
Tumor microenvironment-responsive degradable silica nanoparticles: design principles and precision theranostic applications
Junjie
Zhang†
 *ab,
Kaiyuan
Tang†
a,
Zilu
Liu
a,
Zhijing
Zhang
a,
Shufan
Duan
a,
Hui
Wang
a,
Hui
Yang
a,
Dongliang
Yang
*c and
Wenpei
Fan
*d
*ab,
Kaiyuan
Tang†
a,
Zilu
Liu
a,
Zhijing
Zhang
a,
Shufan
Duan
a,
Hui
Wang
a,
Hui
Yang
a,
Dongliang
Yang
*c and
Wenpei
Fan
*d
aSchool of Fundamental Sciences, Bengbu Medical College, Bengbu 233030, P. R. China. E-mail: zhangjj@bbmc.edu.cn
bState Key Laboratory of Organic Electronics and Information Displays & Institute of Advanced Materials (IAM), Nanjing University of Posts & Telecommunications, 9 Wenyuan Road, Nanjing 210023, P. R. China
cNanjing Polytechnic Institute, Nanjing 210048, P. R. China. E-mail: yangdl1023@njtech.edu.cn
dState Key Laboratory of Natural Medicines and Jiangsu Key Laboratory of Drug Discovery for Metabolic Diseases, Center of Advanced Pharmaceuticals and Biomaterials, China Pharmaceutical University, Nanjing 211198, P. R. China. E-mail: wenpei.fan@cpu.edu.cn
First published on 29th November 2023
Abstract
Silica nanoparticles have emerged as promising candidates in the field of nanomedicine due to their remarkable versatility and customizable properties. However, concerns about their potential toxicity in healthy tissues and organs have hindered their widespread clinical translation. To address this challenge, significant attention has been directed toward a specific subset of silica nanoparticles, namely degradable silica nanoparticles, primarily because of their excellent biocompatibility and responsive biodegradability. In this review, we provide a comprehensive understanding of degradable silica nanoparticles, categorizing them into two distinct groups: inorganic species-doped and organic moiety-doped silica nanoparticles based on their framework components. Next, the recent progress of tumor microenvironment (TME)-responsive degradable silica nanoparticles for precision theranostic applications is summarized in detail. Finally, current bottlenecks and future opportunities of theranostic nanomedicines based on degradable silica nanoparticles in clinical applications are also outlined and discussed. The aim of this comprehensive review is to shed light on the potential of degradable silica nanoparticles in addressing current challenges in nanomedicine, offering insights into their design, applications in tumor diagnosis and treatment, and paving the way for future advancements in clinical theranostic nanomedicines.
 Junjie Zhang | Junjie Zhang received her PhD degree from Nanjing University of Posts and Telecommunications under the supervision of Professor Lianhui Wang in 2020. Currently, she is an assistant professor at the School of Fundamental Sciences, Bengbu Medical College. Her research interest focuses on the design and synthesis of multifunctional nanotheranostics for multimodal imaging guided synergistic therapy. |
 Kaiyuan Tang | Kaiyuan Tang joined Bengbu Medical College in 2020 to pursue a bachelor's degree in Medical Imaging. In 2021, he joined Dr Zhang Junjie's research group, focusing on the design and synthesis of multifunctional nanotherapeutics for multimodal imaging-guided collaborative therapies. |
 Dongliang Yang | Dongliang Yang received his PhD degree from the Nanjing University of Posts and Telecommunications. He is currently an associate professor at the School of Physical and Mathematical Sciences, Nanjing Tech University. His research is primarily focused on nanomedicine, particularly the development of light-functional materials for the treatment of drug-resistant bacterial infections. |
 Wenpei Fan | Prof. Wenpei Fan received his PhD in 2015 from the Shanghai Institute of Ceramics, Chinese Academy of Sciences. During 2015–2019, he worked at the National Institutes of Health (USA) as a postdoctoral fellow. Since November 2019, he has been a full professor at the Center of Advanced Pharmaceuticals and Biomaterials, China Pharmaceutical University. He has published over 80 papers in reputed journals. His research interests focus on the design and synthesis of multifunctional theranostic nanomedicines for drug delivery, multimodal imaging, and precision synergistic therapy. |
1. Introduction
Cancer is a major global cause of death, posing a serious threat to public health and safety.1 It is characterized by abnormal cell proliferation and tumor formation resulting from gene mutations, ultimately leading to secondary metastases. Unlike healthy cells, which maintain a dynamic equilibrium in the surrounding tissue environment through cell proliferation, differentiation, and apoptosis, tumor cells undergo rapid and uncontrolled proliferation, giving rise to a malignant tumor microenvironment (TME).2–6 The TME is a dynamic microenvironment comprising both tumor cells and nontumor cells, including blood vessels, stroma, endothelial cells, and immune cells. It exhibits distinctive features, such as dynamic hypoxia, high concentrations of redox components, elevated intracellular adenosine triphosphate (ATP) and lactate levels, and extracellular acidosis.6–13 Additionally, certain types of tumors possess specific microenvironments. Vascular tumors are characterized by vascular leakage, chaotic branching of blood vessels, uneven blood flow distribution, and high stromal fluid pressure, while stromal tumors present prominent features of abnormal tumor-associated fibroblasts and a dense extracellular matrix.14,15 Unrestrained growth and dissemination of cancer cells lead to their remarkable heterogeneity and resistance, allowing them to evade immune surveillance, rendering conventional cancer treatment approaches (e.g., surgery, chemotherapy (CMT), radiotherapy (RT), etc.) relatively ineffective and often accompanied by severe side effects.16–18 Consequently, there is an urgent need to develop precise cancer-specific treatment methods that not only eradicate tumors but also minimize toxic side effects.In recent years, nanotechnology has garnered growing interest in the field of cancer diagnosis and treatment, leading to a heightened focus on the utilization of nanomaterials in the field of nanomedicine. Nanomedicine utilizes nanomaterials to accumulate in tumor tissues through the enhanced permeability and retention (EPR) effect, leading to enhanced treatment efficacy.19,20 Moreover, certain organic or inorganic nanomaterials, such as silica, polymers, proteins, and metal oxides, are designed to load drugs, proteins, enzymes, RNA, and other molecules, thereby enhancing the utilization and effectiveness of therapeutics.21–23 Although using nanomaterials to deliver small-molecule therapeutic drugs can significantly reduce their interactions with normal tissues and thus decrease toxic side effects, there remains a risk of drug leakage, challenging the safety of the treatment. The proposal of stimuli-responsive therapy has enabled the design of nanomaterials to specifically respond to the TME, leading to various beneficial conformational changes, such as carrier decomposition, ligand exposure, and component disassembly.5,24,25 This unique treatment mechanism leverages nanomaterials to protect drugs, thereby avoiding nonspecific interactions with healthy tissues and achieving excellent diagnostic and therapeutic effects.
The practical clinical applications of nanomaterials are limited by their potential toxicity within the body and their slow biological clearance rate.26 Research has shown that many types of nanomaterials are nondegradable under biological conditions but can be safely excreted through renal glomerular filtration due to their small size below the typical 5.5 nm renal clearance threshold.27–29 Nevertheless, nanomaterials with sizes exceeding 10 nm may be captured and absorbed by the reticuloendothelial system (RES), raising concerns about potential long-term toxicity.30,31 Additionally, most inorganic nanoparticles are nonbiodegradable or have long degradation periods, and some degradation byproducts from heavy metal oxides, such as mercury ions, can cause significant harm to the human body.32,33 The degradation of organic nanoparticles is influenced by factors such as organic components, hydrophobicity, and molecular weight, potentially posing hazards to the body.26 As a result, researchers are focusing on nanomaterials with rapid degradability and biological safety.
Silicon, an abundant element on Earth and a rich trace element in the human body, plays a crucial role in the body's metabolic processes. Silicon substances ingested by the body can circulate in blood plasma, being absorbed in the form of silicic acid and excreted through urine, thus providing robust assurance for human health safety.34,35 As a result, silicon-based nanomaterials have received widespread attention in research. Among these materials, silica nanoparticles, as representative silicon-based nanomaterials, serve as versatile carriers for a wide range of disease diagnosis and treatment applications due to their stable surface silanol groups that facilitate numerous surface chemical reactions.36,37 Silica nanoparticles exhibit various configurations, including nonporous, mesoporous, hollow, and core–shell structures, with abundant surface silanes, significantly aiding drug loading and the construction of TME-responsive systems.38,39 Among these, the degradation ability of silica nanoparticles is of particular attention. This is due to the tightly packed Si–O–Si structure within silica nanoparticles, which leads to slow degradation within the biological system and may result in their accumulation, causing tissue and organ damage and even systemic toxicity.40–43 In recent years, research findings have indicated that silica nanoparticles can be combined with inorganic or organic components, further optimizing their tumor therapeutic and biodegradable capabilities and significantly advancing the development of TME-responsive degradable materials.26,44–46 The outstanding degradability of silica nanoparticles not only enables efficient clearance and low toxicity but also allows precise control of drug release, thereby enhancing therapeutic efficacy.47 These degradable silica nanoparticles can be tailored in terms of particle size and morphology, exhibit excellent biocompatibility, and demonstrate high cellular uptake rates, making them highly attractive for tumor imaging and treatment. Incorporating imaging agents or drugs into degradable silica nanoparticles or surface modification of nanocarriers facilitates the design of TME-responsive nanoprobes, capitalizing on the distinctions between the tumor and normal tissues for precise tumor diagnosis and drug release.48 Furthermore, soft organosilica nanoparticles not only extend the blood circulation time but also facilitate effective drug accumulation within tumor cells.49 Additionally, appropriate surface functionalization prevents interactions between nanocomposites and blood components or the endothelial system, thereby increasing drug utilization.50
Over the last decade, significant advancements have been achieved in the progressive development and fine-tuning of degradable silica nanoparticles meticulously designed for therapeutic and imaging applications, with a specific focus on targeting the intricate and dynamic TME. However, there is still no comprehensive review centered explicitly on TME-responsive precision theranostic applications rooted in the utilization of degradable silica nanoparticles. In this paper, an exhaustive exploration encompassing the synthesis and meticulous classification of degradable silica nanoparticles is undertaken. A meticulous examination of a gamut of TME-specific responses that have been unveiled in recent years is carried out. Simultaneously, sagacious analysis is conducted, delving into the inherent potential opportunities and multifaceted challenges that underlie the strategic harnessing of degradable silica nanoparticles, thereby fostering precision-centric diagnostics and therapeutics within the practical realms of clinical applications (Fig. 1).
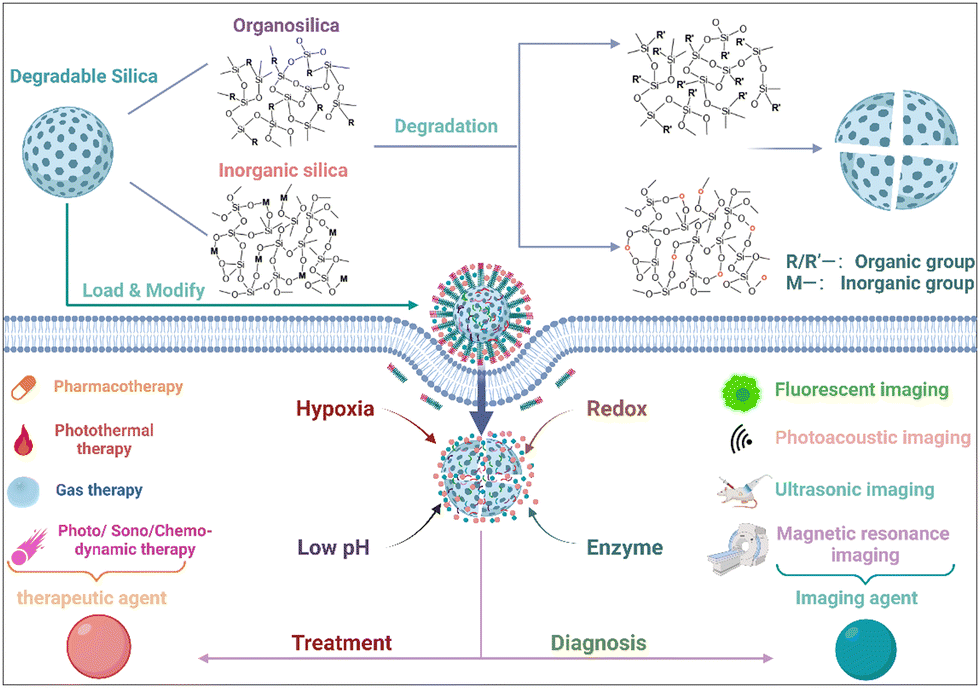 | ||
| Fig. 1 Schematic illustration of TME-responsive precision theranostic applications that are based on the utilization of degradable silica nanoparticles. Copyright from biorender. | ||
2. Classification of degradable silica nanoparticles
Silica nanoparticles have garnered extensive attention and application in biomedical research due to their large surface area and adjustable physical/chemical properties. Nevertheless, the slow degradation and extended degradation period of silica raise concerns about potential long-term and latent pathogenic effects in physiological systems.51,52 Therefore, it is imperative to regulate the degradation of silica nanoparticles to ensure their safe application in living organisms. Studies have reported that silica nanoparticles rapidly dissolve into silicic acid through continuous hydration, hydrolysis, and ion exchange processes, with the dissolution rate dependent on the degradation medium and system saturation.53,54 Thus, reducing the accumulation of silicic acid degradation products can effectively accelerate the degradation of silica nanoparticles in physiological settings.Developing degradable silica nanoparticles that rapidly degrade in the body is a promising approach, enhancing their safety and efficiency for biomedical applications. When inside the body, degradable silica nanoparticles respond to the biological environment, including fluid temperature, pH, and enzyme activity, catalyzing the degradation rate and breaking the material down into small molecular compounds that are eventually absorbed or excreted through metabolic pathways.26,55,56 In this review, we categorize degradable silica based on its framework components into inorganic or organic silica nanoparticles and extensively detail their synthesis methods and degradation behaviours.
2.1. Inorganic species-doped silica nanoparticles
The hydrolysis of silica nanoparticles is a multifaceted process influenced by a plethora of factors, including size, morphology, pore size, and surface functionalization, as well as reaction temperature, concentration, pH, and pressure, all of which collectively contribute to the intricacies of this reaction.26,57–59 Under ideal conditions, the fundamental chemistry of silica nanoparticle hydrolysis in an aqueous environment can be succinctly described as the hydration of silica nanoparticles through interaction with adsorbed water molecules, resulting in the creation of multiple silanol groups. These silanol groups subsequently engage in ion exchange reactions, liberating silicic acid, while the remaining silanol groups persist in their interactions with water, facilitating the ongoing degradation process. As this process unfolds, the strength of silicon–oxygen bonds within the silicon-based nanomaterial gradually diminishes, ultimately leading to the structural collapse of the material.57,60 Consequently, the degradation of the silica framework can be controlled by adjusting the content of degradable organic groups or modifying the strength of the silica framework through a synthetic process.46,61 Introducing metal ions into the framework of silica at the molecular level can alter its inherent physicochemical properties. In physiological environments, metals can interact with specific substances, prompting detachment from the silica framework and resulting in a substantial number of defects in the material structure, which enhances the opportunities for water molecules to react with other silicon atoms, thereby accelerating the aforementioned degradation.62,63 Metals are typically integrated into the silicate network through two distinct mechanisms. In one approach, they establish a direct linkage with silicon hydroxyl groups via Si–O–M bonds, yielding compounds with uniform characteristics. An exemplary illustration of this phenomenon is observed in the diverse array of silicate minerals. The alternative mode of integration entails their incorporation within the network as elements, salts, oxide clusters, or nanoparticles. A wealth of scholarly studies underscores that the introduction of metal ions such as calcium, iron, and manganese not only affords the ability to regulate material stability but also furnishes catalytic and coordination sites that accelerate the hydrolysis of the silica network.63,64 This infusion of supplementary attributes and functionalities enhances the inherent potential of the pristine siloxane matrix, rendering it pertinent across a broad spectrum of domains, including medical imaging and disease treatment (Fig. 2).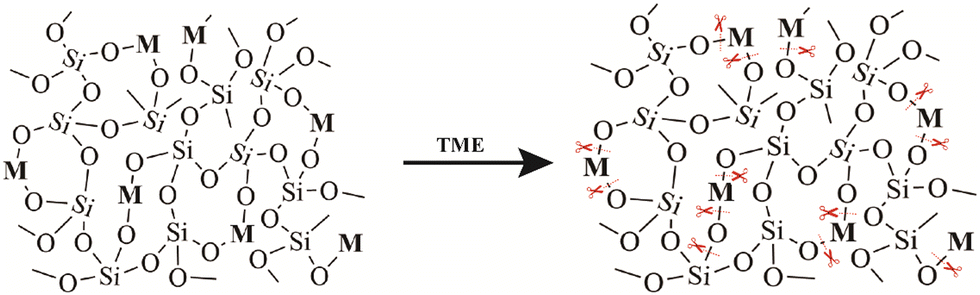 | ||
| Fig. 2 Diagram illustrating the degradation mechanism of the metal ion-doped silica nanoparticles. | ||
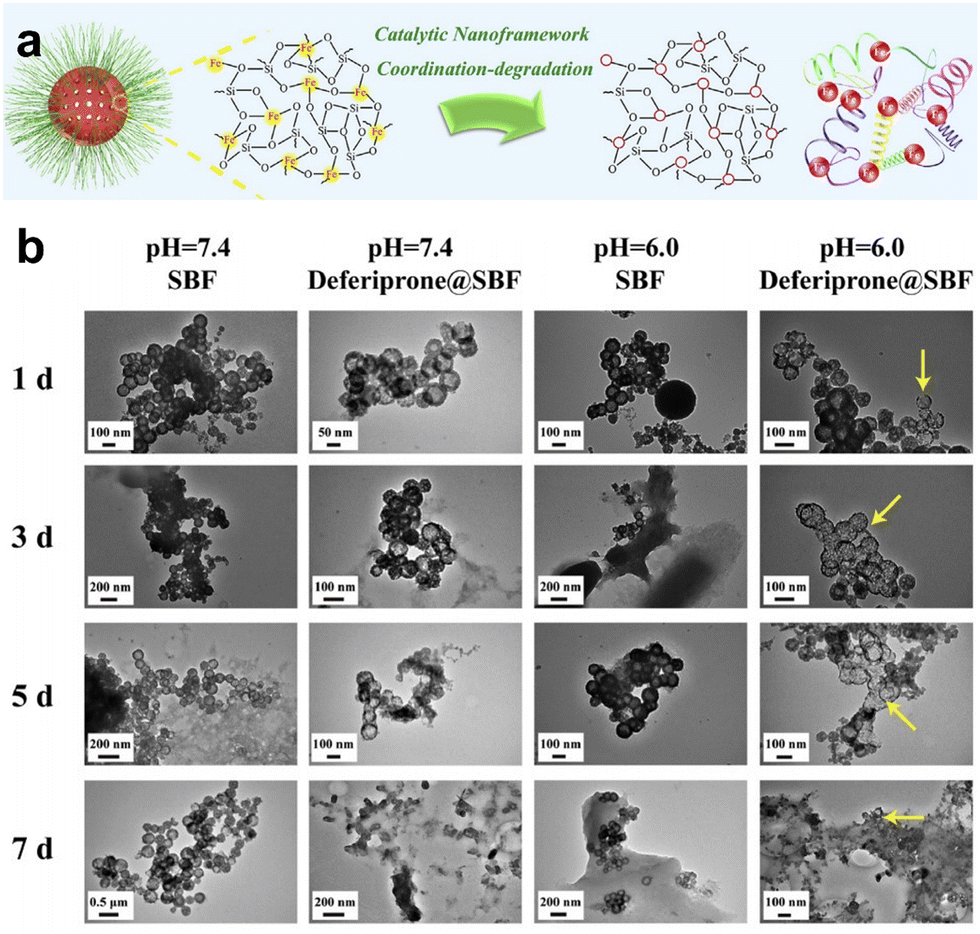 | ||
| Fig. 3 (a) Diagram illustrating the degradation mechanism of the rFeOx-HMSN nanocatalyst. (b) TEM images visually capturing the progressive degradation of rFeOx-HMSNs across diverse simulated body fluid environments. Reproduced from ref. 44 with permission from Elsevier, copyright 2018. | ||
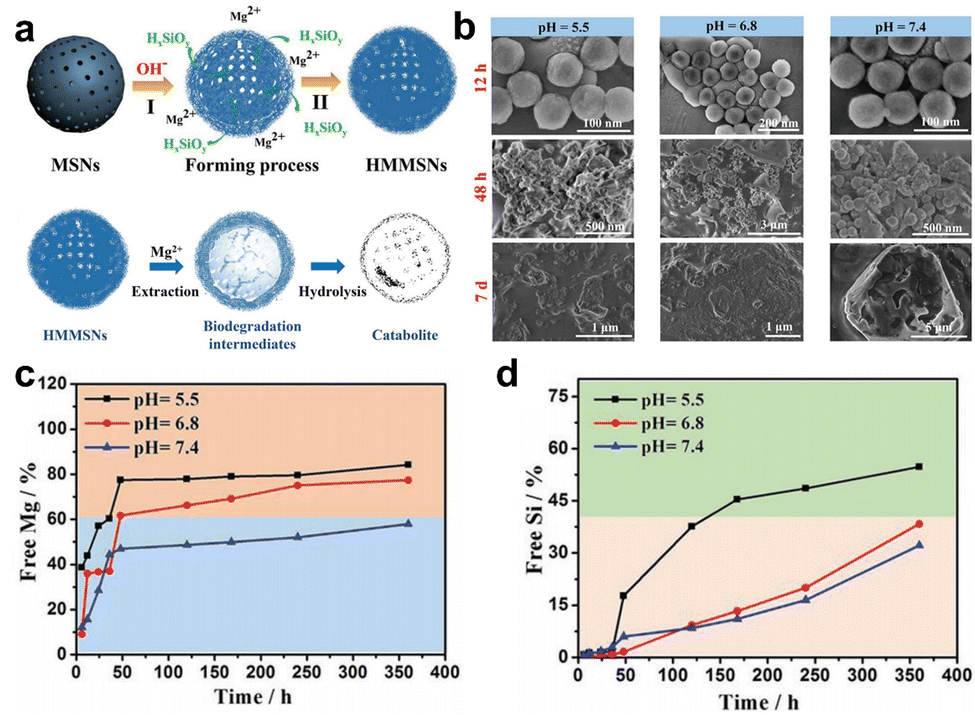 | ||
| Fig. 4 (a) Schematic diagram of the synthesis and degradation of HMMSNs. (b) SEM was utilized for the characterization of the in vitro degradation process of HMMSNs. These images depict HMMSNs following their biodegradation in SBF solutions at three distinct pH levels. The content of (c) Mg and (d) Si components in SBF following exposure to different pH levels. Reproduced from ref. 65 with permission from Wiley, copyright 2018. | ||
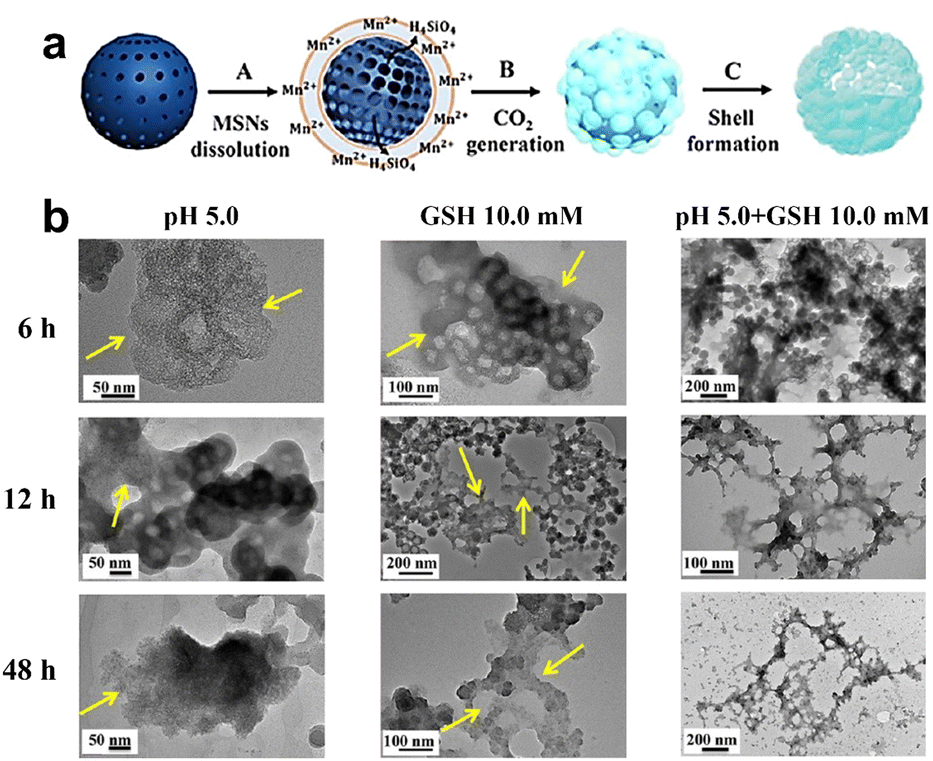 | ||
| Fig. 5 (a) Schematic representation depicting the synthesis process of Mn-HMSNs. (b) Degradation of Mn-HMSNs influenced by pH and GSH concentrations. Reproduced from ref. 66 with permission from ACS, copyright 2016. | ||
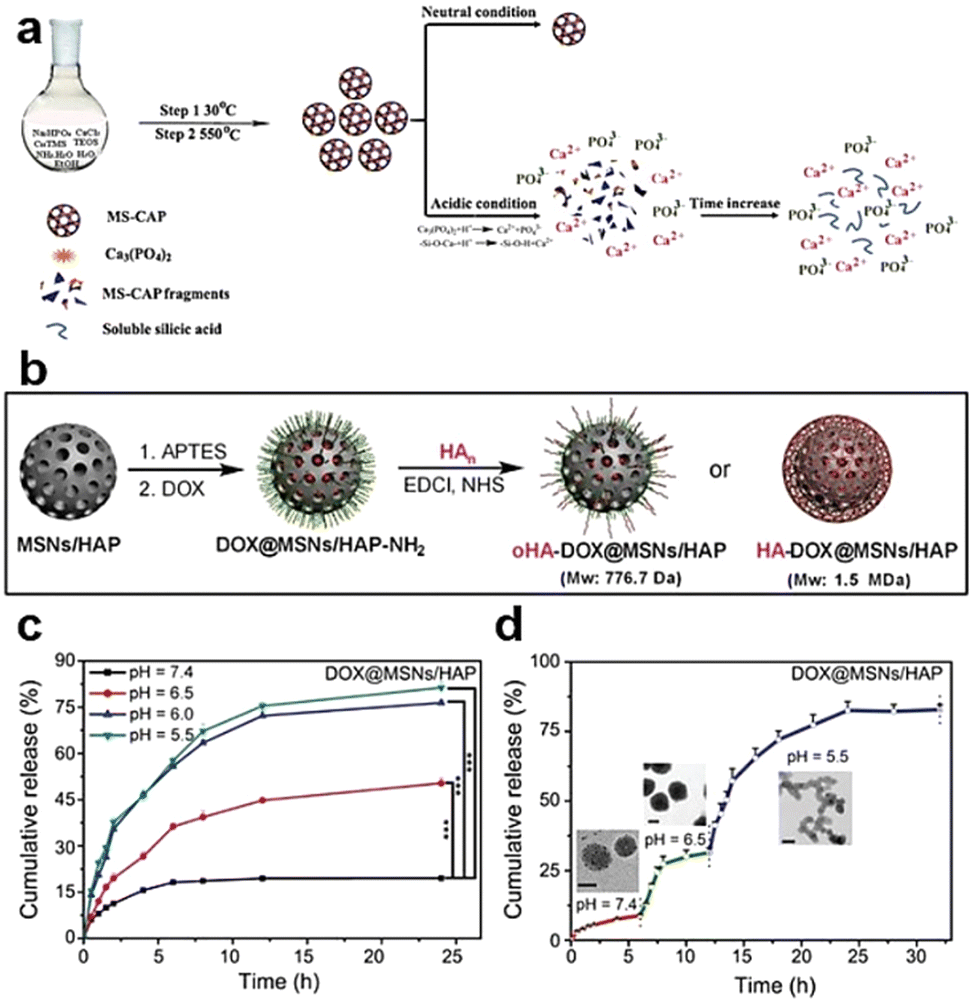 | ||
| Fig. 6 (a) Schematic depiction of the synthesis and degradation process for MS-CAP. Reproduced from ref. 68 with permission from ACS, copyright 2017. (b) Schematic representation of the assembly of MSN/HAP and HA-DOX@MSN/HAP nanoplatforms. (c) Release profiles of DOX from DOX@MSNs/HAP in PBS buffer at varying pH values. (d) DOX release profiles from DOX@MSNs/HAP at different pH values, accompanied by TEM images captured after 4, 10, and 24 h of incubation. Reproduced from ref. 69 with permission from Wiley, copyright 2019. | ||
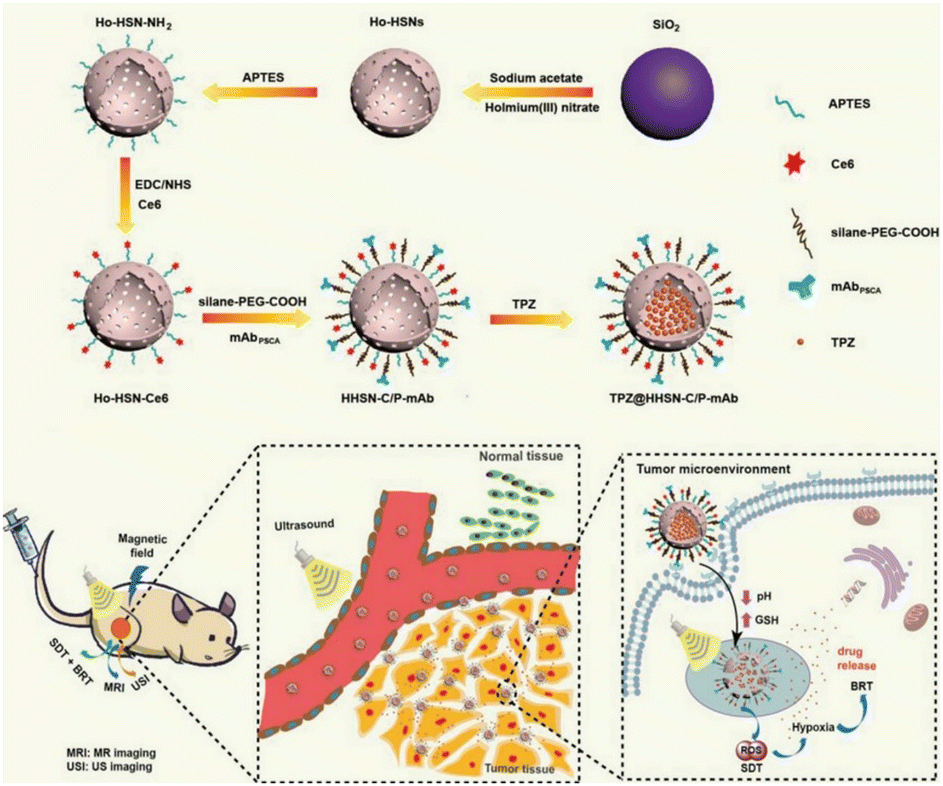 | ||
| Fig. 7 Biological applications of the degradable Ho-HSN nanoplatform. Reproduced from ref. 70 with permission from Wiley, copyright 2018. | ||
2.2. Organic moiety-doped silica nanoparticles
The synthesis of monodisperse silica nanoparticles involves silica precursors such as TEOS and tetramethyl orthosilicate (TMOS), whereas organosilica nanoparticles require additional organosilica precursors during preparation.71 Organosilica nanoparticles can be classified into two types: periodically mesoporous organosilica composed entirely of organosilica precursors and organic–inorganic hybrid mesoporous silica nanoparticles comprising partly organic groups.62,72,73 Both types of organosilica nanoparticles utilize structure-directing agents, such as CTAB and CTAC, during the condensation process of bis-silane units (R′O)3Si–R–Si(OR′)3. Herein, R′O typically represents methoxy or ethoxy, while R denotes organic functional linkages between silicon atoms. The robustness of the silicon–carbon bonds (Si–C) and the hydrophobic interactions stemming from organic moieties collectively contribute to the exceptional stability exhibited by the passive alkane-bridged silica network within aqueous surroundings.26 The introduction of organic bridges could substantially bolster the stability of silica nanoparticles within aqueous phases. However, it concurrently imparts susceptibility to degradation when these silica nanoparticles are subjected to organic solvents. As a result, regulating and controlling the incorporation of organic components into silica nanoparticles allows for degradable organosilica with specific functions. The main methods for achieving this are the incorporation of easily degradable organic covalent groups or cleavable organosiloxanes.26,62,74 Different types of organic bridges (R) containing easily hydrolysed groups and reactive functional groups can be introduced into mesoporous organosilica nanoparticles to significantly affect their physicochemical properties and thus enhance their degradability.71,75,76 The integration of organic moieties, including S–S bonds, selenium–selenium (Se–Se) bonds, and amide bonds, within the framework of silica nanoparticles has paved the way for a highly controlled biodegradation pathway that can adapt to the specific conditions present in the tumor microenvironment. Influenced by specific enzymes, pH levels, and the unique redox environments inherent to tumor tissues, these bonds can rapidly undergo cleavage, leading to the release of therapeutic agents or imaging agents (Fig. 8).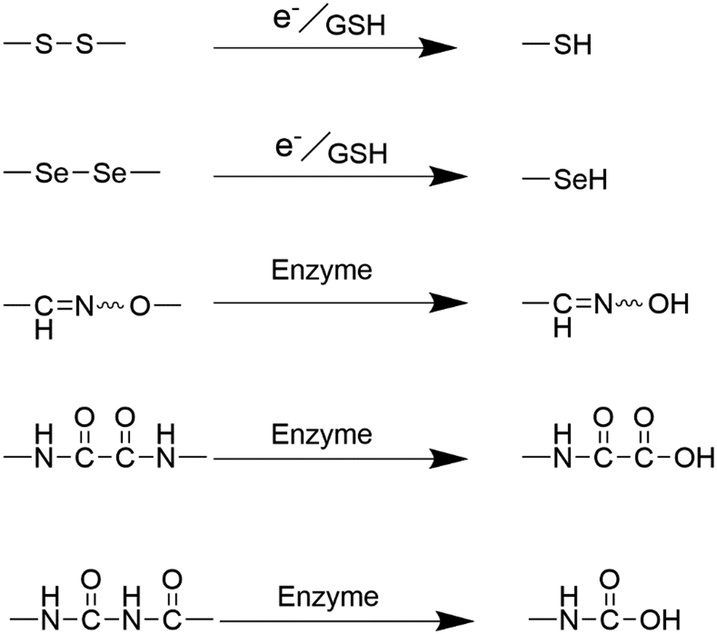 | ||
| Fig. 8 Diagram illustrating the degradation mechanism of the organic moiety-doped silica nanoparticles. | ||
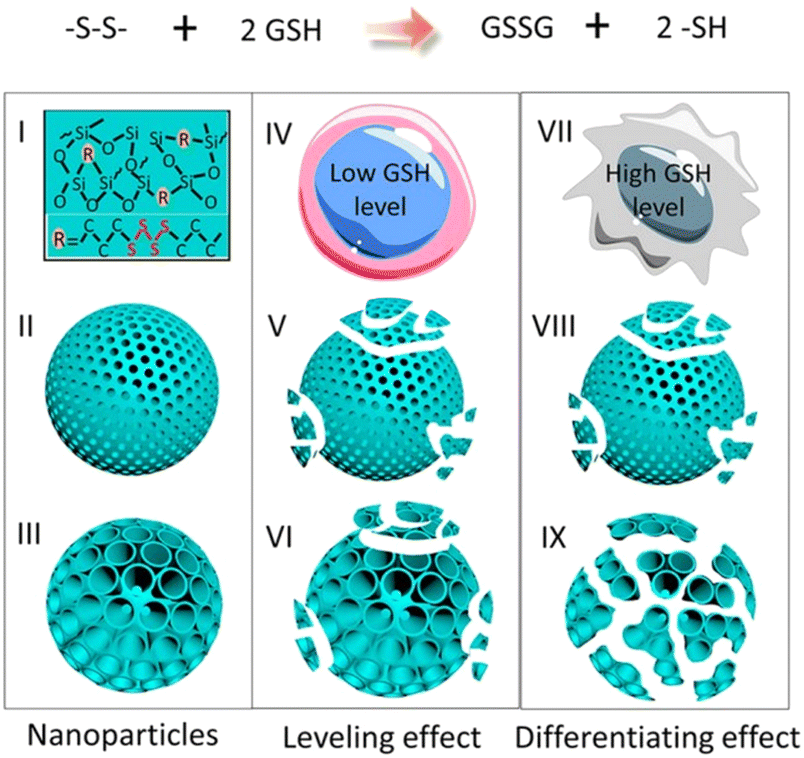
Dendritic mesoporous organosilica nanoparticles exhibit superior degradation performance compared to other silica nanoparticles due to their unique dendritic structure and pore design. The incorporation of disulfide or tetrasulfide bonds into these dendritic mesoporous organosilica nanoparticles enables the specific cleavage of sulfur–thiol bonds under specific conditions, facilitating nanoparticle degradation and enhancing their potential in drug delivery and other biological applications. Yang et al. reported the development of cancer cell-specific degradable dendritic mesoporous organosilica nanoparticles (DDMONs) (Fig. 9).78 The nanoparticles possess well-distributed disulfide bonds within their controllable pores, enabling responsive degradation upon exposure to GSH. Serum incubation of the DDMONs demonstrated a partial structural collapse after 48 h in the presence of 1 mM GSH solution, while the spherical morphology remained largely unchanged. However, an elevated GSH concentration (10 mM) led to premature structural disruption of DDMONs. Furthermore, nanoparticle degradation was assessed by high-performance liquid chromatography (HPLC) and Ellman test across varying GSH concentrations. After 48 h of incubation of nanoparticles with a 1 mM GSH concentration, a modestly elevated oxidized GSH percentage (85%) was observed in DDMONs when compared to MONs (approximately 76%). Additionally, the OD value associated with the generated –SH content in DDMONs (0.20) was slightly greater than the value observed in MONs (0.17). However, at a higher GSH concentration of 10 mM, the percentage of oxidized GSH in DDMONs decreased to approximately 32%, which was three times higher than that of MONs (11%). These results indicated that the porous structure of the DDMONs significantly influenced the degradation of the nanoparticles. To further investigate the cellular-level degradation of the nanomaterials, DDMONs-PEI were incubated in HEK293t normal cells and B16F0 melanoma cancer cells. TEM images unveiled rapid internalization and complete fragmentation of DDMONs-PEI within 4 h in B16F0 cells, whereas degradation was prominently inhibited in normal human cells due to lower GSH levels. Additionally, incubation of DDMONs-PEI with MCF-7 breast cancer cells, Chinese hamster ovary (CHO) normal cells, and RAW264.7 macrophages demonstrated significant degradation only in MCF-7 cancer cells. These findings highlighted the specific degradability of the nanoparticles towards cancer cells. Similarly, GSH-depleted dendritic mesoporous organosilica nanoparticles (GDMON) were ingeniously designed by incorporating embedded tetrasulfide bonds and featuring expansive dendritic mesopores. These nanoparticles demonstrated the capability to encapsulate ovalbumin (OVA) and unmethylated cytosine-phosphate-guanine (CpG).87 Notably, the GDMON-P modified with PEI exhibited remarkable behaviour upon entering antigen-presenting cells (APCs) and experienced decomposition within the cytoplasm, primarily influenced by elevated concentrations of GSH. This process, in turn, triggers the release of antigens and adjuvants, thereby stimulating immune responses. Under the conditions of 2 mM GSH, both GDMON-P and NDMON-P (nonmacroporous) exhibited extremely gradual release of OVA, with only 4.2% and 5.3% of the antigen released after a span of 6 days, respectively. This sustained release profile was attributed to robust electrostatic and hydrophobic interactions between proteins and organosilica frameworks. Conversely, when exposed to 10 mM GSH, GDMON-P demonstrated a substantial enhancement in OVA release (19.8%) over the same 6-day period. In comparison, NDMON-P exhibited a slow release rate, with only 8.6% of the antigen released, which was attributed to GSH-triggered disruption of the pore structure. Further insights into the nanoparticle behavior after coincubation with APCs were gleaned through the utilization of bio-TEM. Within a mere 4 h timeframe, a substantial portion of the nanoparticles had undergone internalization by APCs. Following a 24 h incubation period, GDMON-P revealed partially collapsed structures, while the majority of the alkylated NDMON-P retained their intact architecture, providing valuable observations regarding the interplay between nanoparticles and cells.
 | ||
| Fig. 9 An illustrative portrayal showcasing the structure and functional attributes of DDMONs. Reproduced from ref. 78 with permission from ACS, copyright 2016. | ||
Small silica nanoparticles exhibit substantial potential for various applications within the biomedical sector. They can function as highly effective platforms for drug delivery and therapeutic interventions, thereby contributing to the advancement of precision medicine and tailored treatment approaches. Notably, small-sized organosilica nanoparticles display significant degradation through the degradation of organic functional groups and dissolution of the silica framework. Yu et al. prepared ultrasmall disulfide-bridged organosilica nanoparticles (MONs) with diameters ranging from 15 nm to 45 nm.88 Due to the incorporation of disulfide bonds within the framework of the MONs, a remarkably responsive behaviour to the intracellular GSH environment was exhibited. When placed in an SBF solution containing 10 mM GSH, the MONs underwent biodegradation over a span of 14 days, and subsequent TEM analysis did not reveal any intact spherical MONs. To delve further into the biodegradation dynamics at the cellular level, the MONs were cocultured with breast cancer 4T1 cells. Following a day of coculture, a substantial quantity of MONs was detected within the cytoplasm of the tumor cells. With prolonged incubation, a similar trend of structural collapse and nanoparticle dissolution behaviour emerged within a mere 2 days. On the third day of incubation, an even more pronounced biodegradation pattern was apparent, ultimately culminating in the observation of only a few collapsed MONs after 7 days of cultivation. Similarly, utilizing the “chemical homology” mechanism, Huang et al. ingeniously integrated disulfide bonds into the silica framework, resulting in the creation of scalable organic–inorganic hybrid hollow mesoporous organosilica nanoparticles (HMONs) (Fig. 10).80 The HMONs exhibit appreciable degradability due to the presence of disulfide bonds. The dynamic changes in the microstructural evolution of HMONs in SBF were meticulously examined using TEM. During the initial 7-day period, no significant structural alterations were evident; however, on the 11th day, there was a notable proliferation of structural collapses. Notably, in an SBF solution containing GSH (10 mM), the degradation rate of the HMONs was significantly accelerated, culminating in an almost complete absence of intact spherical HMONs after 14 days. Furthermore, upon uptake by 4T1 cells, a similar trend of structural collapse and dissolution was observed within a mere 2 days, with only a limited number of nanostructured, collapsed HMONs remaining discernible after 7 days. To significantly improve the precision, effectiveness, and safety of cancer therapy, developing a therapeutic that is finely tuned to the TME could achieve enhanced therapeutic outcomes. Building upon this concept, Lu et al. innovatively engineered thioether-bridged organosilica nanoparticles (DOX-CuS@PMOs) via the sol–gel method. The intricate design aimed to synergistically enhance CMT through photothermal effects, capitalizing on a triple response mechanism involving GSH, pH variations, and laser irradiation.45 The CuS@PMOs possessed disulfide bonds that render them degradable in GSH solution. Under conditions of pH 7.4 and 10 mM GSH, DOX-CuS@PMOs exhibited controlled degradation, releasing 20% of DOX within just 1 h and surpassing 40% within 8 h. In the absence of GSH, the release of DOX from DOX-CuS@PMOs was limited to only 13% over 8 h. Moreover, when simulating the conditions of intracellular endosomes/lysosomes (pH 5.5, 10 mM GSH), the nanomaterials showed an accelerated cumulative release amount of 62.5% after 8 h.
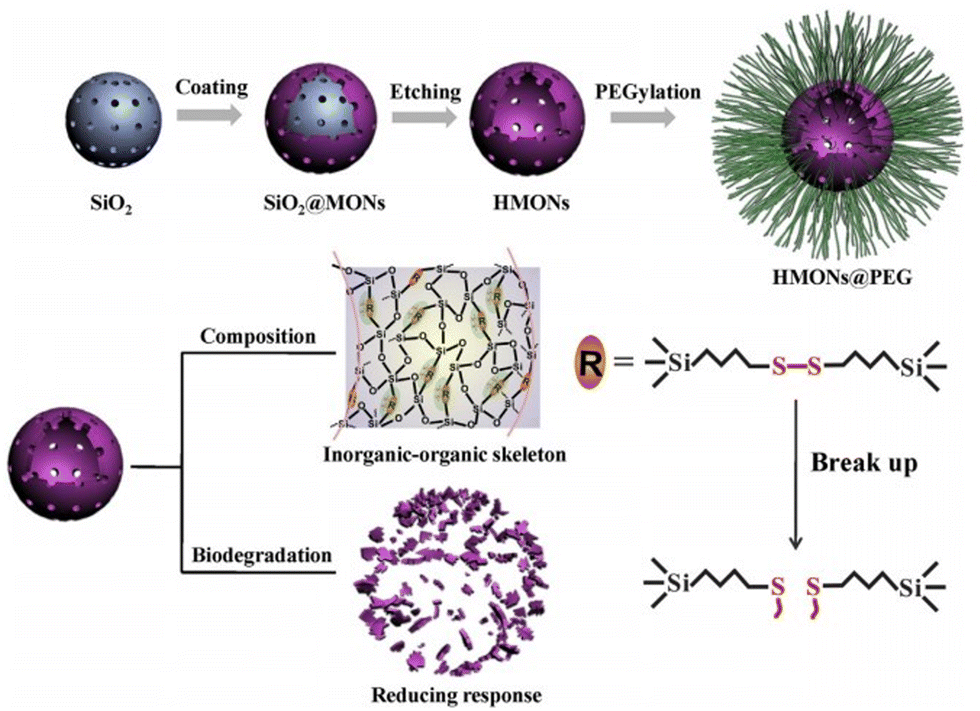 | ||
| Fig. 10 Synthesis and degradation of HMONs. Reproduced from ref. 80 with permission from Elsevier, copyright 2017. | ||
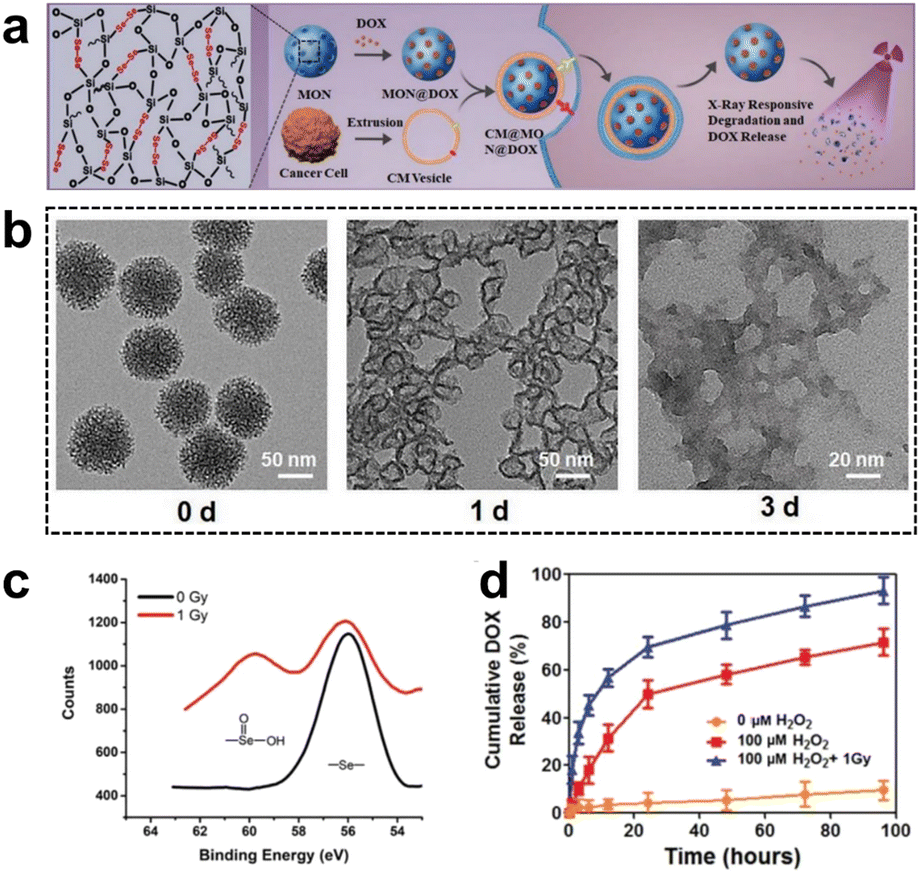 | ||
| Fig. 11 (a) Synthesis and application of selenium-containing organosilica nanoparticles. (b) TEM images of SeMONs subjected to 1 Gy X-ray radiation in the presence of H2O2. (c) XPS analysis of SeMONs before and after radiation. (d) Drug release experiments of SeMONs. Reproduced from ref. 89 with permission from Wiley, copyright 2020. | ||
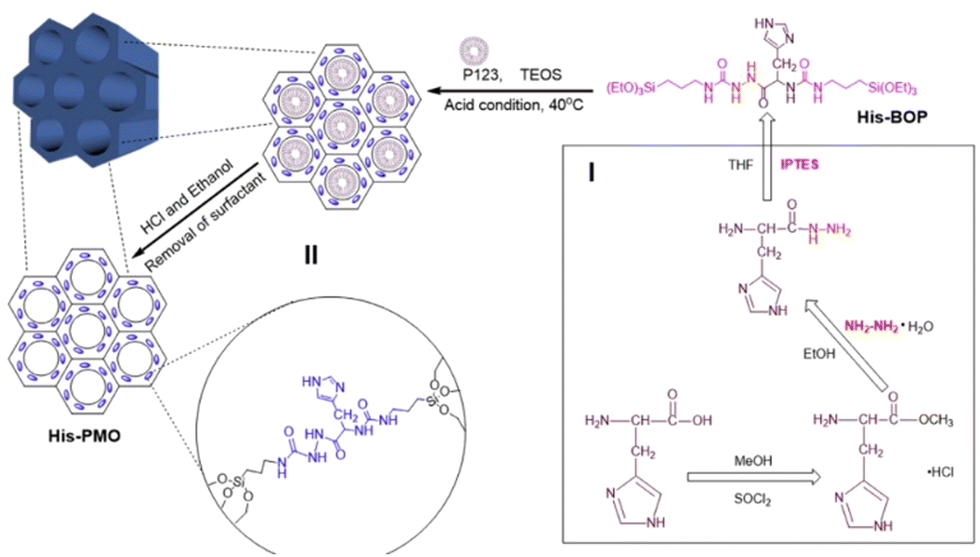 | ||
| Fig. 12 Schematic diagram of the synthesis of His-PMO. Reproduced from ref. 91 with permission from Elsevier, copyright 2019. | ||
Oxamide, an organic compound encompassing an amino group and an acylamide group, serves as a reactive small molecule akin to amino acids. Fatieiev et al. devised a bridged bis-siloxane (BS) nanoplatform with an organic content of approximately 50%. This was achieved by employing oxamide as a bridging agent via a sol–gel reaction mediated by alkoxysilane precursors (Fig. 13a).92 Subsequently, they investigated the biodegradation performance of the BS nanoparticles in simulated biological media. The nanoparticle suspensions were exposed to Tris buffer (pH 4), PBS buffer (pH 7.4), and PBS/pancreatin solution. Experimental findings demonstrated that pancreatin could cleave amino acids into carboxylate and ammonium ion groups, consequently triggering BS nanoparticle degradation solely in the presence of pancreatin. Dynamic light scattering (DLS) measurements exhibited a reduction in particle size after 24 h and 48 h, with an initial mean hydrodynamic diameter of 295 nm dwindling to 122 nm after 48 h of incubation in PBS/pancreatin.
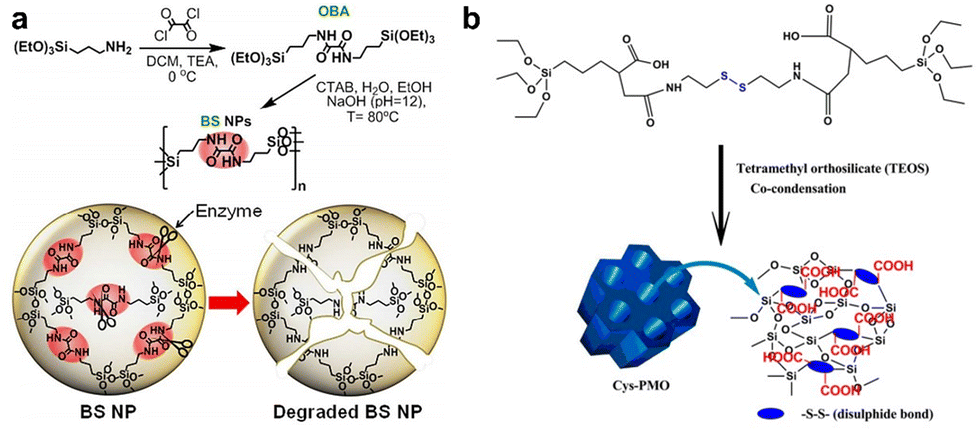 | ||
| Fig. 13 (a) Illustrative depiction of the synthesis and enzyme-triggered degradation process of BS nanoparticles. Reproduced from ref. 92 with permission from The Royal Society of Chemistry, copyright 2015. (b) Schematic outlining the preparation of Cys-PMO hybrid nanoparticles. Reproduced from ref. 93 with permission from ACS, copyright 2018. | ||
Cystamine consists of two amino groups connected by a sulfur atom and is known to possess various physiological and pharmacological activities in biological systems. The thiol group (–SH) in cystamine can react with the silane groups on the oxidized silica reactants to form cystamine disulfide, which imparts the characteristics of organic–inorganic redox-responsive degradation. Based on this, Kummara et al. adopted a cocondensation method to introduce cystamine-modified organosilane functionalities into the POM framework, leading to the creation of Cys-PMO hybrid nanoparticles for loading DOX to combat human cervical cancer (Fig. 13b).93 Notably, this system incorporated disulfide linkages (–S–S–) alongside additional carboxylic acid moieties. As a result, Cys-PMO hybrid nanoparticles manifested a robust drug loading capacity and exhibited pH- and redox-responsive attributes. The outcomes of in vitro release experiments revealed that following 48 h of incubation in PBS (pH 7.4), pH 5.5, and pH 5.5/10 mM DTT solution, the percentages of DOX released from Cys-PMO hybrid nanoparticles were 10%, 56%, and 89%, respectively.
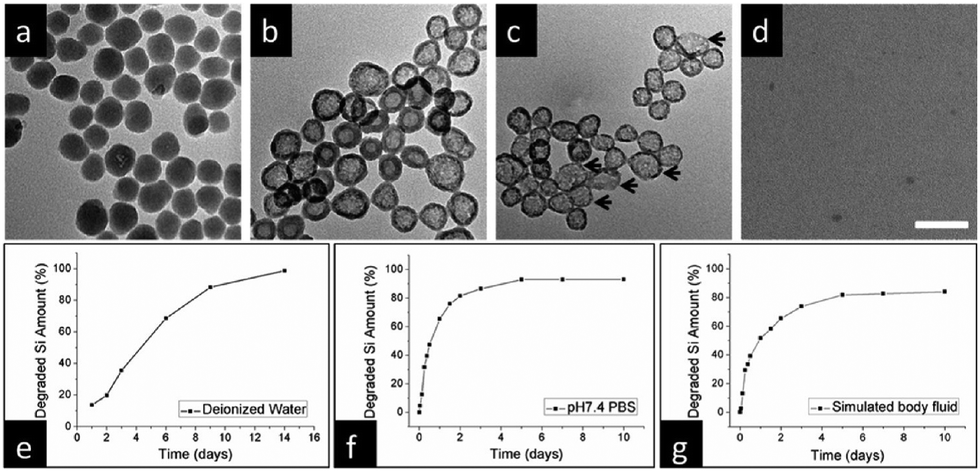 | ||
| Fig. 14 (a) Representative TEM images of SiO2-MB nanoparticles following immersion in deionized water for varying durations: (a) 1 day, (b) 4 days, (c) 9 days, and (d) 14 days. Scale bar, 100 nm. Quantitative analysis using ICP-OES reveals the degradation of silicon content as a function of immersion time across distinct environments: (e) deionized water at room temperature, (f) PBS (pH 7.4), and (g) simulated body fluid (containing 50% FBS) at 37 °C. Reproduced from ref. 94 with permission from ACS, copyright 2013. | ||
3. TME-triggered degradable silica nanoparticles for precision cancer theranostics
Within the confines of the TME, the interplay of tumor cell growth, metabolism, and intricate interactions with adjacent cellular and molecular components culminate in the emergence of distinctive biological and physiological attributes.95,96 These include an acidic environment, heightened redox status, elevated concentrations of specific biomolecules (e.g., growth factors, enzymes, etc.), and varying expression of cell types and surface receptors.9–13,97 TME-responsive nanotherapeutics are diagnostic and therapeutic agents specifically engineered to target the TME in cancer and are capable of responding to specific conditions or signals within the TME, thereby achieving precise diagnosis and treatment.13,98 These nanotherapeutics hold significant promise in the realms of cancer theranostics, as they have the potential to enhance treatment effectiveness, minimize side effects, and propel the advancement of personalized medicine. In this section, we systematically present the latest advancements in the research of TME-responsive degradable silica nanoparticles, elucidating representative types of biological triggers, including pH, redox, hypoxia, and enzymes.3.1. Single biological triggers
In the acidic TME, pH-responsive nanoplatforms can release drugs and achieve targeted therapy for tumor cells. This targeted drug delivery approach enhances drug accumulation in tumor tissues, improving treatment effectiveness while minimizing damage to healthy tissues. Peng et al.107 synthesized rapidly biodegradable nanovectors (DOX@OSNP-PEG1000) by loading DOX into organosilica nanoparticles (OSNPs) and subsequently coating them with poly(ethylene glycol) (PEG) (Fig. 15a and b). The presence of the phenyl group and pH-sensitive imine bonding in the OSNPs facilitated DOX loading through hydrophobic interactions and triggered drug release in response to the acidic environment. In vitro degradation experiments revealed that the weight loss of the nanocarriers was only 10% within 6 h under physiological conditions of pH 7.4, whereas it reached 49% under acidic conditions (pH 6.5). Notably, under simulated lysosomal acidic environmental conditions (pH 5.0), the weight loss of OSNPs was 62% within half an hour and further increased to 90% within 6 h (Fig. 15c). This demonstrated the rapid biodegradability of the OSNPs, particularly in acidic environments, which made them highly suitable for endosomal-triggered drug delivery. Similarly, Zhang et al.91 prepared a histidine periodic mesoporous organosilica nanosystem (His-PMO) for sustained drug release. Since the imidazole moiety of histidine is prone to protonation under acidic conditions, leading to its electrostatic repulsion with drugs, it endowed His-PMO with excellent pH-responsive drug release behavior. To enhance targeted drug delivery, Kang et al.69 developed a multifunctional target nanoplatform (oHA-DOX@MSNs/HAP) coated with oligo hyaluronan (oHA) and containing silica and HAP for targeted cancer treatment. By introducing oHA on the surface of the nanoplatform, oHA-DOX@MSNs/HAP could selectively bind to tumor cells, enabling precise recognition and targeted therapy for tumor cells. Moreover, silica and HAP underwent gradual degradation under weakly acidic conditions and were cleared by metabolism. After exerting their effects on the body, they gradually degrade and release drugs, enabling precise targeted tumor therapy while avoiding adverse effects on healthy tissues.
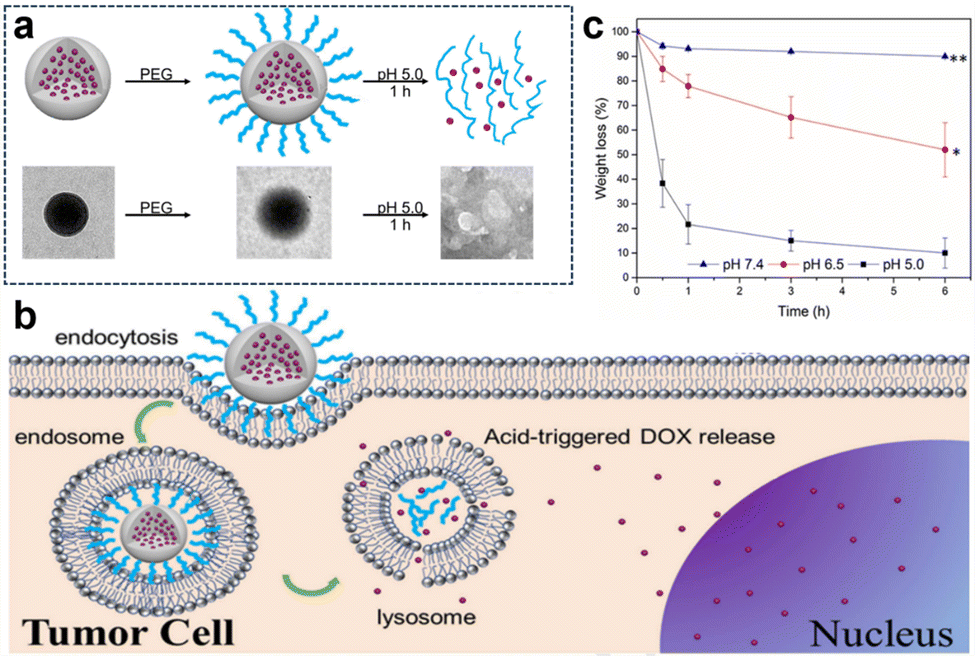 | ||
| Fig. 15 (a) Schematic representation and TEM images of the preparation and degradation of DOX@OSNP-PEG1000. (b) Drug loading and release from biodegradable DOX@OSNP-PEG1000. (c) In vitro degradation of DOX@OSNP-PEG1000 at different pH values. Reproduced from ref. 107 with permission from Elsevier, copyright 2020. | ||
By capitalizing on the unique conditions within tumors, therapeutic agents are activated precisely where needed, optimizing treatment efficacy. Furthermore, the inclusion of external stimuli, such as light or magnetic fields, adds an additional layer of control, allowing for on-demand drug release. Wu et al.108 developed a highly versatile platform that synergistically combined targeted chemo-PTT. This was achieved through the utilization of thioether-bridged mesoporous organosilica nanoparticles (PMOs) as carriers for loading DOX. To facilitate precise targeting, bovine serum albumin-FA-modified MoS2 sheets (MoS2-PEI-BSA-FA) were synthesized and acted as effective capping agents for PMOs (Fig. 16a). Upon cellular uptake through FA receptor-mediated endocytosis, the nanoparticles were successfully encapsulated within endosomes. Under 808 nm laser irradiation, the DOX encapsulated in this nanocomplex exhibited a dual-stimuli responsive release, particularly in the acidic TME, thereby promoting targeted CMT. Additionally, the nanoplatform showed efficient conversion of NIR light energy into thermal energy, thereby enabling photothermal therapy. Furthermore, the release of DOX was expedited in the acidic environment (pH 5.0) compared to pH 7.4, with the maximum cumulative release amount observed at pH 5.0, especially when combined with 808 nm laser irradiation (Fig. 16b). This pH-dependent and NIR-stimulated release of DOX significantly enhanced the synergistic effect of chemo-photothermal therapies, underscoring the enormous potential of this nanoplatform as a highly promising approach for effective cancer treatment.
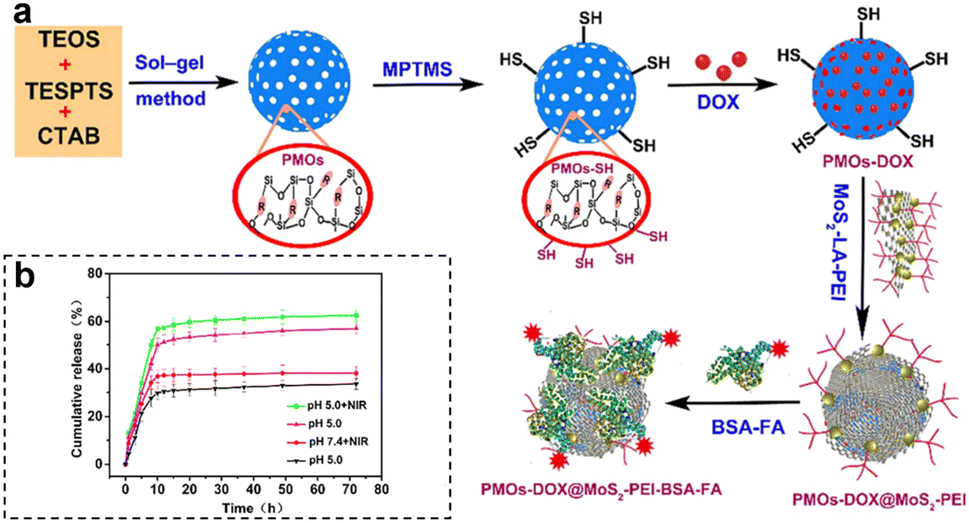 | ||
| Fig. 16 (a) Schematic synthesis and preparation of PMOs-DOX@MoS2-LA-PEI-BSA-FA composites. (b) DOX release profiles of PMOs-DOX@MoS2-PEI-BSA-FA nanocomposites at different pH values, with and without NIR laser irradiation (808 nm, 1 W cm−2). Reproduced from ref. 108 with permission from Elsevier, copyright 2018. | ||
In the field of MR imaging, manganese ions could serve as contrast agents to enhance the visibility of MR images. However, in practical applications, MnO2 is often found in the nanoparticle form. In this state, it is less suitable for direct use as an MR imaging contrast agent due to its limited ability to produce a strong MR imaging signal. This is where GSH plays a pivotal role. GSH can initiate a chemical reaction with MnO2, converting it into soluble Mn2+. These Mn2+ ions can then be sequestered within the body or specific biological tissues, subsequently facilitating their utilization for MR imaging. Zheng et al.111 synthesized a TME-responsive nanotheranostic agent (FMSN-MnO2-BCQ) that integrated precise MR imaging and NIR-II fluorescence imaging with self-reinforcing antitumour therapy (Fig. 17). The core component of this nanotheranostic platform was thioether-doped organosilica. GSH prompted the degradation of FMSN-MnO2-BCQ within the TME, effectively releasing BCQ and MnO2, with their ultrasmall size facilitating tumor penetration. One of the key features of this nanotheranostic platform was its ability to offer precise and high-resolution imaging capabilities. The real-time imaging capabilities provided by MR imaging and NIR-II fluorescence imaging enable precise tumor targeting, ensuring efficient drug delivery and enhancing treatment efficacy. This aspect is crucial for early detection and effective monitoring of treatment response, providing valuable insights for personalized cancer therapy. Moreover, the nanoparticles are engineered to carry therapeutic agents and photothermal agents within their silica framework. When exposed to NIR-II laser irradiation, photothermal agents generate localized heat, triggering drug release and enhancing the therapeutic effect on cancer cells. Furthermore, in the presence of GSH, MnO2 is reduced to Mn2+, acting as a Fenton reagent that converts endogenous H2O2 into highly cytotoxic ·OH, enabling selective chemodynamic therapy (CDT). Additionally, the continuous depletion of endogenous GSH weakens the capacity to clear free radicals, leading to a self-enhancing effect of the CDT.
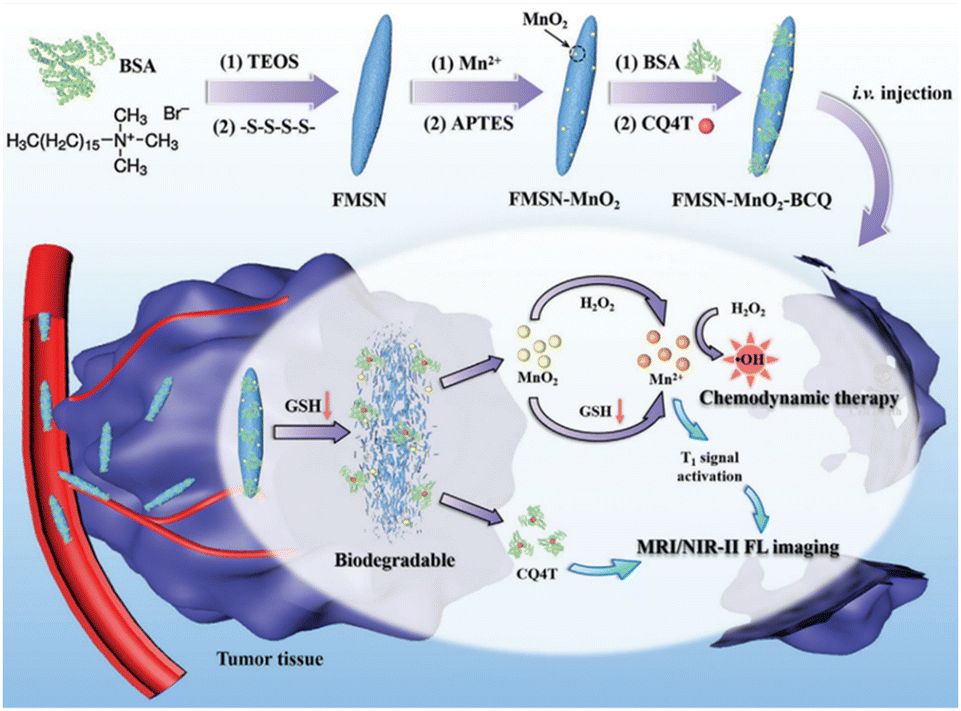 | ||
| Fig. 17 Diagram of the synthesis process of FMSN-MnO2-BCQ and the mechanisms for MR imaging, NIR-II fluorescence imaging and enhanced CDT. Reproduced from ref. 111 with permission from Wiley, copyright 2021. | ||
Similarly, Wei et al.112 designed a virus-like mesoporous silica nanoprobe (VGd@ICG-FA) capable of achieving NIR-II fluorescence imaging and inducing ferroptosis by RT sensitization. The nanoprobe was obtained by utilizing tetrasulfide-bridged silica nanoparticles to load the photosensitizer ICG, which was then coated with gadolinium (Gd) and anchored with FA. VGd@ICG-FA exhibited efficient targeting of tumor cells facilitated by its virus-like morphology and FA anchoring. This nanoprobe was particularly effective at identifying small cancers and guiding precise surgery using NIR-II fluorescence imaging. Once inside the cancer cell cytoplasm, the tetrasulfide linkages in the silica framework were disrupted by GSH. Consequently, the framework exhausted GSH, leading to the disruption of intracellular reactive ROS homeostasis. Additionally, Gd generated more ROS during RT, further triggering ferroptosis and enhancing the effectiveness of RT in breast cancer treatment. Quantitative analysis of fluorescence intensity revealed that VGd@ICG-FA exhibited a fluorescence signal 7.59 times stronger within mammary tumors compared to normal tissues. This substantial contrast was sufficient for the effective differentiation between tumors and normal mammary tissue. Following intravenous administration of VGd@ICG-FA for 36 h, five pairs of mammary tumors were surgically removed in segments under NIR-II fluorescence imaging. The fluorescence intensity of the excised tumors was notably higher than that of the surrounding muscle tissue, yielding a tumor-to-muscle (T/M) ratio of 8.01 ± 0.62. In addition, Guo et al.113 developed a biodegradable nanoplatform (HMON@CuS/Gd) that incorporated Cu nanocrystals and Gd-doped hollow mesoporous organosilica nanotheranostics (HMON) (Fig. 18a). This nanoplatform offered multimode imaging and PTT capabilities. In the TME with elevated GSH concentration, the disulfide bonds within the HMON framework responded to GSH, leading to nanoplatform degradation and subsequent release of CuS and Gd3+. When exposed to NIR light, CuS underwent photothermal conversion, inducing ROS generation and compromising mitochondrial function in tumor cells, resulting in effective photothermal therapeutic effects. Additionally, Gd3+ acted as a radiation sensitizer, enhancing X-ray photon capture and improving MR imaging efficacy. Moreover, the nanoplatform could encapsulate the carbon cyanine dye DIR, enabling NIR fluorescence imaging and facilitating image-guided visualization. Experimental results demonstrated the nanoplatform's effectiveness in accumulating fluorescence signals, significantly enhancing T1-weighted MR imaging signals for improved MR imaging, and substantially inhibiting tumor growth (Fig. 18b).
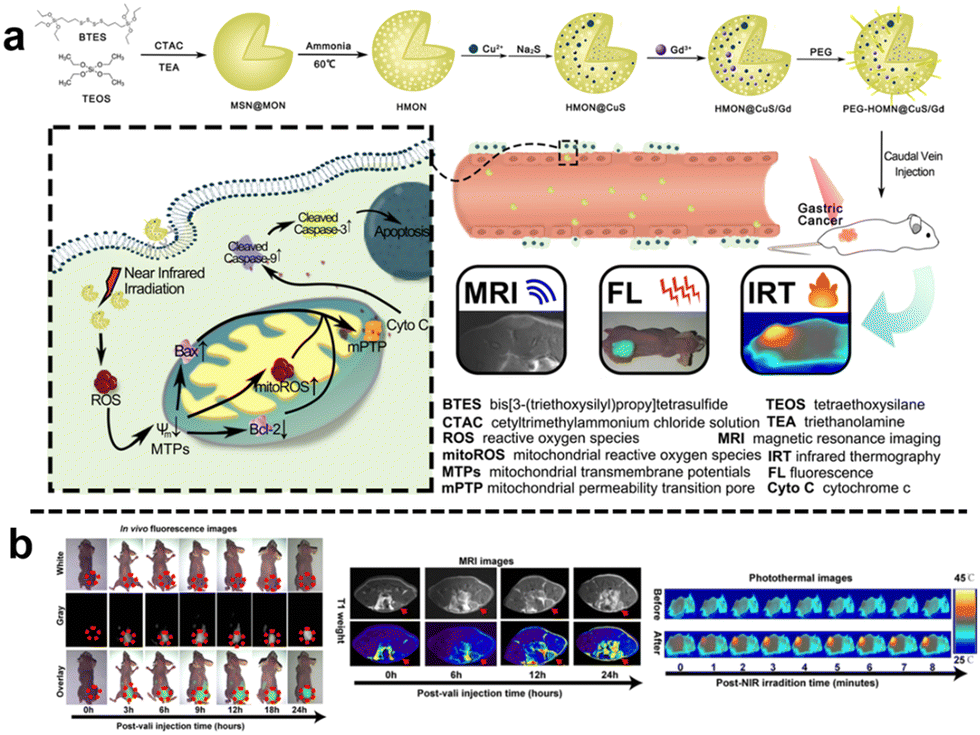 | ||
| Fig. 18 (a) Schematic diagram of the synthesis and mechanism of antitumour action of HMON@CuS/Gd. (b) In vivo FL/MIR/IRT images of HMON@CuS/Gd. Reproduced from ref. 113 with permission from Springer Nature, copyright 2020. | ||
CDT is an innovative therapeutic strategy that employs catalytic reactions to produce ROS from either internal or external sources. This, in turn, triggers oxidative stress within cells, ultimately resulting in cell death. Despite its potential, CDT faces hurdles in fully realizing its benefits, primarily stemming from difficulties in achieving accurate tumor targeting and efficiently disrupting cellular redox homeostasis. To address these issues, Lu et al.114 engineered specialized nanoparticles (nanoMIPs) that could target tumor tissues and disrupt cellular redox homeostasis, which were initially prepared as biodegradable silica nanoparticles loaded with Cu2+ and subsequently subjected to in situ functionalization using Tn antigen (GalNAc) as a template through molecular imprinting technology. The nanoMIP was specifically designed to selectively target tumor cells that overexpressed the Tn antigen, thereby facilitating enhanced cellular uptake. Upon internalization by tumor cells, the nanoMIPs underwent controlled degradation in response to the TME. This degradation process led to the spontaneous release of Cu2+/Cu+ through redox cycles, resulting in highly potent depletion of GSH and triggering ˙OH generation through a Fenton-like reaction. The produced Cu+ could effectively inhibit catalase activity, thereby indirectly elevating the endogenous H2O2 level within the tumor cells. Consequently, the “maladjusted” tumor cells lost their resistance against ˙OH-induced damage, ultimately culminating in tumor cell apoptosis. By interfering with cellular redox processes, the nanoparticles sensitized tumor cells to oxidative stress, making them more susceptible to CDT effects. Both in vitro and in vivo experiments unequivocally demonstrated the exceptional cytotoxicity of nanoMIPs against tumor cells and their remarkable efficacy in inhibiting tumor growth in a xenograft tumor model while simultaneously exhibiting minimal side effects.
Immunotherapy utilizes the body's immune system to precisely target and eliminate cancer cells, potentially achieving long-lasting remission or even a cure. It typically causes fewer side effects than traditional treatments such as CMT, enhancing patient well-being. Incorporating Se–Se moieties into the silica framework not only enables precise control of the degradation rate of silica nanoparticles but also confers GSH responsiveness to them. Zhang et al.115 developed a coordination and redox-responsive nanoplatform (MON@KP1339) for chemoimmunotherapy by utilizing diselenide-bridge mesoporous organosilica nanoparticles (MONs) to load the ruthenium compound KP1339 (Fig. 19a). The incorporation of GSH-responsive diselenide bridges enabled the nanoplatform to achieve controlled degradation, thereby effectively regulating the release of KP1339. KP1339 could promote immunogenic cell death (ICD) in tumor cells, leading to increased antigen release and stimulation of the host immune system's response, consequently bolstering the therapeutic efficacy and ultimately achieving the goal of chemoimmunotherapy. Moreover, the cumulative release amount of KP1339 from MON@KP1339 exceeded 70% after 12 h under simulated tumor cell GSH conditions, whereas the control group without GSH exhibited a cumulative release amount of less than 10% within 96 h (Fig. 19b). This result clearly indicated that the GSH-responsive environment facilitated the controlled release of KP1339. In vivo experiments further demonstrated remarkable tumor growth inhibition by MON@KP1339, indicating outstanding antitumour efficacy (Fig. 19c). In another study, Li et al.116 constructed an in situ therapeutic nanovaccine (HMSsN-ANX5@HOMV) by utilizing diselenide-bridged hollow mesoporous organosilica nanoparticles (HMSeN) to load Annexin A5 (ANX5). These nanoparticles were subsequently enclosed within bacterial outer membrane vesicles (OMVs) and surface-modified with HA. The incorporation of diselenide bridges for ANX5 immobilization not only retained the protein's biological activity but also facilitated the TME-responsive release of ANtheX5. Upon accumulation within the TME, HMSsN-ANX5@HOMV was exposed to elevated levels of GSH, triggering the cleavage of diselenide bridges and facilitating the rapid release of encapsulated annexin A5 protein. Annexin A5 plays a pivotal role in tumor cell apoptosis by selectively binding to phosphatidylserine on the surface of apoptotic cells, effectively impeding their phagocytosis by macrophages and subsequently enhancing the response of cytotoxic T cells, a crucial subset of immune cells responsible for tumor cell recognition and elimination, thereby eliciting robust antitumour effects. By augmenting the activity of cytotoxic T cells, the liberated annexin A5 protein effectively promoted antibody-dependent cellular cytotoxicity and targeted immune responses against the tumor, ultimately restraining tumor growth and metastasis. The in vivo results of the in situ therapeutic vaccine demonstrated the complete eradication of approximately 50% of 4T1 tumors and induced long-term immune memory.
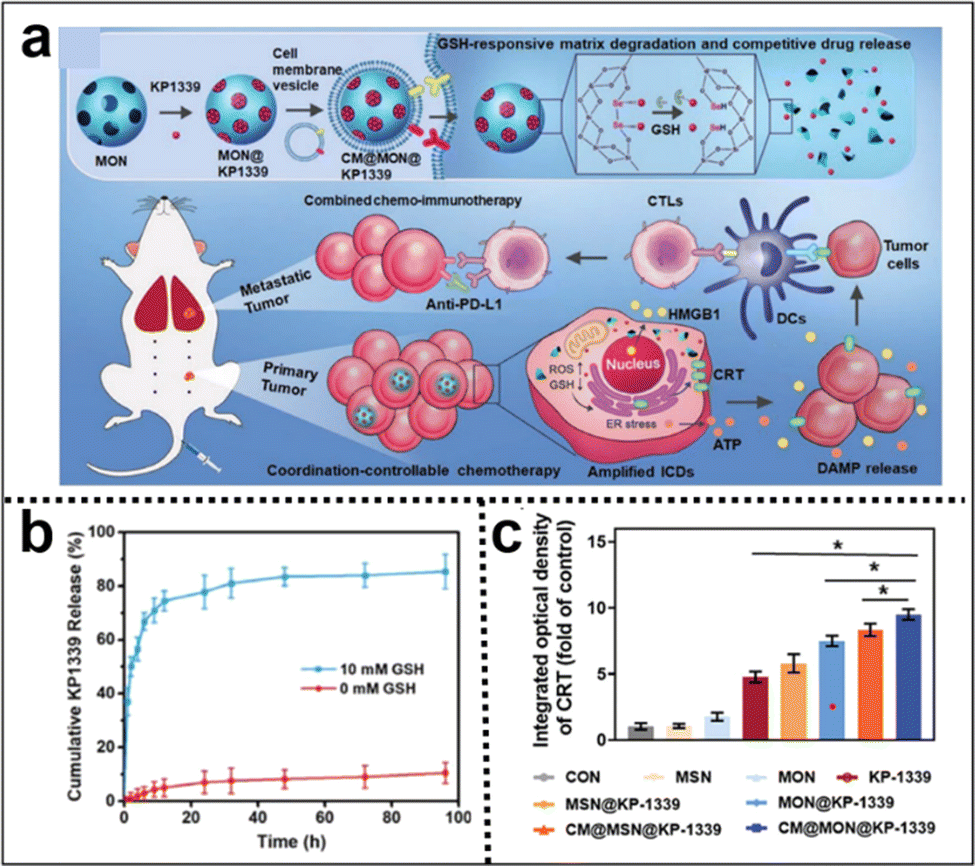 | ||
| Fig. 19 (a) Illustration of the synthesis of diselenide-bridged MONs for responsive drug release and enhanced ICD to facilitate effective and safe cancer chemoimmunotherapy. (b) The cumulative release profiles of KP1339 cells in the presence or absence of GSH. (c) Semiquantitative analysis of the primary tumor's exposure to CRT in each experimental group. Reproduced from ref. 115 with permission from Wiley, copyright 2021. | ||
The integration of diverse therapies in cancer treatment provides several benefits. Synergistic effects can boost treatment efficacy, potentially improving tumor control and response rates. This approach overcomes individual treatment limitations, such as drug resistance or incomplete tumor elimination. In a breaking study by Liu et al.,117 a novel nanocomposite (DCC-HA NCs) was developed that exhibited rapid responsiveness in the TME (Fig. 20a and b). This remarkable nanocomposite comprised tetrasulfide bond-incorporated dendritic mesoporous organosilica (DMOS) as a highly efficient nanocarrier coupled with a newly engineered hydrogen sulfide (H2S) gas generator. Additionally, it was ingeniously loaded with chloroperoxidase (CPO) and CaO2-HA, thus enabling the concurrent application of H2S gas therapy, trimodal-enhanced enzyme dynamic therapy (EDT), and Ca2+-interference therapy. The hallmark feature of this nanocomposite is its dynamic adaptability to the intricate microenvironment of the tumor. Upon entry into the tumor region, the nanocomposite experienced swift decomposition, attributed to the presence of tetrasulfide bonds, culminating in the controlled release of therapeutic H2S gas. Moreover, the strategically incorporated CPO and CaO2-HA synergistically enhanced the efficacy of EDT and Ca2+ interference therapy, respectively, thereby offering a comprehensive and potent treatment strategy. CDT holds great promise as a therapeutic strategy, leveraging ROS generated via Fenton or Fenton-like reactions to induce oxidative stress and trigger tumor cell death.
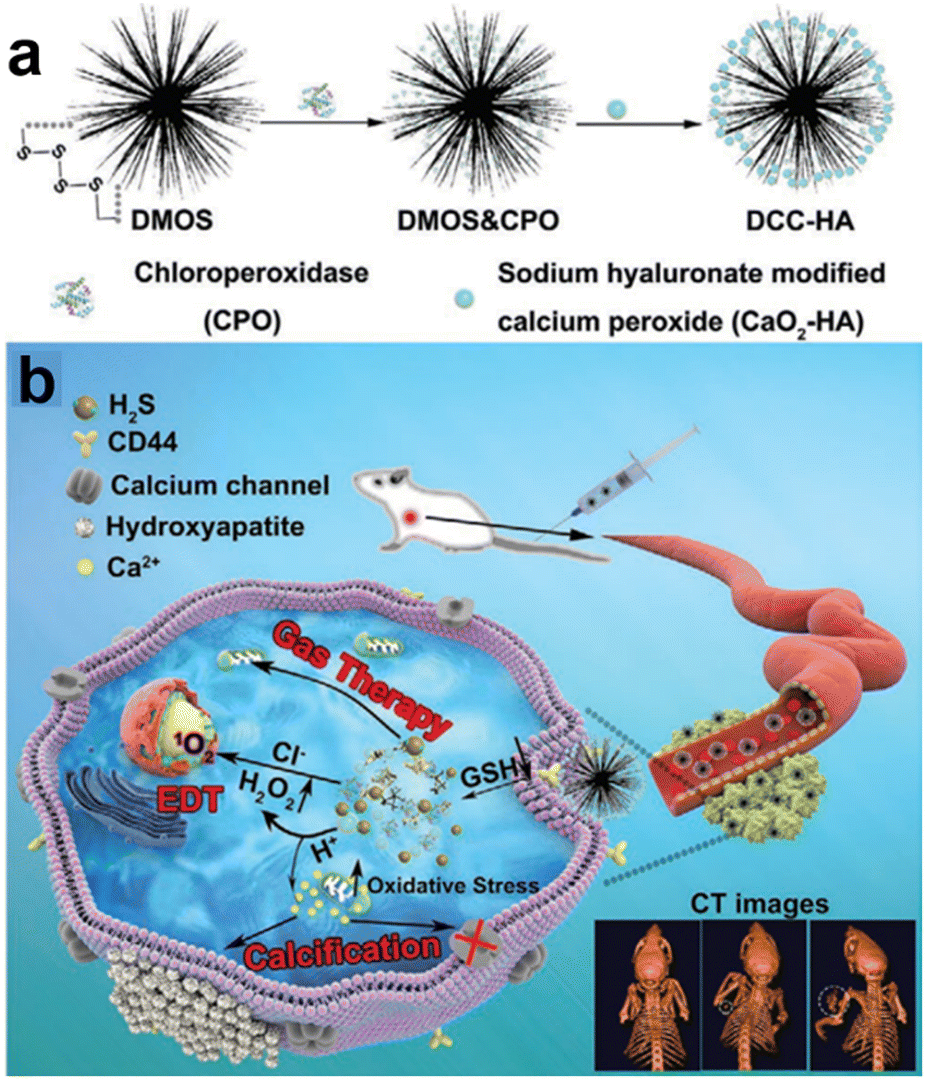 | ||
| Fig. 20 (a) and (b) Schematic diagram of DCC-HA NCs for gas therapy and enzyme dynamic therapy (EDT). Reproduced from ref. 117 with permission from Wiley, copyright 2021. | ||
Hypoxia-responsive fluorescence imaging allows for the specific visualization of regions within tumors that are experiencing low oxygen levels. Gaining insights into the extent of tumor hypoxia can aid medical professionals in anticipating patient responses to these treatments and making necessary adjustments to treatment strategies. This selective targeting enhances the accuracy of disease detection and assessment. Yang et al.127 prepared a nanosensor (HMON-Ce6-[(Ru(dpp)3)]Cl2) for real-time oxygen monitoring during photodynamic therapy (PDT) by synthesizing thioether-bridged deformable hollow mesoporous organosilica nanoparticles and loading an oxygen-sensitive probe [(Ru(dpp)3)]Cl2 and a photosensitizer chlorin e6 (Ce6). As the irradiation time of HMON-Ce6-[(Ru(dpp)3)]Cl2 in the PDT increased, oxygen was transformed into ROS by Ce6. Following excitation at 463 nm, there was a linear increase in fluorescence emission at 620 nm, which correlated with the declining oxygen concentration. This observation demonstrates the nanosensor's sensitive response to changes in oxygen concentration. When HMON-[(Ru(dpp)3)]Cl2 was injected intratumorally into T98G-loaded mice, the nanosensor showed weak red fluorescence before 660 nm laser irradiation, but the fluorescence signal was significantly increased after laser irradiation. The corresponding radiation efficiency of the tumor was significantly increased, which could respond to oxygen consumption in the process of PDT. Similarly, Yuan et al.81 developed a novel nanotheranostic (CPD-protein@BS-qNP), where cetuximab (an FDA-approved antibody drug) was encapsulated within biodegradable silica quenchers (BS-qNP) coated with cell-penetrating poly(disulfide)s (CPDs) (Fig. 21). The silica matrix was further doped with a fluorescent dye (FL-APS 2) and a black hole quencher (BHQ 1-APS 4). By specifically targeting epidermal growth factor receptor (EGFR) on cancer cell surfaces, the loaded cetuximab acted as an effective anticancer drug, avoiding intracellular delivery. The CPD coating prevented internalized cetuximab from lysosomal capture and rapidly disassembled upon exposure to endogenous GSH, maintaining the integrity of the nanotheranostic. Importantly, the inclusion of the fluorescent dye and black hole quencher within Protein@BS-NP enabled the nanoquencher to sense and image cellular hypoxia and protein release. Under hypoxic conditions, the azo portion of BS-NP was reduced by cytochrome P450 reductase, leading to shell disintegration and subsequent release of the antibody and fluorescent dye, thereby efficiently mediating cell state imaging. In fluorescence-triggered testing of the nanotheranostic agent, two control groups were used: FLProtein@BS-NP and FLProtein@BS-qNP, where FITC-labelled protein was directly encapsulated in nanocapsules without or with quencher 4, respectively. Alternatively, the rhodamine dye RB-APS 3 and the quencher BHQ 1-APS 5 were used, resulting in the corresponding nanocapsules (RBProtein@BS-NP or RBProtein@BS-qNP). The experimental results demonstrated that the nanocapsules had excellent hypoxia-responsive fluorescence turn-on properties. Furthermore, the observation experiment of fluorescence signals revealed a significant cytoplasmic fluorescence increase in hypoxic cells treated with CPD-BSA@BS-qNP, while the nondegradable CPD-BSA@NonBS-qNP exhibited negligible fluorescence. This successful imaging after the degradation of hypoxia-responsive nanocapsules indicated the potential of CPD-protein@BS-qNP as an efficient and promising nanotheranostic agent.
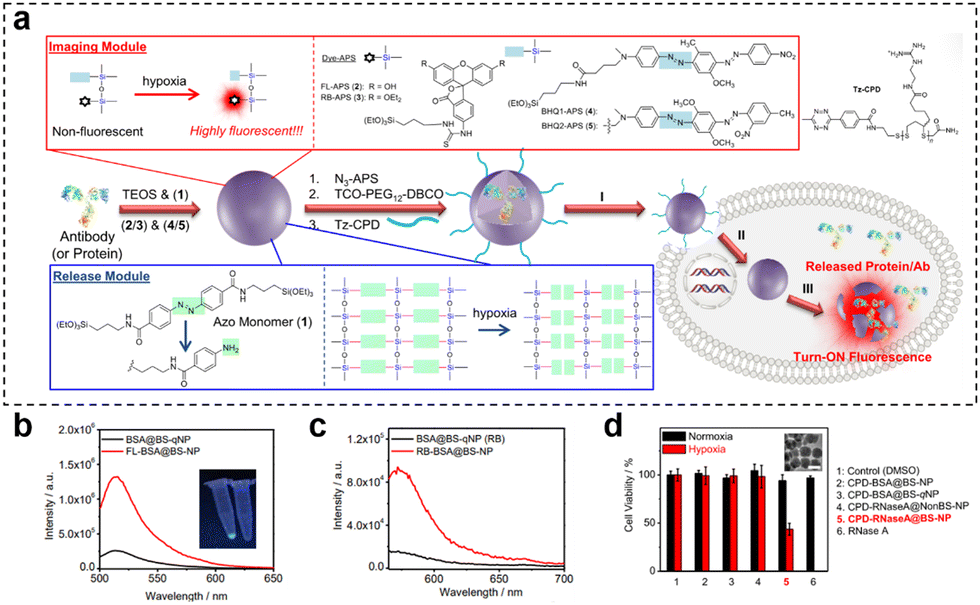 | ||
Fig. 21 (a) Preparation of CPD-protein@BS-qNP and the process of its depolymerization and hypoxia-responsive fluorescence release. (b) Fluorescence spectra of BSA@BS-qNP and FL-BSA@BS-NP (0.01 mg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) mL−1). (c) Fluorescence spectra of BSA@BS-qNP and RB-BSA@BS-NP (0.01 mgmL−1). (d) Cell viability of A549 cells treated with different nanocapsules for 48 h of incubation under hypoxic or normoxic conditions. Reproduced from ref. 81 with permission from Wiley, copyright 2017. mL−1). (c) Fluorescence spectra of BSA@BS-qNP and RB-BSA@BS-NP (0.01 mgmL−1). (d) Cell viability of A549 cells treated with different nanocapsules for 48 h of incubation under hypoxic or normoxic conditions. Reproduced from ref. 81 with permission from Wiley, copyright 2017. | ||
Khashab and coworkers designed enzymatically degradable PSQ nanoparticles using the oxamide bridges present in the organosilica framework.92 The degradability of PSQ nanoparticles was then tested in a simulated biological solution. The results indicated that the PSQ nanoparticles were only degraded in the presence of trypsin enzyme in PBS. Furthermore, the PSQ nanoparticles were effectively rendered fluorescence by incorporating fluorescein dyes, enabling the precise imaging of cancer cells. These PSQ nanoparticles distinctly exhibited the characteristic yellow fluorescence attributed to fluorescein, with a light absorption peak at 505 nm, thus showcasing potential suitability for integration within the domain of biomedical diagnostics. To overcome the low mesoporosity, a groundbreaking approach was introduced in the aforementioned study. Mesoporous organosilica nanoparticles (MONs) based on an oxamide phenylene framework were ingeniously engineered through the expert cocondensation of 1,4-bis(triethoxysilyl)-benzene and (N,N′-bis(3-(triethoxysilyl)-propyl)oxamide) (Fig. 22).134 This innovative design strategy led to the creation of MONs, which exhibited a distinctive and unprecedented capability to accommodate considerably higher drug payloads (84 wt%). This signified a monumental stride forward in the realm of drug delivery systems, allowing for the encapsulation of a substantially larger quantity of therapeutic agents within the intricate mesoporous architecture of MONs. This amplified drug-loading prowess holds considerable promise for enhancing therapeutic efficacy while minimizing adverse effects. In essence, this study not only unveiled the ingenious synthesis of oxamide phenylene-based MONs but also shed light on their distinct ability to be enzymatically degraded, along with their exceptional drug-loading capabilities. These findings collectively contribute to a transformative advancement in the field of nanomedicine, paving the way for novel and potent strategies in drug delivery and cellular targeting.
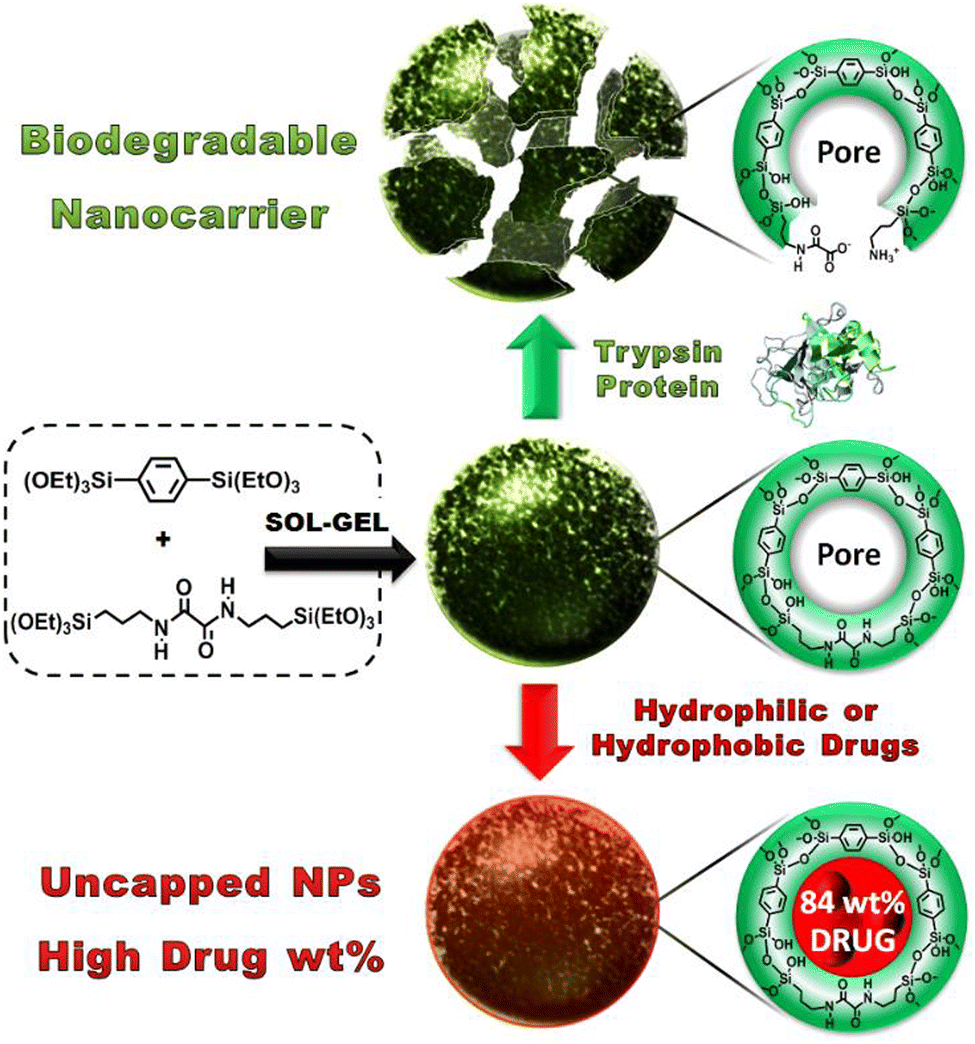 | ||
| Fig. 22 Illustration of the sol–gel synthesis process for MONs and their pore structure before and after protein-mediated degradation (top) or after high drug loading, resulting in nonleaky nanoparticles with uncapped pores (bottom). Reproduced from ref. 134 with permission from Wiley, copyright 2016. | ||
Esterase, a pivotal enzyme responsible for catalyzing the breakdown of ester bonds, has emerged as a significant player within the intricate and ever-changing landscape of the TME.135,136 This enzyme's pronounced upregulation can be attributed to an interplay of factors, encompassing the heightened metabolic demands of cancer cells, the nuanced shifts in pH equilibrium, and the distinctive biochemical makeup characteristic of the TME.5,137 Consequently, the surge in esterase activity presents a captivating avenue for advancing novel cancer therapies and diagnostic methodologies, leveraging its enzymatic prowess to orchestrate targeted and adaptable interventions tailored to the unique intricacies of the tumor milieu. Tsao et al.138 developed enzymatically degradable polymer/silica hybrid microbubbles (SiPCL) for ultrasound (US) imaging. These SiPCL microbubbles were amenable to conjugation with biomolecules for purposes such as tumor cell imaging, drug loading, and targeted drug delivery. The presence of PCL facilitated the degradation of the microbubbles by high levels of lipase in the TME. To evaluate the US imaging capability, they plotted US image intensity response curves for SiPCL microbubbles with varying feed ratios (ranging from 50 to 95 wt%). The results revealed that as SiPCL feed ratios increased, US image intensity also increased, likely attributed to the increasein shell elasticity associated with the initial SiPCL content. However, 95 wt% of the SiPCL microbubbles exhibited a minimal response, possibly due to bubble collapse. Electron micrographs of the 95% SiPCL bubbles unveiled their microbubble shells to be very thin (29 nm) while displaying enhanced elasticity. Both in vitro and in vivo results demonstrated the excellent imaging capabilities of the SiPCL microbubbles, rendering them suitable as ultrasound contrast agents and drug delivery carriers for tumor imaging and treatment.
3.2. Multiple biological triggers
The TME harbours intricate and diverse attributes, encompassing an acidic milieu, elevated redox levels, hypoxia, and aberrant expression of specific enzymes. Through a thorough comprehension and strategic utilization of these distinctive features, the opportunity arises to engineer highly focused nanomedicines or therapeutic systems that are meticulously activated within the TME for precision theranostics. This sophisticated approach not only heightens drug potency but also curtails harm to healthy tissues and alleviates the adverse effects of medications. Furthermore, strategies employing multifaceted triggers inherent to the TME play a pivotal role in surmounting challenges associated with drug resistance. Tumor cells often progressively develop resistance to single therapeutic interventions, leading to a waning efficacy of treatments. By capitalizing on the activation of diverse microenvironmental factors, treatment strategies attain heightened specificity and diversity, ultimately reducing the resistance of tumor cells and amplifying therapy effectiveness. Thus, the advantages derived from harnessing the manifold characteristics of the TME to trigger therapeutic interventions culminate in achieving more accurate and efficient treatment outcomes. This multifaceted approach simultaneously addresses the intricacies of treatment resistance, thereby unveiling the potential for customized therapeutic regimens.3.2.1.1. Redox and pH. Fluorescence imaging possesses high sensitivity, which makes it a valuable tool in various applications. Li et al.139 developed a biodegradable and autofluorescent nanoplatform for pH/redox-responsive drug release by constructing disulfide-bonded bridged organosilica (OS) as a carrier for DOX loading. Subsequently, they modified the surface of the OS carrier with dialdehyde dextrin (DAD) through Schiff base bonding and attached cystamine (Cys) to form a DAD/Cys layer, resulting in the preparation of a biodegradable nanoplatform (Dox-loaded OS-N
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C-DAD/Cys) (Fig. 23a). The DAD/Cys coating effectively prevented DOX leakage, and its S–S and NC bonds could be cleaved in reducing and weakly acidic environments, respectively, leading to pH/redox-responsive drug release. Furthermore, the S–S bonds in the OS framework were also susceptible to reduction, leading to redox-responsive degradation of the OS carrier. Additionally, the Schiff base bond exhibited autofluorescence that responded to changes in acidity, serving as a fluorescent probe for real-time monitoring of drug release. In vitro experiments demonstrated that the DOX accumulation release was only 6.46% within 48 h at pH 7.4, indicating the effective sealing effect of the DAD/Cys coating. Conversely, under acidic pH conditions, the cumulative drug release amount increased as the pH decreased (Fig. 23b). Quantitative analysis of fluorescence intensity revealed a decrease in fluorescence with decreasing pH in PBS. At pH 5.0, after the drug had been released for 4 h, the fluorescence intensity of the carrier was measured at 724. Subsequently, at 12 h and 36 h of release, the fluorescence intensity of the carrier decreased to 521 and 324, respectively (Fig. 23c).
C-DAD/Cys) (Fig. 23a). The DAD/Cys coating effectively prevented DOX leakage, and its S–S and NC bonds could be cleaved in reducing and weakly acidic environments, respectively, leading to pH/redox-responsive drug release. Furthermore, the S–S bonds in the OS framework were also susceptible to reduction, leading to redox-responsive degradation of the OS carrier. Additionally, the Schiff base bond exhibited autofluorescence that responded to changes in acidity, serving as a fluorescent probe for real-time monitoring of drug release. In vitro experiments demonstrated that the DOX accumulation release was only 6.46% within 48 h at pH 7.4, indicating the effective sealing effect of the DAD/Cys coating. Conversely, under acidic pH conditions, the cumulative drug release amount increased as the pH decreased (Fig. 23b). Quantitative analysis of fluorescence intensity revealed a decrease in fluorescence with decreasing pH in PBS. At pH 5.0, after the drug had been released for 4 h, the fluorescence intensity of the carrier was measured at 724. Subsequently, at 12 h and 36 h of release, the fluorescence intensity of the carrier decreased to 521 and 324, respectively (Fig. 23c).
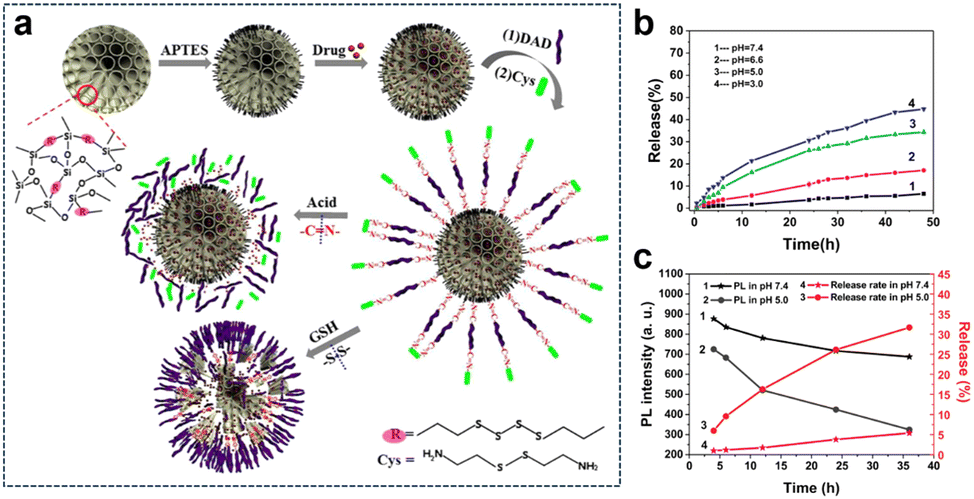 | ||
| Fig. 23 (a) Preparation and degradation of the DOX-loaded OS-NC-DAD/Cys system. (b) Cumulative DOX release from OS-NC-DAD/Cys in PBS under different pH conditions. (c) Relationship between the cumulative release and PL intensity of OS-NC-DAD/Cys excited at 372 nm in PBS with different pH values. Reproduced from ref. 139 with permission from Elsevier, copyright 2020. | ||
Magnetic resonance (MR) imaging is a noninvasive, nonionizing imaging technique that offers high-resolution images, making it a crucial tool in the field of medical imaging. Nonetheless, the clinical utilization of nanoscale contrast agents presents a challenge attributed to their biocompatibility. Premature leakage can potentially cause adverse effects on healthy tissues and compromise the accuracy of imaging results. Therefore, achieving precise delivery and controlled release of these imaging agents is crucial for clinical applications. Zhang et al.140 designed a TME-activated T1–T2 dual-mode MR imaging nanoswitch (PDGFB-FMS). The nanoswitch featured platelet-derived growth factor (PDGFB) conjugated to Fe3O4, which was then coated with Mn-doped silica nanostructures. PDGFB enabled PDGFB-FMS to accumulate in tumors. The acidic and high GSH led to the cleavage of PDGFB-FMS and the release of Fe3O4 and Mn2+. This activated the T1–T2 dual-mode MR imaging signal, resulting in a significant enhancement of T1 and T2 contrasts (Fig. 24). Experimental results demonstrated that PDGFB-FMS achieved a signal-to-noise ratio of 75.6 ± 8.8% in T1-weighted MRI, which was significantly higher than that of the control group. For T2-weighted MR imaging, the maximum signal-to-noise ratios of tumors treated with PDGFB-FMS, FMS, and Fe3O4 were 33.3%, 25.7%, and 39.7%, respectively.
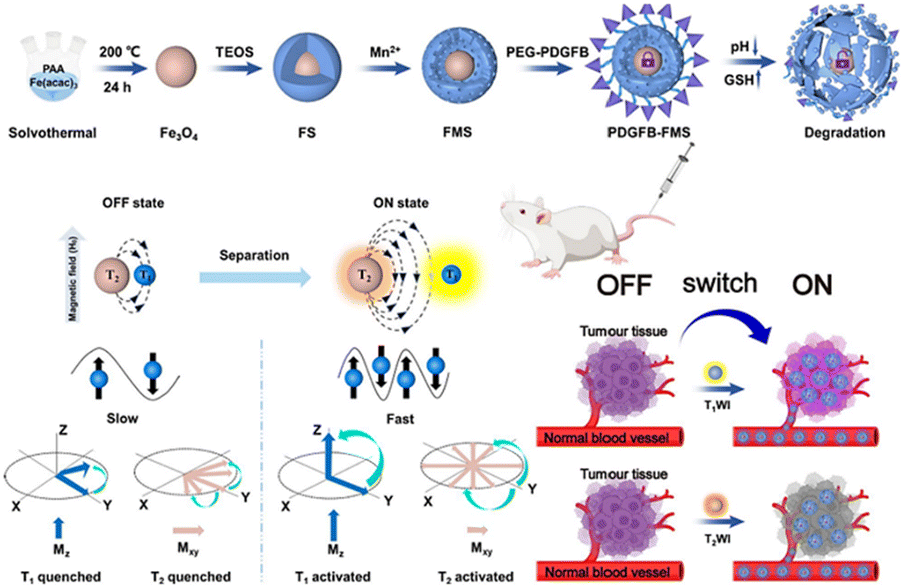 | ||
| Fig. 24 Schematic synthesis of PDGFB-FMS and mechanism of action as a bimodal MR imaging contrast agent. Reproduced from ref. 140 with permission from Springer Nature, copyright 2023. | ||
Similarly, Yu et al.66 developed manganese-doped hollow MSNs (Mn-HMSNs) that harnessed the degradability of –Mn–O– linkages in the TME to improve the biodegradability of MSNs (Fig. 25). For improved stability, Mn-HMSNs were PEGylated to obtain PEG/Mn-HMSNs. The tumor's acidic and reductive conditions triggered a “manganese extraction” response in the TME, where manganese oxides dissolved from the Mn-HMSN framework, promoting decomposition and biodegradation, thereby rapidly triggering the release of anticancer drugs. Additionally, manganese ions acted as effective MR contrast agents, enabling integrated tumor diagnosis and therapy. This innovative approach created new possibilities for tumor theranostics, combining drug delivery and imaging in a single platform. The in vitro cytotoxicity results of PEG/Mn-HMSNs revealed a high cell viability of 94.9% for HepG2 cells, even at a concentration of 200 μg mg−1. Furthermore, the DOX-loaded PEG/Mn-HMSNs demonstrated a significant reduction in the cell survival rate compared to an equal concentration of free DOX, highlighting the enhanced therapeutic effect of DOX on HepG2 cells by PEG/Mn-HMSNs.
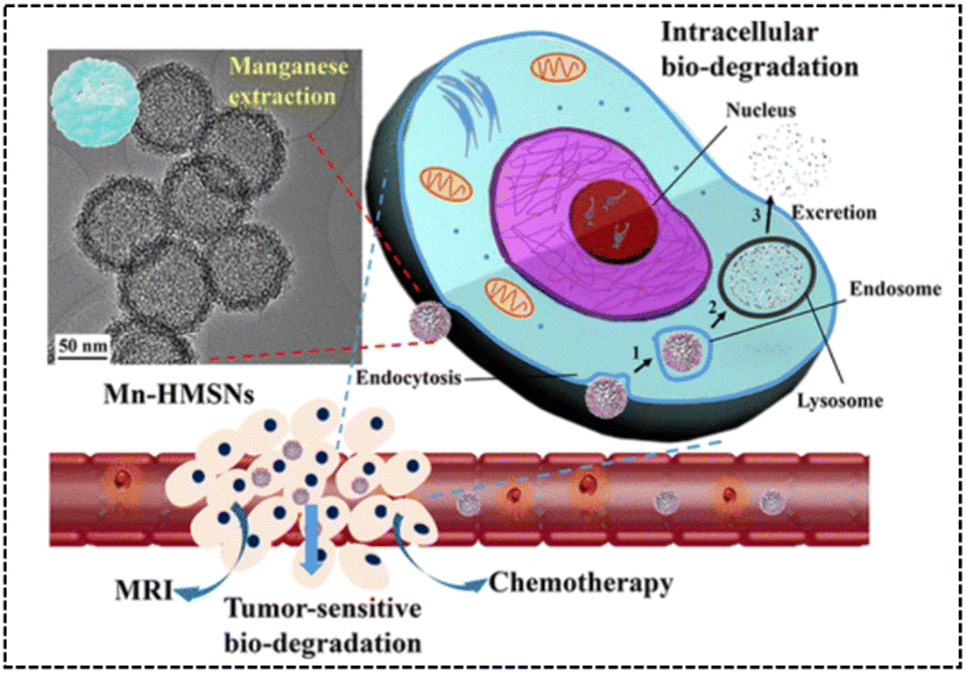 | ||
| Fig. 25 TEM image and intracellular biodegradation of Mn-HMSNs. Reproduced from ref. 66 with permission from ACS, copyright 2016. | ||
Sonodynamic therapy (SDT) utilizes ultrasonic methods with enhanced tissue penetration capabilities. In the SDT, a sonosensitizer can convert oxygen into ROS with potent cytotoxic effects, which leads to further hypoxia in the TME, and hypoxia-activated prodrugs (HAPs) could exert a more potent cytotoxic effect within hypoxic environments. The synergistic combination of these two treatment modalities augments the tumor cell-killing efficacy. Wang et al.70 developed a nanocomposite platform (TPZ@HHSN-C/P-mAb) by synthesizing Ho-doped hollow silica nanospheres sequentially modified with the photosensitizer Ce6 and a monoclonal antibody (mAbPSCA) targeting the prostate stem cell antigen (PSCA). This nanocomposite acted as a carrier for TPZ, effectively delivering it to the tumor region by targeting the overexpressed antigen PSCA on the tumor cell membrane. Additionally, the nanocomposite exhibited ultrasound and MR imaging capabilities. Its hollow structure underwent degradation in the acidic TME. Under ultrasound exposure, HHSN-C/P-mAb consumed oxygen to generate ROS, causing damage to tumor cells, increasing intratumoral hypoxia, and activating the toxicity of TPZ, thus achieving combination therapy with the assistance of imaging (Fig. 26a). The experimental results demonstrated that when the concentration of TPZ@HHSNC/P-mAb is increased to 200 μgmL−1, the cell survival rate under US irradiation decreased significantly to only 13.6% (Fig. 26b). Moreover, incubation of Ho-HSN-PEG in an acidic simulated solution at pH 5.2 for a mere 6 h resulted in the appearance of a substantial amount of flocculent on the surface (Fig. 26c). In contrast, no significant change was observed in Ho-HSN-PEG under neutral conditions, thereby confirming the important role of Ho doping in realizing the degradation of the silica framework in acidic environments.
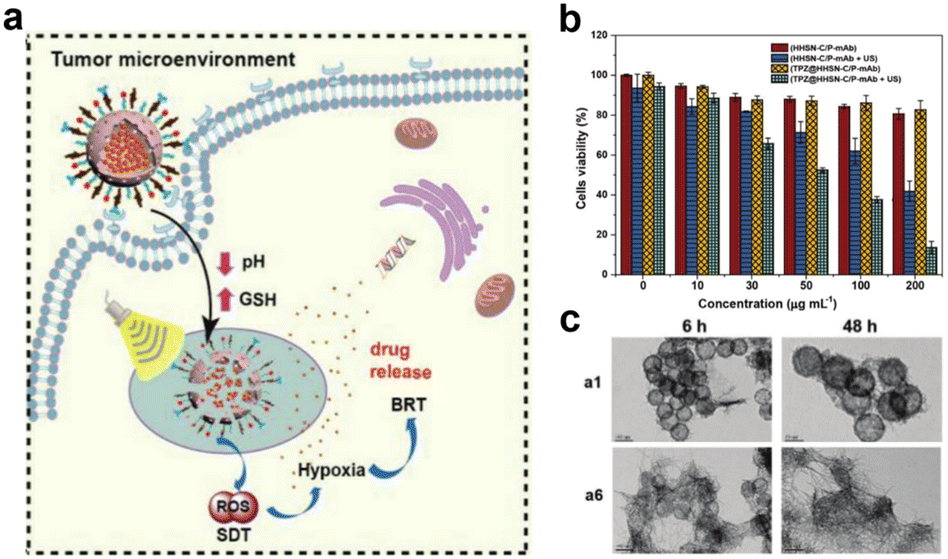 | ||
| Fig. 26 (a) Application of TPZ@HHSN-C/P-mAb for MR/US imaging and SDT/BRT combination therapy. (b) Viabilities of PC-3 cells after different treatments. (c) TEM images of Ho-HSN-PEG (100 μgmL−1) after 6 h and 48 h of incubation with SBFs at different pH and GSH concentrations: (a1) pH = 7.4 (PBS), [GSH] = 0 M; (a6) pH = 5.2 (PBS), [GSH] = 10 × 10−3 M. Reproduced from ref. 70 with permission from Wiley, copyright 2018. | ||
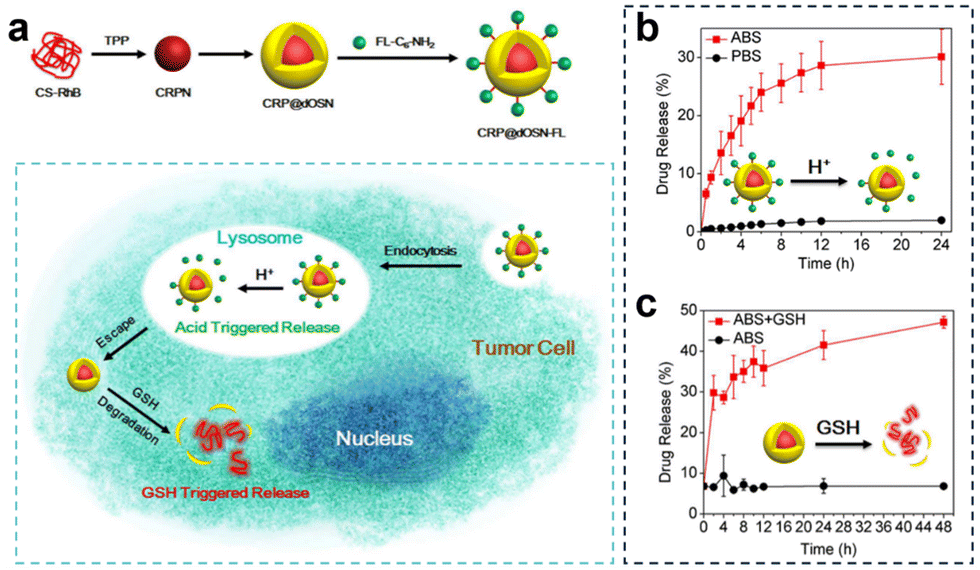 | ||
| Fig. 27 (a) Preparation of CRP@dOSN-FL loaded with two drugs and its responsive drug release and degradation. (b) In vitro release behaviour of FL-C6-NH2 in the presence of PBS (10 mM, pH = 7.4) and ABS (10 mM, pH = 5.0) at 37 °C. (c) In vitro release behaviour of CS-RhB in acidic media (ABS, pH = 5.0) with or without 5 mM GSH. Reproduced from ref. 141 with permission from ACS, copyright 2018. | ||
The dual-responsive approach in the TME offers targeted drug release, reducing effects on healthy tissues. It boosts treatment efficacy by activating agents in specific conditions. Li et al.142 synthesized manganese-doped mesoporous silica (MMS) nanospheres to efficiently load DOX. Subsequently, the entire structure was encapsulated with polydopamine (PDA), resulting in the development of a pH/reduction-responsive biodegradable drug delivery system (DOX-MMS/PDA). The MMS nanocarriers exhibited a substantial specific surface area and porous structure, facilitating a remarkable drug loading capacity for DOX (39.54%). The PDA coating on the external surface of MMS not only effectively prevented the burst drug release but also conferred pH-responsive characteristics onto the nanocarrier, enabling drug release under acidic conditions. Moreover, in the acidic and reducing TME, the Mn–O bond in the MMS nanocarriers underwent cleavage, thereby facilitating the nanocarrier's self-degradation. In vitro experiments demonstrated that when the MMS nanocarriers were immersed in PBS with a pH of 5.4 for 2 h, surface degradation commenced, and within 4 h, all mesopores collapsed. In contrast, the degradation process was notably slower when the samples were immersed in PBS with a pH of 7.4. Furthermore, under neutral pH conditions, the cumulative drug release from DOX-MMS/PDA within 48 h remained below 20%, indicating efficient drug encapsulation. As the acidity increased, the cumulative drug release gradually escalated. At pH 2.2, the cumulative amount of the drug released reached 65.0% within 12 h.
Additionally, to overcome drug resistance, Li et al.141 developed a versatile small-/macromolecule dual drug delivery system based on the biodegradable organosilica core–shell architecture (Fig. 27a). The small molecule drug was immobilized on the surface of the organosilica shell through pH-responsive imine bonds and was released in the acidic lysosomal environment. Subsequently, the elevated GSH levels within tumor cells triggered the degradation of the organosilica shell, leading to the release of the encapsulated large molecule drug. In vitro experiments investigating the acid-responsive drug release of the small molecule FL-C6-NH2 demonstrated that after 4 h, the cumulative drug release amount reached 20% under acidic conditions (pH = 5.0, 10 mM), and after 24 h, it was approximately 4% under acidic conditions (PBS, pH = 7.4, 10 mM), confirming the targeted release of small molecule drugs under acidic conditions (Fig. 27b). Similarly, in the acidic solution (pH = 5.0) containing 5 mM GSH, the initial release percentage was 6.8%. However, this value rapidly escalated, reaching 41% after 24 h, and further increased to 47% after 48 h (Fig. 27c).
3.2.1.2. Redox and hypoxia. Glucose, as a vital nutrient, plays a pivotal role in tumor growth. The Warburg effect highlights the dependence of tumor cell proliferation on aerobic glycolysis, rendering tumor cells highly responsive to fluctuations in glucose concentration within their microenvironment. Glucose oxidase (GOx), an enzyme capable of reacting with intracellular glucose, swiftly converts glucose into H2O2 and gluconic acid, thus disrupting the energy and nutrient supply within the tumor and inhibiting tumor growth. This presents an opportunity for the development of cancer starvation therapy based on GOx. Moreover, the enzymatic conversion of glucose by GOx into gluconic acid and H2O2 consumes oxygen, potentially facilitating a combined approach of hypoxia-based CMT and starvation therapy. Yang et al.143 developed a novel cascade delivery system that synergistically integrated AQ4N with GOx. AQ4N is an oxygen-sensitive topoisomerase II inhibitor that undergoes activation under hypoxic conditions, transforming into its highly effective anticancer form (AQ4). The yolk–shell structure of tetrasulfide-bridged organosilica nanoparticles (YS-DMONs) exhibited GSH-triggered biodegradability while maintaining a higher drug loading capacity due to their hydrophobic nature (Fig. 28a). Within the TME, the YS-DMONs underwent selective disintegration of their large pore shells, releasing GOx. This, in turn, resulted in the exposure of AQ4N to the hypoxic conditions of the TME, converting it into its cytotoxic form (AQ4) for CMT. Notably, the released GOx effectively depleted the tumor's original glucose, thereby starving the tumor cells and catalyzing the production of H2O2, leading to an oxidative cytotoxic effect. This also contributed to exacerbated tumor hypoxia, preventing insufficient AQ4N activation due to uneven oxygen distribution within the tumor tissue. Moreover, the direct depletion of GSH through tetrasulfide bonds within the nanopreparations prevented the additional loss of H2O2, thereby improving the cell death effect. In cell viability assays, treatment with YS-DMONs-AQ4N-GOx under simulated normoxic (20% O2) and hypoxic (2% O2) conditions resulted in cell viabilities of only 9.2% and 5.0%, respectively (Fig. 28b and c). The calculated IC50 of AQ4N in the YS-DMONs-AQ4N-GOx under normoxic conditions (0.96 μg
mL−1) was higher than that under hypoxic conditions (0.59 μgmL−1), further confirming the high efficacy of AQ4N under hypoxic conditions (Fig. 28d). Additionally, in vivo experiments demonstrated that YS-DMON-AQ4N-GOx significantly reduced tumor nodules and exhibited remarkable inhibitory effects on lung metastasis compared to the control groups (YS-DMON-AQ4N, YS-DMON-GOx, and AQ4N + GOx) (Fig. 28e).
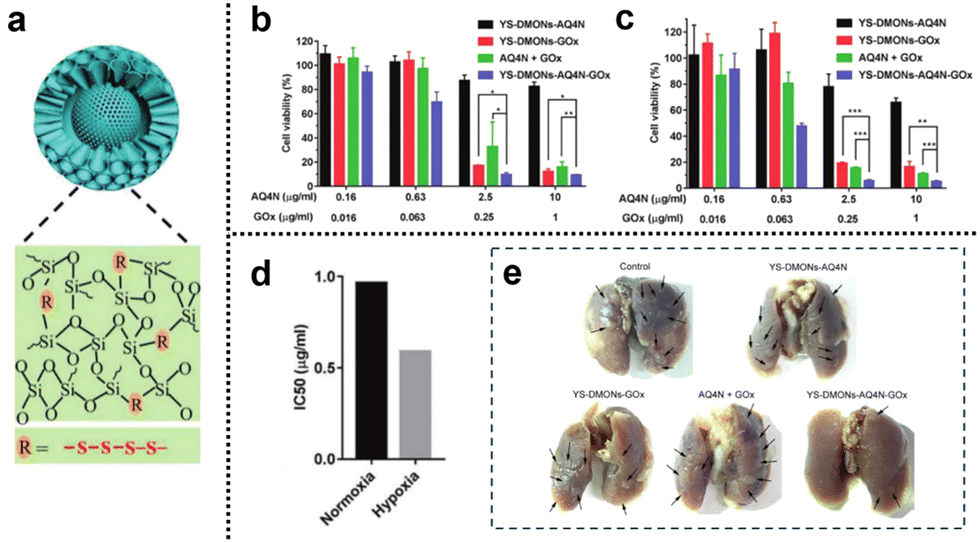 | ||
| Fig. 28 (a) YS-DMONs with a tetrasulfide incorporated framework. (b) and (c) Cell viability of 4T1 cells after different treatments under normoxic and hypoxic conditions for 48 h. (d) IC50 values of YS-DMON-AQ4N-GOx against 4T1 cells under normoxic and hypoxic conditions. (e) Tumor nodules in the lungs after different treatments. Reproduced from ref. 143 with permission from Wiley, copyright 2018. | ||
3.2.1.3. Redox and enzyme. Glutathione S-transferases π (GSTπ), which are highly expressed in hepatocellular carcinoma cells (HCC), are up to 4–5 times higher than those in normal cells.130 Based on this, an enzyme/redox dual-responsive nanodrug (QM-NPQ@PDHN) was developed by Jia et al. through a prodrug loading strategy (Fig. 29a).144 This nanodrug achieved the controlled release of nitric oxide (NO) in response to the activity of GSTπ and GSH within the TME, resulting in effective suppression of tumor growth. Moreover, the nanodrug was thoughtfully designed with switchable size and charge properties, enabling efficient penetration and internalization within the tumor tissue. The integration of the fluorescent molecule QM-2 facilitated real-time tracking and assessment of its targeting efficacy in vivo. The tumor-targeting efficacy of QM-NPQ@PDHN was assessed using fluorescence imaging, and a nude mouse model was established by inoculating SMMC-7721 tumors and administering QM-NPQ@PDHN via the tail vein. The biodistribution of QM-NPQ@PDHN was investigated in vivo through fluorescence imaging employing the IVIS system, revealing robust fluorescence at the tumor site (Fig. 29b). 24 h after QM-NPQ@PDHN injection, the mice were euthanized, and ex vivo fluorescence imaging was conducted on major organs and tumor tissues. The results indicated weak fluorescence in tumor tissues, liver, and kidney (Fig. 29c). Predominant accumulation of QM-NPQ@PDHN in the liver, kidney, and tumor tissues was demonstrated through semiquantitative analysis of the average fluorescence intensity distribution, with significantly higher levels in the tumor tissues than in other organs (Fig. 29d).
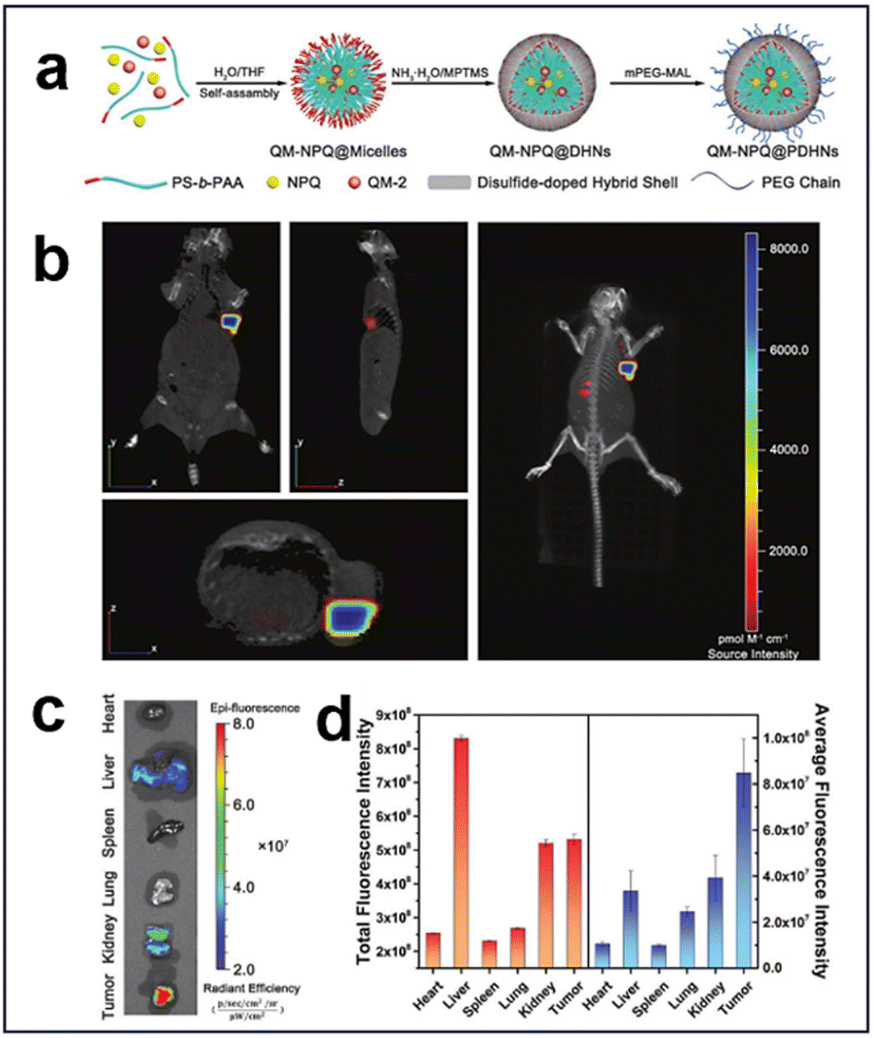 | ||
| Fig. 29 (a) Schematic synthesis of QM-NPQ@PDHNs. (b) Three-dimensional fluorescence images of hormonal mice at 24 h after intravenous injection of QM-NPQ@PDHNs. (c) Ex vivo fluorescence images of each organ at 24 h after intravenous injection of QM-NPQ@PDHNs. (d) Fluorescence intensity of tumors and organs in mice killed at 24 h after intravenous injection of QM-NPQ@PDHNs. Reproduced from ref. 144 with permission from Wiley, copyright 2018. | ||
3.2.2.1. Redox, hypoxia, and enzyme. HAPs represent a novel class of therapeutic agents meticulously designed to target and activate specifically within the hypoxic regions of the TME.145,146 These prodrugs are engineered to remain inactive or minimally toxic in oxygen-rich (normoxic) environments. However, upon encountering the hypoxic TME, they undergo specific chemical transformations or enzymatic reactions that trigger the release of their active drug forms. This activation process occurs exclusively within the hypoxic regions of the tumor, sparing healthy tissues from unnecessary drug exposure. A common strategy employed in HAPs involves incorporating nitroaromatic compounds into the prodrug structure. Inside the hypoxic TME, nitroreductase enzymes present in tumor cells metabolize nitro groups from HAPs, leading to the liberation of potent cytotoxic agents. Consequently, tumor cells are selectively targeted and destroyed, bolstering the effectiveness of cancer treatment while minimizing harm to normal tissues. Currently, a series of HAPs have been developed for cancer treatment, including banoxantrone (AQ4N) and tirapazamine (TPZ). Cheng et al.147 developed a novel nanohybrid (HA-TPZ&IR@GMON) for synergistic cancer therapy (Fig. 30). This nanohybrid involved the incorporation of photosensitizers IR820 and TPZ into GSH-degradable mesoporous organic silica nanoparticles (GMONs), which were then coated with HA. HA played a crucial role as a CD44 receptor-targeting agent, enabling specific and precise targeting of tumor tissues by the nanohybrid. Upon exposure to HAase and GSH, HA and GMON experienced controlled degradation, facilitating the release of IR820 and TPZ. IR820 induced PDT by generating 1O2 upon NIR laser irradiation, concurrently depleting oxygen, thereby intensifying the hypoxic state of the tumor tissue. Moreover, TPZ exhibited enhanced activation under highly hypoxic conditions, thereby augmenting the efficacy of CMT. In vitro experiments demonstrated that treatment with 200 μg
mL−1 HATPZ&IR@GMON for 5 min under NIR laser (l W cm−2) irradiation resulted in a cell survival rate of less than 10%. Additionally, in the absence of NIR laser irradiation, 36% of cells survived after treatment with 200 μgmL−1 HA-TPZ@GMON. Furthermore, under NIR laser irradiation, HA-TPZ&IR@GMONs induced apoptosis in 36.2% of 4T1 cells under normoxic conditions and a remarkable 73.4% of cells in a hypoxic environment.
 | ||
| Fig. 30 Schematic representation of the anticancer mechanism of HA-TPZ&IR@GMONs. Reproduced from ref. 147 with permission from Elsevier, copyright 2021. | ||
3.2.2.2. pH, redox, and enzyme. HAase is highly expressed in the TME, facilitating the degradation of HA to induce the disintegration of nanoplatforms.148,149 Consequently, researchers have actively pursued the development and design of numerous HA-modified nanosystems, strategically targeting tumor cells that exhibit high expression of CD44 molecules on their cell membranes.150,151 Yang et al.152 developed a tumor immunotherapy strategy for triple-negative breast cancer (TNBC), addressing challenges such as high recurrence, poor prognosis, and limited treatment options. Their approach combined mild PTT and nanocatalytic drug action using Fe-loaded mesoporous organosilica (MOF) nanoparticles, with ICG as a photothermal agent and HA as a drug carrier (IMOFH) (Fig. 31). HA incorporation enabled IMOFH to specifically target CD44 molecules on 4T1 breast cancer cells, leading to highly targeted accumulation at the tumor site. The presence of HAase in the TME facilitated drug internalization by promoting HA degradation. Moreover, IMOFH exhibited pH- and GSH-responsive behaviour in the TME. Upon reaching the TME, IMOFH underwent structural degradation, releasing Fe3+ and ICG. Under NIR laser irradiation, ICG demonstrated excellent photothermal conversion ability, enabling mild PTT. Additionally, the Fe3+ Fenton reagent, released from IMOFH, triggered the generation of ROS by exploiting H2O2 within the TME. This ROS generation acted synergistically with mild PTT. This ROS-induced oxidative activity could also trigger immunogenic cell death (ICD) in tumor cells,153 releasing damage-associated molecular patterns (DAMPs) such as ATP and HMGB1. This process activates dendritic cells, preventing T-cell exhaustion and tumor cell immune evasion and increasing T-cell levels in the TME, thereby enhancing the body's immune capacity. The researchers evaluated the pH and GSH responsiveness of IMOFH, noting partial degradation on the third day and nearly complete degradation on the seventh day in an acidic (pH 6.5) and GSH (10 mM) solution. They also assessed the photothermal ability of the IMOFH using thermal imaging technology (808 nm), finding its concentration-dependent and light power-dependent heating abilities. Higher IMOFH concentrations and greater light power resulted in stronger heating effects. To confirm the targeted accumulation of IMOFH in 4T1 breast cancer, Cy5.5-labelled MOFH was intravenously injected into mice bearing 4T1 tumors. The results demonstrated time-dependent enhancement of the fluorescence signal in the 4T1 tumor site, peaking at 8 h after injection. After 24 h, they performed ex vivo fluorescence imaging of major organs and 4T1 tumors, revealing significantly higher signal intensities in the liver and 4T1 tumors than in other tissues. These findings highlighted the excellent tumor accumulation ability of IMOFH, positioning it as a promising therapeutic agent for TNBC.
 | ||
| Fig. 31 Preparation and antitumour scheme of IMOFH. Reproduced from ref. 152 with permission from the Royal Society of Chemistry, copyright 2022. | ||
In addition, Shi et al.154 developed an intelligent pH-responsive nanoplatform (HSPMH-DOX) for MR imaging-guided synergistic chemo-photothermal therapy and CDT (Fig. 32a). Initially, DOX-loaded hollow mesoporous silica nanoparticles (HSP-DOX) were prepared. Then, dopamine, with photothermal conversion performance and pH responsiveness, was used to coat the HSP-DOX and act as a “controlled gate” for drug release. Subsequently, a layer of manganese dioxide nanosheets was grown on the surface of HSP-DOX through in situ potassium permanganate reduction, introducing Mn–O bonds into the –Si–O–Si– framework to alter mesoporous silica biodegradability. Simultaneously, the surface was functionalized with the targeting group HA to form HSPMH-DOX, enabling specific recognition of tumor cells and tissues. In the acidic TME, the MnO2 nanosheets could be converted into Mn2+ through a reduction reaction that provided a higher longitudinal relaxivity (T1), improving the accuracy of tumor localization. The in vitro MR imaging results of HSPMH under varying pH conditions indicated no significant difference in the MR imaging signal at pH 7.4 (Fig. 32b). However, a noticeable signal enhancement was observed at pH 5.5, leading to a considerable improvement in T1-MR contrast within the tumor (Fig. 32c).
 | ||
| Fig. 32 (a) Schematic representation of the preparation of HS@PDA/MnO2/HA-DOX (HSPMH-DOX) and its TME responsive activation for MRI and synergistic therapy. (b) and (c) MR images of the T1 map and T1 reflexivity of HSPMH under different conditions and concentrations. Reproduced from ref. 154 with permission from Elsevier, copyright 2023. | ||
MMPs possess the capacity to degrade extracellular matrix proteins such as collagen and fibronectin, alongside other molecules in the extracellular milieu.155,156 The enzymatic activity of MMPs undergoes meticulous regulation across multiple tiers, encompassing transcription, translation, and activation processes.157–160 Normally, MMPs maintain a subdued activity level, serving to uphold extracellular matrix stability. However, within the TME, numerous factors, including the secretion of growth factors, modifications in the ECM composition, and atypical expression by tumor cells, can culminate in the excessive upregulation and activation of MMPs.161 Hence, the strategic modification of degradable silica nanoparticles with MMP-sensitive peptide sequences enables the effective and targeted delivery of nanodrugs, facilitating precise imaging and treatment of tumors. Xiao et al.162 developed a gelatin-based nanocluster (DOX-icluster) with dual-sized/charge-switchable and multiresponsive characteristics for targeted antitumour therapy (Fig. 33). The formation of the nanocluster involved the electrostatic assembly of FA-modified gelatin (FA-GelDMA). This gelatin was sensitive to MMP and was combined with small-sized NH2-modified hollow mesoporous organosilica nanoparticles loaded with DOX (DOX-HMON-NH2). The distinctive feature of DOX-icluster lies in the incorporation of disulfide bonds into its silica framework, imparting responsiveness to intracellular GSH and leading to rapid degradation, rendering it highly biodegradable.80,85 Additionally, the DOX-icluster demonstrated switchable sizes and charges that facilitated tumor cell internalization. Under neutral conditions, DOX-iclusters exhibited a size of approximately 199 nm. However, in the acidic TME, the dimethylamino methylene bonds within FA-GelDMA were cleaved, resulting in gelatin hydrolysis facilitated by overexpressed MMP. This process led to the release of DOX-HMON-NH2 nanoparticles (approximately 48 nm), which effectively accumulated in tumors and promoted cellular internalization. Intracellularly, DOX-HMON-NH2 was subsequently broken down by intracellular GSH, leading to DOX release at an approximate cumulative release amount of 48.1% and exerting its chemotherapeutic antitumour effect. The tumor inhibition experiments revealed that under conditions of pH = 6.5 and in the presence of MMP-2, the DOX-icluster exhibited the strongest tumor inhibition effect, reducing the tumor volume to 15.1% of the initial volume on the seventh day.
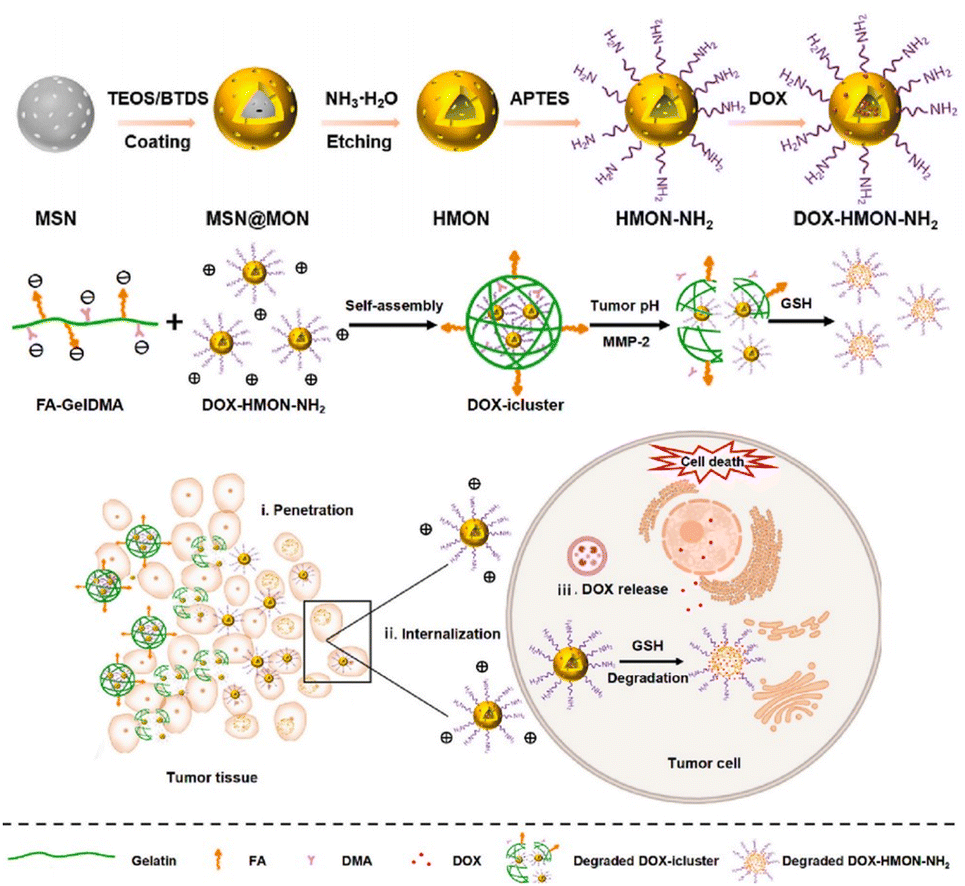 | ||
| Fig. 33 Schematic diagram of the synthesis and antitumour effects of DOX-HMON-NH2. Reproduced from ref. 162 with permission from Elsevier, copyright 2023. | ||
3.2.2.3. pH, redox, and hypoxia. The utilization of HAPs as a promising oncotherapy strategy is impeded by the limitations arising from insufficient tumor hypoxia. Furthermore, standalone prodrugs may not effectively combat the complexity of tumors. To overcome these challenges, Hu et al.163 developed a novel nanosystem (TPZ@FeMSN-GOX) that integrated triple synergistic cancer starvation therapy, hypoxia-activated CMT, and CDT (Fig. 34a). The fabrication of TPZ@FeMSN-GOX involved the synthesis of iron-doped mesoporous silica nanoparticles (FeMSNs), conjugation with GOX on the surface, and subsequent loading with TPZ. The introduction of iron in this nanosystem facilitated its potential for the Fenton reaction, while TPZ underwent transformation into the cytotoxic therapeutic agent TPZ radical under low oxygen conditions, precisely damaging cancer cells and reducing side effects on normal tissues. Upon entering the tumor, TPZ@FeMSN-GOX utilized GOX to induce starvation therapy and produce H2O2, which in turn consumed O2, intensifying the hypoxic environment and thereby enhancing TPZ-mediated CMT. Additionally, the released Fe3+ was reduced to Fe2+ by GSH, further converting H2O2 into highly toxic ˙OH through the Fenton reaction and activating CDT. Moreover, continuous H2O2 production and glucose consumption amplified the acidic environment, ensuring a sustained and effective Fenton reaction. In vitro experiments demonstrated that TPZ@FeMSN-GOX substantially reduced the cell viability of both PANC-1 and 4T1 cells. Even at low concentrations of 2.5 μg
mL−1 and after 24 h of incubation, cell viability was markedly reduced to 45.26% and 48.59%, respectively, underscoring its impressive anticancer efficacy (Fig. 34b and c). In vivo experiments revealed that mice treated with saline and free TPZ exhibited rapid tumor growth, indicating that TPZ alone was not sufficient to yield antitumour effects. In contrast, mice treated with TPZ@FeMSN exhibited partially inhibited tumor growth, whereas TPZ@FeMSN-GOX demonstrated the most substantial effect in suppressing tumor growth in mice. Remarkably, the mice treated with TPZ@FeMSN-GOX survived the longest (25 days) compared to the other groups (Fig. 34d).
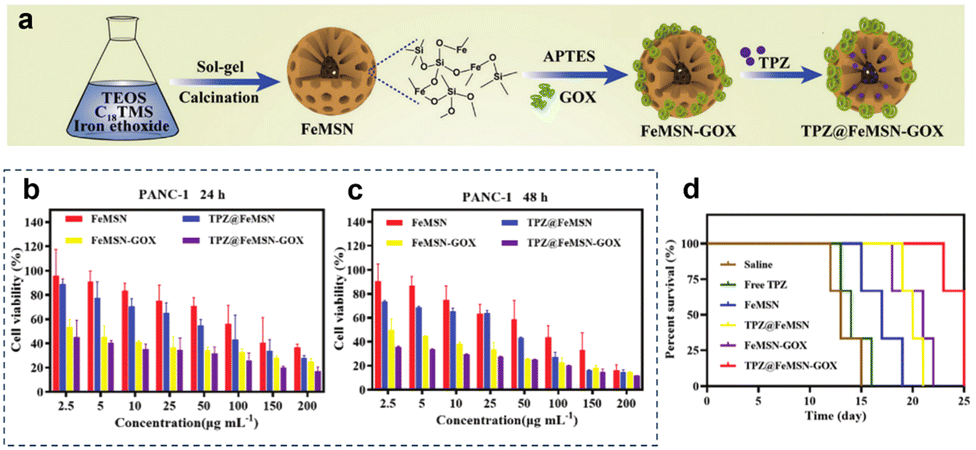 | ||
| Fig. 34 (a) Schematic representation of the preparation process of TPZ@FeMSN-GOx. (b) and (c) Cell viability of PANC-I cells after incubation with different concentrations of samples for 24 h and 48 h. (d) Survival curves of 4T1 tumor-bearing mice after different treatments. Reproduced from ref. 163 with permission from the Royal Society of Chemistry, copyright 2022. | ||
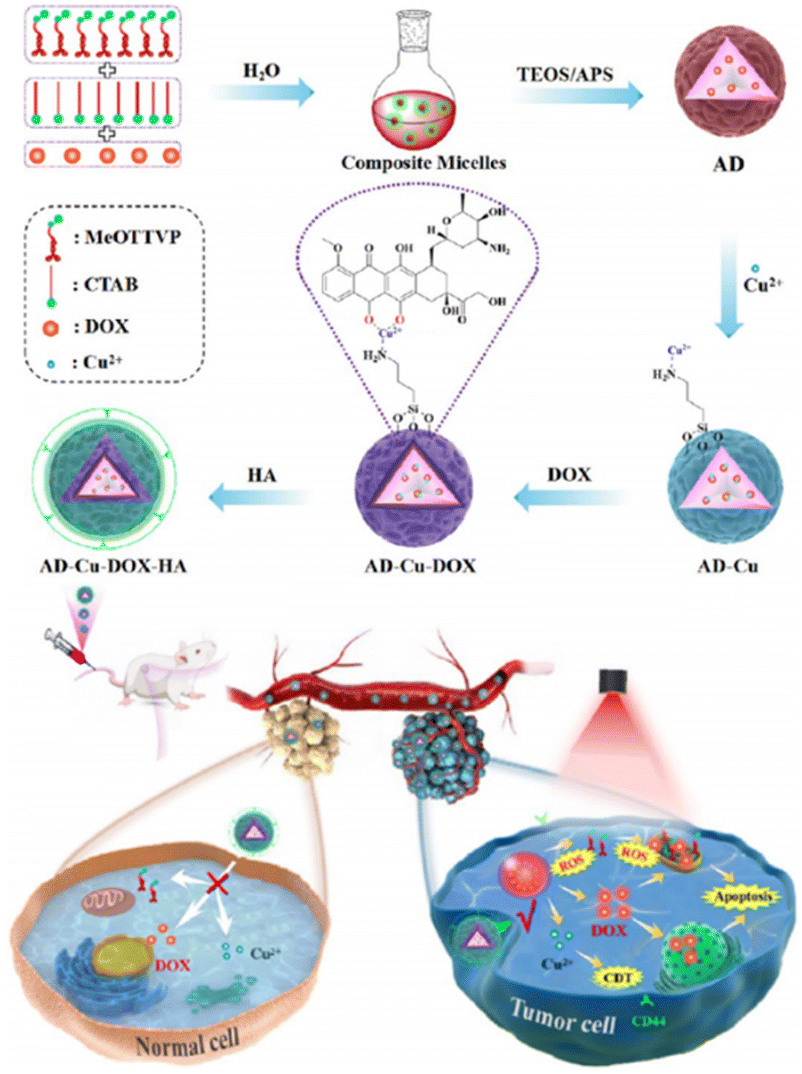 | ||
| Fig. 35 Preparation process of core–shell AD-Cu-DOX-HA and its further application in targeted and tumor-specific therapy. Reproduced from ref. 164 with permission from ACS, copyright 2022. | ||
Wang et al.165 developed synergistic therapeutic nanoplatforms (DCI) for the combination of the PDT and the CDT. The nanoplatforms utilized dithioether-crosslinked dendritic mesoporous organosilica (DMOS) as a carrier to load copper peroxide (CuO2) and a photosensitizer ICG, which was further modified with HA (Fig. 36). HA exhibited specific binding affinity towards CD44 molecules commonly overexpressed on cancer cell membranes. This selective interaction allowed DCI to achieve high accumulation within tumor tissues, thereby facilitating precise and targeted drug delivery. Specifically, under acidic conditions, DCI could effectively release H2O2/O2 while depleting GSH, thus effectively targeting the specific conditions of the TME. Furthermore, the released Cu2+ served as a valuable agent for T1-weighted MR imaging, enabling both in vitro and in vivo imaging for guided therapies. Upon accumulation at the tumor site, DCI adeptly surmounted the challenge of hypoxia and induced a substantial elevation of ROS upon NIR laser irradiation. This ROS generation led to the apoptotic demise of tumor cells, resulting in a significant amplification of therapeutic efficacy against cancer. The results clearly demonstrated the antitumour efficacy of DCI, as evidenced by the DCI + NIR treatment, which exhibited minimal fluctuations in body weight and the smallest tumour volume. The MR imaging performance of DCI was assessed, revealing a significant enhancement of MR imaging signals at the tumor after intravenous administration. The in vitro MR imaging signal intensity exhibited a concentration dependency at acidic pH (pH = 5.5), with higher DCI concentrations resulting in increased intensity. In contrast, the MR imaging signal intensity remained relatively constant at pH = 7.4, regardless of the variations in DCI concentrations.
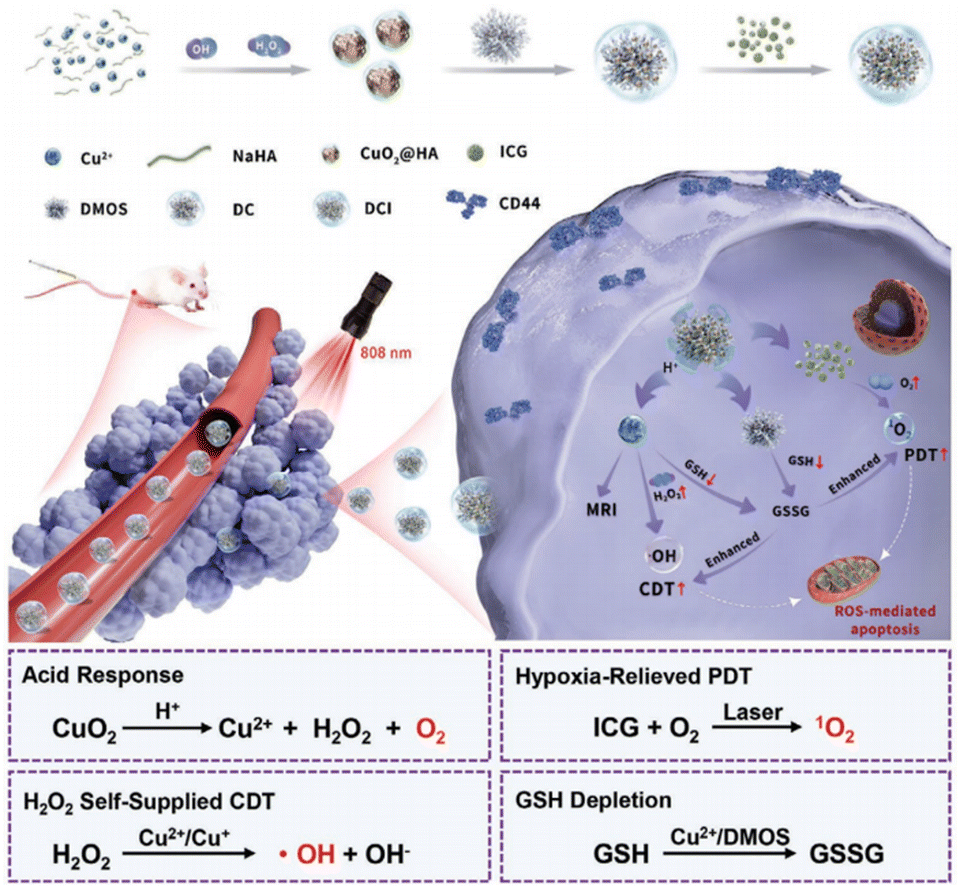 | ||
| Fig. 36 Principled process for MRI-guided PDT combined with CDT of DMOS@CuO2/ICG-HA (DCI). Reproduced from ref. 165 with permission from Wiley, copyright 2022. | ||
4. Conclusions and future perspectives
We have presented a comprehensive overview of the classification and degradation mechanism of degradable silica nanoparticles, and these nanoparticles were designed to respond to specific TME stimuli, including pH, redox, hypoxia, and enzymes, for precision tumor diagnosis and therapy. We emphasize the importance of designing degradable nanoplatforms to mitigate potential toxic side effects in vivo. The degradable silica nanoparticles discussed not only exhibit efficient clearance and low toxicity but also offer controlled drug release, improved imaging capabilities, and enhanced therapeutic effects, thereby presenting promising prospects for cancer theranostics. However, the development of TME-responsive precision diagnosis and therapy based on degradable silica nanoparticles faces several challenges. While the potential of degradable silica nanoparticles for TME-responsive precision theranostics is promising, several challenges must be addressed.First, the preparation of most degradable silica nanoparticles presents challenges, including high difficulty, complex processes, and high costs, which partly restrict their application in the biomedical field. Regarding imaging, there is a lack of rational design to optimize imaging performance and enhance imaging quality in deep tissues. As carriers for drugs, the stability of degradable silica nanoparticles loaded with drugs requires further improvement. Furthermore, to address the TME, the rational design of highly responsive or multiresponsive nanoplatforms, along with the development of intelligent nanomaterials integrating endogenous responses and exogenous stimuli (e.g., light, X-rays, magnetic field, etc.), prove to be valuable in achieving precise tumor therapy, thereby enhancing drug utilization and theranostic efficacy.
Second, the complexity and specificity of the TME often hinder the attainment of optimal therapeutic effects and may even contribute to drug resistance, recurrence, and metastasis. The features of the TME extend beyond those mentioned; researchers have reported immune suppression, inflammation, and cytokine production. The development of more precise TME-responsive strategies and the achievement of specific responses to TME continue to be challenges for researchers. Additionally, the TME closely influences the survival, invasion, and metastasis of tumor cells. Presently, the treatment efficacy for highly resistant tumors remains unsatisfactory. Despite the enhanced penetration and drug accumulation in tumor regions through the use of degradable silica nanoparticles, the high drug resistance and efflux in numerous tumor cells continue to limit treatment effectiveness. Thus, more effective measures are needed to augment drug uptake by tumor cells and prevent tumor cell migration.
Third, the safe application of degradable silica nanoparticles should be ensured. Preliminary results demonstrate the potential of degradable silica nanoparticles for future disease treatment. However, further research is imperative to ensure their safe application in clinical settings. The degradation effects of silica nanoparticles under various physiological conditions within the body need examination. Additionally, the ions generated after material degradation may cause damage to corresponding organs during metabolism. Therefore, systematic and long-term research is required to investigate the biological distribution, circulation, metabolism, degradation, and excretion of these degradable silica nanoparticles.
TME-responsive precision diagnosis and therapy utilizing degradable silica nanoparticles hold immense potential and broad prospects. However, overcoming challenges in their development and understanding their behaviour within the biological system are crucial. Further in-depth research will strengthen the control and modification of degradable silica nanoparticles and enhance our ability to achieve their true potential in tumor diagnosis and treatment, ultimately contributing to advancements in human health.
Author contributions
Junjie Zhang: writing – original draft, supervision, writing – review & editing, funding acquisition. Kaiyuan Tang: investigation, writing – original draft. Zilu Liu: writing – original draft, formal analysis, visualization. Zhijing Zhang: investigation, formal analysis, visualization. Shufan Duan: formal analysis, visualization. Hui Wang: investigation, visualization. Hui Yang: formal analysis, visualization. Dongliang Yang: conceptualization, formal analysis, project administration, writing – review & editing. Wenpei Fan: conceptualization, project administration, writing – review & editing, funding acquisition.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the Department of Education of Anhui Province (KJ2021A0732), the open research fund of State Key Laboratory of Organic Electronics and Information Displays (SKL2023002), the Funding of Double First-Rate Discipline Innovation Team of China Pharmaceutical University (CPUQNJC22_04), the Fundamental Research Funds for the Central Universities of China (2632023TD01), the Science and Technology Innovation Guidance Project of Bengbu City (20220127), and the National College Students’ Innovation and Entrepreneurship Training Program (202210367075).Notes and references
- J. Cortes, J. M. Perez-Garcia, A. Llombart-Cussac, G. Curigliano, N. S. El Saghir, F. Cardoso, C. H. Barrios, S. Wagle, J. Roman, N. Harbeck, A. Eniu, P. A. Kaufman, J. Tabernero, L. Garcia-Estevez, P. Schmid and J. Arribas, Ca-Cancer J. Clin., 2020, 70, 105–124 CrossRef PubMed.
- T. Wu and Y. Dai, Cancer Lett., 2017, 387, 61–68 CrossRef CAS PubMed.
- N. M. Anderson and M. C. Simon, Curr. Biol., 2020, 30, R921–R925 CrossRef CAS PubMed.
- R. Baghban, L. Roshangar, R. Jahanban-Esfahlan, K. Seidi, A. Ebrahimi-Kalan, M. Jaymand, S. Kolahian, T. Javaheri and P. Zare, Cell Commun. Signaling, 2020, 18, 59 CrossRef PubMed.
- F. Gong, N. Yang, X. Wang, Q. Zhao, Q. Chen, Z. Liu and L. Cheng, Nano Today, 2020, 32, 100851 CrossRef CAS.
- Y. Dai, C. Xu, X. Sun and X. Chen, Chem. Soc. Rev., 2017, 46, 3830–3852 RSC.
- M. O. Li, N. Wolf, D. H. Raulet, L. Akkari, M. J. Pittet, P. C. Rodriguez, R. N. Kaplan, A. Munitz, Z. Zhang, S. Cheng and N. Bhardwaj, Cancer Cell, 2021, 39, 725–729 CrossRef CAS PubMed.
- X. Wang, Z. Chen, J. Xu, S. Tang, N. An, L. Jiang, Y. Zhang, S. Zhang, Q. Zhang, Y. Shen, S. Chen, X. Lan, T. Wang, L. Zhai, S. Cao, S. Guo, Y. Liu, A. Bi, Y. Chen, X. Gai, Y. Duan, Y. Zheng, Y. Fu, Y. Li, L. Yuan, L. Tong, K. Mo, M. Wang, S. H. Lin, M. Tan, C. Luo, Y. Chen, J. Liu, Q. Zhang, L. Li and M. Huang, Cell Res., 2022, 32, 638–658 CrossRef CAS PubMed.
- J. Qin, H. Tian, F. Kong, Y. Guo, W. Du, C. Zhang, H. Gu and Y. Li, Sens. Actuators, B, 2022, 371, 132522 CrossRef CAS.
- C. Michiels, C. Tellier and O. Feron, Biochim. Biophys. Acta, Bioenerg., 2016, 1866, 76–86 CAS.
- Y. Luo, C. Li, Y. Zhang, P. Liu, H. Chen, Z. Zhao, Y. Wang, Z. Zhou, H. Song, B. Su, C. Li, X. Li, T. Zhang, H. You, Y. Wu, Z. Tian, S. Zhang, Y. Guo, H. Fan, Q. Chen, C. Jiang and T. Sun, Acta Biomater., 2023, 167, 387–400 CrossRef CAS PubMed.
- R. McShane, S. Arya, A. J. Stewart, P. D. Caie and M. Bates, Biochim. Biophys. Acta, Rev. Cancer, 2021, 1876, 188598 CrossRef CAS PubMed.
- Z. Zhang, C. Ding, T. Sun, L. Wang and C. Chen, Adv. Healthcare Mater., 2023, 12, e2300153 CrossRef PubMed.
- D. A. Snellings, C. C. Hong, A. A. Ren, M. A. Lopez-Ramirez, R. Girard, A. Srinath, D. A. Marchuk, M. H. Ginsberg, I. A. Awad and M. L. Kahn, Circ. Res., 2021, 129, 195–215 CrossRef CAS PubMed.
- B. C. Ozdemir, T. Pentcheva-Hoang, J. L. Carstens, X. Zheng, C. C. Wu, T. R. Simpson, H. Laklai, H. Sugimoto, C. Kahlert, S. V. Novitskiy, A. De Jesus-Acosta, P. Sharma, P. Heidari, U. Mahmood, L. Chin, H. L. Moses, V. M. Weaver, A. Maitra, J. P. Allison, V. S. LeBleu and R. Kalluri, Cancer Cell, 2014, 25, 719–734 CrossRef CAS PubMed.
- S. Turajlic, A. Sottoriva, T. Graham and C. Swanton, Nat. Rev. Cancer, 2019, 20, 404–416 CrossRef CAS PubMed.
- S. Gavas, S. Quazi and T. M. Karpinski, Nanoscale Res. Lett., 2021, 16, 173 CrossRef CAS PubMed.
- B. Peltanova, M. Raudenska and M. Masarik, Mol. Cancer, 2019, 18, 63 CrossRef PubMed.
- J. Fang, W. Islam and H. Maeda, Adv. Drug Delivery Rev., 2020, 157, 142–160 CrossRef CAS PubMed.
- J. K. Tee, L. X. Yip, E. S. Tan, S. Santitewagun, A. Prasath, P. C. Ke, H. K. Ho and D. T. Leong, Chem. Soc. Rev., 2019, 48, 5381–5407 RSC.
- W. Ying, Y. Zhang, W. Gao, X. Cai, G. Wang, X. Wu, L. Chen, Z. Meng, Y. Zheng, B. Hu and X. Lin, ACS Nano, 2020, 14, 9662–9674 CrossRef CAS PubMed.
- L. Gong, Y. Zhang, J. Zhao, Y. Zhang, K. Tu, L. Jiao, Q. Xu, M. Zhang and S. Han, Small, 2022, 18, 2107656 CrossRef CAS PubMed.
- X. Kong, Y. Qi, X. Wang, R. Jiang, J. Wang, Y. Fang, J. Gao and K. C. Hwang, Prog. Mater. Sci., 2023, 101070 CrossRef CAS.
- A. Gulzar, J. Xu, C. Wang, F. He, D. Yang, S. Gai, P. Yang, J. Lin, D. Jin and B. Xing, Nano Today, 2019, 26, 16–56 CrossRef CAS.
- C. S. Sia, H. P. Lim, B. T. Tey, B. H. Goh and L. E. Low, Biochim. Biophys. Acta, Rev. Cancer, 2022, 1877, 188779 CrossRef CAS PubMed.
- J. G. Croissant, Y. Fatieiev and N. M. Khashab, Adv. Mater., 2017, 29, 1604634 CrossRef PubMed.
- J. Liu, M. Yu, C. Zhou and J. Zheng, Mater. Today, 2013, 16, 477–486 CrossRef CAS.
- Y. Tan, M. Chen, H. Chen, J. Wu and J. Liu, Angew. Chem., Int. Ed., 2021, 60, 11713–11717 CrossRef CAS PubMed.
- B. Du, M. Yu and J. Zheng, Nat. Rev. Mater., 2018, 3, 358–374 CrossRef.
- X. Wang, X. Zhong, J. Li, Z. Liu and L. Cheng, Chem. Soc. Rev., 2021, 50, 8669–8742 RSC.
- N. Senthilkumar, P. K. Sharma, N. Sood and N. J. C. C. R. Bhalla, Coord. Chem. Rev., 2021, 445, 214082 CrossRef CAS.
- N. Feliu, D. Docter, M. Heine, P. Del Pino, S. Ashraf, J. Kolosnjaj-Tabi, P. Macchiarini, P. Nielsen, D. Alloyeau, F. Gazeau, R. H. Stauber and W. J. Parak, Chem. Soc. Rev., 2016, 45, 2440–2457 RSC.
- X. Nan, Y. Huyan, H. Li, S. Sun and Y. J. C. C. R. Xu, Coord. Chem. Rev., 2021, 426, 213580 CrossRef CAS.
- H. Ehrlich, K. D. Demadis, O. S. Pokrovsky and P. G. Koutsoukos, Chem. Rev., 2010, 110, 4656–4689 CrossRef CAS PubMed.
- A. Santos, M. Sinn Aw, M. Bariana, T. Kumeria, Y. Wang and D. Losic, J. Mater. Chem. B, 2014, 2, 6157–6182 RSC.
- S. Mohanan, X. Guan, M. Liang, A. Karakoti and A. Vinu, Small, 2023, e2301113 CrossRef PubMed.
- F. Tang, L. Li and D. Chen, Adv. Mater., 2012, 24, 1504–1534 CrossRef CAS PubMed.
- R. S. Guimaraes, C. F. Rodrigues, A. F. Moreira and I. J. Correia, Pharmacol. Res., 2020, 155, 104742 CrossRef CAS PubMed.
- P. Yang, S. Gai and J. Lin, Chem. Soc. Rev., 2012, 41, 3679–3698 RSC.
- Y. Huang, P. Li, R. Zhao, L. Zhao, J. Liu, S. Peng, X. Fu, X. Wang, R. Luo, R. Wang and Z. Zhang, Biomed. Pharmacother., 2022, 151, 113053 CrossRef CAS PubMed.
- J. G. Croissant, K. S. Butler, J. I. Zink and C. J. Brinker, Nat. Rev. Mater., 2020, 5, 886–909 CrossRef CAS.
- Y. Tan, D. Yu, J. Feng, H. You, Y. Bai, J. He, H. Cao, Q. Che, J. Guo and Z. Su, Drug Delivery Transl. Res., 2023, 13, 2213–2238 CrossRef PubMed.
- P. Mohammadi, M. Abbasinia, M. J. Assari and M. Oliaei, Toxicol. Environ. Chem., 2018, 100, 285–316 CrossRef CAS.
- L. Wang, M. Huo, Y. Chen and J. Shi, Biomaterials, 2018, 163, 1–13 CrossRef CAS PubMed.
- N. Lu, P. Huang, W. Fan, Z. Wang, Y. Liu, S. Wang, G. Zhang, J. Hu, W. Liu, G. Niu, R. D. Leapman, G. Lu and X. Chen, Biomaterials, 2017, 126, 39–48 CrossRef CAS PubMed.
- G. Yang, S. Z. F. Phua, A. K. Bindra and Y. Zhao, Adv. Mater., 2019, 31, e1805730 CrossRef PubMed.
- J. Zhang, N. Lu, L. Weng, Z. Feng, J. Tao, X. Su, R. Yu, W. Shi, Q. Qiu, Z. Teng and L. Wang, J. Colloid Interface Sci., 2021, 583, 714–721 CrossRef CAS PubMed.
- Y. Zhang, Z. Teng, Q. Ni, J. Tao, X. Cao, Y. Wen, L. Wu, C. Fang, B. Wan, X. Zhang and G. Lu, ACS Appl. Mater. Interfaces, 2020, 12, 57810–57820 CrossRef CAS PubMed.
- X. Peng, K. Chen, W. Liu, X. Cao, M. Wang, J. Tao, Y. Tian, L. Bao, G. Lu and Z. Teng, Nano-Micro Lett., 2020, 12, 137 CrossRef CAS PubMed.
- M. Manzano and M. Vallet-Regi, J. Mater. Sci., 2018, 29, 65 Search PubMed.
- X. Liu and J. Sun, Biomaterials, 2010, 31, 8198–8209 CrossRef CAS PubMed.
- A. Parodi, M. Evangelopoulos, N. Arrighetti, A. Cevenini, M. Livingston, S. Z. Khaled, B. S. Brown, I. K. Yazdi, F. Paradiso, J. N. Campa-Carranza, A. De Vita, F. Taraballi and E. Tasciotti, Small, 2020, 16, e1907693 CrossRef PubMed.
- J. G. Croissant, Y. Fatieiev, A. Almalik and N. M. Khashab, Adv. Healthcare Mater., 2018, 7, 1700831 CrossRef PubMed.
- X. Du, F. Kleitz, X. Li, H. Huang, X. Zhang and S. Z. Qiao, Adv. Funct. Mater., 2018, 28, 1707325 CrossRef.
- M. Qiu, A. Singh, D. Wang, J. Qu, M. Swihart, H. Zhang and P. N. Prasad, Nano Today, 2019, 25, 135–155 CrossRef CAS.
- S. Liu, S. Han, Y. Song, R. Sun, L. Zhao and C. Yan, Adv. Healthcare Mater., 2023, 12, e2300184 CrossRef PubMed.
- Y. Hu, S. Bai, X. Wu, S. Tan and Y. J. C. I. He, Ceram. Int., 2021, 47, 31031–31041 CrossRef CAS.
- K. Möller and T. J. C. O. M. Bein, Chem. Mater., 2019, 31, 4364–4378 CrossRef.
- B. Kang, M. B. Zheng, P. Song, A. P. Chen, J. W. Wei, J. J. Xu, Y. Shi and H. Y. J. S. Chen, Small, 2017, 13, 1604228 CrossRef PubMed.
- M. B. Gongalsky, A. P. Sviridov, Y. I. Bezsudnova and L. A. Osminkina, Colloids Surf., B, 2020, 190, 110946 CrossRef CAS PubMed.
- V. Poscher and Y. Salinas, Materials, 2020, 13, 3668 CrossRef CAS PubMed.
- B. Yang, Y. Chen and J. Shi, Mater. Sci. Eng., R, 2019, 137, 66–105 CrossRef.
- N. B. Fernandes, Y. Nayak, S. Garg and U. Y. Nayak, Coord. Chem. Rev., 2023, 478, 214977 CrossRef CAS.
- K. Zivojevic, M. Mladenovic, M. Djisalov, M. Mundzic, E. Ruiz-Hernandez, I. Gadjanski and N. Z. Knezevic, J. Controlled Release, 2021, 337, 193–211 CrossRef CAS PubMed.
- L. Yu, Y. Chen, H. Lin, S. Gao, H. Chen and J. Shi, Small, 2018, 14, e1800708 CrossRef PubMed.
- L. Yu, Y. Chen, M. Wu, X. Cai, H. Yao, L. Zhang, H. Chen and J. Shi, J. Am. Chem. Soc., 2016, 138, 9881–9894 CrossRef CAS PubMed.
- J. Tao, Y. Tian, D. Chen, W. Lu, K. Chen, C. Xu, L. Bao, B. Xue, T. Wang, Z. Teng and L. Wang, Angew. Chem., Int. Ed., 2023, 62, e202216361 CrossRef CAS PubMed.
- Y. He, B. Zeng, S. Liang, M. Long and H. Xu, ACS Appl. Mater. Interfaces, 2017, 9, 44402–44409 CrossRef CAS PubMed.
- Y. Kang, W. Sun, S. Li, M. Li, J. Fan, J. Du, X. J. Liang and X. Peng, Adv. Sci., 2019, 6, 1900716 CrossRef PubMed.
- Y. Wang, Y. Liu, H. Wu, J. Zhang, Q. Tian and S. Yang, Adv. Funct. Mater., 2019, 29, 1805764 CrossRef.
- V. Poscher and Y. Salinas, Materials, 2020, 13, 3668 CrossRef CAS PubMed.
- Y. Chen and J. Shi, Adv. Mater., 2016, 28, 3235–3272 CrossRef CAS PubMed.
- C. Urata, H. Yamada, R. Wakabayashi, Y. Aoyama, S. Hirosawa, S. Arai, S. Takeoka, Y. Yamauchi and K. Kuroda, J. Am. Chem. Soc., 2011, 133, 8102–8105 CrossRef CAS PubMed.
- M. Giebler, S. V. Radl, M. Ast, S. Kaiser, T. Griesser, W. Kern and S. Schlögl, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 2319–2329 CrossRef CAS.
- B. Karimi, N. Ganji, O. Pourshiani and W. R. Thiel, Prog. Mater. Sci., 2022, 125, 100896 CrossRef CAS.
- M. H. Lee, Z. Yang, C. W. Lim, Y. H. Lee, S. Dongbang, C. Kang and J. S. Kim, Chem. Rev., 2013, 113, 5071–5109 CrossRef CAS PubMed.
- P. Zhu, Y. Chen and J. Shi, ACS Nano, 2018, 12, 3780–3795 CrossRef CAS PubMed.
- Y. Yang, J. Wan, Y. Niu, Z. Gu, J. Zhang, M. Yu and C. Yu, Chem. Mater., 2016, 28, 9008–9016 CrossRef CAS.
- X. Jia, J. He, L. Shen, J. Chen, Z. Wei, X. Qin, D. Niu, Y. Li and J. Shi, Nano Lett., 2019, 19, 8690–8700 CrossRef CAS PubMed.
- P. Huang, Y. Chen, H. Lin, L. Yu, L. Zhang, L. Wang, Y. Zhu and J. Shi, Biomaterials, 2017, 125, 23–37 CrossRef CAS PubMed.
- P. Yuan, H. Zhang, L. Qian, X. Mao, S. Du, C. Yu, B. Peng and S. Q. Yao, Angew. Chem., Int. Ed., 2017, 56, 12481–12485 CrossRef CAS PubMed.
- S. Quignard, S. Masse, G. Laurent and T. Coradin, Chem. Commun., 2013, 49, 3410–3412 RSC.
- J. L. Vivero-Escoto, W. J. Rieter, H. Lau, R. C. Huxford-Phillips and W. Lin, Small, 2013, 9, 3523–3531 CrossRef CAS PubMed.
- J. G. Croissant, C. Mauriello-Jimenez, M. Maynadier, X. Cattoen, M. Wong Chi Man, L. Raehm, O. Mongin, M. Blanchard-Desce, M. Garcia, M. Gary-Bobo, P. Maillard and J. O. Durand, Chem. Commun., 2015, 51, 12324–12327 RSC.
- J. Croissant, X. Cattoen, M. W. Man, A. Gallud, L. Raehm, P. Trens, M. Maynadier and J. O. Durand, Adv. Mater., 2014, 26, 6174–6180 CrossRef CAS PubMed.
- E. A. Prasetyanto, A. Bertucci, D. Septiadi, R. Corradini, P. Castro-Hartmann and L. De Cola, Angew. Chem., Int. Ed., 2016, 55, 3323–3327 CrossRef CAS PubMed.
- Y. Lu, Y. Yang, Z. Gu, J. Zhang, H. Song, G. Xiang and C. Yu, Biomaterials, 2018, 175, 82–92 CrossRef CAS PubMed.
- L. Yu, Y. Chen, H. Lin, W. Du, H. Chen and J. Shi, Biomaterials, 2018, 161, 292–305 CrossRef CAS PubMed.
- D. Shao, F. Zhang, F. Chen, X. Zheng, H. Hu, C. Yang, Z. Tu, Z. Wang, Z. Chang and J. Lu, Adv. Mater., 2020, 32, 2004385 CrossRef CAS PubMed.
- W. Ratirotjanakul, T. Suteewong, D. Polpanich and P. Tangboriboonrat, Microporous Mesoporous Mater., 2019, 282, 243–251 CrossRef CAS.
- W. Zhang, H. Ma, J. Hua, W. Zhang, C. Guo and J. Wang, J. Solid State Chem., 2019, 277, 761–768 CrossRef CAS.
- Y. Fatieiev, J. G. Croissant, K. Julfakyan, L. Deng, D. H. Anjum, A. Gurinov and N. M. Khashab, Nanoscale, 2015, 7, 15046–15050 RSC.
- K. M. Rao, S. Parambadath, A. Kumar, C. S. Ha and S. S. Han, ACS Biomater. Sci. Eng., 2018, 4, 175–183 CrossRef CAS PubMed.
- S. Zhang, Z. Chu, C. Yin, C. Zhang, G. Lin and Q. Li, J. Am. Chem. Soc., 2013, 135, 5709–5716 CrossRef CAS PubMed.
- G. Yang, J. Ji and Z. Liu, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2021, 13, e1720 CAS.
- E. N. Arner and J. C. Rathmell, Cancer Cell, 2023, 41, 421–433 CrossRef CAS PubMed.
- F. Chen, X. Zhuang, L. Lin, P. Yu, Y. Wang, Y. Shi, G. Hu and Y. Sun, BMC Med., 2015, 13, 45 CrossRef PubMed.
- Y. Yang, H. Wu, B. Liu and Z. Liu, Adv. Drug Delivery Rev., 2021, 179, 114004 CrossRef CAS PubMed.
- Y. Zhang, I. Kim, Y. Lu, Y. Xu, D. G. Yu and W. Song, J. Controlled Release, 2022, 349, 963–982 CrossRef CAS PubMed.
- T. Su, S. Huang, Y. Zhang, Y. Guo, S. Zhang, J. Guan, M. Meng, L. Liu, C. Wang, D. Yu, H. Y. Kwan, Z. Huang, Q. Huang, E. Lai-Han Leung, M. Hu, Y. Wang, Z. Liu and L. Lu, Acta Pharm. Sin. B, 2022, 12, 821–837 CrossRef CAS PubMed.
- R. Zhang, T. Liu, W. Li, Z. Ma, P. Pei, W. Zhang, K. Yang and Y. Tao, J. Nanobiotechnol., 2022, 20, 223 CrossRef CAS PubMed.
- S. Chen, M. Chen, J. Yang, X. Zeng, Y. Zhou, S. Yang, R. Yang, Q. Yuan and J. Zheng, Small, 2021, 17, e2100243 CrossRef PubMed.
- H. Omar, J. G. Croissant, K. Alamoudi, S. Alsaiari, I. Alradwan, M. A. Majrashi, D. H. Anjum, P. Martins, R. Laamarti, J. Eppinger, B. Moosa, A. Almalik and N. M. Khashab, J. Controlled Release, 2017, 259, 187–194 CrossRef CAS PubMed.
- T. Liu, M. Zhu, X. Chang, X. Tang, P. Yuan, R. Tian, Z. Zhu, Y. Zhang and X. Chen, Adv. Mater., 2023, 35, e2300086 CrossRef PubMed.
- L. Dai, X. Li, X. Duan, M. Li, P. Niu, H. Xu, K. Cai and H. Yang, Adv. Sci., 2019, 6, 1801807 CrossRef PubMed.
- C. Li, Y. Wang, Y. Du, M. Qian, H. Jiang, J. Wang, N. Murthy and R. Huang, Biomaterials, 2018, 186, 1–7 CrossRef CAS PubMed.
- S.-Y. Peng, M.-Z. Zou, C.-X. Zhang, J.-B. Ma, X. Zeng and W. Xiao, J. Drug Delivery Sci. Technol., 2020, 55, 101450 CrossRef CAS.
- J. Wu, D. H. Bremner, S. Niu, H. Wu, J. Wu, H. Wang, H. Li and L.-M. Zhu, Chem. Eng. J., 2018, 342, 90–102 CrossRef CAS.
- R. Cheng, F. Feng, F. Meng, C. Deng, J. Feijen and Z. Zhong, J. Controlled Release, 2011, 152, 2–12 CrossRef CAS PubMed.
- M. Wu, Q. Meng, Y. Chen, Y. Du, L. Zhang, Y. Li, L. Zhang and J. Shi, Adv. Mater., 2015, 27, 215–222 CrossRef CAS PubMed.
- Z. Zheng, Z. Jia, C. Qu, R. Dai, Y. Qin, S. Rong, Y. Liu, Z. Cheng and R. Zhang, Small, 2021, 17, e2006508 CrossRef PubMed.
- M. Wei, J. Bai, X. Shen, K. Lou, Y. Gao, R. Lv, P. Wang, X. Liu and G. Zhang, ACS Nano, 2023, 17, 11345–11361 CrossRef CAS PubMed.
- W. Guo, Z. Chen, J. Chen, X. Feng, Y. Yang, H. Huang, Y. Liang, G. Shen, Y. Liang, C. Peng, Y. Li, G. Li, W. Huang, B. Zhao and Y. Hu, J. Nanobiotechnol., 2020, 18, 99 CrossRef CAS PubMed.
- H. Lu, S. Xu, G. Ge, Z. Guo, M. Zhao and Z. Liu, ACS Appl. Mater. Interfaces, 2022, 14, 44098–44110 CrossRef CAS PubMed.
- F. Zhang, F. Chen, C. Yang, L. Wang, H. Hu, X. Li, X. Zheng, Z. Wang, Z. Chang, T. Li, L. Li, M. Ge, J. Du, W. Sun, W. F. Dong and D. Shao, Small, 2021, 17, e2100006 CrossRef PubMed.
- L. Li, J. Zou, Y. Dai, W. Fan, G. Niu, Z. Yang and X. Chen, Nat. Biomed. Eng., 2020, 4, 1102–1116 CrossRef CAS PubMed.
- B. Liu, S. Liang, Z. Wang, Q. Sun, F. He, S. Gai, P. Yang, Z. Cheng and J. Lin, Adv. Mater., 2021, 33, e2101223 CrossRef PubMed.
- Z. Chen, F. Han, Y. Du, H. Shi and W. Zhou, Signal Transduction Targeted Ther., 2023, 8, 70 CrossRef PubMed.
- P. Lee, N. S. Chandel and M. C. Simon, Nat. Rev. Mol. Cell Biol., 2020, 21, 268–283 CrossRef CAS PubMed.
- K. Tang, X. Li, Y. Hu, X. Zhang, N. Lu, Q. Fang, J. Shao, S. Li, W. Xiu, Y. Song, D. Yang and J. Zhang, Biomater. Sci., 2023, 11, 4411–4429 RSC.
- J. Zhang, K. Tang, R. Fang, J. Liu, M. Liu, J. Ma, H. Wang, M. Ding, X. Wang, Y. Song and D. Yang, Front. Pharmacol., 2023, 14, 1140362 CrossRef CAS PubMed.
- M. Z. Zou, W. L. Liu, H. S. Chen, X. F. Bai, F. Gao, J. J. Ye, H. Cheng and X. Z. Zhang, Natl. Sci. Rev., 2021, 8, nwaa160 CrossRef CAS PubMed.
- J. N. Liu, W. Bu and J. Shi, Chem. Rev., 2017, 117, 6160–6224 CrossRef CAS PubMed.
- S. Chen, J. Liu, Y. Li, X. Wu, Q. Yuan, R. Yang and J. Zheng, TrAC, Trends Anal. Chem., 2020, 131, 116010 CrossRef CAS.
- H. B. Cheng, S. Zhang, J. Qi, X. J. Liang and J. Yoon, Adv. Mater., 2021, 33, e2007290 CrossRef PubMed.
- J. Zhu, T. Guo, Z. Wang and Y. Zhao, J. Controlled Release, 2022, 345, 475–493 CrossRef CAS PubMed.
- Q. Tian, Y. Li, S. Jiang, L. An, J. Lin, H. Wu, P. Huang and S. Yang, Small, 2019, 15, e1902926 CrossRef PubMed.
- G. Shim, Q. V. Le, J. Suh, S. Choi, G. Kim, H. G. Choi, Y. B. Kim, R. B. Macgregor, Jr. and Y. K. Oh, J. Controlled Release, 2019, 298, 110–119 CrossRef CAS PubMed.
- X. Jiang, X. Fan, W. Xu, C. Zhao, H. Wu, R. Zhang and G. Wu, J. Controlled Release, 2019, 316, 196–207 CrossRef CAS PubMed.
- J. Fu, L. Liu, Z. Huang, Y. Lai, H. Ji, S. Peng, J. Tian and Y. Zhang, J. Med. Chem., 2013, 56, 4641–4655 CrossRef CAS PubMed.
- W. H. Chen, G. F. Luo, Q. Lei, S. Hong, W. X. Qiu, L. H. Liu, S. X. Cheng and X. Z. Zhang, ACS Nano, 2017, 11, 1419–1431 CrossRef CAS PubMed.
- X. Dong, W. Yin, X. Zhang, S. Zhu, X. He, J. Yu, J. Xie, Z. Guo, L. Yan, X. Liu, Q. Wang, Z. Gu and Y. Zhao, ACS Appl. Mater. Interfaces, 2018, 10, 4271–4284 CrossRef CAS PubMed.
- H. Gong, Y. Chao, J. Xiang, X. Han, G. Song, L. Feng, J. Liu, G. Yang, Q. Chen and Z. Liu, Nano Lett., 2016, 16, 2512–2521 CrossRef CAS PubMed.
- J. G. Croissant, Y. Fatieiev, K. Julfakyan, J. Lu, A.-H. Emwas, D. H. Anjum, H. Omar, F. Tamanoi, J. I. Zink and N. M. Khashab, Chem. – Eur. J., 2016, 22, 14806–14811 CrossRef CAS PubMed.
- N. Qiu, X. Liu, Y. Zhong, Z. Zhou, Y. Piao, L. Miao, Q. Zhang, J. Tang, L. Huang and Y. Shen, Adv. Mater., 2016, 28, 10613–10622 CrossRef CAS PubMed.
- P. Shen, X. Zhang, N. Ding, Y. Zhou, C. Wu, C. Xing, L. Zeng, L. Du, J. Yuan and Y. Kang, ACS Appl. Mater. Interfaces, 2023, 15, 20697–20711 CrossRef CAS PubMed.
- R. Zhang, S. Shao, Y. Piao, J. Xiang, X. Wei, Z. Zhang, Z. Zhou, J. Tang, N. Qiu, X. Xu, Y. Liu and Y. Shen, Adv. Mater., 2023, e2303614, DOI:10.1002/adma.202303614.
- N. H. Tsao and E. A. Hall, Langmuir, 2016, 32, 6534–6543 CrossRef CAS PubMed.
- X. Li, Y. Chen, X. Zhang and Y. Zhao, Mater. Sci. Eng., C, 2020, 112, 110914 CrossRef CAS PubMed.
- Y. Zhang, L. Liu, W. Li, C. Zhang, T. Song, P. Wang, D. Sun, X. Huang, X. Qin, L. Ran, G. Tian, J. Qian and G. Zhang, J. Nanobiotechnol., 2023, 21, 9 CrossRef CAS PubMed.
- J. L. Li, Y. J. Cheng, C. Zhang, H. Cheng, J. Feng, R. X. Zhuo, X. Zeng and X. Z. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 5287–5295 CrossRef CAS PubMed.
- X. Li, X. Zhang, Y. Zhao and L. Sun, J. Inorg. Biochem., 2020, 202, 110887 CrossRef CAS PubMed.
- Y. Yang, Y. Lu, P. L. Abbaraju, I. Azimi, C. Lei, J. Tang, M. Jambhrunkar, J. Fu, M. Zhang, Y. Liu, C. Liu and C. Yu, Adv. Funct. Mater., 2018, 28, 1800706 CrossRef.
- X. Jia, Y. Zhang, Y. Zou, Y. Wang, D. Niu, Q. He, Z. Huang, W. Zhu, H. Tian, J. Shi and Y. Li, Adv. Mater., 2018, 30, e1704490 CrossRef PubMed.
- A. Sharma, J. F. Arambula, S. Koo, R. Kumar, H. Singh, J. L. Sessler and J. S. Kim, Chem. Soc. Rev., 2019, 48, 771–813 RSC.
- Y. Li, L. Zhao and X. F. Li, Front. Oncol., 2021, 11, 700407 CrossRef CAS PubMed.
- D. Cheng, Y. Ji, B. Wang, T. Jin, Y. Xu, X. Qian and W. Zhu, Int. J. Pharm., 2021, 603, 120730 CrossRef CAS PubMed.
- T. Yin, J. Liu, Z. Zhao, Y. Zhao, L. Dong, M. Yang, J. Zhou and M. Huo, Adv. Funct. Mater., 2017, 27, 1604620 CrossRef.
- Y. Zhong, K. Goltsche, L. Cheng, F. Xie, F. Meng, C. Deng, Z. Zhong and R. Haag, Biomaterials, 2016, 84, 250–261 CrossRef CAS PubMed.
- H. Birkedal-Hansen, W. G. Moore, M. K. Bodden, L. J. Windsor, B. Birkedal-Hansen, A. DeCarlo and J. A. Engler, Crit. Rev. Oral Biol. Med., 1993, 4, 197–250 CrossRef CAS PubMed.
- M. Egeblad and Z. Werb, Nat. Rev. Cancer, 2002, 2, 161–174 CrossRef CAS PubMed.
- B. Yang, H. Fu, R. Kong, G. Zheng, X. Wang, Y. Dong and Z. Yang, J. Mater. Chem. B, 2022, 10, 8490–8501 RSC.
- S. J. Park, W. Ye, R. Xiao, C. Silvin, M. Padget, J. W. Hodge, C. Van Waes and N. C. Schmitt, Oral Oncol., 2019, 95, 127–135 CrossRef CAS PubMed.
- Y. Shi, M. Zhou, Y. Zhang, Y. Wang and J. Cheng, Acta Biomater., 2023, 158, 571–582 CrossRef CAS PubMed.
- Y. Fang, X. Luo, Y. Xu, Z. Liu, R. L. Mintz, H. Yu, X. Yu, K. Li, E. Ju, H. Wang, Z. Tang, Y. Tao and M. Li, Adv. Sci., 2023, 10, e2300899 CrossRef PubMed.
- D. Cao, L. Chen, Z. Zhang, Y. Luo, L. Zhao, C. Yuan, J. Lu, X. Liu and J. Li, J. Mater. Chem. B, 2023, 11, 1829–1848 RSC.
- K. Kessenbrock, V. Plaks and Z. Werb, Cell, 2010, 141, 52–67 CrossRef CAS PubMed.
- L. Claesson-Welsh, J. Clin. Invest., 2020, 130, 1093–1095 CrossRef CAS PubMed.
- A. R. Nelson, B. Fingleton, M. L. Rothenberg and L. M. Matrisian, J. Clin. Oncol., 2000, 18, 1135–1149 CrossRef CAS PubMed.
- Y. C. Chan and M. Hsiao, Nanomedicine, 2017, 12, 2153–2159 CrossRef CAS PubMed.
- J. Li, Z. Ge, K. Toh, X. Liu, A. Dirisala, W. Ke, P. Wen, H. Zhou, Z. Wang, S. Xiao, J. F. R. Van Guyse, T. A. Tockary, J. Xie, D. Gonzalez-Carter, H. Kinoh, S. Uchida, Y. Anraku and K. Kataoka, Adv. Mater., 2021, 33, e2105254 CrossRef PubMed.
- R. Xiao, J. Ye, X. Li and X. Wang, Int. J. Biol. Macromol., 2023, 238, 124032 CrossRef CAS PubMed.
- Y. Hu, S. Bai, X. Fan, F. Zhou, B. Chen, S. Tan, H. Xu, A. Pan, S. Liang and Y. He, Biomater. Sci., 2022, 10, 2358–2369 RSC.
- S. Yan, P. Sun, N. Niu, Z. Zhang, W. Xu, S. Zhao, L. Wang, D. Wang and B. Z. Tang, ACS Nano, 2022, 16, 9785–9798 CrossRef CAS PubMed.
- M. Wang, W. Zhang, B. Ding, P. A. Ma and J. Lin, Adv. Opt. Mater., 2023, 11, 2202040 CrossRef CAS.
Footnote |
| † These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |