
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDehydrogenative oxidation of hydrosilanes using gold nanoparticle deposited on citric acid-modified fibrillated cellulose: unveiling the role of molecular oxygen†
Butsaratip Suwattananuruka,
Yuta Uetake *ab,
Rise Ichikawac,
Ryo Toyoshimac,
Hiroshi Kondohc and
Hidehiro Sakurai*ab
*ab,
Rise Ichikawac,
Ryo Toyoshimac,
Hiroshi Kondohc and
Hidehiro Sakurai*ab
aDivision of Applied Chemistry, Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan. E-mail: uetake@chem.eng.osaka-u.ac.jp; hsakurai@chem.eng.osaka-u.ac.jp
bInnovative Catalysis Science Division, Institute for Open and Transdisciplinary Research Initiatives (ICS-OTRI), Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan
cDepartment of Chemistry, Faculty of Science and Technology, Keio University, Kohoku-ku, Yokohama 223-8522, Japan
First published on 29th May 2024
Abstract
Efficient and environmentally friendly synthesis of silanols is a crucial issue across the broad fields of academic and industrial chemistry. Herein, we describe the dehydrogenative oxidation of hydrosilane using a gold nanoparticle catalyst supported by fibrillated citric acid-modified cellulose (F-CAC). Au:F-CAC catalysts with various particle sizes (1.7 nm, 4.9 nm, and 7.7 nm) were prepared using the trans-deposition method, a technique previously reported by our group. These catalysts exhibited significant catalytic activity to produce silanols with high turnover frequency (TOF) of up to 7028 h−1. Recycling experiments and transmission electron microscopy (TEM) observation represented the high durability of Au:F-CAC under the reaction conditions, allowing kinetic studies on size dependency. Mechanistic studies were conducted, including isotope labelling experiments, kinetics, and various spectroscopies. Notably, the near ambient pressure X-ray photoelectron spectroscopy (NAP-XPS) of the model catalyst (Au:PVP) revealed the formation of catalytically active cationic Au sites on the surface through the adsorption of molecular oxygen, providing a new insight into the reaction mechanism.
1. Introduction
Considering the growing demand for silicon-based compounds and materials, silanols serve diverse purposes including excellent building blocks for silicon-based polymeric materials,1 pharmacological functional groups,2 coupling reagents3 and protecting groups4 in organic synthesis. Traditionally, silane oxidation required a stoichiometric amount of oxidants, inevitably producing a stoichiometric amount of by-products.5–10 Therefore, catalytic dehydrogenative oxidation of hydrosilanes has garnered attention in the context of its nontoxic attributes as a reactant, with hydrogen being the only by-product, rendering these processes clean and environmentally benign.11–13 Among them, gold nanoparticles (AuNPs) are well-known catalysts for aerobic oxidation reactions.14–19 Besides, from the first report of the AuNP-catalysed oxidation of hydrosilanes under aerobic conditions by Kaneda et al.,14 various AuNP catalysts stabilised on solid-supports such as hydroxyapatite (HAP),15 carbon nanotubes (CNTs),20–22 cellulose,23 and manganese oxide (MnO2)24,25 have been investigated for the oxidation of hydrosilane. These reports exhibited that solid supports play an important role in catalytic activity; therefore, developing novel support is crucial to enhancing catalytic activity.In the context of promoting sustainable and environmentally friendly science, the use of cellulose-based materials, which are abundant in nature, holds great potential. We have established an easy and scalable method for fabricating citric acid-modified cellulose (F-CAC), easily defibrated into a fluffed nanostructure using a commercially available mixer.26,27 The fluffed nanostructure and high tolerance against organic solvents of F-CAC make it suitable as a heterogeneous catalyst for reaction in organic solvents. Our earlier work demonstrated the size-selective preparation of Au:F-CAC catalysts through the trans-deposition method (Fig. 1).28–30 Using the size-selectively prepared Au:PVP(K-15) (PVP: poly(N-vinyl-2-pyrrolidone)),31,32 Au:F-CAC with various particle sizes (1.7–8.2 nm) can be prepared, enabling investigation of size effects. The thus-prepared Au:F-CACs have been applied to the oxidation of alcohols30 and intramolecular cyclisation reaction of amines, showing good catalytic activity.33 Besides, AuNPs in Au:F-CAC are stable enough under the reaction conditions, possibly due to the benefits of introduced coordinative carboxy groups, allowing the size-selective preparation and recycling.
 | ||
| Fig. 1 Citric acid-modification of cellulose and size-selective preparation of Au:F-CAC. | ||
In this study, we investigated the catalytic activity of Au:F-CAC on the dehydrogenative oxidation of hydrosilane. The size-selectively prepared Au:F-CAC catalysts were subjected to kinetic studies, unveiling the size effect that has not been considered. In addition, the labelling experiments and the detailed spectroscopic analyses of the model AuNP catalyst using near-ambient pressure X-ray photoelectron spectroscopy (NAP-XPS) and solution-state X-ray absorption spectroscopy (XAS) were conducted to uncover the effect of molecular oxygen on the AuNP under aerobic conditions.
2. Experimental section
Preparation of Au:F-CAC catalyst
Au:PVP(K-15) and Au:PVP(K-30) were prepared according to the literature procedure.31,32 F-CAC33 and Au:F-CAC30,33 were prepared according to the literature procedure. In a reaction tube (φ = 3 cm) equipped with a magnetic stir bar, Au:PVP(K-15) (5.1 × 10−3 mmol of Au) and F-CAC (600 mg) were mixed in EtOH (30 mL), and then the pH was adjusted to 4 using aqueous hydrochloric acid solution (0.1 mol L−1). After stirring at 27 °C (1300 rpm) for 90 min, the solid was separated from the supernatant by centrifugation (7500 rpm) at room temperature and washed with ethanol (ca. 30 mL × 3). The remaining powder was dried under vacuum at 45 °C for 12 h to afford Au:F-CAC (1.7 × 10−3 wt%).General procedure for dehydrogenative oxidation of hydrosilane
To a reaction tube equipped with a magnetic stir bar, Au:F-CAC, hydrosilane (0.50 mmol), water, and solvent (3 mL) were added. The mixture was stirred at 27 °C under an ambient atmosphere. After stirring for a specific period, the catalyst was removed by filtration and washed with diethyl ether (ca. 5 mL × 3). The filtrate was concentrated under reduced pressure. To the residue was added 1,1,2,2-tetrachloroethane (52.7 μL, 0.50 mmol) and CDCl3 (ca. 1 mL), and then 1H NMR analysis was conducted using a portion of this solution. The yields were determined by comparison of an integrated value of the peak that corresponds to a proton of silanol with that corresponding to two protons of 1,1,2,2-tetrachloroethane (δ 5.98 ppm).Recycling run experiment
To a reaction tube equipped with a magnetic stir bar was added Au:F-CAC (0.01 atom%), 1a (0.50 mmol), water (400 mol%), and THF (3 mL). The mixture was stirred at 27 °C under an ambient atmosphere. After stirring for 5 h, the catalyst was removed by filtration and washed with diethyl ether (ca. 5 mL × 3). The spent catalyst was dried at 45 °C under reduced pressure for 12 h, and this was used for the next recycling run.Near ambient pressure X-ray photoelectron spectroscopy experiment of Au:PVP under oxygen/water atmosphere
The near ambient pressure X-ray photoelectron spectroscopy (NAP-XPS) experiments were performed at the BL13B beamline of photon factory (PF) equipped with an APPLE II type undulator34 under the ring-conditions of 2.5 GeV and 450 mA. X-ray beam energy was tuned to be 630 eV by a Monk-Gillieson-type monochromator.35 NAP-XPS data was collected at Au 4f and Si 2p core levels with constant analyser energy (CAE) mode with the energy step size of 0.05 eV at room temperature. The binding energy was corrected using the Si 2p3/2 signal of a Si substrate to 99.1 eV. The spectra were normalised to exclude the attenuation caused by the introduced gases.A silicon substrate (P-type, low conductivity, Si(100) facet, Nilaco Co., SI-500440, ca. 10 mm × ca. 10 mm × 0.5 mm) was cleaned by ultrasound irradiation in ethanol for 15 min and dried under ambient atmosphere. 6.8 mg of Au:PVP(K-30) was dissolved in ethanol (20 mL) using a volumetric flask to prepare 3.0 mmol L−1 Au:PVP(K-30) solution. 0.1 mL of the solution was diluted with 0.9 mL ethanol to afford 0.3 mmolPVP L−1 Au:PVP(K-30) solution. 5 μL of the Au:PVP(K-30) solution (0.3 mmolPVP L−1) was applied on a Si substrate. After drying, the sample was attached to a sample holder. The sample holder was fixed on a sample bank, and then this was connected to a linear and rotatable manipulator. After the pressure in the load-lock chamber reached less than 1 × 10−7 Torr, the sample was transferred to a main chamber. XPS data were collected under the pressure of 6.8 × 10−9 Torr. Then, oxygen gas was slowly introduced to the main chamber from a gas cylinder connected through a variable leak valve. After the pressure of the main chamber reached 0.1 Torr, NAP-XPS experiments were performed under an oxygen atmosphere. H2O was purified by the freeze–thaw-pumping method and slowly introduced by a variable leak valve to the main chamber. After the pressure of the main chamber reached 0.2 Torr (poxygen = 0.1 Torr, pwater = 0.1 Torr), NAP-XPS experiments were performed under the oxygen/water atmosphere.
Solution-state X-ray absorption spectroscopy of Au:PVP in water
Au L3-edge X-ray absorption spectroscopy (XAS) experiments were performed at the BL14B2 beamline of SPring-8 using Si(111) double-crystal monochromatised synchrotron radiation under the ring-conditions of 8.0 GeV and 100 mA. All experiments were carried out using the fluorescent method with quick scan technique (QXAFS) at room temperature, otherwise noted. Ionisation chambers were used to measure the intensities of the incident (I0) and transmitted (I1) X-ray. X-ray fluorescence was monitored using a 19-element Ge solid-state detector (SSD). XAS analysis was conducted using the Demeter package, a comprehensive system for processing and analysing XAS data.36–38 Background removal and normalisation of raw data were performed using the cubic spline method using Athena software. E0 was defined as photon energy at the absorption edge where μT = 0.5 in the normalised XAS spectrum. E0 values of Au foil were set to 11919 eV for photon energy calibration. Reference samples (Au foil and Au2O3) were measured using the transmission method.Au:PVP(K-30) was dissolved in H2O to prepare 8.0 mmolAu L−1 solution. The solution was introduced in a polyethylene bag and subjected to an XAS experiment. Then, the solution was bubbled with N2 gas at the flow rate of 60 mL min−1 for 15 min to degas the dissolved oxygen. After sealing, the XAS experiment was again conducted.
3. Results and discussion
The Au:F-CAC catalyst with the particle size of 1.7 ± 0.8 nm (1.7 × 10−3 wt%) was prepared according to the reported method,30 and the dehydrogenative oxidation of hydrosilane was investigated using triphenylsilane (1a) as a substrate. In the presence of Au:F-CAC (0.5 atom%) and H2O (1000 mol%) in n-hexane, the oxidation of hydrosilane proceeded to give triphenylsilanol (2a) in 58% yield after 15 min (Table 1, entry 1). Hexaphenyldisiloxane (3a) was not detected. The use of polar solvents, such as EtOAc and tetrahydrofuran (THF), increases the yield slightly (entries 2 and 3). The complete conversion of 1a was achieved by elongating the reaction time to 120 min, giving 2a in quantitative yield (entry 4). Eventually, the amount of Au loading could be reduced to 0.01 atom% without a decrease in the yield of 2a, although a slight extension of the reaction time was required (entries 5–7). The reaction did not proceed in the absence of Au:F-CAC (entry 8). In addition, reducing the amount of water requires a longer reaction time (entries 6 and 7).
Recycling experiments evaluated the durability of the Au:F-CAC catalyst. Fig. 2a presents the result of recycling experiments using 0.01 atom% of Au-F-CAC in THF. The spent catalyst was easily recovered through filtration and reused without additional treatment. Notably, Au:F-CAC exhibited a consistently high activity without substantial loss up to the sixth recycling experiment. The transmission electron microscopy (TEM) images of fresh and reused Au:F-CAC are shown in Fig. 2b. Although slight aggregation of Au nanoparticles was observed after the sixth cycle, the mean diameter of Au NPs was 2.5 ± 1.0 nm, which is within the error of the fresh catalyst (1.7 ± 0.8 nm). The sustained catalytic activity signifies the high stability of this catalyst, likely attributed to the presence of carboxylic acid attached to cellulose. The notable stability of Au:F-CAC allowed us to investigate the size dependency study in this reaction (vide infra). The hot filtration of the Au:F-CAC catalyst was conducted to separate the catalyst from the reaction mixture when the yield of 2a reached 56% (ca. after 3 h, Fig. 2c). The resulting filtrate was further treated under the same reaction conditions in the absence of the filtered catalyst, and no increase in the yield of 2a was observed. In addition, the inductively coupled plasma spectroscopy-atomic emission spectroscopy (ICP-AES) of the filtrate confirmed the absence of Au species (detection limit: 10 ppb). These results indicated that the reaction takes place on the surface of Au nanoparticles in a heterogeneous fashion. Other substrates, such as methyldiphenylsilane (1b) and dimethylphenylsilane, were also subjected to the reaction conditions to give the corresponding silanols 2b and 2c quantitatively (Table S4†).
 | ||
| Fig. 2 Catalytic performance of Au-F-CAC catalyst for oxidation of hydrosilane. (a) Recycling run experiment. (b) TEM images of the fresh and spent catalyst after the sixth run. Mean diameter and standard deviation are based on the average 300 particles. (c) Hot filtration experiment. | ||
Thanks to the high durability of the Au:F-CAC catalyst under the reaction conditions, our attention was then focused on the size dependency study. Au:F-CAC catalysts with the sizes of 1.7 ± 0.8 nm, 4.9 ± 0.7 nm, and 7.7 ± 1.4 nm were prepared according to the originally developed trans-deposition method for F-CAC,30 and the kinetic studies were carried out using 0.01 atom% catalysts at 27 °C. In all catalysts, the oxidation reaction proceeded to give 2a. It should be noted that the severe aggregation of AuNPs was not observed after the reaction, even in the cases of 4.9 nm and 7.7 nm (Fig. S1†). Fig. 3 displays the time-course plots of the concentration of 1a. The order of the reaction rate was 1.7 nm > 4.9 nm > 7.7 nm. Although the induction period was observed in the case of Au:F-CAC with the size of 7.7 nm (ca. 2 h), these plots could be fitted with linear functions, showing the zeroth-order kinetics for 1a. The zeroth-order kinetics signifies that the catalytically active surface of Au was thoroughly saturated with reactants, which is due to the small amount of Au used ([1a]/[Nsurface Au] > 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000). Therefore, the zeroth-order kinetic constant (k, mmol L−1 h−1) and turnover frequency (TOF) based on total loaded Au atoms (TONtotal Au, h−1) were evaluated using the data after the induction period (Table 2) as follows,
000). Therefore, the zeroth-order kinetic constant (k, mmol L−1 h−1) and turnover frequency (TOF) based on total loaded Au atoms (TONtotal Au, h−1) were evaluated using the data after the induction period (Table 2) as follows,
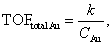
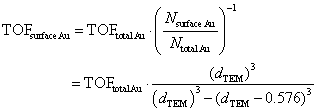
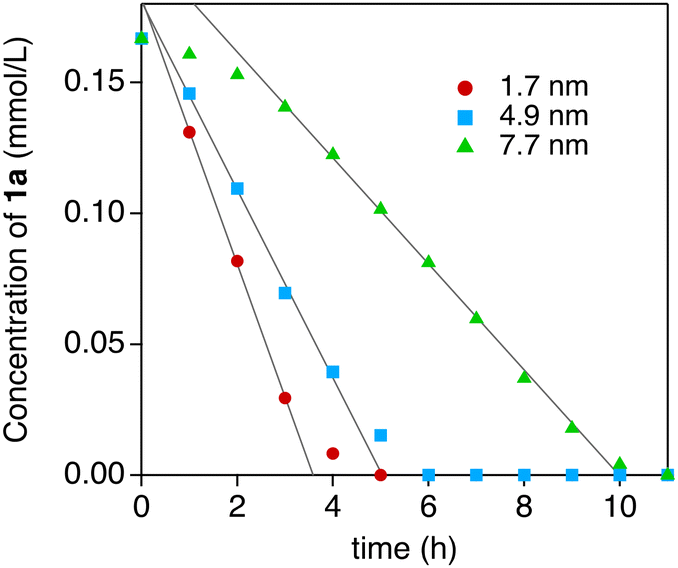 | ||
| Fig. 3 Plots of the concentration of 1a after the specific time. The reaction was performed under air using 0.01 atom% Au:F-CAC and H2O (400 mol%) in THF (3 mL) at 27 °C. | ||
The distance between Au atoms in the particles is assumed to be 0.288 nm from the crystal structure of Au. The TOFsurface Au values of Au:F-CAC of 1.7 nm, 4.9 nm, and 7.7 nm were calculated to be 4304 h−1, 7028 h−1, and 5976 h−1, respectively. These results indicated that Au:F-CAC, with a size of 4.9 nm, exhibited the highest catalytic activity, showing explicit size dependency on the oxidation of hydrosilane. The catalytic activity of previously reported AuNP catalysts, including Au:F-CAC, has been compiled in Table S4.† While the catalytic activity, based on TOF, was found to be moderate compared to other catalysts, it is important to note that Au:F-CAC offers distinct advantages. Unlike some alternatives, Au:F-CAC does not necessitate expensive materials or intricate synthetic procedures. In fact, the preparation of F-CAC can be scaled up to 30 grams using straightforward protocols and inexpensive, naturally derived starting materials. This accessibility renders Au:F-CAC a highly convenient nanomaterial option.
The effect of the reaction atmosphere was investigated using 0.01 atom% of Au:F-CAC catalyst at 27 °C and evaluated by the yield of 2a after 2 h. The yields of 2a in oxygen, air, and argon atmosphere were 93%, 29%, and 6%, respectively (Table 3). This result clearly indicated that the oxygen partial pressure affects the reaction rate, and the adsorption of molecular oxygen on the surface of Au NP and/or such adsorbed Au species is involved in the rate-determining step. An isotope labelling experiment and theoretical calculations were conducted to understand the reaction mechanism of hydrosilane oxidation. First, the reaction was carried out using [18O]H2O under ambient aerobic conditions to track the origin of the oxygen atom of silanol.39 The reaction was completed after 4 h using the optimised reaction conditions. Fourier transform infrared (FT-IR) absorption spectroscopy of the thus-synthesized 2a was measured (Fig. 4a). A peak corresponding to the Si−O vibration appeared at 845 cm−1, red-shifted by 12 cm−1 from that of non-labelled 2a (857 cm−1). This result indicated the formation of [18O]2a, and the oxygen atom introduced in 2a was derived from H2O rather than molecular oxygen. The theoretical calculations were conducted using the density functional theory (DFT) method at the B3LPY-D3BJ/6-31G(d) level of theory in the gas phase to assign the vibration mode. The Si–18O vibration was shifted from the Si–16O vibration by 11 cm−1, showing good agreement with the experimental data (Fig. S4†). Therefore, molecular oxygen accelerates the reaction by adsorbing it on the surface of Au NPs. Previously, we reported that AuNP-catalysed intramolecular cyclisation of amines and/or alcohols proceeded under an aerobic atmosphere using AuNP catalysts, such as Au:PVP40 and Au:F-CAC.33 In addition, theoretical calculations were also investigated to suggest that a cationic Au site that catalyses the intramolecular cyclisation was expected to be formed on their surface through the adsorption of molecular oxygens.41 Given this, the acceleration effect on the oxygen in this reaction would be attributed to the effective formation of the cationic Au species. Camargo et al. reported that the higher number of cationic Au sites formed through Au-support interactions is key for high catalytic activity on the oxidation of hydrosilane, suggesting a good coincidence.24
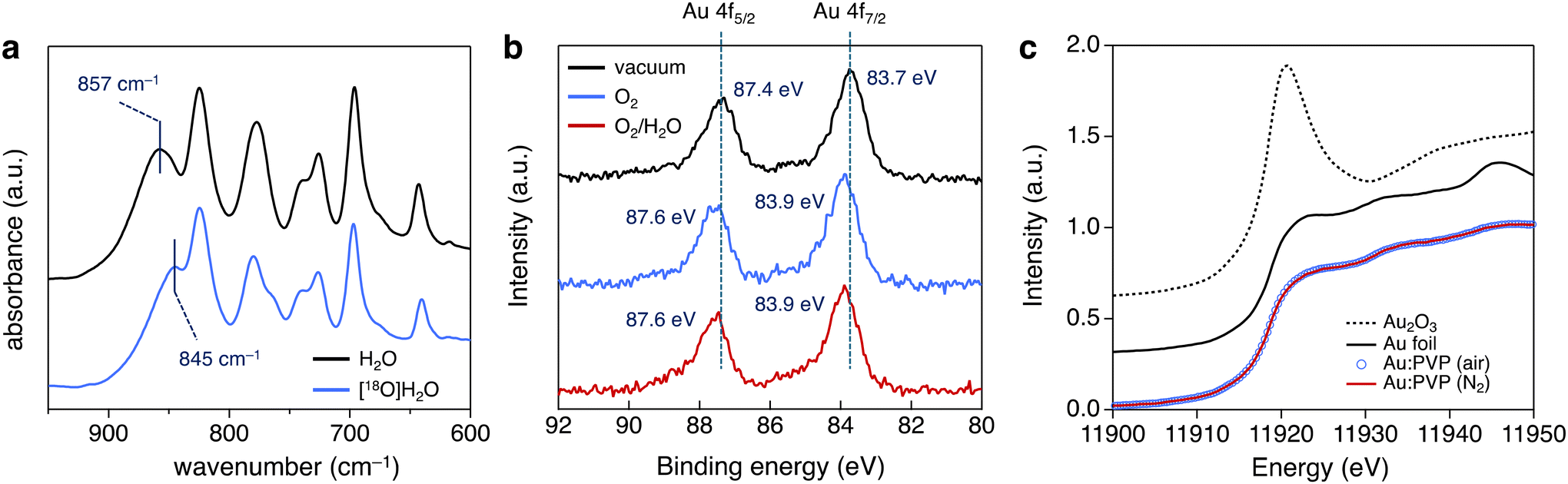 | ||
| Fig. 4 Mechanistic studies. (a) Isotope labelling experiment using [18O]H2O and FT-IR spectra of 2a. Au:F-CAC (0.01 atom%), [18O]H2O (400 mol%), and THF (3 mL) were used. (b) Au 4f NAP-XPS data of Au:PVP under vacuum (black line), poxygen = 0.1 Torr (bule line), and poxygen = 0.1 Torr, pH2O = 0.1 Torr (red line). (c) Au L3-edge XAS data of Au:PVP in H2O. | ||

To confirm the formation of cationic Au site experimentally, NAP-XPS experiments were conducted using Au:PVP(K-30, 1.7 nm) as a model catalyst. Au 4f NAP-XPS experiments were conducted at a poxygen of 0.1 Torr along with ultrahigh vacuum condition (6.8 × 10−9 Torr) using the same sample, where p denotes the partial pressure of gaseous molecules. XP spectra at the Au 4f core level are shown in Fig. 4b. Under ultrahigh vacuum conditions, the peaks corresponding to Au 4f7/2 and Au 4f5/2 core levels were observed at 83.7 eV and 87.4 eV, respectively, indicating a shift toward lower binding energy relative to bulk Au (84.0 eV for Au 4f7/2, 87.7 eV for Au 4f5/2).42 This shows the formation of slightly anionic Au(0) species. Meanwhile, Au 4f XPS signals appeared at 83.9 eV and 87.6 eV under the O2 atmosphere (poxygen = 0.1 Torr). The 0.2 eV shift is attributed to the adsorption of molecular oxygen on the surface of Au NPs, representing experimental evidence for the formation of cationic Au sites. In addition, the NAP-XPS experiment was also conducted under gaseous O2/H2O co-existing condition (poxygen = 0.1 Torr, pwater = 0.1 Torr). However, no further peak shift was observed. These results suggested that molecular oxygen strongly influences the electronic state of AuNPs, while the influence of water is small. To sum up, the NAP-XPS result clearly shows that molecular oxygen plays an essential role in generating active cationic Au sites, explaining the effect of the atmosphere. Hence, this result provides a new aspect of the generally accepted reaction mechanism for the oxidation of hydrosilane.
In addition to the NAP-XPS experiment, the Au L3-edge solution state XAS experiments were also conducted to gain an experimental insight into the electronic state of Au NPs. Au:PVP(K-30) was dissolved in H2O (Au concentration: 8.0 mmol L−1), and the solution was bubbled with N2 gas for 15 min. Then, the solution that was prepared was packed in a gas barrier bag and subjected to the XAS experiment using the fluorescent method. However, the XAS spectra were almost the same before and after bubbling N2 gas due to the existence of a solvent amount of H2O, which adsorbs on the Au surface before the gaseous oxygen (Fig. 4c).
To clarify the detailed mechanism, the kinetic study was further investigated. The reaction rate was plotted against the concentration of H2O, revealing an increase in reaction rate with higher H2O concentration, indicating the participation of H2O in the rate-determining step (RDS) (Fig. S2 and S3†). Next, the reaction rate was measured using D2O as an additive instead of H2O, revealing the presence of a secondary kinetic isotope effect (KIE) (kH/kD = 1.17, n = 3), suggesting that the O–H bond cleavage occurs as a pre-equilibrium process before the RDS (Fig. S5†).43,44 This implies that processes such as dissociative adsorption of H2O or proton transfer are not involved. Based on these experimental findings, a possible reaction mechanism is shown in Fig. 5. The reaction initiates with oxygen adsorption to form cationic Au surface species such as A. Subsequently, dissociative adsorption of H2O and hydrosilane occurs through Si–H and/or O–H bond cleavage, yielding intermediate B and/or C, which exist in equilibrium. Following this, Si–O bond formation is postulated to occur via an SN2 type mechanism, akin to the previously reported PdNP-catalysed dehydrogenative oxidation of hydrosilanes,45 resulting in the formation of silanol and intermediate D. Finally, reductive elimination from D occurs on the Au surface to regenerate A or B with expelling H2 thus completing the catalytic cycle. Although the role of the cationic Au sites generated through the adsorption of O2 remains unclear, further investigations utilizing theoretical calculations are warranted to gain insights into the precise reaction mechanism. It is noteworthy that the actual reaction mechanism is likely more complex than the proposed model mechanism due to the presence of multiple molecules on the surface under experimental conditions.
 | ||
| Fig. 5 Possible reaction mechanism. | ||
4. Conclusions
In summary, our study presents the dehydrogenative oxidation of hydrosilane using size-selectively prepared Au:F-CAC catalysts. These catalysts demonstrated notable catalytic activity in terms of TOF and exhibited high stability under the reaction conditions, enabling the exploration of size dependency. Since the first finding of Au NP-catalysed oxidation of hydrosilanes in 2009, the role of oxygen has remained unclear despite its significant influence on reaction facilitation. This study, especially the mechanistic studies, sheds light on a new aspect of the role of oxygen: the formation of cationic Au sites by adsorption. Under such aerobic conditions, it is well known that Au NPs show high catalytic activity in various oxidation reactions, but at the same time, they also possess cationic properties, which is another crucial factor in reaction development and mechanistic studies.Author contributions
H. S. and Y. U. conceived and designed the experiments. B. S. performed the preparation and characterizations of catalysts and chemical reactions. Y. U., R. I., R. T., and H. K. performed NAP-XPS experiments and analysis. Y. U. conducted XAS experiments and analysis. B. S. and Y. U. prepared an earlier version of the manuscript, and all authors discussed the results and edited the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
We thank H. Uyama (Osaka Univ.) for the preparation of F-CAC. We also thank K. Mase (High Energy Accelerator Research Organization) for support of NAP-XPS experiments at the photon factory and T. Honma (JASRI) for support of XAS experiments at SPring-8. NAP-XPS experiments were performed at the BL13B beamline of KEK under the approval of the Photon Factory Program Advisory Committee (proposal no. 2020G548). Au L3-edge XAS measurements were performed at the BL14B2 beamline of SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (proposal no. 2021B1941 and 2022B1890). The theoretical calculations were performed using Research Center for Computational Science, Okazaki, Japan (Project: 22-IMS-C068). This research was supported by the JST-Mirai Program (JPMJMI18E3) and JSPS KAKENHI grant no. JP19K22187 (H. S.), JP20K15279 (Y. U.) and JP22K05095 (Y. U.). B.S. acknowledges JSPS for the scholarship.References
- R. Pietschnig, Advances and Properties of Silanol-Based Materials, in Main Group Strategies towards Functional Hybrid Materials, 2018, pp. 141–162 Search PubMed.
- A. K. Franz and S. O. Wilson, J. Med. Chem., 2013, 56, 388 CrossRef CAS PubMed.
- S. E. Denmark and S. Fujimori, J. Am. Chem. Soc., 2005, 127, 8971 CrossRef CAS PubMed.
- P. W. Long, X. F. Bai, F. Ye, L. Li, Z. Xu, K. F. Yang, Y. M. Cui, Z. J. Zheng and L. W. Xu, Adv. Synth. Catal., 2018, 360, 2825 CrossRef CAS.
- M. Jeon, J. Han and J. Park, ACS Catal., 2012, 2, 1539 CrossRef CAS.
- P. D. Lickiss and R. Lucas, J. Organomet. Chem., 1996, 521, 229 CrossRef CAS.
- K. Valliant-Saunders, E. Gunn, G. R. Shelton, D. A. Hrovat, W. T. Borden and J. M. Mayer, Inorg. Chem., 2007, 46, 5212 CrossRef CAS PubMed.
- B. Yang and Z. X. Wang, Org. Lett., 2019, 21, 7965 CrossRef CAS PubMed.
- L. H. Sommer, L. A. Ulland and G. A. Parker, J. Am. Chem. Soc., 1972, 94, 3469 CrossRef CAS.
- L. Spialter and J. D. Austin, J. Am. Chem. Soc., 1965, 87, 4406 CrossRef CAS.
- J. M. Asensio, D. Bouzouita, P. W. N. M. van Leeuwen and B. Chaudret, Chem. Rev., 2020, 120, 1042 CrossRef CAS PubMed.
- H. Yamagishi, J. Shimokawa and H. Yorimitsu, ACS Catal., 2023, 13, 7472 CrossRef CAS.
- D. Limnios and C. G. Kokotos, ACS Catal., 2013, 3, 2239 CrossRef CAS.
- T. Mitsudome, A. Noujima, T. Mizugaki, K. Jitsukawa and K. Kaneda, ChemComm, 2009, 5302 RSC.
- T. Urayama, T. Mitsudome, Z. Maeno, T. Mizugaki, K. Jitsukawa and K. Kaneda, Chem. Lett., 2015, 44, 1062 CrossRef CAS.
- L. Ma, W. Leng, Y. Zhao, Y. Gao and H. Duan, RSC Adv., 2014, 4, 6807 RSC.
- T. Mitsudome, Y. Yamamoto, A. Noujima, T. Mizugaki, K. Jitsukawa and K. Kaneda, Chem. – Eur. J., 2013, 19, 14398 CrossRef CAS PubMed.
- N. Asao, Y. Ishikawa, N. Hatakeyama, Menggenbateer, Y. Yamamoto, M. Chen, W. Zhang and A. Inoue, Angew. Chem., Int. Ed., 2010, 49, 10093 CrossRef CAS PubMed.
- V. Gitis, R. Beerthuis, N. R. Shiju and G. Rothenberg, Catal. Sci. Technol., 2014, 4, 2156 RSC.
- T. Liu, F. Yang, Y. Li, L. Ren, L. Zhang, K. Xu, X. Wang, C. Xu and J. Gao, J. Mater. Chem., 2014, 2, 245–250 RSC.
- G. M. A. Rahman, D. M. Guldi, E. Zambon, L. Pasquato, N. Tagmatarchis and M. Prato, J. Nanotechnol., 2005, 1, 527 CAS.
- J. John, E. Gravel, A. Hagège, H. Li, T. Gacoin, T. Gacoin and E. Doris, Angew. Chem., Int. Ed., 2011, 50, 7533 CrossRef CAS PubMed.
- Z. T. Xie, T. Asoh, Y. Uetake, H. Sakurai and H. Uyama, Carbohydr. Polym., 2020, 247, 116723 CrossRef CAS PubMed.
- A. G. M. da Silva, C. M. Kisukuri, T. S. Rodrigues, E. G. Candido, I. C. de Freitas, A. H. M. da Silva, J. M. Assaf, D. C. Oliveira, L. H. Andrade and P. H. C. Camargo, Appl. Catal., 2016, 184, 35 CrossRef CAS.
- H. T. Tang, H. Y. Zhou, Y. M. Pan, J. L. Zhang, F. H. Cui, W. H. Li and D. Wang, Angew. Chem., Int. Ed., 2024, 63, e202315032 CrossRef CAS PubMed.
- X. Cui, A. Ozaki, T. Asoh and H. Uyama, Polym. Degrad. Stab., 2020, 175, 109118 CrossRef CAS.
- X. Cui, T. Honda, T. Asoh and H. Uyama, Carbohydr. Polym., 2020, 230, 115662 CrossRef CAS PubMed.
- S. Haesuwannakij, T. Poonsawat, M. Noikham, E. Somsook, Y. Yakiyama, R. N. Dhital and H. Sakurai, J. Nanosci. Nanotechnol., 2017, 17, 4649 CrossRef CAS.
- S. Haesuwannakij, Y. Yakiyama and H. Sakurai, ACS Catal., 2017, 7, 2998 CrossRef CAS.
- T. Chutimasakul, Y. Uetake, J. Tantirungrotechai, T. Asoh, H. Uyama and H. Sakurai, ACS Omega, 2020, 5, 33206 CrossRef CAS PubMed.
- H. Tsunoyama, H. Sakurai, Y. Negishi and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 9374 CrossRef CAS PubMed.
- H. Tsunoyama, H. Sakurai and T. Tsukuda, Chem. Phys. Lett., 2006, 429, 528 CrossRef CAS.
- Y. Uetake, B. Suwattananuruk and H. Sakurai, Sci. Rep., 2022, 12, 20602 CrossRef CAS PubMed.
- S. Sasaki, K. Kakuno, T. Takada, T. Shimada, K. Yanagida and Y. Miyahara, Nucl. Instrum. Methods Phys. Res., Sect. A, 1993, 331, 763 CrossRef.
- K. Amemiya and T. Ohta, J. Synchrotron Radiat., 2004, 11, 171 CrossRef PubMed.
- J. J. Rehr and R. C. Albers, Rev. Mod. Phys., 2000, 72, 621 CrossRef CAS.
- M. Newville, J. Synchrotron Radiat., 2001, 8, 322 CrossRef CAS PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537 CrossRef CAS PubMed.
- Q. Zhang, M. Peng, Z. Gao, W. Guo, Z. Sun, Y. Zhao, W. Zhou, M. Wang, B. Mei, X. L. Du, Z. Jiang, W. Sun, C. Liu, Y. Zhu, Y. M. Liu, H. Y. He, Z. H. Li, D. Ma and Y. Cao, J. Am. Chem. Soc., 2023, 145, 4166 CrossRef CAS PubMed.
- H. Kitahara and H. Sakurai, Chem. Lett., 2009, 39, 46 CrossRef.
- K. Bobuatong, H. Sakurai and M. Ehara, ChemCatChem, 2017, 9, 4450 CrossRef CAS.
- H. Tsunoyama, N. Ichikuni, H. Sakurai and T. Tsukuda, J. Am. Chem. Soc., 2009, 131, 7086 CrossRef CAS PubMed.
- E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066 CrossRef CAS PubMed.
- N. J. F. Christensen, Synlett, 2015, 508 CAS.
- T. Kamachi, K. Shimizu, D. Yoshihiro, K. Igawa, K. Tomooka and K. Yoshizawa, J. Phys. Chem. C, 2013, 117, 22967 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr01184h |
| This journal is © The Royal Society of Chemistry 2024 |