz, M. G. Garnier and P. Oelhafen, Adv. Funct. Mater., 2003, 13, 359–364 CrossRef CAS.
- H. G. Boyen, K. Fauth, B. Stahl, P. Ziemann, G. Kästle, F. Weigl, F. Banhart, M. Hessler, G. Schütz, N. S. Gajbhiye, J. Ellrich, H. Hahn, M. Büttner, M. G. Garnier and P. Oelhafen, Adv. Mater., 2005, 17, 574–578 CrossRef CAS.
- C. Chen, B. Liang, D. Lu, A. Ogino, X. Wang and M. Nagatsu, Carbon, 2010, 48, 939–948 CrossRef CAS.
- C.-H. Tseng, C.-C. Wang and C.-Y. Chen, Chem. Mater., 2007, 19, 308–315 CrossRef CAS.
- C. Chen, B. Liang, A. Ogino, X. Wang and M. Nagatsu, J. Phys. Chem. C, 2009, 113, 7659–7665 CrossRef CAS.
- J. Wu, Thesis for the degree of Master of Science in the Department of Metallurgical and Materials Engineering in the Graduate School of the University of Alabama, 2011 Search PubMed.
- D. Takagi, Y. Kobayashi, H. Hibino, S. Suzuki and Y. Homma, Nano Lett., 2008, 8, 832–835 CrossRef CAS PubMed.
- N. Chopra, L. G. Bachas and M. R. Knecht, Chem. Mater., 2009, 21, 1176–1178 CrossRef CAS.
- S. Rosler, J. Obenauf and R. Kempe, J. Am. Chem. Soc., 2015, 137, 7998–8001 CrossRef PubMed.
- Z. J. Jiang and Z. Jiang, Sci. Rep., 2016, 6, 27081 CrossRef CAS PubMed.
- S. Z. Islam, A. Reed, D. Y. Kim and S. E. Rankin, Microporous Mesoporous Mater., 2016, 220, 120–128 CrossRef CAS.
- S. Z. Islam, A. Reed, N. Wanninayake, D. Y. Kim and S. E. Rankin, J. Phys. Chem. C, 2016, 120, 14069–14081 CrossRef CAS.
- T. Morikawa, R. Asahi, T. Ohwaki, K. Aoki and Y. Taga, Jpn. J. Appl. Phys., 2001, 40, L561–L563 CrossRef CAS.
- I. Banerjee, V. Kumaran and V. Santhanam, Nanotechnology, 2016, 27, 305401 CrossRef PubMed.
- J. A. Bau, E. J. Luber and J. M. Buriak, ACS Appl. Mater. Interfaces, 2015, 7, 19755–19763 CrossRef CAS PubMed.
- Z. Wang, Y. Zhang, E. C. Neyts, X. Cao, X. Zhang, B. W.-L. Jang and C.-j. Liu, ACS Catal., 2018, 8, 2093–2110 CrossRef CAS.
- I. Banerjee, V. Kumaran and V. Santhanam, J. Phys. Chem. C, 2015, 119, 5982–5987 CrossRef CAS.
- J. A. Bau, P. Li, A. J. Marenco, S. Trudel, B. C. Olsen, E. J. Luber and J. M. Buriak, Chem. Mater., 2014, 26, 4796–4804 CrossRef CAS.
- Z. Zhang, L. Zhang, S. Senz and M. Knez, Chem. Vap. Deposition, 2011, 17, 149–154 CrossRef CAS.
- N. Inagaki, Plasma Surface Modification and Plasma Polymerization, CRC Press, Lancaster, PA, 1996 Search PubMed.
- S. Shaw, J. L. Colaux, J. L. Hay, F. C. Peiris and L. Cademartiri, Adv. Mater., 2016, 28, 8900–8905 CrossRef CAS PubMed.
- S. Shaw, T. F. Silva, P. Mohapatra, D. Mendivelso-Perez, X. Tian, F. Naab, C. L. Rodrigues, E. A. Smith and L. Cademartiri, Phys. Chem. Chem. Phys., 2019, 21, 1614–1622 RSC.
- J. M. Grace and L. J. Gerenser, J. Dispersion Sci. Technol., 2003, 24, 305–341 CrossRef CAS.
- H. Mistry, A. S. Varela, C. S. Bonifacio, I. Zegkinoglou, I. Sinev, Y. W. Choi, K. Kisslinger, E. A. Stach, J. C. Yang, P. Strasser and B. R. Cuenya, Nat. Commun., 2016, 7, 12123 CrossRef PubMed.
- D. Gao, I. Zegkinoglou, N. J. Divins, F. Scholten, I. Sinev, P. Grosse and B. Roldan Cuenya, ACS Nano, 2017, 11, 4825–4831 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |