
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStimuli-responsive nanoparticle self-assembly at complex fluid interfaces: a new insight into dynamic surface chemistry
Jieun
Heo
 a,
Seunghwan
Seo
a,
Hongseok
Yun
*b and
Kang Hee
Ku
*a
a,
Seunghwan
Seo
a,
Hongseok
Yun
*b and
Kang Hee
Ku
*a
aSchool of Energy and Chemical Engineering, Ulsan National Institute of Science and Technology (UNIST), Ulsan 44919, Republic of Korea. E-mail: kangheeku@unist.ac.kr
bDepartment of Chemistry and Research Institute for Convergence of Basic Science, Hanyang University, Seoul 04763, Republic of Korea. E-mail: yunhs@hanyang.ac.kr
First published on 30th January 2024
Abstract
The self-assembly of core/shell nanoparticles (NPs) at fluid interfaces is a rapidly evolving area with tremendous potential in various fields, including biomedicine, display devices, catalysts, and sensors. This review provides an in-depth exploration of the current state-of-the-art in the programmed design of stimuli-responsive NP assemblies, with a specific focus on inorganic core/organic shell NPs below 100 nm for their responsive adsorption properties at fluid and polymer interfaces. The interface properties, such as ligands, charge, and surface chemistry, play a significant role in dictating the forces and energies governing both NP–NP and NP–hosting matrix interactions. We highlight the fundamental principles governing the reversible surface chemistry of NPs and present detailed experimental examples in the following three key aspects of stimuli-responsive NP assembly: (i) stimuli-driven assembly of NPs at the air/liquid interface, (ii) reversible NP assembly at the liquid/liquid interface, including films and Pickering emulsions, and (iii) hybrid NP assemblies at the polymer/polymer and polymer/water interfaces that exhibit stimuli-responsive behaviors. Finally, we address current challenges in existing approaches and offer a new perspective on the advances in this field.
 Jieun Heo | Jieun Heo received her B.S. in Energy Engineering at the Ulsan National Institute of Science and Technology (UNIST). She is pursuing a combined MS/Ph.D. program in the School of Energy and Chemical Engineering at UNIST, guided by Prof. Kang Hee Ku. With an interest in the field of colloid interface engineering, her academic pursuits are centered on nanoparticle assembly at fluid interfaces, aiming to fabricate thin films that can be utilized in batteries, catalysts, and sensors. |
 Seunghwan Seo | Seung Hwan Seo is currently serving as a master under the guidance of Prof. Kang Hee Ku in the School of Energy and Chemical Engineering at UNIST. He graduated from the Department of Chemical Engineering and Applied Chemistry at Chungnam National University in 2023. His research focuses on nanoparticle self-assembly at air/liquid and liquid/liquid interfaces and their transfer to solids. He aims to devise a simple method for creating nanoparticle thin films, with potential applications in electrodes and catalysts. |
 Hongseok Yun | Hongseok Yun is an assistant professor in the department of chemistry at Hanyang University. He received his B.S. and M.S. degrees in chemistry from University of Chicago in 2010. He completed his Ph.D. research under the guidance of Prof. Christopher B. Murray from University of Pennsylvania in 2015. After the Ph.D. degree, he worked as a postdoctoral researcher in Prof. Bumjoon Kim's group at KAIST until 2020. His current research interests include the synthesis and self-assembly of inorganic nanoparticle–polymer hybrid materials for stimuli-responsive smart materials and energy storage/harvesting applications. |
 Kang Hee Ku | Kang Hee Ku is currently serving as an assistant professor in the School of Energy and Chemical Engineering at UNIST. She completed her doctorate in 2018 under the guidance of Prof. Bumjoon Kim at KAIST and then worked with Prof. Timothy Swager at MIT as a postdoctoral researcher until 2020. Her research focuses on understanding the physical and chemical behavior of soft matter based on polymer science and colloid interface engineering to apply them in real-word life. Her current interests lie in the development of bio-inspired photonic crystals and intelligent colloids for microdisplays, sensors, and photonic cosmetics. |
1. Introduction
The self-assembly of core/shell nanoparticles (NPs) at a fluid interface represents a captivating realm within the field of nanotechnology, offering numerous opportunities for the design and creation of functional materials with tailored properties.1–3 For example, the high mobility of NPs at fluid interfaces enables rapid self-assembly or directed assembly into two- or three-dimensional colloidal arrays with unique optical, magnetic, or electronic properties.4–6 When NPs attach to an interface, they lower the Gibbs free energy, which depends on the square of the particle's radius, thereby stabilizing the overall system.7 These NPs can selectively attach to or detach from the interface if the energy supplied is enough to overcome certain thermodynamic barriers. This process allows for a dynamic rearrangement of the NPs at the interface.8 Therefore, a soft and adaptable fluid interface is key to effectively positioning NPs in a dynamic way. Particularly fascinating in this area of study is how NPs organize themselves in response to different triggers, such as temperature shifts, pH changes, light exposure, or external fields. The intricate interplay between various colloidal interactions (e.g., van der Waals force, electrostatic interactions, and capillary force) influences the NP assemblies at the fluid interface and their functionalities.9–11 The stimuli-responsiveness of NP arrays is attributed to the characteristics of either the organic ligands adsorbed on NP surfaces or the inorganic cores of the NPs.12,13 Achieving neutral wetting conditions for different fluids at the NP interface is crucial for ensuring the long-term stability of the NP array, although it can be a complex task. In this context, there is a growing interest in designing ligand materials that facilitate fluid–NP interactions.14–16 These ligands are typically organic molecules, polymers, and oligomers, covalently linked to the surface of NPs.17 The key to achieving the responsive assembly and disassembly of NPs is dynamic reversibility in the intermolecular interactions between the fluid and organic materials that serve as ligands through external stimuli.Over the decades, numerous reviews covering theoretical and experimental studies of the self-assembly of NPs at liquid/liquid or liquid/air interface have been published.18–22 The majority of these reviews have primarily emphasized the thermodynamic interpretation of the core–shell structures that achieve static equilibrium for NP adsorption. However, the significance of designing organic ligands, particularly in accordance with the physical principles that govern the dynamic NP interactions, has been relatively unexplored in achieving responsive NP arrays. This review aims to present the current state-of-the-art in the programmed design of stimuli-responsive NP assemblies. We narrow our scope to inorganic core/organic shell NPs with sizes below a few tens of nanometers for two main reasons. First, these NPs have a lower adsorption barrier and are more responsive to external stimuli compared to larger particles, while their surface properties can be finely tuned through organic ligands.23 Second, their size aligns well with the domain size in polymer/NP hybrid systems, typically in the tens of nanometers range, ensuring compatibility and effective interaction.24,25 These two factors together enable the NPs to assemble responsively at interfaces, even in a reversible manner. Our primary emphasis is on elucidating the principles that govern the surface chemistry of NPs in a reversible manner, followed by detailed experimental examples. The stimuli-responsive NP assembly covered in this review comprises four key aspects: (i) stimuli-triggered assembly at the air/liquid interface; (ii) dynamic behavior at the liquid/liquid interface, encompassing films and Pickering emulsions; hybrid NP assembly (iii) at the polymer/polymer interface and (iv) polymer/water interfaces that exhibit stimuli-responsive behaviors (Scheme 1). We introduce a new perspective on utilizing specific polymer interfaces in NP assemblies, exploring their similarities and differences with air/liquid and liquid/liquid interfaces. Furthermore, we will address emerging trends and challenges, and suggest future directions, with a strong emphasis on the importance of interdisciplinary collaboration and the need for innovative solutions.
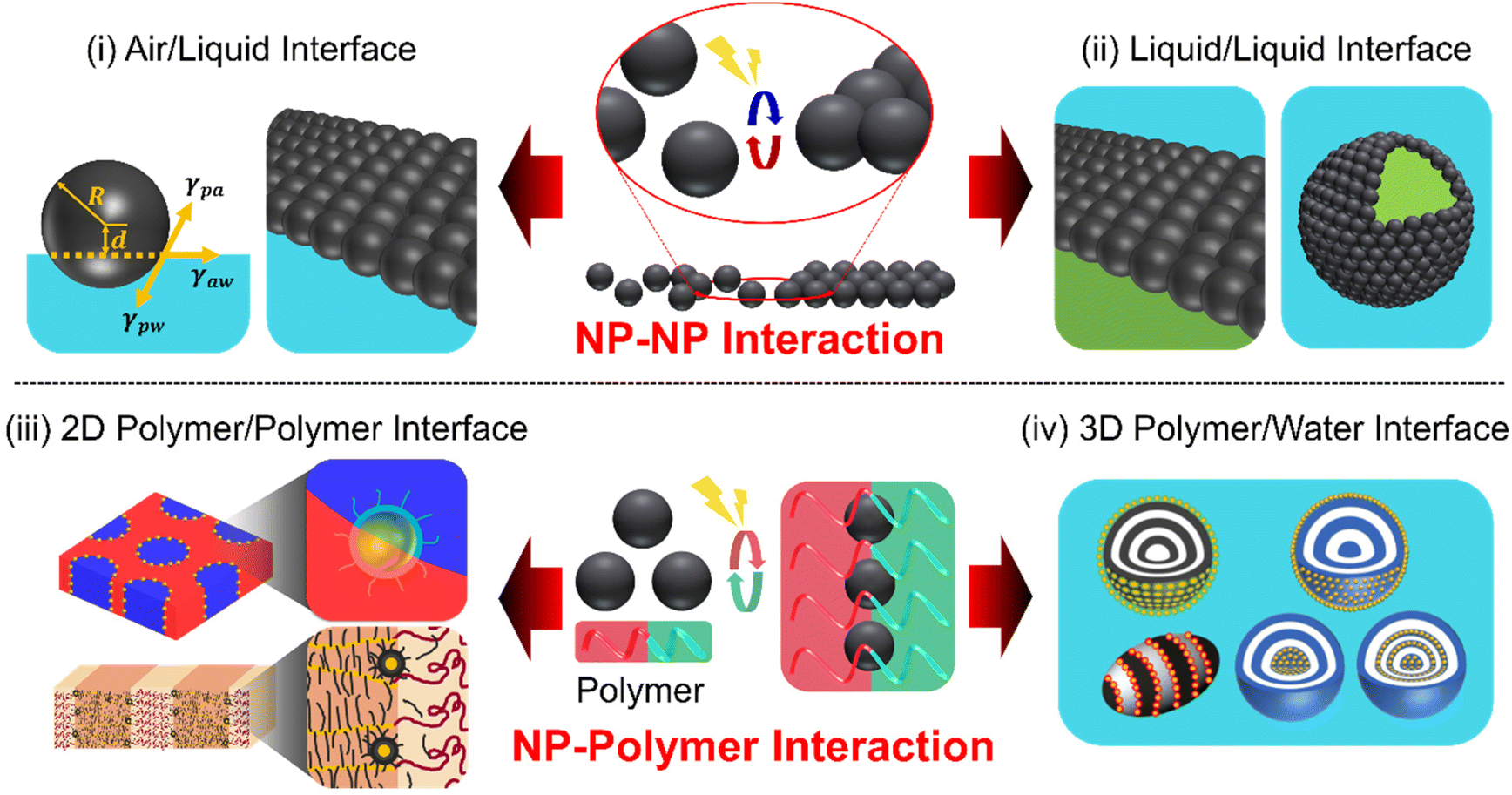 | ||
| Scheme 1 Schematic illustration of stimuli-responsive self-assembly of NPs in complex fluids in this minireview: (i) NPs at air/liquid interface, (ii) NPs at liquid/liquid interface, (iii) NPs at polymer/polymer interface, and (iv) NPs at polymer/water interface. | ||
2. Self-assembly mechanism: NPs at fluid interfaces
At soft air/liquid or liquid/liquid interfaces, NPs exhibit high mobility, allowing them to rapidly form dense assemblies. When NPs are trapped at a fluid interface, their assembly is primarily driven by the reduction of interfacial energy, as described by Gibbs free energy. To obtain an ordered NP array from a loose packing structure through external stimuli, it is essential to consider additional interactions that can influence the alignment of neighboring NPs.7 In this context, we will briefly introduce the thermodynamic and kinetic approaches for NP assembly with particular emphasis on NP–NP interactions triggered by external stimuli.2.1. Thermodynamic and kinetic approaches
When spherical particles with a radius r are present at the interface between adjacent α and β fluid phases, they occupy an area equivalent to the interfacial area. The adsorption of particles at the interface reduces Gibbs free energy, expressed by the following equation:ΔE = −πr2γαβ(1 ± cos![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ)2 θ)2 |
2.2. Stimuli-responsive NP interaction
Besides the decrease in interface energy achieved by NP adsorption, forming an ordered NP array requires specific interactions that can counterbalance the entropy penalty associated with NP assembly. The dominant interactions between a pair of NPs are van der Waals and electrostatic interactions as described in the Derjaguin–Landau–Verwey–Overbeek theory.39 van der Waals interactions typically result in attraction between similar particles within the same medium,40 and when this attraction exceeds electrostatic repulsion, it promotes the self-assembly of NPs. Therefore, external stimuli capable of modulating or overcoming electrostatic repulsion are crucial for forming well-aligned NP arrays. The organic ligand design plays a key role in controlling NP interactions, including their responsiveness to external stimuli and wettability. Fig. 1 summarizes the NP interactions induced by chemical (e.g., pH) and physical (e.g., light, temperature, and electromagnetic fields) stimuli.41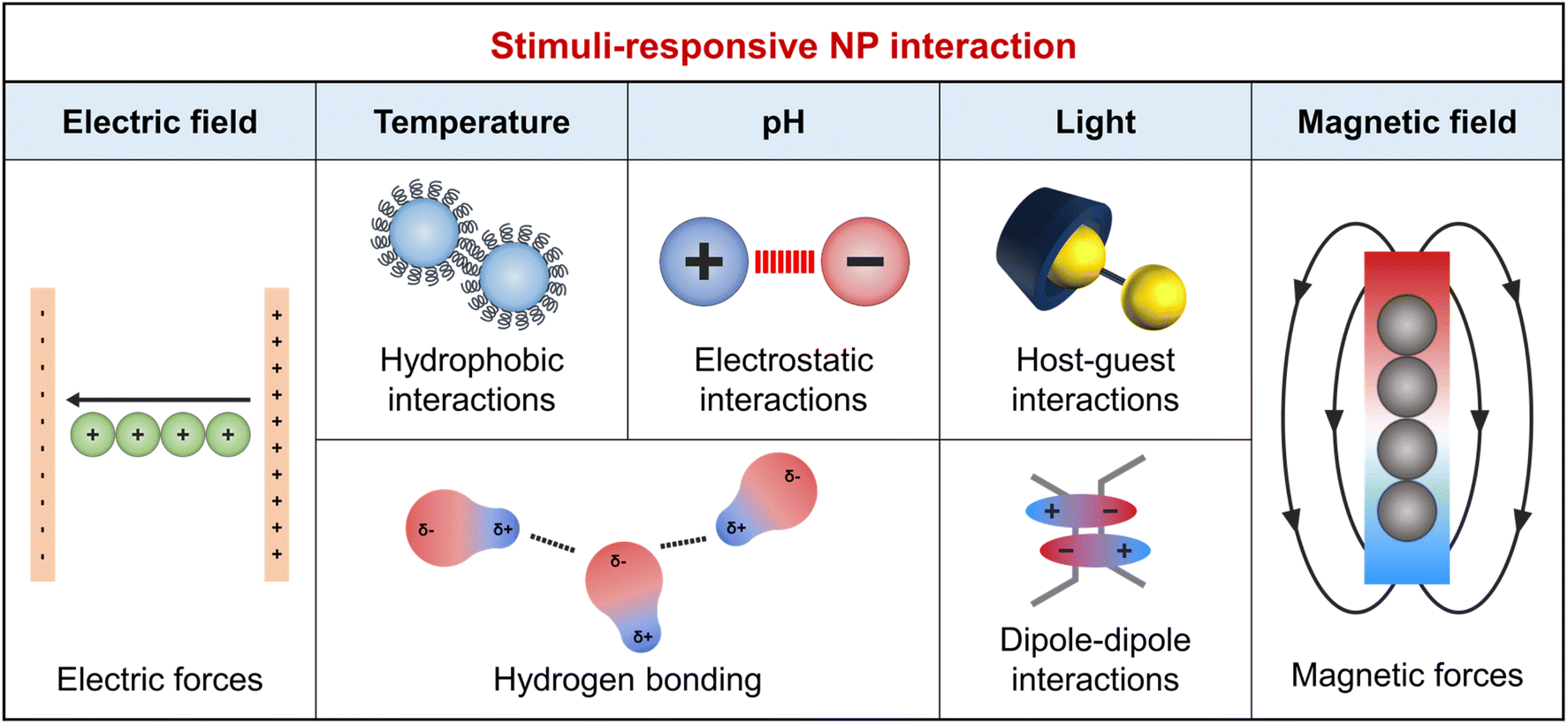 | ||
| Fig. 1 Summary of the external stimuli and corresponding physical interactions to induce self-assembly of NPs at fluid interface. | ||
Chemical stimuli, such as changes in pH due to acid, base, or CO2 addition, adjust the surface charge of organic ligands. For instance, the addition of a base increases the pH, leading to increased electrostatic repulsion between acidic functional groups like COOH and enhanced NP dispersion in solvents. Conversely, low pH weakens this repulsion and enhances van der Waals forces, promoting NP assembly.42 Additionally, pH affects hydrogen bond strength, varying with the density of surface anchoring groups (i.e., –OH, –COOH, or –NH2).43 Physical stimuli include light, temperature, and electromagnetic fields. Light-responsive groups, such as nitrobenzyl and coumarin esters, break down into smaller segments upon light absorption.44 Photoisomerizable groups, such as azobenzene, undergo structural transformations under specific light wavelengths, affecting molecular volume and hydrophilicity, crucial for NP wetting properties and host–guest chemistry.45 Dipole–dipole interactions occur between molecules that have a permanent electric dipole moment. Alkanethiol ligands significantly increase the dipole moment of Au NPs. Furthermore, the introduction of azobenzene to these ligands allows for interaction switching under different light wavelengths through photoisomerization. UV light activates these interactions, while visible light deactivates them.46 These enhanced attractive forces overcome the electrostatic repulsion between NPs, facilitating the attachment of adjacent particles47 and thereby enabling the photo-responsive assembly of NPs.48 Photo-initiators, such as acrylate-based ligands, induce the conversion of monomers into a polymer upon exposure to specific wavelengths of light.49 Hydrogen bonds, essential in host–guest chemistry, exhibit temperature sensitivity. Higher temperatures break hydrogen bonds due to increased kinetic energy, altering molecular structures. Lower temperatures favor hydrogen bond reformation.50 This behavior is key for thermoresponsive polymer ligands on NPs, affecting solubility and surface interactions. Notably, around specific temperature thresholds, such as the lower critical solution temperature (LCST), these polymers lose hydrogen bonds with water molecules and become hydrophobic, promoting clustering and reducing interaction with polar solvents.51 In external electric fields, the polarization of ligands causes their reorientation, affecting NP arrangement. Magnetic NPs align and magnetize in magnetic fields, enabling directional alignment.52
3. NP arrays at fluid interfaces
3.1. Self-assembly of NPs at an air/liquid interface
For the air/liquid interface system, the physicochemical stimuli can be directly applied, taking advantage of the direct contact with the air interface. In some cases, when NPs are assembled at the air/water interface, they form loosely packed structures, requiring additional effort to create a more organized array. The conventional interfacial tension gradient method employs volatile liquids such as ethanol and ethyl acetate to achieve a well-ordered NP array by reducing interfacial energy.53,54 The method involves using volatile liquids with different surface tensions to create a capillary force, pulling NPs towards higher surface tension regions. Careful control of solvent addition allows manipulation of these forces, compressing interparticle distance and facilitating self-assembly into a well-ordered array. Alternatively, specific external stimuli can replace volatile liquids, when these stimuli can trigger the manipulation of the interparticle distance between NPs. In this section, we present recent experimental progress in stimuli-triggered NP assembly at an air/liquid interface.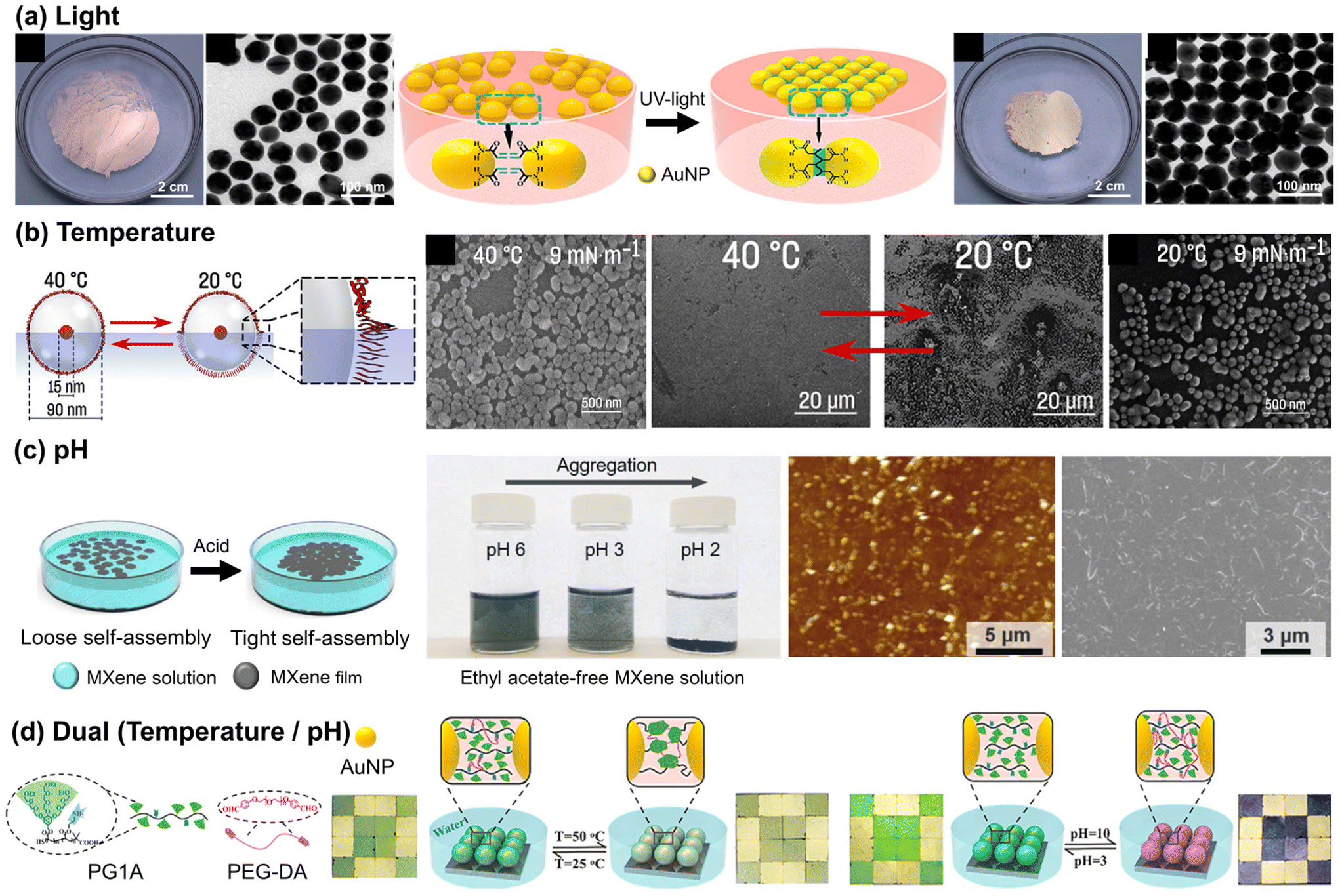 | ||
| Fig. 2 Stimuli-responsive self-assembly of NP at air/liquid interface: (a) assembly of the Au NP monolayer film by UV-induced interfacial crosslinking at the air/water interface. Reproduced from ref. 31 with permission from American Chemical Society 2018; (b) temperature-responsive assembly of NPs decorated with PNIPAM. Reproduced from ref. 32 with permission American Chemical Society 2023; (c) pH-responsive arrangement of the MXene film between loose and dense packing structures. Reproduced from ref. 37 with permission from Wiley 2022; (d) temperature- and pH-dual responsive NP array with an oligoethylene-glycol-based dendronized copolymer (PG1A) and polyethylene glycol dibenzyl aldehyde (PEG-DA). Reproduced from ref. 64 with permission American Chemical Society 2018. | ||
Another effective approach for temperature adjustment is the photothermal method, wherein light irradiation increases temperature near NPs through the conversion of absorbed photons into heat energy (phonons), thereby facilitating the thermally-driven assembly of NPs. As an example, the Deng group achieved reversible assembly of Au NPs by using light-induced thermal evaporation of liquid. This method, based on the manipulation of surface tension gradients, allows efficient control over NP assembly without affecting surface charge or ligand molecule structure. During the synthesis of oleylamine-coated Au NPs, some of the oleylamine molecules on the Au NPs were oxidized into carbonyl groups, allowing hydrogen bonds with the remaining unoxidized oleylamine groups.59 The presence of an excess amount of oleylamine in the chloroform solution of Au NPs led to the formation of oleylamine double layers. This occurred through the hydrogen bonding between the excess oleylamine and the grafted oleylamine on the surface of Au NPs, stabilizing the Au NP dispersion and thus preventing the self-assembly of Au NPs. In the absence of excess oleylamine or with the replacement of surface ligands by n-hexadecane thiol, the NPs were assembled through photothermal-driven evaporation of chloroform. The reversible assembly behavior was governed by the metastable state, with films coated with both oleylamine and alkyl thiol groups exhibiting negative thermodynamic potential energy changes. In this state, the film can be easily disassembled by an external force and subsequently reassembled through a light-irradiation plasmonic effect.60 In a similar way, the Pillai group reported a method by utilizing DMSO hygroscopicity and sunlight-driven water evaporation as driving forces. Thymine thiol-functionalized Au NPs dispersed in DMSO formed stable 2D assemblies due to moisture absorption, while photothermal-induced temperature increase to 80 °C resulted in disassembly of the NPs through water evaporation.61
3.2. Self-assembly of NPs at a liquid/liquid interface
Self-assembly of NPs at liquid/liquid interface has proven to be a cheap, convenient, and efficient route to obtain densely packed layers of NPs with minimal interparticle distances. When NPs assemble at the liquid/liquid interface, they can form either a two-dimensional monolayer film or a three-dimensional structure, called Pickering emulsions.65–69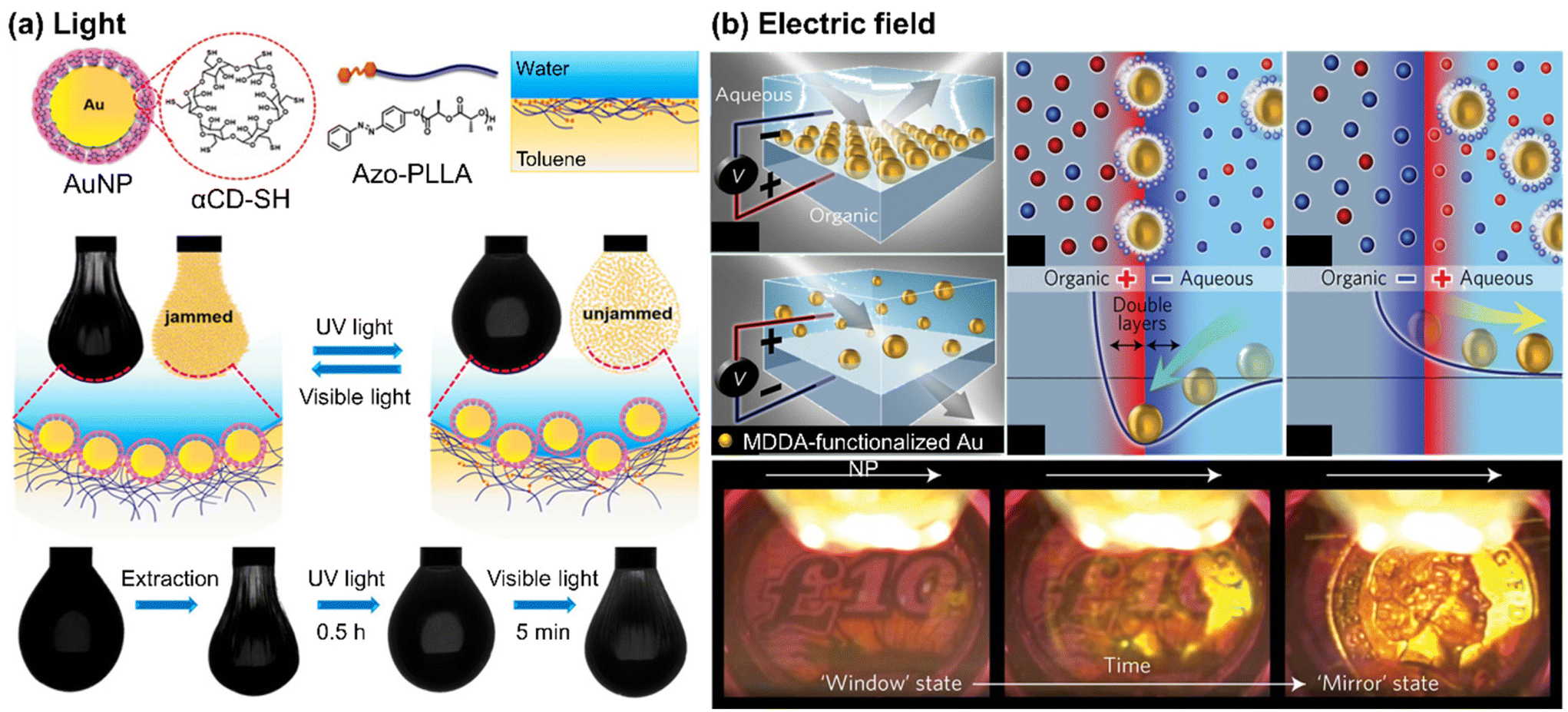 | ||
| Fig. 3 Stimuli-responsive self-assembly of NPs at liquid/liquid interface to form a monolayer film: (a) photo-switchable jamming-to-unjamming transition of Au NPs at oil/water interface, driven by host–guest molecular recognition. Reproduced from ref. 70 with permission from American Chemical Society 2020; (b) reversible electro-tunable liquid mirror based on voltage-controlled self-assembly/disassembly of plasmonic NPs at the interface between two immiscible electrolyte solutions. Reproduced from ref. 74 with permission from Nature Publishing Group 2017. | ||
When the organic ligand of NPs carries a surface charge, it becomes possible to control the assembly of these NPs using an external electric field.72 For example, in one approach, the positively charged trimethylammonium-terminated Au NPs were utilized to facilitate their transport from the aqueous phase to the organic phase in two immiscible electrolyte solutions when subjected to an electric field. By adjusting the voltage, a dense array of NPs was achieved.73 Kornyshev and co-workers developed a voltage-controlled liquid mirror by assembling negatively charged Au NPs, functionalized with 12-mercapto dodecanoic acid (MDDA). As depicted in Fig. 3(b), the Au NPs were effectively adsorbed or desorbed at the interface, depending on the direction of the electric field. Applying a positive voltage to the aqueous solution dispersed the NPs, while a negative voltage led to the NPs being most stably located at the interface with the organic electrolyte.74
To facilitate denser arrangements of NPs, it is essential to diminish electrostatic repulsion. A prevalent approach to overcome this challenge involves the binding of electrically neutral organic ligands to the NP surface. Notably, the modification of NPs with a metal core using charge-neutral thiols is a conventional method for generating NP monolayers at the liquid/liquid interface. For example, by reducing the surface charge of hydrophilic Au NPs through acetone introduction and subsequent agitation, thiolates replaced citrate anions as ligands.75,76 This allowed for the creation of ordered arrays of Au NPs with neutral ligands, mitigating electrostatic repulsion. At the hexane/water interface, these NPs assembled into a densely packed monolayer film.77 In a similar manner, perfluorodecanethiol (PFT) was introduced as an in situ NP surfactant. Initially, Au NPs were dispersed in water, and hexane with PFT was added. By increasing the PFT content, they enhanced the hydrophobicity of the NPs, thereby reducing the kinetic energy barrier associated with interface assembly.78,79
Ding and colleagues demonstrated a thermo-sensitive Pickering emulsion stabilized by hydrophilic silica NPs and poly(ethylene oxide-block-propylene oxide-block-ethylene oxide) (PEO-b-PPO-b-PEO) (Fig. 4(a)). At room temperature, stable paraffin oil-in-water Pickering emulsions were obtained using the silica NPs with surface-adsorbed PEO-b-PPO-b-PEO through hydrogen bonds. Upon heating to 55 °C, phase separation occurred due to disruption of the hydrogen bond between the PEO and the silanol group on the silica NP. Subsequent cooling and homogenization allowed for the polymer re-adsorption onto the silica surface through hydrogen bond formation, enabling the re-emulsification of the Pickering emulsion. The length of the PEO group capable of forming hydrogen bonds with silanol groups played a critical role in determining the temperature tolerance of the Pickering emulsion.82 Similarly, the Palmqvist group modified the surface of silica NP by combining hydrophilic methyl poly(ethylene) glycol (mPEG) and hydrophobic propyl groups to attain reversible phase inversion between oil-in-water and water-in-oil configurations. Salt concentration, pH, and heat were the crucial factors that influenced phase inversion. Temperature changes can cause phase inversion in emulsions stabilized by nonionic PEG-based surfactants. This happens due to the conformational changes in the polyoxyethylene chains. At lower temperatures, polar conformations dominate, but as temperature increases, nonpolar conformations prevail, leading to a phase inversion from oil-in-water to water-in-oil at the phase inversion temperature. Lowering pH facilitated the adsorption of the PEG chain onto the silica, reducing electrostatic repulsion, whereas an increased salt concentration caused interfacial NP adsorption, both of which consequently decreased the temperature required for the phase inversion.83
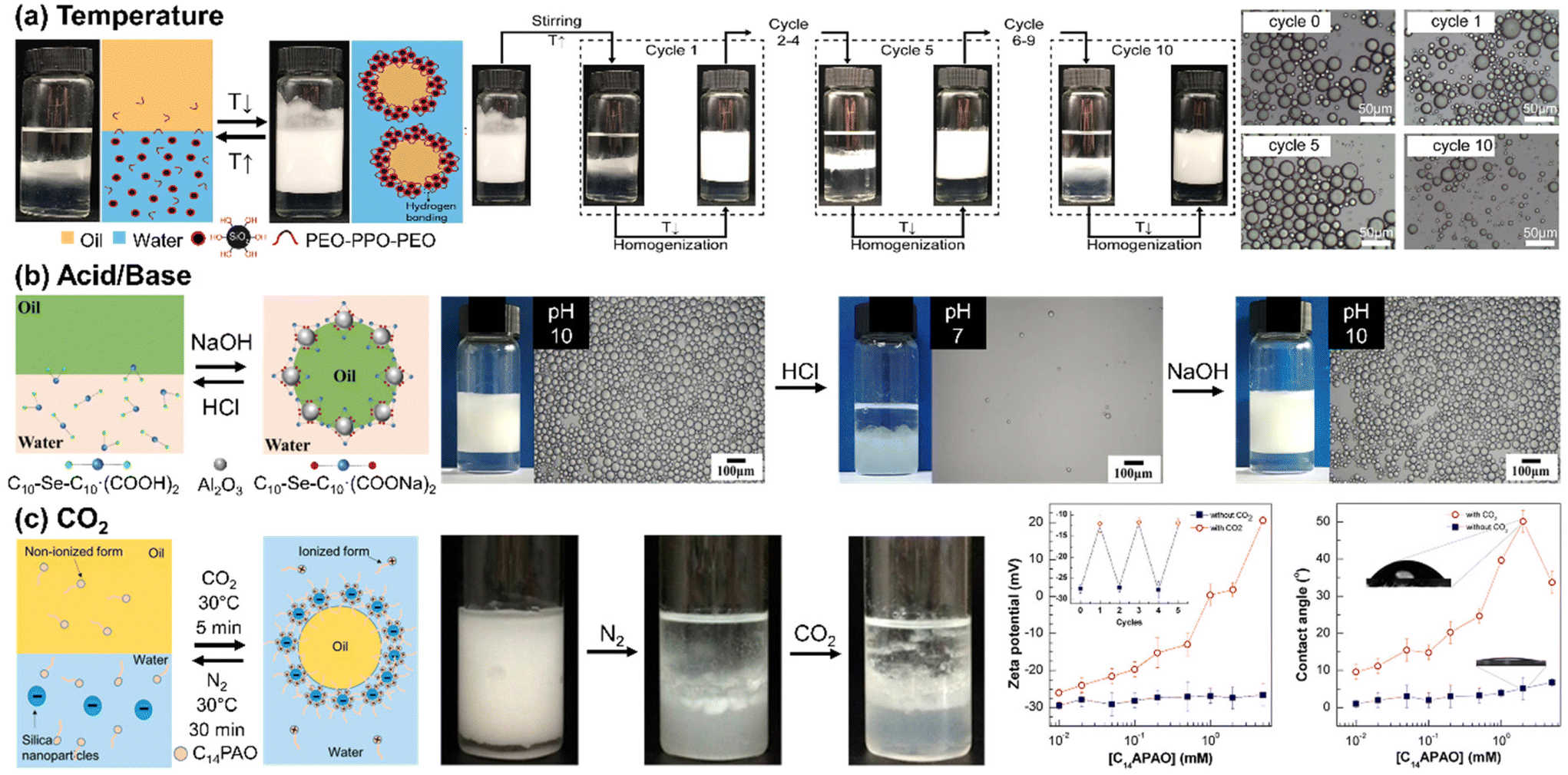 | ||
| Fig. 4 Stimuli-responsive self-assembly of NPs at liquid/liquid interface to form Pickering emulsions. (a) Temperature-responsive emulsification and demulsification of Pickering emulsion droplets stabilized by silica NPs modified by PEO-b-PPO-b-PEO. Reproduced from ref. 82 with permission from Elsevier 2022; (b) pH-induced Pickering emulsion stabilized by alumina NPs and sodium carboxylate-derived selenium surfactant. Reproduced from ref. 84 with permission from American Chemical Society 2021; (c) CO2-switchable emulsification and demulsification of Pickering emulsions. Reproduced from ref. 86 with permission from American Chemical Society 2018. | ||
Typically, pH-responsive Pickering emulsions are stimulated by either acid/base addition or CO2 bubbling. Acid–base addition strategies involve the carboxylate group either on the NP surface or within the surfactant molecules. The Cui group presented a Pickering emulsion employing positively charged alumina NPs and sodium carboxylate-derived selenium surfactants as illustrated in Fig. 4(b). The carboxylic sodium groups in the surfactant could be anionic or nonionic based on pH levels. At a high pH (around 10.29), the anionic carboxylate surfactant and alumina NPs worked together to stabilize the Pickering emulsion. However, lowering the pH led to the protonation of carboxylate surfactants, transforming them into nonionic carboxylic acid. This change reduced the electrostatic attraction between the NPs and the surfactants, consequently destabilizing the Pickering emulsions.84 Furthermore, they also used zwitterionic carboxyl betaine surfactants and negatively charged silica NPs to create pH-responsive Pickering emulsions. These emulsions remained stable only at a pH of 5 or lower, as this pH range facilitated electrostatic attraction between the negatively charged NPs and the positively charged carboxyl betaine surfactants.85
When CO2 is used to trigger pH variation, no hazardous byproducts are produced, thus reversible CO2-induced Pickering emulsions have the advantages of biocompatibility and reuse. The Fang group reported myristylamidopropyl amine oxide (C14PAO), a CO2-responsive surfactant that can switch between cation and non-ion forms reversibly, as summarized in Fig. 4(c). The highly polar head group of C14PAO allows for a transition between nonionic and cationic states in response to pH changes. Therefore, a CO2-driven increase in pH led to the conversion of C14PAO to C14PAOH+, which was effectively adsorbed onto negatively charged silica NPs through electrostatic attraction. Consequently, by CO2 bubbling, the C14PAOH+-functionalized silica NPs formed stable Pickering emulsions, while demulsification occurred when N2 bubbling removed the CO2.86 Wang and co-workers utilized negatively charged silica NPs and ionic liquids, N-alkylimidazole bicarbonates. CO2 enables the formation of carbonic acid (H2CO3), which protonates N-alkylimidazole and converts to N-alkylimidazole bicarbonates. This molecule was ionized in an aqueous solution, and the N-alkylimidazole cations were adsorbed on the surface of the silica NPs to form Pickering emulsions.87 In a similar manner, Jiang and colleagues employed surfactant ionization to enable adsorption and desorption on NPs. They utilized 11-(N,N-dimethylamino)sodium undecanoate (NCOONa), featuring both anion carboxylate and tertiary amine groups, to bind to positively charged alumina NPs through electrostatic forces, thereby stabilizing the emulsion.88 This stability was reversed by CO2 bubbling, which made the surfactant more hydrophilic, causing it to desorb from the NPs and move into the water phase, aiding in demulsification. The Cui group developed a biphasic biocatalyst system using a CO2-responsive Pickering emulsion. They loaded the enzyme lipase, N,N-dimethyldocylamine, which was converted to ammonium bicarbonate by CO2 bubbling. These cationic ammonium surfactants were adsorbable onto negatively charged silica NPs, thereby facilitating the process of Pickering emulsification. Triggered by N2 bubbling, the demulsification process effectively separated the oil and water phases: the product was isolated in the oil phase, while the enzymes and NPs remained in the aqueous phase, facilitating their subsequent separation and recycling.89
3.3. Self-assembly of NPs at polymer interfaces
In Sections 3.1 and 3.2, we explored the assembly of NPs at fluid (i.e., liquid and air) interface systems. Advancing this discussion, we now turn our attention to the crucial role of polymer interfaces in facilitating and directing these assemblies. Polymer chains, depending on their fluidic nature, serve as flexible and dynamic scaffolds for NP assembly, mirroring the behavior observed at air/liquid and liquid/liquid interfaces. This flexibility, combined with the inherent capacity to precisely control the spatial arrangement of the constituents, significantly enhances the functionality of the resulting nanocomposites, including optical, electronic, magnetic, and catalytic properties.90,91 It is noted that the integration of NPs into the polymer matrix results in entropy penalties due to the elongation and compression of the surrounding polymer chains. Consequently, there is an entropy-driven preference for NPs to localize at the interface rather than within the polymer chain matrix, a process analogous to their adsorption at both air/liquid and liquid/liquid interfaces.92 Distinct from air or liquid interfaces, polymer matrices offer an added advantage: their susceptibility to external stimuli manipulation.93 Controlling properties of air and liquid interfaces typically demands an environment where pressure and temperature are tightly regulated, a process often marred by inefficiency. By contrast, polymer matrices exhibit responsive behaviors such as swelling/deswelling in response to physicochemical stimuli or undergoing structural modifications when exposed to specific substances.94,95 Moreover, polymer interfaces afford an exceptional degree of control over the positioning of NPs. This is achieved through meticulously calibrating the size ratio between the NP and the NP–hosting polymer domain, as well as fine-tuning the interactions between NP ligands and polymer chains. In this section, we categorize polymer interfaces into two distinct types: two-dimensional polymer/polymer interfaces and three-dimensional polymer/water interfaces.| ΔG = (ΔHligand–BCP) − T(ΔScon + ΔStrans) |
In this section, we first discuss the experimental developments in crafting controlled NP arrays at the BCP interface. Specifically, we emphasize recent efforts to engineer the chemical structure of core/shell NPs to fine-tune BCP–NP interactions, thus establishing an innovative hybrid BCP/NP system through interfacial assembly of NPs at the BCP interface. We also highlight the design rules of stimuli-responsive assemblies for these hybrid BCP/NP systems, with specific attention to the surface chemistry of the NPs and their responsive interaction with the BCP domains. Furthermore, we will expand our insight into three-dimensional hybrid systems involving colloidal particles where emulsion droplets serve as flexible and dynamic templates for guiding the polymer assembly.
In the BCP-guided self-assembly of NPs, the key factors dictating their location can be classified based on surface selectivity, which pertains to enthalpic considerations, and NP size dependence, affecting entropic considerations.101,108 In particular, NPs that possess a non-selective preference for both blocks can assemble at the interface due to favorable enthalpic interactions.109,110 For instance, when PS-coated Au NPs were co-assembled with polystyrene-block-poly(2-vinyl pyridine) (PS-b-P2VP) BCPs, the NPs located either at PS/P2VP interface or selective BCP domain depending on the PS grafting density. Specifically, when the NPs were partially shielded by PS ligand (<1.3 chains per nm2), they located at the interface due to strong favorable interactions between the bare Au surface and the P2VP domain.111 Similar behavior was observed when Au NPs were coated with a mixture of PS and P2VP, with the assembly outcome dictated by the ratio between the two components.112
Recent advancements involve modifying the NP surface with ligands that strongly interact with one of the polymer blocks, such as hydrogen bonding. For example, the Hawker group synthesized Au NPs with thiol-terminated BCP ligands, forming a polyisoprene (PI) inner shell, and a PS outer shell (Fig. 5(b)).113 These core–shell Au NPs were located within the PS domain due to the hydrophobic interaction between the ligand and PS domain. In contrast, the hydroxylated core–shell Au NPs were segregated to the PS/P2VP interface due to hydrogen bonding between the hydroxylated inner shell and the P2VP. Precise control over the number of hydroxyl groups enabled fine-tuning of the segregation behavior of the Au NPs.114 Also, they investigated the interplay between core–shell Au–Pt NPs coated with the same BCP ligands and PS-b-P2VP. The Pt-shell was formed through the cross-linking of the PI brush of ligand in the presence of Pt catalyst. Prior to the cross-linking process, the NPs resided within the PS domain, primarily due to the enthalpic interaction between the outer PS brush of the ligand and the PS domain. However, following the cross-linking, NPs were located at the PS/P2VP interface due to the increased interaction between the P2VP domain and the residual Pt catalyst in the PI inner shell, particularly due to the reduced effective areal chain density of the PS brush on the PI shell compared to that on the NP surface.115
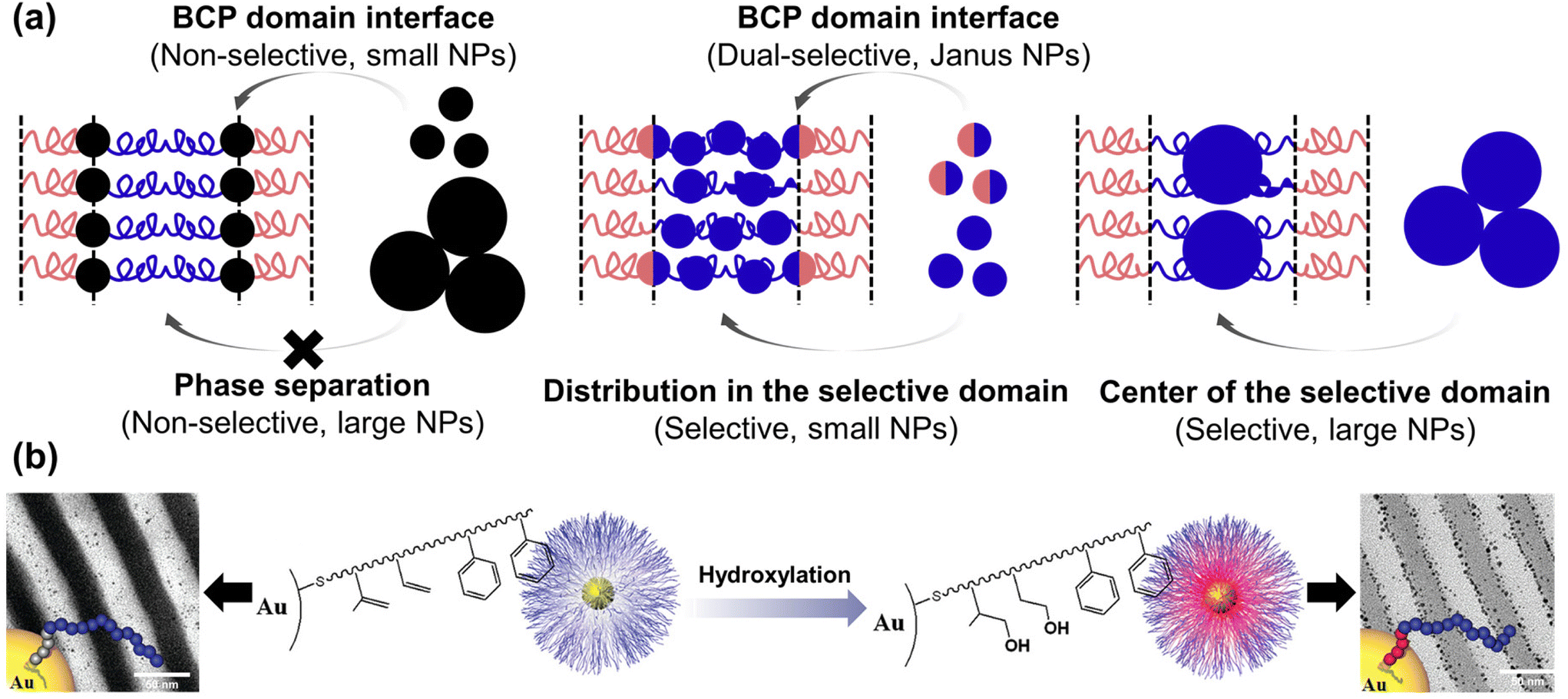 | ||
| Fig. 5 BCP-guided self-assembly of NPs at the BCP interface. (a) Schematics for the selective localization of NPs within the BCP domains or at the BCP interface. Reproduced from ref. 101 with permission from Elsevier 2014; (b) controlled supramolecular assembly of core–shell Au NPs in PS-b-P2VP by hydrogen bonding. Reproduced from ref. 113 with permission from American Chemical Society 2011. | ||
To achieve responsive transformation of NP assembly at the BCP interface, the enthalpic interactions between BCPs and NPs should be tunable in response to stimuli, either through responsive NP ligands or the polymer matrix, or both. The Watkins group employed UV-responsive ligands, poly(styrene)-block-poly(o-nitrobenzene acrylate) (PS-b-PNBA), for Au NPs to control the selectivity of the NPs towards different blocks of BCPs.116 Initially, the ligands had neutral interactions with the matrix BCP blocks, segregating the Au NPs to the PS/P2VP interface within the PS-b-P2VP film. However, exposure to UV light resulted in the photocleavage of the ester group in PNBA, converting it into hydrophilic PAA. Consequently, the NPs became uniformly distributed in the P2VP domain due to the hydrogen bonding between P2VP and PAA.
Another representative example can be found in the small molecule-directed approach, where small molecules serve as non-covalent linkers between BCPs and NP ligands. The assembly process is guided by the energetic cost arising from defects, the chain mobility, and the activation energy for inter-domain diffusion.117 Their interactions can be modulated by external stimuli, either strengthened or weakened as needed. The Xu group exploited 3-n-pentadecylphenol (PDP) as a small molecule to create a supramolecular system with polystyrene-block-poly(4-vinyl pyridine) (PS-b-P4VP).118 The hydrogen bonding between PDP and P4VP is weakened in response to temperature above 100 °C. Strong hydrogen bonding induced the selective localization of CdSe NPs within the P4VP domain at low temperatures (i.e., 50 °C). With increasing temperature to around 110 °C, PDP molecules became soluble in the PS domain, resulting in the segregation of the NPs at the PS/P4VP interface to minimize unfavorable interactions and entropic penalties. As the temperature was further increased to 150 °C, the breaking of the hydrogen bonds reduced the stiffness in the P4VP domain, decreasing the entropic driving force for the NPs to be located at the PS/P4VP interface. Consequently, a uniform distribution of NPs in the P4VP domain was achieved.
In contrast to the production of polymer films on a solid substrate, polymer composite films formed at a fluid interface offer higher mobility for both polymer chains and NPs, making it easier to achieve external field-induced assembly. The Park group reported the self-assembly of iron oxide NPs and conjugated BCPs, poly(3-hexyl thiophene)-block-poly(ethylene glycol) (P3HT-b-PEG), at the air/water interface under a magnetic field (Fig. 6(c)).119 Due to the rod-coil configuration of P3HT-b-PEG, short polymer nanowires decorated with the NPs were created through the crystallization of P3HT followed by the growth of these magnetic subunits into macroscopically aligned nanowire arrays through the magnetic interaction. The NPs preferred to be located at the interface between P3HT and PEG due to the hydrophobic oleic acid on the NP surface and the strong crystallization of P3HTs. P3HT has a strong tendency to crystallize, which results in the expulsion of NPs out of the P3HT domain. As a result, these hydrophobic NPs tend to position themselves around polymer nanowires at the interface between P3HT and PEG to avoid contact with the water subphase. Under the magnetic field, the orientation of the array was aligned with the direction of the magnetic field. The key to achieving this lies in precise control of NP size, as the NPs should be neither too large to be affected by the entropy effect nor too small to enable effective magnetic dipole interactions.
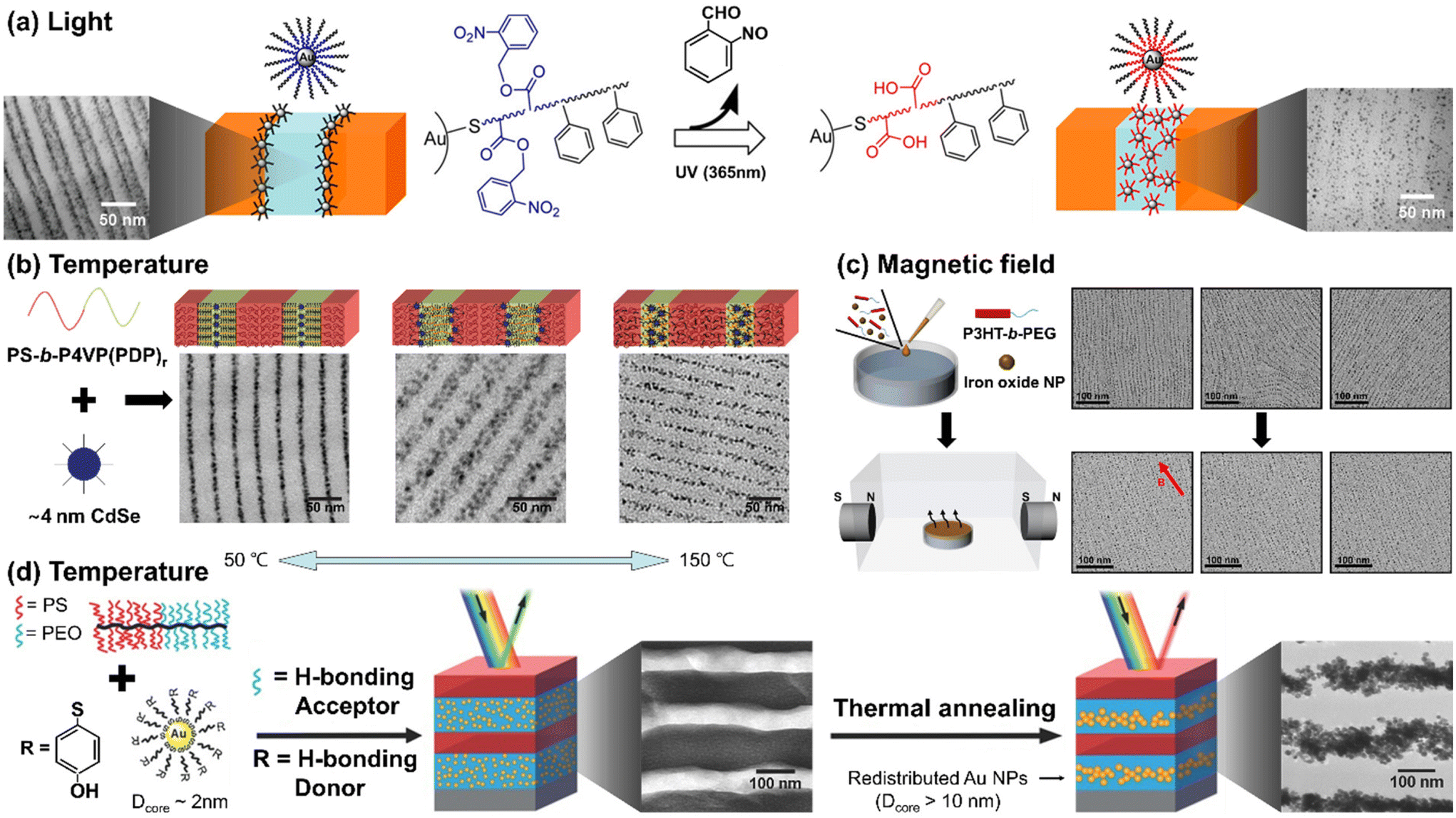 | ||
| Fig. 6 Stimuli-responsive NP arrays within BCP matrices. (a) UV-responsive localization of core–shell Au NPs within the PS-b-P2VP. Reproduced from ref. 116 with permission from American Chemical Society 2014; (b) small-molecule-directed CdSe NP assembly in response to temperature. Reproduced from ref. 118 with permission from Nature Publishing Group 2009; (c) magnetic field-induced self-assembly of P3HT-b-PEG and iron oxide NPs at the air/water interface. Reproduced from ref. 119 with permission from American Chemical Society 2022; (d) thermally tunable Au NPs assembly in brush BCPs driven by the change of NPs size due to the ripening of NPs. Reproduced from ref. 122 with permission from Wiley 2015. | ||
While distinct from the principles underlying the enthalpy contributions discussed in this paper, it is worth noting that recent reports have demonstrated changes in NP assembly driven by stimuli-induced alterations in entropy contributions. For example, the Watkins group reported temperature-responsive growth of Au NPs capped with 4-mercaptophenol ligands. The Au NPs, which can grow at high temperatures due to thermally induced ripening,120,121 were incorporated into brush block copolymers composed of poly(ethylene oxide) (PEO) and PS (Fig. 6(d)).122 Initially, 2 nm Au NPs were selectively distributed in the PEO domain through hydrogen bonds between PEO and 4-mercaptophenol ligands on the Au surface. Upon annealing at 120 °C, however, their size increased to over 10 nm, resulting in the segregation of the NPs to the center of the PEO domain to reduce the entropy penalty associated with accommodating larger NPs.
Self-assembly of NPs at the interface of the BCP microparticles and the surrounding medium was first reported by the Hawker group. As shown in Fig. 7(a), a crosslinked polymer shell was formed at the surface of Au NPs through the grafting of PS-b-PI. When these NPs were co-assembled with PS-b-P2VP during solvent evaporation, they were localized at the interface of the microparticles.134 As a result, a remarkable change in shape and morphology from sphere to ellipsoid was achieved because the Au-based NPs could adsorb at the emulsion surface and highly affect the interfacial interactions between the BCPs and the surrounding media. In particular, the entropic contribution plays an important role in such NP surfactant systems, because the NPs should possess a preference for segregating to the particle surface rather than positioning within the BCP domain. At the same time, they must selectively favor interaction with the BCPs to prevent aggregation due to strong NP–NP interaction. In this context, a similar approach was also reported by the Kim group using a series of size-controlled Au NPs or length-controlled Cu Pt nanorods for PS-b-P4VP microparticles.24,135 The small molecule linker, PDP was used to induce selective interaction of NPs with the P4VP block. Especially, the size- and length-dependent positioning of NPs in the BCP particles enabled the systematic modulation of the interfacial properties and induced the formation of anisotropic PS-b-P4VP particles such as prolate and oblate particles (Fig. 7(b)).135 The ratio of the NP length to the size of the NP–hosting domain (i.e., P4VP(PDP)) was key to determining the segregation of the NPs within the PS-b-P4VP(PDP) particles, allowing morphological transitions within critical ranges.
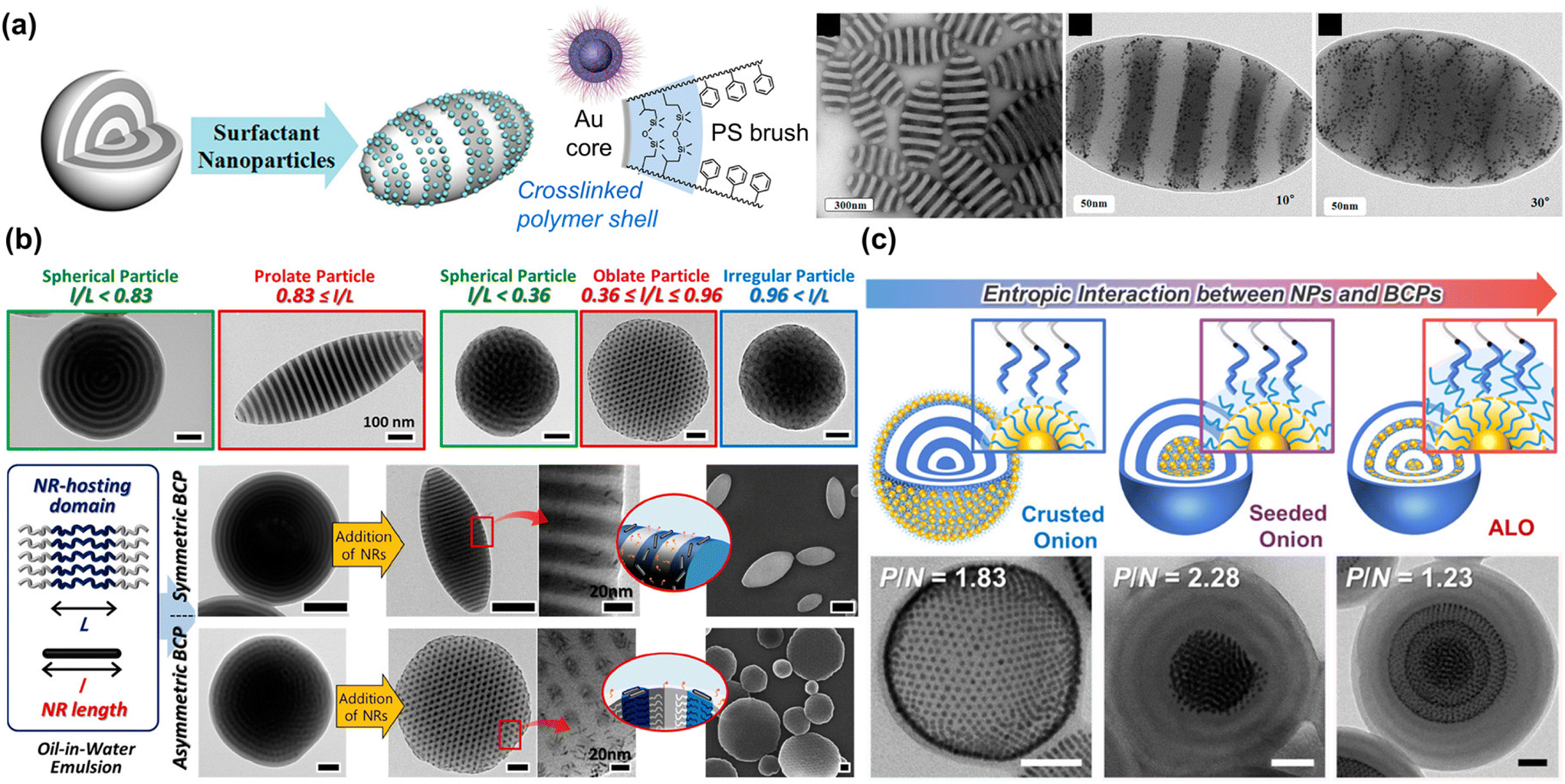 | ||
| Fig. 7 Controlled assembly of BCPs and NPs within 3D microparticles to modulate overall particle shape or NP array. (a) Production of striped ellipsoids of PS-b-P2VP by using Au NPs as surfactants. Reproduced from ref. 134 with permission from American Chemical Society 2013; (b) shape transition of PS-b-P4VP particles driven by length-controlled CuPt nanorod surfactants. Reproduced from ref. 135 with permission from American Chemical Society 2018; (c) control of entropic interaction between PS-grafted Au NPs and PS-b-P4VP to form crusted onion, seeded onion, and alternated layered onion (ALO). Reproduced from ref. 139 with permission from American Chemical Society 2021. | ||
The entropy-driven transition of particle morphologies was further investigated either by positioning surface-modified Au NPs at the PS/P4VP interface or inducing macrophase separation between polystyrene-block-poly(vinylpyridine) (PS-b-PVP) and Au NPs. Zhu and Jiang group reported Janus hybrid microparticles with a variety of shapes: ellipsoids with concentrated Au NPs at one pole, spheres with AuNPs enriched in a bulge on the surface, and more complex shapes like gourd-like, clover-like, and four-leaf-clover-like particles, which resulted from a hierarchical assembly of these Janus hybrid microparticles.136,137 In these studies, adept regulation of enthalpy and entropy was achieved through strategic chemical modification of NP surface and careful control of NP size and volume fraction, efficiently guiding spatial arrangement and interaction at BCP interfaces. More recently, the Kim group reported unique BCP/NP hybrid particles with well-ordered hexagonal NP superlattices by introducing an important parameter known as the swelling ratio (Fig. 7(c)). This ratio is defined as the number-average molecular weight (Mn) of the polymer matrix relative to the Mn of the polymer ligand on the NP surface. Upon co-assembly of symmetric PS-b-P4VP and PS-coated Au NPs, Au NPs with low Mn ligands segregated either to the surface (i.e. crusted onion) or to the center (i.e., seeded onion) of the spherical particles with radially stacked lamellae. In contrast, when Au NPs with higher Mn ligands were co-assembled with the same PS-b-P4VP, the NPs aligned at the center of the PS domains, forming alternate-layered, hexagonally packed superlattices within the spherical particles.138 These phenomena were attributed to the autophobic dewetting behavior of NPs based on their swelling ratio with BCPs. With low Mn ligands, NPs were pushed to the surface or to the center due to the large entropic penalty on BCP domains. On the other hand, longer PS ligands could strongly interact with the PS chains of the BCPs, promoting the selective localization of the NPs with high Mn ligands in the PS domains. Further elaboration by systematic control of the Au NP diameter, grafting density, and Mn of the PS ligand was conducted to correlate morphological transitions of the hybrid particles to conformational changes of the PS ligands.139
To render these hybrid microparticles responsive to stimuli, dynamic positioning of NPs at the emulsion interface should be feasible, while maintaining the hydrophilic–lipophilic balance (HLB) required to form stable oil-in-water emulsions. However, once NPs are adsorbed on the particle surface, achieving reversibility becomes a challenge. Reversibility in polymer particles requires a solvent-mediated particle reconstruction process to provide sufficient mobility for the polymer chains.140,141 Only a limited number of examples of this process have been reported to date. Several examples involve the use of pH-responsive NPs with polystyrene-block-poly(dimethyl siloxane) (PS-b-PDMS) particles as the matrix. The Zhu group utilized iron oxide (Fe3O4) NPs coated with pH-responsive PAA-b-PS as co-surfactants for emulsions to modulate the shape of PS-b-PDMS particles (Fig. 8(a)). The addition of pH-responsive Fe3O4 NPs rendered the shape of pH-inert PS-b-PDMS microparticles sensitive to pH variations. This led to a change in the particle shape, transforming from an elongated Janus pupa-like structure to a bud-like and onion-like structure when the pH of the aqueous medium was increased.142 At lower pH values, Janus pupa-like structures emerged, driven by the entropy penalty between the PS chains and NPs, resulting in NP aggregation at the end of the microparticles. With an increase in pH, the Fe3O4@PAA-b-PS NPs became amphiphilic, leading to the formation of a bud-like and an onion-like structure. Similar pH-responsive behavior was also observed in core-cross-linked PS-b-P4VP NPs.143 As an example of structural changes in BCP particles in response to external stimuli, the Kim group demonstrated light-triggered shape changes in PS-b-P2VP particles by employing azobenzene-grafted Au NPs, whose ligands undergo photoisomerization as depicted in Fig. 8(b). Since the NPs were designed to be located at the BCP–water interface, the polarity of the NPs influenced the wetting layer on the BCP particle surface. When exposed to visible light, onion-like PS-b-P2VP particles with a PS outer layer formed, attributed to the nonpolar nature of trans-Azo ligands on the NP surface. In contrast, UV-driven trans-to-cis isomerization of ligands shifted the polarity of NPs to be more hydrophilic, transforming these particles into ellipsoids with both PS and P2VP exposed on their surfaces.144
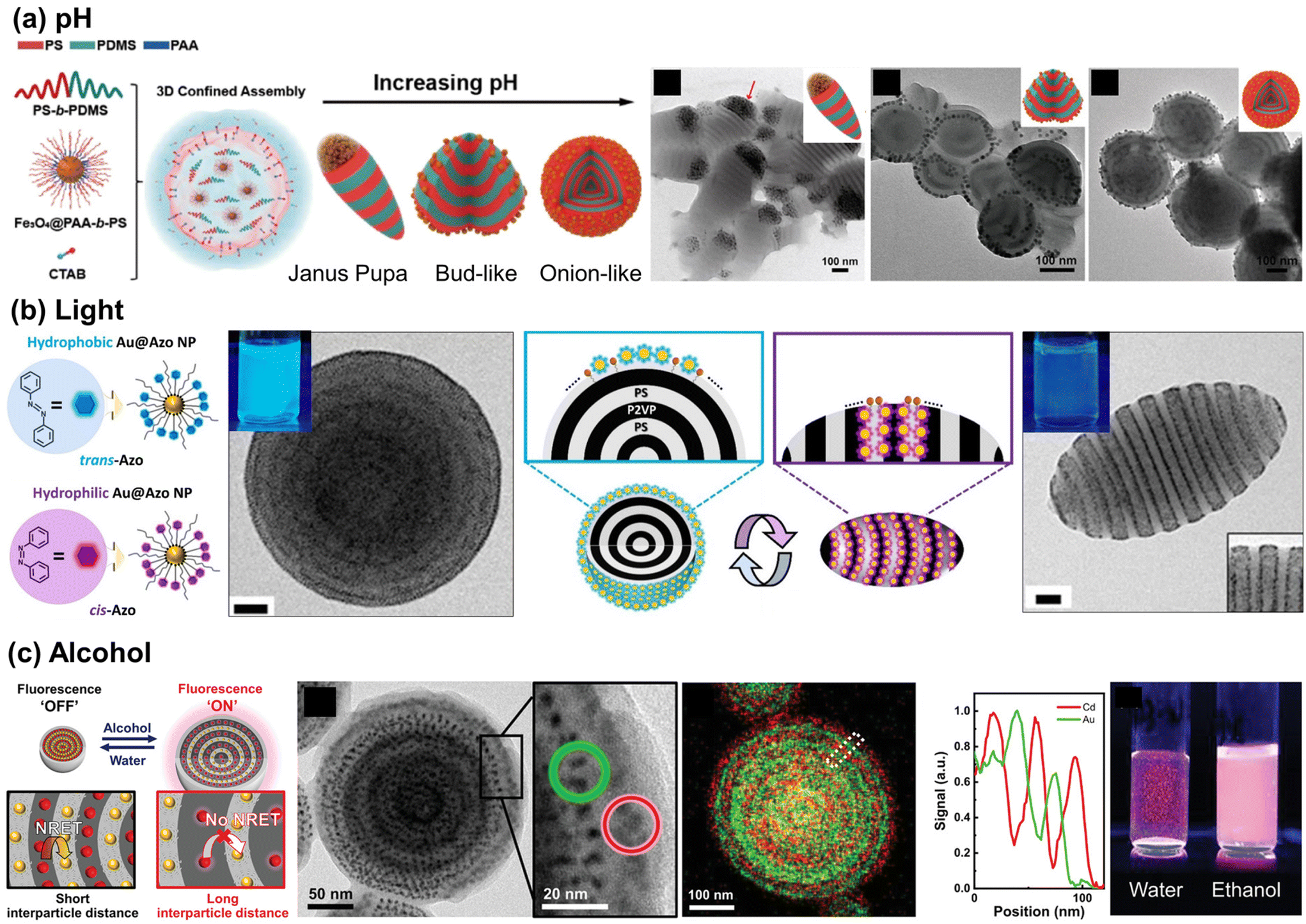 | ||
| Fig. 8 Stimuli-responsive NP arrays within the polymer matrix. (a) pH-Responsive assembly of Fe3O4 NPs coated with PAA-b-PS within PS-b-PDMS particles. Reproduced from ref. 142 with permission from American Chemical Society 2021; (b) photo-switchable Au NP surfactants and their responsive assembly within BCP particles. Reproduced from ref. 144 with permission from American Chemical Society 2021; (c) solvent-responsive distance between QDs and Au NPs in doubly alternate-layered NP array and corresponding fluorescence switching behavior. Reproduced from ref. 145 with permission from Wiley 2021. | ||
In addition to the examples of NPs acting as surfactants, another intriguing discovery involves utilizing a responsive BCP domain to exert precise control over the intermolecular NP distance in a reversible manner. This was achieved through the co-assembly of PS-grafted Au NPs (as quenchers) and CdSe/ZnS core/shell quantum dots (QDs, as emitters) within symmetric PS-b-P4VP particles. This process led to the creation of hybrid BCP particles with doubly alternating Au NP and QD layers due to the enthalpically favorable interaction with PS and P4VP domains, respectively, as depicted in Fig. 8(c).145 When these hybrid particles were dispersed in water, the proximity of the Au NPs to the QD arrays resulted in quenching of the fluorescence of the QDs, due to strong nonradiative energy transfer from the QDs to Au NPs. Conversely, upon the addition of ethanol, the interparticle distance significantly increased due to the swelling of P4VP domains, effectively inhibiting nonradiative energy transfer and thereby preserving the fluorescence of the particles. These examples highlight how structural changes in BCP particles, responsive to external stimuli, can be utilized to induce their functional property changes. This opens up significant possibilities for developing responsive smart colloidal materials.
4. Perspectives and conclusions
In this review, we have discussed the physical principles and recent strategic advances in the development of stimuli-responsive assembly of NPs at complex fluid interfaces. Current progress in creating responsive NP arrays at air/liquid, liquid/liquid, and polymer interfaces has primarily focused on experimental parameters to control dynamic packing structures and NP interactions. The key driving force to obtain a self-assembled NP array is the reduction in interfacial energy. On the nanoscale, particles face a balance between thermal fluctuations and interfacial energy, rendering self-assembly size dependent. In designing a responsive NP assembly system, the ability to control the NP–NP interactions, NP–hosting fluid interaction, and their entropic contribution in response to stimuli is crucial. In the air/liquid interface systems, two primary strategies have been employed to address the entropy penalties associated with ordered arrays: (1) light- and temperature-induced structural transformations of functional groups, and (2) adjusting pH to control electrostatic repulsion. In the case of liquid/liquid interface systems, the surface charge of the NP ligands for electrostatic repulsion control can be tailored to respond to external stimuli such as pH, light, electric field, and heat. In the polymer/NP hybrid system, achieving reversible interfacial assembly relies on controlling the interaction between the NP and polymer interface, balancing between neutral selectivity and selective preference in response to stimuli. Novel approaches have emerged, including the introduction of small molecule linkers, grafting a mixture shell onto the NP core, and utilizing a controlled three-phase interaction (i.e., water, polymer, and NP) for three-dimensional assembly.Despite the numerous efforts to achieve stimuli-responsive NP assembly at complex fluid interfaces, several challenges should be addressed. First, NP assembly at the air/liquid interface often involves volatile liquids or surface tension gradients induced by solvent evaporation. To leverage external stimuli as a driving force, resolving issues like particle aggregation and the presence of voids in the array, as well as the generation of sufficient alteration in interfacial properties, requires a strategic design of inorganic core/organic shell structures. In addition, when employing polymers as NP ligands, the fluidity of the polymer chains limits the NP mobility, emphasizing the necessity for a polymer matrix design that allows faster NP movement. While a well-established thermodynamic theory addresses NP assemblies responsive to stimuli, a kinetic understanding is lacking, especially regarding the rate at which NPs assemble. Lastly, most of the research concerning NPs at the interface of two immiscible liquids has spotlighted dynamic transformations between stable Pickering emulsions and inverse structures. In addition, studies on the NPs at the air/liquid interface have focused on dynamic behaviors between ordered and disordered arrays. However, the design of responsive NP assembly across multiple interfaces remains unexplored. For example, starting at the liquid/liquid interface by forming a Pickering emulsion, it is conceivable to induce the migration of the NPs in the Pickering emulsion to the air/liquid interface with close-packed structures. Such multiple-phase transition becomes feasible with robust stimuli that detach the NPs from the liquid/liquid interface, ensuring efficient adsorption kinetics at the air/liquid interface. In the case of BCP systems, achieving responsive behavior has proven challenging, primarily due to difficulties in ensuring polymer chain mobility in the absence of a solvent. As a result, only a limited number of examples of polymer-mediated stimuli-responsive NP assembly have been developed to date. One potential avenue for achieving dynamic NP arrays in the absence of a solvent is the use of low Tg polymer or leveraging pH-driven swelling/deswelling behavior in a polymer matrix. Photoacid, which can convert the light response into the chemical property, also can be utilized in the design of core/shell NP ligands, following a similar principle. From a broader materials science perspective, dynamic systems and networks comprising NPs and functional surface ligands represent an excellent starting point for designing and realizing dynamic assemblies that would perform programmed tasks such as sensing and spatiotemporal catalysis.
Author contributions
The manuscript was written using contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This research was supported by the Korea Research Foundation Grant, funded by the Korean Government (2022R1C1C1006324).References
- A. Böker, J. He, T. Emrick and T. P. Russell, Soft Matter, 2007, 3, 1231–1248 RSC.
- S. Kinge, M. Crego-Calama and D. N. Reinhoudt, ChemPhysChem, 2008, 9, 20–42 CrossRef CAS PubMed.
- L. Duan, C. Wang, W. Zhang, B. Ma, Y. Deng, W. Li and D. Zhao, Chem. Rev., 2021, 121, 14349–14429 CrossRef CAS PubMed.
- M. Grzelczak, J. Vermant, E. M. Furst and L. M. Liz-Marzán, ACS Nano, 2010, 4, 3591–3605 CrossRef CAS PubMed.
- M. D. Scanlon, E. Smirnov, T. J. Stockmann and P. Peljo, Chem. Rev., 2018, 118, 3722–3751 CrossRef CAS PubMed.
- X. Li, L. Chen, Y. Ma, D. Weng, Z. Li, L. Song, X. Zhang, G. Yu and J. Wang, Adv. Funct. Mater., 2022, 32, 2205462 CrossRef CAS.
- Y. Lin, A. Böker, H. Skaff, D. Cookson, A. Dinsmore, T. Emrick and T. P. Russell, Langmuir, 2005, 21, 191–194 CrossRef CAS PubMed.
- Z. Niu, J. He, T. P. Russell and Q. Wang, Angew. Chem., Int. Ed., 2010, 49, 10052–10066 CrossRef CAS PubMed.
- D. Liu, W. Cai, M. Marin, Y. Yin and Y. Li, ChemNanoMat, 2019, 5, 1338–1360 CrossRef CAS.
- J. Forth, P. Y. Kim, G. Xie, X. Liu, B. A. Helms and T. P. Russell, Adv. Mater., 2019, 31, 1806370 CrossRef PubMed.
- M. Z. Iqbal, I. Ali, W. S. Khan, X. Kong and E. Dempsey, Mater. Des., 2021, 205, 109694 CrossRef.
- M. Grzelczak, L. M. Liz-Marzán and R. Klajn, Chem. Soc. Rev., 2019, 48, 1342–1361 RSC.
- W. Lee, D. Kim, S. Lee, J. Park, S. Oh, G. Kim, J. Lim and J. Kim, Nano Today, 2018, 23, 97–123 CrossRef CAS.
- S. Hou, L. Bai, D. Lu and H. Duan, Acc. Chem. Res., 2023, 56, 740–751 CrossRef CAS PubMed.
- S. Shi and T. P. Russell, Adv. Mater., 2018, 30, 1800714 CrossRef PubMed.
- S. K. Ghosh and A. Böker, Macromol. Chem. Phys., 2019, 220, 1900196 CrossRef.
- J. Owen, Science, 2015, 347, 615–616 CrossRef CAS PubMed.
- J. J. Giner-Casares and J. Reguera, Nanoscale, 2016, 8, 16589–16595 RSC.
- A. J. Zarbin, Mater. Horiz., 2021, 8, 1409–1432 RSC.
- R. Dong, T. Zhang and X. Feng, Chem. Rev., 2018, 118, 6189–6235 CrossRef CAS PubMed.
- F. Bresme and M. Oettel, J. Phys.: Condens. Matter, 2007, 19, 413101 CrossRef CAS PubMed.
- L. Hu, M. Chen, X. Fang and L. Wu, Chem. Soc. Rev., 2012, 41, 1350–1362 RSC.
- B. P. Binks, Curr. Opin. Colloid Interface Sci., 2002, 7, 21–41 CrossRef CAS.
- K. H. Ku, J. M. Shin, M. P. Kim, C.-H. Lee, M.-K. Seo, G.-R. Yi, S. G. Jang and B. J. Kim, J. Am. Chem. Soc., 2014, 136, 9982–9989 CrossRef CAS PubMed.
- K. H. Ku, J. M. Shin, H. Yun, G. R. Yi, S. G. Jang and B. J. Kim, Adv. Funct. Mater., 2018, 28, 1802961 CrossRef.
- L. Xu, G. Han, J. Hu, Y. He, J. Pan, Y. Li and J. Xiang, Phys. Chem. Chem. Phys., 2009, 11, 6490–6497 RSC.
- O. S. Deshmukh, D. van den Ende, M. C. Stuart, F. Mugele and M. H. Duits, Adv. Colloid Interface Sci., 2015, 222, 215–227 CrossRef CAS PubMed.
- J. Vialetto and M. Anyfantakis, Langmuir, 2021, 37, 9302–9335 CrossRef CAS PubMed.
- V. Sashuk, K. Winkler, A. Zywocinski, T. Wojciechowski, E. Gorecka and M. Fiałkowski, ACS Nano, 2013, 7, 8833–8839 CrossRef CAS PubMed.
- V. Garbin, J. C. Crocker and K. J. Stebe, J. Colloid Interface Sci., 2012, 387, 1–11 CrossRef CAS PubMed.
- X. Lu, Y. Huang, B. Liu, L. Zhang, L. Song, J. Zhang, A. Zhang and T. Chen, Chem. Mater., 2018, 30, 1989–1997 CrossRef CAS.
- R. Zbonikowski, M. Iwan and J. Paczesny, ACS Omega, 2023, 8, 23706–23719 CrossRef CAS PubMed.
- M. Zhang, Z. Hou, H. Wang, L. Zhang, J. Xu and J. Zhu, Langmuir, 2020, 37, 454–460 CrossRef PubMed.
- J. K. Stolarczyk, A. Deak and D. F. Brougham, Adv. Mater., 2016, 28, 5400–5424 CrossRef CAS PubMed.
- G. Zhu, Z. Huang, Z. Xu and L.-T. Yan, Acc. Chem. Res., 2018, 51, 900–909 CrossRef CAS PubMed.
- C. Huang, X. Chen, Z. Xue and T. Wang, Acc. Chem. Res., 2020, 54, 35–45 CrossRef PubMed.
- S. J. Kim, J. Choi, K. Maleski, K. Hantanasirisakul, H.-T. Jung, Y. Gogotsi and C. W. Ahn, ACS Appl. Mater. Interfaces, 2019, 11, 32320–32327 CrossRef CAS PubMed.
- S. Kutuzov, J. He, R. Tangirala, T. Emrick, T. Russell and A. Böker, Phys. Chem. Chem. Phys., 2007, 9, 6351–6358 RSC.
- D. A. Walker, B. Kowalczyk, M. O. de La Cruz and B. A. Grzybowski, Nanoscale, 2011, 3, 1316–1344 RSC.
- J. N. Israelachvili, Contemp. Phys., 1974, 15, 159–178 CrossRef CAS.
- M. Liu, M. Yang, X. Wan, Z. Tang, L. Jiang and S. Wang, Adv. Mater., 2023, 35, 2208995 CrossRef CAS PubMed.
- D. Wang, B. Kowalczyk, I. Lagzi and B. A. Grzybowski, J. Phys. Chem., 2010, 1, 1459–1462 CAS.
- X. Li, X. Liu and X. Liu, Chem. Soc. Rev., 2021, 50, 2074–2101 RSC.
- G. Pasparakis, T. Manouras, P. Argitis and M. Vamvakaki, Macromol. Rapid Commun., 2012, 33, 183–198 CrossRef CAS PubMed.
- X. Yao, T. Li, J. Wang, X. Ma and H. Tian, Adv. Opt. Mater., 2016, 4, 1322–1349 CrossRef CAS.
- R. Klajn, K. J. Bishop, M. Fialkowski, M. Paszewski, C. J. Campbell, T. P. Gray and B. A. Grzybowski, Science, 2007, 316, 261–264 CrossRef CAS PubMed.
- H. Zhang and D. Wang, Angew. Chem., 2008, 120, 4048–4051 CrossRef.
- R. Klajn, K. J. Bishop and B. A. Grzybowski, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10305–10309 CrossRef CAS PubMed.
- E. Blasco, M. Wegener and C. Barner-Kowollik, Adv. Mater., 2017, 29, 1604005 CrossRef PubMed.
- J. Zhang, P. J. Santos, P. A. Gabrys, S. Lee, C. Liu and R. J. Macfarlane, J. Am. Chem. Soc., 2016, 138, 16228–16231 CrossRef CAS PubMed.
- W. Blokzijl and J. B. Engberts, Angew. Chem., Int. Ed. Engl., 1993, 32, 1545–1579 CrossRef.
- Y. Sahoo, M. Cheon, S. Wang, H. Luo, E. Furlani and P. Prasad, J. Phys. Chem. B, 2004, 108, 3380–3383 CrossRef CAS.
- D. Liu, C. Li, F. Zhou, T. Zhang, G. Liu, W. Cai and Y. Li, Adv. Mater. Interfaces, 2017, 4, 1600976 CrossRef.
- T. Zhao, P. Xie, H. Wan, T. Ding, M. Liu, J. Xie, E. Li, X. Chen, T. Wang and Q. Zhang, Nat. Photonics, 2023, 17, 622–628 CrossRef CAS.
- B. Kim, S. L. Tripp and A. Wei, J. Am. Chem. Soc., 2001, 123, 7955–7956 CrossRef CAS PubMed.
- N. J. Brewer, R. E. Rawsterne, S. Kothari and G. J. Leggett, J. Am. Chem. Soc., 2001, 123, 4089–4090 CrossRef CAS PubMed.
- T. Nishimura, N. Ito, K. Kinoshita, M. Matsukawa, Y. Imura and T. Kawai, Small, 2020, 16, 1903365 CrossRef CAS PubMed.
- F. Nakagawa, R. Kichijo, M. Matsukawa, K.-H. Wang, Y. Imura and T. Kawai, Colloids Surf., A, 2023, 656, 130499 CrossRef CAS.
- X. Liu, M. Atwater, J. Wang, Q. Dai, J. Zou, J. P. Brennan and Q. Huo, J. Nanosci. Nanotechnol., 2007, 7, 3126–3133 CrossRef CAS PubMed.
- F. Zheng, Y. Zhang, L. Dong, D. Zhao, R. Feng, P. Tao, W. Shang, B. Fu, C. Song and T. Deng, Nanoscale, 2021, 13, 20521–20530 RSC.
- S. Roy, R. K. Kashyap and P. P. Pillai, J. Phys. Chem. C, 2023, 127, 10355–10365 CrossRef CAS.
- H. Zhang, S. Nayak, W. Wang, S. Mallapragada and D. Vaknin, Langmuir, 2017, 33, 12227–12234 CrossRef CAS PubMed.
- F. Reincke, S. G. Hickey, W. K. Kegel and D. Vanmaekelbergh, Angew. Chem., Int. Ed., 2004, 43, 458–462 CrossRef CAS PubMed.
- B. Liu, X. Lu, Z. Qiao, L. Song, Q. Cheng, J. Zhang, A. Zhang, Y. Huang and T. Chen, Langmuir, 2018, 34, 13047–13056 CrossRef CAS PubMed.
- K. H. Ku, J. Li, K. Yoshinaga and T. M. Swager, Adv. Mater., 2019, 31, 1905569 CrossRef CAS PubMed.
- A. Dinsmore, M. F. Hsu, M. Nikolaides, M. Marquez, A. Bausch and D. Weitz, Science, 2002, 298, 1006–1009 CrossRef CAS PubMed.
- J. Wu and G. H. Ma, Small, 2016, 12, 4633–4648 CrossRef CAS PubMed.
- H. Jiang, Y. Sheng and T. Ngai, Curr. Opin. Colloid Interface Sci., 2020, 49, 1–15 CrossRef CAS PubMed.
- R. Aveyard, B. P. Binks and J. H. Clint, Adv. Colloid Interface Sci., 2003, 100, 503–546 CrossRef.
- H. Sun, L. Li, T. P. Russell and S. Shi, J. Am. Chem. Soc., 2020, 142, 8591–8595 CrossRef CAS PubMed.
- H. Sun, M. Li, L. Li, T. Liu, Y. Luo, T. P. Russell and S. Shi, J. Am. Chem. Soc., 2021, 143, 3719–3722 CrossRef CAS PubMed.
- K. Watanabe, T. A. Welling, S. Sadighikia, H. Ishii, A. Imhof, M. A. van Huis, A. van Blaaderen and D. Nagao, J. Colloid Interface Sci., 2020, 566, 202–210 CrossRef CAS PubMed.
- M. K. Bera, H. Chan, D. F. Moyano, H. Yu, S. Tatur, D. Amoanu, W. Bu, V. M. Rotello, M. Meron and P. Král, Nano Lett., 2014, 14, 6816–6822 CrossRef CAS PubMed.
- Y. Montelongo, D. Sikdar, Y. Ma, A. J. McIntosh, L. Velleman, A. R. Kucernak, J. B. Edel and A. A. Kornyshev, Nat. Mater., 2017, 16, 1127–1135 CrossRef CAS PubMed.
- M. N. Martin, J. I. Basham, P. Chando and S.-K. Eah, Langmuir, 2010, 26, 7410–7417 CrossRef CAS PubMed.
- M. J. Junk, W. Li, A. D. Schlüter, G. Wegner, H. W. Spiess, A. Zhang and D. Hinderberger, J. Am. Chem. Soc., 2011, 133, 10832–10838 CrossRef CAS PubMed.
- H. Tian, H. Li and Y. Fang, ACS Appl. Mater. Interfaces, 2019, 11, 16207–16213 CrossRef CAS PubMed.
- L. Song, B. B. Xu, Q. Cheng, X. Wang, X. Luo, X. Chen, T. Chen and Y. Huang, Sci. Adv., 2021, 7, eabk2852 CrossRef CAS PubMed.
- Y.-K. Park and S. Park, Chem. Mater., 2008, 20, 2388–2393 CrossRef CAS.
- Z. Li, Y. Shi, A. Zhu, Y. Zhao, H. Wang, B. P. Binks and J. Wang, Angew. Chem., 2021, 133, 3974–3979 CrossRef.
- S. Acter, M. L. P. Vidallon, J. P. King, B. M. Teo and R. F. Tabor, J. Mater. Chem. B, 2021, 9, 8962–8970 RSC.
- L. Zhang, G. Zhang, J. Ge, P. Jiang and L. Ding, J. Colloid Interface Sci., 2022, 616, 129–140 CrossRef CAS PubMed.
- S. Björkegren, M. C. A. Freixiela Dias, K. Lundahl, L. Nordstierna and A. Palmqvist, Langmuir, 2020, 36, 2357–2367 CrossRef PubMed.
- S. Yu, M. Lv, G. Lu, C. Cai, J. Jiang and Z. Cui, Langmuir, 2021, 37, 10683–10691 CrossRef CAS PubMed.
- K. Liu, J. Jiang, Z. Cui and B. P. Binks, Langmuir, 2017, 33, 2296–2305 CrossRef CAS PubMed.
- Y. Zhang, X. Ren, S. Guo, X. Liu and Y. Fang, ACS Sustainable Chem. Eng., 2018, 6, 2641–2650 CrossRef CAS.
- Y. Shi, D. Xiong, Y. Chen, H. Wang and J. Wang, J. Mol. Liq., 2019, 274, 239–245 CrossRef CAS.
- H. Li, Y. Liu and J. Jiang, Green Chem., 2022, 24, 8062–8068 RSC.
- S. Yu, D. Zhang, J. Jiang, Z. Cui, W. Xia, B. P. Binks and H. Yang, Green Chem., 2019, 21, 4062–4068 RSC.
- A. Alvarez-Fernandez, C. Cummins, M. Saba, U. Steiner, G. Fleury, V. Ponsinet and S. Guldin, Adv. Opt. Mater., 2021, 9, 2100175 CrossRef CAS.
- A. C. Balazs, T. Emrick and T. P. Russell, Science, 2006, 314, 1107–1110 CrossRef CAS PubMed.
- C. Yi, Y. Yang, B. Liu, J. He and Z. Nie, Chem. Soc. Rev., 2020, 49, 465–508 RSC.
- L. Zhai, Chem. Soc. Rev., 2013, 42, 7148–7160 RSC.
- C. Xu, B. B. Wayland, M. Fryd, K. I. Winey and R. J. Composto, Macromolecules, 2006, 39, 6063–6070 CrossRef CAS.
- I. Tokarev and S. Minko, Adv. Mater., 2009, 21, 241–247 CrossRef CAS.
- J. Wang, W. Li and J. Zhu, Polymer, 2014, 55, 1079–1096 CrossRef CAS.
- J. Kao, K. Thorkelsson, P. Bai, B. J. Rancatore and T. Xu, Chem. Soc. Rev., 2013, 42, 2654–2678 RSC.
- W. A. Lopes and H. M. Jaeger, Nature, 2001, 414, 735–738 CrossRef CAS PubMed.
- J. Huang, A. Hall, I. Jayapurna, S. Algharbi, V. Ginzburg and T. Xu, Macromolecules, 2021, 54, 1006–1016 CrossRef CAS.
- K. H. Ku, H. Yang, S. G. Jang, J. Bang and B. J. Kim, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 228–237 CrossRef CAS.
- B. Sarkar and P. Alexandridis, Prog. Polym. Sci., 2015, 40, 33–62 CrossRef CAS.
- Y. Gai, Y. Lin, D.-P. Song, B. M. Yavitt and J. J. Watkins, Macromolecules, 2016, 49, 3352–3360 CrossRef CAS.
- S.-W. Hsu and T. Xu, Macromolecules, 2019, 52, 2833–2842 CrossRef CAS.
- M. Konefał, P. Černoch, V. Patsula, E. Pavlova, J. Dybal, K. Załęski and A. Zhigunov, ACS Appl. Mater. Interfaces, 2021, 13, 9195–9205 CrossRef PubMed.
- D.-P. Song, C. Li, W. Li and J. J. Watkins, ACS Nano, 2016, 10, 1216–1223 CrossRef CAS PubMed.
- D.-P. Song, S. Shahin, W. Xie, S. Mehravar, X. Liu, C. Li, R. A. Norwood, J.-H. Lee and J. J. Watkins, Macromolecules, 2016, 49, 5068–5075 CrossRef CAS.
- L. Yao, Y. Lin and J. J. Watkins, Macromolecules, 2014, 47, 1844–1849 CrossRef CAS.
- J. Kao, P. Bai, J. M. Lucas, A. P. Alivisatos and T. Xu, J. Am. Chem. Soc., 2013, 135, 1680–1683 CrossRef CAS PubMed.
- K. C. Bryson, T. I. Löbling, A. H. E. Müller, T. P. Russell and R. C. Hayward, Macromolecules, 2015, 48, 4220–4227 CrossRef CAS.
- S. Kim, M. Yoo, N. Kang, B. Moon, B. J. Kim, S.-H. Choi, J. U. Kim and J. Bang, ACS Appl. Mater. Interfaces, 2013, 5, 5659–5666 CrossRef CAS PubMed.
- B. J. Kim, J. Bang, C. J. Hawker, J. J. Chiu, D. J. Pine, S. G. Jang, S.-M. Yang and E. J. Kramer, Langmuir, 2007, 23, 12693–12703 CrossRef CAS PubMed.
- B. J. Kim, J. Bang, C. J. Hawker and E. J. Kramer, Macromolecules, 2006, 39, 4108–4114 CrossRef CAS.
- S. G. Jang, E. J. Kramer and C. J. Hawker, J. Am. Chem. Soc., 2011, 133, 16986–16996 CrossRef CAS PubMed.
- S. G. Jang, A. Khan, M. D. Dimitriou, B. J. Kim, N. A. Lynd, E. J. Kramer and C. J. Hawker, Soft Matter, 2011, 7, 6255–6263 RSC.
- S. G. Jang, B. J. Kim, C. J. Hawker and E. J. Kramer, Macromolecules, 2011, 44, 9366–9373 CrossRef CAS.
- D.-P. Song, X. Wang, Y. Lin and J. J. Watkins, J. Phys. Chem. B, 2014, 118, 12788–12795 CrossRef CAS PubMed.
- J. Kao, K. Thorkelsson, P. Bai, Z. Zhang, C. Sun and T. Xu, Nat. Commun., 2014, 5, 4053 CrossRef CAS PubMed.
- Y. Zhao, K. Thorkelsson, A. J. Mastroianni, T. Schilling, J. M. Luther, B. J. Rancatore, K. Matsunaga, H. Jinnai, Y. Wu, D. Poulsen, J. M. J. Fréchet, A. Paul Alivisatos and T. Xu, Nat. Mater., 2009, 8, 979–985 CrossRef CAS PubMed.
- M. Park, S. Kang, C. Nam, K. Narasimha, W. B. Lee and S.-J. Park, ACS Appl. Mater. Interfaces, 2022, 14, 8266–8273 CrossRef CAS PubMed.
- N. G. Bastús, J. Comenge and V. Puntes, Langmuir, 2011, 27, 11098–11105 CrossRef PubMed.
- T. Shimizu, T. Teranishi, S. Hasegawa and M. Miyake, J. Phys. Chem. B, 2003, 107, 2719–2724 CrossRef CAS.
- D.-P. Song, C. Li, N. S. Colella, X. Lu, J.-H. Lee and J. J. Watkins, Adv. Opt. Mater., 2015, 3, 1169–1175 CrossRef CAS.
- D. Hu, Y. Wang, J. Liu, Y. Mao, X. Chang and Y. Zhu, Nanoscale, 2022, 14, 6291–6298 RSC.
- S.-J. Jeon, S.-M. Yang, B. J. Kim, J. D. Petrie, S. G. Jang, E. J. Kramer, D. J. Pine and G.-R. Yi, Chem. Mater., 2009, 21, 3739–3741 CrossRef CAS.
- K. H. Ku, J. M. Shin, H. Yun, G.-R. Yi, S. G. Jang and B. J. Kim, Adv. Funct. Mater., 2018, 28, 1802961 CrossRef.
- J. M. Shin, Y. Kim, H. Yun, G.-R. Yi and B. J. Kim, ACS Nano, 2017, 11, 2133–2142 CrossRef CAS PubMed.
- C. K. Wong, X. Qiang, A. H. E. Müller and A. H. Gröschel, Prog. Polym. Sci., 2020, 102, 101211 CrossRef CAS.
- N. Yan, Y. Zhu and W. Jiang, Chem. Commun., 2018, 54, 13183–13195 RSC.
- J. Kim, H. Yun, Y. J. Lee, J. Lee, S.-H. Kim, K. H. Ku and B. J. Kim, J. Am. Chem. Soc., 2021, 143, 13333–13341 CrossRef CAS PubMed.
- K. H. Ku, M. P. Kim, K. Paek, J. M. Shin, S. Chung, S. G. Jang, W.-S. Chae, G.-R. Yi and B. J. Kim, Small, 2013, 9, 2667–2672 CrossRef CAS PubMed.
- K. H. Ku, J. M. Shin, D. Klinger, S. G. Jang, R. C. Hayward, C. J. Hawker and B. J. Kim, ACS Nano, 2016, 10, 5243–5251 CrossRef CAS PubMed.
- D. Klinger, C. X. Wang, L. A. Connal, D. J. Audus, S. G. Jang, S. Kraemer, K. L. Killops, G. H. Fredrickson, E. J. Kramer and C. J. Hawker, Angew. Chem., Int. Ed., 2014, 53, 7018–7022 CrossRef CAS PubMed.
- K. H. Ku, Y. J. Lee, Y. Kim and B. J. Kim, Macromolecules, 2019, 52, 1150–1157 CrossRef CAS.
- S. G. Jang, D. J. Audus, D. Klinger, D. V. Krogstad, B. J. Kim, A. Cameron, S.-W. Kim, K. T. Delaney, S.-M. Hur, K. L. Killops, G. H. Fredrickson, E. J. Kramer and C. J. Hawker, J. Am. Chem. Soc., 2013, 135, 6649–6657 CrossRef CAS PubMed.
- K. H. Ku, J. H. Ryu, J. Kim, H. Yun, C. Nam, J. M. Shin, Y. Kim, S. G. Jang, W. B. Lee and B. J. Kim, Chem. Mater., 2018, 30, 8669–8678 CrossRef CAS.
- N. Yan, H. Liu, Y. Zhu, W. Jiang and Z. Dong, Macromolecules, 2015, 48, 5980–5987 CrossRef CAS.
- N. Yan, Y. Zhang, Y. He, Y. Zhu and W. Jiang, Macromolecules, 2017, 50, 6771–6778 CrossRef CAS.
- M. Xu, K. H. Ku, Y. J. Lee, J. J. Shin, E. J. Kim, S. G. Jang, H. Yun and B. J. Kim, Chem. Mater., 2020, 32, 7036–7043 CrossRef CAS.
- M. Xu, K. H. Ku, Y. J. Lee, T. Kim, J. J. Shin, E. J. Kim, S.-H. Choi, H. Yun and B. J. Kim, Macromolecules, 2021, 54, 3084–3092 CrossRef CAS.
- J. M. Shin, Y. J. Lee, M. Kim, K. H. Ku, J. Lee, Y. Kim, H. Yun, K. Liao, C. J. Hawker and B. J. Kim, Chem. Mater., 2019, 31, 1066–1074 CrossRef CAS.
- R. Deng, F. Liang, W. Li, Z. Yang and J. Zhu, Macromolecules, 2013, 46, 7012–7017 CrossRef CAS.
- Z. Hou, M. Ren, K. Wang, Y. Yang, J. Xu and J. Zhu, Macromolecules, 2020, 53, 473–481 CrossRef CAS.
- M. Zhang, Z. Hou, H. Wang, L. Zhang, J. Xu and J. Zhu, Langmuir, 2021, 37, 454–460 CrossRef CAS PubMed.
- S. H. Kwon, M. Xu, J. Kim, E. J. Kim, Y. J. Lee, S. G. Jang, H. Yun and B. J. Kim, Chem. Mater., 2021, 33, 9769–9779 CrossRef CAS.
- T. Kim, M. Xu, Y. J. Lee, K. H. Ku, D. J. Shin, D. C. Lee, S. G. Jang, H. Yun and B. J. Kim, Small, 2021, 17, 2101222 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |