
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLight concentration and electron transfer in plasmonic–photonic Ag,Au modified Mo-BiVO4 inverse opal photoelectrocatalysts†
Martha
Pylarinou
a,
Elias
Sakellis
ab,
Polychronis
Tsipas
b,
Spiros
Gardelis
 a,
Vassilis
Psycharis
b,
Athanasios
Dimoulas
b,
Thomas
Stergiopoulos
b and
Vlassis
Likodimos
*a
a,
Vassilis
Psycharis
b,
Athanasios
Dimoulas
b,
Thomas
Stergiopoulos
b and
Vlassis
Likodimos
*a
aSection of Condensed Matter Physics, Department of Physics, National and Kapodistrian University of Athens, University Campus, 15784, Greece. E-mail: vlikodimos@phys.uoa.gr
bInstitute of Nanoscience and Nanotechnology, National Center for Scientific Research “Demokritos”, 15341 Agia Paraskevi, Athens, Greece
First published on 30th April 2024
Abstract
Plasmonic photocatalysis based on metal–semiconductor heterojunctions is considered a key strategy to evade the inherent limitations of poor light harvesting and charge separation of semiconductor photocatalysts. It can be profitably combined with three-dimensional photonic crystals (PCs) that offer an ideal scaffold for loading plasmonic nanoparticles and a unique architecture to intensify photon capture. In this work, Mo-doped BiVO4 inverse opals were applied as visible light-responsive photonic hosts of Ag and/or Au plasmonic nanoparticles in order to exploit the synergy of plasmonic and photonic amplification effects with interfacial charge transfer for the photoelectrocatalytic degradation of recalcitrant pharmaceutical contaminants under visible light. Photoelectrochemical evaluation indicated a major contribution from hot spot-assisted local field enhancement, most pronounced for Ag/Mo-BiVO4 PCs due to the spectral overlap of the localized surface plasmon resonance with the electronic absorption and blue-edge slow photon region of Mo-BiVO4 PCs, in contrast to weak plasmonic sensitization effects for the Au-modified PCs. The diverse band alignment at the metal–semiconductor interfaces resulted in the enhanced photoelectrocatalytic degradation of tetracycline broad spectrum antibiotic by Ag/Mo-BiVO4 and the refractory ibuprofen drug by (Ag,Au)/Mo-BiVO4, attributed to the enhanced charge separation by electron transfer toward Ag nanoparticles. Combination of visible light activated semiconductor PCs and plasmonic nanoparticles with suitable band alignment and photonic band gap may provide a versatile approach for the rational design of efficient plasmonic–photonic photoeletrocatalysts.
Introduction
Plasmonic metal–semiconductor heterostructures have emerged as highly promising materials to alleviate the major limitations of weak visible light harvesting as well as poor charge carrier separation and transport that compromise the efficiency of semiconductor photocatalysts.1,2 Radiative and non-radiative decay of the localized surface plasmon resonance (LSPR) of subwavelength metallic nanoparticles (NPs), the most common being Au and Ag, provides distinct interaction mechanisms for improving photocatalytic performance via the size-dependent local electromagnetic field enhancement and scattering3,4 as well as electron transfer, mostly by means of indirect hot electron injection,5–7 plasmon-induced resonant energy transfer8 and plasmonic heating2 to adjacent semiconductor substrates. These effects have been intensely investigated and established for wide band gap metal oxide photocatalysts coupled with plasmonic NPs, the most prominent system being Au–TiO2, whose photocatalytic response can be extended to the visible range via hot-electron injection primarily over the Schottky barrier at the Au–TiO2 interface.9,10 However, relatively fewer efforts have been devoted to heterojunctions between plasmonic NPs and narrow band gap semiconductors11,12 where bi-directional electron flow between metallic and semiconductor NPs may occur depending on the interfacial band-alignment and Schottky barrier height,13 resulting in enhanced charge separation and plasmon resonant energy transfer.8,14An advanced approach to enhance photochemical reactions was introduced by the seminal works of Ozin's group15,16 to synergistically amplify the performance of TiO2 photocatalysts, based on the utilization of photonic crystals (PCs) loaded with metallic NPs. In that case, fine tuning of PCs structural periodicity can selectively increase light harvesting at frequencies of weak materials’ electronic absorption by means of slow light propagation at the edges of the photonic bandgap (PBG).17,18 Apart from extending the optical path of incident photons, PCs’ periodic refractive index modulation on the scale of visible light's wavelength provides an interconnected macroporous structure for molecular adsorption and transport during photocatalytic reactions, especially in the form of bottom-up assembled inverse opals.19 The open inverse opal architecture also offers an excellent scaffold for loading plasmonic NPs with a high density of hot spots that may intensify electromagnetic field enhancement at the metal–semiconductor interface.20 Besides TiO2 PC photocatalysts incorporating Au and Ag NPs,21–25 the synergy of plasmonic and photonic amplification has been pursued for visible light activated (VLA) photonic photocatalysts by spectral matching the slow-photon high/low frequency regions to the LSPR absorption of metallic NPs.26 Surface modification of VLA photocatalysts by noble metal NPs has been mainly investigated on bismuth vanadate (BiVO4), the benchmark metal oxide photoanode for photoelectrochemical (PEC) water splitting,27 where significant activity improvements have been reported for PBG engineered BiVO4 inverse opals related to the slow-photon enhanced LSPR excitation and hot-electron injection to the BiVO4 conduction band.28–32 Moreover, spectral overlap of the electronic band gap with the LSPR of Au NPs and the incident light wavelength resulted in markedly enhanced performance on 4-nitrophenol reduction for Au–V2O5 inverse opals over homologous Au–TiO2 PC films,33 where electron transfer from the V2O5 conduction band to the Au NPs was favoured under simultaneous visible light excitation of the semiconductor band gap and LSPR.
In this work, PBG engineered molybdenum (Mo)-doped BiVO4 inverse opal photoelectrodes were selected as VLA photonic hosts for grafting plasmonic Ag and Au NPs both as single and dual co-catalysts aiming to the development of plasmonic–photonic photoelectrocatalysts for pharmaceuticals degradation. Aside from enhanced PEC activity due to improved visible light harvesting, electron transport and charge separation by Mo-doping, the selection of Mo-BiVO4 PCs as semiconductor photonic substrate, which has a work function straddling those of the two noble metal NPs, allowed investigating the variation of the metal–semiconductor band alignment and thus the interplay of different plasmonic enhancement mechanisms in the metal–semiconductor heterostructure performance. Comparative evaluation of the (Ag,Au)/Mo-BiVO4 PC films was carried out by PEC measurements and visible light photoelectrocatalytic degradation of two pharmaceutical emerging contaminants in water bodies, namely tetracycline (TC) broad spectrum antibiotic34 and the highly refractory ibuprofen (IBU) nonsteroidal anti-inflammatory drug,35 whose removal by immobilized photocatalysts is currently the focus of significant research efforts for water remediation.36,37 Although a major contribution from local field enhanced electron–hole generation was inferred from the PEC response of the metal-decorated Mo-BiVO4 PC photoelectrodes, different degradation mechanisms for the two pharmaceutical contaminants probed by scavenger tests, indicate additional contributions from interfacial electron transfer especially for the Ag–Au co-decorated plasmonic–photonic photoelectrocatalysts.
Experimental methods
Ag,Au modified Mo-BiVO4 inverse opal film fabrication
Inverse opal films were fabricated by the liquid infiltration of self-assembled colloidal templates prepared by the horizontal deposition method (Fig. 1) using monodisperse polystyrene (PS) spheres of 239, 287, 340 and 418 nm diameter (Microparticles GmbH, standard deviation SD = 6–11 nm, 2.6–3.0% CV), in the form of 5% solids (w/v) colloidal dispersions in deionized (DI) water. Fluorine-doped tin oxide (FTO, thickness 2.2 mm, surface resistivity 7 Ω sq−1, Sigma Aldrich) glass were cleaned by ultrasound sonication in Hellmanex™ III (Helma Analytics) and propanol-2 (HPLC, 99.8%) followed by dipping in water and blowing with nitrogen gas. The films hydrophilicity was enhanced by dipping in H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HNO3 (5:1) for several minutes. The sacrificial PS sphere opal templates were prepared by the material-saving convective evaporation-induced self-assembly of PS spheres on horizontal substrates.38 To this aim, 100 μl of 1.25 wt% PS sphere suspension were spread on a horizontal FTO glass slide followed by drying at 20 °C. Then, liquid infiltration of the opal films was carried out by dip-coating in a complex metal salt precursor based on ammonium vanadate (NH4VO3) (ACS, 99.0% min) and bismuth(III) nitrate pentahydrate, Bi(NO3)3·5H2O (99.999% trace metals basis) with the addition of ammonium molybdate tetrahydrate (NH4)6Mo7O24·4H2O (BioUltra, ≥99.0%) as source of molybdenum (Mo) dopants at Mo:V molar ratios of 3%, followed by drying for 1 h at 70 °C.39 After repeating the dip-coating process for three times, calcination was performed at 400 °C to remove the PS opal template and crystallize the amorphous precursor in the Mo-BiVO4 inverse opal structure. The fabricated PC films were labelled as Mo-BiVO4 PCXXX with XXX being the templating sphere diameter.
:HNO3 (5:1) for several minutes. The sacrificial PS sphere opal templates were prepared by the material-saving convective evaporation-induced self-assembly of PS spheres on horizontal substrates.38 To this aim, 100 μl of 1.25 wt% PS sphere suspension were spread on a horizontal FTO glass slide followed by drying at 20 °C. Then, liquid infiltration of the opal films was carried out by dip-coating in a complex metal salt precursor based on ammonium vanadate (NH4VO3) (ACS, 99.0% min) and bismuth(III) nitrate pentahydrate, Bi(NO3)3·5H2O (99.999% trace metals basis) with the addition of ammonium molybdate tetrahydrate (NH4)6Mo7O24·4H2O (BioUltra, ≥99.0%) as source of molybdenum (Mo) dopants at Mo:V molar ratios of 3%, followed by drying for 1 h at 70 °C.39 After repeating the dip-coating process for three times, calcination was performed at 400 °C to remove the PS opal template and crystallize the amorphous precursor in the Mo-BiVO4 inverse opal structure. The fabricated PC films were labelled as Mo-BiVO4 PCXXX with XXX being the templating sphere diameter.
 | ||
| Fig. 1 Schematic illustration of the fabrication of (Ag,Au)/Mo-BiVO4 inverse opal films. | ||
The Mo-BiVO4 PCs were decorated with 10 nm Ag NPs (Thermo Scientific, 0.02 mg ml−1 suspension, supplied in 2 mM sodium citrate, 4 × 1012 NP per ml) or 5 nm Au NPs (Sigma Aldrich, stabilized suspension in citrate buffer, 5.5 × 1013 NPs per ml) by dropping 15 μL of suspension on the films’ surface, which were kept at 50 °C for 60 min, followed by washing with DI water and ethanol, yielding the Ag/Mo-BiVO4 and Au/Mo-BiVO4 inverse opals, respectively (Fig. 1).
This process was also applied for the fabrication of co-decorated (Ag,Au)/Mo-BiVO4 films using a mixture of 15 μL with equal amounts of the plasmonic NP suspensions.
Materials characterization and photocatalytic evaluation
The (Ag,Au)/Mo-BiVO4 PCs were characterized by scanning and transmission electron microscopies (SEM-TEM) coupled with energy-dispersive X-ray spectroscopy (EDX), X-ray diffraction (XRD) micro-Raman, diffuse and specular reflectance, X-ray photoelectron (XPS) and photoluminescence (PL) spectroscopies (detailed description in S1, ESI†). Electrochemical impedance spectroscopy (EIS) and PEC evaluation were carried out in a three-electrode configuration using the PC photoanodes, whose performance was evaluated on the photoelectrocatalytic degradation of TC and IBU pharmaceuticals under visible light, as detailed in S2, ESI.†Results and discussion
Structural and optical properties
SEM images (Fig. 2a–d) indicate that crystallization of the metal salt precursor in the interstices of the PS colloidal template after calcination, resulted in well-ordered, interconnected inverse opal structures with macropore void diameter (D) determined from that of the PS templating spheres and approximately 1.2 μm film thickness (Fig. 2e). XRD measurements on the Mo-BiVO4 PC films before and after surface modification with the metallic NPs (Fig. 2f), showed that the PC films crystallize in the monoclinic scheelite (ms) BiVO4 phase (space group I2/a, JCPDS 014-0688), identified by the splitting of the (002)/(200) diffraction peaks at 34.5° and 35.2° as well as the (240)/(042) ones at about 46.9° and 47.3°. However, the observed splitting was considerably moderated and less resolved for all Mo-BiVO4 films, indicating that the introduction of molybdenum dopants substituting for vanadium in the BiVO4 lattice, leads to the averaging of the ms low-symmetry toward the tetragonal scheelite structure.39 In addition, no sign of metallic Au and Ag NPs could be traced in the XRD patterns of the Ag,Au-modified PC films, indicative of their relatively low loading amount.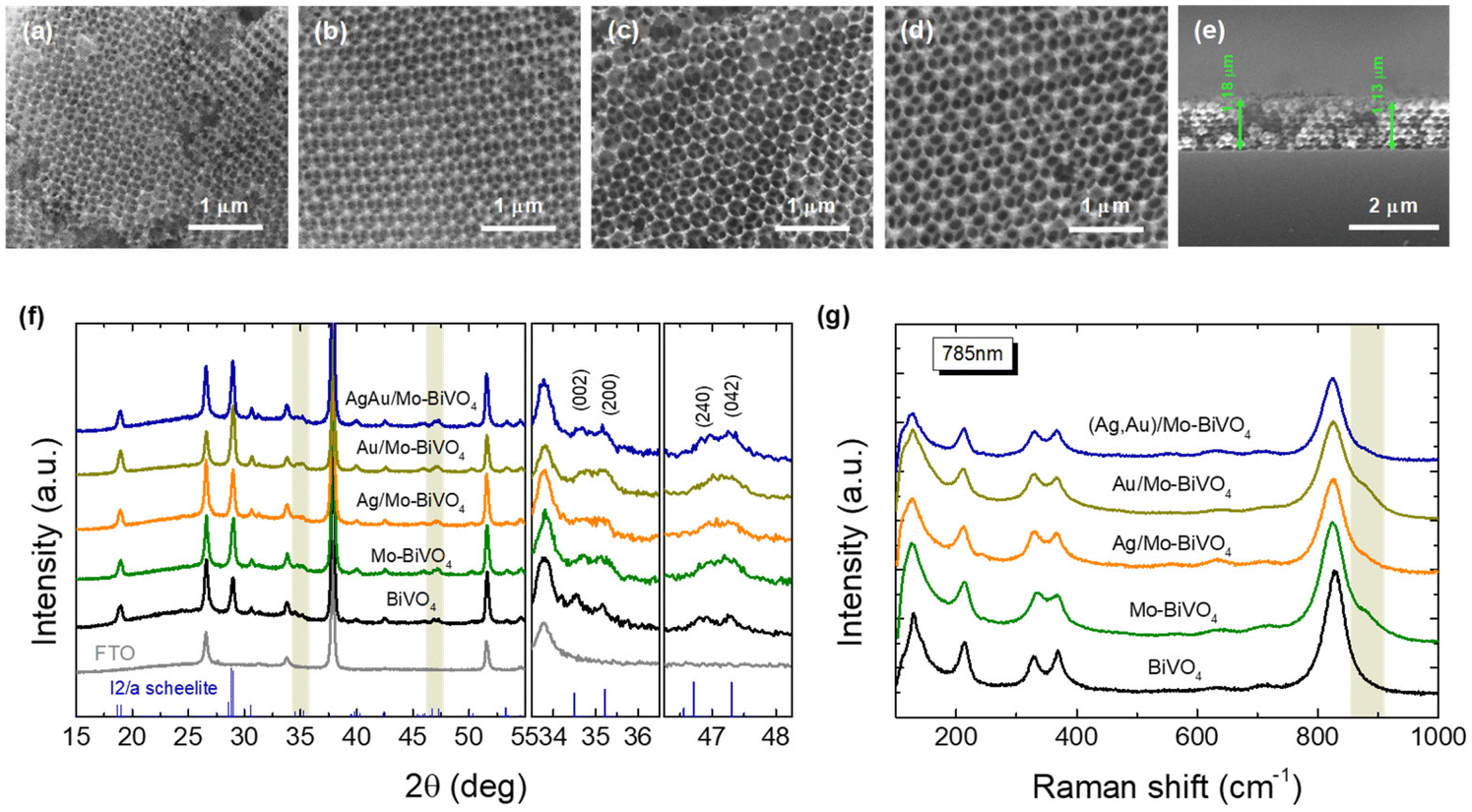 | ||
| Fig. 2 Plan view SEM images for (a) PC239, (b) PC287, (c) PC340, (d) PC418, and cross section for (e) PC340 inverse opal films. (f) XRD patterns and (g) Raman spectra of Ag,Au-modified Mo-BiVO4 PC340 films compared to the pristine Mo-doped and undoped BiVO4. The XRD patterns of the FTO substrate and the ms phase (JCPDS 014-0688) are also displayed, while the right panels in (f) depict in detail the shaded areas of the XRD patterns. | ||
Micro-Raman spectra for the Ag,Au-modified Mo-BiVO4 films at 785 nm, displayed in all cases the characteristic vibrational modes of single-phase monoclinic scheelite (ms) BiVO4 with no sign of the tetragonal scheelite or zircon polymorphic phases.40 Specifically, the strong symmetric (νs) along with the weak antisymmetric (νas) V–O stretching modes were identified at 824 and 712 cm−1, respectively.41 The monoclinic distortion of the scheelite structure was identified by the observation of two separate modes at 366 and 331 cm−1 (Fig. S1, ESI†) arising from the symmetric (δs) and antisymmetric (δas) bending modes of the VO4 tetrahedra, as well as the two intense external (rotation/translation) lattice modes at 210 and 124 cm−1.40 The presence of Mo6+ dopants substituting for V5+ cations in BiVO4,42 was traced by the observation of an additional weak shoulder at about 882 cm−1 related to the corresponding MoO4 stretching vibration,43 as well as the shift of the Raman peaks in comparison to the characteristic vibrational modes of pristine BiVO4 PCs (Fig. 2c). Both the symmetric νs and antisymmetric νas stretching bands as well as the δs and δas bending modes of the VO4 tetrahedra approached each other for the Mo-doped PCs, indicating the reduction of the ms lattice distortion towards the tetragonal phase. Using the exponential function ν = 21349e−1.9176R, which relates the V–O stretching frequencies (cm−1) to V–O bond lengths (Å) in vanadium oxides,44 the values of 1.697 and 1.773 Å, were derived for the two V–O tetrahedral bond lengths in Mo-BiVO4 in comparison to 1.694 and 1.775 Å, for the pristine BiVO4 PCs, corroborating the averaging of the VO4 tetrahedral deformation after Mo-doping. On the other hand, surface decoration of Mo-BiVO4 inverse opals by Ag and/or Au NPs had no influence on the Raman frequencies apart from a weak intensity variation, most likely due to the reflectance of the metallic NPs.
The chemical composition of the PC films consisting of Bi, V, O and Mo elements, and their surface modification by Au and Ag NPs was investigated by EDX elemental maps of scanning TEM (STEM) images. The uniform coverage and infiltration of the inverse opal skeleton by metallic NPs was thereby verified for the co-deposited (Ag,Au)/Mo-BiVO4 (Fig. 3a–d) as well as the singly-decorated Ag/Mo-BiVO4 (Fig. S2, ESI†) and Au/Mo-BiVO4 (Fig. S3, ESI†) PC340 films. The formation of high surface density of metallic NPs on the Mo-BiVO4 walls with numerous clusters was directly observed in higher magnification TEM images (Fig. 3e). The individual Ag and Au NPs of different sizes could be identified in the fast Fourier transform (FFT) patterns of high-resolution TEM images on the (Ag,Au)/Mo-BiVO4 PC walls (Fig. 3f and g), where the d-spacing of 2.05 Å from the (200) fcc planes of ∼10.5 nm Ag NPs was derived in contrast to the 2.36 Å interplanar spacing of (111) fcc planes for ∼6 nm Au NPs.
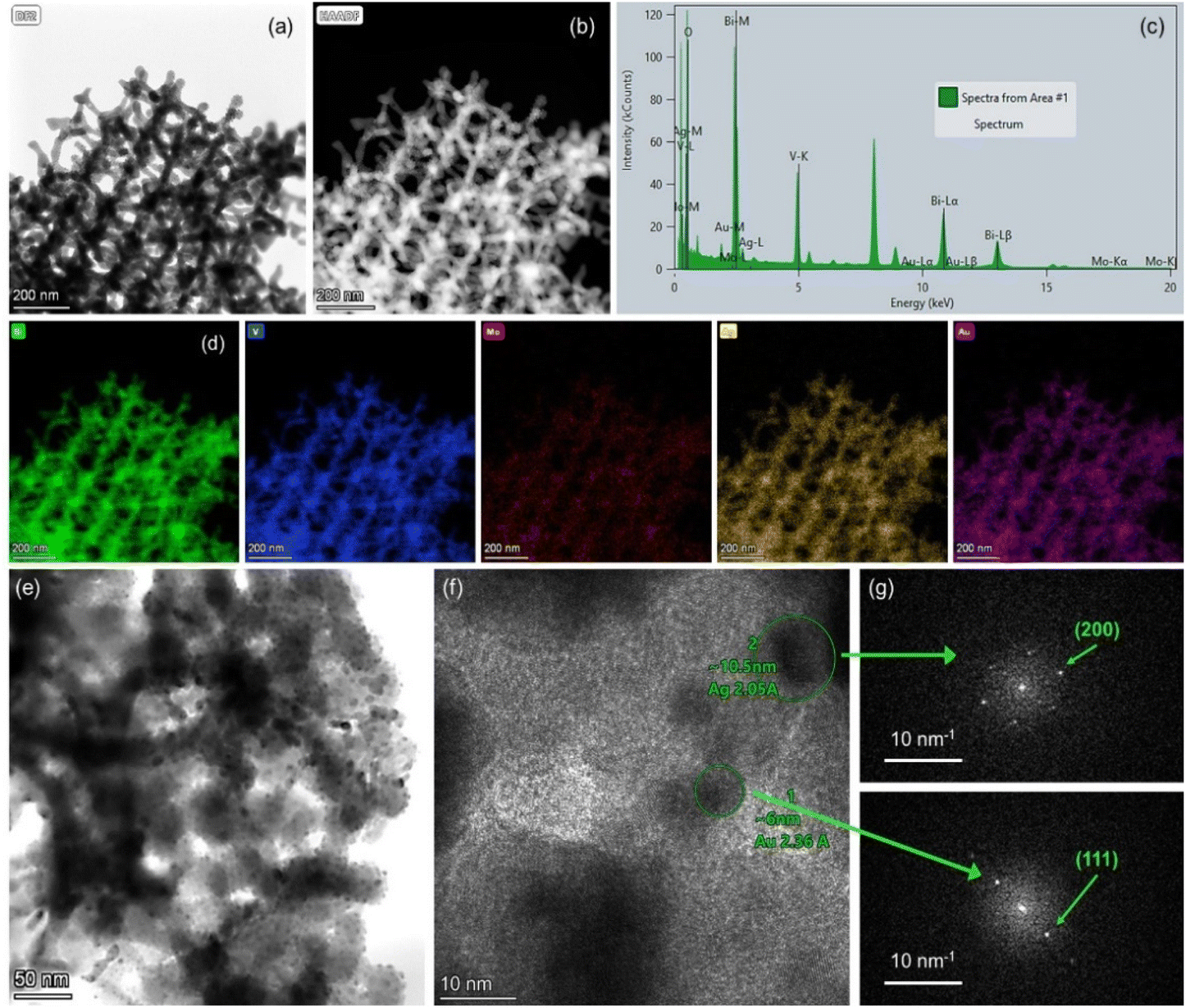 | ||
| Fig. 3 (a) and (b) TEM images of (Ag,Au)/Mo-BiVO4 PC340 films and (c) the corresponding local EDX spectrum and (d) Bi, V, O, Mo, Ag and Au elemental maps obtained from (e and f) higher magnification TEM images of (Ag,Au)/Mo-BiVO4 and (g) the FFT patterns of the encircled areas in (f), where individual Ag and Au NPs can be identified. | ||
The surface elemental composition of the plasmonic NP-modified Mo-BiVO4 PCs films was determined by XPS (Fig. 4). The Bi 4f core level spectra for the Mo-BiVO4 photonic substrate showed two distinct peaks at binding energies (BEs) of 159.3 and 164.5 eV (splitting Δ = 5.2 eV), which correspond to the 4f7/2–4f5/2 spin–orbit doublet of Bi3+ ions.45 The V 2p spectra presented the characteristic V 2p3/2 peak of V5+ ions at 516.9 eV, while the O 1s spectra showed an asymmetric broad peak at 530.1 eV arising from lattice oxygen and adsorbed oxygen species.46 In addition, the presence of Mo6+ dopants was confirmed by the 3d5/2–3d3/2 spin–orbit doublet at BEs of 232.4 and 235.6 eV (splitting Δ = 3.2 eV) in the Mo 3d core level region.42 The deposition of metallic NPs at the Mo-BiVO4 inverse opal walls was identified in the respective Au 4f and Ag 3d core level spectra. The observed peaks at BEs of 368.1 and 374.2 eV (splitting Δ = 6.1 eV) correspond to the 3d5/2–3d3/2 spin–orbit doublet of metallic Ag,47 while the peaks at 87.3 and 83.6 eV (splitting Δ = 3.7 eV) correspond to the 4f5/2 and Au 4f7/2 spin–orbit components of metallic Au.48 Moreover, the Bi 4f, V 2p and O 1s peaks for the plasmonic modified PC films shifted to lower BE's by about 0.2–0.4 eV compared to the unmodified ones, implying different Fermi level shifts toward Mo-BiVO4 valence band maximum, especially for the (Ag,Au)/Mo-BiVO4 PCs.
 | ||
| Fig. 4 Bi 4f, V 2p, O 1s, Mo 3d, Au 4f and Ag 3d core level XP spectra for (1) Mo-BiVO4, (2) Ag/Mo-BiVO4, (3) Au/Mo-BiVO4, and (4) (Ag,Au)/Mo-BiVO4 PC340 films. | ||
Specular reflectance (R%) measurements at 15° angle of incidence showed the characteristic Bragg reflections due to the incomplete PBG (stop band) formation along the [111] direction in the Mo-BiVO4 inverse opal films (Fig. 5a).17,18 The stop band positions shifted to higher wavelengths with the increase of the inverse opal void diameter allowing PBG engineering with respect to the Ag and Au NPs LSPR absorption peaks, which were determined from the extinction spectra of the corresponding NP dispersions at 406 and 520 nm, respectively (Fig. 5b). The R% Bragg intensities decreased with the decrease of PC macrospore size because of absorption losses that arise from the inherent electronic band gap absorption of Mo-BiVO4 (vide infra). Applying modified Bragg's law for the obtained stop band wavelengths at 15°,17,18 which were not modified by the Ag and Au NP deposition (Fig. S4, ESI†), together with the values of the PC macropore diameters D determined from SEM and the refractive indices of BiVO4 (nBiVO4 = 2.4) and air (nair = 1.0),39 provided an estimate for the variation of the effective refractive index neff and the solid filling fraction 1 − f of the inverse opals. The obtained 1 − f values (Table S1†) were smaller than the theoretical value of 0.26 for complete filling of the inverse fcc lattice indicative of the formation of nanocrystalline walls. Moreover, using the values for 1 − f and nH2O = 1.33, the stop band positions were estimated after PC infiltration with water, where the photocatalytic reaction takes place.
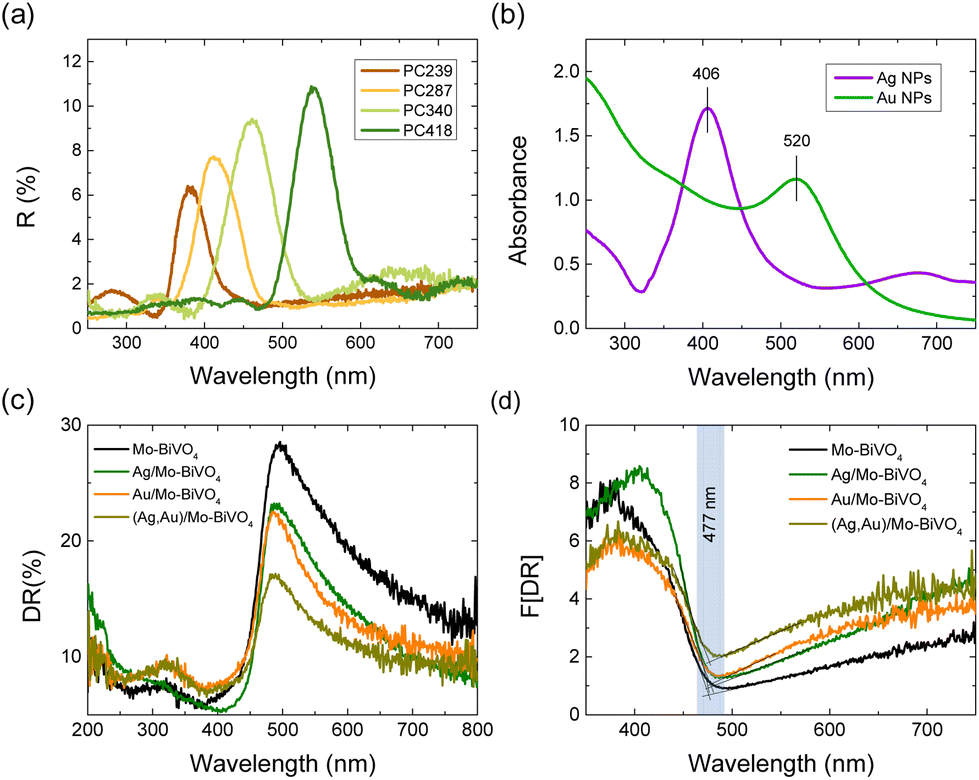 | ||
| Fig. 5 (a) Specular reflectance (R%) at 15° incidence (R%) for the Mo-BiVO4 PC films and (b) LSPR absorbance of Ag and Au NP suspensions (c) diffuse reflectance (DR%) and the corresponding (d) Kubelka–Munk transforms F(DR) for the Ag,Au-modified PC340 films. | ||
Diffuse reflectance (DR%) spectra showed a nearly constant edge for the Ag,Au-modified PC340 films (Fig. 5c), independent of the plasmonic NPs presence with no evident contribution of the Bragg reflection, which appears in the DR% spectra only for higher PC diameters where the spectral overlap of the stop band positions with the materials’ electronic absorption is reduced (Fig. S5, ESI†).39 An absorption edge of about 477 nm was accordingly derived from the Kubelka–Munk transforms F(DR) of the DR% spectra (Fig. 5d), while the electronic band gap Eg of 2.57 eV was estimated from the corresponding indirect band gap Tauc plots for the pristine and Ag,Au-modified Mo-BiVO4 PC films (Fig. S6, ESI†), in agreement with previous works on BiVO4 inverse opals.46,49 Nevertheless, the DR% of the plasmonic NP-modified PCs decreased appreciably above the electronic absorption edge of Mo-BiVO4, especially after surface modification by Au NPs (Fig. 5c), showing the appearance of visible light absorption extending beyond the narrow band LSPR bands of dispersed Ag and Au NPs (Fig. 5b). This indicates the excitation of higher-order plasmonic modes at longer wavelengths due to interparticle coupling effects in clusters consisting of closely spaced or touching metallic NPs on the BiVO4 surface.50,51
Photoelectrocatalytic evaluation
The Ag,Au-modified PC films were comparatively evaluated on the photoelectrocatalytic degradation of TC and IBU pharmaceutical contaminants in 0.1 M NaHCO3 supporting electrolyte at applied potential +1.0 V vs. Ag/AgCl under visible light. Fig. 6a and b compare the corresponding degradation kinetics for the Ag,Au-modified PC340 films. Dark adsorption of TC molecules on the PC films reached approximately 12% after 30 min, while IBU adsorption was negligible. Blank tests without PC films, showed negligible TC or IBU degradation under visible light, whereas illumination in their presence led to continuous temporal decrease of their concentration (C), monitored spectrophotometrically by the TC and IBU absorption bands at 370 and 225 nm, respectively (Fig. S7, ESI†). The ln(C/C0) vs. t plots, where C0 is the initial TC or IBU concentration after dark adsorption, varied linearly with time (Fig. 6b and e), indicating pseudo first-order kinetics, with kinetic constants k (min−1) derived from the corresponding linear slopes. The reaction rates rvis were calculated as rvis = kC0, independent of concentration variations arising from dark adsorption. In the case of TC degradation, Ag/Mo-BiVO4 showed the highest performance (Fig. 6c), while Au NPs deposition resulted in slight deterioration of photoelectocatalytic activity toward TC degradation. The obtained kinetic constant k of 0.0271 min−1 exceeded that (0.01901 min−1) of pristine Mo-BiVO4 PC340 photoelectrodes by about 43%, and it is among the highest reported for TC photoelectrocatalytic degradation by BiVO4-based photoanodes.39,52 In the case of IBU degradation, plasmonic NPs deposition on Mo-BiVO4 photonic films resulted in enhanced photoelectocatalytic performance with the kinetic constants increasing from 1.2471 min−1 for Mo-BiVO4 to 1.7686 and 1.7918 min−1 for Ag/Mo-BiVO4 and Au/Mo-BiVO4, while it reached an almost two-fold increase of 2.466 min−1 in the case of dual-deposited (Ag,Au)/Mo-BiVO4 PC photoelectrodes.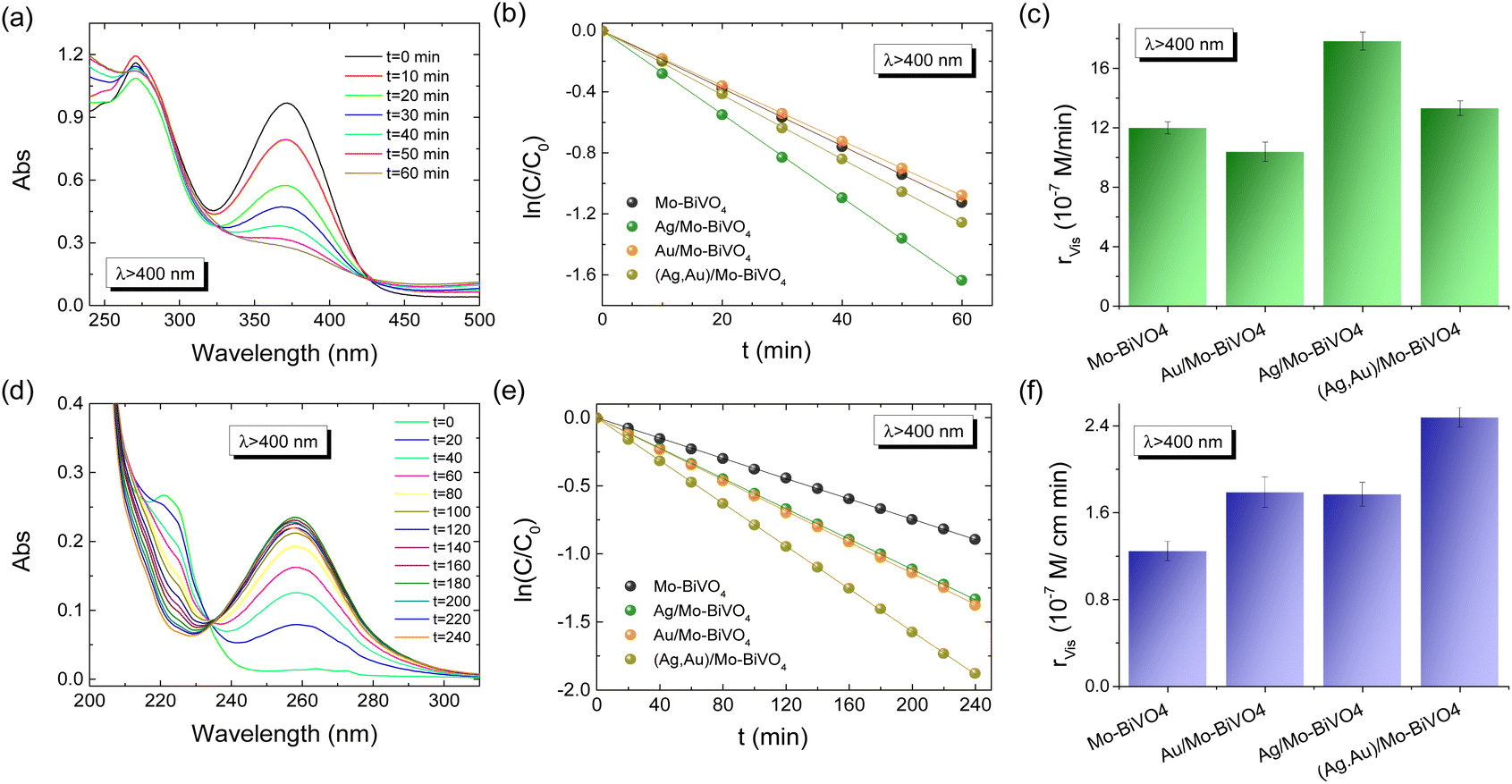 | ||
| Fig. 6 TC (a–c), IBU (d–f), absorbance spectra evolution, photodegradation kinetics, and reaction rates for the plasmonic modified Mo-BiVO4 PC340 photoelectrodes at +1.0 V vs. Ag/AgCl under visible light irradiation. | ||
The reaction mechanisms for the best-performing PC films on pharmaceuticals TC and IBU degradation were investigated in comparison to the pristine Mo-BiVO4 PCs by trapping experiments of superoxide radical anions (˙O2−), holes (h+) and hydroxyl radicals (˙OH), by the addition of 1 mM 1,4 benzoquinone (Bq), 5 mM formic acid (FA) and 5 mM isopropanol (IPA) radical scavengers in the reaction solution, respectively. In the case of TC degradation by Mo-BiVO4 PCs, rvis was considerably reduced by the addition of Bq and FA (rvis reduction by approximately 46% in both cases), whereas weaker decrease of rvis (by about 21%) was observed in the presence of IPA (Fig. S8a and b, ESI†).
This indicates that h+ and ˙O2− radicals are the major species involved in the VLA TC degradation, whereas ˙OH radicals play a minor role. In the case though of Ag/Mo-BiVO4 PCs that presented the highest rvis for TC degradation (Fig. 6c), the addition of both FA and IPA resulted in significant inhibition of the photoelectrocatalytic reaction (rvis decreased by 57% and 54% for FA and IPA, respectively), whereas the influence of Bq was significantly moderated (rvis decreased by about 25%) (Fig. S8c and d, ESI†). This indicates a change of the nature of reactive species underlying TC degradation upon Ag NPs deposition, which caused an increased contribution of ˙OH against ˙O2− radicals to the photocatalytic reaction, besides direct TC oxidation by photogenerated h+. In the case of IBU degradation, FA's addition caused a drastic rvis reduction by 57% for the unmodified Mo-BiVO4 PC films (Fig. S9, ESI†), whereas Bq and IPA scavengers had much weaker effects in the photocatalytic performance (rvis decreased by about 15%), indicating that h+ are the major reactive species.53,54 Similar behavior was observed for the dual-modified (Ag,Au)/Mo-BiVO4 PCs showing the highest performance on IBU degradation, where FA produced the most significant inhibitory effect (rvis decrease by 63%), indicating that photogenerated h+ remain the dominant reactive species.
The combination of LSPR with photonic amplification was explored by comparative photoelectrocatalytic tests on TC degradation for the Ag- and Au-modified Mo-BiVO4 PC films in 0.1 M NaHCO3 at +1.0 V vs. Ag/AgCl under visible light (Fig. S10, ESI†). The TC degradation rates showed a distinct size-dependence with respect to the PC periodicity with Ag/Mo-BiVO4 PC340 presenting the highest rvis value, followed by the PC239 films, whereas the PC418 and PC287 films showed much weaker activity (Fig. S10c, ESI†). This variation can be related to the optimal spectral overlap of the Ag NP LSPR and the Mo-BiVO4 electronic absorption edge with the slow photons of the supporting PC340 scaffold, whose stop band is expected at ≈540 nm in water (Table S1†). Assuming that the stop band spectral width is about 60 nm, i.e. the full width at half-maximum of the corresponding Bragg reflection (Fig. 5a), the narrower blue-edge slow photon region will be roughly expected below 510 nm, approaching both the Mo-BiVO4 absorption edge (≈480 nm) and the extended LSPR absorption of the Ag NP aggregates, observed in the DR% spectra (Fig. 5c). On the other hand, the detrimental matching of the PC287 Bragg reflection in water at ≈480 nm (Table S1†) with the Mo-BiVO4 band gap complies with the lowest rvis value, while PC418's stop band is expected at relatively high wavelengths (≈646 nm) to trigger slow photon effects. Significant overlap of red-edge slow photons with Ag LSPR and the semiconductor absorption edge is also expected for the ≈440 nm stop band (in water) of Ag/Mo-BiVO4 PC239 (Table S1†), whose performance may be compromised by absorption losses in the Mo-BiVO4 band gap in comparison to the best performing PC340 films. Similar size-selective activity was observed for the Au-modified Mo-BiVO4 PCs (Fig. S10d–f, ESI†) but with less pronounced variations of the rvis values compared to the Ag-modified PCs, which can be associated with the extended electronic absorbance of the Au NPs that moderate photonic effects.
Furthermore, considering the modest uniformity of metal oxide inverse opals fabricated by wet chemical methods,55 compared to more robust plasmonic–photonic structures,56,57 the activity of Ag- and Au-modified Mo-BiVO4 PC340 on TC degradation was investigated for films of the same composition but higher thickness of ≈2.3 μm (Fig. S11, ESI†). The obtained results reproduced closely the relative variation of rvis for the thin Ag- and Au-modified Mo-BiVO4 PCs but with overall weaker photoelectrocatalytic performance, indicative of higher recombination losses for the thicker films.58
PEC performance
The PEC response of the plasmonic modified Mo-BiVO4 PC340 photoelectrodes was comparatively evaluated by using linear sweep voltammetry at a potential scan rate of 10 mV s−1 both in the dark and under back-side visible and UV-Vis illumination in a 0.5 M NaHCO3 aqueous electrolyte (Fig. 7a and b). Illumination from the back-side led to much higher photocurrent density compared to front-side illumination (Fig. S12a, ESI†), indicative of the slow transport of electrons photogenerated at the semiconductor/electrolyte interface across the film thickness to reach the FTO back contact.58,59 Significant photocurrent enhancement was also observed for the Mo-BiVO4 PCs compared to approximately 500 nm thick-planar films deposited by dip-coating on FTO substrates without the colloidal template (Fig. S12b, ESI†),39 which confirms the advantage of the inverse opal photonic structure. Decoration of plasmonic Ag and Au NPs on the inverse opal skeleton resulted in marked improvements of the photocurrent densities by about 44% (53%) and 94% (90%) at 1.23 V vs. RHE potential under visible (UV-Vis) back-side illumination for the Au/Mo-BiVO4 and Ag/Mo-BiVO4 PCs, respectively, while a relatively weaker increase of approximately 20% (28%) was observed for the co-deposited (Ag,Au)/Mo-BiVO4 PCs, verifying the presence of plasmonic amplification effects. | ||
| Fig. 7 Current density–potential curves under (a) visible and (b) UV-Vis light back-side illumination (solid lines) for the Ag and Au modified Mo-BiVO4 PC340 photoelectrodes. Dashed lines display the corresponding dark response. (c) IPCE spectra at 1.23 V vs. RHE potential (the inset shows the IPCE enhancement factors with respect to the unmodified Mo-BiVO4 PC340). EIS Nyquist plots under (d) visible and (e) UV-Vis light, and (f) PL spectra. Solid lines depict the best fit curves to the modified Randles equivalent circuit. All PEC measurements were performed in 0.5 M NaHCO3 aqueous electrolyte. | ||
The enhanced PEC activity of the Ag and Au modified Mo-BiVO4 PC photoelectrodes was spectrally analyzed through measurements of incident photon-to-current efficiency (IPCE) in 0.5 M NaHCO3 aqueous electrolyte under back-side illumination at 1.23 V vs. RHE potential (Fig. 7c). Consistently with the LSV results, plasmonic NPs grafting on Mo-BiVO4 PCs resulted in significant enhancement of the IPCE spectra. The highest value was obtained for Ag/Mo-BiVO4 followed by Au/Mo-BiVO4 PCs, while the co-modified (Ag,Au)/Mo-BiVO4 showed relatively weaker increase compared to the bare Mo-BiVO4 photoelectrode. Moreover, the rise of IPCE was mainly observed below the absorption edge of Mo-BiVO4 (<490 nm), as evidenced in the corresponding enhancement factors calculated with respect to the unmodified Mo-BiVO4 PC (inset of Fig. 7c). In particular, a rapid decrease of ICPE was observed above 490 nm for Ag/Mo-BiVO4, whereas the relatively lower ICPE values for Au/Mo-BiVO4 and (Ag,Au)/Mo-BiVO4 PCs extended spectrally up to longer wavelengths of about 530 nm. Considering the weak light scattering efficiency of 10 nm Ag and 5 nm Au NPs,51,60 the enhanced PEC response within the absorption range of Mo-BiVO4 points to a major amplification by the local electromagnetic field enhancement from the metallic NPs that promotes electron–hole generation at the interface with the BiVO4 inverse opal skeleton. The near-field enhanced photocarrier production is more pronounced for Ag than Au NPs (Fig. 7c), as the Ag LSPR absorption overlaps entirely with the BiVO4 electronic absorption, while it can be further intensified by the hot spots between the metallic NP aggregates. On the other hand, the relatively small ICPE enhancement above the absorption edge of BiVO4 up to 530 nm for the Au/Mo-BiVO4 and (Ag,Au)/Mo-BiVO4 PCs, implies a weak plasmonic “sensitization” effect by hot-electron injection from Au NPs to the conduction band of Mo-BiVO4.60
In order to investigate charge separation at the photoelectrode/electrolyte interface for the Ag and Au modified Mo-BiVO4 PCs, EIS measurements were performed under visible (Fig. 7d) and UV-vis (Fig. 7e) light as well in the dark (Fig. S13, ESI†). The obtained Nyquist plots were fitted to a Randles equivalent circuit consisting of series resistance RS, the charge-transfer resistance RCT and a constant phase element CPE (Table S2†). Light irradiation caused the reduction of the capacitive arc radius in the EIS plane, i.e. the charge transfer resistance, resulting in enhanced separation of photo-induced charge carriers. Moreover, infiltration of the Mo-BiVO4 PC films with plasmonic NPs resulted in significant reduction of RCT, with (Ag,Au)/Mo-BiVO4 presenting the smallest value and consequently the lowest electron–hole recombination, especially under UV-Vis light. This was supported by PL measurements (Fig. 7f). A broad emission peak around 585 nm (approximately 2.12 eV) was observed, which is well above Mo-BiVO4 PC's absorption edge, confirming the presence of indirect band gap. The PL intensity was reduced when metallic NPs were decorated on Mo-BiVO4, indicating a decrease in electron–hole recombination with (Ag,Au)/Mo-BiVO4 PCs presenting the weakest PL signal in agreement with the EIS analysis.
Depending on the difference between the metal work function ϕM and the semiconductor ϕS, the formation (ϕM − ϕS > 0) or not (ϕM − ϕS < 0) of a Schottky barrier ϕB = ϕM − χS, where χS is the semiconductor electron affinity, is essential to the electron transfer path at the metal–semiconductor interface.61 Using the ϕM values of 4.26 and 5.1 eV (absolute vacuum scale) for the Ag and Au NPs,62 the Eg value of 2.57 eV (Fig. 5d) and χS of 4.68 eV recently determined for Mo-BiVO4 PCs,39 the energy band diagrams can be derived for Au/Mo-BiVO4 (Fig. 8a), Ag/Mo-BiVO4 (Fig. 8b) and (Ag,Au)/Mo-BiVO4 (Fig. 8c) heterojunctions. In the case of Ag NPs, the metal work function is lower than that of Mo-BiVO4 (ϕS = 5.01 eV) precluding Schottky barrier formation (ϕB = −0.42 eV < 0) and favoring electron flow from the semiconductor to the Ag NPs under visible light excitation. The scavenging of photogenerated electrons by the metallic NPs may accordingly rationalize the suppression of ˙O2− radicals from the observed reactive species in TC degradation for Ag/Mo-BiVO4 (Fig. S8, ESI†). On the other hand, the work function ϕM for Au NPs is higher than ϕS of Mo-BiVO4 leading to a Schottky barrier height ϕB = 0.42 eV, which is considerably lower than the corresponding ϕB ≈ 1.1 eV for Au–TiO2.9 Hot electron flow from Au NPs to the semiconductor will be accordingly favored, which, however, according to the IPCE results produces a relatively weak PEC response at longer wavelengths compared to the Mo-BiVO4 absorption edge (Fig. 7c). In the case of dual-decorated Mo-BiVO4 nanocrystals by both Ag and Au NPs, the injection of a relatively small fraction energetic hot electrons from Au NPs to the conduction band of Mo-BiVO4 will be followed by intensified electron flow to the Ag NPs, leading to cascade charge transfer in the corresponding heterojunctions for (Ag,Au)/Mo-BiVO4 PCs under visible light and UV-Vis excitation (Fig. 8c). This can greatly reduce electron–hole recombination and promote IBU degradation beyond the light concentration effects of plasmonic NPs.
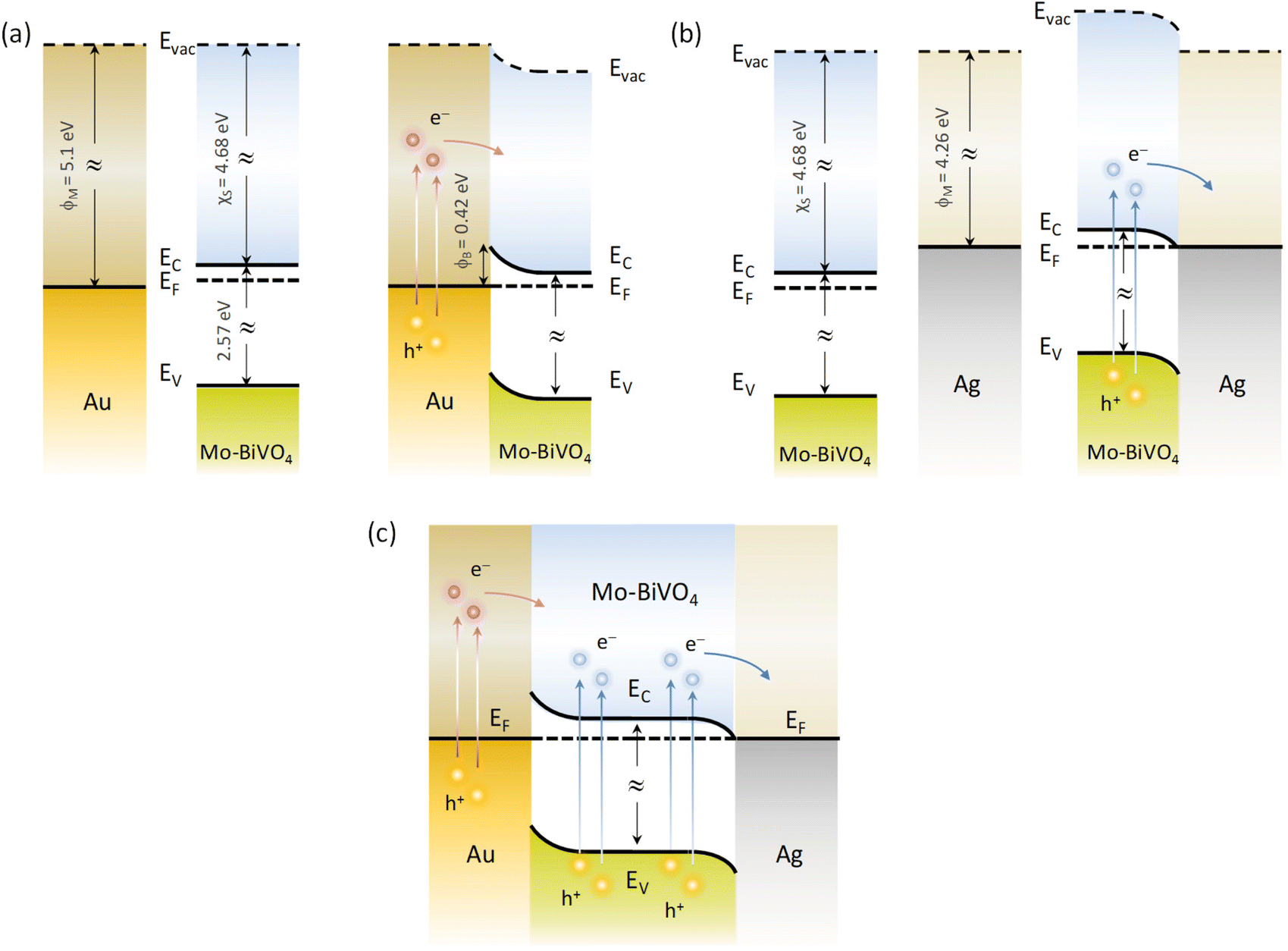 | ||
| Fig. 8 Schematic of the dominant charge transfer routes for (a) Au/Mo-BiVO4, (b) Ag/Mo-BiVO4 and (c) (Ag,Au)/Mo-BiVO4. | ||
In order to explore plasmonic size effects, comparative photoelectrocatalytic experiments were carried out for Au-modified Mo-BiVO4 PC340 films using 10, 50 and 80 nm Au NP citrate stabilized dispersions (Sigma Aldrich) on TC degradation at +1.0 V vs. Ag/AgCl under visible light (Fig. 9 and Fig. S14, ESI†). The obtained results show that Mo-BiVO4 PC films decorated by 10 nm Au NPs (Fig. S15, ESI†) present similar, slightly higher, rvis rates to that of 5 nm Au NPs. Conversely, the TC degradation rates decreased for the surface modified BiVO4 PCs with larger Au NPs, especially for the 50 nm diameter. This variation can be related to the dominant local field enhancement for the smaller Au NPs characterized by large absorption cross sections,51,60,63 along with the concomitant high surface coverage of the BiVO4 PC walls that favors NP aggregation and hot spot formation, leading to enhanced electron–hole generation and electron transfer at the abundant metal–semiconductor interfaces. On the other hand, the enhanced scattering cross section of larger diameter Au NPs leads to appreciably higher extinction,60,63 which underlies the decrease of DR% above the absorption edge of Mo-BiVO4 with increasing NP size (Fig. 9d), most pronounced for the 80 nm Au/Mo-BiVO4. The preferential light scattering toward BiVO4 by the large Au NPs is, though, not sufficient to promote the photocatalytic activity for the successively lower surface coverage of the PC skeleton by 50 nm (Fig. S16, ESI†) and 80 nm (Fig. S17, ESI†) Au NPs that also reduces the number of plasmonic metal–semiconductor heterojunctions.
 | ||
| Fig. 9 TEM images and the corresponding V and Au EDX elemental maps for Mo-BiVO4 PC340 films surface modified by Au NPs of (a) 50 and (b) 80 nm diameters. (c) Size dependent TC degradation rates at +1.0 V vs. Ag/AgCl under visible light and (d) DR (%) spectra for the Au/Mo-BiVO4 PC340 photoelectrodes with Au NPs of different diameters. | ||
Conclusions
In conclusion, surface decoration of Mo-BiVO4 inverse opal photoelectrodes by single and dual plasmonic Ag and/or Au NPs is shown as a competent approach for the photoelectrocatalytic degradation of TC and IBU pharmaceutical contaminants. A major contribution from local field enhancement was concluded from the PEC response of the metal-decorated Mo-BiVO4 PCs. This was most prominent for Ag/Mo-BiVO4 due to the optimal spectral overlap of the LSPR absorption of Ag NPs with the semiconductor band gap. It was further assisted by slow photon propagation in the inverse opal structure and the formation of hot spots by the NP aggregates, whereas a weak sensitization effect by hot electrons was derived for the Au-modified PCs. More importantly, the variable band alignment at the metal–semiconductor interfaces resulted in different electron transfer pathways from Mo-BiVO4 to Ag NPs and from Au NPs to Mo-BiVO4, which in the case of co-decorated (Ag,Au)/Mo-BiVO4 PCs may lead to cascade charge transfer in the corresponding heterojunctions. The former was found to intensify TC degradation for Ag/Mo-BiVO4, while the latter underlined the enhanced IBU degradation via photogenerated holes for the (Ag,Au)/Mo-BiVO4 PCs. Judicious combination of VLA semiconductor inverse opals and metallic NPs with favourable band alignment and PBG engineering is accordingly proposed as a versatile route for the rational design of efficient plasmonic–photonic photoelectrocatalysts.Author contributions
Martha Pylarinou performed experimental investigations, formal analysis, methodology and writing of the original draft. Elias Sakellis and Polychronis Tsipas contributed to the experimental investigations and formal analyses. Spiros Gardelis, Vassilis Psycharis, Athanasios Dimoulas and Thomas Stergiopoulos contributed to the experimental investigations, resources and results validation. Vlassis Likodimos contributed to the conceptualization, methodology, supervision, resources, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research work was supported by the Hellenic Foundation for Research and Innovation (H.F.R.I.) under the “First Call for H.F.R.I. Research Projects to support Faculty members and Researchers and the procurement of high-cost research equipment grant” (Project Number: 543).References
- H. A. Atwater and A. Polman, Nat. Mater., 2010, 9, 205–213 CrossRef CAS PubMed.
- L. Mascaretti, A. Dutta, Š. Kment, V. M. Shalaev, A. Boltasseva, R. Zbořil and A. Naldoni, Adv. Mater., 2019, 31, 1–23 Search PubMed.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS PubMed.
- N. Wu, Nanoscale, 2018, 10, 2679–2696 RSC.
- C. Clavero, Nat. Photonics, 2014, 8, 95–103 CrossRef CAS.
- A. Furube and S. Hashimoto, NPG Asia Mater., 2017, 9, e454 CrossRef CAS.
- Y. Zhang, S. He, W. Guo, Y. Hu, J. Huang, J. R. Mulcahy and W. D. Wei, Chem. Rev., 2018, 118, 2927–2954 CrossRef CAS PubMed.
- J. Li, S. K. Cushing, F. Meng, T. R. Senty, A. D. Bristow and N. Wu, Nat. Photonics, 2015, 9, 601–607 CrossRef CAS.
- D. C. Ratchford, A. D. Dunkelberger, I. Vurgaftman, J. C. Owrutsky and P. E. Pehrsson, Nano Lett., 2017, 17, 6047–6055 CrossRef CAS PubMed.
- Y. Shiraishi, N. Yasumoto, J. Imai, H. Sakamoto, S. Tanaka, S. Ichikawa, B. Ohtani and T. Hirai, Nanoscale, 2017, 9, 8349–8361 RSC.
- J. W. Ha, T. P. A. Ruberu, R. Han, B. Dong, J. Vela and N. Fang, J. Am. Chem. Soc., 2014, 136, 1398–1408 CrossRef CAS PubMed.
- X. Yu, A. Shavel, X. An, Z. Luo, M. Ibanez and A. Cabot, J. Am. Chem. Soc., 2014, 136, 9236–9239 CrossRef CAS PubMed.
- S. K. Dutta, S. K. Mehetor and N. Pradhan, J. Phys. Chem. Lett., 2015, 6, 936–944 CrossRef CAS PubMed.
- J. K. Kim, X. Shi, M. J. Jeong, J. Park, H. S. Han, S. H. Kim, Y. Guo, T. F. Heinz, S. Fan, C.-L. Lee, J. H. Park and X. Zheng, Adv. Energy Mater., 2018, 8, 1701765 CrossRef.
- J. I. Chen, G. von Freymann, S. Y. Choi, V. Kitaev and G. A. Ozin, Adv. Mater., 2006, 18, 1915–1919 CrossRef CAS.
- J. I. L. Chen, E. Loso, N. Ebrahim and G. A. Ozin, J. Am. Chem. Soc., 2008, 130, 5420–5421 CrossRef CAS PubMed.
- G. von Freymann, V. Kitaev, B. V. Lotschz and G. A. Ozin, Chem. Soc. Rev., 2013, 42, 2528–2554 RSC.
- V. Likodimos, Appl. Catal., B, 2018, 230, 269–303 CrossRef CAS.
- K. R. Phillips, G. T. England, S. Sunny, E. Shirman, T. Shirman, N. Vogel and J. Aizenberg, Chem. Soc. Rev., 2016, 45, 281–322 RSC.
- J. Wang, P. W. H. Pinkse, L. I. Segerink and J. C. T. Eijkel, ACS Nano, 2021, 15, 9299–9327 CrossRef CAS PubMed.
- Y. Lu, H. Yu, S. Chen, X. Quan and H. Zhao, Environ. Sci. Technol., 2012, 46, 1724–1730 CrossRef CAS PubMed.
- Z. Zhang, L. Zhang, M. N. Hedhili, H. Zhang and P. Wang, Nano Lett., 2013, 13, 14–20 CrossRef CAS PubMed.
- X. Zhang, Y. Liu, S.-T. Lee, S. Yang and Z. Kang, Energy Environ. Sci., 2014, 7, 1409–1419 RSC.
- F. Temerov, K. Pham, P. Juuti, J. M. Mäkelä, E. V. Grachova, S. Kumar, S. Eslava and J. J. Saarinen, ACS Appl. Mater. Interfaces, 2020, 12, 41200–41210 CrossRef CAS PubMed.
- T. Raja-Mogan, A. Lehoux, M. Takashima, E. Kowalska and B. Ohtani, Chem. Lett., 2021, 50, 711–713 CrossRef CAS.
- T. Raja-Mogan, B. Ohtani and E. Kowalska, Catalysts, 2020, 10, 827 CrossRef CAS.
- J. H. Kim and J. S. Lee, Adv. Mater., 2019, 31, 1806938 CrossRef PubMed.
- L. W. Zhang, C. Y. Lin, V. K. Valev, E. Reisner, U. Steiner and J. J. Baumberg, Small, 2014, 10, 3970–3978 CrossRef CAS PubMed.
- L. Fang, F. Nan, Y. Yang and D. Cao, Appl. Phys. Lett., 2016, 108, 093902 CrossRef.
- K. M. Ji, H. Arandiyan, P. Liu, L. Zhang, J. H. Han, Y. C. Xue, J. G. Hou and H. X. Dai, Nano Energy, 2016, 27, 515–525 CrossRef CAS.
- S. Zhou, R. Tang, L. Zhang and L. Yin, Electrochim. Acta, 2017, 248, 593–602 CrossRef CAS.
- H. Zhao, C.-F. Li, Z.-Y. Hu, J. Liu, Y. Li, J. Hu, G. Van Tendeloo, L.-H. Chen and B.-L. Su, J. Colloid Interface Sci., 2021, 604, 131–140 CrossRef CAS PubMed.
- G. Collins, A. Lonergan, D. McNulty, C. Glynn, D. Buckley, C. Hu and C. O'Dwyer, Adv. Mater. Interfaces, 2020, 7, 1901805 CrossRef CAS.
- S. Wu and Y. Hu, Chem. Eng. J., 2021, 409, 127739 CrossRef CAS.
- J.-y. Lin, Y. Zhang, Y. Bian, Y.-x. Zhang, R.-z. Du, M. Li, Y. Tan and X.-s. Feng, Sci. Total Environ., 2023, 904, 166897 CrossRef CAS PubMed.
- N. Jallouli, L. M. Pastrana-Martínez, A. R. Ribeiro, N. F. F. Moreira, J. L. Faria, O. Hentati, A. M. T. Silva and M. Ksibi, Chem. Eng. J., 2018, 334, 976–984 CrossRef CAS.
- K. R. Davies, M. G. Allan, S. Nagarajan, R. Townsend, T. Dunlop, J. D. McGettrick, V. S. Asokan, S. Ananthraj, T. Watson, A. R. Godfrey, J. R. Durrant, M. M. MarotoValer, M. F. Kuehnel and S. Pitchaimuthu, J. Environ. Chem. Eng., 2023, 11, 110256 CrossRef CAS.
- Q. Yan, Z. Zhou and X. S. Zhao, Langmuir, 2005, 21, 3158–3164 CrossRef CAS PubMed.
- M. Pylarinou, E. Sakellis, P. Tsipas, G. Em. Romanos, S. Gardelis, A. Dimoulas and V. Likodimos, ACS Appl. Nano Mater., 2023, 6, 6759–6771 CrossRef CAS.
- J. Pellicer-Porres, D. Vázquez-Socorro, S. López-Moreno, A. Muñoz, P. Rodríguez-Hernández, D. Martínez-García, S. N. Achary, A. J. E. Rettie and C. B. Mullins, Phys. Rev. B, 2018, 98, 214109 CrossRef CAS.
- F. D. Hardcastle, I. E. Wachs, H. Eckert and D. A. Jefferson, J. Solid State Chem., 1991, 90, 194–210 CrossRef CAS.
- W. Zhang, F. Wu, J. Li, D. Yan, J. Tao, Y. Ping and M. Liu, ACS Energy Lett., 2018, 3, 2232–2239 CrossRef CAS.
- D. Zhou, L.-X. Pang, J. Guo, H. Wang, X. Yao and C. Randall, Inorg. Chem., 2011, 50, 12733–12738 CrossRef CAS PubMed.
- F. F. D. Hardcastle and I. E. Wachs, J. Phys. Chem., 1991, 95, 5031–5041 CrossRef CAS.
- Y. Hermans, A. Klein, K. Ellmer, R. van de Krol, T. Toupance and W. Jaegermann, J. Phys. Chem. C, 2018, 122, 20861–20870 CrossRef CAS.
- Y. X. Liu, H. X. Dai, J. G. Deng, L. Zhang and C. T. Au, Nanoscale, 2012, 4, 2317–2325 RSC.
- J. Ginter and I. Piwoński, Mater. Res. Bull., 2018, 107, 100–110 CrossRef CAS.
- T. G. U. Ghobadi, A. Ghobadi, M. C. Soydan, M. B. Vishlaghi, S. Kaya, F. Karadas and E. Ozbay, ChemSusChem, 2020, 13, 2577–2588 CrossRef CAS PubMed.
- H. Wu, R. Irani, K. Zhang, L. Jing, H. Dai, H. Y. Chung, F. F. Abdi and Y. H. Ng, ACS Energy Lett., 2021, 6, 3400–3407 CrossRef CAS.
- N. Stefanou and A. Modinos, J. Phys.: Condens. Matter, 1991, 3, 8149–8157 CrossRef CAS.
- M. Valenti, E. Kontoleta, I. A. Digdaya, M. P. Jonsson, G. Biskos, A. Schmidt-Ott and W. A. Smith, ChemNanoMat, 2016, 2, 739–747 CrossRef CAS.
- B. O. Orimolade and O. A. Arotiba, J. Electroanal. Chem., 2020, 878, 114724 CrossRef CAS.
- X. Zhang, Y. Ma, L. Xi, G. Zhu, X. Li, D. Shi and J. Fan, Sci. Total Environ., 2019, 647, 245–254 CrossRef CAS PubMed.
- M. Jiménez-Salcedo, M. Monge and M. T. Tena, Materials, 2021, 14, 3912 CrossRef PubMed.
- K. R. Phillips, T. Shirman, E. Shirman, A. V. Shneidman, T. M. Kay and J. Aizenberg, Adv. Mater., 2018, 30, 1706329 CrossRef PubMed.
- L. M. Otto, E. A. Gaulding, C. T. Chen, T. R. Kuykendall, A. T. Hammack, F. M. Toma, D. F. Ogletree, S. Aloni, B. J. H. Stadler and A. M. Schwartzberg, Sci. Rep., 2021, 11, 7656 CrossRef CAS PubMed.
- H. N. Q. Tran, K. N. Tran, S. Gunenthiran, J. Wang, C. S. Law, S. Y. Lim, Y. C. G. Lim, A. D. Abell, L. F. Marsal and A. Santos, ACS Appl. Mater. Interfaces, 2024, 16, 11787–11799 CrossRef CAS PubMed.
- L. Zhang, E. Reisner and J. J. Baumberg, Energy Environ. Sci., 2014, 7, 1402–1408 RSC.
- Y. Liang, T. Tsubota, L. P. A. Mooij and R. van de Krol, J. Phys. Chem. C, 2011, 115, 17594–17598 CrossRef CAS.
- L. Zhang, L. O. Herrmann and J. J. Baumberg, Sci. Rep., 2015, 5, 16660 CrossRef CAS PubMed.
- Z. Zhang and J. T. Yates Jr., Chem. Rev., 2012, 112, 5520–5551 CrossRef CAS PubMed.
- A. Meng, L. Zhang, B. Cheng and J. Yu, Adv. Mater., 2019, 31, 1807660 CrossRef PubMed.
- A. Derkachova, K. Kolwas and I. Demchenko, Plasmonics, 2016, 11, 941–951 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details on materials characterization and photoelectrochemical performance. Spectral deconvolution of Raman spectra for single-, Ag-, Au- and (Ag,Au)-modified Mo BiVO4 PC films. Bi, V, O, Mo, and Ag elemental EDX maps for Ag/Mo-BiVO4 PCs. Bi, V, O, Mo, and Au elemental EDX maps for Au/Mo-BiVO4 PCs. Specular (R%), diffuse (DR%) and Tauc plots for the Ag,Au-modified Mo-BiVO4 PCs. TC and IBU photodegradation kinetics for the Ag,Au-modified Mo-BiVO4 PC photoelectrodes at +1.0 V vs. Ag/AgCl under visible light. TC and IBU photoelectrocatalytic degradation kinetics for Mo-BiVO4, Ag/Mo-BiVO4 and (Ag,Au)/Mo-BiVO4 PCs in the presence of scavengers. TC photodegradation kinetics and reaction rates for the Ag- and Au-modified Mo-BiVO4 as a function of PC macropore diameter. TC photodegradation kinetics and reaction rates for thick and thin Ag- and Au-modified Mo-BiVO4 PCs. Current density–potential curves for Mo-BiVO4 PCs and planar films under front and back side light illumination. EIS Nyquist plots for the Ag and Au-modified Mo-BiVO4 PCs in the dark. TC photodegradation kinetics for Au/Mo-BiVO4 PC340 photoelectrodes with Au NPs of different diameters. TEM images and EDX elemental maps of 10, 50 and80 nm Au/Mo-BiVO4 PC340 films. See DOI: https://doi.org/10.1039/d3nr06407g |
| This journal is © The Royal Society of Chemistry 2024 |