
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanostructured electrocatalysts for organic synthetic transformations
Francesco
Mancuso
 a,
Paolo
Fornasiero
ab,
Maurizio
Prato
acd,
Michele
Melchionna
*ab,
Federico
Franco
*a and
Giacomo
Filippini
*a
a,
Paolo
Fornasiero
ab,
Maurizio
Prato
acd,
Michele
Melchionna
*ab,
Federico
Franco
*a and
Giacomo
Filippini
*a
aDepartment of Chemical and Pharmaceutical Sciences University of Trieste via Licio Giorgieri 1, 34127 Trieste, Italy. E-mail: melchionnam@units.it; federico.franco@units.it; gfilippini@units.it
bCenter for Energy, Environment and Transport Giacomo Ciamician and ICCOM-CNR Trieste Research Unit University of Trieste, via Licio Giorgieri 1, 34127 Trieste, Italy
cCenter for Cooperative Research in Biomaterials (CIC BiomaGUNE) Basque Research and Technology Alliance (BRTA), Paseo de Miramón 194, 20014, Donostia San Sebastián, Spain
dBasque Foundation for Science Ikerbasque, 48013 Bilbao, Spain
First published on 20th February 2024
Abstract
Organic chemists have made and are still making enormous efforts toward the development of novel green catalytic synthesis. The necessity arises from the imperative of safeguarding human health and the environment, while ensuring efficient and sustainable chemical production. Within this context, electrocatalysis provides a framework for the design of new organic reactions under mild conditions. Undoubtedly, nanostructured materials are under the spotlight as the most popular and in most cases efficient platforms for advanced organic electrosynthesis. This Minireview focuses on the recent developments in the use of nanostructured electrocatalysts, highlighting the correlation between their chemical structures and resulting catalytic abilities, and pointing to future perspectives for their application in cutting-edge areas.
 Giacomo Filippini | Giacomo Filippini obtained his master's degree in industrial chemistry from the University of Bologna (Italy). In 2013 he joined the group of Prof. Paolo Melchiorre at ICIQ in Tarragona (Spain), where he undertook his doctoral studies. In 2017, he started a postdoctoral appointment in the group of Prof. Maurizio Prato at the University of Trieste (Italy), where he is currently working as an assistant professor, investigating the use of carbon-based nanomaterials to design novel organic transformations. |
1. Introduction
Electrochemistry has been gaining great momentum in the area of industry-relevant organic synthesis.1 The basic concept is to exploit the electrochemical potential to form highly reactive intermediates, such as radicals and radical ions, from suitable organic precursors through single electron transfer (SET) processes.2,3–6 In principle, these SET events occur under mild operating conditions and in a controlled fashion, thus avoiding the use of potentially harmful reagents (e.g. a stoichiometric amount of oxidants/reductants), or harsh conditions as in the case of conventional thermal processes.7 As a consequence, electrochemical synthetic strategies are extremely attractive with regard to the sustainability of a chemical process. The exploitation of electrochemical potential as the energy source can also enable reaction pathways and/or intermediates which would be difficult to reach with classical thermal methods, paving the way to novel synthetic routes.8 An additional advantage of electrosynthesis includes the feasibility of process scaling-up at limited cost and time, favoring their exploitation in the industrial synthesis of bulk and fine chemicals.A central element of the electrochemical device is represented by the catalytic material, a species able to harness the electrochemical potential and open specific synthetic pathways for the controlled evolution of reagents to products. Therefore, a correct design and assembly of the electrocatalytic material holds the key to improved efficiency and selectivity. Within this framework, nanostructured electrocatalysts are particularly attractive in view of their high reactivity, which originates from the abundance of low-coordination active sites. In addition, their large electrocatalytic surface area ensures high current densities.9,10 A clear demonstration of the central importance of electrocatalysts, and in particular nanostructured catalysts, is the fact that many industrial systems rely on the use of nanometric active particles dispersed on high-surface-area supports, ensuring the minimum required energy efficiency.11 The support also deserves some consideration as its role goes beyond the mere stabilization of the nanoparticles, and in several cases it exhibits active participation in the process. In fact, the support is able to efficiently “wire” the nanoparticles to the electrode's surface or even assist the catalytic sites in generating key intermediates.12
The vision of decarbonization of the chemical industry must take into account the potentiality of organic electrosynthesis powered by renewable electricity, which surely represents an emerging sustainable alternative route to thermal processes for the production of value-added and fine chemicals.13–16 Conventional synthetic processes are typically energy-intensive and suffer from CO2 emissions, with consequent environmental issues related to the CO2 greenhouse effect. At the same time, CO2 is an attractive low-cost abundant carbon source and can be in principle used as a C1 feedstock or building block for the preparation of organic molecules.17 The electrochemical CO2 reduction reaction (CO2RR) catalyzed by nanostructured materials has been widely explored as a promising strategy to produce C1–C2+ fuels and carbonaceous products, including carbon monoxide, oxygenates and hydrocarbons.18,19 However, coupling the CO2RR with organic electrosynthesis presents an opportunity to expand the scope of the process to synthesize a large variety of organic products. According to this approach, CO2RR products can be used in situ or ex situ as building blocks for organic synthesis. For instance, the CO2RR-derived CO was recently used to promote organic carbonylation reactions for the scalable synthesis of pharmaceutically relevant compounds.20 Furthermore, reactive key intermediates involved in the multi-electron CO2RR have been employed as reactants to drive C–N or C–S coupling reactions in combination with small-molecule N or S-containing precursors.21,22 Hence, the CO2RR selectivity towards a specific target product is the key to achieving efficient organic electrosynthesis. In this regard, the mechanistic elucidation of the catalytic pathways is an essential tool to understand the factors affecting the selectivity, which include not only the reaction conditions but also the intrinsic properties of the catalyst. For Cu-based CO2RR nanostructured electrocatalysts, structural features such as particle size/shape dependence,23–26 morphological restructuring27 and oxidation state28 are known to strongly impact the observed selectivity.
Together with CO2 reduction, the production of green hydrogen by water electrolysis also represents a milestone to realize the energetic transition towards a more sustainable chemical production. In this regard, hydrogenation reactions are extremely important in organic synthesis and typically require elevated temperatures and pressures to occur.29 Recently, electrochemical hydrogenation (ECH) of organic substrates in aqueous electrolytes has proven to be a suitable alternative approach to hydrogenate molecules using water as the hydrogen source. Compared to traditional hydrogenation reactions, ECH not only reduces the carbon footprint for hydrogenation reactions, but also promotes an in situ utilization of hydrogen to prepare a wide variety of organic molecules, thus preventing the safety issues related to the transport, handling and storage of H2.30
This minireview aims at highlighting the most recent advances in the production of electrocatalytically active nanomaterials and their applications in organic catalysis. In particular, in Section 2, the review will focus on electrocarboxylation processes, based on the concomitant activation of organic substrates and CO2. Direct ECH of organic substrates will be discussed in Section 3, focusing on structural and functional aspects of the state-of-the-art nanocatalysts in relation to the efficiency and selectivity of the process. Finally, future prospects offered by this thriving research field will be proposed, and how they may contribute to addressing the current challenges in several organic reactions.
2. Reductive electrochemical organic transformations with CO2 as a substrate
In this section, we critically rationalize the state-of-the-art results in the field of reductive electrocatalytic conversion of organic substrates in combination with the CO2 reduction reaction (CO2RR), in which CO2 is used as a carbon source to produce value-added products and fine chemicals. We primarily focus on the electrocarboxylation reactions of organic halides (section 2.1) and ketones (section 2.3), leading to carboxylic acids and their hydroxy derivatives, respectively. In section 2.2 we discuss recent findings about the reductive electrochemical processes of nitrogenated substrates with CO2 based on carbon–nitrogen coupling reactivity, including urea electrosynthesis, N-methylation of amines and the synthesis of carbamates. The reactions herein discussed are mostly reported in organic non-aqueous media, due to the solubility limitations of CO2 and organic substrates/products in water. One further limitation of these types of processes is the concomitant CO2 reduction reaction (CO2RR), which typically leads to the formation of oxalate, carbon monoxide and carbonate in anhydrous non-aqueous media. On one hand, the parasitic CO2RR current contributes to the reduction in the overall faradaic efficiency (FE) for the desired organic product, on the other hand the CO2RR products may also contribute to drastically affect the durability and stability of the carboxylation process, for instance carbonate precipitation leading to cathode passivation.31,32 Moreover, the electrocarboxylation processes typically occur via radical-based mechanisms involving extremely reactive radical intermediates which may undergo several competitive coupling side reactions.2.1 Electrocarboxylation of organic halides
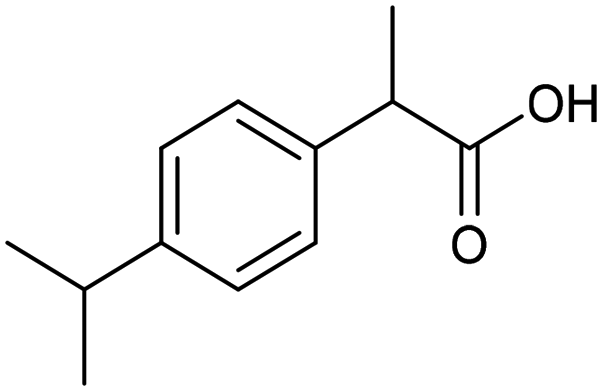
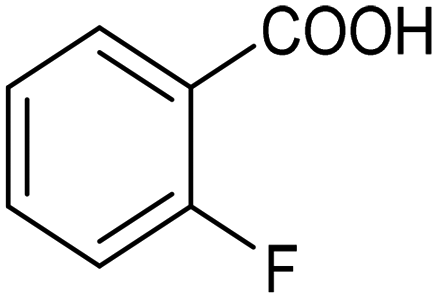
The electrochemical fixation of CO2 into organic halides stands out as a prominent sustainable strategy to produce carboxylic acids, which are essential chemicals for the polymer and pharmaceutical sectors. The traditional approach for the synthesis of carboxylic acids is based on the reaction between a Grignard reagent and CO2 to form a carboxylate salt which, upon protonation, yields carboxylic acids. The electrochemical pathways instead are built on a completely different sequence of events: the electrochemical potential pumps electrons at the cathode, where the reduction of the organic halide occurs on the electrode's surface; concomitantly, a CO2 coupling step leads to the formation of the carboxylate product. Considering the differences between the two approaches, we must emphasize the absence of a chemical reducing species in the electrochemical approach, which implies better atom economy as well as reduced cost and impact on the environment.The dehalogenation of organic halides coupled with carboxylation at metal electrodes has shown good versatility, being applicable for a large variety of substrates.33–35 Metal-based nanostructures are the most suitable cathode materials, often guaranteeing a good level of conversion selectivity into the corresponding carboxylic acids, occurring with high efficiencies. Pt nanoparticles (NPs) embedded into N-doped carbon/carbon cloth (Pt-NP@NCNF@CC) prepared by the electrospinning technique could drive the electrocarboxylation of 1-phenylethyl bromide with CO2 to yield the corresponding 2-phenylpropionic acid, a crucial intermediate for the synthesis of non-steroidal anti-inflammatory drugs, with an efficiency of 99% (entry 1 in Table 1).36 The same group also developed a core–shell composite material, labelled AgNW/NC700, based on the encapsulation of silver nanowires (Ag NWs) within an N-doped carbon shell followed by thermal treatment at 700 °C.37 Under the optimized conditions, AgNW/NC700 could efficiently catalyze the production of ibuprofen, a widely used anti-inflammatory drug, in 95% chemical yield and >99% selectivity using the corresponding benzyl bromide and CO2 as substrates (entry 2 in Table 1 and Fig. 1). Interestingly, the same catalyst provided good-to-excellent 59–91% yields for the CO2 electrocarboxylation of a series of other benzylic and aliphatic halides. Control experiments suggested that the catalytic activity had to be attributed to the Ag active sites, whereas the porous N-doped carbon shell served to increase the local CO2 concentration at the Ag sites, thus boosting the carboxylation process. Despite the encouraging results, a drawback in the above examples is the reliance on precious metals, which could be problematic for translation to larger scale set-ups.
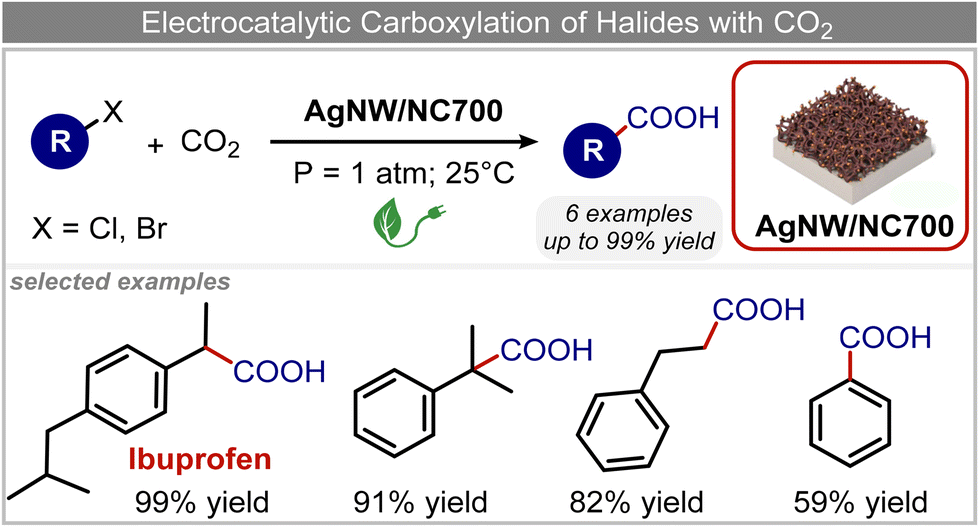 | ||
| Fig. 1 Electrocatalytic carboxylation of organic halides.37 | ||
| Catalyst | Substrate | Product | Yield (FE) | E (V) | Electrolyte |
|---|---|---|---|---|---|
| a j = 3 mA cm−2. b j = 120 mA cm−2. c j = 10 mA cm−2. | |||||
Pt-NP@NCNF@CC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 36 36 |
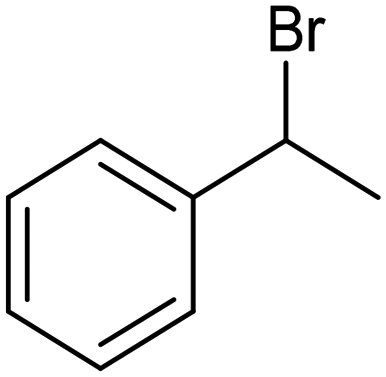
|
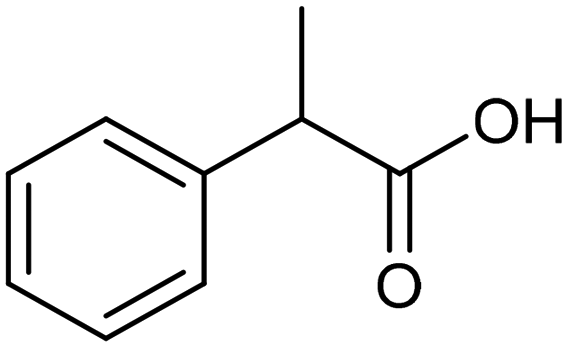
|
99% | —a | 0.1 M TEAI/MeCN |
| AgNW/NC70037 |
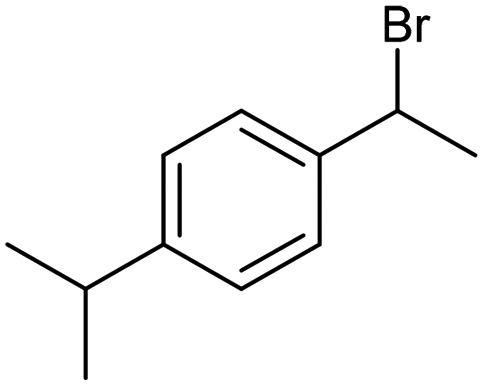
|
|
96% | −0.8 VRHE | 0.1 M TEAI/MeCN |
| Cu@Cz-POP39 |
|
|
65% | —b | 0.1 M TBABF4/MeCN |
| CuAg NWs47 |
|
|
67% (98%) | −2.4 VSCE | 0.1 M TBAPF6/DMF |
| CoPc/CNTs61 |
|
|
(7.5%) | −0.93 VRHE | 0.1 M KHCO3 |
| Pd2.2/Co–N/carbon62 |
|
|
92% | −2.3 VAg/Ag+ | 0.5 M ([Bmim]Tf2N)/MeCN |
| Cu–N–C-364 |
|
|
71% | —c | 0.1 M TEABr/MeCN |
| Ag/BDD68 |
|
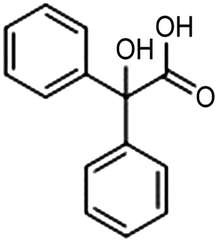
|
87% | −2.22 VSCE | 0.1 M TBABF4/DMF |
| Ag-CeO270 |
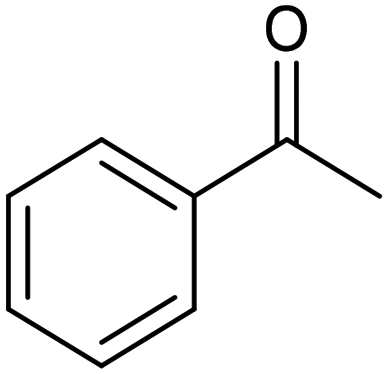
|
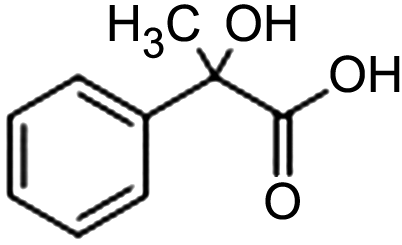
|
91% | −1.8 VAg/agI | 0.2 M TBABr/MeCN |
| La0.7Sr0.3FeO371 |
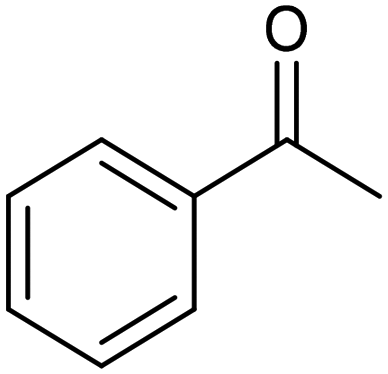
|
|
68% | −2.0 VAg/AgI/I− | 0.1 M TBAI/DMF |
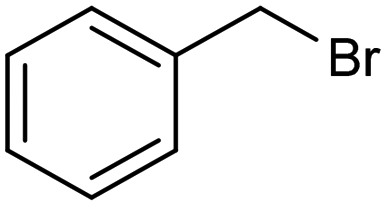
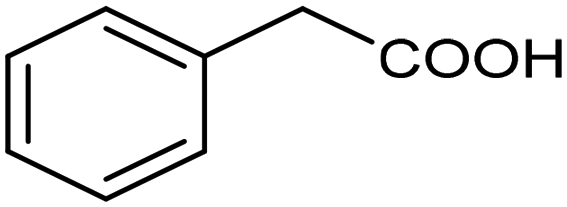
The inclusion of more abundant metals (and the concomitant reduction in precious metal use) is therefore extensively explored, with copper arguably being the most popular transition metal, however, it is challenging to match its activity to those of noble metals. Nevertheless, some notable examples are available, such as a trimetallic AgxCuyNiz alloy electrodeposited on glassy carbon (GC), which could catalyze the conversion of benzyl bromide to phenylacetic acid with >95% yield in 0.1 M TBABF4/DMF.38 Here, it is important to emphasize that while the catalytic material is made of more abundant elements, and therefore at lower cost, the sustainability is partly compromised by the electrolyte system, which is based on more toxic DMF and high priced TBABF4. The formulation of an electrocatalyst employing only copper as the metal element has been recently proposed, featuring Cu embedded into a carbazole-derived porous organic polymer to form the Cu@Cz-POP nanohybrid. This system catalyzed electrocarboxylation of benzyl bromide in a CO2-saturated 0.1 M TBABF4/MeCN electrolyte, therefore implying better sustainability of the process.39 On the other hand, the yield was not quantitative, reaching a value of 65% for the production of phenylacetic acid (entry 3 in Table 1), whose formation was proposed on the basis of a previously hypothesized mechanism involving the carboxylation of the electrogenerated benzyl radical.40 Benzyl 2-phenylacetate (ester) and 1,2-diphenylethane (radical dimerization) were identified to be the major side-products limiting the selectivity of the reaction towards carboxylic acids.
In addition to the intrinsic structural and functional properties of the nanostructured catalyst, the efficiency and selectivity of the CO2 electrocarboxylation of halides is dependent on other factors such as the electrolyte medium and the configuration of the electrochemical reactor. Simple undivided electrochemical setups are typically used for electrolysis experiments, without dividing the cathode/anode compartments by a membrane separator and using a sacrificial metal anode (usually Mg or Al) to avoid undesired oxidation of the substrate or products. The presence of sacrificial anodes is often beneficial for improving the selectivity of CO2 electrocarboxylation towards carboxylic acids.41–43 The cations produced upon oxidation at the anode coordinate the carboxylate species and prevent side nucleophilic reactions with the organic halide substrate leading to the formation of undesired products, such as esters, alcohol and carbonates.41 On the other hand, the utilization of sacrificial anodes drastically limits the sustainability of the process, due to the additional energy consumption required for anode regeneration.44 Moreover, the anionic products typically formed by the parallel CO2 reduction reaction (CO2RR) in aprotic media (e.g., carbonates and oxalates)45 may form insoluble salts with the cations derived from the sacrificial anode during the process, leading to cathode passivation or uncontrolled increase of the cell voltage during operation. These limitations have been recently mitigated by performing the reaction in a two-compartment electrochemical cell with a membrane separator and by adding a soluble inorganic salt (anhydrous MgBr2) to the catholyte, which mimics the beneficial role played by the sacrificial anode in an undivided configuration.46 A sacrificial anode-free system was recently tested for the carboxylation of a variety of aliphatic, benzylic and aromatic halides, providing moderate-to-good yields (34–78%) of carboxylic acids.
By using an H-type electrochemical cell with a Nafion membrane, Cao et al. developed an efficient system for the highly selective electrocarboxylation of aryl bromides coupled with the anodic oxygen evolution reaction (OER).47 CuAg nanowires featured as the cathodic catalysts, displaying remarkably high selectivity for the production of carboxylic acids (the maximum FE of 98%) and a good tolerance to mono-substituted aryl bromides (including naphthalene and heteroaromatic ones). Using bromobenzene as a model substrate, benzoic acid was obtained with 90% FE in a flow cell reactor at −2.4 VSCE (entry 4 in Table 1). The proposed mechanism for the carboxylation reaction involves an initial single-electron reduction of the halide, leading to the formation of a radical intermediate which then undergoes CO2 coupling to yield the carboxylate product. Competitive proton trapping by the organic radical may lead to the hydrogenation side product. Here, the bifunctionality of the heterobimetallic CuAg nanocatalyst appears to be the decisive feature, with Cu activating and reducing CO2 and Ag promoting the dehalogenation reaction. As a result, the competitive hydrogenolysis and reductive coupling reactions of aryl bromides could be effectively suppressed. Notably, a thorough mechanistic study of the electrocarboxylation of aryl bromides recently revealed that the hydrogenolysis side reaction involves a solvent deprotonation step.48 Hence, it was demonstrated that solvent optimization is an important parameter in controlling the selectivity of carboxylation by adjusting the rate of the competitive hydrogenolysis pathway.
2.2 Carbon–nitrogen coupling with CO2
Due to its versatility, the utilization of CO2 electroreduction to build new C–N bonds is a platform for the synthesis of a large variety of organic nitrogenated compounds with high selectivity according to the N-precursor and the CO2RR catalyst. Once again, the catalyst design is critical for selectivity, since reactive intermediates generated at the catalyst's surface during the CO2RR may in situ react with the N-precursor, directing the selectivity towards the formation of specific C–N products.49Urea is one of the typical targets of electrosynthesis, on account of its use as a fertilizer. Finding a more convenient and less environment-impacting synthetic method than the traditional energy-intensive processes from CO2 and Haber–Bosch-derived ammonia is highly attractive. The electrocatalytic approach is based on coupling the CO2RR with the co-reduction of abundant low-cost inorganic nitrogen sources, such as NO3−, NO2− and N2.50–58 Since the seminal work by Shibata et al. in 1995,50 several nanostructured electrocatalysts have been reported for urea electrosynthesis in aqueous media from CO2 and NO3−/NO2− with variable FEs and different proposed mechanisms for C–N coupling.51,52 Among them, the use of an In(OH)3 catalyst at an applied potential of −0.6 VRHE afforded 53.4% FEurea from the co-reduction of NO3− and CO2, for which single {100} facets displayed a low energy barrier for the key coupling step between the *NO2 and *CO2 intermediates.53 More recently, a FEurea of 27.7% has been reported for the same reaction catalyzed by MoOx nanoclusters anchored on carbon black at −0.6 VRHE.58 For these systems, the low-valent highly unsaturated Mo sites were proposed to be beneficial for promoting the catalytic C–N coupling process. In a few cases, dual-site catalysts led to improved urea selectivity; for example, in the case of core–shell bimetallic catalysts based on Cu@Zn nanowires, a very high urea selectivity (9.28% FE) from the co-reduction of CO2 and NO3− evolved from a more favorable coupling of the key *CO and *NH2 intermediates compared to the single metal components.56 Other examples include PdCu alloy nanoparticles on TiO2 nanosheets, where despite a relatively low FE (8.92% at −0.4 VRHE), the work is notable because urea is prepared in an aqueous electrolyte by the direct coupling between CO2 and N2 under ambient conditions.54
In addition to urea, in a few cases the co-electroreduction of CO2 and NOx− led to the formation of added-value organic amines. The first example of cascade electrochemical conversion of CO2 and NO3− precursors to methylamine in aqueous media under ambient conditions has been reported only recently, with FE = 13% and a partial current density of 3.4 mA cm−2 at −0.94 VRHE.57 In this work, the cobalt β-tetraaminophthalocyanine molecular complex supported on carbon nanotubes (CNT) functioned as the catalytic ensemble and it is noteworthy that this is one of the few systems reported to date capable of converting CO2 into CH3OH with good efficiencies.59 The key step is the condensation between formaldehyde (HCHO) and hydroxylamine (NH2OH), which are the critical intermediates formed along the pathways for electrocatalytic CO2-to-CH3OH and NO3−-to-NH3 reactions, respectively. The condensation reaction occurred under ambient conditions leading to the formation of formaldoxime which, in turn, was ultimately converted into methylamine upon further electrochemical reduction. Interestingly, an analogous mechanism involving the spontaneous reaction between acetaldehyde and hydroxylamine intermediates was proposed to explain the electrochemical formation of ethylamine in small amounts (0.15% FE) by the co-reduction of CO2 and NO3− catalyzed by a CuO-based material.60
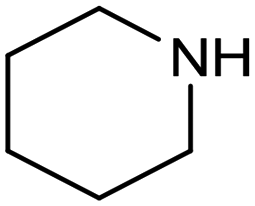
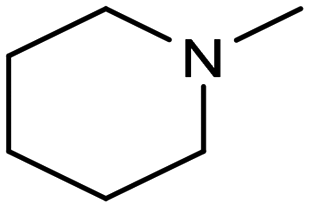
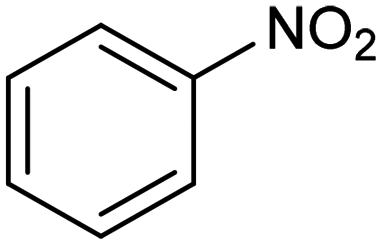
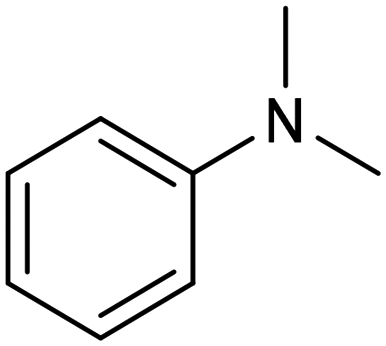
In a follow-up work, the same group extended the substrate scope to a large variety of nitrogen nucleophiles, based on their fast reactivity with the in situ generated HCHO from CO2 electroreduction. This represents the first electrocatalytic approach for the direct N-methylation of amino derivatives with CO2 in aqueous media (Fig. 2), obtained by using a hybrid catalyst based on cobalt phthalocyanine supported on CNTs (CoPc/CNTs).61 The heterogenized molecular catalyst is able to catalyze the four-electron reduction of CO2 to *HCHO, which then reacts with the nucleophilic amine substrate providing the corresponding imines/iminium ion intermediates in solution. Under the catalytic regime, the CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds within these intermediates may be reduced to yield the desired N-methylated products. The reductive N-methylation reaction with CO2 was tested for a variety of aliphatic and aromatic primary and secondary amines. For the model piperidine substrate, the N-methylpiperidine (NMP) product was formed with an FE value of 7.5% at −0.93 VRHE (entry 5 in Table 1). The efficiency of the C–N coupling reaction was partially limited by the competition with the concomitant CO2-to-CH3OH reaction catalyzed by CoPc/CNTs, therefore, an increased nucleophilicity of the amine precursor tends to favor the formation of the N-methylated product over the CO2RR. The formation of N-methylamines was also attempted by starting directly from nitro-compounds as precursors, resulting, however, in very low FE (6.0%) at −0.93 VRHE.61 Nevertheless, selective electrochemical N,N′-dimethylation of nitrobenzenes under CO2 using water as the hydrogen source was successfully attained with Pd NPs supported on Co–N/carbon as the catalyst.62 By using a H-cell configuration, nitrobenzene was converted into N,N-dimethylaniline with a 92% of yield at an applied potential of −2.3 VAg/Ag+ (entry 6 in Table 1), and a decisive role was played by 1-aminomethylphosphonic acid (AMPA) as the co-catalyst which could promote the methylation step of aniline upon reaction with the *HCHO intermediate derived from the CO2RR.
N bonds within these intermediates may be reduced to yield the desired N-methylated products. The reductive N-methylation reaction with CO2 was tested for a variety of aliphatic and aromatic primary and secondary amines. For the model piperidine substrate, the N-methylpiperidine (NMP) product was formed with an FE value of 7.5% at −0.93 VRHE (entry 5 in Table 1). The efficiency of the C–N coupling reaction was partially limited by the competition with the concomitant CO2-to-CH3OH reaction catalyzed by CoPc/CNTs, therefore, an increased nucleophilicity of the amine precursor tends to favor the formation of the N-methylated product over the CO2RR. The formation of N-methylamines was also attempted by starting directly from nitro-compounds as precursors, resulting, however, in very low FE (6.0%) at −0.93 VRHE.61 Nevertheless, selective electrochemical N,N′-dimethylation of nitrobenzenes under CO2 using water as the hydrogen source was successfully attained with Pd NPs supported on Co–N/carbon as the catalyst.62 By using a H-cell configuration, nitrobenzene was converted into N,N-dimethylaniline with a 92% of yield at an applied potential of −2.3 VAg/Ag+ (entry 6 in Table 1), and a decisive role was played by 1-aminomethylphosphonic acid (AMPA) as the co-catalyst which could promote the methylation step of aniline upon reaction with the *HCHO intermediate derived from the CO2RR.
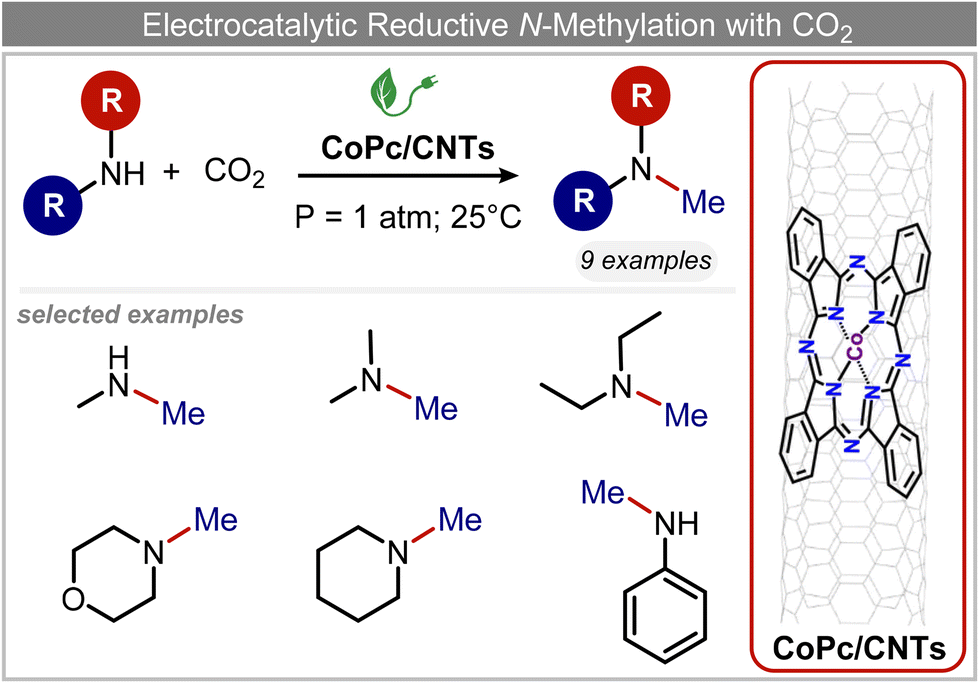 | ||
| Fig. 2 Electrocatalytic reductive methylation of amines.61 | ||
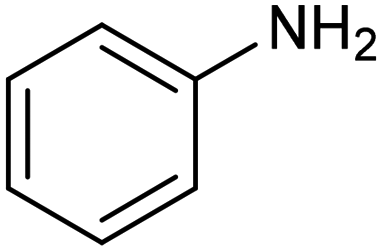
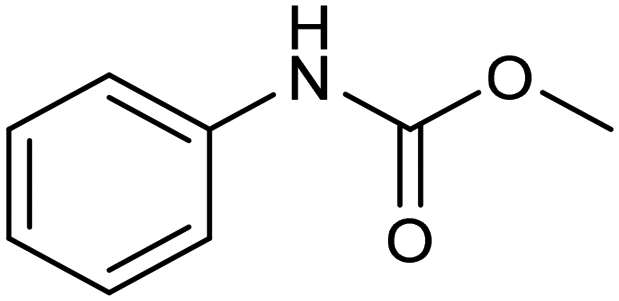
Another interesting class of nitrogenated compounds is carbamates, which find extensive applications in medicinal, polymer and pesticide chemistry.63 Remarkably, traditional thermal synthetic processes mostly employ toxic gases, including phosgene and CO. From this perspective, the electrochemical approach, applicable by using CO2 and amines as C and N sources, offers a significant increase in sustainability. As an example, a Cu catalyst dispersed on N-doped carbon nanosheets obtained by pyrolysis of a metal–organic framework (MOF) proved to be efficient for the electrosynthesis of carbamates.64 Under optimized conditions, the catalyst could promote the conversion of aniline and CO2 to form the methyl N-phenylcarbamate in high yields (71%) (entry 7 in Table 1). The high content and atomic dispersion of Cu on the N-doped carbon nanosheets with large specific surface area make Cu–N–C materials rich in active sites, leading to improved catalytic performance in comparison with bulk Cu electrodes.
2.3 Electrocarboxylation of aromatic ketones
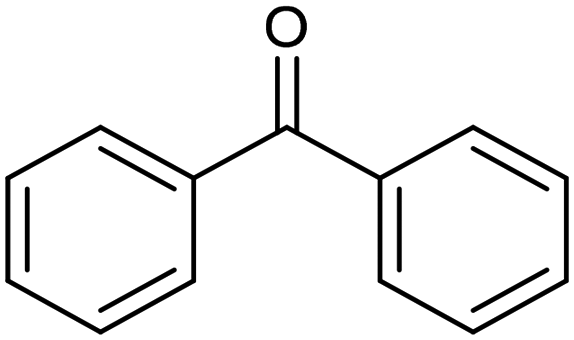
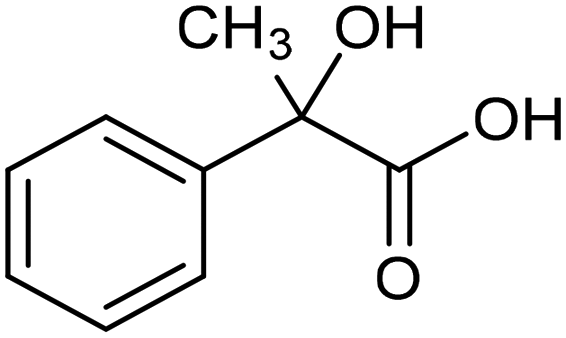
The electrochemical reduction of aromatic ketones in non-aqueous solvents containing carbon dioxide (CO2) gives access to the preparation of hydroxycarboxylic acids, which are intermediates in the production of non-steroidal anti-inflammatory drugs.65,66 Similarly to that discussed for the electrocarboxylation of organic halides, the carboxylation of aromatic ketones involves an initial single-electron reduction of the substrate to form a ketyl radical anion, which reacts with CO2 at the activated carbon atom of the carbonyl group to yield the desired hydroxycarboxylic acid product.67 Direct experimental evidence for radical formation was provided by in situ electron spin resonance (ESR) spectroscopy for the electrocarboxylation of benzophenone catalyzed by Ag NPs deposited on the boron-doped diamond electrode (entry 8 in Table 1).68 However, in analogy with the electrocarboxylation of halides, the extreme reactivity of the ketyl radical may lead to the formation of undesired by-products (e.g., pinacol and alcohol), thus reducing the selectivity of the process. In particular, the selectivity for carboxylic acids is extremely sensitive to the CO2 concentration.67 In this regard, the uniform dispersion of NiC2O4 NPs on ordered mesoporous carbon with a high specific surface area and CO2 adsorption properties can be beneficial for the selectivity towards the formation of carboxylic acids.69The regulation of the nanocatalyst's structural properties is fundamental to control selectivity. Guan et al. showed that double activation by the Ag-CeO2 NW catalyst led to the electrocarboxylation of acetophenone with CO2 to form 2-phenyllactic acid.70 The dual-site Ag-CeO2 NWs exhibited the ability to activate both CO2 and acetophenone concurrently through cathodic activation, resulting in the synergistic generation of CO2˙− and ketyl radical anions, thus favoring the electrocarboxylation process. Notably, the desired 2-phenyllactic acid product was produced with a maximum FE = 91% at −1.8 VAg/AgI (entry 9 in Table 1). Combining a heterogeneous La1−xSrxFeO3 perovskite catalyst with a homogeneous chiral auxiliary CoII-(salen) complex proved to be a powerful strategy to drive the enantioselective electrocarboxylation of acetophenone.71 The reported results indicate that the perovskite catalyst facilitates the electrocarboxylation of acetophenone, thanks to the Sr doping-induced accumulation of strong Lewis acidic Fe species and O2− species on the surface, which tend to stabilize the ketyl radical intermediate. On the other hand, the chiral Co complex is essential for obtaining optically active products, as CO2 coordinates with the reduced CoI state so that the enantioselective carboxylation step is facilitated by interaction with the ketyl intermediate. This proposed cooperative mechanism resulted in the synthesis of 2-hydroxy-2-phenylpropionic acid with a 68% yield and 94% enantiomeric excess at an applied potential of −2.0 VAg/AgI (entry 10, Table 1).
3. Electrochemical hydrogenation and functionalization of organic substrates
In this section, we will summarize the most relevant recent findings on the ECH of organic substrates, including unsaturated hydrocarbons (alkynes and alkenes), carbonyl compounds such as aldehydes and ketones, nitrogenated substrates and halides. In ECH processes, the electrolyzer provides the reducing equivalents necessary to reduce the substrate, whereas water functions as the hydrogen source. For this reason, unlike CO2 carboxylation processes discussed in section 2 which are usually carried out in aprotic non-aqueous electrolytes, ECHs are typically carried out in aqueous media. The lack of CO2 in the reaction scheme avoids the issues related to poor CO2 solubility in water. On the other hand, while enabling easier workup for product analysis, aqueous media pose significant challenges related to the limited solubility of the organic substrates and/or products. Moreover, in aqueous electrolytes they are prone to undergoing the competitive hydrogen evolution reaction (HER) as a side reaction, which limits ECH efficiency. In fact, the main challenges in this field are mainly related to the need for developing effective strategies for suppressing the HER (via catalyst design, technological modifications of the electrochemical reactor, etc.) in order to improve ECH selectivity.3.1 Hydrogenation of unsaturated hydrocarbons
Electrochemical semihydrogenation (ECSH) of alkynes to alkenes provides an essential and well-established process to access fine chemicals and useful building blocks. Highly chemo- and regioselective semihydrogenation of alkynes is a challenging but extremely important process for the synthesis of functionalized alkenes, which have widespread applications in pharmaceutical and polymer industries.72 Most of the reported catalytic systems for electrocatalytic alkyne reduction are based on noble transition metals and specifically palladium.73–75 It has been demonstrated that heteroatom-doping is an effective strategy to tune the electronic properties of Pd nanostructures, dramatically affecting the ECSH efficiency and selectivity. In particular, the design of S-rich isolated Pd sites within amorphous PdSx nanocapsules enabled excellent yields and selectivity for the conversion of 4-ethynylaniline to alkene over a wide potential window in aqueous media, outperforming reference Pd NPs which suffer from selectivity loss due to alkyne over-hydrogenation (entry 1, Table 2).76 Detailed mechanistic studies highlighted the unique structural features of isolated PdSx moieties, which favor substrate adsorption via the terminal alkyne group over the commonly reported flat adsorption mode, thus favoring the ECSH mechanism as further confirmed by in situ Raman spectroscopy. In the ECSH mechanism, electron paramagnetic resonance (EPR) spectroscopy and high-resolution mass spectrometry (HR-MS) indicate the involvement of key carbon and hydrogen radicals, the latter originating from H2O electrolysis. Importantly, the PdSx catalyst provided excellent chemo- and regioselectivity for ECSH of functionalized alkynes, also displaying a high chemical tolerance to the presence of easily reducible and Pd passivating groups. Importantly, the catalytic system was proven to be effective for the gram-scale synthesis of 4-bromostyrene and for the production of extremely expensive deuterated alkenes. Sulfur modification was also applied to Pd nanotips, resulting in a highly efficient and selective ECSH of alkynes, reaching a maximum 97% conversion yield, 96% selectivity and 75% FE toward alkene production (entry 2, Table 2).77 In this study, deuterium labelling experiments confirmed that H2O is the unique H source in the reaction and the kinetic isotope effect suggests the H2O dissociation step to be the rate-determining step for ECSH. The authors highlighted the crucial role played by the surface thiolate groups in promoting water splitting to produce surface-adsorbed hydrogen (H*ads), which is the key reactant for the following hydrogenation step.| Catalyst | Substrate | Product | Yield (FE) | E (V) | Electrolyte |
|---|---|---|---|---|---|
| a i = 13 mA. | |||||
| PdSx ANCs76 |
|
|
93% (28%) | −1.05 VHg/HgO | 1.0 M KOH/diox. |
| ArS-Pd4S NTs77 |
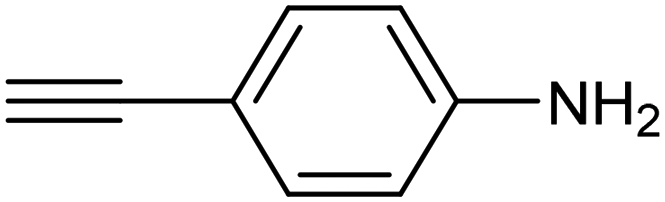
|
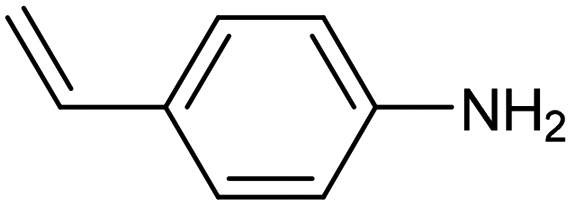
|
96% (75%) | −1.24 VHg/HgO | 1.0 M KOH/diox. |
| PdB78 |
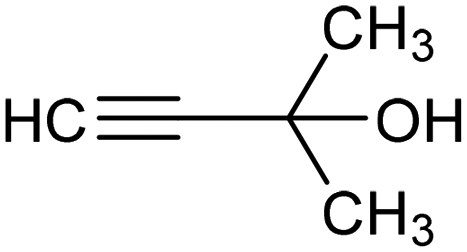
|
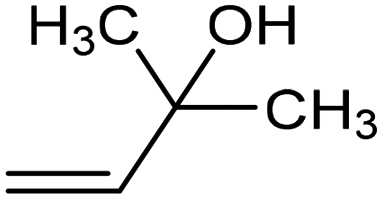
|
94.5% (39%) | −1.0 VRHE | 0.5 M K2CO3 |
| PdRuCu alloy80 |
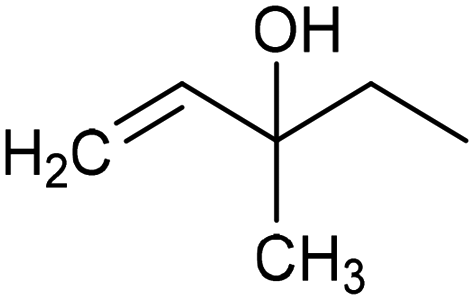
|
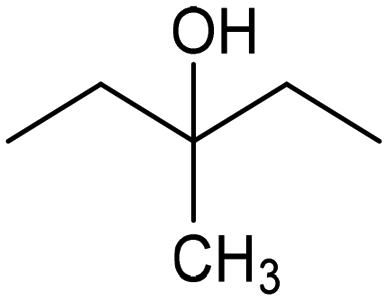
|
93% (22.3%) | −0.3 VRHE | 0.1 M HClO4/EtOH |
| Pd/CNTs81 |
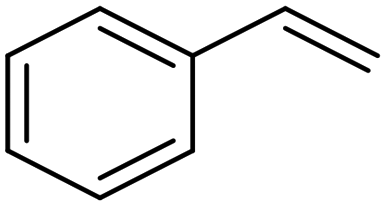
|
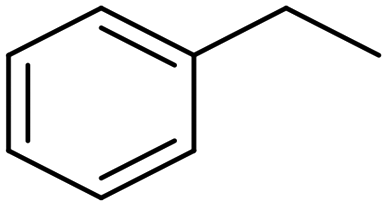
|
(95%) | −0.65 VRHE | 0.5 M H2SO4 |
| Ag/C NP86 |
|
|
98% (96.2%) | −1.3 VAg/AgCl | Sodium borate buffer 0.5 M |
| Ru/RGO87 |
|
|
91% (95%) | −1.25 VAg/AgCl | 1 M H2SO4 |
| PdCu NBWs91 |
|
|
54.8% | −0.3 VRHE | 0.2M KH2PO4 + 1 M KOH |
| CoFe-SSM93 |
|
|
98% (32.4%) | −0.7 VRHE | 0.1 M HCl |
| Fe94 |
|
|
100% (20.1%) | −0.3 VRHE | 0.5 M K2CO3 |
| Al-NFM96 |
|
|
92.1% (49.8%) | —a | 0.1 M KOH |
| Pd–Mo metallenes98 |
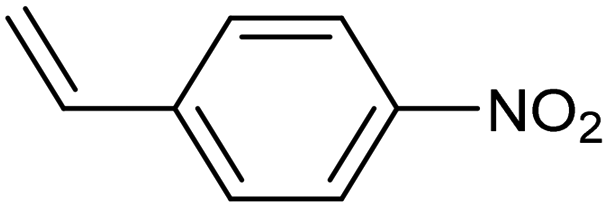
|
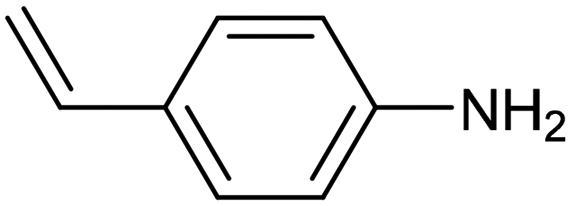
|
90% (78.3%) | −0.25 VRHE | 0.1 M LiClO4 |
| CuNiAl alloy99 |
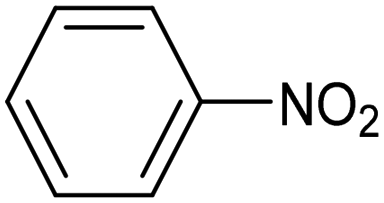
|
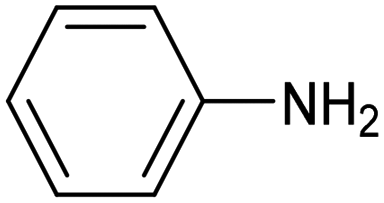
|
88.5% (82.3%) | −0.07 VRHE | 1 M KOH |
| Co–F NW101 |
|
|
94% | −1.1 VHg/HgO | 1 M KOH/diox. |
| Pd@Ni-foam107 |
|
|
(82.97 ± 2.44%) | −1.2 VAg/AgCl | 1 M KOH |
| RuTiO2112 |
|
|
(41%) | −0.1 VRHE | 0.2 M phosphate buffer |
| CoPc/CNT113 |
|
|
(100%) | −0.2 VRHE | 0.1 M KHCO3 |
The fine tuning of the catalyst's electronic structure may be combined with the rational design of the electrode–electrolyte interface to further improve the FE for ECSH in aqueous media. For example, boron-doping was recently reported to alter the electronic structure of a Pd catalyst, enhancing the semi-hydrogenation rate and ensuring >90% selectivity for the conversion of alkynols to the corresponding alkene derivative (entry 3, Table 2).78 Moreover, the addition of quaternary ammonium cationic surfactants to the catholyte induced a hydrophobic microenvironment at the electrode–electrolyte interface, causing further enhancement of the FE for ECSH due to the suppression of competitive HER.
The facet dependence on the ECSH performances in Pd nanocatalysts has also been investigated recently. In particular, Xu and co-workers studied the role played by low-index Pd (hkl) surfaces in ECSH of alkynes by systematically comparing Pd NPs with different shapes, including Pd nanocubes (NCs), nano-octahedra and nanorhombic dodecahedrons.79 As a result, they found that the alkyne conversion rate and alkene selectivity follow the order Pd (100) > Pd (111) > Pd (110), indicating that Pd NCs achieve the highest ECSH activity and alkene selectivity in the series and demonstrating that the interfacial water structure is crystal plane-dependent.
In addition to ECSH, the electrocatalytic hydrogenation of unsaturated CC chemical bonds has attracted growing interest in the last few years as a promising strategy to produce high-value chemicals from unsaturated hydrocarbons using water as the hydrogen source. Recently, Zhu et al. have reported a tensile-strained PdRuCu alloy, which was used as a model system to explore the influence of both tensile strain and alloy structure on the ECH of alkenes.80 It was revealed that tensile strain enhances the adsorption of CC, while the incorporation of Ru and Cu into the Pd lattice facilitates hydrogenation by weakening the Pd–H bond. Hence, the PdRuCu icosahedra exhibited outstanding ECH performances in the conversion of 2-methyl-3-buten-2-ol (entry 4, Table 2). However, the low solubility of the unsaturated hydrocarbons in water represents a strong limitation to an efficient aqueous ECH of alkenes. In order to address this challenge, Han and co-workers developed a novel Pickering emulsion system based on compartmentalization of organic substrates and aqueous electrolytes in different phases, thus enabling an efficient ECH at the interface (entry 5, Table 2, Fig. 3).81 The reported composite material based on Pd NPs immobilized on positively charged CNTs (Pd/CNTs) played the role of both a stabilizer for Pickering emulsion and an electrocatalyst for olefin ECH at oil–water interfaces. This system allowed the direct hydrogenation of a variety of olefins with high current densities and FEs, outperforming the state-of-the-art Pd membrane reactors.82
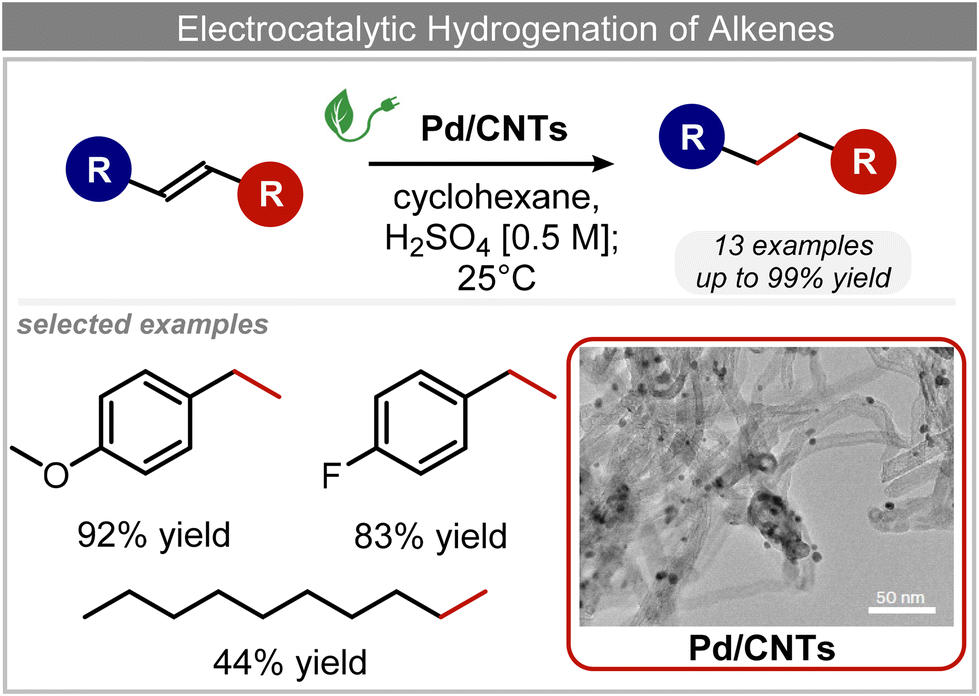 | ||
| Fig. 3 Electrocatalytic hydrogenation of alkenes.81 | ||
Although in ECH the majority of reports feature Pd-based systems, recent findings have highlighted the possibility of using non-Pd nanostructures as competent catalysts for the ECH of olefins. Among them, a hybrid cathode based on CuxO quantum dots supported on graphdiyne efficiently promoted the selective hydrogenation of olefins in unsaturated carboxylic acids as substrates using H2O as the hydrogen source, exhibiting remarkable activity and selectivity.83 Maleic acid as a model substrate could be fully converted into succinic acid after 10 hours of reaction. The reported system also displayed high selectivity for cis/trans isomerism, with a preference for the cis-structure when both cis-and trans-structures are present in solution.
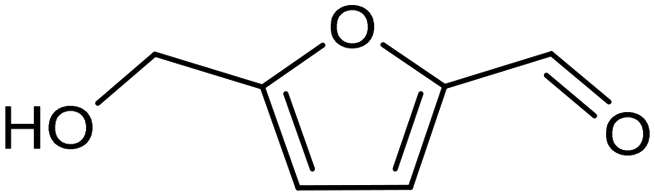
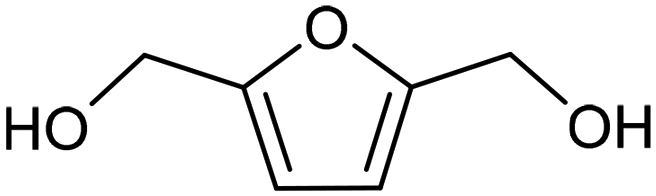
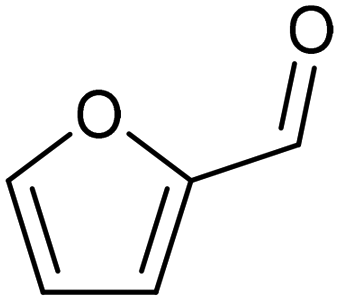
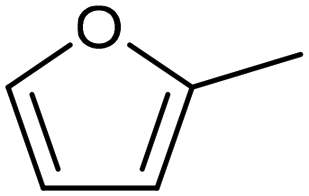
3.2 Hydrogenation and functionalization of carbonyl compounds
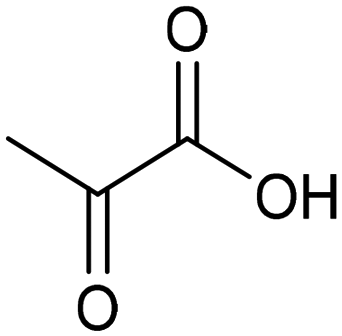
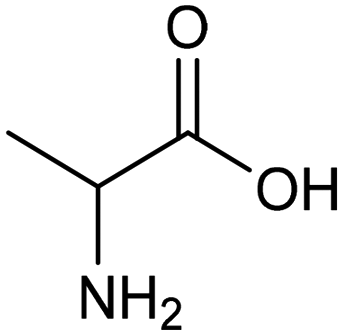
The electrocatalytic hydrogenation of biomass-derived carbonyl compounds, such as furfural derivatives, have garnered significant interest in the green synthesis of various products, including lubricants, polymers, and plasticizers.84 Among them, 5-(hydroxymethyl)furfural (HMF), which is obtained from biomass through dehydration of fructose or glucose, is a promising renewable feedstock and building block for a variety of value added chemicals and fuels. In particular, 2,5-bis(hydroxymethyl)furan (BHMF) is an interesting potential target product of ECH of HMF, due to its widespread applications in the polymer industry.85 Recently, Chadderdon and co-workers developed a highly efficient electrolyzer by pairing HMF hydrogenation to BHMF and HMF oxidation to 2,5-furandicarboxylic acid (FDCA).86 At the cathodic side, carbon-supported Ag nanoparticles (Ag/C) served as a catalyst for ECH of HMF to BHMF under mild conditions (entry 6, Table 2). The selectivity towards BHMF production versus undesired HER and HMF hydrodimerization reactions was found to be extremely potential-dependent, affording the optimal 96.2% FEBHMF at an applied potential of −1.3 VAg/AgCl. Simultaneously, the oxidation of HMF to 2,5-furandicarboxylic acid (FDCA) was achieved in a divided cell, reaching overall yields of 85% for BHMF and 98% for FDCA, respectively. Paired electrolysis was also demonstrated as an effective electrosynthetic approach for producing high-value chemicals using furfural as a biomass-derived precursor. In particular, a Ru/reduced graphene oxide (Ru/RGO) nanocomposite was recently employed as a bifunctional catalyst for simultaneous furfural electrohydrogenation and electrooxidation in aqueous media, leading to the production of furfuryl alcohol and 2-methylfuran at the cathode.87 The selectivity of the Ru/RGO cathode towards 2-methylfuran formation was significantly higher in a paired electrolyzer (91% yield, 95% FE) compared to the ECH half-cell reaction (entry 7, Table 2).Other interesting derivatives from lignocellulosic biomass that can be exploited as precursors for organic synthesis are α-ketoacids.88 In recent years, the electrochemically driven reductive amination of α-ketoacids has been shown as a viable green synthetic route for the production of amino acids, an alternative to the current methods based on microbial fermentation.89 The electrochemical process is based on a general two-step mechanism, in which the condensation between the carbonyl precursor and the nitrogenous source (e.g., NH3 or NH2OH) leads to the formation of an intermediate imine or oxime intermediate, which subsequently undergoes reductive hydrogenation to yield the desired amino acid product. Following this strategy, a TiO2-based electrode promoted the synthesis of 7 kinds of amino acids with FEs of 77–99% from the corresponding α-ketoacids and NH2OH.90 Using a flow-type reactor, continuous production of alanine was afforded from pyruvic acid and NH2OH with a 77% FE in aqueous media. According to the general paradigm of turning waste into value, the active NH2OH reactant can also be generated in situ by electroreduction of a more oxidized nitrogen source, such as NOx contaminants. In this context, a tandem electrochemical reactor was recently reported for the synthesis of alanine by coupling the electroreduction of pyruvic acid and the electrochemical removal of NO3− from wastewater, catalyzed by bimetallic PdCu nano-beadwires (PdCu NBWs).91In situ X-ray absorption spectroscopy (XAS), Raman spectroscopy and attenuated total reflectance surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS) studies shed light on a cascade process in which the *NH2OH intermediate generated from in situ NO3− electroreduction, undergoes spontaneous condensation with pyruvic acid to form pyruvic oxime, which is then reduced to yield the final alanine product. Owing to the dual nature of the catalyst, copper was proposed to favor the rate-determining initial NO3− activation step, while palladium acts as a promoter of the reductive hydrogenation of the oxime intermediate. The optimal Pd/Cu ratio in the sample led to a maximum 54.8% yield of produced alanine (entry 8, Table 2). Spectroscopy data also highlighted the impact of the copper oxidation state on the efficiency of the reaction, with electrogenerated Cu+ sites playing a crucial role in the coupling process. Notably, continuous alanine production at a 400 μmol h−1 cm−2 rate was demonstrated using a flow membrane electrode assembly (MEA) reactor at 1.7 V.
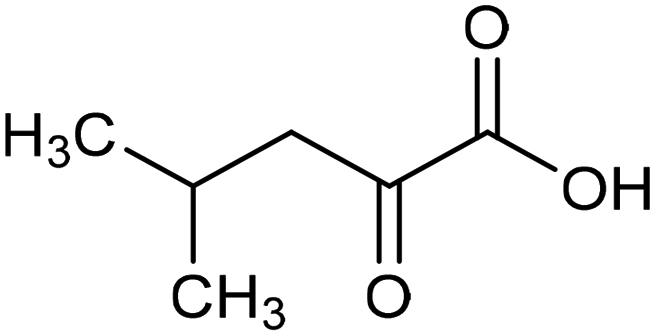
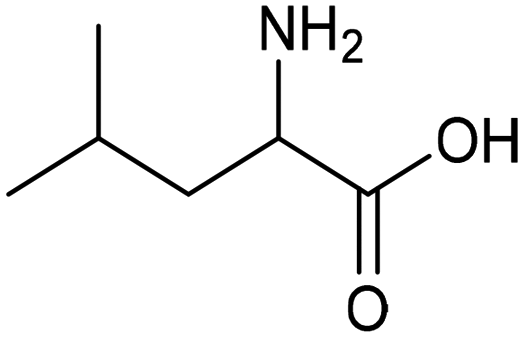
Other nitrogenous compounds can be also used in place of NO3− for amino acid electrosynthesis, e.g. gaseous nitrogen oxides which are common air pollutants and industrial exhaust gases. As a proof of concept, valine was synthesized through the electrochemical co-reduction of NO and 3-methyl-2-oxobutanoic acid in H2O using an atomically dispersed Fe catalyst with a selectivity of 11.3%.92 In follow-up work, the same group reported an analogous setup for the electrosynthesis of 13 types of amino acids using a self-standing carbon fiber membrane with a Co–Fe alloy cathode (CoFe-SSM, Fig. 4).93 Specifically, leucine synthesis was demonstrated from co-reduction NO and 4-methyl-2-oxovaleric acid as precursors, resulting in a 32.4% FE at −0.7 VRHE (entry 9 in Table 2). Remarkably, the catalytic system enabled gram-scale synthesis of leucine and displayed stability for over 24 hours, while maintaining the main morphological and structural features of the original material. In situ X-ray absorption fine structure (XAFS) measurements showed that the metallic Fe and Co sites act synergistically under operating conditions, promoting the adsorption and reactivity of the key intermediates involved in the reductive amination process.
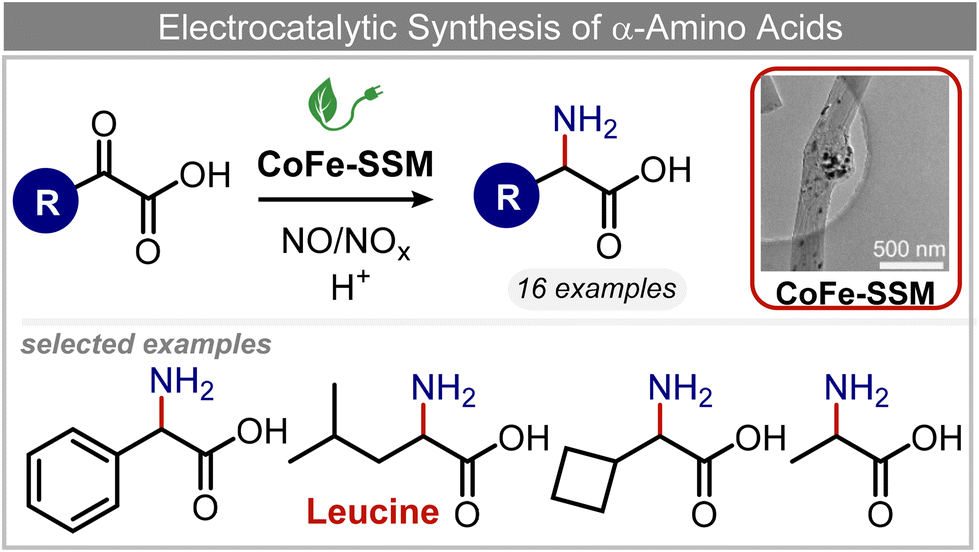 | ||
| Fig. 4 Electrocatalytic synthesis of amino acids.93 | ||
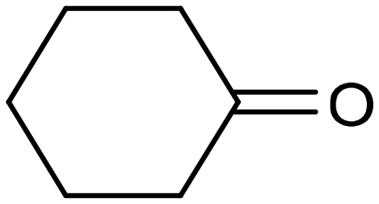
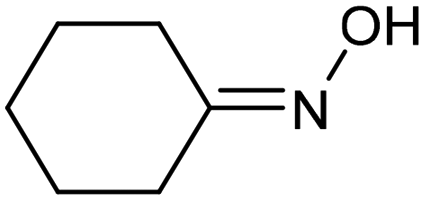
A similar approach for the electrochemical synthesis of oximes can be extended to a wide range of carbonyl precursors, offering the opportunity to expand potential applications. For instance, a novel gram-scale electrosynthesis of cyclohexanone oxime was recently obtained through the coupling of electroreduction of nitrogen oxides and cyclohexanone as starting materials (Fig. 5).94 As in the above discussed mechanism, the reaction between *NH2OH and the adsorbed cyclohexanone intermediates was identified as the key C–N coupling step. Interestingly, compared to a Pd catalyst which tends to favor the direct ketone hydrogenation side reaction, a Fe electrocatalyst could efficiently promote selective formation of the ketoxime product with a close to 100% yield in a flow cell (entry 10 in Table 2). The resulting cyclohexanone oxime subsequently underwent a mild thermochemical Beckmann rearrangement step to form caprolactam, a key precursor involved in the industrial preparation of nylon-6.
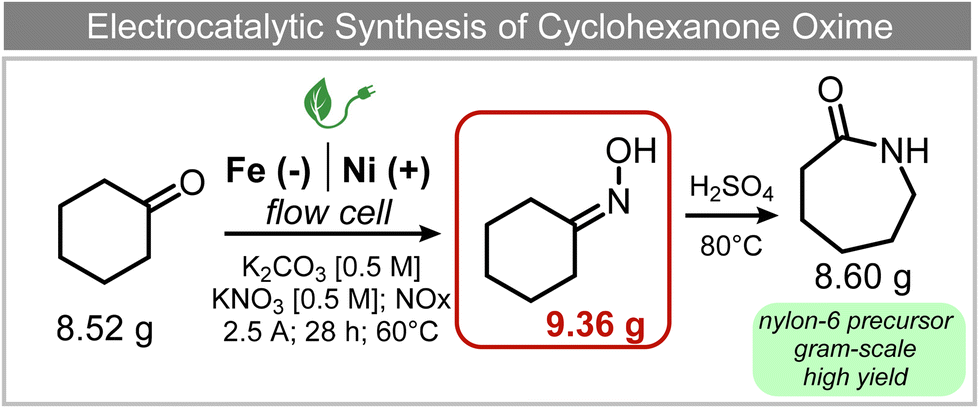 | ||
| Fig. 5 Electrocatalytic gram-scale synthesis of a nylon-6 precursor.94 | ||
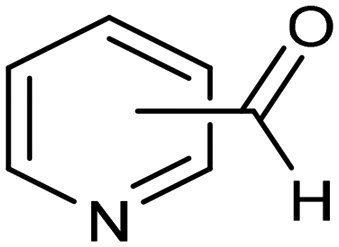
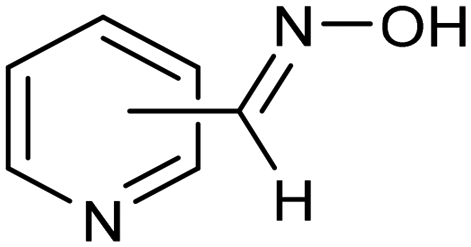
The same type of approach can be also applied to the synthesis of highly valuable pyridine oximes, which have extensive applications in pharmaceuticals, enzymology, and sterilization.95 These oximes are normally synthesized through nucleophilic addition of hydroxylamine (NH2OH) or its salts to the corresponding aldehyde. To avoid issues related to storage and handling the corrosive and unstable NH2OH raw material, an alternative strategy was recently proposed for the synthesis of pyridine oximes using NH2OH in situ generated from NO electrocatalytic reduction with an aldehyde over a nanofiber membrane prepared through electrospinning and calcination from NH2-MIL-53(Al) (Al-AFM).96 The catalyst promoted the production of 2-pyridinealdoxime with an FE value of up to 49.8% and a yield of 92.1% in aqueous electrolyte (entry 11, Table 2) and was demonstrated to be effective for the synthesis of a wide variety of functionalized pyridine oximes.
3.3 Hydrogenation of nitrogenated substrates
Amine compounds, essential for drug, dye, and pharmaceutical production, can be selectively electrocatalytically hydrogenated, converting nitro-organic compounds from –NO2 to –NH2 and yielding high-value chemical products. This process utilizing water (H2O) as the hydrogen source, provides an efficient and sustainable approach for synthesizing hydrogenated products under ambient conditions. Palladium (Pd)-based electrocatalysts capable of adsorbing/activating protons and water are typically highly active for cathodic hydrogenation.97 Recently, Zhang and colleagues reported for the first time a two-dimensional Pd-based metallenes as electrocatalysts for the ECH of nitroarenes.98 Pd–Mo metallenes, characterized by a highly exposed active surface and strong electronic interaction between Pd and Mo atoms, demonstrated exceptional chemoselectivity in the hydrogenation of 4-nitrostyrene to produce high-value 4-vinylaniline with a remarkable selectivity (>90%) and FE (78.3%) (entry 12, Table 2). For nitrobenzene hydrogenation to aniline, a self-supported electrode based on the CuNiAl alloy with a bimodal nanoporous structure displayed a remarkable selectivity to aniline (FE = 82.3%, 98% selectivity) at a very low potential (−0.07 VRHE) (entry 13, Table 2).99The electrocatalysts for ECH of nitriles to valuable amines via hydrogenating C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N have also been studied. Recently, Ao et al. introduced a study employing an in situ restoration strategy to synthesize ultrathin Pd–Ni(OH)2 nanosheets.100 The study showcased exceptional activity and selectivity in the reversible electrochemical reforming of ethylamine and acetonitrile. The Pd–Ni(OH)2 nanosheets served as efficient catalysts in the acetonitrile reduction process, utilizing Pd as the reaction center and Ni(OH)2 for proton generation through water dissociation. Additionally, the oxidation process of ethylamine took place on the surface of the heterostructured nanosheets, capitalizing on the abundant Ni(II) defects.
N have also been studied. Recently, Ao et al. introduced a study employing an in situ restoration strategy to synthesize ultrathin Pd–Ni(OH)2 nanosheets.100 The study showcased exceptional activity and selectivity in the reversible electrochemical reforming of ethylamine and acetonitrile. The Pd–Ni(OH)2 nanosheets served as efficient catalysts in the acetonitrile reduction process, utilizing Pd as the reaction center and Ni(OH)2 for proton generation through water dissociation. Additionally, the oxidation process of ethylamine took place on the surface of the heterostructured nanosheets, capitalizing on the abundant Ni(II) defects.
1,2,3,4-Tetrahydroquinoline (THQ) skeletons, inherent in bioactive compounds and natural products, pose a significant and challenging aspect in achieving room temperature and selective hydrogenation of quinolines to 1,2,3,4-tetrahydroquinolines, utilizing a safe and clean hydrogen donor catalyzed by cost-effective materials, mainly due to the intricate activation of quinolines and H2. Guo et al. electrochemically synthesized a fluorine-modified cobalt catalyst from a Co(OH)F precursor. This catalyst demonstrated remarkable activity in electrocatalytic hydrogenation of quinolines using H2O as the hydrogen source, yielding 1,2,3,4-tetrahydroquinolines with an exceptional selectivity of up to 99% and an isolated yield of 94% under ambient conditions (entry 14, Table 2).101
3.4 Hydrodechlorination processes
Electrocatalytic hydrodechlorination is a promising technology for the swift and efficient detoxification of halogenated antibiotic pollutants in environmental remediation. Although traditional methods like microbial dehalogenation, zero-valent iron reduction, and Fenton reactions are common, they suffer from drawbacks such as low efficiency, secondary pollution, and high energy consumption. Electrochemical reductive dehalogenation is gaining prominence due to its high efficiency, absence of extra reducing agents and limited generation of toxic byproducts.102 Palladium (Pd) stands out as a highly efficient electrocatalyst for dechlorination reactions. This efficacy arises from its capability to efficiently accept protons, leading to the adsorption of active atomic hydrogen on the Pd surface and subsequent incorporation into the Pd lattice by forming Pd-H bonds. The former can efficiently cleave C–Cl bonds.103 Jiang and collaborators observed that the electrocatalytic hydrochlorination reaction on Pd NPs exhibits facet-dependent behavior, with catalytic activity decreasing in the sequence {111} > {110} > {100}.104 Actually several studies demonstrated that high-index faceted Pd nanocrystals, characterized by abundant step sites on the surface, consistently exhibit higher catalytic activity in comparison to Pd nanocrystals encapsulated by low-index facets.103,105Among the halogenated antibiotics, chloramphenicol (CAP) is frequently misused in the treatment of bacterial infections in both humans and poultry, contributing to a growing environmental risk posed by antibiotic-resistant bacteria and genes. Recently, ultra-small Pt NPs loaded onto Ti3C2Tx MXene nanosheets were found to effectively catalyze electrochemical CAP degradation, removing 98.7% of CAP within 90 minutes.106 Florfenicol (FLO) is also known as one class of broad-spectrum antimicrobial agents commonly employed as an alternative to CAP and widely used in veterinary medicine. Yang et al. reported a novel Ni foam electrode decorated with ultrafine Pd NPs (Pd@Ni-foam) for electrocatalytic FLO dechlorination.107 The catalyst demonstrated excellent electrochemical dechlorination performance, achieving a remarkable 99.5% removal efficiency. The dechlorination rate was notably carried out at 16.58 mg min−1 under a cathode potential of −1.2 VAg/AgCl (entry 15, Table 2), outperforming the reference Ni foam and commercial Pd/C, respectively.
Several strategies have also been proposed in the last few years to improve the electrochemical degradation of chlorophenols, which represent another notorious class of organic pollutants due to their wide distribution, bioaccumulation and high toxicity.108 As a result, they have been classified as priority toxic pollutants for environmental protection and human health. In order to achieve the complete mineralization of halogenous pollutants, a novel reduction–oxidation mediated heterogeneous electro-Fenton technology based on a PdFe nanoalloy has been proposed.109 While the PdFe nanoalloy was found to promote the generation of active *H sites and improve electron transfer, the carbon defects selectively facilitated the two-electron oxygen reduction pathway. These mechanisms resulted in the successful achievement of complete total organic carbon removal and dechlorination of the model 3-chlorophenol substrate. The rational tuning of the interfacial microenvironment also plays a crucial role in enhancing the dechlorination efficiency. For instance, the addition of a hydrophilic polyethylene glycol-based polymer to a Pd/C electrode was shown to facilitate *H generation and mass transfer, thus leading to a remarkable improvement in the electrochemical hydrochlorination performance of 2,4-dichlorophenol.110 Furthermore, the dispersion of a Pd nanocatalyst into a porous architecture with enhanced adsorption properties for the halogenated contaminant was also found to be beneficial for dechlorination efficiency, as recently reported for the electrochemical degradation of 2,4-dichlorophenoxyacetic acid catalyzed by a Pd catalyst supported on a Ni-based layered MOF.111
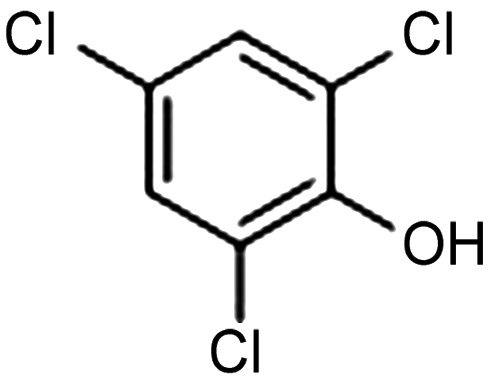
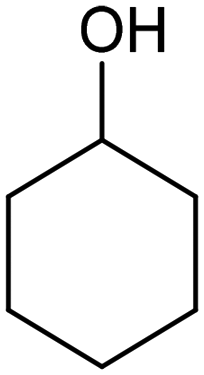
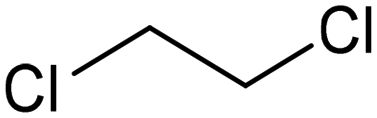
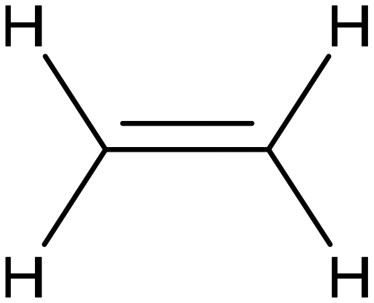
Extensive recent research has focused on finding more cost-effective alternatives to Pd to promote dechlorination reactions. Moreover, Pd also displays other specific limitations including the production of toxic phenol as the primary product and a remarkable susceptibility to poisoning by reactive species. From this perspective, Gu and colleagues recently demonstrated the potential of Ru in serving as a Pd substitute for the hydrogenation of 2,4,6-trichlorophenol (TCP).112 By utilizing a Ru catalyst, an impressive 99.8% TCP removal efficiency was obtained, with a 99% selectivity for cyclohexanol (entry 16, Table 2). In contrast, the Pd electrode exhibited a lower TCP removal efficiency (66%) as well as the formation of phenol as the primary hydrogenation product. For the Ru catalyst, a primary reaction mechanism was proposed based on direct electron transfer, unlike the indirect *H-mediated pathway occurring on Pd. Interestingly, Wang's group has recently developed a highly efficient and cost-effective system for valorization of the electrocatalytic degradation of common chlorinated organic water pollutants.113 In particular, the authors designed a novel electrocatalytic approach that can directly convert 1,2-dichloroethane (DCA), an extremely toxic and harmful environmental pollutant upon inhalation or ingestion, into ethylene, that is widely used in a variety of industrial sectors.113 To perform this electrochemical transformation, a new catalyst comprising cobalt phthalocyanine molecules assembled on multiwalled carbon nanotubes (CoPc/CNTs) was developed (Fig. 6). This catalyst can electrochemically decompose aqueous DCA with high current and energy efficiencies. Ethylene was obtained at high rates of production with unprecedented ∼100% FE across wide electrode potential and reactant concentration ranges (entry 17, Table 2).
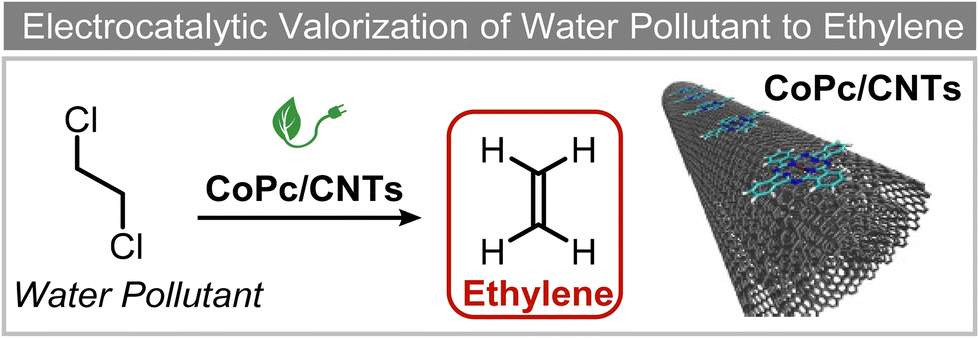 | ||
| Fig. 6 Electrocatalytic valorization of 1,2-dichloroethane to ethylene.113 | ||
4. Conclusions and future perspectives
In this minireview, we have summarized the most recent advances in electrochemical reductive organic transformations catalyzed by nanostructured electrocatalysts. The results herein presented highlight the extreme versatility and the huge potential of electrochemical methods for organic chemists, offering a playground for developing sustainable synthesis. We have shown that the selectivity and the efficiency of the electrosynthesis processes discussed in this work are highly dependent not only on the electronic and structural properties of the nanocatalyst but also on the design of the electrode–electrolyte interface and even on technological aspects related to the employed electrochemical setup. We have first discussed reductive transformations of organic substrates coupled with the CO2RR, including electrocarboxylation and methylation reactions among others, which utilize CO2 as an abundant and low-cost C1 feedstock to produce value-added chemicals for the pharmaceutical and polymer industry. In the second part of the work, we have revised the most significant findings recently reported for ECH of organic substrates in aqueous media and discussed the main strategies used to improve the ECH selectivity over competing HER, mostly based on tailoring the properties of the nanostructured catalyst and the electrode–electrolyte interface. We have specifically focused on the hydrogenation of such substrates as unsaturated hydrocarbons, carbonyl-based biomass-derivatives, heterocyclic compounds and halides, for several applications, ranging from the synthesis of industrially relevant value-added and fine chemicals to environmental remediation.Despite the significant progress made in the last few years, there is still huge room left for improvement in the field, especially regarding a comprehensive mechanistic understanding of the catalytic pathways. From this perspective, the development of in situ/operando spectroscopic and microscopic techniques will be of crucial relevance, allowing us to investigate the nature of the catalytically active sites as well as the structural, compositional and morphological dynamics of the catalyst during the reaction. Moreover, these techniques will provide experimental evidence for key surface intermediates involved in the process, contributing to establishing a more precise structure–activity correlation. In parallel, careful analytical quantification of the secondary products obtained during electrolysis experiments will also help to rationalize the underlying mechanisms. Based on the fundamental understanding of the reaction, alternative approaches for rational catalyst design hold promise as effective strategies to improve the efficiency and selectivity of electrocatalytic organic transformations (e.g. dual-site catalytic systems). It is worth mentioning that the traditional and most used nanostructured electrocatalysts are generally based on expensive and potentially harmful metal-containing species (such as Pd, Ru, and Pt, among others). However, in recent years, reliance on noble metal-based catalytic systems has constantly declined because of their cost, toxicity and poor availability. Thus, the use of metal-based catalysts has been discouraged by the modern guidelines towards the implementation of sustainable chemical productions. As a new trend of research, we hope that cheaper, abundant and less harmful classes of metal-free electrocatalytically active nanostructures (e.g., carbon nitrides, carbon nanodots, etc.) will make their way as valuable alternatives for achieving green synthesis of valuable organic molecules. Moreover, enantioselective organic catalysis could be accomplished by using chiral nano-electrocatalysts. To this aim, careful control of the chiral catalytic surface will be pivotal to capitalize on the reactive features of its functionalities. We therefore foresee that forthcoming investigations on enantioselective nano-electrocatalysis will be carried out leading to the sustainable production of enantioenriched valuable organic molecules.
In addition to the catalyst design, further research should also address the challenges related to the limited solubility of several organic substrates in water as well as the limited compatibility of modern flow reactors with non-aqueous environments. In this regard, the choice of the oxidative reaction is also crucial for the design of efficient electrolyzers. Finally, we are confident that this minireview will positively contribute to further development of the field.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the University of Trieste, INSTM, and the European Commission (H2020 – RIA-CE-NMBP-25 Program, Grant No. 862030). P. F. and M. M. acknowledge the COST Action 21101. M. M. acknowledges the project P2022WANKS funded by the Ministero dell'Università e della Ricerca (MUR). P. F., M. M. and G. F. acknowledge PRIN 2022 Project funded by the Italian Ministry MUR Italy (project acronym: SYSSY-CAT). F. M. kindly acknowledges the PhD scholarship co-funded under the National Recovery and Resilience Plan (NRRP). G. F., M. M. and F. F. kindly acknowledge FRA2023 funded by the University of Trieste. M. P. is the AXA Chair for Bionanotechnology (2016–2023). Part of this work was performed under the Maria de Maeztu Units of Excellence Program – Grant MDM-2017-0720. M. P. kindly acknowledges PRIN 2022 Project funded by the Italian Ministry MUR Italy (n. 20228YFRNL) funded by the European Union – Next Generation EU.References
- L. F. T. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt and S. Lin, Chem. Soc. Rev., 2021, 50, 7941–8002 RSC.
- J.-M. Savéant, Chem. Rev., 2008, 108, 2348–2378 CrossRef PubMed.
- G. Sportelli, G. Grando, M. Bevilacqua, G. Filippini, M. Melchionna and P. Fornasiero, Chem. – Eur. J., 2023, 29, e202301718 CrossRef CAS PubMed.
- M. Marchi, E. Raciti, S. M. Gali, F. Piccirilli, H. Vondracek, A. Actis, E. Salvadori, C. Rosso, A. Criado, C. D'Agostino, L. Forster, D. Lee, A. C. Foucher, R. K. Rai, D. Beljonne, E. A. Stach, M. Chiesa, R. Lazzaroni, G. Filippini, M. Prato, M. Melchionna and P. Fornasiero, Adv. Sci., 2023, 10, 2303781 CrossRef CAS PubMed.
- F. Longobardo, G. Gentile, A. Criado, A. Actis, S. Colussi, V. Dal Santo, M. Chiesa, G. Filippini, P. Fornasiero, M. Prato and M. Melchionna, Mater. Chem. Front., 2021, 5, 7267–7275 RSC.
- G. Filippini, F. Longobardo, L. Forster, A. Criado, G. Di Carmine, L. Nasi, C. D'Agostino, M. Melchionna, P. Fornasiero and M. Prato, Sci. Adv., 2023, 6, eabc9923 CrossRef PubMed.
- D. H. Evans, Chem. Rev., 2008, 108, 2113–2144 CrossRef CAS PubMed.
- K. D. Moeller, Chem. Rev., 2018, 118, 4817–4833 CrossRef CAS PubMed.
- N. R. Shiju and V. V. Guliants, Appl. Catal., A, 2009, 356, 1–17 CrossRef CAS.
- M. N. Jackson and Y. Surendranath, Acc. Chem. Res., 2019, 52, 3432–3441 CrossRef CAS PubMed.
- A. T. Bell, Science, 2003, 299, 1688–1691 CrossRef CAS PubMed.
- D. Astruc, Chem. Rev., 2020, 120, 461–463 CrossRef CAS PubMed.
- C. Zhu, N. W. J. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Cent. Sci., 2021, 7, 415–431 CrossRef CAS PubMed.
- T. Ali, H. Wang, W. Iqbal, T. Bashir, R. Shah and Y. Hu, Adv. Sci., 2023, 10, 2205077 CrossRef CAS PubMed.
- D. Pollok and S. R. Waldvogel, Chem. Sci., 2020, 11, 12386–12400 RSC.
- Y. Li, S. Dana and L. Ackermann, Curr. Opin. Electrochem., 2023, 40, 101312 CrossRef CAS.
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS PubMed.
- F. Franco, C. Rettenmaier, H. S. Jeon and B. Roldan Cuenya, Chem. Soc. Rev., 2020, 49, 6884–6946 RSC.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- M. T. Jensen, M. H. Rønne, A. K. Ravn, R. W. Juhl, D. U. Nielsen, X.-M. Hu, S. U. Pedersen, K. Daasbjerg and T. Skrydstrup, Nat. Commun., 2017, 8, 489 CrossRef PubMed.
- X. Peng, L. Zeng, D. Wang, Z. Liu, Y. Li, Z. Li, B. Yang, L. Lei, L. Dai and Y. Hou, Chem. Soc. Rev., 2023, 52, 2193–2237 RSC.
- J. Li, H. Al-Mahayni, D. Chartrand, A. Seifitokaldani and N. Kornienko, Nat. Synth., 2023, 2, 757–765 CrossRef.
- A. Loiudice, P. Lobaccaro, E. A. Kamali, T. Thao, B. H. Huang, J. W. Ager and R. Buonsanti, Angew. Chem., Int. Ed., 2016, 55, 5789–5792 CrossRef CAS PubMed.
- R. Reske, H. Mistry, F. Behafarid, B. Roldan Cuenya and P. Strasser, J. Am. Chem. Soc., 2014, 136, 6978–6986 CrossRef CAS PubMed.
- P. Grosse, D. Gao, F. Scholten, I. Sinev, H. Mistry and B. Roldan Cuenya, Angew. Chem., Int. Ed., 2018, 57, 6192–6197 CrossRef CAS PubMed.
- G. L. De Gregorio, T. Burdyny, A. Loiudice, P. Iyengar, W. A. Smith and R. Buonsanti, ACS Catal., 2020, 10, 4854–4862 CrossRef CAS PubMed.
- R. M. Arán-Ais, R. Rizo, P. Grosse, G. Algara-Siller, K. Dembélé, M. Plodinec, T. Lunkenbein, S. W. Chee and B. R. Cuenya, Nat. Commun., 2020, 11, 3489 CrossRef PubMed.
- J.-J. Velasco-Vélez, T. Jones, D. Gao, E. Carbonio, R. Arrigo, C.-J. Hsu, Y.-C. Huang, C.-L. Dong, J.-M. Chen, J.-F. Lee, P. Strasser, B. Roldan Cuenya, R. Schlögl, A. Knop-Gericke and C.-H. Chuang, ACS Sustain. Chem. Eng., 2019, 7, 1485–1492 CrossRef.
- M. R. Friedfeld, H. Zhong, R. T. Ruck, M. Shevlin and P. J. Chirik, Science, 2018, 360, 888–893 CrossRef CAS PubMed.
- S. A. Akhade, N. Singh, O. Y. Gutiérrez, J. Lopez-Ruiz, H. Wang, J. D. Holladay, Y. Liu, A. Karkamkar, R. S. Weber, A. B. Padmaperuma, M.-S. Lee, G. A. Whyatt, M. Elliott, J. E. Holladay, J. L. Male, J. A. Lercher, R. Rousseau and V.-A. Glezakou, Chem. Rev., 2020, 120, 11370–11419 CrossRef CAS PubMed.
- O. Scialdone, A. Galia, C. Belfiore, G. Filardo and G. Silvestri, Ind. Eng. Chem. Res., 2004, 43, 5006–5014 CrossRef CAS.
- B. M. Setterfield-Price and R. A. W. Dryfe, J. Electroanal. Chem., 2014, 730, 48–58 CrossRef CAS.
- M. Mazzucato, A. A. Isse and C. Durante, Curr. Opin. Electrochem., 2023, 39, 101254 CrossRef CAS.
- A. A. Isse, C. Durante and A. Gennaro, Electrochem. Commun., 2011, 13, 810–813 CrossRef CAS.
- A. A. Isse and A. Gennaro, Chem. Commun., 2002, 2798–2799 RSC.
- H.-P. Yang, Q. Lin, H.-W. Zhang, G.-D. Li, L.-D. Fan, X.-Y. Chai, Q.-L. Zhang, J.-H. Liu and C.-X. He, Chem. Commun., 2018, 54, 4108–4111 RSC.
- H. Yang, H. Zhang, Y. Wu, L. Fan, X. Chai, Q. Zhang, J. Liu and C. He, ChemSusChem, 2018, 11, 3905–3910 CrossRef CAS PubMed.
- V. Rajagopal, P. Manivel, N. Nesakumar, M. Kathiresan, D. Velayutham and V. Suryanarayanan, ACS Omega, 2018, 3, 17125–17134 CrossRef CAS PubMed.
- B. Boro, P. Kalita, A. Vijayaprabhakaran, D. Q. Dao, S. Nandy, K. H. Chae, Y. Nailwal, M. Kathiresan and J. Mondal, ACS Appl. Nano Mater., 2023, 6, 11788–11801 CrossRef CAS.
- S. Anandhakumar, R. Sripriya, M. Chandrasekaran, S. Govindu and M. Noel, J. Appl. Electrochem., 2009, 39, 463–465 CrossRef CAS.
- J. Chaussard, J.-C. Folest, J.-Y. Nedelec, J. Perichon, S. Sibille and M. Troupel, Synthesis, 1990, 369–381 CrossRef CAS.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed.
- G. Silvestri, S. Gambino, G. Filardo, M. Tiitta, M. Sjöström, S. Wold, R. Berglind and B. Karlsson, Acta Chem. Scand., 1991, 45, 987–992 CrossRef CAS.
- H. Li, W. Zhang, Q. Li and B. Chen, Resour., Conserv. Recycl., 2015, 100, 41–48 CrossRef.
- A. Gennaro, A. A. Isse, M.-G. Severin, E. Vianello, I. Bhugun and J.-M. Savéant, J. Chem. Soc., Faraday Trans., 1996, 92, 3963–3968 RSC.
- N. Corbin, D.-T. Yang, N. Lazouski, K. Steinberg and K. Manthiram, Chem. Sci., 2021, 12, 12365–12376 RSC.
- Y. Cao, D. Li, C. Ding, S. Ye, X. Zhang, H. Chi, L. Liu, Y. Liu, J. Xiao and C. Li, ACS Catal., 2023, 13, 11902–11909 CrossRef CAS.
- N. Corbin, G. P. Junor, T. N. Ton, R. J. Baker and K. Manthiram, J. Am. Chem. Soc., 2023, 145, 1740–1748 CrossRef CAS PubMed.
- Z. Tao, C. L. Rooney, Y. Liang and H. Wang, J. Am. Chem. Soc., 2021, 143, 19630–19642 CrossRef CAS PubMed.
- M. Shibata, K. Yoshida and N. Furuya, J. Electroanal. Chem., 1995, 387, 143–145 CrossRef.
- Electrochemical Synthesis of Urea at Gas-diffusion Electrodes V. Simultaneous Reduction of Carbon Dioxide and Nitrite Ions with Various Boride Catalysts, https://www.jstage.jst.go.jp/article/kogyobutsurikagaku/66/6/66_584/_article, (accessed 14 November 2023).
- M. Shibata and N. Furuya, J. Electroanal. Chem., 2001, 507, 177–184 CrossRef CAS.
- C. Lv, L. Zhong, H. Liu, Z. Fang, C. Yan, M. Chen, Y. Kong, C. Lee, D. Liu, S. Li, J. Liu, L. Song, G. Chen, Q. Yan and G. Yu, Nat. Sustain., 2021, 4, 868–876 CrossRef.
- C. Chen, X. Zhu, X. Wen, Y. Zhou, L. Zhou, H. Li, L. Tao, Q. Li, S. Du, T. Liu, D. Yan, C. Xie, Y. Zou, Y. Wang, R. Chen, J. Huo, Y. Li, J. Cheng, H. Su, X. Zhao, W. Cheng, Q. Liu, H. Lin, J. Luo, J. Chen, M. Dong, K. Cheng, C. Li and S. Wang, Nat. Chem., 2020, 12, 717–724 CrossRef CAS PubMed.
- N. Meng, Y. Huang, Y. Liu, Y. Yu and B. Zhang, Cell Rep. Phys. Sci., 2021, 2, 100378 CrossRef CAS.
- N. Meng, X. Ma, C. Wang, Y. Wang, R. Yang, J. Shao, Y. Huang, Y. Xu, B. Zhang and Y. Yu, ACS Nano, 2022, 16, 9095–9104 CrossRef CAS PubMed.
- Y. Wu, Z. Jiang, Z. Lin, Y. Liang and H. Wang, Nat. Sustain., 2021, 4, 725–730 CrossRef.
- M. Sun, G. Wu, J. Jiang, Y. Yang, A. Du, L. Dai, X. Mao and Q. Qin, Angew. Chem., Int. Ed., 2023, 62, e202301957 CrossRef CAS PubMed.
- Y. Wu, Z. Jiang, X. Lu, Y. Liang and H. Wang, Nature, 2019, 575, 639–642 CrossRef CAS PubMed.
- Z. Tao, Y. Wu, Z. Wu, B. Shang, C. Rooney and H. Wang, J. Energy Chem., 2022, 65, 367–370 CrossRef CAS.
- C. L. Rooney, Y. Wu, Z. Tao and H. Wang, J. Am. Chem. Soc., 2021, 143, 19983–19991 CrossRef CAS PubMed.
- X. Sun, Q. Zhu, J. Hu, X. Kang, J. Ma, H. Liu and B. Han, Chem. Sci., 2017, 8, 5669–5674 RSC.
- L. Wang, C. Qi, W. Xiong and H. Jiang, Chin. J. Catal., 2022, 43, 1598–1617 CrossRef CAS.
- S. Li, Y. Shi, J. Zhang, Y. Wang, H. Wang and J. Lu, ChemSusChem, 2021, 14, 2050–2055 CrossRef CAS PubMed.
- A. A. Isse, A. Galia, C. Belfiore, G. Silvestri and A. Gennaro, J. Electroanal. Chem., 2002, 526, 41–52 CrossRef CAS.
- O. Scialdone, M. A. Sabatino, C. Belfiore, A. Galia, M. P. Paternostro and G. Filardo, Electrochim. Acta, 2006, 51, 3500–3505 CrossRef CAS.
- O. Scialdone, C. Amatore, A. Galia and G. Filardo, J. Electroanal. Chem., 2006, 592, 163–174 CrossRef CAS.
- K. R. Saravanan, M. Chandrasekaran and V. Suryanarayanan, J. Electroanal. Chem., 2015, 757, 18–22 CrossRef CAS.
- Y. Shi, Y. Hou, Y. Wang, J.-J. Zhang, H. Wang and J.-X. Lu, Microporous Mesoporous Mater., 2021, 323, 111174 CrossRef CAS.
- A. Guan, Y. Quan, Y. Chen, Z. Liu, J. Zhang, M. Kan, Q. Zhang, H. Huang, L. Qian, L. Zhang and G. Zheng, Chin. J. Catal., 2022, 43, 3134–3141 CrossRef CAS.
- L.-R. Yang, J.-J. Zhang, Y.-J. Zhao, Z.-L. Wang, H. Wang and J.-X. Lu, Electrochim. Acta, 2021, 398, 139308 CrossRef CAS.
- C. Oger, L. Balas, T. Durand and J.-M. Galano, Chem. Rev., 2013, 113, 1313–1350 CrossRef CAS PubMed.
- C. W. A. Chan, A. H. Mahadi, M. M.-J. Li, E. C. Corbos, C. Tang, G. Jones, W. C. H. Kuo, J. Cookson, C. M. Brown, P. T. Bishop and S. C. E. Tsang, Nat. Commun., 2014, 5, 5787 CrossRef CAS PubMed.
- D. Teschner, J. Borsodi, A. Wootsch, Z. Révay, M. Hävecker, A. Knop-Gericke, S. D. Jackson and R. Schlögl, Science, 2008, 320, 86–89 CrossRef CAS PubMed.
- D. Albani, M. Shahrokhi, Z. Chen, S. Mitchell, R. Hauert, N. López and J. Pérez-Ramírez, Nat. Commun., 2018, 9, 2634 CrossRef PubMed.
- H. Li, Y. Gao, Y. Wu, C. Liu, C. Cheng, F. Chen, Y. Shi and B. Zhang, J. Am. Chem. Soc., 2022, 144, 19456–19465 CrossRef CAS PubMed.
- Y. Gao, R. Yang, C. Wang, C. Liu, Y. Wu, H. Li and B. Zhang, Sci. Adv., 2022, 8, eabm9477 CrossRef CAS PubMed.
- Y. Zhao, J. Xu, K. Huang, W. Ge, Z. Liu, C. Lian, H. Liu, H. Jiang and C. Li, J. Am. Chem. Soc., 2023, 145, 6516–6525 CrossRef CAS PubMed.
- X. Xu, J. Ma, B. Kui, G. Zhu, G. Jia, F. Wu, P. Gao and W. Ye, ACS Appl. Nano Mater., 2023, 6, 5357–5364 CrossRef CAS.
- K. Zhu, X. Xu, M. Xu, P. Deng, W. Wu, W. Ye, Z. Weng, Y. Su, H. Wang, F. Xiao, Z. Fang and P. Gao, ChemElectroChem, 2021, 8, 3855–3862 CrossRef CAS.
- C. Han, J. Zenner, J. Johny, N. Kaeffer, A. Bordet and W. Leitner, Nat. Catal., 2022, 5, 1110–1119 CrossRef CAS.
- A. Kurimoto, R. P. Jansonius, A. Huang, A. M. Marelli, D. J. Dvorak, C. Hunt and C. P. Berlinguette, Angew. Chem., Int. Ed., 2021, 60, 11937–11942 CrossRef CAS PubMed.
- C. Xing, Y. Xue, X. Zheng, Y. Gao, S. Chen and Y. Li, Angew. Chem., Int. Ed., 2023, 62, e202310722 CrossRef CAS PubMed.
- W. Deng, Y. Feng, J. Fu, H. Guo, Y. Guo, B. Han, Z. Jiang, L. Kong, C. Li, H. Liu, P. T. T. Nguyen, P. Ren, F. Wang, S. Wang, Y. Wang, Y. Wang, S. S. Wong, K. Yan, N. Yan, X. Yang, Y. Zhang, Z. Zhang, X. Zeng and H. Zhou, Green Energy Environ., 2023, 8, 10–114 CrossRef CAS.
- C. Moreau, M. N. Belgacem and A. Gandini, Top. Catal., 2004, 27, 11–30 CrossRef CAS.
- X. H. Chadderdon, D. J. Chadderdon, T. Pfennig, B. H. Shanks and W. Li, Green Chem., 2019, 21, 6210–6219 RSC.
- G. Bharath and F. Banat, ACS Appl. Mater. Interfaces, 2021, 13, 24643–24653 CrossRef CAS PubMed.
- C.-H. Zhou, X. Xia, C.-X. Lin, D.-S. Tong and J. Beltramini, Chem. Soc. Rev., 2011, 40, 5588–5617 RSC.
- J. Becker and C. Wittmann, Curr. Opin. Biotechnol, 2012, 23, 718–726 CrossRef CAS PubMed.
- T. Fukushima and M. Yamauchi, Chem. Commun., 2019, 55, 14721–14724 RSC.
- J. Wu, L. Xu, Z. Kong, K. Gu, Y. Lu, X. Wu, Y. Zou and S. Wang, Angew. Chem., Int. Ed., 2023, 62, e202311196 CrossRef CAS PubMed.
- J. Xian, S. Li, H. Su, P. Liao, S. Wang, Y. Zhang, W. Yang, J. Yang, Y. Sun, Y. Jia, Q. Liu, Q. Liu and G. Li, Angew. Chem., Int. Ed., 2023, 62, e202304007 CrossRef CAS PubMed.
- J. Xian, S. Li, H. Su, P. Liao, S. Wang, R. Xiang, Y. Zhang, Q. Liu and G. Li, Angew. Chem., 2023, 62, e202306726 CrossRef CAS PubMed.
- Y. Wu, W. Chen, Y. Jiang, Y. Xu, B. Zhou, L. Xu, C. Xie, M. Yang, M. Qiu, D. Wang, Q. Liu, Q. Liu, S. Wang and Y. Zou, Angew. Chem., Int. Ed., 2023, 62, e202305491 CrossRef CAS PubMed.
- S. M. Soliman, J. Albering and M. A. M. Abu-Youssef, J. Mol. Struct., 2017, 1139, 17–30 CrossRef CAS.
- R. Xiang, S. Wang, P. Liao, F. Xie, J. Kang, S. Li, J. Xian, L. Guo and G. Li, Angew. Chem., 2023, 62, e202312239 CrossRef CAS PubMed.
- F. Nosheen, N. Wasfi, S. Aslam, T. Anwar, S. Hussain, N. Hussain, S. N. Shah, N. Shaheen, A. Ashraf, Y. Zhu, H. Wang, J. Ma, Z. Zhang and W. Hu, Nanoscale, 2020, 12, 4219–4237 RSC.
- W. Zhang, W. Zhang, J. Tan, D. Pan, Y. Tang and Q. Gao, J. Mater. Chem. A, 2023, 11, 7505–7512 RSC.
- R. Yao, Y. Li, X. Zhang, Y. Zhao, Y. Wang, X. Lang, Q. Jiang, H. Tan and Y. Li, Chem. Eng. J., 2023, 471, 144487 CrossRef CAS.
- W. Ao, H. Ren, C. Cheng, Z. Fan, Q. Qin, P. Yin, Q. Zhang and L. Dai, Angew. Chem., Int. Ed., 2023, 62, e202307924 CrossRef CAS PubMed.
- S. Guo, Y. Wu, C. Wang, Y. Gao, M. Li, B. Zhang and C. Liu, Nat. Commun., 2022, 13, 5297 CrossRef CAS PubMed.
- S. Wu and Y. H. Hu, Chem. Eng. J., 2021, 409, 127739 CrossRef CAS.
- Y.-Y. Lou, C. Xiao, J. Fang, T. Sheng, L. Ji, Q. Zheng, B.-B. Xu, N. Tian and S.-G. Sun, Phys. Chem. Chem. Phys., 2022, 24, 3896–3904 RSC.
- G. Jiang, X. Li, Y. Shen, X. Shi, X. Lv, X. Zhang, F. Dong, G. Qi and R. Liu, J. Catal., 2020, 391, 414–423 CrossRef CAS.
- C. Xiao, B.-A. Lu, P. Xue, N. Tian, Z.-Y. Zhou, X. Lin, W.-F. Lin and S.-G. Sun, Joule, 2020, 4, 2562–2598 CrossRef CAS.
- L.-X. Li, G.-C. Zhang, W.-J. Sun, H.-Y. Zhang, S.-X. Wang, J.-L. Wei, J.-H. He, K. Song and J.-M. Lu, Chem. Eng. J., 2022, 433, 134415 CrossRef CAS.
- L. Yang, Z. Chen, D. Cui, X. Luo, B. Liang, L. Yang, T. Liu, A. Wang and S. Luo, Chem. Eng. J., 2019, 359, 894–901 CrossRef CAS.
- S. Pei, Y. Wang, S. You, Z. Li and N. Ren, Engineering, 2022, 9, 77–84 CrossRef CAS.
- X. Shen, F. Xiao, H. Zhao, Y. Chen, C. Fang, R. Xiao, W. Chu and G. Zhao, Environ. Sci. Technol., 2020, 54, 4564–4572 CrossRef CAS PubMed.
- Z. Fan, H. Zhao, K. Wang, W. Ran, J.-F. Sun, J. Liu and R. Liu, Environ. Sci. Technol., 2023, 57, 1499–1509 CrossRef CAS PubMed.
- Y. Shen, Y. Tong, J. Xu, S. Wang, J. Wang, T. Zeng, Z. He, W. Yang and S. Song, Appl. Catal., B, 2020, 264, 118505 CrossRef CAS.
- Z. Gu, N. Ni, G. He, Y. Shan, K. Wu, C. Hu and J. Qu, Environ. Sci. Technol., 2023, 57, 16695–16706 CrossRef CAS PubMed.
- C. Choi, X. Wang, S. Kwon, J. L. Hart, C. L. Rooney, N. J. Harmon, Q. P. Sam, J. J. Cha, W. A. Goddard, M. Elimelech and H. Wang, Nat. Nanotechnol., 2023, 18, 160–167 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |