
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStrategies to alleviate distortive phase transformations in Li-ion intercalation reactions: an example with vanadium pentoxide
Muhammad Waseem
Yaseen
 a,
Manju P.
Maman
a,
Shashank
Mishra
a,
Ibrahim
Mohammad
b and
Xuefei
Li
*a
a,
Manju P.
Maman
a,
Shashank
Mishra
a,
Ibrahim
Mohammad
b and
Xuefei
Li
*a
aDepartment of Chemistry, Georgia State University, Atlanta, Georgia 30302, USA. E-mail: xli81@gsu.edu
bChattahoochee High School, Johns Creek, Georgia 30022, USA
First published on 29th April 2024
Abstract
Chemical and electrochemical Li-ion insertion in transition metal oxides, either via a phase transformation reaction (ions insert into specific crystallographic sites of the host lattice) or a solid solution insertion (ions distribute uniformly throughout the host lattice), enables high energy density electrochemical energy storage. Many phase transformation cathode materials, that undergo two-phase reactions, exhibit high theoretical capacities arising from multi-electron redox reactions. However, challenges in distortive phase transformations and uncontrolled phase nucleation, propagation, segregation, and co-existence continue to limit the energy density, (dis)charging rate performances, and cycling stability of available phase transformation cathode materials. Vanadium pentoxide (V2O5), a classical layered intercalation host material with high theoretical capacity, undergoes irreversible structural changes and capacity fading when intercalating more than one lithium ion per V2O5 unit in its thermodynamically stable phase. Here, we review recent synthetic strategies to alter the V–O connectivity, thereby alleviating distortive phase transformations and promoting solid solution-based Li-ion insertion in V2O5. We also summarize several widely accessible and classical molecular-based analytical tools that can provide local structural dynamics and phase transformation mechanism information on the lithiation of V2O5, including single-crystal X-ray diffraction, infrared and Raman spectroscopy, electron paramagnetic resonance, and nuclear magnetic resonance spectroscopy.
 Muhammad Waseem Yaseen | Muhammad Waseem Yaseen is currently a research graduate student at Georgia State University. He obtained his M.S. from the National University of Sciences and Technology, Islamabad. His current research focuses on the development of new ion storage materials for electrochromic devices and batteries. |
 Manju P Maman | Manju. P. Maman is currently a post-doctoral fellow at Georgia State University. She obtained her Ph.D. from the Indian Institutes of Science Education and Research at Thiruvananthapuram (IISER-TVM). Her research focuses on the synthesis and characterization of atomically precise nanoclusters. |
 Shashank Mishra | Shashank Mishra is currently a research graduate student at Georgia State University. Shashank obtained his M.S. from the Indian Institute of Technology Delhi (IIT Delhi) and he currently focuses on synthesizing transition metal oxides and chalcogenide nanomaterials for electrochemical applications. |
 Ibrahim Mohammad | Ibrahim Mohammad is currently a senior student at Chattahoochee High School. Ibrahim was a recipient of National Honors for US Chemistry Olympiad in 2023 and joined Dr Li's lab in May 2023, where he worked on the synthesis and electrochemical characterization of transition metal oxides and sulfides. |
 Xuefei Li | Prof. Xuefei Li has been an Assistant Professor in Chemistry at Georgia State University since August 2022. She obtained her Ph.D. in Chemistry from the Pennsylvania State University in 2019 with Prof. Ray Schaak and did her postdoc work with Prof. Jianguo Mei at Purdue. Her research group focuses on the synthesis and characterization of nanomaterials for electrochemical applications, covering materials systems including atomically precise nanoclusters, transition metal oxides, and chalcogenides, with application interests in electrochemical energy storage, conversion, and ion enrichment. She received the ACS PRF Doctoral New Investigator Grant in 2023 and is a Scialog fellow for Sustainable Minerals, Metals, and Materials in 2024. |
1 Introduction
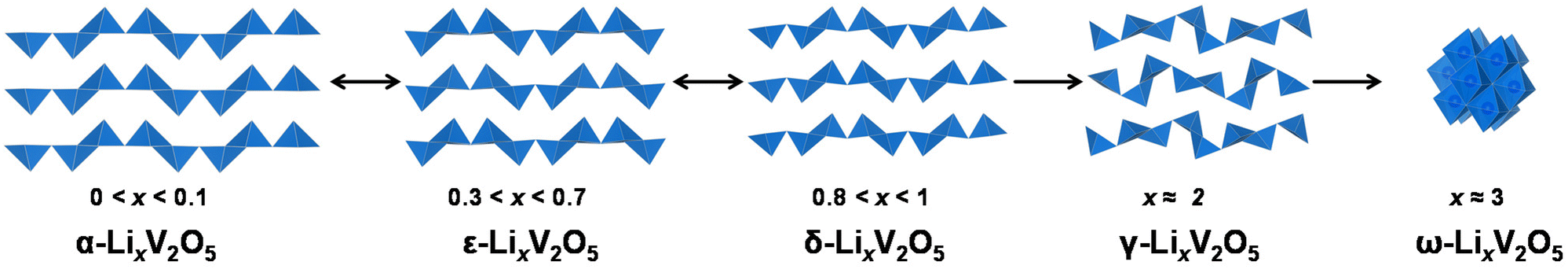
Vanadium pentoxide (V2O5), a highly oxidizing layered oxide, reacts readily with lithium through multi-electron redox reactions and accommodates Li ions within its interlayer spacings, making it a promising cathode material for Li-ion batteries with high voltage and high theoretical capacity.1–8 The theoretical capacity for full lithiation of V2O5 is 442 mA h g−1 and the practical capacity without distortive phase transformation is ∼147 mA h g−1 at about one Li ion per V2O5, which is comparable to commercialized lithium iron phosphate (LiFePO4) and nickel manganese cobalt (LiNixMnyCozO2, x+y+z=1) type Li-ion battery cathode materials.9,10 Commercially, V2O5 is utilized as a cathode material in small-size Li-ion batteries.11 Additionally, V2O5 exhibits multi-electrochromic color switching and in the thin film form, it is a good candidate material for charge-balancing counter electrodes in electrochromic windows.3,12–15 An understanding in extending the reversible ion insertion region underpins the design of reversible energy storage materials with higher capacity and better rate performances.Thermodynamically stable orthorhombic α-V2O5 consists of two-dimensional single-polyhedron thick layers at an interlayer distance of 4.38 Å.16 In each polyhedron (VO5 square pyramid), the vanadium atom forms a short vanadyl bond with an apical oxygen (V–OA at 1.54 Å), a V–O bond with doubly coordinated oxygen between two corner-shared pyramids (bridge V–OB–V at 1.77 Å), and three V–O bonds with three triply coordinated edge-shared oxygen (3V–OC at 1.88, 1.88, and 2.02 Å).17 Edge- and corner-shared VO5 square pyramids within each layer form “up–up–down–down” zig-zag chains and the sixth V⋯O weak electrostatic interaction between layers allows interlayer intercalation of ions and small molecules (Fig. 1 and 8).18
α-V2O5 has an open circuit potential of 3.7 V vs. Li/Li+ and can readily react with lithium via xLi+ + xe− + V2O5 = LixV2O5 (0 < x < 3). The electrode capacity, in practice, depends on the structural reversibility, which in turn is limited by the extent of lithiation, x, in LixV2O5.19 As shown in Fig. 1, pristine α-V2O5 transforms to α-LixV2O5 at x < 0.1 with only increased interlayer spacing and then to ε-LixV2O5 at 0.3 < x < 0.7 with increased interlayer spacing and puckering. A δ-LixV2O5 phase is formed when Li intercalation approaches x ≈ 0.8–1 at around 3.1–3.2 V vs. Li/Li+. This first-order transition proceeds by gliding of alternating layers for half a unit cell along the b-axis and can result in hysteresis in charge and discharge profiles. At a higher Li concentration (x > 1), the layered structures undergo irreversible distortive phase transformation to γ- and ω-LixV2O5 phases, which have prohibited the full theoretical capacity of V2O5 from being realistically achieved (∼442 mA h g−1 for three Li ions). δ-LixV2O5 structurally rearranges to distinctly different γ-LixV2O5 phases at around 2.3–2.4 V vs. Li/Li+. Instead of the “up–up–down–down” arrangement of VO5 square pyramids in the α-, ε-, and δ-phases, the VO5 square pyramids in γ-LixV2O5 are arranged in a highly puckered “up–down–up–down” manner. Once the γ-LixV2O5 phase forms, complete delithiation of γ-LixV2O5 no longer restores the original layered α-V2O5. Instead, metastable γ′-V2O5 forms, with retained “up–down–up–down” structural motifs. At deeper discharging potentials of around 2.0–2.1 V vs. Li/Li+, a highly disordered rock-salt ω-LixV2O5 forms, typically with a significant loss of long-range ordering.20–24
 | ||
| Fig. 1 Lithiation-induced phase transformation in thermodynamically stable bulk α-V2O5, the lithium atoms are omitted to highlight the structural transformation of the V2O5 host. | ||
At a higher Li-ion concentration (x > 1 per LixV2O5), while abovementioned distortive structural transformations effectively accommodate increasing Li-ion flux with minimized cationic repulsions, incommensurate lattices at the boundaries of co-existing phases that have different Li concentrations present challenges in Li ion diffusion and mechanical stability. Phase boundaries within particles and at interfaces also strongly modulate ion diffusion between the two phases and experimental diffusivities can be orders of magnitude lower than theoretical values.25 Lattice mismatch at phase boundaries also induces dislocations and stress build-up, which may propagate and aggregate to cause cracks. Materials that lost electrical contact due to the crack become electrochemically inactive, accelerating capacity fading. In addition, as the phase transformation requires a first-order reaction with the nucleation of new phases from existing phases, the electrode particle size, geometry, and connectivity strongly influence the spatiotemporal dynamics of lithiation, potentially leading to inconsistent observations of the electrochemical performances due to variations in materials processing and packing during device fabrication.
Despite the problems with distortive phase transformation at higher Li concentrations, certain highly lithiated phases can be topochemically deintercalated to obtain metastable V2O5 polymorphs that have the same composition and theoretical capacity, but different atomic connectivities and Li-ion insertion pathways. γ-LixV2O5 with a highly puckered “up–down–up–down” arrangement of VO5 square pyramids can reversibly cycle 1.5 Li ions per V2O5via solid solution-based Li-ion insertion (0 < x < 1.5).26 The disordered rock-salt ω-Li3V2O5 can reversibly cycle through a solid solution mechanism without phase transformation.27 A deeply discharged disordered rock-salt Li3+xV2O5 (0 < x < 2) anode material obtained from reducing V2O5 down to 0.01 V vs. Li/Li+ allows fast and reversible insertion of two Li ions with a specific capacity of 266 mA h g−1. The fast Li diffusion through a redistributive lithium insertion mechanism along concerted tetrahedra–octahedra–tetrahedra sites enables over 1000 stable charge–discharge cycles with negligible decay.28
From the solid solution-based lithiation mechanism observed in γ- and ω-LixV2O5, altering atomic connections without changing the V2O5 stoichiometry retains the thermodynamic driving forces; therefore the high voltage and high capacitance of V2O5 remain. However, reconfigured atomic connections can change the kinetics of Li-ion diffusion, which is governed by interlayer and interfacial migration barriers. The change in structures potentially induces a solid solution-based lithiation mechanism instead of phase transformation, thereby enhancing ion diffusion kinetics and cycling reversibility. We review the synthetic strategies for structures that retain the high voltage and high capacity of V2O5, but with an altered atomic connectivity, increased Li-ion diffusivity, and structural, electrochemical, and mechanical stability (Fig. 2). We also discuss several less-explored but commonly accessible molecular-based analytical tools that play unique roles and can provide dynamics of structural transformation during (de)lithiation.29–31 Such tools are complementary to synchrotron-based X-ray techniques that provide atomistic information on lattice transformation with high temporal or spatiotemporal resolution, but are not universally available.32,33 Widely accessible molecular-based tools like single-crystal X-ray diffraction (XRD), infrared (IR) spectroscopy, Raman spectroscopy, electron paramagnetic resonance (EPR), and nuclear magnetic resonance (NMR) spectroscopy are highlighted for their potential applications in differentiating phase transformation mechanisms during (de)intercalation.34–38 Single-crystal XRD, with its precise determination of Li atom positions for millimetre scale single crystals, identifies Li positions for differently lithiated V2O5 in addition to lattice transformation and captures diffusion pathways.16,36 IR spectroscopy, sensitive to V–O bond stretching and bending, implicates lithiation-induced structural changes in V2O5, while Raman spectroscopy reveals local disorder and ion coordination dynamics during electrochemical reactions.38,39 EPR that measures unpaired electrons (V4+, 3d1) distinguishes several crystalline phases during lithiation with high sensitivity.37 NMR spectroscopy that employs isotopes including 7Li and 51V offers valuable information on local environments such as the Li environment and hydration sites.34,40,41 The collaborative use of these techniques holds immense potential for advancing the design and optimization of V2O5 materials for electrochemical energy storage applications.
 | ||
| Fig. 2 Structural modification strategies that alleviate distortive phase transformations in α-V2O5. | ||
2 Strategies
2.1 Theoretical considerations
The thermodynamically stable α-V2O5 exhibits an interlayer distance of 4.38 Å, and the diffusion of Li ions in bulk α-V2O5 is sluggish at around 10−12 cm s−1. At a higher Li-ion concentration of x > 1 for LixV2O5, the Li-ion diffusion through the framework and the diffusion coefficient decreases to 10−15 cm s−1.24 Additionally, high local coordination of the Li ions and polaron–ion interactions further increase the migration barriers and impede Li-ion diffusion.36,42,43 Due to sluggish diffusion, even in the reversible lithiation region of 0 < x < 1, intercalation of lithium into bulk α-V2O5 often results in the co-existence of multiple lithiated phases.20 The phase coexistence further slows down the overall lithium diffusion.Li-ion migration energetics and diffusion pathways in various lithiated phases of V2O5 have been systematically calculated using the Density Functional Theory with Nudged Elastic Band (DFT NEB) method at the atomistic scale. The Li-ion diffusion coefficient shows an exponential dependence on the activation energy barrier, a 0.06 eV change in the migration barrier correlates with a one-order-of-magnitude change in the diffusion rate. In α-V2O5, Li ions encounter a 0.17 eV energy barrier per unit cell while diffusing between cubooctahedral coordination environments. The transformation to the ε-LixV2O5 phase results in a migration barrier of 0.29 eV, attributed to the further constriction of the trigonal site where ions reside. In the δ-phase, Li ions encounter a migration barrier of 0.20 eV along a distinctive diffusion pathway. The γ-phase displays the lowest migration barrier at 0.13 eV, attributed to the presence of a relatively spacious square-planar transition state between tetrahedral Li-ion sites.25 Between different V2O5 metastable phases, NEB calculations show that lithium ions exhibit suitable migration barriers (<0.525 eV for micrometer-sized particles, <0.650 eV for nanometer-sized particles) in multiple phases of V2O5 with minimal differences due to the limited ability of Li ions to polarize the anion framework. Conversely, larger cations, such as sodium, magnesium, and aluminum, display substantial variations in migration barriers across different metastable phases of V2O5, arising from their better ability to polarize the anion framework.25
As DFT NEB is an atomistic method without consideration of defects and phase boundaries, the roles of electrode particle size, geometry, grain boundaries, and stacking pressure during cell assembly further lead to a deviation of experimentally measured macroscopic ion diffusivity with theoretically predicted results. Nevertheless, the calculation of Li-ion diffusion and migration energetics in different diffusion pathways helps in rationalizing and designing materials with optimized lattice-ion interaction, facilitating fast and solid solution-based lithiation. The design rules include decreasing Li-ion coordination with lattice oxygens, reducing lattice polarization, and expanding the interlayer distance, which will be introduced in the following sections.20,25
2.2 Nanosizing
Nanosized α-V2O5 enhances the accessible electrode volume for reversible ion storage, reduces lithiation heterogeneity, and improves electrochemical reversibility.44–46 Crystallite size and geometry alter the local electrochemical potential at surfaces and energy penalty for phase boundary formation within the particle and at interfaces. Meanwhile, nanostructures with a high surface-to-volume ratio offer improved contact with the electrolyte, shorter Li-ion diffusion paths, and efficient strain relaxation.25Early work by Chan and Cui et al. reported fast and completely reversible Li insertion in V2O5 nanoribbons from the deeply discharged, disordered rock salt ω-LixV2O5 back to the original single-crystalline α-V2O5 without the formation of phase boundaries. Meanwhile, the bulk deeply discharged α-V2O5 retains the disordered rock-salt structure during delithiation and cannot fully reverse back to the original α-V2O5, but instead remains in the ω-V2O5. The authors also observed that the Li diffusion in nanoribbons surpasses that of the bulk material by three orders of magnitude, requiring less time for complete lithiation.24 Luo and Banerjee et al. systematically compared the phase transformation dynamics in bulk and nanoscale α-V2O5 under otherwise identical packing and device assembly methods. The irregularly shaped α-V2O5 microparticles and α-V2O5 nanospheres (∼50 nm) were investigated within a voltage range of 2.8 V–4.0 V vs. Li/Li+ (Fig. 3). The α-V2O5 microparticle shows severe phase coexistence during lithiation, where δ-LiV2O5 co-exists with ε- and ε′-LixV2O5 at around 2.8 V vs. Li/Li+. In contrast, ∼50 nm α-V2O5 is stabilized by the metastable ε′-LixV2O5 that is structurally similar to ε-LixV2O5 but accommodates Li flux through solid solution-based Li re-ordering with minimized structural rearrangement and less mechanical stress build-up. Because of the suppressed phase transformation and controlled coexistence through the metastable ε′ phase, the nanosphere V2O5 exhibits improved Li-ion diffusivity, substantially decreased charge-discharge hysteresis, and enhanced capacity retention.47
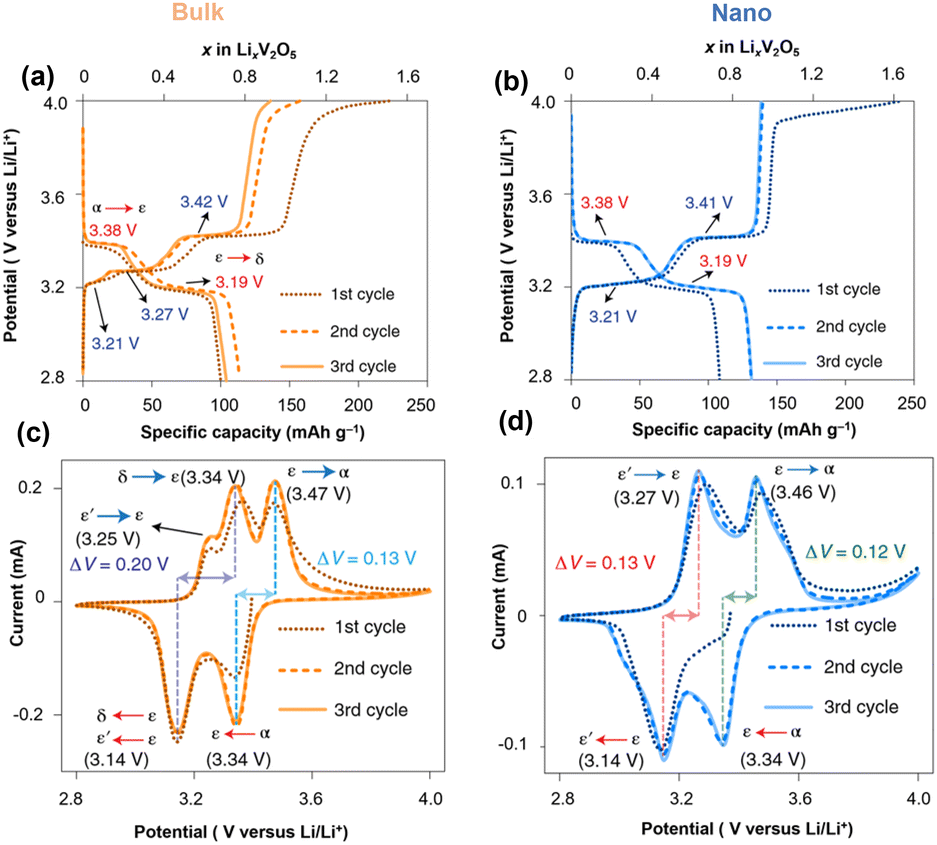 | ||
| Fig. 3 Charging/discharging profile and cyclic voltammetry data of (a and c) α-V2O5 microparticle and (b and d) α-V2O5 nanosphere. Reproduced from ref. 47 with permission from Springer Nature. Copyright 2021. | ||
The change in the lithiation mechanism by the nanosizing effect – from distortive phase transformation to the solid solution mechanism – has been extensively explored in LiFePO4-based cathode materials. In large single crystals at room temperature, there is a miscibility gap between FePO4 and LiFePO4.48 Below a particle size of ∼50–100 nm, the miscibility gap decreases, therefore the single-phase regions increase. As smaller nanoparticle sizes show increased energy penalty for phase boundary formation, phase boundaries cannot be stabilized within a particle and each crystallite below 15–25 nm is a single phase.49 The solid solution lithiation mechanism minimizes restricted ionic diffusion observed in bulk electrode materials in two-phase reactions, ultimately enhancing the rate performance, reversibility, and active material utilization of cathode materials.10
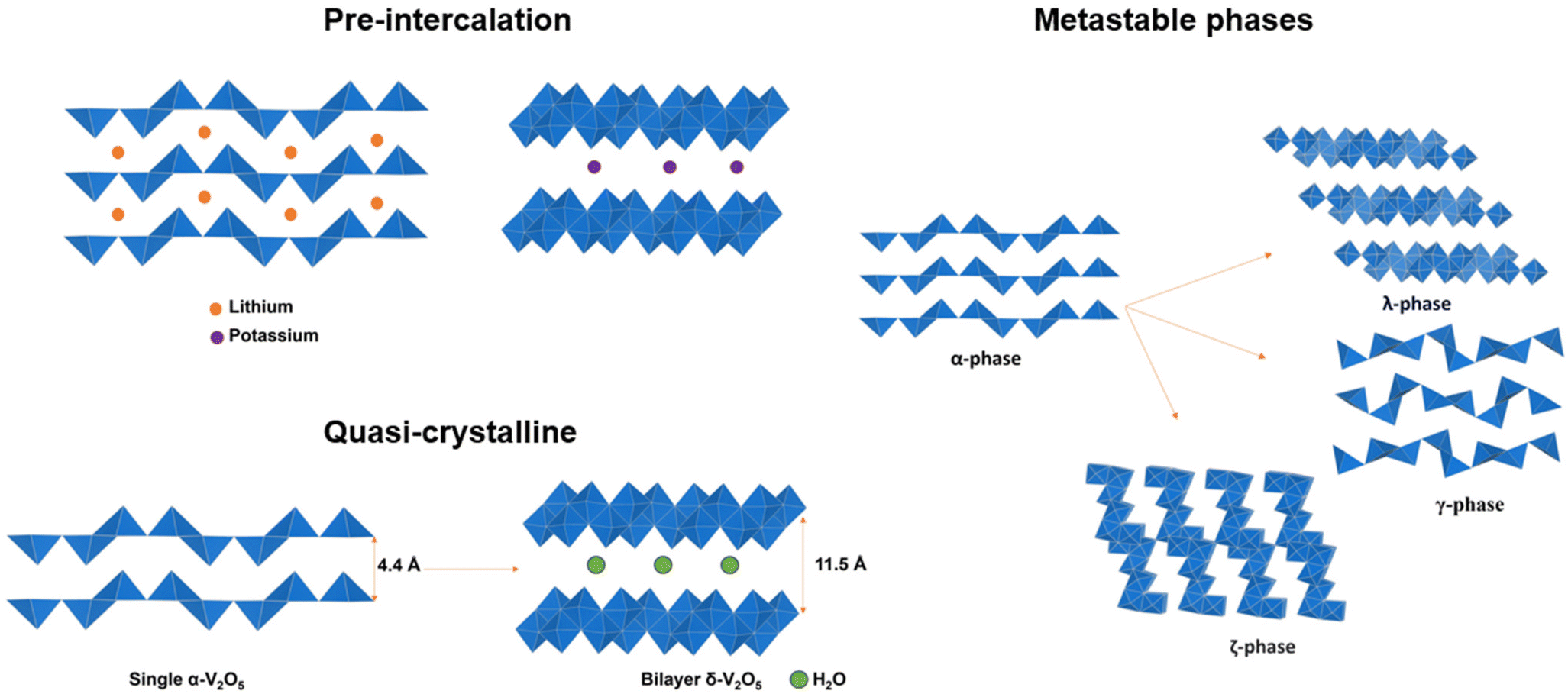
2.3 Pre-intercalation
Distortive phase transformation in α-V2O5 involves the gliding of alternating single-polyhedron thick layers. To circumvent distortive phase transformation, one effective strategy is to expand the interlayer spacing and “hinge” the layers by pre-intercalation of metal ions before the primary intercalation process, thereby stabilizing V2O5 for an extended lithiation region.50–52 Zhong, Huang and Liu et al. demonstrated the pre-intercalation of α-V2O5 through the hydrothermal reaction into lithium-ordered Li0.0625V2O5 nanobelts with superstructures (Fig. 4a). This superstructure consists of eight V2O5 unit cells, with about 1% expansion along the c-axis. The VO5 square pyramids are distorted and compressed, and the pre-intercalated lithium-ions form LiO6 triangular prisms located between the layers of VO5. The pre-intercalation leads to additional pseudocapacitive reactions in a low voltage range of 2.5–3.2 V vs. Li/Li+, and the reversible capacity is significantly increased to 215 mA h g−1 at 1.5 Li ions per V2O5 within the voltage range of 2.5–4 V vs. Li/Li+. By pre-intercalation, the superstructure is stable for 1000 cycles without capacity fade, while pure α-V2O5 cycles 1 Li ion per V2O5 in the same voltage range and lost more than 60% capacity within 100 cycles under the same cycling conditions. In addition to the enhanced cycling stability and increased reversible capacity, the rate performance of Li0.0625V2O5 is also much higher than α-V2O5 with a similar size and geometry.53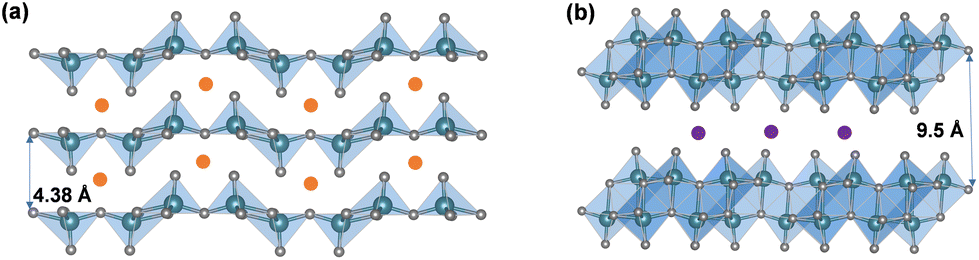 | ||
| Fig. 4 Pre-intercalated α-V2O5 into (a) "hinged" structure and (b) bilayered structures with expanded interlayer distance. Reproduced with adaption from ref. 58 with permission from Elsevier. Copyright 2019. | ||
The pre-intercalation strategy to stabilize α-V2O5 is applicable to ions other than lithium. Pre-intercalation of larger yttrium Y3+ ions form yttrium-ordered Y0.02V2O5 structures, with YO6 octahedra pillars that together with the role of V4+ formed during pre-intercalation, effectively enhanced Na+ diffusion and stabilized the layer structure during discharge and charge cycles.54 The pre-intercalation of Na+ in V2O5 into NaxV2O5 (x = 0.00, 0.005, 0.01, or 0.02) “hinges” the VO5 layers, while providing more space for hosting the lithium ions with lower insertion energy, therefore enhancing the cycling stability and rate capabilities for Li intercalation.55 Li and Cao et al. reported Sn-doped α-V2O5, where introduction of Sn4+ in between VO5 slabs leads to a slightly expanded lattice along the a- and c axes and compression along the b axis. The SnO6 octahedra stabilized the VO5 polyhedron layers and enhanced the capacity and cycling stability of α-V2O5.56 Theoretical calculations reported by Suthirakun et al. rationalized the pre-intercalation effect, where Sn intercalation is energetically preferred than the substitution of vanadium and that the Sn insertion increased the number of charge carriers to improve electronic conductivity, while the lattice distortion introduced diffusion pathways with lower-lying energy levels, thereby facilitating the Li intercalation.57
In addition to the “hinging” effect, pre-intercalation can also lead to expanded bilayered structures. Zhu, Yan, and Jiang et al. demonstrated potassium pre-intercalated double-layered δ-K0.51V2O5 nanobelts with greatly enhanced capacity and faster ion diffusion for K-ion storage than pristine α-V2O5. The double-layered structure with reconstructed two single sheets of VO6 octahedra within each layer, has significantly larger interlayer spacing, 9.5 Å, compared to 4.38 Å in α-V2O5 and the increased interlayer distance mitigated the high diffusion barriers for K ions (Fig. 4b).58 Due to their expanded interlayer distance and optimized growth orientation, the nanobelt structures exhibit extended single-phase reactions regime upon insertion and extraction of K ions. The primary single-phase mechanism was confirmed by ex situ XRD from subtle shifts in peak positions across wide voltage windows. The pre-intercalated δ-K0.51V2O5 exhibits a high and completely reversible specific capacity of 131 mA h g−1 for reversible K-ion insertion, compared to 14 mA h g−1 for α-V2O5.58
The pre-intercalation strategy, where guest metal ions are introduced in between the layers before primary electrochemical (de)intercalation, is distinct from doping, where guest ions can substitute vanadium from the host lattice. Examples of metal doping in introducing “pre-transformation” have also been demonstrated, which suppresses the distortive phase transformation and extends the solid-solution regime of intercalation, thereby enhancing the reversibility of V2O5 lithiation.52
2.4 Metastable structures
Metastable polymorphic V2O5 structures with substantially different atomic connectivities and bonding motifs from the thermodynamically stable α-V2O5 phase provide a comparable theoretical capacity but offer a range of alternative ion diffusion pathways and potentially minimized distortion in phase transformations.42,59–62 Topochemical deintercalation of alkali and transition metal ions, such as Li, Ag, and Cu, from ternary bronze phases can introduce new and otherwise inaccessible phases under mild conditions with preserved key structural features that are distinct from thermodynamically stable V2O5.16,26,63 A well-explored example is the metastable γ′-V2O5 from the chemical or electrochemical deintercalation of the γ′-LiV2O5 bronze phase.26 Such metastable structures present a diverse range of alternative diffusion pathways, along with an abundance of interstitial sites that have relatively shallow potential energies in between layers and channels, which can accommodate Li ions with minimized distortion of the host structure upon (de)intercalation.20,78,79 Reported metastable V2O5 structures with different V-O atomic arrangements include tunnel (ζ-V2O5) and layered (ρ, γ, δ, λ-V2O5) structures, as shown in Table 1.80| Polymorph | Space group | Crystal system | Structure | Synthesis | Interlayer distance (Å) | Diffusion barrier (eV) |
|---|---|---|---|---|---|---|
| α-V2O5 | Pmmn 16 | Orthorhombic16 | 2D-layered16 | Direct chemical16 | 2.75326 4.464 | 0.3473 |
| β-V2O5 | P21/m65 | Tetragonal66 | Layered66 | Direct chemical (high pressure)68 | 7.1269 | 0.4570 |
| δ-V2O5 | Cmcm 71 | Monoclinic67 | 3D-layered71 | Direct chemical (high pressure)72 | 11.5–1364 | 0.07–0.1920(δ′) |
| Orthorhombic71 | ||||||
| ε-V2O5 | C2/m73 | Monoclinic67 | 2D-layered73 | Cu leaching from ε -CuxV2O520,74 | 4.15936 | 0.3773 |
| λ-V2O5 | C2/m75 | Monoclinic36 | 2D-layered36 | Cu leaching from Cu0.85V2O536 | 4.1336 | — |
| γ-V2O5 | Pnma 71 | Orthorhombic67 | 2D-layered26,76 | Leaching from γ-LixV2O526,76 | 5.32526 | 0.1–0.1820 (γ′) |
| Direct chemical synthesis | ||||||
| ζ-V2O5 | C2/m77 | Monoclinic16 | Tunnel16 | Leaching from β-AgxV2O516 | — | 0.2473 |
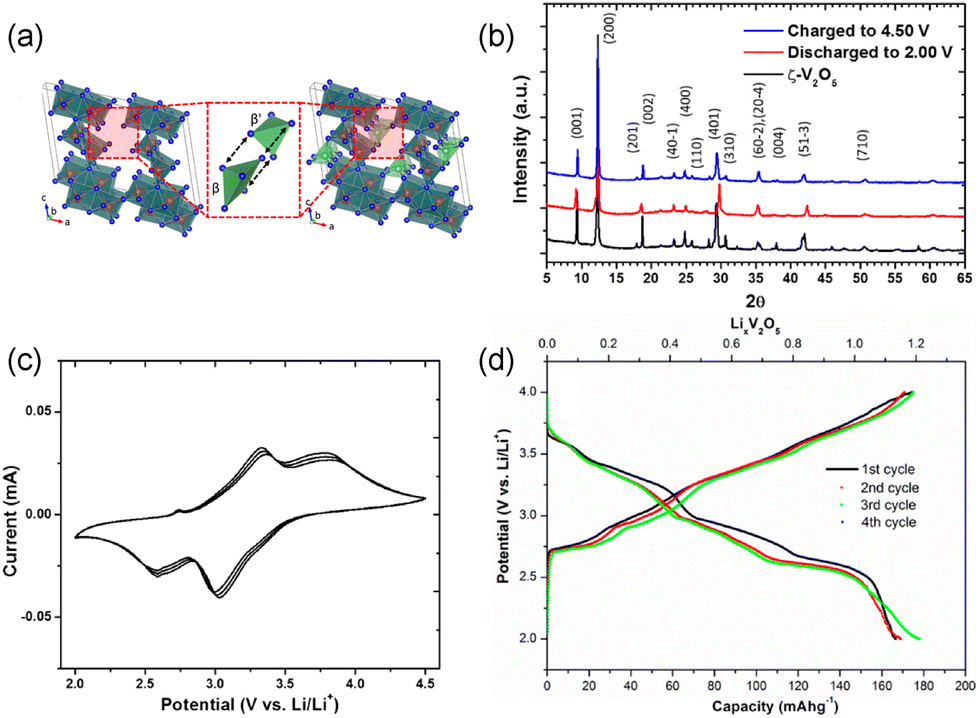 | ||
| Fig. 5 Structural and electrochemical data of metastable ζ-V2O5. (a) crystal structures and (b) XRD patterns of ζ-V2O5 and lithiated ζ-V2O5. (c) cyclic voltammogram and (d) galvanostatic charge-discharge curves of ζ-V2O5. Reproduced with adaptation from ref. 43 with permission from American Chemical Society. Copyright 2017. | ||
At the atomic level, higher ionic diffusion in ζ-V2O5 than α-V2O5 can be attributed to different electronic structures and Li-ion coordinations arising from the reconfigured atomic connectivity.26 Altered V–O bonding, revealed by the oxygen K-edge in the X-ray absorption spectroscopy measurement and electron density calculations, increases bond covalency and facilitates ionic diffusion through the tunnel.43 The degeneracy of vanadium 3d states at the bottom of the conduction band destabilizes polarons and decreases electron localization, thereby improving cation mobility. Moreover, ζ-V2O5 exhibits a less drastic change in Li-ion coordination during ion hopping, 4 → 3 → 5 → 3 → 4, leading to faster ion diffusion compared to α-V2O5, 8 → 3 → 8.16,43,73,82 These structures feature a significantly lower migration barrier than α-V2O5, making ζ-V2O5 a promising candidate host material for not just Li-ion energy storage, but also larger cations such as Na and Mg.
Topochemical deintercalation of two-dimensional bronze phases can also provide new metastable V2O5 with interlayer distances and Li-ion coordination environments that are different than α-V2O5. For instance, the leaching of Cu from ε-Cu0.85V2O5 produced layered λ-V2O5. This λ-V2O5 maintains a preserved V–O connectivity but a shift of layers compared to the original ε-Cu0.85V2O5, and has substantial void space from the original Cu positions between layers that can accommodate Li ions.75,84,85 Chemical lithiation with dilute n-butyllithium transforms the metastable layered λ-V2O5 to λ-LiV2O5, where Li preferentially occupies 5-coordinated sites that are distinct from the 6-coordinated sites originally occupied by Cu, before residing in sites that are originally occupied by Cu at higher lithiation levels.26,36 The metastable λ-V2O5 transforms to a thermodynamically stable phase at 350–400 °C.
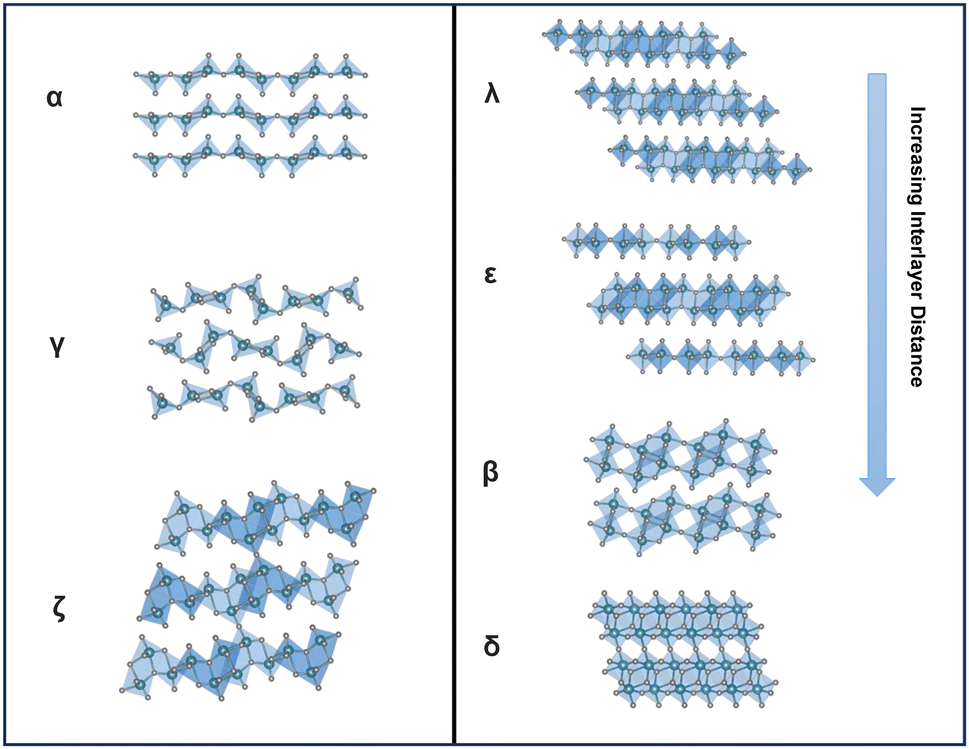 | ||
| Fig. 6 Structural comparison between several metastable structures. | ||
2.5 V2O5·nH2O
Unlike crystalline V2O5 which relies on multi-electron redox processes for its energy storage capacity,90,91 quasi-crystalline hydrated V2O5·nH2O stores potentially more ions through a pseudocapacitive mechanism that involves both redox and capacitive processes. In electrochemical reactions, crystalline V2O5 displays distinct voltammogram peaks and multiple charge–discharge plateaus corresponding to a sequence of redox events along with phase transformation and co-existence, whereas quasi-crystalline V2O5·nH2O shows a featureless and smooth voltammogram and charge–discharge profile (Fig. 7).92–95 Many reports show that, Li-ion intercalation in quasi-crystalline or amorphous V2O5 exhibits higher lithium intercalation capacity ranging from 3.3 to more than 5 Li ions per V2O5.96–100 However, there are reports where the high surface area V2O5·nH2O gel experiences rapid capacity loss during extended cycling due to the accumulation of electrochemically inactive LiOH.101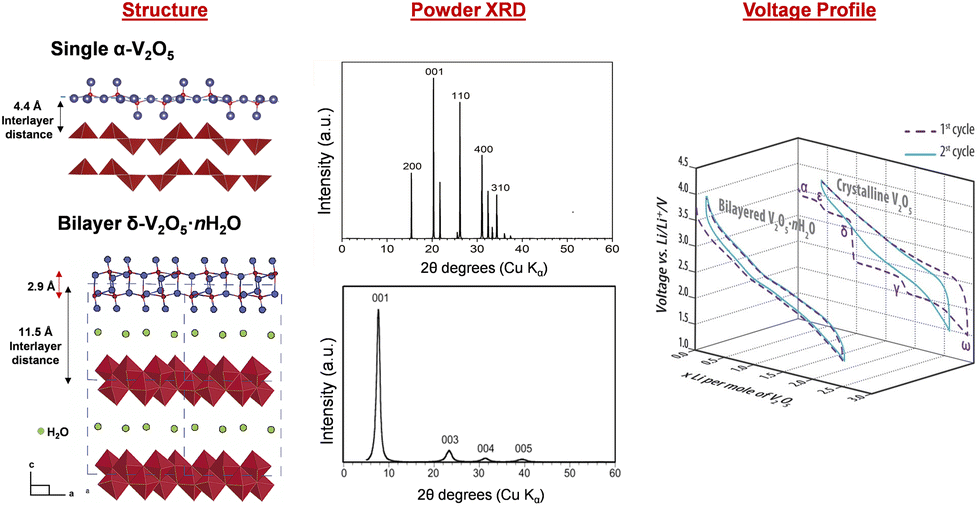 | ||
| Fig. 7 Structural and electrochemical comparison between crystalline V2O5 and bilayered δ-V2O5·nH2O. Reproduced and adapted from ref. 23, 102 and 103, with permission from Elsevier, copyright 1994, John Wiley & Sons, copyright 2016, American Chemical Society, copyright 2002. | ||
The hydrated V2O5·nH2O was primarily identified as amorphous V2O5 in early literature. More recent studies, however, have shown ordering in the quasi-crystalline, bilayer δ-V2O5·nH2O with layers of double polyhedra stacked along the c axis and a large interlayer spacing of 9–13 Å. The removal of interlayer water causes a smaller interlayer spacing. For example, the interlayer spacing is 8.7 Å for V2O5·0.5H2O and 11.6 Å for V2O5·1.6H2O. Broad XRD reflections for the (00l) planes corresponding to the large interlayer spacings facilitate structural identification resolved by the atomic pair distribution function and density functional theory calculation.104–106 The bilayer δ-V2O5·nH2O is synthesized through methods including (1) sol–gel processes, where organometallic precursors such as pervanadate ions condense under controlled pH;107,108 (2) cooling of molten V2O5 (splat cooling);109,110 and (3) electrochemical deposition.111,112 These methods preserve structural water between the layers, which reside in the layers in two forms: intercalated (removable at 120 °C) and tightly bound (removable at above 200 °C), respectively. The structural water coordinates with incoming ions, shielding positive charges and enhancing diffusion.113–115 The bilayer δ-V2O5·nH2O is the precursor of high surface area V2O5 xerogel and aerogel structures, which are produced by freeze drying (xerogel) and supercritical CO2 drying (aerogel) of δ-V2O5·nH2O obtained from sol–gel methods.116
The expanded interlayer distance allows the quasi-crystalline bilayer δ-V2O5·nH2O, or its dried derivative aerogel and xerogel, to accommodate for the intercalation of larger ions such as Na+, K+, Zn2+, and Mg2+ in organic and aqueous electrolytes. Wei, He, and Mai et al. reported reversible Na-ion intercalation in the V2O5·nH2O xerogel with significant interlayer spacing shrinkage and decreased crystallinity upon intercalation, which partially reverses upon deintercalation.116 Tepavcevic, Stamenkovic, Johnson, and Rajh et al. reported ordering upon Na-ion intercalation and disordering upon Na-ion extraction from bilayered V2O5. The different trends in ordering upon intercalation from different quasi-crystalline or amorphous V2O5 systems suggest a potential structural diversity produced from different synthetic methods.117,118 As an example, an aerogel has larger interlayer spacing, higher porosity, and a smaller ion diffusion pathway than a xerogel; therefore it has a significantly higher specific capacity and better reversibility. Despite the possible structural variations of quasi-crystalline or amorphous V2O5, certain trends are consistent with its crystalline counterparts, for example, oxygen defects in amorphous V2O5 demonstrated enhanced performance in aqueous Zn-ion batteries and the same trend has been observed in crystalline V2O5 as well.119,120
3 Characterization by conventional molecular tools
Characterizing different V2O5 polymorphs and probing their in situ phase transformation pathways upon lithiation requires multiple corroborative structural analysis tools. Techniques such as synchrotron-based X-ray diffraction, absorption, and scattering reveal in situ and in operando structural information with high temporal resolution, including phase inhomogeneities, Li gradient, phase co-existence, local structural environments, structural defects, and impurities.92 However, the advanced characterization tools are not universally available in many research institutions and data interpretation is not trivial. Therefore, we highlight several widely available molecular-based tools that are more commonly found in institutional chemistry departments and shared research facilities, including single-crystal X-ray diffraction (XRD), infrared (IR) spectroscopy, Raman spectroscopy, electron paramagnetic resonance (EPR), and nuclear magnetic resonance (NMR) spectroscopy. Each of the techniques has its own set of advantages and limitations, therefore needs to be used corroboratively, as shown in Fig. 8.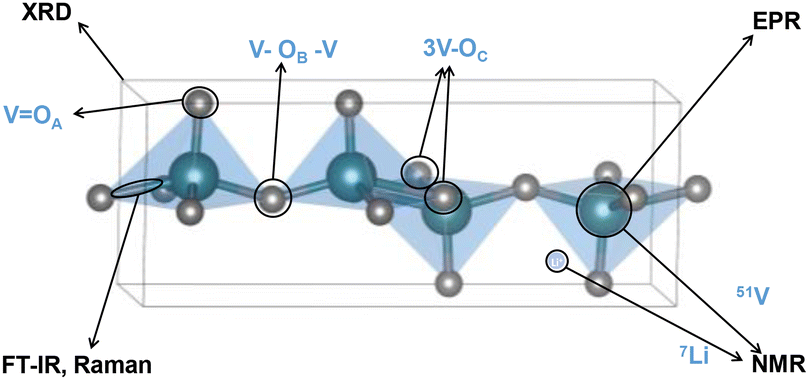 | ||
| Fig. 8 Overview of structural motifs that can be resolved by molecular-based characterization techniques. | ||
3.1 Single-crystal XRD
Operando and series of ex situ powder X-ray diffraction can provide information on the phase transformation pathways of electrode materials at the lattice level.121 However, its ability to directly image the positions of low-Z atoms and resolve their site occupancies, which is crucial for extrapolating Li diffusion pathways, is limited, with exceptions corroborated by neutron diffraction. In contrast, single-crystal X-ray diffraction with a Mo target (λ = 0.71 Å), typically used for identifying molecular single crystals, provides precise determination of low-Z atoms at the Angstrom level by accurately mapping their electron densities. The detailed depiction of Li atomic positions by single-crystal XRD, which captures approximately 1000 times more raw reflections than powder XRD, illustrates diffusion pathways through snapshots at different intercalation concentrations and guides metastable cathode material design.26,36,81,123 For instance, single-crystal XRD was utilized by the Banerjee group to examine millimeter-sized 2D layered single-crystal λ-V2O5 formed from topochemical leaching of ε-Cu0.85V2O5. The single-crystal XRD traced the sites occupied by Li ions in sequence during different stages of chemical lithiation in λ-V2O5, capturing sub-Å resolution positions of Li ions as they diffuse across the 2D host lattice.36,75 Similarly, structural motifs and atomistic diffusion pathways were reconstructed by high-resolution “snapshots” of a chemical lithiation–delithiation cycle for ζ-V2O5 with 1D tunnels. The “snapshot” approach revealed a series of Li-ion accommodation sites, including those originally occupied by Cu atoms, and reconstructed a cation reordering process in tunnel-structured ζ-V2O5 in contrast to phase coexistence in the thermodynamically stable α-V2O5.16,81In addition to single-crystal X-ray diffraction, a few studies combined operando X-ray diffraction, which monitors the change in diffraction patterns, peaks shift or broadening as lattice parameters change during (de)intercalation, and neutron diffraction to locate cation sites.124,125 These work illustrated the structural dynamics of host upon (de)intercalation,47,69,126 and we refer the readers to review papers focusing on the advanced synchrotron techniques.121 Advanced apparatus revealed phase transformations at high temporal or spatiotemporal resolution, such as AMPIX cell (Argonne's multi-purpose in situ X-ray cell) that applies uniform stack pressure for various X-ray scattering and spectroscopic measurements to be performed in operando with high temporal resolution.47,127 Synchrotron-based microscopy methods such as scanning transmission X-ray microscopy (STXM) can collect near-edge fine structure with sub-micron resolution for both chemically (de)lithiated samples and samples from electrochemically (de)lithiated and disassembled coin cells.16 High spatiotemporal resolution can be achieved by operando synchrotron energy dispersive XRD (EDXRD), which can map phase inhomogeneity across the thickness of the electrode in a coin cell during electrochemical cycling.47 Compared with the synchrotron-based measurements, the single-crystal XRD method is uniquely advantaged to identify Li positions and occupancy for different V2O5 phases at varying stages of lithiation, while in operando synchrotron diffraction and absorption spectroscopy do not resolve the Li positions. However, synchrotron-based diffraction resolves the positions of larger cations with higher electron density, such as Mg2+.122,128
3.2 Infrared (IR) spectroscopy
Infrared (IR) spectroscopy is an important method to analyze the structural modifications of lithiated metal oxides in both inorganic glasses and crystalline solids with peak splitting, shifting, and intensity change, and can measure both ex situ and in situ (e.g. a sputtered film in a three-electrode configuration with a thin layer of electrolyte).129 In crystalline V2O5, signals for V–O bond stretching, bending, and the relative motion of structural units can be divided into three distinct regions.38,39,130,131 The bands between 1020 and 950 cm−1 are associated with the stretching mode of the short vanadyl V–OA bond, a structural feature that is generally observed in both crystalline V2O5 and quasi-crystalline V2O5·nH2O gels. The bridging V–OB–V stretching between 700 and 900 cm−1 corresponds to oscillation along the a-axis. The bands below 600 cm−1 correspond primarily to the bridge V–OB–V deformation and 3-fold-coordinated 3V–OC stretching modes.109,132 The 3-fold-coordinated triple bridging 3V–OC modes are typically absent in disordered V2O5, suggesting a disruption in the linkage between edge-shared VO5 square pyramids (Fig. 9).107,130,133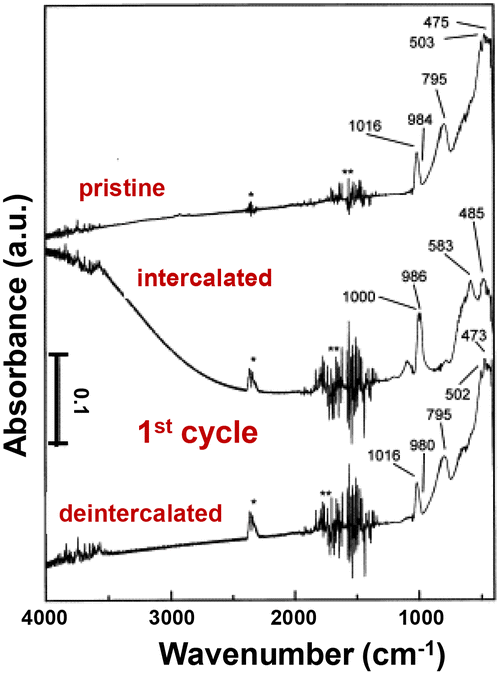 | ||
| Fig. 9 IR absorption spectra of the V2O5 crystalline film at pristine, intercalated, and de-intercalated states. Reproduced with adaption from ref. 129 with permission from Elsevier. Copyright 1999. | ||
During lithiation in the range of 0 < x < 1, polaron absorption from electronic transitions emerges in charged crystalline V2O5, extending from near-infrared to 2000 cm−1. The polaron intensity is correlated with the amount of intercalated Li+ under applied potentials. The V–OA stretching modes shift to 1000 and 986 cm−1, indicating the presence of V4+–O group modes and interactions with lithium ions. The stretching mode for V–OB–V at 795 cm−1 and 3V–OC at 503 cm−1 completely diminished. At more reductive potentials for x > 1, the polaron absorption disappears, concomitant with the loss of long-range ordering during irreversible phase transformations (Fig. 9).129,134
The limitations of the IR spectroscopy technique include the sensitivity to surface species and limited penetration depth. The absence of certain vibrational modes for thin film samples with preferred orientation, and band overlap and broadening upon lithiation further complicates the interpretation of change in structural motifs. In addition, the accurate interpretation of IR modes requires comparison with reference data, which may not be available for certain new metastable phases or lithiation dynamics.39
3.3 Raman spectroscopy
Raman spectroscopy measurements provide an understanding of local disorder, bond length, and ion coordination dynamics during electrochemical reactions. As shown in Fig. 10a, Raman modes of crystalline α-V2O5 can be categorized into three main peak groups: a high-frequency mode at around 1000 cm−1 related to vanadyl V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond (V–OA) stretching, several bands in the 500–1000 cm−1 range for stretching and in the 200–500 cm−1 range for bending, and the low-frequency translation mode at 145 cm−1 for the shear motion and rotations of the ladders between VO5 square pyramids. Disordered and oxygen-deficient V2O5 exhibits broadened Raman peaks with an additional peak at around 932 cm−1, indicating the V4+O double bond and a diminished translational band at 145 cm−1 due to the lack of long-range order (shown in Fig. 10a).135,136
O bond (V–OA) stretching, several bands in the 500–1000 cm−1 range for stretching and in the 200–500 cm−1 range for bending, and the low-frequency translation mode at 145 cm−1 for the shear motion and rotations of the ladders between VO5 square pyramids. Disordered and oxygen-deficient V2O5 exhibits broadened Raman peaks with an additional peak at around 932 cm−1, indicating the V4+O double bond and a diminished translational band at 145 cm−1 due to the lack of long-range order (shown in Fig. 10a).135,136
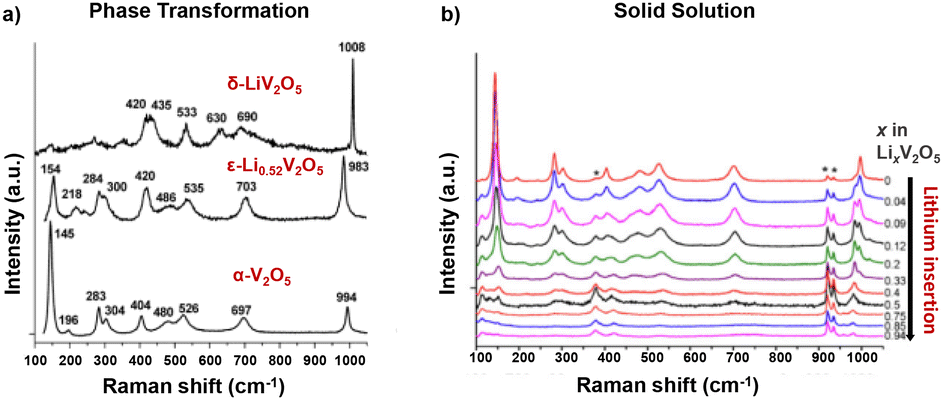 | ||
| Fig. 10 Raman spectra for V2O5 at varying extents of lithiation with (a) the phase transformation in chemically lithiated bulk V2O5 and (b) the solid solution mechanism in V2O5 nano particle thin films. Reproduced with adaption from ref. 137 and 139 with permission from Elsevier, copyright 2009 and American Chemical Society, copyright 2006. | ||
In situ Raman microspectrometry during electrochemical lithiation has been reported to distinguish between the solid solution or phase transformation Li-ion insertion mechanisms, where V2O5 is sputtered onto quartz optical windows with Au wire current collectors for in situ electrochemical reactions. The solid solution mechanism has been observed in nanostructured α-V2O5 thin films, which exhibits good electrochemical reversibility in the range of 0 < x < 0.94 for LixV2O5 with complete recovery of the Raman spectrum after the de-intercalation of Li ions. As shown in Fig. 10b, during intercalation in the range of 0 < x < 0.5, the vanadyl stretching mode at 997 cm−1 gradually disappears, while a new peak at 985 cm−1 keeps increasing in intensity and shifts to 980 cm−1. A stiffening of the lattice along the a-axis is reflected in the shift of several stretching and bending modes at 404 cm−1 and 527 cm−1 toward higher wavenumbers (V–OB–V). An increase in disorder between the square pyramids (3V–OC) is reflected by progressively quenched and shifted translational mode as the peak shift from 145 cm−1 to 153 cm−1.137
At x ≥ 0.75, the vanadyl peak at 980 cm−1 remained unchanged, while a new peak emerged at 958 cm−1, indicating the formation of Li-rich ε-Li0.94V2O5 without the formation of δ-LixV2O5 as observed in bulk V2O5.137 The Raman mode shift for the solid solution mechanism, corroborated by a continuous shift in XRD peaks, is distinct from the fingerprint patterns for distinct lithiated LixV2O5 phases obtained through ex situ measurement of chemically (de)lithiated V2O5 that is shown in Fig. 10b.138,139
At more reductive potentials (2.0 V vs. Li/Li+) and over cycles, most of the Raman signals rapidly diminish for the electrochemically obtained γ- and ω-phases, indicating the distortion of crystallinity and near-amorphous nature of the highly lithiated phases. The Raman band at around 980 cm−1 broadens and shifts to lower wavenumbers, indicating the elongation and disordering of the vanadyl bond upon further lithiation, which is correlated with capacity fading.31 Well-defined Raman shifts have been reported from chemically prepared γ- and ω-LixV2O5 in contrast to electrochemically obtained near amorphous structures.
3.4 Electron paramagnetic resonance (EPR)
Electron paramagnetic resonance (EPR), also known as electron spin resonance, is a technique used for studying materials with unpaired electrons. Key parameters analyzed in EPR include the “g tensor”, “signal width”, and “A factor”, which resolve electrons’ response to magnetic fields, the width of the signal, and interactions with nearby atomic nuclei, respectively. In vanadium oxides, EPR reveals paramagnetic local environments, such as V4+ and vanadyl VO2+ ions, for ex situ measurement of samples ranging from single crystals, powder, and cathode materials disassembled from a coin cell.132,140EPR effectively differentiates various crystalline phases of LixV2O5 during the lithiation, as shown in Fig. 11. In the topotactic lithiation range (0 < x < 1), the EPR signal primarily reflects the interactions between V4+ ions. While the EPR features of the ε-LixV2O5 phase closely resemble that of α-V2O5, substantial shifts emerge at x > 0.75, indicating the nucleation onset of the δ-LixV2O5 phase. Low-temperature EPR helps in differentiating signals for some of the distinct phases, such as for ε-LixV2O5 where the signal width linearly varies with temperature to be differentiated with α-LixV2O5.37 When x > 1, the EPR signals exhibit entirely different features, indicating significant changes in the interactions of unpaired electrons coupled with major phase transformations. This change is manifested by the emergence of new EPR signals from liberated VO2+ ions, leading to irreversible phase transformations when x > 1.141
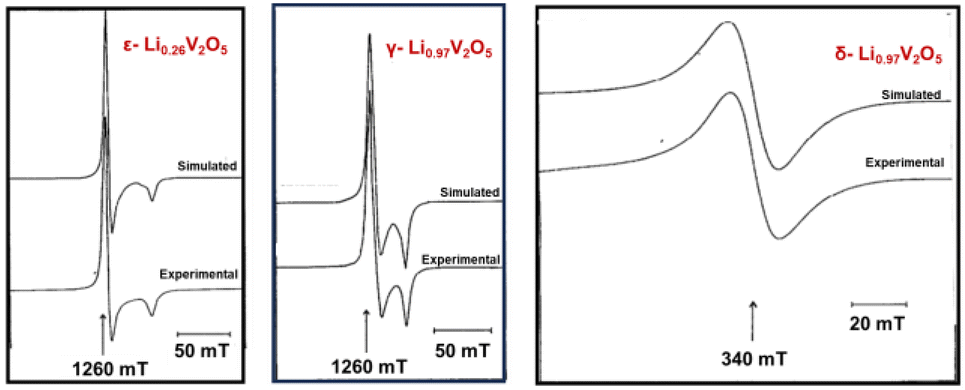 | ||
| Fig. 11 Experimental and calculated EPR spectra of ε-Li0.26V2O5, γ-Li0.97V2O5, and δ-Li0.97V2O5. Reproduced with adaption from ref. 141 with permission from Elsevier. Copyright 1995. | ||
One advantage of EPR in identifying phase co-existence is the sensitivity. The EPR study reveals that both α-LixV2O5 and ε-LixV2O5 phases coexist at x = 0.01 in powder samples undergoing electrochemical lithiation.141 For metastable polymorphs of V2O5, a broader range of electronic states in the EPR spectra indicates increased lithiation levels and a non-uniform pattern of site filling.36,142 The limitation of EPR is that EPR is most effective on materials with well-ordered structures, as it detects magnetic moments associated with unpaired electrons within a crystalline lattice. In quasi-crystalline or amorphous V2O5, the lack of long-range order results in more complicated EPR spectra that are difficult to interpret. Additionally, only a subset of vanadium ions has unpaired electrons, leading to low signal concentrations.
3.5 Nuclear magnetic resonance (NMR)
Nuclear magnetic resonance (NMR) spectroscopy, employing nuclei such as 7Li, 17O, 19F, 31P, and 51V, provides important information regarding the local environments and chemical structures of transition metal oxides. 51V and 17O NMR magic-angle spinning (MAS) spectroscopy, using VOCl3 and H2O as references, shows a clear distinction between crystalline V2O5 and quasi-crystalline V2O5·nH2O and can characterize single crystalline and powder samples that are either directly synthesized, chemically (de)lithiated, or electrochemically (de)lithiated and disassembled from a coin cell.34,41 Crystalline V2O5 shows a single resonance at −620 ppm in 51V NMR, representing the V1 site, whereas layered V2O5·nH2O gels show superimposed patterns of spinning sidebands representing multiple distinct sites at −580 ppm, −593 ppm, −597 ppm, −620 ppm, and −663 ppm (Fig. 12a), which suggest different coordination of vanadium in the presence of water in the gel. Upon dehydration at increasing temperature up to 350 °C (the temperature that converts V2O5·nH2O gels to crystalline V2O5), the resonances of the V1 site gradually emerge, while the sites coordinated with water disappear (Fig. 12b). While crystalline V2O5 does not undergo re-hydration, the re-hydration of the partially dehydrated structure V2O5·0.06H2O revealed preferred initial hydration at sites that are trans to vanadyl oxygen, which is then readily exchanged into all types of oxygen sites.34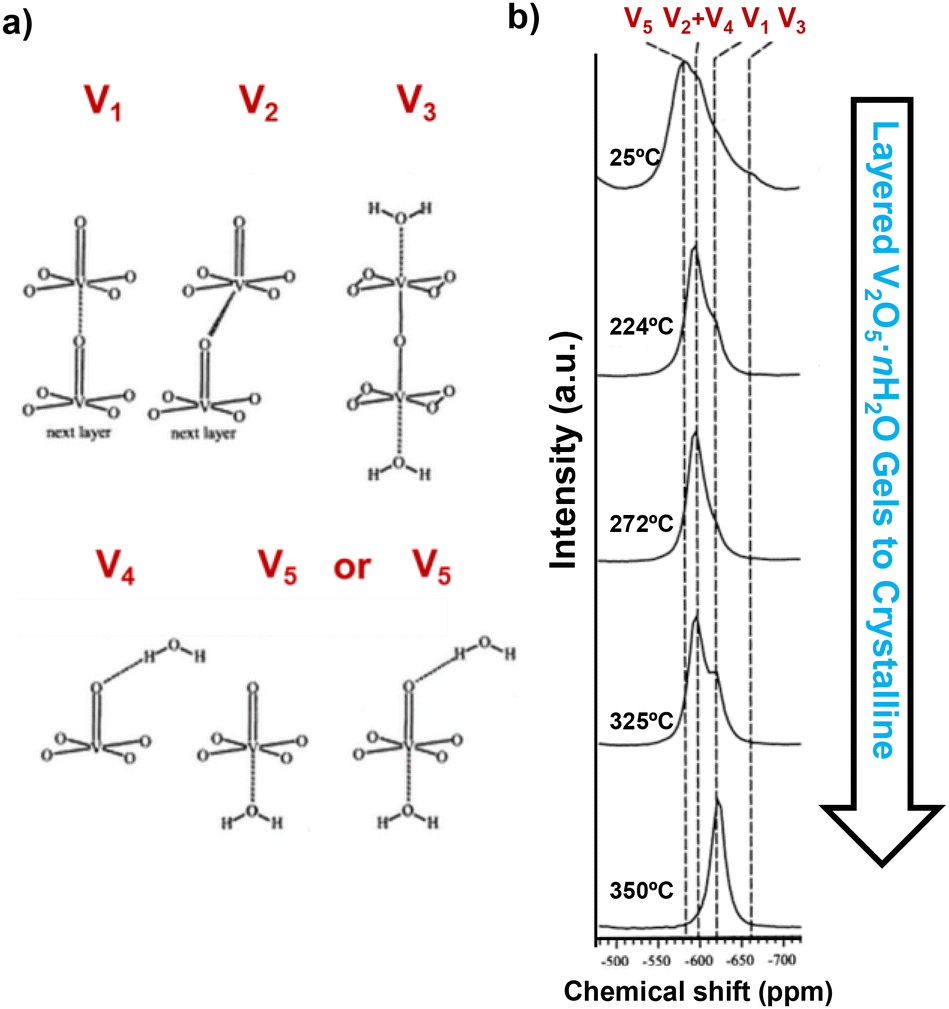 | ||
| Fig. 12 (a) Different coordination configurations of vanadium sites with water and (b) 51V MAS NMR spectra of V2O5·nH2O gels upon drying. Reproduced with adaption from ref. 34 with permission from American Chemical Society. Copyright 2002. | ||
Holland et.al. used 7Li MAS NMR spectroscopy to investigate the local environments of lithium ions as x increases in LixV2O5·0.5H2O xerogels and resolved two distinct lithium environments (interfacial sites, shown in Fig. 13a (A) and intercalated sites, shown in Fig. 13a (B)) in comparison with crystalline LixV2O5, which has only one intercalated site. For both xerogel and crystalline LixV2O5, as the Li-ion content increases, a broad upfield resonance continues to shift further to a lower frequency. The upfield shift is ascribed to the enhanced hyperfine interaction between intercalated lithium nuclei and paramagnetic V4+ sites on the lattice, as the number of paramagnetic V4+ ions increases when the Li content increases (Fig. 13a). The difference between crystalline V2O5 and LixV2O5·0.5H2O is that the xerogel exhibits a narrow resonance positioned close to 0 ppm, which is assigned to surface-bound Li+ sites at interfaces within the xerogel material (Fig. 13b). From 7Li MAS NMR, upon initial lithiation, the majority of Li ions were loaded onto the interface of the xerogel. At x = 0.17 in LixV2O5, the lithium ions occupy both the interfacial (capacitive) and interlayer (intercalation) at equal percentages. At x ≥ 0.55, the interfacial capacitive sites are saturated and only the interlayer intercalation sites continue to fill. The continuous upfield shift in the lithiation of LixV2O5·0.5H2O xerogels is distinct from the chemical shift in lithiated crystalline LixV2O5, where certain downfield shifts correspond to phase transformation and the change in Li coordination, such as from the trigonal prism in ε-LixV2O5 to tetrahedral in δ-LixV2O5.40
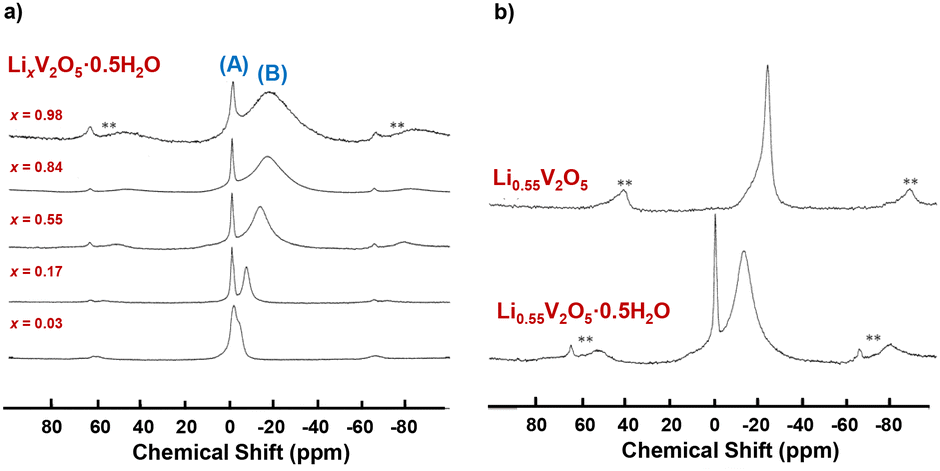 | ||
| Fig. 13 7Li MAS NMR spectra of (a) LixV2O5·0.5H2O xerogels upon lithiation and (b) crystalline Li0.55V2O5 and Li0.55V2O5·0.5H2O xerogel. Reproduced with adaption from ref. 40 with permission from American Chemical Society. Copyright 2002. | ||
Challenges using NMR spectroscopy to study V2O5 materials include the broad NMR signals from 51V nuclei, the availability of a magic angle spinning probe, and data interpretation. To fully understand the structure of V2O5 using NMR, complementary techniques including XRD and EPR can provide corroborative information.
4 Conclusion and outlook
In summary, materials design and synthesis strategies including nanosizing, pre-intercalation, and crystal engineering for metastable and quasi-crystalline structures effectively alleviate distortive phase transformations in V2O5 and promote alternative solid solution mechanisms, thereby enhancing the Li-ion diffusion and reversible capacity. Building on the recent progress in the field, we envision that future research efforts in the directions below will contribute to further understanding and development of reversible V2O5 energy storage materials.Substantial progress in understanding the lithiation mechanism has been made by either high spatial resolution methods using free-standing electrode materials or high temporal resolution in operando electrochemical setups or operating devices. For example, ex situ diffraction and imaging at sub-Angstrom resolution for free-standing particles or millimeter scale single crystals reveal atomistic information for "relaxed" structures, while operando measurement of cathode materials reveal real-time structural dynamics and phase coexistence with tens of hundreds of nanometers spatial resolution. While characterization techniques with higher spatiotemporal resolution and three-dimensional reconstruction keep advancing (such as synchrotron-based tomography and Bragg coherent diffractive imaging, and electron-beam based 4D scanning transmission electron microscopy), one should take into consideration the different electrochemical and mechanical conditions and parameters when comparing and correlating the data from ex situ diffraction with the ones from operando conditions. Meanwhile, the identification of distortive phase transformations and solid solution mechanisms, especially in early literature, has relied on structural identification tools such as powder XRD, which has relatively low detection limits and can be complicated by heterogeneous Li gradient, phase coexistence, and amorphization. Mechanisms such as non-distortive Li reordering require techniques with an enhanced signal-to-noise ratio and high spatio-temporal resolution. We expect opportunities to arise from “rediscovering” solid solution and Li-ion reordering mechanisms that continue to guide the design and synthesis of host materials without distortive phase transformations, and understanding of the roles of phase boundary-induced modifications of Li diffusion, especially for larger and more convoluted transport pathways involving dislocations and extended defects, that is beyond the computational power of atomistic calculation. Future advancements and better understanding of electrochemical phase transformation pathways should empower the design of new phases that facilitate developments in diverse fields such as energy storage materials and electrochromics.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
X. L. acknowledges the startup fund from Georgia State University and Department of Energy, Basic Energy Science DE-SC0024457.References
- N. A. Chernova, M. Roppolo, A. C. Dillon and M. S. Whittingham, Layered Vanadium and Molybdenum Oxides: Batteries and Electrochromics, J. Mater. Chem., 2009, 19(17), 2526, 10.1039/b819629j.
- X. Xu, F. Xiong, J. Meng, X. Wang, C. Niu, Q. An and L. Mai, Vanadium-Based Nanomaterials: A Promising Family for Emerging Metal-Ion Batteries, Adv. Funct. Mater., 2020, 30(10), 1904398, DOI:10.1002/adfm.201904398.
- P. Hu, P. Hu, T. D. Vu, M. Li, S. Wang, Y. Ke, X. Zeng, L. Mai and Y. Long, Vanadium Oxide: Phase Diagrams, Structures, Synthesis, and Applications, Chem. Rev., 2023, 123(8), 4353–4415, DOI:10.1021/acs.chemrev.2c00546.
- T. K. Le, P. V. Pham, C.-L. Dong, N. Bahlawane, D. Vernardou, I. Mjejri, A. Rougier and S. W. Kim, Recent Advances in Vanadium Pentoxide (V2O5) towards Related Applications in Chromogenics and beyond: Fundamentals, Progress, and Perspectives, J. Mater. Chem. C, 2022, 10(11), 4019–4071, 10.1039/D1TC04872D.
- Y. Wang and G. Cao, Synthesis and Enhanced Intercalation Properties of Nanostructured Vanadium Oxides, Chem. Mater., 2006, 18(12), 2787–2804, DOI:10.1021/cm052765h.
- X. Liu, J. Zeng, H. Yang, K. Zhou and D. Pan, V2O5-Based Nanomaterials: Synthesis and Their Applications, RSC Adv., 2018, 8(8), 4014–4031, 10.1039/C7RA12523B.
- Q. Wang, J. Xu, W. Zhang, M. Mao, Z. Wei, L. Wang, C. Cui, Y. Zhu and J. Ma, Research Progress on Vanadium-Based Cathode Materials for Sodium Ion Batteries, J. Mater. Chem. A, 2018, 6(19), 8815–8838, 10.1039/C8TA01627E.
- H. T. Tan, X. Rui, W. Sun, Q. Yan and T. M. Lim, Vanadium-Based Nanostructure Materials for Secondary Lithium Battery Applications, Nanoscale, 2015, 7(35), 14595–14607, 10.1039/C5NR04126K.
- M. S. Whittingham, Lithium Batteries and Cathode Materials, Chem. Rev., 2004, 104(10), 4271–4302, DOI:10.1021/cr020731c.
- M. S. Whittingham, Ultimate Limits to Intercalation Reactions for Lithium Batteries, Chem. Rev., 2014, 114(23), 11414–11443, DOI:10.1021/cr5003003.
- Y. Zhang, K. S. R. Chandran and H. Z. Bilheux, Imaging of the Li Spatial Distribution within V2O5 Cathode in a Coin Cell by Neutron Computed Tomography, J. Power Sources, 2018, 376, 125–130, DOI:10.1016/j.jpowsour.2017.11.080.
- X. Li, K. Perera, J. He, A. Gumyusenge and J. Mei, Solution-Processable Electrochromic Materials and Devices: Roadblocks and Strategies towards Large-Scale Applications, J. Mater. Chem. C, 2019, 7(41), 12761–12789, 10.1039/C9TC02861G.
- X. Li, Z. Wang, K. Chen, D. Y. Zemlyanov, L. You and J. Mei, Stabilizing Hybrid Electrochromic Devices through Pairing Electrochromic Polymers with Minimally Color-Changing Ion-Storage Materials Having Closely Matched Electroactive Voltage Windows, ACS Appl. Mater. Interfaces, 2021, 13(4), 5312–5318, DOI:10.1021/acsami.0c19685.
- X. Li, X. Wang, L. You, K. Zhao and J. Mei, Improving Electrochemical Cycling Stability of Conjugated Yellow-to-Transmissive Electrochromic Polymers by Regulating Effective Overpotentials, ACS Mater. Lett., 2022, 4(2), 336–342, DOI:10.1021/acsmaterialslett.1c00760.
- C. G. Granqvist, Electrochromics for Smart Windows: Oxide-Based Thin Films and Devices, Thin Solid Films, 2014, 564, 1–38, DOI:10.1016/j.tsf.2014.02.002.
- Y. Luo, S. Rezaei, D. A. Santos, Y. Zhang, J. V. Handy, L. Carrillo, B. J. Schultz, L. Gobbato, M. Pupucevski, K. Wiaderek, H. Charalambous, A. Yakovenko, M. Pharr, B.-X. Xu and S. Banerjee, Cation Reordering Instead of Phase Transitions: Origins and Implications of Contrasting Lithiation Mechanisms in 1D ζ- and 2D α-V2O5, Proc. Natl. Acad. Sci. U. S. A., 2022, 119(4), e2115072119, DOI:10.1073/pnas.2115072119.
- J. Yao, Y. Li, R. C. Massé, E. Uchaker and G. Cao, Revitalized Interest in Vanadium Pentoxide as Cathode Material for Lithium-Ion Batteries and Beyond, Energy Storage Mater., 2018, 11, 205–259, DOI:10.1016/j.ensm.2017.10.014.
- M. S. Whittingham, The Role of Ternary Phases in Cathode Reactions, J. Electrochem. Soc., 1976, 123(3), 315–320, DOI:10.1149/1.2132817.
- M. S. Whittingham, Electrical Energy Storage and Intercalation Chemistry, Science, 1976, 192(4244), 1126–1127, DOI:10.1126/science.192.4244.1126.
- A. Parija, D. Prendergast and S. Banerjee, Evaluation of Multivalent Cation Insertion in Single- and Double-Layered Polymorphs of V2O5, ACS Appl. Mater. Interfaces, 2017, 9(28), 23756–23765, DOI:10.1021/acsami.7b05556.
- D. Koch, V. V. Kulish and S. Manzhos, A First-Principles Study of Potassium Insertion in Crystalline Vanadium Oxide Phases as Possible Potassium-Ion Battery Cathode Materials, MRS Commun., 2017, 7(4), 819–825, DOI:10.1557/mrc.2017.107.
- J. M. Cocciantelli, J. P. Doumerc, M. Pouchard, M. Broussely and J. Labat, Crystal Chemistry of Electrochemically Inserted LixV2O5, J. Power Sources, 1991, 34(2), 103–111, DOI:10.1016/0378-7753(91)85029-V.
- C. Delmas, H. Cognac-Auradou, J. M. Cocciantelli, M. Ménétrier and J. P. Doumerc, The LixV2O5 System: An Overview of the Structure Modifications Induced by the Lithium Intercalation, Solid State Ionics, 1994, 69(3), 257–264, DOI:10.1016/0167-2738(94)90414-6.
- C. K. Chan, H. Peng, R. D. Twesten, K. Jarausch, X. F. Zhang and Y. Cui, Fast, Completely Reversible Li Insertion in Vanadium Pentoxide Nanoribbons, Nano Lett., 2007, 7(2), 490–495, DOI:10.1021/nl062883j.
- Y. Luo, L. R. De Jesus, J. L. Andrews, A. Parija, N. Fleer, D. J. Robles, P. P. Mukherjee and S. Banerjee, Roadblocks in Cation Diffusion Pathways: Implications of Phase Boundaries for Li-Ion Diffusivity in an Intercalation Cathode Material, ACS Appl. Mater. Interfaces, 2018, 10(36), 30901–30911, DOI:10.1021/acsami.8b10604.
- J. V. Handy, J. L. Andrews, B. Zhang, D. Kim, N. Bhuvanesh, Q. Tu, X. Qian and S. Banerjee, Topochemical Stabilization and Single-Crystal Transformations of a Metastable 2D γ′-V2O5 Intercalation Cathode, Cell Rep. Phys. Sci., 2022, 3(1), 100712, DOI:10.1016/j.xcrp.2021.100712.
- C. Delmas, S. Brèthes and M. Ménétrier, ω-LixV2O5—a New Electrode Material for Rechargeable Lithium Batteries, J. Power Sources, 1991, 34(2), 113–118, DOI:10.1016/0378-7753(91)85030-Z.
- H. Liu, Z. Zhu, Q. Yan, S. Yu, X. He, Y. Chen, R. Zhang, L. Ma, T. Liu, M. Li, R. Lin, Y. Chen, Y. Li, X. Xing, Y. Choi, L. Gao, H. S. Cho, K. An, J. Feng, R. Kostecki, K. Amine, T. Wu, J. Lu, H. L. Xin, S. P. Ong and P. Liu, A Disordered Rock Salt Anode for Fast-Charging Lithium-Ion Batteries, Nature, 2020, 585(7823), 63–67, DOI:10.1038/s41586-020-2637-6.
- A. Mukherjee, H. A. Ardakani, T. Yi, J. Cabana, R. Shahbazian-Yassar and R. F. Klie, Direct Characterization of the Li Intercalation Mechanism into α-V2O5 Nanowires Using in situ Transmission Electron Microscopy, Appl. Phys. Lett., 2017, 110(21), 213903, DOI:10.1063/1.4984111.
- S. Ki Park, P. Nakhanivej, J. Seok Yeon, K. Ho Shin, W. M. Dose, M. De Volder, J. Bae Lee, H. Jin Kim and H. S. Park, Electrochemical and Structural Evolution of Structured V2O5 Microspheres during Li-Ion Intercalation, J. Energy Chem., 2021, 55, 108–113, DOI:10.1016/j.jechem.2020.06.028.
- G. Zhang, T. Xiong, X. Pan, Y. Zhao, M. Yan, H. Zhang, B. Wu, K. Zhao and L. Mai, Illumining Phase Transformation Dynamics of Vanadium Oxide Cathode by Multimodal Techniques under Operando Conditions, Nano Res., 2019, 12(4), 905–910, DOI:10.1007/s12274-019-2321-z.
- F. Lin, Y. Liu, X. Yu, L. Cheng, A. Singer, O. G. Shpyrko, H. L. Xin, N. Tamura, C. Tian, T.-C. Weng, X.-Q. Yang, Y. S. Meng, D. Nordlund, W. Yang and M. M. Doeff, Synchrotron X-Ray Analytical Techniques for Studying Materials Electrochemistry in Rechargeable Batteries, Chem. Rev., 2017, 117(21), 13123–13186, DOI:10.1021/acs.chemrev.7b00007.
- Q. Fu, X. Wu, X. Luo, Z. Ding, S. Indris, A. Sarapulova, Z. Meng, M. Desmau, Z. Wang, W. Hua, C. Kübel, B. Schwarz, M. Knapp, H. Ehrenberg, Y. Wei and S. Dsoke, Ca2+ Pre-Intercalated Bilayered Vanadium Oxide for High-Performance Aqueous Mg-Ion Batteries, Energy Storage Mater., 2024, 66, 103212, DOI:10.1016/j.ensm.2024.103212.
- C. J. Fontenot, J. W. Wiench, G. L. Schrader and M. Pruski, 17O MAS and 3QMAS NMR Investigation of Crystalline V2O5 and Layered V2O5·nH2O Gels, J. Am. Chem. Soc., 2002, 124(28), 8435–8444, DOI:10.1021/ja0265254.
- Y. Xu, Z. Wang, Z. Yang, J. Na, A. Azhar, S. Wang, J. Yu and Y. Yamauchi, New Insights into the Lithium-Ion Diffusion Mechanism in Vanadate Compounds, ACS Energy Lett., 2021, 6(3), 886–892, DOI:10.1021/acsenergylett.0c02586.
- J. V. Handy, J. L. Andrews, S. Perez-Beltran, D. R. Powell, R. Albers, L. Whittaker-Brooks, N. Bhuvanesh and S. Banerjee, A “Li-Eye” View of Diffusion Pathways in a 2D Intercalation Material from Topochemical Single-Crystal Transformation, ACS Energy Lett., 2022, 7(6), 1960–1962, DOI:10.1021/acsenergylett.2c00739.
- D. Gourier, A. Tranchant, N. Baffier and R. Messina, EPR Study of Electrochemical Lithium Intercalation in V2O5 Cathodes, Electrochim. Acta, 1992, 37(15), 2755–2764, DOI:10.1016/0013-4686(92)85203-W.
- E. Culea, A. Nicula and I. Eratu, An Infrared Study of xV2O5 · (1 − x)B2O3 Glasses, Phys. Status Solidi A, 1984, 83(1), K15–K18, DOI:10.1002/pssa.2210830152.
- C. Sanchez, J. Livage and G. Lucazeau, Infrared and Raman Study of Amorphous V2O5, J. Raman Spectrosc., 1982, 12(1), 68–72, DOI:10.1002/jrs.1250120110.
- G. P. Holland, D. A. Buttry and J. L. Yarger, 7Li NMR Studies of Electrochemically Lithiated V2O5 Xerogels, Chem. Mater., 2002, 14(9), 3875–3881, DOI:10.1021/cm020260a.
- D. Rehder, T. Polenova and M. Bühl, Vanadium-51 NMR, in Annual Reports on NMR Spectroscopy, ed. G. A. Webb, Academic Press, 2007, vol. 62, pp. 49–114. DOI:10.1016/S0066-4103(07)62002-X.
- J. L. Andrews, A. Mukherjee, H. D. Yoo, A. Parija, P. M. Marley, S. Fakra, D. Prendergast, J. Cabana, R. F. Klie and S. Banerjee, Reversible Mg-Ion Insertion in a Metastable One-Dimensional Polymorph of V2O5, Chem, 2018, 4(3), 564–585, DOI:10.1016/j.chempr.2017.12.018.
- G. A. Horrocks, A. Parija, L. R. De Jesus, L. Wangoh, S. Sallis, Y. Luo, J. L. Andrews, J. Jude, C. Jaye, D. A. Fischer, D. Prendergast, L. F. J. Piper and S. Banerjee, Mitigating Cation Diffusion Limitations and Intercalation-Induced Framework Transitions in a 1D Tunnel-Structured Polymorph of V2O5, Chem. Mater., 2017, 29(24), 10386–10397, DOI:10.1021/acs.chemmater.7b03800.
- J. L. Andrews, P. Stein, D. A. Santos, C. J. Chalker, L. R. De Jesus, R. D. Davidson, M. A. Gross, M. Pharr, J. D. Batteas, B.-X. Xu and S. Banerjee, Curvature-Induced Modification of Mechano-Electrochemical Coupling and Nucleation Kinetics in a Cathode Material, Matter, 2020, 3(5), 1754–1773, DOI:10.1016/j.matt.2020.08.030.
- D. Fraggedakis, N. Nadkarni, T. Gao, T. Zhou, Y. Zhang, Y. Han, R. M. Stephens, Y. Shao-Horn and M. Z. Bazant, A Scaling Law to Determine Phase Morphologies during Ion Intercalation, Energy Environ. Sci., 2020, 13(7), 2142–2152, 10.1039/D0EE00653J.
- M. G. Boebinger, O. Yarema, M. Yarema, K. A. Unocic, R. R. Unocic, V. Wood and M. T. McDowell, Spontaneous and Reversible Hollowing of Alloy Anode Nanocrystals for Stable Battery Cycling, Nat. Nanotechnol., 2020, 15(6), 475–481, DOI:10.1038/s41565-020-0690-9.
- Y. Luo, Y. Bai, A. Mistry, Y. Zhang, D. Zhao, S. Sarkar, J. V. Handy, S. Rezaei, A. C. Chuang, L. Carrillo, K. Wiaderek, M. Pharr, K. Xie, P. P. Mukherjee, B.-X. Xu and S. Banerjee, Effect of Crystallite Geometries on Electrochemical Performance of Porous Intercalation Electrodes by Multiscale Operando Investigation, Nat. Mater., 2022, 21(2), 217–227, DOI:10.1038/s41563-021-01151-8.
- A. Yamada, H. Koizumi, S. Nishimura, N. Sonoyama, R. Kanno, M. Yonemura, T. Nakamura and Y. Kobayashi, Room-Temperature Miscibility Gap in LixFePO4, Nat. Mater., 2006, 5(5), 357–360, DOI:10.1038/nmat1634.
- N. Meethong, H.-Y. S. Huang, W. C. Carter and Y.-M. Chiang, Size-Dependent Lithium Miscibility Gap in Nanoscale Li1−xFePO4, Electrochem. Solid-State Lett., 2007, 10(5), A134, DOI:10.1149/1.2710960.
- E. Pomerantseva, Chemical Preintercalation Synthesis of Versatile Electrode Materials for Electrochemical Energy Storage, Acc. Chem. Res., 2023, 56(1), 13–24, DOI:10.1021/acs.accounts.2c00193.
- M. Clites and E. Pomerantseva, Bilayered Vanadium Oxides by Chemical Pre-Intercalation of Alkali and Alkali-Earth Ions as Battery Electrodes, Energy Storage Mater., 2018, 11, 30–37, DOI:10.1016/j.ensm.2017.09.005.
- P. Schofield, Y. Luo, D. Zhang, W. Zaheer, D. Santos, G. Agbeworvi, J. D. Ponis, J. V. Handy, J. L. Andrews, E. J. Braham, A. R. Balakrishna and S. Banerjee, Doping-Induced Pre-Transformation to Extend Solid-Solution Regimes in Li-Ion Batteries, ACS Energy Lett., 2022, 7(10), 3286–3292, DOI:10.1021/acsenergylett.2c01868.
- W. Zhong, J. Huang, S. Liang, J. Liu, Y. Li, G. Cai, Y. Jiang and J. Liu, New Prelithiated V2O5 Superstructure for Lithium-Ion Batteries with Long Cycle Life and High Power, ACS Energy Lett., 2020, 5(1), 31–38, DOI:10.1021/acsenergylett.9b02048.
- C. Liu, J. Yao, Z. Zou, Y. Li and G. Cao, Boosting the Cycling Stability of Hydrated Vanadium Pentoxide by Y3+ Pillaring for Sodium-Ion Batteries, Mater. Today Energy, 2019, 11, 218–227, DOI:10.1016/j.mtener.2018.12.003.
- X. Li, C. Liu, C. Zhang, H. Fu, X. Nan, W. Ma, Z. Li, K. Wang, H. Wu and G. Cao, Effects of Preinserted Na Ions on Li-Ion Electrochemical Intercalation Properties of V2O5, ACS Appl. Mater. Interfaces, 2016, 8(37), 24629–24637, DOI:10.1021/acsami.6b08052.
- Y. Li, J. Yao, E. Uchaker, M. Zhang, J. Tian, X. Liu and G. Cao, Sn-Doped V2O5 Film with Enhanced Lithium-Ion Storage Performance, J. Phys. Chem. C, 2013, 117(45), 23507–23514, DOI:10.1021/jp406927m.
- S. Suthirakun, S. Jungthawan and S. Limpijumnong, Effect of Sn-Doping on Behavior of Li-Intercalation in V2O5 Cathode Materials of Li-Ion Batteries: A Computational Perspective, J. Phys. Chem. C, 2018, 122(11), 5896–5907, DOI:10.1021/acs.jpcc.7b12321.
- Y.-H. Zhu, Q. Zhang, X. Yang, E.-Y. Zhao, T. Sun, X.-B. Zhang, S. Wang, X.-Q. Yu, J.-M. Yan and Q. Jiang, Reconstructed Orthorhombic V2O5 Polyhedra for Fast Ion Diffusion in K-Ion Batteries, Chem, 2019, 5(1), 168–179, DOI:10.1016/j.chempr.2018.10.004.
- L. R. De Jesus, J. L. Andrews, A. Parija and S. Banerjee, Defining Diffusion Pathways in Intercalation Cathode Materials: Some Lessons from V2O5 on Directing Cation Traffic, ACS Energy Lett., 2018, 3(4), 915–931, DOI:10.1021/acsenergylett.8b00156.
- A. J. Martinolich and J. R. Neilson, Toward Reaction-by-Design: Achieving Kinetic Control of Solid State Chemistry with Metathesis, Chem. Mater., 2017, 29(2), 479–489, DOI:10.1021/acs.chemmater.6b04861.
- A. Parija, G. R. Waetzig, J. L. Andrews and S. Banerjee, Traversing Energy Landscapes Away from Equilibrium: Strategies for Accessing and Utilizing Metastable Phase Space, J. Phys. Chem. C, 2018, 122(45), 25709–25728, DOI:10.1021/acs.jpcc.8b04622.
- W. Sun, S. T. Dacek, S. P. Ong, G. Hautier, A. Jain, W. D. Richards, A. C. Gamst, K. A. Persson and G. Ceder, The Thermodynamic Scale of Inorganic Crystalline Metastability, Sci. Adv., 2016, 2(11), e1600225, DOI:10.1126/sciadv.1600225.
- L. T. Alameda, P. Moradifar, Z. P. Metzger, N. Alem and R. E. Schaak, Topochemical Deintercalation of Al from MoAlB: Stepwise Etching Pathway, Layered Intergrowth Structures, and Two-Dimensional MBene, J. Am. Chem. Soc., 2018, 140(28), 8833–8840, DOI:10.1021/jacs.8b04705.
- M. Clites, W. B. Byles and E. Pomerantseva, Bilayered Vanadium Oxide as the Host Material for Reversible beyond Lithium Ion Intercalation, Adv. Mater. Lett., 2017, 8(6), 679–688, DOI:10.5185/amlett.2017.1536.
- J. M. Gallardo-Amores, N. Biskup, U. Amador, K. Persson, G. Ceder, E. Morán and M. E. Arroyo y de Dompablo, Computational and Experimental Investigation of the Transformation of V2O5 Under Pressure, Chem. Mater., 2007, 19(22), 5262–5271, DOI:10.1021/cm071360p.
- R. Baddour-Hadjean, M. B. Smirnov, K. S. Smirnov, V. Y. Kazimirov, J. M. Gallardo-Amores, U. Amador, M. E. Arroyo-de Dompablo and J. P. Pereira-Ramos, Lattice Dynamics of β-V2O5: Raman Spectroscopic Insight into the Atomistic Structure of a High-Pressure Vanadium Pentoxide Polymorph, Inorg. Chem., 2012, 51(5), 3194–3201, DOI:10.1021/ic202651b.
- Q. Fu, H. Zhao, A. Sarapulova and S. Dsoke, V2O5 as a Versatile Electrode Material for Postlithium Energy Storage Systems, Appl. Res., 2023, 2(3), e202200070, DOI:10.1002/appl.202200070.
- D. Vernardou, M. Apostolopoulou, D. Louloudakis, N. Katsarakis and E. Koudoumas, Hydrothermally Grown β-V2O5 Electrode at 95 °C, J. Colloid Interface Sci., 2014, 424, 1–6, DOI:10.1016/j.jcis.2014.03.004.
- R. Córdoba, J. Goclon, A. Sarapulova, Q. Fu, J. Maibach, S. Dsoke, F. Fauth, A. Kuhn and F. García-Alvarado, From High-Pressure β-V2O5 to κ-NaxV2O5 (x = 0.4 − 0.55): A Structural, Chemical, and Kinetic Insight into a Sodiated Phase with a Large Interlayer Space, Appl. Res., 2023, 2(1), e202200052, DOI:10.1002/appl.202200052.
- J. Jiang, Z. Wang and L. Chen, Structural and Electrochemical Studies on β-LixV2O5 as Cathode Material for Rechargeable Lithium Batteries, J. Phys. Chem. C, 20071112810707–10711, DOI:10.1021/jp071635o.
- G. Sai Gautam, P. Canepa, A. Abdellahi, A. Urban, R. Malik and G. Ceder, The Intercalation Phase Diagram of Mg in V2O5 from First-Principles, Chem. Mater., 2015, 27(10), 3733–3742, DOI:10.1021/acs.chemmater.5b00957.
- A. M. Kannan and A. Manthiram, Synthesis and Electrochemical Properties of High Capacity V2O5 - δ Cathodes, J. Electrochem. Soc., 2003, 150(7), A990, DOI:10.1149/1.1581261.
- A. Parija, Y. Liang, J. L. Andrews, L. R. De Jesus, D. Prendergast and S. Banerjee, Topochemically De-Intercalated Phases of V2O5 as Cathode Materials for Multivalent Intercalation Batteries: A First-Principles Evaluation, Chem. Mater., 2016, 28(16), 5611–5620, DOI:10.1021/acs.chemmater.6b01006.
- T. M.Tolhurst, B. Leedahl, J. L. Andrews, S. Banerjee and A. Moewes, The Electronic Structure of ϵ′-V2O5: an Expanded Band Gap in a Double-Layered Polymorph with Increased Interlayer Separation, J. Mater. Chem. A, 2017, 5, 23694–23703, 10.1039/C7TA05066F.
- P. Rozier, M. Dollé and J. Galy, Ionic Diffusion Mastering Using Crystal-Chemistry Parameters: τ-Cu1/2Ag1/2V2O5 Structure Determination and Comparison with Refined δ-AgxV2O5 and ε-CuxV2O5 Ones, J. Solid State Chem., 2009, 182(6), 1481–1491, DOI:10.1016/j.jssc.2009.03.017.
- M. V. Ganduglia-Pirovano and J. Sauer, Reduction of the (001) Surface of γ-V2O5 Compared to α-V2O5, J. Phys. Chem. B, 2005, 109(1), 374–380, DOI:10.1021/jp046233k.
- P. M. Marley, T. A. Abtew, K. E. Farley, G. A. Horrocks, R. V. Dennis, P. Zhang and S. Banerjee, Emptying and Filling a Tunnel Bronze, Chem. Sci., 2015, 6(3), 1712–1718, 10.1039/C4SC03748K.
- A. J. Mannix, B. Kiraly, M. C. Hersam and N. P. Guisinger, Synthesis and Chemistry of Elemental 2D Materials, Nat. Rev. Chem., 2017, 1(2), 1–14, DOI:10.1038/s41570-016-0014.
- M. Q. Arguilla, J. Katoch, K. Krymowski, N. D. Cultrara, J. Xu, X. Xi, A. Hanks, S. Jiang, R. D. Ross, R. J. Koch, S. Ulstrup, A. Bostwick, C. Jozwiak, D. W. McComb, E. Rotenberg, J. Shan, W. Windl, R. K. Kawakami and J. E. Goldberger, NaSn2As2: An Exfoliatable Layered van Der Waals Zintl Phase, ACS Nano, 2016, 10(10), 9500–9508, DOI:10.1021/acsnano.6b04609.
- R. Baddour-Hadjean, M. Safrany Renard and J. P. Pereira-Ramos, Unraveling the Structural Mechanism of Li Insertion in γ′-V2O5 and Its Effect on Cycling Properties, Acta Mater., 2019, 165, 183–191, DOI:10.1016/j.actamat.2018.11.043.
- J. V. Handy, Y. Luo, J. L. Andrews, N. Bhuvanesh and S. Banerjee, An Atomic View of Cation Diffusion Pathways from Single-Crystal Topochemical Transformations, Angew. Chem., Int. Ed., 2020, 59(38), 16385–16392, DOI:10.1002/anie.202005513.
- R. Shepard and M. Smeu, EvaluationAb initio Investigation of α- and ζ-V2O5 for Beyond Lithium Ion Battery Cathodes, J. Power Sources, 2020, 472, 228096, DOI:10.1016/j.jpowsour.2020.228096.
- D. A. Santos, J. L. Andrews, Y. Bai, P. Stein, Y. Luo, Y. Zhang, M. Pharr, B.-X. Xu and S. Banerjee, Bending Good Beats Breaking Bad: Phase Separation Patterns in Individual Cathode Particles upon Lithiation and Delithiation, Mater. Horiz., 2020, 7(12), 3275–3290, 10.1039/D0MH01240H.
- J. Galy, D. Lavaud, A. Casalot and P. Hagenmuller, Les Bronzes Oxygénés de Vanadium de Formule CuxV2O5: I. Structure Cristalline Des Phases CuxV2O5β et CuxV2O5∈, J. Solid State Chem., 1970, 2(4), 531–543, DOI:10.1016/0022-4596(70)90048-4.
- J. L. Andrews, L. R. De Jesus, T. M. Tolhurst, P. M. Marley, A. Moewes and S. Banerjee, OnesIntercalation-Induced Exfoliation and Thickness-Modulated Electronic Structure of a Layered Ternary Vanadium Oxide, Chem. Mater., 2017, 29(7), 3285–3294, DOI:10.1021/acs.chemmater.7b00597.
- I. Loa, A. Grzechnik, U. Schwarz, K. Syassen, M. Hanfland and R. K. Kremer, Vanadium Oxides V2O5 and NaV2O5 under High Pressures: Structural, Vibrational, and Electronic Properties, J. Alloys and Compd., 2001, 317–318, 103–108, DOI:10.1016/S0925-8388(00)01404-3.
- J. M. Cocciantelli, P. Gravereau, J. P. Doumerc, M. Pouchard and P. Hagenmuller, On the Preparation and Characterization of a New Polymorph of V2O5, J. Solid State Chem., 1991, 93(2), 497–502, DOI:10.1016/0022-4596(91)90323-A.
- V. P. Filonenko, M. Sundberg, P.-E. Werner and I. P. Zibrov, Structure of a High-Pressure Phase of Vanadium Pentoxide, β-V2O5, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60(4), 375–381, DOI:10.1107/S0108768104012881.
- I. P. Zibrov, V. P. Filonenko, S. G. Lyapin and V. A. Sidorov, The High Pressure Phases β- and δ-V2O5: Structure Refinement, Electrical and Optical Properties, Thermal Stability, High Pressure Res., 2013, 33(2), 399–408, DOI:10.1080/08957959.2013.796375.
- B. E. Conway, Transition from “Supercapacitor” to “Battery” Behavior in Electrochemical Energy Storage, J. Electrochem. Soc., 1991, 138(6), 1539, DOI:10.1149/1.2085829.
- M. Clites, B. W. Byles and E. Pomerantseva, Bilayered Vanadium Oxide as the Host Material for Reversible beyond Lithium Ion Intercalation, Adv. Mater. Lett., 2017, 8(6), 679–688, DOI:10.5185/amlett.2017.1536.
- O. B. Chae, J. Kim, I. Park, H. Jeong, J. H. Ku, J. H. Ryu, K. Kang and S. M. Oh, Reversible Lithium Storage at Highly Populated Vacant Sites in an Amorphous Vanadium Pentoxide Electrode, Chem. Mater., 2014, 26(20), 5874–5881, DOI:10.1021/cm502268u.
- M. Nabavi, C. Sanchez, F. Taulelle, J. Livage and A. de Guibert, Electrochemical Properties of Amorphous V2O5, Solid State Ionics, 1988, 28–30, 1183–1186, DOI:10.1016/0167-2738(88)90353-0.
- J. Livage, Sol-Gel Chemistry and Electrochemical Properties of Vanadium Oxide Gels, Solid State Ionics, 1996, 86–88, 935–942, DOI:10.1016/0167-2738(96)00336-0.
- W. Dong, J. S. Sakamoto and B. Dunn, Electrochemical Properties of Vanadium Oxide Aerogels, Sci. Technol. Adv. Mater., 2003, 4(1), 3–11, DOI:10.1016/S1468-6996(03)00012-3.
- S. Wu, Y. Ding, L. Hu, X. Zhang, Y. Huang and S. Chen, Amorphous V2O5 as High Performance Cathode for Aqueous Zinc Ion Battery, Mater. Lett., 2020, 277, 128268, DOI:10.1016/j.matlet.2020.128268.
- H. Y. Lee and J. B. Goodenough, Ideal Supercapacitor Behavior of Amorphous V2O5·nH2O in Potassium Chloride (KCl) Aqueous Solution, J. Solid State Chem., 1999, 148(1), 81–84, DOI:10.1006/jssc.1999.8367.
- Y. Sakurai, S. Okada, J. Yamaki and T. Okada, Electrochemical Behaviour of Amorphous V2O5(-P2O5) Cathodes for Lithium Secondary Batteries, J. Power Sources, 1987, 20(3), 173–177, DOI:10.1016/0378-7753(87)80108-8.
- M. Chiku, H. Takeda, S. Matsumura, E. Higuchi and H. Inoue, Amorphous Vanadium Oxide/Carbon Composite Positive Electrode for Rechargeable Aluminum Battery, ACS Appl. Mater. Interfaces, 2015, 7(44), 24385–24389, DOI:10.1021/acsami.5b06420.
- D. Kim and J. H. Ryu, Amorphous V2O5 Positive Electrode Materials by Precipitation Method in Magnesium Rechargeable Batteries, Electron. Mater. Lett., 2019, 15(4), 415–420, DOI:10.1007/s13391-019-00138-7.
- L. W. Wangoh, Y. Huang, R. L. Jezorek, A. B. Kehoe, G. W. Watson, F. Omenya, N. F. Quackenbush, N. A. Chernova, M. S. Whittingham and L. F. J. Piper, Correlating Lithium Hydroxyl Accumulation with Capacity Retention in V2O5 Aerogel Cathodes, ACS Appl. Mater. Interfaces, 2016, 8(18), 11532–11538, DOI:10.1021/acsami.6b02759.
- S. Correal, D. Hernández-Gómez, A. S. Esquivel, A. Cardona-Rodríguez, A. Reiber, Y. Hernandez, R. González-Hernández and J. G. Ramírez, Tuning Electronic and Magnetic Properties through Disorder in V2O5 Nanoparticles, Sci. Rep., 2023, 13(1), 6752, DOI:10.1038/s41598-023-32642-0.
- V. Petkov, P. N. Trikalitis, E. S. Bozin, S. J. L. Billinge, T. Vogt and M. G. Kanatzidis, Structure of V2O5·nH2O Xerogel Solved by the Atomic Pair Distribution Function Technique, J. Am. Chem. Soc., 2002, 124(34), 10157–10162, DOI:10.1021/ja026143y.
- A. Moretti and S. Passerini, Bilayered Nanostructured V2O5·nH2O for Metal Batteries, Adv. Energy Mater., 2016, 6(23), 1600868, DOI:10.1002/aenm.201600868.
- V. Petkov, V. Parvanov, P. Trikalitis, C. Malliakas, T. Vogt and M. G. Kanatzidis, Three-Dimensional Structure of Nanocomposites from Atomic Pair Distribution Function Analysis: Study of Polyaniline and (Polyaniline)0.5V2O5·1.0H2O, J. Am. Chem. Soc., 2005, 127(24), 8805–8812, DOI:10.1021/ja051315n.
- H. H. Kristoffersen and H. Metiu, Structure of V2O5·nH2O Xerogels, J. Phys. Chem. C, 2016, 120(7), 3986–3992, DOI:10.1021/acs.jpcc.5b12418.
- E. Uchaker, Y. Z. Zheng, S. Li, S. L. Candelaria, S. Hu and G. Z. Cao, Better than Crystalline: Amorphous Vanadium Oxide for Sodium-Ion Batteries, J. Mater. Chem. A, 2014, 2(43), 18208–18214, 10.1039/C4TA03788J.
- C. J. Fontenot, J. W. Wiench, M. Pruski and G. L. Schrader, Vanadia Gel Synthesis via Peroxovanadate Precursors. 1. In Situ Laser Raman and 51V NMR Characterization of the Gelation Process, J. Phys. Chem. B, 2000, 104(49), 11622–11631, DOI:10.1021/jp0021897.
- L. Rivoalen, A. Revcolevschi, J. Livage and R. Collongues, Amorphous Vanadium Pentoxide, J. Non-Cryst. Solids, 1976, 21(2), 171–179, DOI:10.1016/0022-3093(76)90039-9.
- M. Gotić, S. Popović, M. Ivanda and S. Musić, Sol–Gel Synthesis and Characterization of V2O5 Powders, Mater. Lett., 2003, 57(21), 3186–3192, DOI:10.1016/S0167-577X(03)00022-3.
- Y. Song, T.-Y. Liu, B. Yao, T.-Y. Kou, D.-Y. Feng, X.-X. Liu and Y. Li, Amorphous Mixed-Valence Vanadium Oxide/Exfoliated Carbon Cloth Structure Shows a Record High Cycling Stability, Small, 2017, 13(16), 1700067, DOI:10.1002/smll.201700067.
- S. Liu, Z. Tong, J. Zhao, X. Liu, J. Wang, X. Ma, C. Chi, Y. Yang, X. Liu and Y. Li, Rational Selection of Amorphous or Crystalline V2O5 Cathode for Sodium-Ion Batteries, Phys. Chem. Chem. Phys., 2016, 18(36), 25645–25654, 10.1039/C6CP04064K.
- G. Sai Gautam, P. Canepa, W. D. Richards, R. Malik and G. Ceder, Role of Structural H2O in Intercalation Electrodes: The Case of Mg in Nanocrystalline Xerogel-V2O5, Nano Lett., 2016, 16(4), 2426–2431, DOI:10.1021/acs.nanolett.5b05273.
- R. Y. Wang, B. Shyam, K. H. Stone, J. N. Weker, M. Pasta, H.-W. Lee, M. F. Toney and Y. Cui, Reversible Multivalent (Monovalent, Divalent, Trivalent) Ion Insertion in Open Framework Materials, Adv. Energy Mater., 2015, 5(12), 1401869, DOI:10.1002/aenm.201401869.
- M. Yan, P. He, Y. Chen, S. Wang, Q. Wei, K. Zhao, X. Xu, Q. An, Y. Shuang, Y. Shao, K. T. Mueller, L. Mai, J. Liu and J. Yang, Water-Lubricated Intercalation in V2O5·nH2O for High-Capacity and High-Rate Aqueous Rechargeable Zinc Batteries, Adv. Mater., 2018, 30(1), 1703725, DOI:10.1002/adma.201703725.
- Q. Wei, J. Liu, W. Feng, J. Sheng, X. Tian, L. He, Q. An and L. Mai, Hydrated Vanadium Pentoxide with Superior Sodium Storage Capacity, J. Mater. Chem. A, 2015, 3(15), 8070–8075, 10.1039/C5TA00502G.
- D. B. Le, S. Passerini, F. Coustier, J. Guo, T. Soderstrom, B. B. Owens and W. H. Smyrl, Intercalation of Polyvalent Cations into V2O5 Aerogels, Chem. Mater., 1998, 10(3), 682–684, DOI:10.1021/cm9705101.
- S. Tepavcevic, H. Xiong, V. R. Stamenkovic, X. Zuo, M. Balasubramanian, V. B. Prakapenka, C. S. Johnson and T. Rajh, Nanostructured Bilayered Vanadium Oxide Electrodes for Rechargeable Sodium-Ion Batteries, ACS Nano, 2012, 6(1), 530–538, DOI:10.1021/nn203869a.
- S.-H. Cho, J.-S. Park, J. H. Kim, Y.-H. Chang, J. Ahn, J. S. Nam, J.-W. Jung, I.-D. Kim and H.-S. Kim, Oxygen-Related Defect Engineering of Amorphous Vanadium Pentoxide Cathode for Achieving High-Performance Thin-Film Aqueous Zinc-Ion Batteries, ACS Appl. Energy Mater., 2023, 6(5), 2719–2727, DOI:10.1021/acsaem.2c03055.
- J. Cao, D. Zhang, Y. Yue, T. Pakornchote, T. Bovornratanaraks, M. Sawangphruk, X. Zhang and J. Qin, Revealing the Impacts of Oxygen Defects on Zn2+ Storage Performance in V2O5, Mater. Today Energy, 2021, 21, 100824, DOI:10.1016/j.mtener.2021.100824.
- Q. He, B. Sheng, K. Zhu, Y. Zhou, S. Qiao, Z. Wang and L. Song, Phase Engineering and Synchrotron-Based Study on Two-Dimensional Energy Nanomaterials, Chem. Rev., 2023, 123(17), 10750–10807, DOI:10.1021/acs.chemrev.3c00389.
- Q. Fu, A. Sarapulova, V. Trouillet, L. Zhu, F. Fauth, S. Mangold, E. Welter, S. Indris, M. Knapp, S. Dsoke, N. Bramnik and H. Ehrenberg, In Operando Synchrotron Diffraction and in Operando X-Ray Absorption Spectroscopy Investigations of Orthorhombic V2O5 Nanowires as Cathode Materials for Mg-Ion Batteries, J. Am. Chem. Soc., 2019, 141(6), 2305–2315, DOI:10.1021/jacs.8b08998.
- X. Yao, Y. Zhao, F. A. Castro and L. Mai, Rational Design of Preintercalated Electrodes for Rechargeable Batteries, ACS Energy Lett., 2019, 4(3), 771–778, DOI:10.1021/acsenergylett.8b02555.
- R. Wang, Y. Sun, A. Brady, S. Fleischmann, T. B. Eldred, W. Gao, H.-W. Wang, D. Jiang and V. Augustyn, Fast Proton Insertion in Layered H2W2O7 via Selective Etching of an Aurivillius Phase, Adv. Energy Mater., 2021, 11(1), 2003335, DOI:10.1002/aenm.202003335.
- N. Sharma, W. K. Pang, Z. Guo and V. K. Peterson, In Situ Powder Diffraction Studies of Electrode Materials in Rechargeable Batteries, ChemSusChem, 2015, 8(17), 2826–2853, DOI:10.1002/cssc.201500152.
- A. H. McCarthy, K. Mayilvahanan, M. R. Dunkin, S. T. King, D. Quilty, L. M. Housel, J. Kuang, K. J. Takeuchi, E. S. Takeuchi, A. C. West, L. Wang and A. C. Marschilok, Lithium Vanadium Oxide (Li1.1V3O8) Thick Porous Electrodes with High Rate Capacity: Utilization and Evolution upon Extended Cycling Elucidated via Operando Energy Dispersive X-Ray Diffraction and Continuum Simulation, Phys. Chem. Chem. Phys., 2021, 23(1), 139–150, 10.1039/D0CP04622A.
- O. J. Borkiewicz, B. Shyam, K. M. Wiaderek, C. Kurtz, P. J. Chupas and K. W. Chapman, The AMPIX Electrochemical Cell: A Versatile Apparatus for in Situ X-Ray Scattering and Spectroscopic Measurements, J. Appl. Crystallogr., 2012, 45(6), 1261–1269, DOI:10.1107/S0021889812042720.
- Q. Fu, A. Sarapulova, L. Zhu, G. Melinte, A. Missyul, E. Welter, X. Luo, M. Knapp, H. Ehrenberg and S. Dsoke, In Operando Study of Orthorhombic V2O5 as Positive Electrode Materials for K-Ion Batteries, J. Energy Chem., 2021, 62, 627–636, DOI:10.1016/j.jechem.2021.04.027.
- A. Šurca and B. Orel, IR Spectroscopy of Crystalline V2O5 Films in Different Stages of Lithiation, Electrochim. Acta, 1999, 44(18), 3051–3057, DOI:10.1016/S0013-4686(99)00019-5.
- T. R. Gilson, O. F. Bizri and N. Cheetham, Single-Crystal Raman and Infrared Spectra of Vanadium(V) Oxide, J. Chem. Soc., Dalton Trans., 1973, 3, 291–294, 10.1039/DT9730000291.
- L. Abello, E. Husson, Y. Repelin and G. Lucazeau, Vibrational Spectra and Valence Force Field of Crystalline V2O5, Spectrochim. Acta, Part A, 1983, 39(7), 641–651, DOI:10.1016/0584-8539(83)80040-3.
- C. Sanchez, J. Livage, J. P. Audiere and A. Madi, Influence of the Quenching Rate on the Properties of Amorphous V2O5 Thin Films, J. Non-Cryst. Solids, 1984, 65(2), 285–300, DOI:10.1016/0022-3093(84)90053-X.
- N. Özer, Electrochemical Properties of Sol-Gel Deposited Vanadium Pentoxide Films, Thin Solid Films, 1997, 305(1), 80–87, DOI:10.1016/S0040-6090(97)00086-2.
- A.Š Vuk, S. Benčič, B. Orel and F. Decker, UV-Visible and, IR Spectroelectrochemical Properties of V2O5 Crystalline Films Charged/Discharged in Extended Potential Range, J. Sol-Gel Sci. Technol., 2002, 23(1), 53–66, DOI:10.1023/A:1013375803983.
- C. V. Ramana, O. M. Hussain, B. Srinivasulu Naidu and P. J. Reddy, Spectroscopic Characterization of Electron-Beam Evaporated V2O5 Thin Films, Thin Solid Films, 1997, 305, 219–226, DOI:10.1016/S0040-6090(97)00141-7.
- S.-H. Lee, H. M. Cheong, M. Je Seong, P. Liu, C. E. Tracy, A. Mascarenhas, J. R. Pitts and S. K. Deb, Microstructure Study of Amorphous Vanadium Oxide Thin Films Using Raman Spectroscopy, J. Appl. Phys., 2002, 92(4), 1893–1897, DOI:10.1063/1.1495074.
- R. Baddour-Hadjean, C. Navone and J. P. Pereira-Ramos, In Situ Raman Microspectrometry Investigation of Electrochemical Lithium Intercalation into Sputtered Crystalline V2O5 Thin Films, Electrochim. Acta, 2009, 54(26), 6674–6679, DOI:10.1016/j.electacta.2009.06.052.
- R. Baddour-Hadjean and J.-P. Pereira-Ramos, Raman Microspectrometry Applied to the Study of Electrode Materials for Lithium Batteries, Chem. Rev., 2010, 110(3), 1278–1319, DOI:10.1021/cr800344k.
- R. Baddour-Hadjean, E. Raekelboom and J. P. Pereira-Ramos, New Structural Characterization of the LixV2O5 System Provided by Raman Spectroscopy, Chem. Mater., 2006, 18(15), 3548–3556, DOI:10.1021/cm060540g.
- J. Livage and R. Collongues, Semiconducting Properties of Amorphous V2O5 Prepared by Splat Cooling, Mater. Sci. Eng., 1976, 23(2), 297–299, DOI:10.1016/0025-5416(76)90213-5.
- B. Pecquenard, D. Gourier and N. Baffier, EPR Identification of LixV2O5 Phases Generated by Chemical and Electrochemical Lithium Intercalation in V2O5, Solid State Ionics, 1995, 78(3–4), 287–303, DOI:10.1016/0167-2738(95)00099-R.
- C. Szczuka, R.-A. Eichel and J. Granwehr, Exploring the Solvation Sphere and Spatial Accumulation of Dissolved Transition-Metal Ions in Batteries: A Case Study of Vanadyl Ions Released from V2O5 Cathodes, ACS Appl. Energy Mater., 2022, 5(1), 449–460, DOI:10.1021/acsaem.1c02979.
| This journal is © The Royal Society of Chemistry 2024 |