
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing paracellular and transcellular permeability using nanotechnological approaches for the treatment of brain and retinal diseases
Asmaa
Khalil
a,
Alexandre
Barras
a,
Rabah
Boukherroub
 a,
Ching-Li
Tseng
bc,
David
Devos
d,
Thierry
Burnouf
bc,
Winfried
Neuhaus
ef and
Sabine
Szunerits
*a
a,
Ching-Li
Tseng
bc,
David
Devos
d,
Thierry
Burnouf
bc,
Winfried
Neuhaus
ef and
Sabine
Szunerits
*a
aUniv. Lille, CNRS, Univ. Polytechnique Hauts-de-France, UMR 8520 – IEMN, F-59000 Lille, France. E-mail: sabine.szunerits@univ-lille.fr
bTaipei Medical University, Graduate Institute of Biomedical Materials and Tissue Engineering (GIBMTE), New Taipei City 235603, Taiwan
cTaipei Medical University, International PhD Program in Biomedical Engineering (IPBME), New Taipei City 235603, Taiwan
dUniversity Lille, CHU-Lille, Inserm, U1172, Lille Neuroscience & Cognition, LICEND, Lille, France
eAIT – Austrian Institute of Technology GmbH, Center Health and Bioresources, Competence Unit Molecular Diagnostics, 1210 Vienna, Austria
fLaboratory for Life Sciences and Technology (LiST), Faculty of Medicine and Dentistry, Danube Private University, 3500 Krems, Austria
First published on 13th September 2023
Abstract
Paracellular permeability across epithelial and endothelial cells is, in large part, regulated by apical intercellular junctions also referred to as tight junctions (TJs). These junctions contribute to the spatial definition of different tissue compartments within organisms, separating them from the outside world as well as from inner compartments, with their primary physiological role of maintaining tissue homeostasis. TJs restrict the free, passive diffusion of ions and hydrophilic small molecules through paracellular clefts and are important for appropriate cell polarization and transporter protein localisation, supporting the controlled transcellular diffusion of smaller and larger hydrophilic as well as hydrophobic substances. This traditional diffusion barrier concept of TJs has been challenged lately, owing to a better understanding of the components that are associated with TJs. It is now well-established that mutations in TJ proteins are associated with a range of human diseases and that a change in the membrane fluidity of neighbouring cells can open possibilities for therapeutics to cross intercellular junctions. Nanotechnological approaches, exploiting ultrasound or hyperosmotic agents and permeation enhancers, are the paradigm for achieving enhanced paracellular diffusion. The other widely used transport route of drugs is via transcellular transport, allowing the passage of a variety of pro-drugs and nanoparticle-encapsulated drugs via different mechanisms based on receptors and others. For a long time, there was an expectation that lipidic nanocarriers and polymeric nanostructures could revolutionize the field for the delivery of RNA and protein-based therapeutics across different biological barriers equipped with TJs (e.g., blood–brain barrier (BBB), retina–blood barrier (RBB), corneal TJs, etc.). However, only a limited increase in therapeutic efficiency has been reported for most systems until now. The purpose of this review is to explore the reasons behind the current failures and to examine the emergence of synthetic and cell-derived nanomaterials and nanotechnological approaches as potential game-changers in enhancing drug delivery to target locations both at and across TJs using innovative concepts. Specifically, we will focus on recent advancements in various nanotechnological strategies enabling the bypassing or temporally opening of TJs to the brain and to the retina, and discuss their advantages and limitations.
1. Challenges in delivering therapeutics across tight junctions and biological barriers
While small molecule drugs (<900 Daltons) currently still dominate the pharmaceutical drug market, advancements in recombinant DNA technology and solid-phase synthesis of peptides and proteins since the 1980s have enabled large-scale production of therapeutic peptides and proteins. A prime example of this success is the availability of recombinant human insulin as a protein-based therapy for diabetes mellitus.1,2 Another would be the use of monoclonal antibodies (mAbs) and engineered antibodies for the treatment of various auto-immune diseases (inflammatory, neurological, etc.).3,4 Peptides and proteins are believed to be more specific to their target sites than small drugs and show in general reduced interference with the patient's normal physiology. As a result, the search for efficient administration strategies for proteins and peptides5,6 alongside small therapeutic molecules has emerged as a crucial research target in recent years.Administration of macromolecular therapeutics commonly relies on intravenous (I.V), intraperitoneal (I.P.), and intramuscular (I.M.) injections. While oral administration remains the preferred route for patients, as it avoids the pain and discomfort associated with injections, the development of orally available protein and peptide formulations has been proven to be extremely challenging. This challenge arises from factors including the limited water solubility of macromolecular drugs, susceptibility to physiological enzymatic degradation and to the acidic stomach environment, and short biological half-life. More specifically, when targeting the brain or retina through oral delivery of peptide/protein drugs, additional barriers, such as the intestinal epithelial barrier (IEB), with its protective mucus layer, followed by entry into the peripheral bloodstream, and subsequent crossing of the blood–brain barrier (BBB) for brain targeting or the blood–retinal barrier (BRB) for reaching the retina must be overcome (Fig. 1).
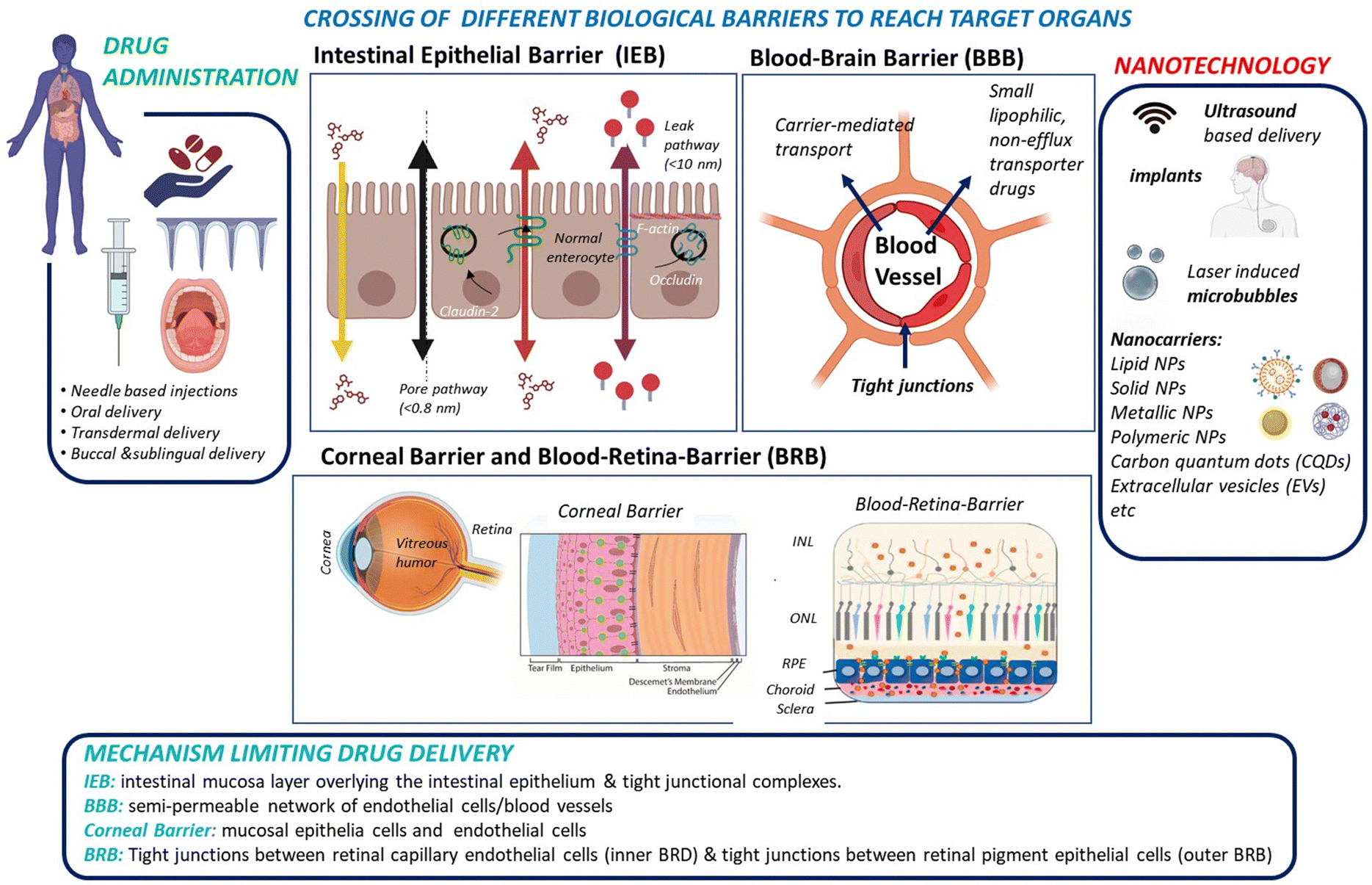 | ||
| Fig. 1 From drug administration to delivery of therapeutics to the intestine, brain and eye via crossing of biological tight junctions and biological barriers: drug administration can take various forms, including needle-based administration as well as non-invasive approaches, such as oral, transdermal, and buccal routes. For some modes of administration, notably oral, the intestinal epithelia barrier might be an important barrier for drug crossing. Other approaches, such as needles, transdermal, and buccal delivery systems directly introduce the drug into the blood stream. Drug delivery to the brain requires crossing of the blood–brain barrier (BBB), or the delivery via the nose-to-brain route to by-pass the BBB hurdles. Drug delivery to the posterior segment of the eye, specifically the retina, implies crossing the blood–retina barrier (BRB). The use of eye droplets applied to the cornea is an attractive alternative, but it necessitates overcoming the corneal barriers as well as the vitreous humour to reach the different layers of the retina: inner nuclear layer (INL), outer nuclear layer (ONL), and retinal pigment epithelium (RPE). Various synthetic and natural nanotechnological approaches, using nanoparticle formulations either independently or in combination with implants and physical techniques enhancement, have been explored to enhance crossing of biological barriers. | ||
To date, progress in developing orally administered pharmaceuticals and parentally administered peptide and protein drugs capable of reaching the brain and the retina has been slow. This limitation is largely attributed to the poor stability within the gastrointestinal tract for oral administration, and it is also generally due to the low permeability across barrier membranes such as the IEB, BBB, BRB and corneal epithelium for topical drug application. However, these shortcomings have been partially addressed through the development of novel formulation technologies based on nanocarriers and other nanotechnological approaches, which allow to enhance drug uptake by these barriers (Fig. 1).
The aim of this review is to assess the promising nanoparticle-based drug delivery systems (DDS) proposed in the last 5 years to administer drugs to the brain, retina or cornea. Remarkably, the eye can be regarded as an anatomical extension of the brain, sharing similarities in terms of neurons, vasculature and immune response. Therefore, multidisciplinary research investigating both organs can provide mutual insights, especially valuable in the understanding and treatment of neurodegenerative diseases. A comprehensive understanding of the physical barriers that must be overcome and the various strategies evaluated to penetrate these barriers is crucial for nanotechnological approaches. Before discussing in depth the potential and limitations of different nanocarriers and nanotechnological approaches for delivering drugs across the BBB and BRB, this review will first provide a thorough comparison of cellular barrier systems.
2. Paracellular and transcellular permeability concepts
In order to protect the organism and organs from toxins and pathogens, the body has evolved intricate cellular and molecular mechanisms for each of its barrier. In all cases, a physical barrier made of a cellular layer tightly regulates the movement of ions, molecules, and cells between two tissue compartments. For instance, in the case of the intestinal epithelial barrier (IEB) (Fig. 1),7 intestinal epithelial cells separate the gut lumen from the internal space. For the BBB, brain capillary endothelial cells separate the lumen of blood vessels from the CNS parenchyma.8 An important distinction between the IEB and the BBB lies in the microenvironment to which these barriers are exposed. Unlike the BBB, which is only occasionally exposed to microorganisms, the IEB is in constant contact with the microbiota, which plays a beneficial role in digestion and protection. As a result, one notable difference between the two barriers is the presence of a mucous layer in the IEB, which physically separates the microbiota from the epithelial barrier. The intestinal epithelial cells, coated with a mucosa film, are themselves interconnected by tight junctions (TJs). These TJs are instrumental in restricting the passage of molecules larger than 22–30 Å (or 2–2.3 nm) in diameter, with drugs over 100–200 Daltons (Da) typically being excluded from paracellular transport via TJs.9 TJs are composed of several proteins, whose prominence and expression levels depend on the specific tissue or cell type. For instance, occludin and claudin TJ proteins directly control the paracellular permeability of ions in the brain (Fig. 2). Another notable TJ associated protein is zona occludens-1 (ZO-1) in the RPE layer, which ensures the selective transport of substances between the retina and the choroidal blood supply10(Fig. 2). While the mucus mesh space (20–200 nm) connected to the IEB is large enough to allow for the diffusion of smaller biological entities, such as human papillomavirus (HPV; 55 nm), and small globular proteins, it effectively traps larger macromolecules, preventing their access to the underlying epithelial cells. To overcome this barrier, low molecular weight polyethylene glycol (PEG) mucoadhesive polymer nanoparticles with a net neutral charge and hydrophilic nature have been developed, enabling them to evade entrapment in the mucus layer.11 However, even after passing the mucus layer, the TJs of the IEB still block the passive paracellular diffusion of molecules to the bloodstream.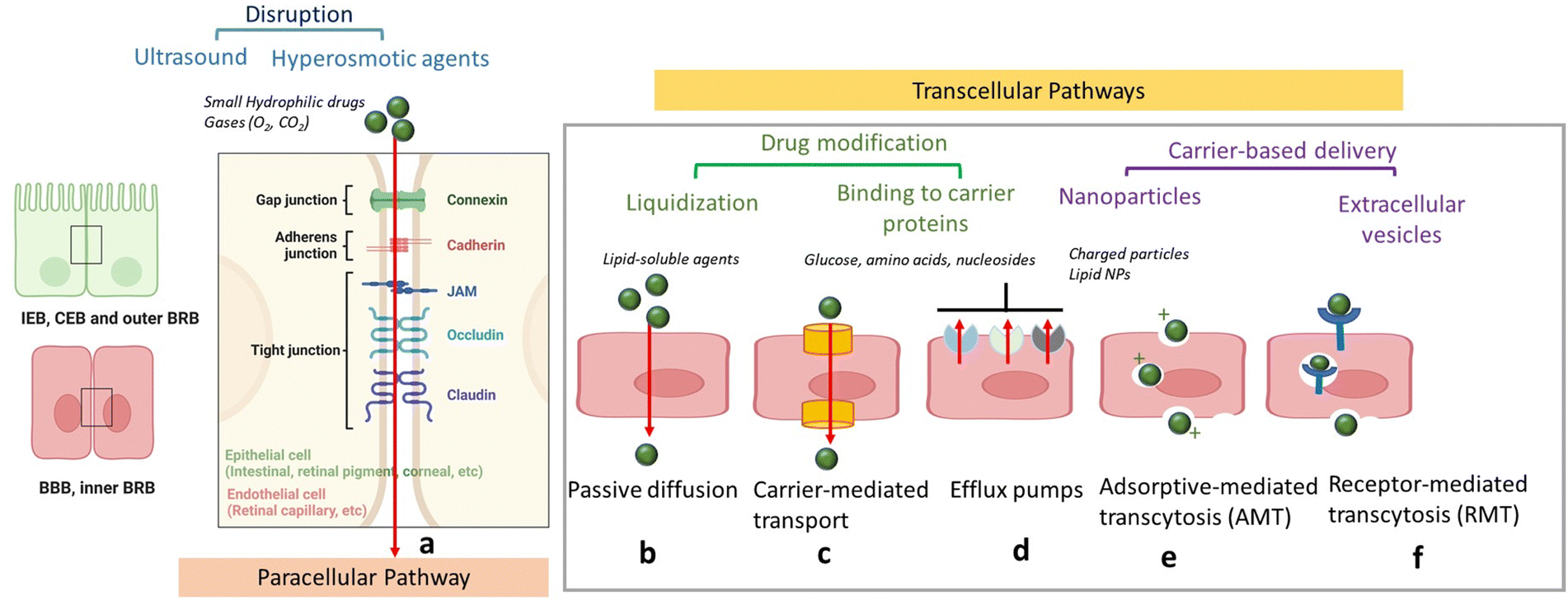 | ||
| Fig. 2 Schematics of drug delivery via paracellular and transcellular transport routes: (a) paracellular pathway with tight junctions preventing passage of hydrophilic compounds (inset: zoom on TJs proteins) could be opened through chemicals, such as mannitol, ultrasound application or treatment with hyperosmotic agents. (b) Passive diffusion of lipid-soluble agents. (c) Transport of small molecules, such as glucose and amino acids via carrier proteins. (d) Efflux via efflux pumps such as ATP-binding cassette (ABC) – transporters ABCB1 (P-glycoprotein), ABCCs (MRPs) or ABCG2 (BCRP). (e). Adsorption-mediated transcytosis (AMT) of protein species, such as albumin as well as cationic nanoparticles and lipid-based particles. (f) Receptor-mediated transcytosis (RMT) notably for particles with targets, such as transferrin, insulin, etc. | ||
Blood–brain barrier
Similar to other cell layers of biological barriers, the intercellular gaps between brain capillary endothelial cells are closed by tight junctions. The protein composition of TJs depends on the tissue, for example, the main claudin of the BBB is claudin-5, whereas in classical epithelial layers, claudin-3 and -4 are very often found. The paradigm of the BBB was fully established in 1967, when the accumulation of horseradish peroxidase exclusively in the lumens of brain capillaries was observed, indicating that endothelial TJs confer unique barrier properties to the BBB.12 It was postulated that the smaller gaps between endothelial cells impede paracellular transport at the BBB, compared to other endothelial barriers. The semipermeable character of the BBB allows for the unrestricted passage of O2, CO2, and water from the blood into the brain. However, passive paracellular diffusion of larger drugs is limited, with even less than 2% of small drug molecules bypassing the BBB.13 This defence system is crucial, serving to protect the brain against harmful neurotoxic compounds, bacteria, viruses and other parasites that could infiltrate the brain and generate neuroinflammation. Most molecules, apart from lipophilic molecules and nutrients, cannot freely cross the BBB, which is one of the most selective endothelial barriers.14 Delivery of therapeutics across the BBB is thus challenging due to limited paracellular transport, which necessitates the exploration of other regulated mechanisms to transport drugs across the BBB. Furthermore, although transcytosis mechanisms utilizing receptors, e.g., transferrin or insulin have been identified, the transcytosis across the BBB is in general very low in comparison to endothelia of peripheral tissues.One way to enhance paracellular drug transport of drugs involves the use of active excipients that modulate TJs such as permeation enhancers (PEs), including methanol, bilobalide or latrunculin. These compounds act through unspecific interactions to target TJs and promote paracellular drug transport.15 Other examples are toxins or their engineered derivatives derived from, e.g., Clostridium perfringens, which bind to claudins to open the paracellular clefts and enable increased permeation of small compounds.16 Although these compounds are interesting from a biological point of view for TJ modulation, to date merely a few of these novel active excipients have advanced to clinical trials, primarily due to lack of documentation on safety and effectiveness in vivo, which impedes their clinical relevance.
The lipophilic surface area of the endothelial layer in the BBB should offer, in principle, an ideal pathway for passive transport of small lipophilic molecules (MW < 400 Da) into the BBB. The octanol/buffer partition coefficient is an established method used to predict the likelihood of small molecules crossing the BBB through passive diffusion.17 Nevertheless, many lipophilic compounds, including vincristine, and cyclosporine A, show much lower brain penetration than expected based on this calculation. This discrepancy can be partially attributed to the presence of transmembrane efflux pump mechanisms.18 These transporters are instrumental in protecting neural cells against naturally occurring damaging toxins by restricting their entry into the brain and facilitating their removal. However, they can also result in the efflux of therapeutic compounds from the CNS. Efflux is mediated by proteins referred to as multi-drug resistance (MDR) proteins, most of which belong to the ATP-binding cassette (ABC) family19 of transporters. These integral membrane proteins possess multiple domains and use the energy generated by ATP hydrolysis to transport solutes across cellular membranes. One well-studied efflux transporter, P-glycoprotein 1 (P-gp), is encoded by the human MDR-1 (ABCB1) gene and expressed at the BBB, and is one of most extensively described hurdles for improved drug delivery to the brain. In addition, other ABC-transporters, such as ABCG2 (BCRP) or ABCCs (MRPs), have overlapping substrate specificities and contribute to the transport barrier function of the BBB.20
The transcellular transport mechanism (Fig. 2) is the most widely used approach for facilitating drug transport across biological membranes. This mechanism plays a crucial role in shuttling larger hydrophilic drugs, peptides, proteins, and nanoparticles across the BBB; such transcellular transport is facilitated by a process known as receptor mediated transcytosis (RMT) where an “active” transport via transcytosis is mediated by insulin and transferrin receptors, located on the apical blood-facing BBB side (Fig. 2). RMT receptors allow for highly selective binding and internalization of macromolecules within vesicles, enabling their transport across the BBB and into the brain. In contrast to RMT, another transcellular transport route is adsorptive-mediated transcytosis (AMT), which involves positively charged molecules, such as immunoglobulins, that associate with the negatively charged endothelial surface, are internalized via endocytosis, and follow a similar transport mechanism as RMT. However, it has to be emphasized that the transcytosis rate at the BBB is proposed to be significantly lower than that across endothelia at the periphery (Table 1).
| Different barrier | Commonalities | Diver points | Characteristics for penetration of nano-agent |
|---|---|---|---|
| Intestinal epithelial barrier (IEB) | Consists of intestinal epithelial cells, separating the gut lumen from the internal space | Paracellular barrier properties are conserved throughout the whole intestine | Mucosal penetrating nano-agents |
| Presence of a mucous layer forming a first layer of protection between the gut and the external world by physically separating the microbiota from the epithelial barrier | Transcellular barrier properties differ according to the section of the intestine that is considered | ||
| Blood–brain barrier (BBB) | Present at the capillary endothelium of cerebral blood vessels possessing strong TJs | Larger and lipophilic drugs can pass via transcellular pathways, hydrophilic compounds might enter by means of carrier proteins. | PS80 addition to nanoparticles is the gold standard for increasing the BBB making PLGA-PEG polysorbate 80 particles most promising |
| Semi-permeable character: Gases such as O2 and CO2 or EtOH pass from blood into the brain via passive diffusion | Efflux pumps such as P-glycoprotein and BCRP are key elements of the molecular machinery that confers special permeability properties to the BBB | Chitosan based structures able to be retained in the mucus layer, and then undergo transcellular passage are the ideal structures currently | |
| TJs can be altered upon inflammation, neurological and neurodegenerative diseases | |||
| Blood–cerebrospinal fluid barrier (BCSFB) | Created by a layer of a modified cuboidal epithelium, that secretes cerebrospinal fluid | Next to the BBB, the largest interface between blood and brain extracellular fluids | as structures similar to BBB the same considerations for nanoparticles apply |
| Large similarities with the BBB in regard to expression of solute carrier (SLC) and ATP-binding case (ABC) transporter families | |||
| Blood–leptomeningeal barrier | The outer blood–cerebrospinal fluid barrier is formed by leptomeningeal cells of the arachnoidea | Altered leptomeningeal blood barrier may be accompanied by intraparenchymal blood–brain barrier disruption | As structures similar to BBB the same considerations for nanoparticles apply |
| Structures underlying this barrier are tight junctions | |||
| Blood–retina barrier (BRB) | Divided into an inner and outer barrier composed of retinal endothelial cells, retinal pigment | Lipophilic molecules can pass through retinal capillaries | Targeting of retinal photoreceptors with PEG-liposomes claudin-5 targeting structures |
| Epithelial cells | Break down results in macular oedema, diabetic retinopathy retinal pigment and epithelial cells | Blood-circulating EVs are believed to cross the BRB. Enriched with anti-VEGF and steroid they might be ideal for therapy | |
| Corneal barriers | Complex structure: corneal epithelium, Bowman's layer, corneal stroma, descemet's membrane and corneal endothelium | Corneal endothelium allows the diffusion of drugs with molecular dimensions up to about 20 nm | Nanostructures of 1–2 nm sch as carbon quantum dots and gold nanoparticles |
| Presence of intercellular TJs acting as a selective barrier for small molecules, completely hindering the diffusion of macromolecules via the paracellular route | Tear films and lacrimal system covering corneal epithelium results in drug outflow into the blood circulation to large extent | Chitosan-based structures due to mucoadhesive nature and positive charge | |
| Corneal stroma is a highly hydrophilic tissue allowing the diffusion of hydrophilic drugs up to 500 kDa |
Blood–retina barrier
The barrier in the posterior segment (retina/choroid) of the eye, the blood–retina barrier (BRB) (Fig. 1) is located in the posterior part of the eye. Its barrier properties were recognized in 194721 using intravenous trypan blue administration to rabbits, which stained all organs except the CNS and the retinal tissue. Both the BBB and BRB tightly control the neuronal environment, regulating the flux of blood borne substances into the neural parenchyma. These barriers play a crucial role in maintaining neural homeostasis and safeguarding neural tissue from potential blood-borne toxicity.Unlike the BBB, the BRB is composed of two distinct barriers; the outer BRB (oBRB) and the inner BRB (iBRB). The oBRB, an intercellular junction complex, is created by TJs of retinal pigment epithelial cells (RPE) separating the neurosensory retina from the choroid. The RPE regulates transport between the choriocapillaris and the retina, and the inner BRB (iBRB) regulates transport across retinal capillary endothelial cells. The adenosine tetraphosphate (ATPP) may enhance the transport and permeation of NPs across the retina via mediating the P2Y receptor on the apical plasma membrane of the RPE.22 Being a tight ion-transporting barrier, the oBRB restricts paracellular transport of polar solutes from the choroidal side. The integrity of the oBRD is crucial for the health and integrity of the inner retina. Breakdown of the BRB may lead to macular edema and various ocular disorders. The endothelial cells of retinal vessels, similar to the BBB, possess TJs, adherens junctions, and gap junctions. Alterations in the oBRB have been associated with neovascular age-related macular degradation (AMD). Similar to the BBB, permeability depends on drug characteristics, as lipophilic molecules generally pass through retinal capillaries and RPE. Due to the limited blood flow to the posterior segment of the eye, high drug doses are often needed, increasing the risk of adverse effects and hampering the use of drugs with narrow therapeutic ranges.
The method for delivery of medications plays a crucial role in influencing the therapeutic effect for posterior eye drug delivery. The main method uses intravitreal injection (IVT), where drugs are injected into the vitreous humour.23 Upon injection, active compounds diffuse through the vitreous humour, reach the inner limiting membrane (ILM) of the stratified retina (Fig. 1), and ultimately traverse the multilayers of the retina. As a result of IVT, the vitreous humour, a highly hydrated matrix of >98% water, with 15–20 wt% of total water bound to proteins (collagen II, IX V/XI, IV) and glycosaminoglycans (GAGs), is the first barrier that drugs must overcome to reach the retina. The major GAGs found in the vitreous humour is hyaluronic acid (HA). The negatively charged HA and anionic collagens form a network gel that can cause the aggregation or precipitation of large or charged molecules within the vitreous humour, making it especially challenging for the drugs to be transported to the retina.23–25 Furthermore, positively charged molecules tend to clump together in the vitreous humour, hindering their diffusion.
Corneal barriers
The corneal epithelial barrier (Fig. 1) acts as a natural protection against microorganisms and confers mechanical stability to the eye; it is a highly complex structure made of different layers.26 The cornea, a transparent and avascular tissue with an average thickness of 500 μm in humans consists of several layers: (i) the corneal epithelium consisting of flattened superficial cells, wing cells, and columnar basal cells, (ii) Bowman's layer, (iii) the corneal stroma, (iv) the Descemet's membrane and (v) the corneal endothelium. Drug delivery is restricted in the cornea due to the presence of intercellular TJs between epithelial and endothelial cells, completely hindering the diffusion of macromolecules via the paracellular route. However, penetration enhancers, such as cyclodextrins (CD), cyclic oligosaccharide with a lipophilic central cavity where the drug is loaded and a hydrophilic outer surface, have been shown to increase ocular permeability, as demonstrated using riboflavin loaded hydroxypropyl-β-cyclodextrin.27 The negative charge of the cornea makes positively charged (cationic) polymers like chitosan, gelatine, poly-L-arginine ideal as penetration enhancers, exhibiting increased corneal retention.The corneal stroma, consisting of closely arranged collagen fibres, is a highly hydrophilic tissue with an open structure, allowing the diffusion of hydrophilic drugs up to 500 kDa. However, it is a rate-limiting barrier for most lipophilic drugs. The innermost layer of the cornea is the corneal endothelium, made of a monolayer of hexagonal endothelial cells, which adjusts water influx into the cornea and acts as a barrier between the cornea and the aqueous humour. The corneal endothelium maintains physiological corneal hydration and allows the diffusion of drugs up to about 20 nm. Drug delivery in the eye is further challenged by the presence of a tear film, which includes lipid/aqueous/mucin layers, covering the cornea and the conjunctiva. The conjunctiva, a mucous membrane, consists of outer epithelial and goblet cell layers, with blood and lymphatic capillaries present. Drugs can take this route to outflow into the blood circulation or the lacrimal system, making, as a result, drug permeation of eye drops through the conjunctival route generally unreliable.
The above description underscores the heterogeneity in the function and composition of epithelial and endothelial barriers, which distinctly impacts drug permeability and the permeation of therapeutic nanostructures. The following sections will explore the potential of nanoparticles and nanotechnological approaches, as well as their advantages and limitations for delivering therapeutics via paracellular and transcellular pathways.
3. Nanoparticles
A key focus has been placed on using carrier-mediated drug delivery systems in the form of nanoparticles (NPs) or extracellular vesicles (EVs) to address the challenges associated with drug transport and targeting, especially to the brain.28 NPs (Fig. 3) have attracted considerable attention in the biomedical field as their interaction with cells and/or tissues can be customized through nanoparticle design involving parameters such as size, charge, shape and surface chemistry.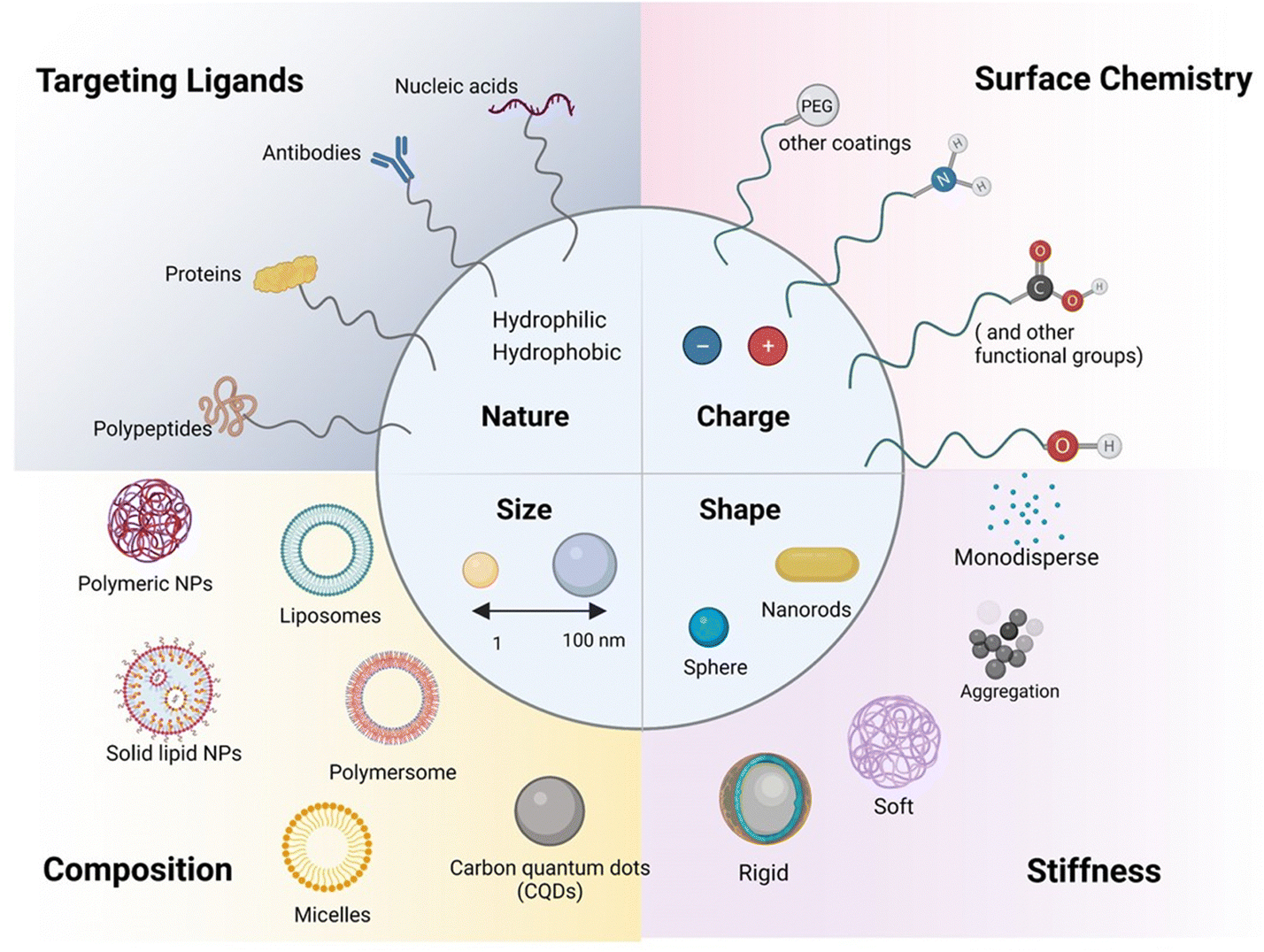 | ||
| Fig. 3 Schematic illustration of the range of available nanoparticles for paracellular and transcellular pathways: considerations such as size, shape, surface charge (defined by the zeta potential, ζ (in mV) of the nanostructures), porosity, and roughness are important factors guiding the para and transcellular penetration capability. The composition, notably the presence of targeting elements (antibodies, proteins, nucleic acids such as aptamers, peptides), as well as the coating (PEG and protein corona protein), are in addition important physio-chemical parameters to be fine-tuned. | ||
Importance of nanoparticle size
The small and tuneable size of NPs plays a major role in their drug delivery efficiency, as it may improve blood circulation time and contact time with the BBB and BRB, thanks to reduced clearance by the reticuloendothelial system (RES). Indeed, NPs of less than 200 nm have slower clearance and accumulation in organs, such as the liver and spleen, where macrophages are found and can actively uptake particles through phagocytosis, eliminating them from the blood stream. NP size is also influential when considering the size of TJs in the BBB. With TJs typically measuring 200–500 nm in length and 11–15 nm in width, with the intermembrane space at these junctions being only 10–20 nm,29 passive diffusion through TJs is limited to nanostructures smaller than 10 nm. This might explain in part the failure seen in clinical trials with the delivery of an anti-amyloid-beta antibody, such as gantenerumab, which has a molecular mass of 146 kDa corresponding to ca. 12–15 nm.30 However, the delivery of gantenerumab across the BBB was made possible for the phase I clinical trial by fusing it to a single chain Fab antibody against the human transferrin receptor, utilizing receptor-mediated transport.31 This achievement underlines the potential to overcome size dependent issues by favoring a transcellular targeted approach over a passive TJ pathway. In addition, the BBB impermeability is altered in neurodegenerative diseases such as Alzheimer.Importance of surface charge
Next to size, the second important aspect to improve nanoparticle-based drug delivery lies in the surface chemistry and surface coating employed. The basic composition of NPs comprises the surface layer, the shell layer, and the core, which is fundamentally the central portion of the NP and is usually termed as the NP itself. Surface functionalization has, however, been shown to be important to improve nanoparticle blood circulation time and tissue transport ability.32 Delivery across cellular barriers is facilitated by coupling targeting receptors to the nanostructure, allowing specific binding to cell-surface markers involved in transport, such as transferrin, lactoferrin, insulin, taking the examples for various receptors for RMT across the BBB. The encapsulation of resveratrol in solid–liquid nanoparticles and modification with an antibody specific for the transferrin receptor target at the BBB33 resulted in improved drug permeation. With respect to TJs, coating nanoparticles with polysorbates has been studied for opening TJs in the BBB to increase access to the brain tissue.34 Coating with hydrophilic polymers allows bypassing RES clearance. This highlights that the surface chemistry and thus the surface layer of NPs most strongly impacts NPs' bioavailability and delivery capacity. Next to the chemical nature, charge is the third essential parameter. Positively charged particles were found to be better delivered to the brain than negatively charged ones, owing to the cationic surface interactions with the anionic terminal groups of phospholipids on the cell wall through electrostatic interactions. It is the cationic nature of chitosan that mediates electrostatic interactions with the negatively charged nasal endothelium and sialic acid-contained mucins within the mucous, resulting in longer residence time of these particles. Cornea and conjunctiva have a negative charge; thus, cationic polymers are also used as penetration enhancers, such as chitosan, gelatin, poly-L-arginine, to increase ocular and transepithelial drug absorption. The negatively charged hyaluronic acid and anionic collagen network gel, present in the vitreous humour, make drug transport of positively charged nanoparticles however challenging as they tend to get clumped in the vitreous humour, without diffusing. Therefore, negative nanoparticles delivered by IVT are preferred for effectively delivering drugs to the retina via diffusion through the vitreous humour to the retina.35Importance of nanoparticles' shape
Along with size and charge, the particle shape and composition strongly influence cellular uptake. The vast majority of nanoparticles developed for drug delivery have a spherical shape, but other forms such as cube-shaped, cylindrical, ellipsoids, and disks have recently been proposed as new drug nanocarriers.36–38 When considering the passage through junctions, there is not yet a general tendency to prefer one shape over others, and the study of how nanoparticle geometry affects their ability to pass through junctions has been rather limited. Recently, Nowak et al.37 reported that spherical particles associate more with the endothelium compared to rod-shaped particles, with rod-shaped particles exhibiting higher BBB transport.Importance of elasticity and composition of nanoparticles
The interest in assessing the impact of particle elasticity on nanoparticle delivery is a relatively recent development.39,40 It has been demonstrated that softer nanoparticles offer enhanced blood circulation and subsequently enhanced targeting compared to harder nanoparticles in vivo. Softer nanoparticles exhibit significantly reduced cellular uptake in immune cells (J774 macrophages), endothelial cells (bEnd.3), and cancer cells (4T1). Only one study has investigated the role of nanoparticles' flexibility in their interactions with, and penetration across, the BBB.37 Hard particles associate more with the endothelium compared to soft particles, and soft particles exhibit comparable transport via the BBB to hard particles.The composition has a strong influence on the overall behavior of the nanostructures. For example, muco-adhesive NPs and tight junction opening NPs (such as chitosan) can be retained in the mucus layer and then undergo transcellular passage. Mitragoti et al. recently underlined the profound effect of particle composition on particle transport across the BBB.38 As the composition is of high importance, the classification of NPs will be based on composition rather than charge and size in the following sections.
Lipid-based nanoparticles
Lipid-based nanocarriers are a large class of nanostructures with liposomes, lipid nanoparticles (LNPs), lipid nanocapsules (LNCs), and solid lipid nanoparticles (SLNs) being at the forefront of attention. The development of LNPs dates back to the work of the Cullis group in the 1990s on pH-sensitive LNPs.41 Principally, they found that the neutral lipid dioleoyl-phosphatidylethanolamine (DOPE) adopts a hexagonal IIn (HII) phase42 under physiological conditions, promoting fusion of lipid bilayers.43 The concept of “hexagonal HII phase” formation inspired the design of the first LNP for the delivery and intercellular release of short interfering RNA (siRNA). Since then, LNPs have become one of the most promising technologies for in vivo delivery of siRNA and mRNA. Several review articles are also devoted to the use of lipid-based nanoparticles for the delivery of drugs to the brain underlining the strong focus on these structures for drug delivery.44,45 Scalable manufacturing of LNPs has been achieved lately and played an important part in the large-scale provision of the COVID-19 mRNA vaccine for systemic administration via intramuscular injection. While low encapsulation efficiency and rapid leaking were pendulous issues for the liposomal delivery of many amphipathic basic drugs, doxorubicin was encapsulated into unilamellar vesicles with over 90% trapping efficiency using the concept of drug diffusion driven by a pH gradient across the lipid membranes for drug loading. LNPs are clinically approved for several applications46 notably for the treatment of various cancers, leukaemia and infections.Visudyne by Bausch and Lomb, a verteporfin loaded liposomal formulation, remains one of the rare examples for the treatment of an eye disorder, macular degradation, causing blurred or reduced central vision due to the breaking down of the inner layers of the macula, the part of the retina that gives the eye clear vision in the direct line of sight. In 2020, KPI-121 mucus-penetrating particles for enhanced penetration of loteprednol etabonate were FDA approved for the temporary relief of signs and symptoms of dry eye disease and for the prevention of cornea transplant rejection.47 Numerous considerations still need to be addressed before repurposing LNPs for brain, retinal and corneal delivery, such as drug loading capacity, toxicity, fate of the nanoparticles once crossed the brain and retinal blood barriers. One of the main open questions often revolves around the uptake mechanisms of lipidic nanostructures by the different biological barriers and their ability to pass through TJs. Indeed, knowing that TJs are about 10–20 nm in size makes paracellular transport of 10–200 nm large lipid nanostructures restricted to transcellular routes (Fig. 2).
A large number of attempts and concepts have been described in connection with lipid-based nanoparticles for drug delivery to the brain, but none of the formulations have aroused wider interest. The transferrin receptor is of special interest for brain drug delivery since its expression is higher in the brain endothelium than in endothelia at the periphery. The uptake of fluorescence oxaliplatin-loaded, transferrin-targeting immunoliposomes (OX 26 immunoliposomes) in brain capillaries was evaluated in vivo in mice using spinning disk confocal microscopy.48 While the association of these liposomes with the rat brain capillaries was validated, no evidence of transcytosis into the brain parenchyma was seen nor was there any evidence of BBB crossing.
Low-density lipoprotein receptors are present on the BBB and have been the target in a number of works via apolipoprotein E (apoE) modified LNP-driven BBB entry, with, however, controversial findings.49 Other targeting strategies using the nonapeptide RMP-7, a bradykinin analogue, or lactoferrin (Lf), a member of the transferrin family with an Lf receptor on human brain microvascular endothelial cells activating receptor-mediated transcytosis for the BBB, for the delivery of quercetin-loaded liposomes have improved neuronal viability in vitro by reducing Aβ-induced neurotoxicity.50 A promising approach might be the use of lipid nanocapsules loaded with anti-inflammatory prostaglandin D2-glycerol ester (PGD2-G) lipid and coated with a cell-penetrating protein.51 These particles showed increased brain administration after nose-to-brain delivery with reduced expression of proinflammatory cytokines in the CNS.51
The reformulation of mRNA-loaded LNPs conjugated to receptor-specific monoclonal antibodies for RMT might be a promising approach for crossing brain cell member–brain cell nuclear membranes (Fig. 4A) like demonstrated in adult rhesus monkeys, following IV administration of plasmid DNA encapsulated within human insulin receptor monoclonal antibody-(HIRMAB) targeted lipid nanoparticles.52–54 Expression of the lacZ transgene throughout the whole primate brain could be observed 24 h after LNP administration.
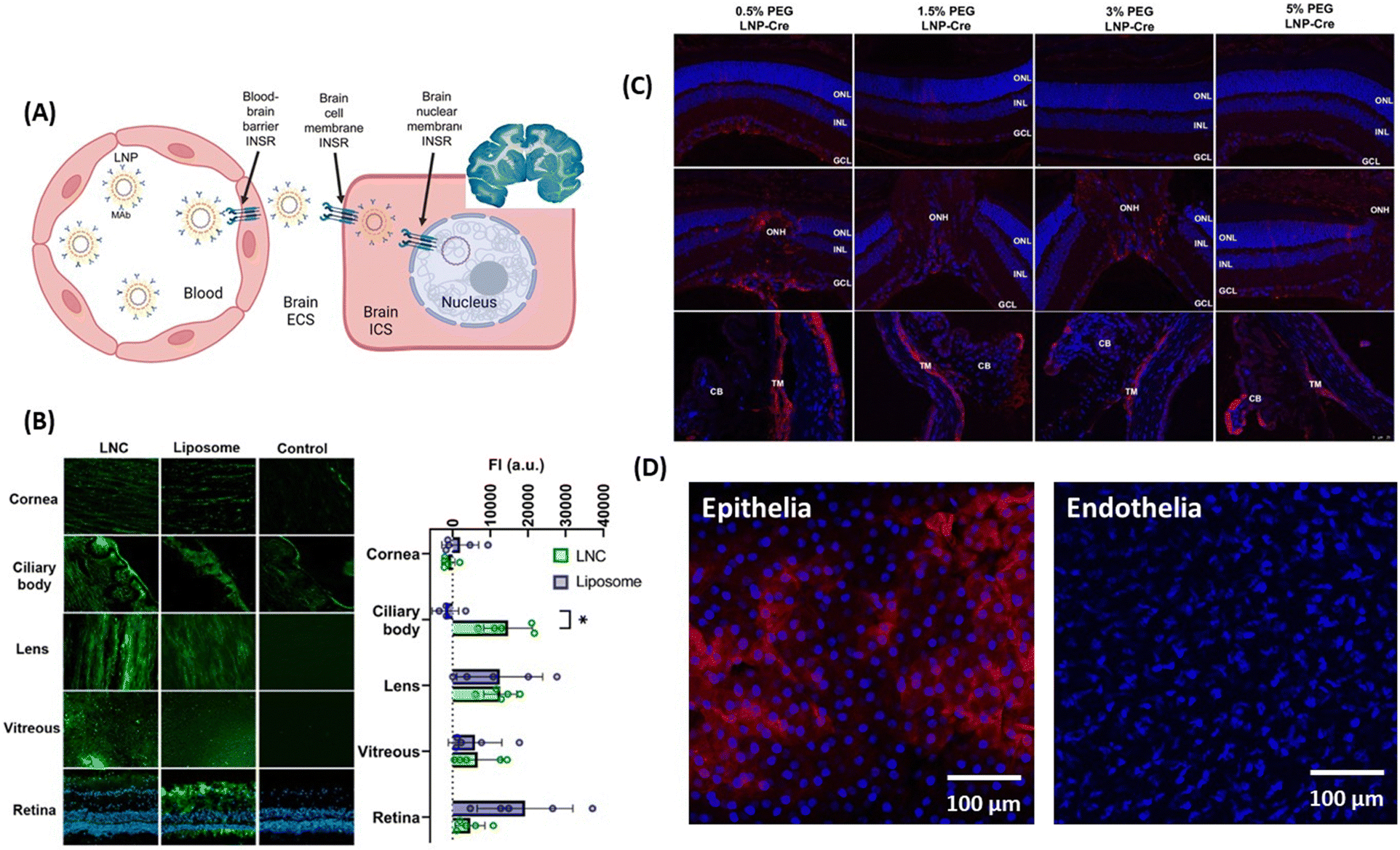 | ||
| Fig. 4 Lipid-based nanoparticles for drug delivery to the brain and eye: (A) (left) action of LNPs loaded with plasmid DNA and modified with monoclonal antibodies against HIR to pass the BBB via RMT, followed by uptake into the brain cellular membrane through endocytoses, and plasmid DNA delivery to the nucleus. (right) The coronal section of the primate brain removed 48 h after LNP administration showing global expression of the SV40-lacZ transgene throughout the primate brain. This figure has been adapted from ref. 54 with permission from Cell Press, copyright 2023. (B) Biodistribution of fluorescently-labeled LNCs and liposomes after 24 h of INV administration in whole porcine eyes determined from microscopy images of different tissue sections and measured fluorescence intensity. This figure has been adapted from ref. 55 with permission from Elsevier, copyright 2023. (C) Effect of PEG amount present in LNPs on particle delivery after post-INV injection evaluated through confocal imaging of immunohistochemistry showing red fluorescence protein (RFP) expression in the Muller glia, the optical nerved head (ONH) and the trabecular network (TM): ONL = outer nuclear layer, INL = inner nuclear layer, GCL = ganglion cell layer, and CB ciliary body. This figure has been adapted from ref. 56 with permission from PMOS One, copyright 2020. (D) Confocal images of corneal penetration of 3,3′-dioctadecyloxacarbocyanine perchlorate (Dil) loaded LNC25 (1 mg mL−1) investigated on ex vivo pig eye models showing penetration of the epithelial, but not the endothelial layer (unpublished results). | ||
Solid lipid nanoparticles (SLNs), stable spherical nanocarriers comprising a solid hydrophobic core of lipids coated with a monolayer of phospholipids as emulsifiers loaded with β-elemene,57 curcumin58 and resveratrol59 represented promising treatments for neurodegenerative disorders. However, these systems face restricted stability and rapid clearance. In the case of SLN-β-elemene, a 2-fold higher brain concentration compared to free β-elemene was observed, which is a practically irrelevant increase.57 Transcytosis based delivery of resveratrol loaded apoE-SLNs showed a 1.8-fold higher BBB permeability compared to the non-functionalized ones on hCMEC/D3 cells.59 Again, the penetration profile of these lipidic particles is limited for practical considerations. SLNs loaded with BACE1 siRNA60 showed improved penetration across a Caco-2 monolayer model; however, the amount of siRNA delivered to brain tissue was not investigated and its therapeutic potential was never assessed further. It was found that the olfactory epithelial cells or the trigeminal nerve ends in the respiratory epithelium transport siRNA into the cerebrospinal fluid.
Surprisingly, the literature on LNPs for ocular permeability and retinal drug delivery is very limited. The team of Schipper and Paquet-Durand used LNCs to deliver CN03, a cGMP analogue, for the treatment of retinal degeneration,55 but the drug permeation through the cornea proved to be too low for further clinical considerations. However, INV application of 2 mg mL−1 of fluorescently labeled LNCs (d = 48–72 nm, ζ = −0.3 to −11 mV) and liposomes showed promising improvements (Fig. 4B). Using ex vivo porcine eyes, it was demonstrated that 10% of the LNCs reached the retina, with 40% accumulating in the ciliary bod, 17% in the vitreous and 33% in the lens. The team of Gaurav Sahay at Oregon State University reported encouraging results upon injection of mRNA-LNP with 0.5% PEG (d = 150 nm) into the vitreous chamber via IVT, with expression of luciferase activity being observed in the Muller glia, the optic nerve head and the trabecular meshwork, but it failed to reach the retinal pigment epithelium (Fig. 4C).56 While targeting the corneal epithelia and endothelia still remains difficult, Dil-loaded LNCs of d = 25 nm and ζ = 4 mV accumulated clearly in the epithelium layer of cow eyes (Fig. 4D), but could not reach the endothelial of the cornea.
Polymer-based nanocarriers
Polymeric nanocarriers have attracted wider interest for the shuttling of drugs across the BBB and the eye due to their low-cost and easy preparation methods and the high flexibility in integrating pH and temperature triggerable drug-releasing parameters. Chitosan, a hemi-synthetic cationic linear polysaccharide made of randomly distributed D-glucosamine and N-acetyl-D-glucosamine units,61 gelatine and sodium alginate, remain the most widely used natural building blocks with synthetic polymers such as poly-lactide co-glycosides, polyvinyl alcohol being often considered in parallel.62 The non-toxic nature of chitosan has led to its approval for medical applications and has demonstrated significant improvements in delivering pharmacological compounds across the BBB63 as well for ocular drug delivery,64 even though none of the formulations has passed clinical trial phase evaluation.Chitosan nanoparticles
The positive charge of chitosan nanoparticles favors electrostatic interaction with the negatively charged capillary endothelium membrane of the BBB. In work dating back to 2005, the group of Patrick Couvreur designed PEG-modified chitosan nanostructures with attached monoclonal antibody OX26 for the delivery of a caspase inhibitor, peptide Z-DEVD-FMK, to the brain.65 This work showed the translocation of an important number of nanoparticles into the brain tissue after I.V. administration, a finding confirmed through electron microscopic examination of the brain tissue. Chrysin-loaded chitosan NPs (Chr-Chi NPs) were lately evaluated for their neuroprotection efficacy66 using zebrafish as a model after oral administration. Notably, brain sections analyzed through immunohistochemistry data revealed that Chr-Chi NPs disintegrate amyloid aggregation, whereas chrysin reduces neuronal cell death and protects synaptic integrity. Indeed, protein aggregation and deposition of uniformly arranged amyloid fibrils in the form of plaques or amorphous aggregates is characteristic of amyloid diseases. Despite the strenuous effort to find a suitable treatment option for these amyloid disorders, very few compounds have made it through unsuccessful clinical trials. It has become a compelling challenge to understand and manage amyloidosis with the increased life expectancy and ageing population and such a dual action strategy might be the future in this field. More recently, Chuang and co-workers have advanced the field by proposing multi-stimuli-responsive curcumin-fucoidan/chitosan nanocarriers formed through the self-assembly and strong ionic interactions between the negatively-charged sulphated fucoidan polysaccharide and positively-charge chitosan.67 Upon intranasal delivery to the brain, curcumin-containing chitosan/fucoidan nanocarriers (d = 170 nm, ζ = 25 mV, and encapsulation efficiency 88%) (Fig. 5A) accumulated strongly in the brain in comparison to the free drug. The endothelial tumor vasculature was targeted by the teams of Daniel Heller and Praveen Raju using vismodegib-loaded fucoidan-based nanocarriers (d = 80 nm), which were transported to the brain using caveolin-1 dependent transcytosis. In combination with radiation treatment, striking efficacy was observed in a Sonic hedgehog medulloblastoma animal model.68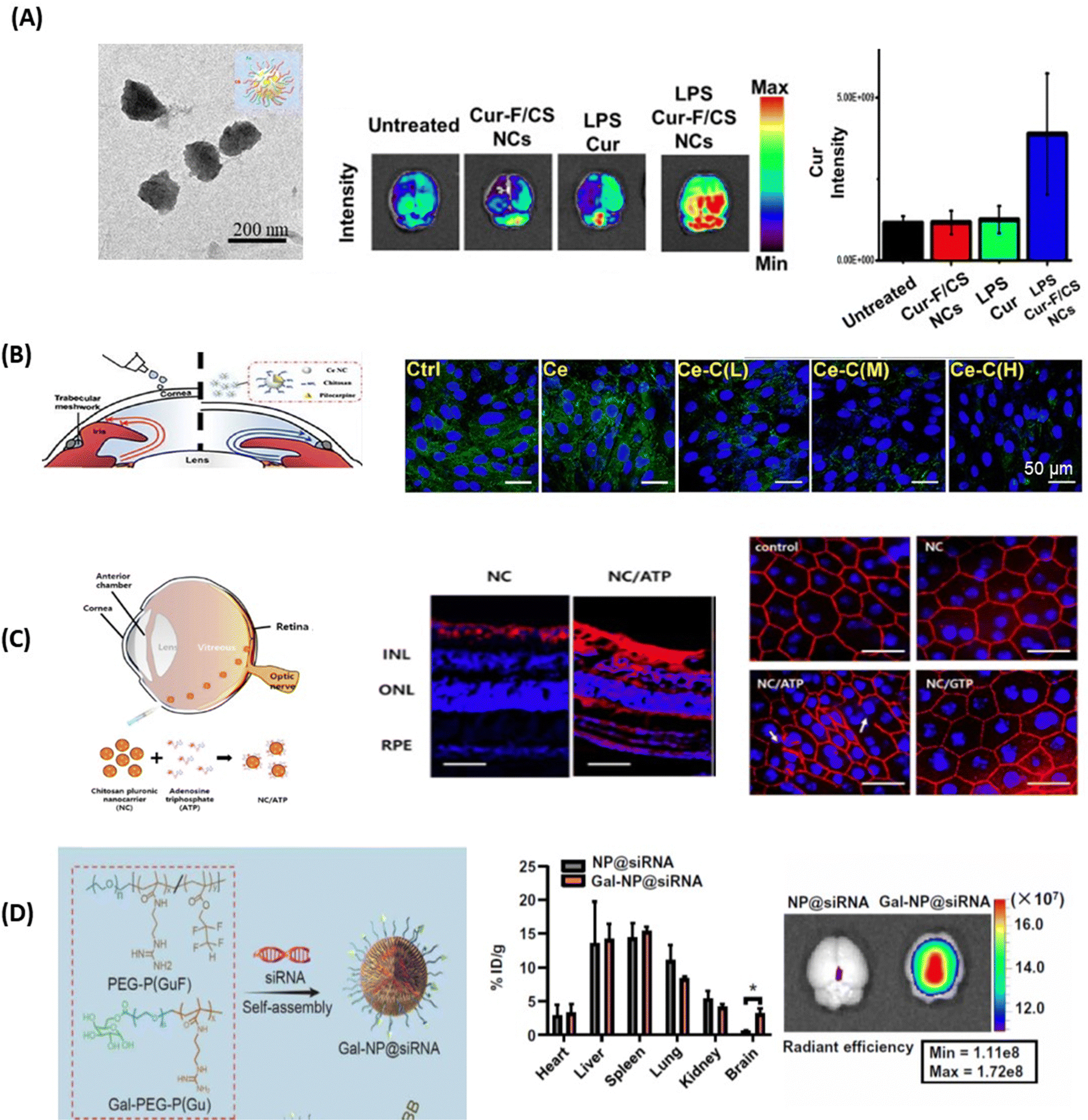 | ||
| Fig. 5 Polymeric nanoparticles for drug delivery to the brain and eye: (A) (left) TEM image of curcumin-containing chitosan/fucoidan nanocarriers and (middle) qualitative and (right) quantitative fluorescence data of curcumin distribution in brain tissues after animals were treated with different formulations via intranasal administration (this figure has been adapted from ref. 67 with permission from Elsevier, copyright 2021). (B) Confocal microscopy images of Statens Serum Institut rabbit cornea cell layers stained with DAPI (blue) and ZO-1 (green) after 4h of incubation with Ce-NC particles alone or in the presence of chitosan coating of low (L), medium (M) and high (H) content of amino groups: Scale bars: 50 μm. This figure has been adapted from ref. 69 with permission from Elsevier, copyright 2023. (C) (left) Chitosan-functionalized pluronic-based nanocarrier with ATP modification penetrating different retinal layers after IVT together with (right) fluorescence images of Cy5.5 labeled nanocarriers (red) showing that NC/ATP can cross the retina and affect TJ ZO-1 in RPE (red staining) (this figure has been adapted from ref. 70 with permission from MDPI, copyright 2021). (D) (left) Gal-NP@siRNA fabrication together with (right) quantification of Cy5-SiRNA accumulation in different organs through fluorescence spectroscopy 1 h after tail vein injection of particles (this figure has been adapted from ref. 71 with permission from American Association for the Advancement of Science, copyright 2020). | ||
The mucoadhesive nature of chitosan also makes it an ideal nanomaterial for corneal permeability studies. Ionic interactions between the positively charged amino groups of chitosan and the negatively charged sialic acid residues in the corneal mucus help to retain chitosan on the tear film. Chitosan can also disrupt TJs, thus increasing the permeability of the cornea epithelium.72 This was validated by examining the transendo/epithelial electrical resistance (TEER) of human corneal epithelium cells (HCECs) treated with chitosan NPs, where TEER resistance was reduced by 70% compared to the control, along with a parallel decrease in ZO-1 expression.73 In general, TEER is a measure of barrier integrity and depends on the flux of ions across the biological barrier layers. The higher the TEER value, the fewer ions can permeate, indicating a tighter barrier. It was shown that TEER corresponds to the tightness of TJs. Similar results were obtained by Schuerer et al. using TEER measurements on human conjunctival epithelial (HCjE) cells.74 It could also be shown that FITC-labelled chitosan NPs were present within guinea pig conjunctival epithelial cells 120 min after topical administration. These observations together indicate that chitosan nanoparticles can increase the drug concentration in the cornea/conjunctiva via trans- and paracellular routes. Increased concentrations of cyclosporin A (CyA) in the cornea/conjunctiva/aqueous humor after the topical administration of CyA-loaded chitosan NPs rather than chitosan alone are in line with the advantageous nature of chitosan for crossing corneal barriers.75 Chitosan coated ceria nanocapsules (Ce-NCs) were consequently proposed for the release of pilocarpine for glaucoma treatment (Fig. 5B).69 Treating Statens Serum Institut rabbit cornea cells with these nanostructures revealed a loss in TJ integrity and an increase in permeability when a high number of amino groups are present. While Ce-NCs without amino group coating (Ce only) could not open TJs, a single dose topical instillation of pilocarpine-loaded chitosan-coated Ce-NCs could effectively reduce high intraocular pressures to normal and inhibit retinal degeneration. These findings show a great promise for the development of nano eye-drops for efficient management of acute glaucoma.
Chitosan NPs have also garnered interest for overcoming barriers in the posterior segment of the eye. Chitosan-coated pluronic-based nanocarriers (NCs) functionalized with ATP demonstrated facilitated diffusion in the vitreous humour and reached the retina via IVT.70 The fluorescence images revealed that positively charged NCs with ATP (NC/ATP) facilitated transport through the vitreous humour, reaching the posterior region of eyes (Fig. 5C). Indeed, NCs accumulated on the surface of the retina, but did not penetrate across the retinal layers; NC/ATP particles (red signal) were observed throughout the whole retina from the INL (inner nuclear layer) and ONL (outer nuclear layer) to the RPE (retinal pigment epithelium). In addition, the ZO-l staining of tight junctions in the RPE showed that TJ disruption was observed in the NC/ATP group, indicating that NC/ATP has significant potential for applications in retina-associated diseases.
Other polymers
Polymeric micelles, spherical shaped nanostructures formed by the self-assembly of amphiphilic block copolymers in an aqueous medium with a size between 10 and 100–200 nm were proposed by Shao et al. to deliver the antifungal agent amphotericin (AmB) to treat fungal infection of the CNS.76 Angiopep-2 surface-modified polymeric micelles loaded with AmB showed higher penetration efficiency across the BBB with higher brain accumulation compared to the unmodified system. Yu et al. conjugated PEG-PLGA polymersomes (self-assembled structures having different building block types with improved solubility over liposomes) with lactoferrin, loaded with the S14G-humanin neuroprotective peptide (SHN-Lf-POS)77 and could validate a protective effect on neurons by inhibiting the overexpression of Bax and caspase-3 neuron-dependent apoptotic factors. BBB penetrating siRNA nanostructures for AD therapy based on complexation of siRNA with a galactose-modified PEG-block-poly[(N-(3-methacrylamidopropyl)guanidium/PEG-block-poly[(N-(3methacrylamidopropyl)guanidium-co-2,2,3,3-tetrafluoropropylmethacrylate polymer mixture (Fig. 5D) were proposed lately to restore cognitive capacity in AD mice without notable side effects.71 The particles featured a better blood circulation stability relative to conventional cationic polymer-based approaches. A biodegradable poly(lactic-co-glycolic acid)(PLGA)-block-(b)-PEG functionalized with pending lipophilic triphenylphosphine (TPP) cations and embedded siRNA created a hydrophobic cationic surface, playing a significant role in particle accumulation in the brain.78 Fluorescence spectroscopy after 1 h tail vein injection of these particles revealed predominant accumulation in the cerebral cortex and to a lesser extent in the hypothalamus, hippocampus and thalamus. A study using poloxamer 188 modified PLGA-PEG NPs, conducted by Joseph et al., also demonstrated enhanced permeability across the BBB with 19-fold higher uptake in the brain parenchyma compared to non-coated NPs79 due to a combined effect of PLGA-PEG polysorbate 80, with reported enhanced permeability across the BBB.80,81 These particles thus seem to be highly promising and more detailed investigation should be carried out in this direction to validate the therapeutic interest.The main method for posterior drug delivery is intravitreal injection where the drug is injected into the vitreous humor, and then it diffuses through the vitreous to reach the inner limiting membrane of the stratified retina where it finally passes through the multilayers of the retina (Fig. 1). The first barrier of IVT for drug delivery to the retina remains the vitreous humour. The negatively charged HA and anionic collagen network gel can aggregate or precipitate positively charged particles in the vitreous humour, making drug transport to the retina challenging.23 Koo et al. showed that polyethyleneimine (PEI) NPs with strong positive charges (ζ = +33.5 mV) aggregate spontaneously before reaching the retina, while negatively charged HA-based NPs (ζ = −26.2 mV) did not form aggregations in the vitreous due to their firm surfaces, and most of these HA NPs penetrate the retina and enter the RPE cell layer.82 Various kinds of polybutylcyanoacrylate (PBCA) NPs were delivered in mice via IVI.83,84 Middle-sized PBCA NPs (272 nm) with ζ = 5 mV resulted in a highly efficient BRB passage but did not open the BRB, while PBCA NPs of reduced size (d = 172 nm) showed reduced passage,85 as confirmed by in vivo confocal neuroimaging (ICON). The difference is believed to be due to the different uptake mechanism, with particles below 200 nm in diameter being taken up through clathrin-mediated endocytosis, whereas larger particles up to a size of 500 nm utilize caveolae-mediated uptake.86
Inorganic nanocarriers
The interest in applying gold nanoparticles (Au NPs) to cross the BBB started with some work on the trafficking of Au NPs coated with the 8D3 anti-transferrin receptor antibody (anti-TfR mAb 8D3) across the mouse BBB after intravenous injection.87 It is probably the work on the use of L- or D-glutathione modified Au NPs of 3.3 nm to inhibit Aβ42 aggregation and to pass the BBB which has induced increased interest88 (Fig. 6A). In vivo mice experiments demonstrated that these Au NPs can be transported from the blood circulation into the brain with the amount of Au in the brain, 6h after intravenous post-injection, being higher for D-glutathione-modified Au NPs compared to the L-glutathione analogue. Importantly, a 4-week intravenous administration of the D-glutathione modified Au NPs decreased Aβ42 plaque deposition in the brain, demonstrating, for the first time, the potential of a chiral agent in the treatment of neurodegenerative diseases.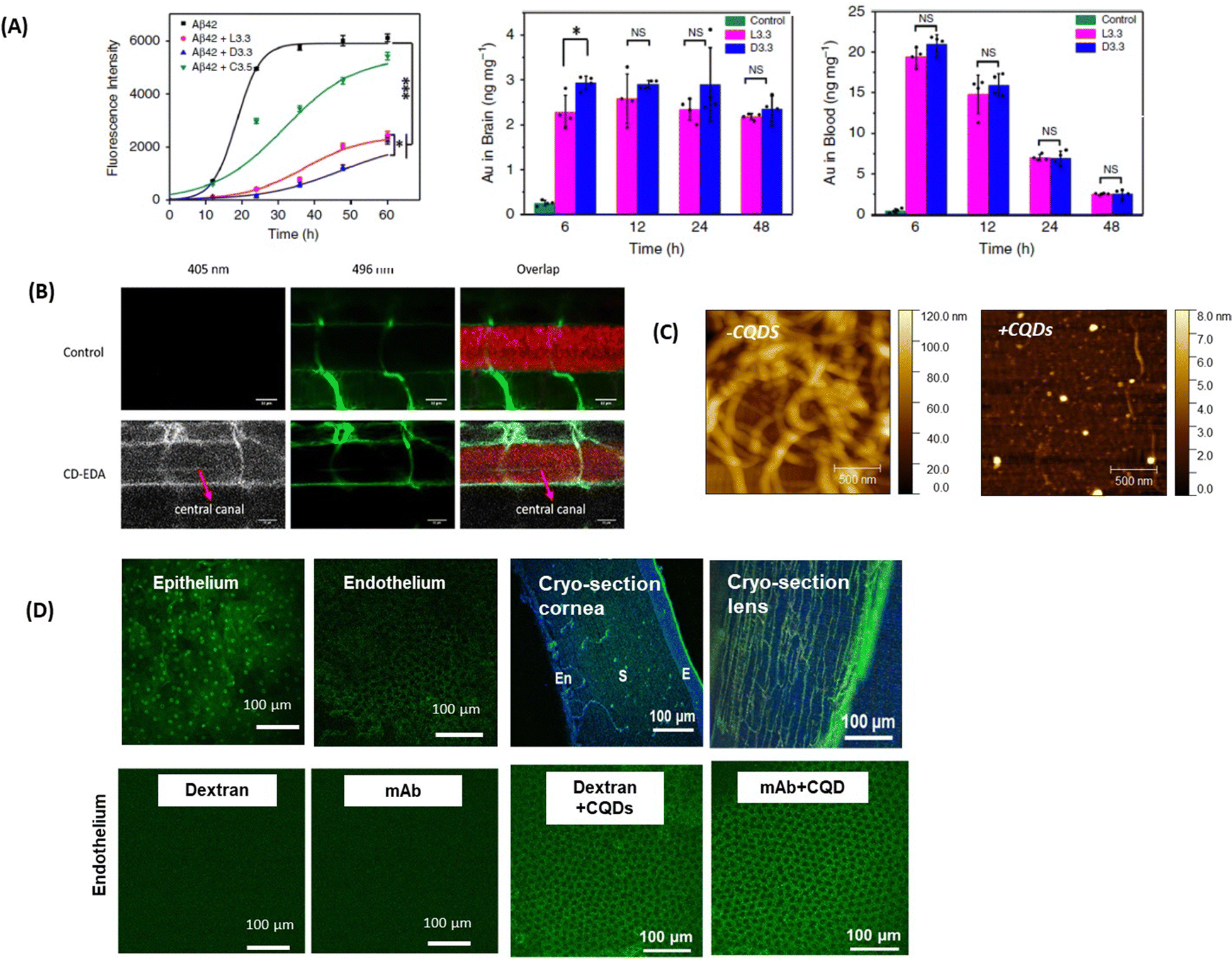 | ||
| Fig. 6 Inorganic- and carbon-based nanocarriers for drug delivery to the brain and eye: (A) (left) effect of the presence of L- or D-glutathione on Au NPs (L3.3 and D3.3) on the aggregation of Aβ42 using ThT fluorescence assay together with (left) the biodistribution of L3.3 and D3.3 in the brain and blood after 6, 12, 24 and 48 h post-injection. This figure has been adapted from ref. 88 with permission from Springer Nature, copyright 2020. (B) Confocal microscopy images of a six-day-old, transgenic zebrafish larva expressing Mcherry (585 nm) in the CNS. The larvae were treated with 10 kDa fluorescein-dextran (496 nm) (control, top row) or a combination of fluorescein-dextran and CQDs (second row). The crossing of the BBB is seen in the central canal due to the fluorescence of the CQDs (405 nm) and is highlighted with the red arrow (this figure has been adapted from ref. 89 with permission from Elsevier, copyright 2019. (C) AFM images of collagen I (0.3 mg mL−1) in the absence and presence of 100 μg mL−1 CQDs formed from glucosamine hydrochloride and ethylenediamine as a passivating agent. This figure has been adapted from ref. 90 with permission from Royal Society of Chemistry, copyright 2021. (D) (top) Fluorescence confocal microscopy images of the ex vivo bovine cornea before and after topical application of CQDs of size 1–2 nm for 1 h (reprinted with permission from Rf.91 together with cryo-sections of the cornea and the lens: E = epithelium, S = stroma, and En = endothelium (Sauvage, de Smedt, S. Szunerits unpublished results). (below) Confocal microscopy images showing the bovine corneal endothelium after 1 h of incubation with fluorescent model drugs of different molecular weights (unpublished results Szunerits@Sauvage). | ||
For improving the penetration into the anterior segment of the eye, silver nanoparticles (Ag NPs) were proposed for topical delivery,92 notably 1%-Ag/SiO2 NPs (d =10 nm).92 The mechanism of penetration is believed to be via disruption of TJs in-line with a report by Mortensen et al., revealing that Ag/SiO2 NPs could increase the permeability coefficient of the intestinal epithelium by disrupting tight junction integrity.93 While these works are of academic interest, the uncertainty of the fate of Au and Ag NPs and their eventual toxicity over time might not make them the ideal candidates for the future.
The biodegradable nature of silicate nanoparticles (Si NPs) might make them more important as it was revealed that Si NPs of 40 nm in size can pass through the corneal epithelium and go deeper to the corneal stroma.94 These structures were even found in the anterior chamber and vitreous using inductively coupled plasma atomic emission spectrometry (ICP-AES).94 As Si NPs also enable the inhibition of phosphorylation of ERK 1/2, a signaling molecule of the Mitogen-activated protein kinase (MAK) pathway, suppressing new vessel formation and vascular leakage,94 their use for the treatment of ocular neovascularization might be ideal.
Light-sensitive TiO2 NPs have aroused in parallel some interest.95 Treatment with TiO2 NPs reduced the claudin-5 protein level in endothelial cells with a decline in the TEER value, leading to an increase in paracellular permeability confirmed by fundus images of mice eyes treated with TiO2 NPs via IVI. The alleviated ocular blood flow indeed indicated a clear damage to the TJs of endothelial cells at the inner BRB induced by the injection of TiO2 NPs, making the TiO2 NPs of limited use due to their hazardous nature.
The potential to track magnetic nanoparticles (MNPs) through magnetic resonance imaging (MRI) and gain information about the distribution of these particles in ocular structures, was lately assessed using iron oxide nanoparticles coated with human serum albumin (HSA). These particles could be observed in the entire back part of the eye, notably in the photoreceptor outer nuclear layer (ONL), subretinal debris zone layer, choroid and sclera, from 1–4 weeks after suprachoroidal injection. Local hyperthermia, which can be induced by the MNPs themselves, was used by Tabatabaei et al. to overcome BRB restriction96 without exceptional results, questioning the use of this approach in the context of the BRB.
Carbon-based nanostructures
Carbon quantum dots (CQDs) and graphene quantum dots (GQDs)97 have received significant attention for nanomedical application and have been more widely considered as penetration enhancers, allowing drugs to cross the BBB. Not only their small size, but also the ease of synthesis from all kinds of carbon-based precursors via carbonization approaches and the resulting wide range of properties have motivated the team of Roger Leblanc to investigate the potential of these nanostructures in more detail.89,98–100 Still, the mechanism of crossing the BBB by CQDs remains poorly understood.101,102 The anionic area on the TJs was indeed one of the first targets of positively charged CQDs, measuring 2.6 nm in size, which were shown to pass the BBB.103 3 nm sized amphiphilic CQDs formed from citric acid and o-phenylenediamine were not only able to cross the lipid barrier of the BBB passively, but could also move through the brain fluids due to their hydrophilic character.100 Receptor-mediated crossing was reported on the other hand with CQDs conjugated to human transferrin104 or with glucose derived CQDs via GLUT-1 mediated transport.101L-Type amino acid transporter (LAT-1) mediated transport and BBB uptake of tryptophan-derived CQDs were validated on transgenic zebrafish expressing mCherry (Fig. 6B).89 However, no drug was delivered with this approach. Conjugation of glycine–proline–glutamate (Gly-Pro-Glu, GPE), the N-terminal tripeptide of insulin-like growth factor-1, onto graphene quantum dots105 resulted in neuroprotective action via the inhibition of the aggregation of Aβ1–42 and decreasing the expression of proinflammatory factors as validated in vivo studies in mice. The use of C60 fullerene (CF) rather than CQDs carrying monomethyl fumarate (MMF), an active drug that stimulates the lysis of tumor cells,106 caused also enhanced permeability of MMF into the brain.Due to their tunable physicochemical parameters, the use of CQDs in the context of ocular nanomedicine represents an interesting platform to enhance the efficiency of topical treatment for corneal diseases. Recently, the teams of Szunerits and de Smedt showed the potential of positively charged CQDs (ξ = 32.5 mV, d = 16 nm) in inhibiting fibrillation of type I collagen (Fig. 6C).90 Interestingly, the diffusion coefficients in water and vitreous are comparable (31.4 ± 7.4 μm2 s−1 and 32.9 ± 13.2 μm2 s−1, respectively) for these nanostructures suggesting that, despite a positive charge, CQDs retain their mobility in the vitreous probably due to their small size allowing diffusion through the meshes of the collagen network whose sizes are range between 500 and 1000 nm. The possibility to destroy type I collagen aggregates and vitreous opacities (as obtained from patients after vitrectomy) was investigated in combination with pulsed-laser illumination (see Section 5).90
Size is a critical parameter for designing nanoparticles for corneal delivery and according to the literature data, nanoparticles with a size <200 nm can permeate the corneal epithelium.107 The interest in positively charged CQDs has, in this context, to be underlined by the potential of spermidine-derived CQDs to reversibly open the TJs of the corneal epithelium,108 allowing these CQDs to reach the corneal stroma, which were used for the treatment of S. aureus induced ocular infections. Some of us showed lately that CQDs, synthesized via a microwave-assisted synthesis method at 180 °C from spermidine and glucosamine, resulted in positively charged ultra-small CQDs (1–2 nm in diameter) with corneal penetration until the endothelium mostly via the paracellular transport route (Fig. 6D). The particles were also present within the stroma as well as in the lens when performing staining experiments (Fig. 6D). The permeation-enhancing ability of these CQDs was demonstrated on model drugs such as FITC-labeled dextran 150 kDa (no corneal crossing alone) or an ATTO-488-labeled monoclonal antibody of 47 kDa (Fig. 6D). Indeed, while dextran and the mAb fragment could not cross the epithelium, in the presence of CQDs, this was possible as seen from the fluorescence image. The mechanism for the crossing remains to be evaluated in depth, but it is possible that TJs proteins occludin and ZO-1 might be involved in the passage.
Willner and Mandel109 took this idea of CQDs being TJ penetration enhancers further and integrated vascular endothelial growth factor (VEGF) aptamer modified CQDs. The hybrid CQDs effectively inhibited VEGF-stimulated angiogenesis in choroidal blood vessels, achieving results comparable to two commercially available anti-VEGF drugs, bevacizumab and aflibercept, making the nanoparticle approach as a versatile nanomaterial to treat age-related macular degeneration and diabetic retinopathy.109
4. Extracellular vesicles as alternatives to nanoparticles as therapeutics and drug-carriers
Extracellular vesicles (EVs) (Fig. 7A) have emerged as key players in cellular communication, both under physiological and pathological conditions.110–113 In contrast to solid NPs, such as metallic NPs, CQDs and polymeric based nanostructures, EVs are a heterogeneous population of lipid bilayer membrane-enclosed nanostructures. They are released by cells and found in several, if not all, bodily fluids such as CSF, blood, tears, or saliva.114 While liposomes and lipid nanocapsules are artificial vesicles composed of lipid bilayers, capable of encapsulating both hydrophilic and hydrophobic substances, EVs are natural vesicles secreted by cells. These lipid-bilayer enclosed vesicles encompass diverse populations, including exosomes (typically 30–150 nm in diameter), microvesicles (50–1000 nm), and apoptotic bodies (1–5 μm).115 Each type of EV has unique biogenesis pathways and functions,115,116 carrying an array of different molecular cargoes, from proteins and lipids to different types of RNA and/or DNA, representing a snapshot of the cells’ current physiological state. Proteomic analysis has enabled a more detailed study of EV composition, leading to increasing interest in leveraging EVs for therapeutic purposes.117–119 EVs, like NPs, can be loaded with drugs. One of the most distinguishing features of EVs compared to synthetic NPs is that their membrane expresses proteins (such as integrins and scavenging receptors) from the parent cells. This unique characteristic enables EVs to possess specific functions that facilitate barrier crossing, as well as targeting and retention to particular tissues or cells. This natural expression of parental proteins endows EVs with a tailored ability for interaction and integration with biological systems, and for crossing tissue barriers, setting them apart from undecorated synthetic NPs.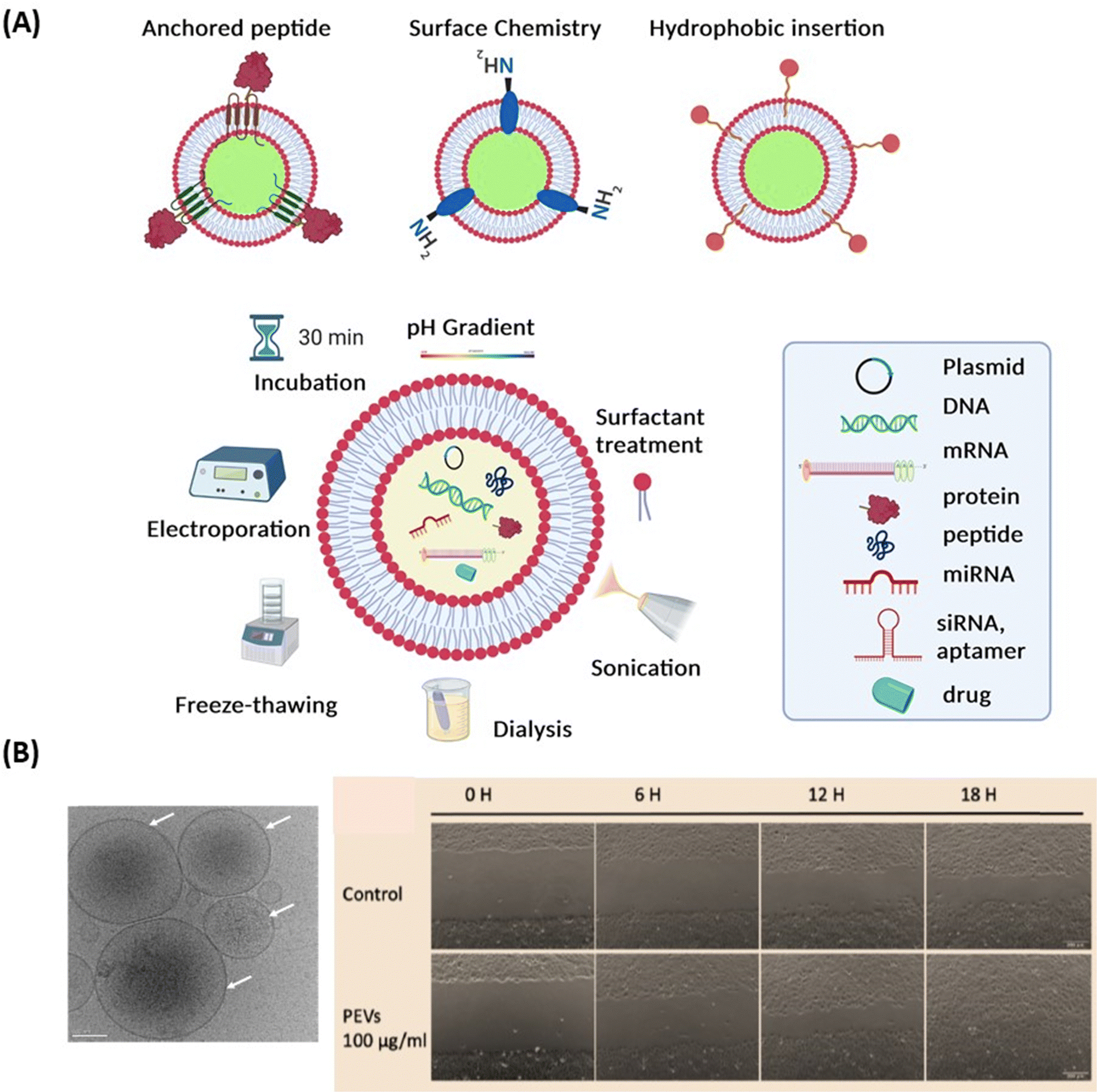 | ||
| Fig. 7 Extracellular vesicles: (A) surface engineering strategies of EVs and drug loading possibilities. (B) (left) Cryo-electron microscopy observation of pEVs (concentration of 6.95 × 1011 particles per mL determined by nanoparticle tracking analysis) isolated from clinical-grade apheresis human platelet concentrates. Scale bare: 50 nm (unpublished results, H-T Chien & T Burnouf), (right) image of B4G12 cell wound healing during various time periods following scratching after treatment without and with 100 μg pEVs (20 × 108 EVs). | ||
The preparation methods of EVs greatly differ from those of lipid nanocapsules since they mainly rely on extraction and purification, using procedures such as centrifugation, chromatography, tangential flow filtration, or filtration,118 whereas lipid nanocapsules are synthesized from their basic ingredients. Similarly, drug loading methods into/onto EVs are different from those used for lipid nanocapsules. While lipid nanoparticles are loaded using passive or active approaches, various therapeutic agents – from small molecule drugs, antioxidants, neurotrophic factors, to RNA-based therapeutics – can be loaded into EVs. Techniques used for loading include freeze–thaw cycles, sonication/cooling phases, electroporation to permeabilize the cell membrane of EVs, extrusion via 200 nm pores, or simple mixing and incubation for various time lengths118,119 (Fig. 7A). Compared to synthetic NPs, EVs exhibit specific challenges that may encompass the need for GMP-grade cell culture facility when using cells that need to be expanded ex vivo, the control of microbial sterility, and the guarantee of batch-to-batch consistency.118
The nature of the EV membranes and their content provide unique capabilities to EVs as therapeutic agents. In particular, their demonstrated ability to cross the BBB during CNS diseases, position EVs as a potentially powerful tool for the treatment of a variety of brain disorders, where conventional therapeutic approaches often fail to yield satisfactory outcomes and may exert side-effects.120 Delivery of EVs and EVs loaded with drugs has been investigated in preclinical models for various brain diseases, providing experimental evidence of uptakes by neurons, microglia, and other cells in the brain. However, the distribution in the brain depends strongly on the EVs’ size and composition. Various preclinical studies have explored the use of naïve (i.e., unloaded) EVs and pEVs for treating brain disorders. The administration of stem cell-derived EVs has been evaluated in several animal models of neurological diseases, as reviewed recently.121 Examples include the use of mesenchymal stromal cells or other stem cell-derived EVs to deliver neuroprotective molecules to the brain in rodent models of ischemic stroke,122,123 neonatal hypoxic-ischemic brain injury124,125 and Parkinson's disease.126 Similarly, pEVs that are rich in anti-inflammatory molecules and antioxidants might be used to facilitate brain repair after injury,127,128 possibly explaining the benefits of platelet lysates rich in pEVs in rescuing brain cells and improving behavior in two mice models of TBI.129 However, it is important to note that there is a lack of data tracking single EVs across the BBB to confirm their transcytosis. Although the detection of radioactive signals of labeled injected EVs suggested quite fast permeation of, e.g., cancer-derived EVs across the mouse in vivo BBB, this did not prove permeation of the single, same EVs.130 Thus, alternative fates of EVs and their cargo after their uptake in brain endothelial cells should not be neglected such as degradation of the EVs, repackaging of EVs and their cargo or intracellular release of the cargo initiating the release of secondary EVs. In this regard, the status of the BBB should also be considered. EV permeation might be enhanced across a disrupted BBB during diseases130 since it is known that both paracellular pathways due to TJ opening, and transcellular pathways due to an increase in the transcytosis rate might enable EV transport during diseases.
Intercellular communication between various retinal cell-types (e.g., neurons, glia, endothelial cells, pericytes, and immune cells) is essential for maintaining normal tissue function and physiology. Studies have focused on identifying and characterizing EVs in the retina, and investigating their pathogenic role in retinal degenerative diseases.131 The therapeutic applications of EVs in the retina have been explored in several studies, notably as blood-circulating EVs are believed to cross the BRB, most likely due to receptor-mediated endocytosis. However, our understanding of this aspect is limited, and further work is much needed to understand the mechanism of EV uptake.
Thierry Burnouf's team also demonstrated the therapeutic potential of human blood platelet-derived EVs (pEVs), isolated from therapeutic-grade platelet concentrates or present in platelet lysates, for corneal endothelial cell protection from oxidative stress and regeneration.132,133 The pEVs exhibiting a regular, fairly rounded shape, with an average size of <200 nm, were present at a concentration of approximately 1011/mL133 (Fig. 7B) and expressed characteristic membrane markers such as CD9, CD41, CD61 and CD63. ELISA and LC-MS/MS proteomic analyses revealed that the pEVs contained mixtures of growth factors and multiple other trophic factors, as well as proteins related to extracellular exosomes with functional activities associated with cell cadherin and adherens pathways. Corneal endothelial cells treated with pEVs exhibited increased viability, an enhanced wound-healing rate (Fig. 7B) and did not exert cellular toxicity, as evidenced by the maintenance of cellular morphology and preservation of corneal endothelial proteins.133 These data open the perspectives of using pEVs as eye drop formulations for the repair of the corneal endothelium. As for other EVs, it is not yet established whether pEVs, either “naïve” or loaded with drugs, could be used to cross the BRB and exert beneficial effects.
5. Other nanotechnological concepts
Intrathecal, intracerebroventricular, and intraparenchymal administration
In addition to the use of nanoparticles and EVs carrier for drugs for enabling the crossing of the BBB, the BRB and corneal TJs, several other methods (Fig. 8A) have been developed to circumvent these biological barriers. In the case of the BBB, the methods include intrathecal administration (IT) into the lumbosacral subarachnoid space of the spinal cord, intracerebroventricular administration (ICV) into the ventricular system and intraparenchymal administration, which is called convection-enhanced delivery and is more dedicated to maximizing the local concentration for brain tumor.134 This could be either single or intermittent injections through ports or pumps. The pump approach should be preferred when it is necessary to consider continuous administration (i.e., restoration of a neurotransmitter, hormone, etc.) or very high drug doses that are incompatible with other administration routes.135 The feasibility and safety have been demonstrated with the brain infusion of growth factors.136 This approach is currently considered for dopamine administration.137,138 With such procedures, high local concentrations of drugs and personalized concentrations can be achieved with minimal or no systemic adverse effects. This strategy can be also combined with NPs.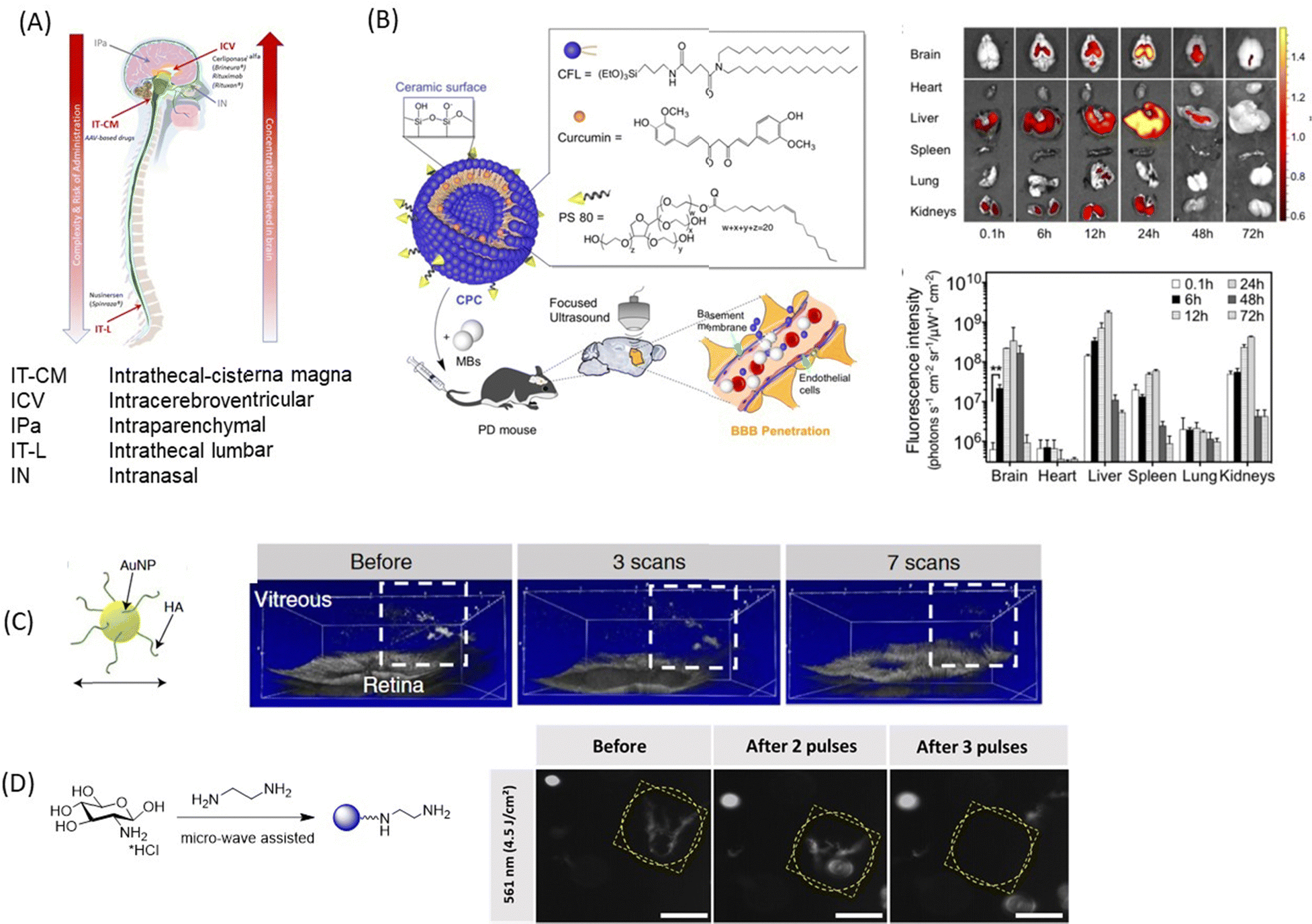 | ||
| Fig. 8 A selection of nanotechnological concepts other than nanoparticle delivery alone: (A) schematics of novel technologies for drug delivery to the brain using intracerebroventricular, intrathecal-cisterna magna, intrathecal-lumbar, intraparenchymal, and intranasal approaches. (B) (left) Curcumin loaded cerasomes coated with polysorbate80 for focused ultrasound based BBB penetration together with (right) fluorescence images and quantification of curcumin in major organs (this figure has been adapted from ref. 139 with permission from Ivyspring International Publisher, copyright 2018.) (C) Laser-induced ablation of vitreous opacities from HA-coated AuNPs validated through optical coherence tomography (OCT) on rabbit eyes. This figure has been adapted from ref. 140 with permission from American Chemical Society, copyright 2019. (D) Dark-field microscopy images of water-dispersed type I collagen fibres before and after light illumination with 561 nm laser in the presence of positively charged CQDs (0.4 mg mL−1). Scale bar: 100 μm. This figure has been adapted from ref. 141 with permission from Royal Society of Chemistry, copyright 2021. | ||
Prodrug design via lipidization process
The use of prodrugs is another widely used strategy based on increasing the lipophilic nature of neuroactive agents using receptor- and carrier-mediated BBB transport.142,143 Hydrophilic drugs are chemically transformed into lipophilic prodrugs by masking polar functional groups with nonpolar and lipophilic substituents. The prodrugs obtained through such a “lipidization” process are inactive agents in vivo, but can easily access the brain where their conversion to parent active agents is induced by enzymatic or chemical processes. This approach was successfully applied to morphine a long time ago; morphine was transformed into its lipophilic prodrug heroin, through double acetylation, where the prodrug increased BBB permeability up to 100-fold in comparison to morphine.144 Once in the brain, heroin was enzymatically converted to morphine, allowing its interaction with the opioid receptor. Another example is that of dopamine, which does not cross the BBB because of its hydrophilic properties. L-Dopa in contrast enters easily the brain due to the substrate-mediated transport processes via LAT-1. To understand the advances in the field, selective inhibition of glycine transporter 1 (GlyT1) by the prodrug [18F]ALX5406 has emerged as a potential approach to alleviate N-methyl-D-aspartate receptor (NMDAR) hypofunction in patients with schizophrenia and cognitive decline.145 A comparable approach was used by the team of Couvreur,146 showing that conjugation of adenosine to the lipid squalene and the subsequent formation of nanoassemblies allow prolonged circulation of this nucleoside, providing neuroprotection in mouse stroke and rat spinal cord injury.Laser light and ultrasound-initiated drug delivery process
The use of low-frequency focused ultrasound is an emerging technology for drug delivery to the brain.139,147,148 Such an approach represents an interesting alternative for intermittent administration of drugs with long lasting effects. The BBB is disrupted transiently and reversibly, allowing mechanically stress induced drug delivery. The temperature increases by ≈5 °C and there is a possibility of producing transient microbubbles, which raises safety concerns. The effect of the size of Au NPs in focused ultrasound-induced BBB opening has been investigated and revealed that 15 nm Au NPs allowed the highest delivery efficiency.148 Curcumin loaded PS 80-modified cerasome nanoparticles (CPC NPs) achieved interesting drug delivery ability using the ultrasound-targeted approach (Fig. 8B).139 The team of De Smedt has recently shown that Au NPs can also form vapor nanobubbles under pulsed-laser irradiation, an approach used for the destruction of collagen floaters140 (Fig. 8D). Indeed, following IVI, hyaluronic acid-coated Au NPs and indocyanine green, widely used as a dye in vitreoretinal surgery, spontaneously accumulate on collagenous vitreous opacities in the eyes of rabbits. Applying nanosecond laser pulses generates vapour nanobubbles that mechanically destroy the opacities in rabbit eyes. This original approach offers a safe and efficient treatment to millions of patients suffering from debilitating vitreous opacities and paves the way for a highly safe use of pulsed lasers in the posterior segment of the eye. The Szunerits team elaborated on this work lately and showed that positively charged CQDs had similar effects as hyaluronic acid-coated Au NPs where irradiation with short light pulses (<3 ns) at 561 nm (4.5 J cm−2) destroyed the collagen fibres (Fig. 8E).90Alternative non-invasive delivery via nose-to-brain delivery
The possibility of reaching the CNS without crossing the BBB conquered a remarkable interest among researchers. The nose, providing a direct connection into the brain via the olfactory region of the nasal mucosa (Fig. 9A) is ideally suited for drug delivery to the brain. Intranasal (IN) drug delivery offers the following advantages for direct brain delivery:149–151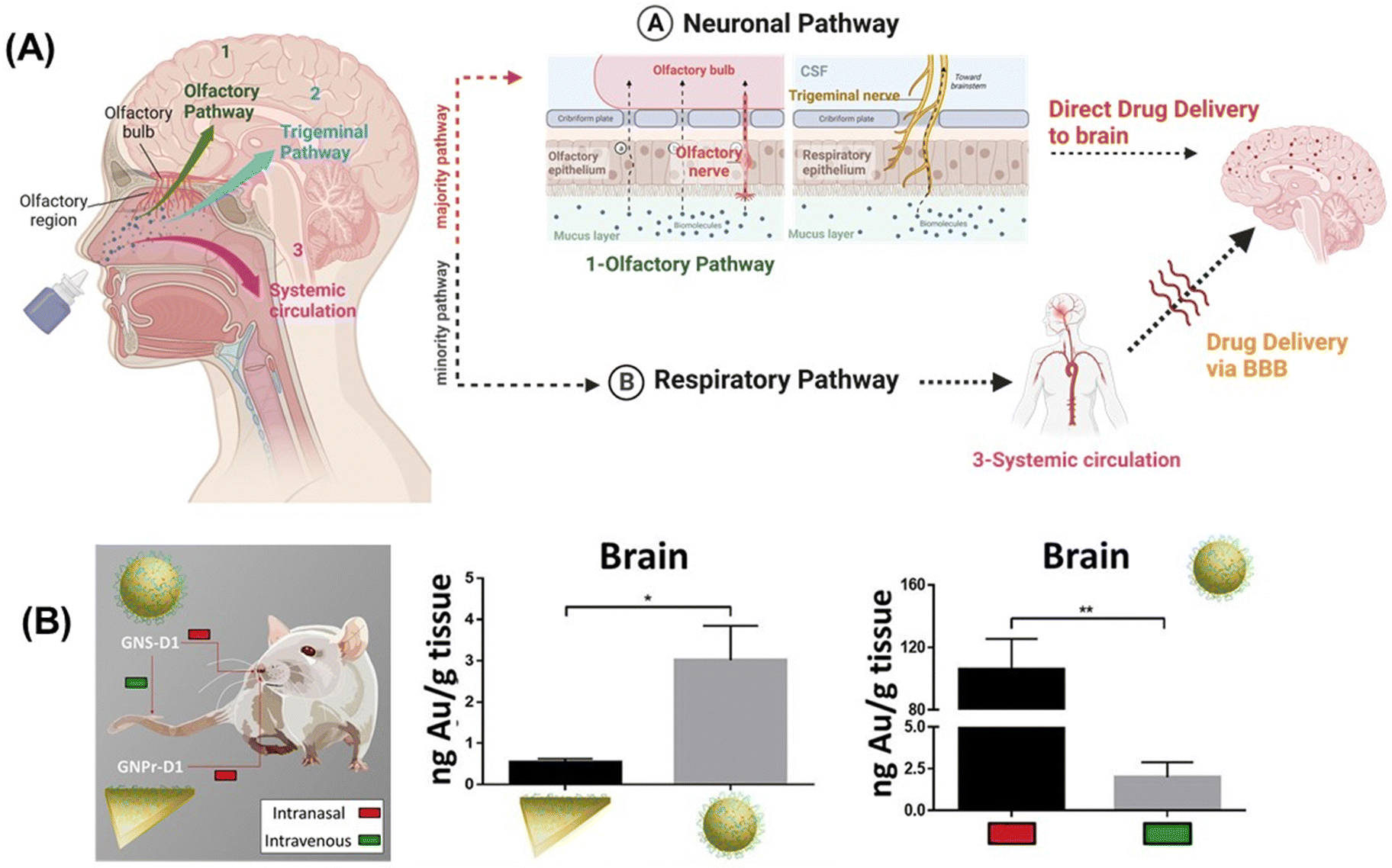 | ||
| Fig. 9 Nose-to-brain transport option: (A) schematics of nose-to-brain delivery approach of drug microemulsions formed by nasal sprays and drug delivery via the neuronal pathway (majority route) and respiratory pathway (minority route). (B) Gold quantification after 30 min of IN and IV administration of gold nanoprisms modified with D1 peptides or gold nanospheres modified with D1 in the brain. This figure has been adapted from ref. 152 with permission from Elsevier, copyright 2020. | ||
(1) A non-invasive delivery route,
(2) Overcomes the challenge to deliver the drug selectively with higher accumulation in CNS tissues,
(3) Adapted for intermittent administration of small molecules and small volumes of drugs,
(4) Improved comfort for patients compared to direct injection into the brain or systemic injection,
(5) Reduced risk of unwanted systemic side effects, and
(6) Rapid onset of action as compared to systemic administration.
However, compared to ICV administration, the control of the effective dose reaching the targeted brain location is challenging. Moreover, rapid mucocilliary clearance in the nasal cavity poses a challenge for the IN route due to limited residence time in the nasal cavity and the quick delivery of drugs through the nasal mucosa.
Polymeric nanoparticles, notably chitosan NPs are the most popular nanocarriers used in nose-to-brain delivery and are at the forefront of neuropharmacological treatments due to their mucoadhesive properties and the ability of CS NPs to open the TJ existing within the nasal epithelial barrier.153,154 Wilson et al. reported the delivery of sitagliptin (SIT) into the brain of rats using SIT-loaded chitosan particles of 188.4 ± 48.1 nm mean size and ζ = +20.8 mV.155 Gabold et al. studied the passage of β-galactosidase through human nasal epithelial cells (RPMI 2650) using transferrin-decorated chitosan NP carriers.153 Readers are referred to the excellent review by Donnelly and Paredes.154 Galladro-Tolers delivered peptide modified Au NPs IN (Fig. 9B) and the detected Au amount gives an idea about the concentration of the structures reaching the brain compared to I.V. injections. About 55 times higher Au NP load was detected in the brain after IN administration compared to I.V. administration. However, no significant difference was observed between the IN and IV routes in the distribution of the nanostructure in various brain areas. The IN-delivery method is also an attractive approach for the efficient delivery of EVs, pEVs and drug loaded variants. IN delivery of stem cell-EVs was investigated in preclinical models for various brain diseases. Data provide experimental evidence that IN-EVs can target regions of injury or inflammation, be taken up by neurons, microglia, and other cells in the brain and exert therapeutic effects.156–158 This demonstrates that EVs administered intranasally can be efficiently delivered to the brain in rodent models. Once various challenges are circumvented,159,160 naïve or drug-loaded IN-EVs could potentially be used to transport therapeutic agents for the treatment of neurological diseases in humans (Table 2).
| Nanocarrier | Compound | Size/nm charge/mV | Disease target | Test model | Targeting performance | Ref. |
|---|---|---|---|---|---|---|
| AD = Alzheimer. BCEC = brain capillary endothelial cell. CEC = corneal endothelial cell. CS = chitosan. CQDs = carbon quantum dots. HEK = human embryonic kidney cell. HCjE = human conjunctival epithelial cell. HA = human astrocytes. HBMEC = human brain microvascular endothelial cell. HREC = human retinal microvascular endothelial cell. HTCEi = immortalized human corneal epithelial cell. LNC = lipid nanocapsule. PEG = poly(ethylene glycol). PGD2-G = D2-glycerol ester. pEVs = platelet-derived extracellular vesicles. RCFs = primary rabbit corneal fibroblasts. RSV = resveratrol. SLN = solid nanoparticle. SK-N-MC = neuroblastoma cells. TAT = cell-penetrating peptide transactivator of transcription. VEGF = vascular endothelial growth factor | ||||||
| Lipidic nanoparticles | ||||||
| TAT-LNC | PGD2-G lipid | d = 60 ± 1 ξ = −7 ± 1 | Neuro-inflammation | In vitro: BV2 cells (Murine microglial cell line) olfactory cell monolayers (extracted rat olfactory mucosa primary cells) | Enhance brain uptake through via nasal administration route anti-inflammatory effect by reducing pro-inflammatory cytokine expression in the olfactory bulb in LPS-treated mice | 51 |
| In vivo: LPS-induced mice | ||||||
| RMP-7 and lactoferrin grafted liposome | Quercetin | d = 90–170 ξ = 7–35 | AD | In vitro: artificial BBB layer on transwell (HBMECs and HAs) | reduced Aβ-induced neurotoxicity protection of SK-N-MC cells against apoptosis | 50 |
| SK-N-MC (neuroblastoma cells) | ||||||
| SLN-β | β-Elemene | d = 129, ξ = −3.07 | Glioblastoma | In vitro: U87, C6, GL261 glioma cells | SLN-β show similar performance to free β-elemene in vitro, Improved stability of β-elemene in SNL formulation &enhance brain drug accumulation in vivo | 57 |
| In vivo: ICR mice, SD rats, and BALB/c nude mice | ||||||
| (U87M6-Luc cells inoculated into brain BALBlc nude mice to establish brain glioma model) | ||||||
| Curcumin-loaded SLN and NLC | Curcumin | SLNs: d = 205 | AD & CNS | In vitro: mouse fetal fibroblast cells | Enhanced curcumin brain uptake with curcumin-loaded NLCs | 58 |
| NLCs: d = 117 | In vivo: SD rats | |||||
| ApoE-SLNs | Resveratrol (RSV) | d = 168–217, ξ = −13.05 | Neurological-disorders | In vitro: transwell BBB model (hCMEC/D3 cell line) | 1.8-fold higher RSV uptake with ApoE-SLNs compared to non-functionalized ones | 59 |
| Chitosan-SLNs | RVG-9R/BACE1 siRNA complex | d = 358 ± 25, ξ = 10.5 ± 0.6 | AD | In vitro: Caco-2 monolayer model (human epithelial colorectal adenocarcinoma cells) | Enhanced permeability through intracellular nerve pathway (nose-to-brain administration) | 60 |
| LNC | CN03 (cGMP analogues) | d = 72 ± 1 ξ = −11.3 ± 0.8 | Retinal degeneration (intravitreal injections) | Ex vivo: porcine eyeballs retinal explants prepared from wild-type (WT) and retinal degeneration rd1 mouse | Higher LNCs permeation in the ciliary body while poor uptake by the retina | 55 |
| PEG-LNPs | mRNA | d = 150 | Retinal degeneration | In vivo: Ai9 mice, Albino BALB/c, apoE−/− and Mertk−/−, C57BL6 mice | The lowest content of PEG (0.5%) in the NPs reveals highest luciferase activity and expression in the Müller glia, the trabecular meshwork, and the optic nerve head, but without reaching the retina | 56 |
| Polymeric nanoparticles | ||||||
| CS-PEG-BIO-SA/OX26 | Z-DEVD-FMK caspase inhibitor | d = 637 ± 3, ξ = 18.2 ± 4.0 | Cerebral ischemia | In vivo: mice | Crossing of the BBB, inhibition of caspase activity with subsequent neuroprotection | 65 |
| CS | Chrysin | d = 100–120, ξ = 22.4 | AD | In vivo: zebrafish | Reduced amyloid-β aggregate formation Improved neuronal protection, reduction of ROS production | 66 |
| Maintaining of synaptic integrity, improved cognitive abilities | ||||||
| Fucoidan-CS | Curcumin | d = 172, ξ = 25.5 ± 2.4 | Neuroinflammation and neurological disorders | In vitro: BV-2 murine microglial cell line | Improved curcumin accumulation in inflammatory brain lesions and reduce brain inflammation | 67 |
| In vivo: ICR mice | ||||||
| Fucoidan | Vismodegib | d = 80 ± 10 | Medulloblastoma brain tumour | In vivo: mice | Enhanced transendothelial transport by targeting P-selectin on tumor vasculature reduce bone toxicity and drug exposure to healthy brain tissues | 68 |
| CS and CS-N-acetylcysteine | Ocular diseases | In vitro: HCjE cells | CS promotes drug permeability by affecting the tight junctions | 74 | ||
| In vivo: guinea pigs | C-NAC appears to be a good candidate for ocular formulations that must resist for long time on the ocular surface without cellular uptake | |||||
| CS | Cyclosporine A | d = 293 ± 9, ξ = 37.5 ± 0.9 | Dry eye syndrome | In vivo: rabbits | Improved drug delivery to the external ocular tissues with negligible drug exposure to the inner ocular tissues, plasma, and blood | 75 |
| CS-coated ceria NC | Pilocarpine | d = 70–80, ξ = 18–28 | Acute glaucoma | In vitro: rabbit corneal epithelial cell line and bovine corneal endothelium | Reduction of high intraocular pressure | 69 |
| In vivo: rabbit | Prevention of endothelium damage and inhibit retinal degeneration | |||||
| CS-pluronic-NC | ATP | — | Intravitreal injection retinal diseases | In vitro: ARPE-19 (human retinal pigment epithelial cells) | Improved transport and penetration of NC across retina | 70 |
| Ex vivo: fresh porcine eyes | ||||||
| In vivo: BALB/C male mice | ||||||
| Angiopep-2 PE-PEG polymeric micelles | Amphotericin (AmB) | d = 13.78 ± 1.48 (DLS) | Fungal infections of CNS | In vitro: BCECs | Improved AmB solubilization | 76 |
| d = 4.07 (AFM), ξ = 24.74 | In vivo: ICR mice SD-rats | Enhanced BBB permeability | ||||
| Reduced toxicity to mammalian cells | ||||||
| PEG-PLGA polymersome + lactoferrin | S14G-peptide | d = 126 ± 20, ξ = −2.54± 0.05 | AD & CNS | In vivo: Kunming mice, BALB/c mice SD rats | Attenuated expression of Bax and caspase-3 | 77 |
| Increased choline acetyltransferase (ChAT) activity | ||||||
| Inorganic nanostructures | ||||||
| 8D3-AuNP | d = 46.7± 0.1, ξ = −11.2 ± 0.69 | Brain endothelium | In vivo: male ICR-CD1 mice | Receptor-mediated transcytosis (TfR-mediated) and clathrin-dependent internalization | 87 | |
| Most AuNPs remained attached to the abluminal membrane without reaching the brain parenchyma | ||||||
| Au NP | L & D glutathione | d = 3–7, ξ = −42 to −43 | AD | In vitro: human neuroblastoma cell line (SH-SY5Y) | Inhibition of Aβ42 aggregation | 88 |
| In vivo: AD mice | D-Glutathione AuNPs reveals better performance and higher brain biodistribution | |||||
| Ag/SiO2 NP (1%) | d = 10.6 ± 7.1 | Corneal wound | In vitro: hTCEi RCFs | Penetration of all the corneal layer without causing histopathological changes in vivo | 92 | |
| In vivo: rabbit model | No alteration in corneal epithelial wound healing in in vivo rabbit model | |||||
| Reduction of keratocyte-fibroblast-myofibroblast transformation in vitro | ||||||
| TiO2 NPs | d = 42 ± 3 (TEM), d = 184 ± 64 (DLS), ξ = −14.2 ± 7.4 | Retinal degeneration | In vitro: bEnd.3 cell line (an immortalized mouse cerebral vascular endothelial cell) ARPE-19 cell line (human retinal pigment epithelial cells) HRECs | Damages the inner blood–retinal barrier | 95 | |
| In vivo: C57BL/6 mice | Affects the retinal electrophysiology | |||||
| Fe3O4 NP | Transferrin glycoproteins | d = 8–20 | Retinoblastoma and retinal degeneration | In vivo: rat model | Enhanced permeability across the BRB upon applying hyperthermia of MNPs | 96 |
| Carbon-based nanostructures | ||||||
| PEI-N-CQD | — | d = 2.6 | Brain disorders | In vitro: isolated primary rat microvascular endothelial cells and astrocytes | BBB crossing | 103 |
| Traceable drug delivery in brain disorders | ||||||
| Amphiphilic CQD | — | d = 3.4 ± 1.0, ξ = −15.3 | AD | In vitro: pediatric glioblastoma cell lines (SJGBM2, CHLA200) HEK293 Chinese hamster ovary cells | BBD crossing | 100 |
| In vivo: old wild-type zebrafish | Prevention of overexpression of Aβ42 | |||||
| Transferrin-CQD | Human transferrin | — | CNS | In vivo: zebrafish | BBB crossing | 104 |
| GQDs | Glycine-proline-glutamate peptide | d = 18 | AD | In vitro: Aβ1–42 samples | Enhanced delivery of neuroprotective peptides to the CNS | 105 |
| In vivo: APP/PS1 transgenic mice | Inhibition of Aβ1–42 aggregation improved learning and memory abilities | |||||
| Decreased pro-inflammatory cytokines levels, protection of the synapse | ||||||
| Promotion of neurogenesis | ||||||
| Lysine-based C60 | Monomethyl fumarate | d = 137± 2 ξ = −12.2 ± 1.7 | Brain tumor | In vitro: SH-SY5Y neuroblastoma cells of human origin | pH-dependent drug release with maximum release at cancer cell pH, Improved pharmacokinetic of drug | 106 |
| Ex vivo: healthy human blood | Hemo-compatibility | |||||
| CQDs | — | d = 16.8 ± 6.7, ξ = 32.5 ± 0.8 | Vitreous opacities | In vitro: collagen type I from rat tail ARPE-19 spontaneously immortalized cells of human retinal pigment epithelium MIO-M1 Müller progenitor cell line HeLa cell line derived from cervical carcinoma from a 31-year-old female U-87 MG cell line derived from a malignant glioma | Cationic CQDs prevent the fibrillation of type I collagen | 90 |
| Ex vivo: human vitreous containing opacities | Destruction of protein aggregates in human vitreous | |||||
| Spermidine-derived CQDs | — | d = 6 nm, ξ = 45 mV | Bacterial keratitis | In vitro: Bacterial cultures: S. aureus, S. enteritidis, MRSA, E. coli, and P. aeruginosa | Super-cationic CQD induce opening of TJs for paracellular transport across corneal epithelial cells | 108 |
| Cell cultures: rabbit corneal keratocytes (RCKs) and human red blood cells (RBCs) | Antibacterial agent against multi- and non-multi-drug-resistant bacteria | |||||
| In vivo: white rabbits | ||||||
| CQDs-aptamer | VEGF | Ocular vascular disorders | In vitro: human embryonic stem cells, ARPE19, fibroblast cells, Y79 cells | therapeutic levels in intraocular structures achieved | 109 | |
| In vivo: murine animal model (Long-Evans pigmented rats) | Inhibits VEGF-stimulated angiogenesis in choroidal blood vessels | |||||
| Other nanostructures | ||||||
| pEVs | d < 200 nm | Corneal endothelial regeneration | In vitro: CECs | Enhanced wound healing rate | 132 | |
| Improved adhesion rate | ||||||
6. Models providing insight into the transport mechanism
In vivo studies currently provide most comprehensive data sets for a complete understanding of drug penetration, biodistribution and behavior of therapeutics. Animal experiments represent intact organisms necessary to simulate the complex interplay of different processes, which is crucial for studying drug delivery and action. They are widely used despite being expensive, time consuming and often poorly correlating with humans. The existence of interspecies differences is one of the reasons why they often fail as a robust platform for result translation into clinical outcomes. Furthermore, the extensive use of animals in research raises general ethical concerns, which are nowadays addressed by weighing the potential harm to animals against the potential benefit to society. The main goal of 3Rs (replace, reduce and refine) principles is to change traditional animal testing practices so that animal experimentation is kept to a minimum whenever possible. In the European Union, according Directive 63/2010 on the protection of animals used for scientific purposes, it is mandatory to use an available, appropriate alternative method instead of the animal experiment.A number of alternative approaches are indeed available for predicting drug permeability across biological barriers (Fig. 10A). Developments in molecular and cellular biology resulted in powerful cell-based in vitro models to study the behavior of drugs in the context of biological barriers reducing the number of laboratory animals and associated costs. The first models, e.g., those for the BBB focused on reproducing TJs between endothelial cells and were grown on traditional plastic culture dishes. While the 2D in vitro model is a simplified and cost-effective option, the 3D in vitro models provide more complex and realistic representation of the barrier. Further developments incorporating milli-fluidics (hollow-fiber models) or microfluidics enabled the inclusion of the role of constant nutrient supply and the effects of shear stress on barrier properties. Exploiting the potential of microfluidic models and constructing multi-compartment systems together with spheroid or organoid cultivation techniques, novel devices allow the co-culture of different cell types in three dimensions in different compartments linked via microfluidic channels, mimicking living tissue microarchitecture.161–163 In the following, models for the different barriers will be discussed (Table 3).
 | ||
| Fig. 10 From in vivo to ex vivo and in vitro cellular models: (A) from animal models to traditional cell culture systems and their limitations (created with https://BioRender.com). (B) Permeability percentage and apparent permeability coefficient of free fluorescein (black), chitosan NPs (green) and dextran coated chitosan NPs loaded with fluorescein as a model drug using RPMI 2650 cell culture onto transwell insert plates. (C) Use of pig or cow eyes as models for the permeation of fluorescein labelled drugs or nanostructures combined with confocal imaging and staining. | ||
| Barrier | Models |
|---|---|
| Intestinal epithelial barrier (IEB) | Cell types: Caco-2 human colon carcinoma cells, Caco-2/HT29 co-cultures, adult colon stem cell-based models with up to 5 different cell types, hiPSCs-based epithelial cells |
| Model types: transwell, organoids, microfluidic models, organomimetic gut-on-a-chip approach, gut slices | |
| Blood–brain barrier (BBB) | Cell types: monocultures primary, immortalized, tumour or hiPSCs-derived brain microvascular endothelial cells, and co-cultures with mainly primary astrocytes and pericytes, but sometimes also with microglia, neuroprogenitor cells or neurons |
| Model types: transwell, multicellular spheroids, millifluidic hollow-fibre or microfluidic models with channel systems implementing shear stress, more complex multi-tissue/organ chips connecting BBB with neuronal tissue or neurospheres, lung, gut, kidney and/or liver organoids or cell layers. Brain slices, isolated brain capillaries | |
| Blood–cerebrospinal fluid barrier (BCSFB) | Cell types: mainly monolayers of choroid plexus epithelial cells (often immortalized) or hiPSC-derived multicellular complex models |
| Model types: transwell, multicellular organoids, brain slices | |
| Blood–retina barrier (BRB) | Cell types: in vitro using retinal pericytes, retinal astrocytes, and retinal endothelial cells, mainly primary and immortalized cells |
| Model types: transwell, microfluidic models, excised animal tissue such as bovine and porcine eyes | |
| Corneal barriers | Cell types: epithelial cell culture models (CEPI, SIRC and HCE-T cell lines), primary cultures of human corneal epithelium (HCEpiC) commercially available models (RHC and epiocular) |
| Model types: transwell, organotypic human cornea constructs (HCC, HCC-HCE-T), excised animal tissue such as bovine and porcine eyes |
In vitro BBB models
There is a plethora of different in vitro models for the blood–brain barrier that represent the complexity of the BBB in varying detail. Comprehensive descriptions can be found in many reviews.161 In general, for the BBB in vitro model selection process, one can follow the principle “as simple as possible, as complex as necessary”. Depending on the research question, the models can also consist only of brain endothelial cells grown in 2D formats. If one wants to take into account the known influences of shear stress generated by the blood flow164 or the neighbouring cells of the microenvironment (astrocytes or pericytes are most often added, although other cell types of the CNS such as microglia, oligodendrocytes or even neurons can also have an influence), more complex models are required. In order to measure the transport of particles through the brain endothelial layer, models with at least two compartments (apical for the blood side and basolateral for the CNS side) are used. The majority of BBB in vitro models used for drug transport studies are the so-called transwell-based models (Fig. 10A) and can include more than one cell type. This device contains two chambers, apical and basolateral, separated by a porous membrane, where endothelial cells are seeded on the apical side of the membrane, while further cells from the neurovascular unit can be added to the basolateral chamber.141,165 The selection of the transwell model is crucial for the success of the experiments. Especially for nanoparticle or EV studies, the pore size is a crucial parameter. Most plastic membranes for transwell models are available with pore sizes of 0.4, 1, 3 or 8 μm. For larger particles or EVs in the range of 100–200 nm, the pore size of 0.4 μm can be a critical parameter, even though this pore size is preferred because the tightest barriers can be produced with it. Besides the pore size, the material (its lipohphilicity), the porosity and the coating of the plastic also play a role. In addition, the size of the format must be considered, as the smaller the format (e.g., 24-well), the smaller the circumference around which the cells will not form TJs with the plastic, thus resulting in the formation of a tight layer on average. In addition, the surface area is considered when calculating the TEER value, which can also lead to format-dependent values, which can be highly relevant for the comparison of studies in which the TEER is determined as the main parameter for barrier integrity. Another influencing factor is the medium in which the tests are carried out. The simpler the experimental medium (often an aqueous buffer) and the more dissimilar it is to the growth medium, the higher the likelihood that the barrier function will be altered during the experiment. Therefore, appropriate control experiments must be included. The presence of serum or proteins in the medium can also have an immense impact, considering the lipophilicity of some nanoparticles or EVs that may interact either with these proteins or directly also with the plastic of the models.166,167 The choice of the cell source is key to the development of in vitro BBB models. In general, primary cells, immortalized cells or cells from tumors can be used. In addition, since species differences are known at the BBB, the question arises from which species the cells should be used for the models. Access to animal brains (porcine, bovine, rat, mouse, etc.) is much easier compared to human brains. Therefore, there are also a number of protocols to isolate primary cells from animal brains and to build corresponding BBB models in vitro with the brain endothelial cells. In addition to increased ethical concerns, access to human brain endothelial cells is much more difficult; small quantities can be obtained, for example, from tumor or epilepsy surgeries. Since human cells are preferred to avoid possible species differences, but primary cells are a limited resource, immortalised human cell lines are often used (e.g., hCMEC/D3). Although these have significantly weaker barriers and lower TEER values (leakier TJs), they do exhibit BBB relevant transcellular transport mechanisms (SLC-, ABC-transporter, RMT, and AMT). In recent years, the use of human induced pluripotent stem cells to differentiate brain capillary endothelial-like cells became more popular, since these cells can form very tight barriers (in vivo similar) and retain BBB transport properties, although current differentiation protocols should still be improved to achieve endothelial features of a mature BBB.168,169 Because of functional differences due to species (MDCK), organ origin (Caco-2) or endothelial tissue origin (HUVEC), it is highly recommended to apply endothelial cells from the brain microvasculature to mimic the BBB in vitro.Transwell models are of huge importance in this field not only for drug development, but also for a more conceptual understanding of the possibility to bypass and permeate across the paracellular sealings. The general consideration presented above is valid for a wide range of biological barrier models, including nasal, retina or cornea epithelial models. When used for nanoparticle-based permeability studies, fluorescent labelled or fluorescent dye loaded nanostructures are mostly used. Fig. 10B shows the permeability efficiency of FITC-labelled chitosan NPs and fluorescein-loaded chitosan NPs coated with dextran using a widely used cell line employed to study the permeability of drugs for intranasal delivery, the RPMI 2650 cell line isolated from squamous cell carcinoma of a human nasal septum. These cells were seeded and cultured onto transwell insert plates to perform an artificial permeation mimicking the nasal epithelial barrier and the integrity of the formed cell layer was assessed by measuring the transepithelial electrical resistance (TEER) which reached 150 Ω cm2. While fluorescein still diffuses rather freely through this model barrier, chitosan NPs have a significantly decreased permeability, which can however be improved by post-coating with a dextran film.
While transwell-based model can be rather easily installed in any research laboratory, the drawbacks of the model are that it does not incorporate flow, and consequently, endothelial cells are not exposed to shear stress. In this sense, transwell models of the BBB represent a step towards mimicking the human in vivo BBB, although they still display some limitations. Dynamic 3D organ-on-a chip models integrating real-time readouts are more accurate representation of the BBB in healthy and disease states and have evolved in a research field of its own with the advantage that such models offer greater physiological relevance.170
In vitro BRB models
Any model for retinal disease research struggles with reproducing the highly specific adaptations of the human eye and retina. Animal experimentation remains an essential part in the research and development of ocular drugs and delivery systems. The applications of animal experiments include pharmacokinetic and pharmacodynamic studies, and toxicity evaluations (it should be noted that as an alternative to using animals especially for chemical toxicity testing such as for the Draize test, cell culture-based methods have already been accepted by the OECD, see test guideline TG492). With the rabbit being the most commonly used animal model as the small eye size of mice and rats limits their value in ocular studies. However, there are morphological and biochemical differences between the rabbit eye and the human eye. The most obvious disadvantage of rabbits as a animal model for ocular pharmacokinetic studies is their infrequent blinking rate. The low blinking frequency 5/h decreases precorneal drainage of topically applied solutions in comparison to humans with a blinking frequency of 6–7/min. Ocular pharmacokinetics are also influenced by the 1.5- to 2-times larger corneal surface area of rabbits. In addition, the conjunctival surface area of the rabbit is 9 times larger than its corneal surface area. In humans, the difference is 17-fold. As a consequence, the ocular bioavailability of topically applied drugs in the rabbit is less influenced by the non-productive absorption through the conjunctiva. There are many important differences between the human situation and that of almost any other mammalian species – except higher primates – that the idea of investigating a human retinal disease in, for example, a mouse may sound unrealistic.Ex vivo models have large similarity with in vivo models, making them viable alternative approaches, provided that tissue viability and integrity are maintained. Nowadays, the ocular field uses excised animal tissue such as bovine and porcine eyes (Fig. 10C) as the model of choice to study corneal penetration and promising advancements have been reported over the years.26 An ex vivo bovine whole eye was used lately as model for corneal penetration studies of CQDs, in conjunction with confocal microscopy91 (Fig. 6D). This model is indeed a suitable tool for screening corneal penetration of compounds of interest. The drawback of this model is that the quantification of compounds at each layer is based on the fluorescence intensity. In addition, inter-species characteristics must be for sure considered in details when using such models before any conclusion to humans can be drawn.
The use of both primary and secondary cell culture systems for modelling the ocular barriers is described in the literature. Immortalized human cell lines, derived from corneal epithelial, conjunctival epithelial and retinal pigment epithelial cells, are currently available.26,171 The use of retinal microvascular endothelial cells (HRMVEC/ACBRI181, cell systems), retinal pigment epithelium cells (RPE/ARPE-19, ATCC) and Müller glial cells (Moorfield Institute of Ophthalmology-Müller 1, UCL) is common for in vitro studies and considered promising for studying the drug transport into ocular tissues.
7. Perspectives
While significant progress has been made in using nanotechnological approaches to overcome biological barriers, their efficacy remains limited, despite several advantages over more traditional methods (Table 4). The field has primarily evolved through the input of chemistry and biotechnological engineers, focusing on enhancing drug stability via nanoparticle encapsulation, and achieving efficient target-driven drug delivery. While targeting molecules such as transferrin has demonstrated significant benefits for the penetration of BBB cell membranes, unfortunately, the predominant focus has often been on particle formulation, with little or no consideration given to the choice of drugs and reporting of pharmacokinetic profiles. In this regard, the influence of the corona on the particles formed in different artificial liquids, cell culture media or bodily fluids is sometimes an understudied parameter, which can significantly affect the particle's pharmacokinetics. This lack of comprehensive evaluation makes it hard to validate the eventual success of the proposed nanostructures for enhanced drug delivery to organs such as the brain, retina or cornea. Despite achieving 2–5-fold higher drug concentration in the BBB compared to free drugs in most cases, the amounts delivered remain too low for reaching therapeutic effectiveness and applicability. The demonstration of the efficiency of chiral Au NPs in decreasing Aβ42 plaque deposition in the brain in mice, is one of the highly appealing approaches for the treatment of neurodegenerative diseases, in particular Alzheimer disease. The toxic nature of Au NPs will not make clinical developments of this approach possible. If the concepts would be translated however to polymeric- or lipid-based formulations with similar results, this might be one of the formulations of the future. The combination of ultrasound and photothermal vapor nanobubble formation has shown promise for eye floater destruction and increased drug delivery to the brain. However, the viability of these concepts beyond the academic demonstrations is much questionable. Although large-scale devices for clinical applications are under development, ultrasound remains quite unspecific and poses safety risks. Moreover, the effects of ultrasound are very transient, while treatments during chronic diseases often need repeated administration of large amounts of drug.| Method | Advantages | Limitations |
|---|---|---|
| Intracerebroventricular administration (ICV) | High and controlled drug concentration over time in cerebrospinal fluid | Invasive procedure (less risky than deep brain stimulation) |
| High level of control of drug biodistribution in the CNS | Requires neurosurgery expertise | |
| Allow continuous administration and thus opotherapy | ||
| Intracerebral administration (i.e. intra-parenchymal) | High and controlled drug concentration over time | Highly invasive |
| High level of control of drug biodistribution in a very focal brain area | Requires neurosurgery expertise | |
| Intrathecal administration (IT) | High and controlled drug concentration over time in cerebrospinal fluid allowing dorso-lumbar spinal cord distribution | Mildly invasive |
| Relatively focal distribution (lower part of the spinal cord) | ||
| Requires neurosurgery expertise | ||
| Intranasal administration (IN) | Non-invasive | Unpredicted or less predictable drug distribution |
| Brain administration possibility | Only a part of drug reaches the brain | |
| Can be repeated over time for intermittent administration | ||
| Pro-drugs via different administration routes (IN, IV, etc.) | Non-invasive | Requires specific metabolization and thus variability of active product concentration |
| Increases BBB permeation | Higher risk of reaching the targets and thus less efficacy and maybe more side effects | |
| Eye droplets | Non-invasive | Unpredicted drug distribution |
| Treatment on demand | Only a small percentage of drug reaches posterior eye segments | |
| Painless | ||
| Nanoparticles | Can be administrated by different means (ICV, IT, eye droplets, etc.) | Unknown fate of the particles |
| Improved drug stability | Often only 2- to 5-fold increase in concentration administered compared to the free drug | |
| Improved membranes transport | ||
| BBB permeation possibility | Often unknown cytotoxicity | |
| Hydrophobic drugs can be better administered | Limited drug concentration in eye and brain until now | |
| Engineering of release strategies, targeting | Need rigorous and complex pharmacokinetics studies | |
| Surface engineering possibilities to make particles mucoadhesive | ||
| Extracellular vesicles (EVs) | Same advantages as nanoparticles, but being natural compounds | Same limitations as nanoparticles with lack/extreme difficulty to measure them in humans (proper labelling necessary) |
Using drug formulations with NPs and EVs is a highly appealing approach, but it is crucial that manufacturing aspects are carefully designed to make them viable for commercial production. Lipid NPs have indeed overcome this barrier, as evidenced by the marketing of liposomal doxorubicin against various tumours, and mRNA-based vaccines against COVID-19. However, for other nanostructures, this hurdle remains unresolved, necessitating urgent synergetic interactions between academia and commercial partners to advance this field. It will be beneficial to establish standardized criteria for biocompatibility and toxicity to enable meaningful comparisons of nanostructures performance, which is essential for expanding their application in clinical trials.47 While the future of brain- and eye-related diseases remains unclear, physics, chemistry, cell biology, and nanotechnology have emerged as key players in overcoming persistent hurdles and facilitating the development of promising novel therapeutic concepts.
When it comes to EVs, achieving accurate control of the effective dose targeting the desired brain location is challenging. This is especially true due to their potential quick clearance from the nasal cavity or diffusion in the blood circulation upon intravenous administration, thereby possibly limiting their uptake into the brain. Additionally, parameters such as size, surface properties, surface protein expression, and formulation may influence EVs’ absorption and distribution in the brain. However, their physiological origin and particle-type specific somewhat longer circulation times make them valid candidates for further pre-clinical and clinical evaluations in some indications including brain and eye disorders.
In recent years, there has been a shift toward exploring innovative and neuroprotective therapies through IN administration. Recent preclinical evidence has demonstrated that the IN-administration route can achieve drug concentrations close to those provided by invasive intrathecal infusions for therapeutic antibodies. Therefore, the IN route should be considered as a valuable approach for delivering drugs or trophic factors to the brain. Similar considerations apply to crossing the BRB, where uncertainties surrounding the technical capacity to develop nanoformulations that can cross the BRB have stimulated interest in using the IN route as a means of delivering drugs to the retina.172 Retrospectively, these attempts can be traced back to the progressive understanding of the connection between the nasal cavity and the retina with a link found between the IN administration of prednisolone and the occurrence of retinal vein thrombosis173 or the observation that IN administration of corticosteroids induced retinal and choroidal microvascular embolism.174 Furthermore, a series of studies in the 1990s identified sudden retinal manifestations associated with intranasal abuse of cocaine and methamphetamine.175,176 This marked the beginning of exploration into the effects of IN applications on the eye like the IN administration of the secretome of amnion-derived multipotent progenitor cells, rich in growth factors and anti-inflammatory cytokines, found to attenuate visual dysfunction and prevent retinal ganglion cell (RGC) loss177 or IN administration of erythropoietin to rescue photoreceptors in the degenerative retina, highlighting the benefits of this noninvasive and efficient approach172 It can be expected that further studies will evaluate the capacity of various intranasal nanoformulations, including naïve and drug-loaded EVs, to exert beneficial effects on the retina. Improving the efficiency of the IN administration of EVs will hopefully benefit from the experience gained with other drugs or NPs. For instance, clinical trials have explored the IN delivery of insulin in patients with AD and mild cognitive impairment, evidencing some improvements in memory and cognition, but without confirmation of neuroprotective effects.178,179 Also, IN administration of antioxidant glutathione could mitigate oxidative stress in PD preclinical animal models, suggesting a potential value in disease management.180–183 Additionally, in an APPxPS1 mice model of Alzheimer's disease, IN delivery of an NGF mutant was found to prevent neurodegeneration and behavioral deficits.184 Integrating such free drugs into nanotechnological formulations is expected to further contribute to these promising advancements by allowing better control over mucoadhesive character, drug retention time and mucosal clearance, thus optimizing the amount of drug delivered to the brain.
8. Conclusions
The use of drug-loaded nanocarriers has attracted much attention as a potential means for overcoming highly selective and semipermeable barriers, such as the BBB, BRB, and TJs in corneal epithelial and endothelial cells. However, so far, only a handful of nanocarriers have progressed into clinical trials. There are several pharmacokinetic and pharmacodynamic challenges to overcome before successfully implementing this therapeutic concept. One of the pharmacokinetic challenges is ensuring sufficient diffusion of the nanocarrier and, above all, delivering an effective quantity of the drug. The competitive and variable drug penetration of drugs into the brain parenchyma leads to low and unpredictable bio-distribution in the CNS. Thus, rigorous pharmacokinetic studies are needed to advance the clinical implementation of the developed approaches. While there is a strong general interest in understanding the pharmacology of administered nanocarriers and their associated drugs, the issue is complex because repetitive access to the cerebrospinal fluid, which results in a biased measurement of drug concentration in the targeted brain region, is not feasible. In terms of pharmacodynamics, using nanocarriers for neuroprotection remains very challenging due to the lack of validated neuroprotective treatments. While this therapeutic concept seems relevant in preclinical settings, it faces sincere pharmacokinetic issues associated with cerebral administration and complex pharmacodynamic issues of efficacy.The treatment of eye diseases shares similar challenges to brain diseases in the search to move away for injection-based therapies towards easily applicable topical eye droplets for restoring retina- and cornea-based conditions. We believe that CQDs present unique potential for such applications. CQDs have demonstrated extraordinary ability to cross the BBB and cornea, offering new possibilities for treatment. The availability of various carbon sources for their fabrication makes CQDs an interesting approach for recycling carbon materials. However, it is premature to conclude that CQDs will be the future remedy for brain- and eye-related diseases. The field of nanomedicine, specifically therapeutic CQD formulations, is still in its early stages of development and faces a range of issues that must be addressed before wider clinical applications. Ethical considerations, biological concerns, and cost-related issues need to be carefully addressed before more commercially-oriented developments. Ensuring the short-term and long-term safety of these nanocolloids for humans and the environment is crucial and must be established under controlled conditions. Additionally, the potential of utilizing different carbon sources for CQD fabrication adds to their appeal as an approach for recycling carbon materials.
Conflicts of interest
The authors declare that they have no competing financial interest or personal relationships that would have appeared to influence the work reported.Acknowledgements
Financial supports from the Centre National de la Recherche Scientifique (CNRS) and the University of Lille are acknowledged. We want to thank the financial support from EuroNanoMed III (GSkin) and the National Science and Technology Council (NSTC), Taiwan, under grant 112-2923-E-038-001 and 110-2314-B-038 -044 -MY2.References
- L. Wang, N. Wang, W. Zhang, X. Cheng, Z. Yan, G. Shao, X. Wang, R. Wang and C. Fu, Signal Transduction Targeted Ther., 2022, 7, 48 CrossRef CAS PubMed.
- J. L. Lau and M. K. Dunn, Bioorg. Med. Chem., 2018, 26, 2700–2707 CrossRef CAS PubMed.
- R.-M. Lu, Y.-C. Hwang, I.-J. Liu, C.-C. Lee, H.-Z. Tsai, H.-J. Li and H.-C. Wu, J. Biomed. Sci., 2020, 27, 1 CrossRef CAS PubMed.
- Q. Pagneux, N. Garnier, M. Fabregue, S. Sharkaoui, S. Mazzoli, I. Engelmann, R. Boukherroub, M. Strecker, E. Cruz, P. Ducos, A. Zarubica, R. Suderman and S. Szunerits, BioRxiv, 2023 DOI:10.1101/2023.04.03.535401.
- P. Barman, S. Joshi, S. Sheetal, S. Preet, S. Sharma and A. Saini, Int. J. Pept. Res. Ther., 2023, 29, 61 CrossRef CAS PubMed.
- S. Szunerits, S. Melinte, A. Barras, Q. Pagneux, A. Voronova, A. Abderrahmani and R. Boukherroub, Chem. Soc. Rev., 2021, 50, 2102–2146 RSC.
- C. Pellegrini, V. D’Antongiovanni, F. Miraglia, L. B. L. Rota, C. Di Salvo, G. Testa, S. Capsoni, G. Carta, L. Antonioli, A. Cattaneo, C. Blandizzi, E. Colla and M. Fornai, npj Parkinson's Disease, 2022, 8, 9 CrossRef CAS PubMed.
- E. G. Knox, M. R. Aburto, G. Clarke and J. F. C. C. M. O’Driscoll, Mol. Psychiatry, 2022, 27, 2659–2673 CrossRef CAS PubMed.
- A. Monaco, B. Ovryn, J. Axis and J. Amsler, Int. J. Mol. Sci., 2021, 22, 7677 CrossRef CAS PubMed.
- S. Yang, J. Zhou and D. Li, Front. Pharmacol., 2021, 12, 727870 CrossRef CAS PubMed.
- S. K. Lai, Y. Y. Wang and J. Hanes, Adv. Drug Delivery Rev., 2009, 61, 158–171 CrossRef CAS PubMed.
- T. S. Reese and M. J. Karnovsky, J. Cell Biol., 1967, 34, 207–217 CrossRef CAS PubMed.
- W. M. Pardridge, Drug transport across the Blood–brain barrier, J. Cereb. Blood Flow Metab., 2012, 32, 1959–1972 CrossRef CAS PubMed.
- W. M. Pardridge, NeuroRx, 2005, 2, 3–14 CrossRef PubMed.
- J. Brunner, S. Ragupathy and G. Brochared, Adv. Drug Delivery Rev., 2021, 171, 266–288 CrossRef CAS PubMed.
- W. Neuhaus, A. Piontek, J. Protze, M. Eichner, A. Mahringer, E. A. Subileau, I. M. Lee, J. D. Schulzke, G. Krause and J. Piontek, Biomater., 2012, 161, 129–143 CrossRef PubMed.
- D. van der Spoel, S. Manzetti, H. Zhang and A. Klamt, ACS Omega, 2019, 4, 13772–13781 CrossRef CAS PubMed.
- H. Kusuhara and Y. Sugiyama, Drug Discovery Today, 2001, 6, 206–212 CrossRef CAS PubMed.
- A. Catalano, D. Iacopetta, J. Ceramella, D. Scumaci, F. Giuzio, C. Saturnino, S. Aquaro, C. Rosano and M. S. Sinicropi, Molecules, 2022, 27, 616 CrossRef CAS PubMed.
- W. Neuhaus and C. R. Noe, in Transporters as drug carriers, ed. G. F. Ecker and P. Chiba, Whiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2009 Search PubMed.
- E. Palm, Acta Ophthalmol., 1947, 25, 29–35 CrossRef CAS PubMed.
- L. Erb and G. A. Weisman, Rev. Membr. Transp. Signaling, 2012, 1, 789–803 CrossRef CAS PubMed.
- X. Huang and Y. Chau, Drug Discovery Today, 2019, 24, 1–14 CrossRef CAS PubMed.
- P. Cai and X. Chen, ACS Mater. Lett., 2019, 8(1), 285–289 CrossRef.
- M. Thacker, C.-L. Tseng and F.-H. Lin, Polymers, 2021, 13, 1–15 Search PubMed.
- I. De Hoon, R. Boukherroub, S. De Smedt, S. Szunerits and S. Felix, Mol. Pharmaceutics, 2023, 20, 3298–3319 CrossRef CAS PubMed.
- A. C. Penedo, V. D. Tomé, A. F. Ferreiro, M. G. Barcia and F. J. O. Espinar, Eur. J. Pharm. Biopharm., 2021, 162, 12–22 CrossRef PubMed.
- V. Agrahari, P. A. Burnouf, T. Burnouf and V. Agrahari, Adv. Drug Delivery Rev., 2019, 148, 146–180 CrossRef CAS PubMed.
- M. G. Farquhar and G. E. Palade, J. Cell. Biol., 1963, 17, 375–412 CrossRef CAS PubMed.
- https://www.fiercebiotech.com/biotech/roches-anti-amyloid-antibody-gantenerumab-fails-phase-3-alzheimers-trials, acessed 18-06-2023.
- W. M. Pardridge, Pharmaceuticals, 2020, 13, 394 CrossRef CAS PubMed.
- S. Z. Alshawwa, A. A. Kassem, R. M. Farid, S. Khamis Mostafa and a G. S. Labib, Pharmaceutics, 2022, 14, 883 CrossRef CAS PubMed.
- J. A. Loureiro, S. Andrade, A. Duarte, A. R. Neves, J. Queiroz, C. Nunes, E. Sevin, L. Fenart, F. Gosselet, M. A. N. Coelho and M. C. Pereir, Molecules, 2017, 22, 277 CrossRef PubMed.
- T. M. Goppert and R. H. Muller, J. Drug Targeting, 2002, 13, 179–187 CrossRef PubMed.
- H. Koo, H. Moon, H. Han, J. H. Na, M. S. Huh, J. H. Park, S. J. Woo, K. H. Park, I. C. Kwon, K. Kim and H. Kim, Biomaterials, 2012, 33, 3485–3493 CrossRef CAS PubMed.
- H. Hillaireau and P. Couvreur, Cell. Mol. Life Sci., 2009, 66, 2873–2896 CrossRef CAS PubMed.
- M. Nowak, T. D. Brown, A. Graham, M. E. Helgeson and S. Mitragotri, Bioeng. Transl. Med., 2020, 5, e10153 CrossRef CAS PubMed.
- T. D. Brown, N. Habibi, D. Wu, J. Lahann and S. Mitragotri, ACS Biomater. Sci. Eng., 2020, 6, 4916–4928 CrossRef CAS PubMed.
- A. Anselmo and S. Mitragotri, Adv. Drug Delivery Rev., 2017, 108, 51–67 CrossRef CAS PubMed.
- A. Anselmo, M. Zhang, S. Kumar, D. R. Vogus, S. Menegatti, M. E. Helgeson and S. Mitragotri, ACS Nano, 2015, 9, 3169–3177 CrossRef CAS PubMed.
- I. M. Hafez and P. R. Cullis, Adv. Drug Delivery Rev., 2001, 47, 139–148 CrossRef CAS PubMed.
- J. Jouhet, Front. Plant Sci., 2013, 4, 1–5 Search PubMed.
- R. N. Lewis, D. A. Mannock, R. N. McElhaney, D. C. Turner and S. M. Gruner, Biochemistry, 1989, 28, 541–548 CrossRef CAS PubMed.
- P. Khare, S. X. Edgecomb, C. M. Hamadani, E. E. L. TAnner and D. S. Manickam, Adv. Drug Delivery Rev., 2023, 197, 114861 CrossRef CAS PubMed.
- A. C. Correira, A. R. Monteiro, R. Silva, J. N. Moreira, J. M. Sousa Lobo and A. C. Silva, Adv. Drug Delivery Rev., 2022, 189, 114485 CrossRef PubMed.
- A. C. Anselmo and S. Mitragotri, Bioeng. Transl. Med., 2019, 4, e10143 CrossRef PubMed.
- E. D. Namiot, A. V. Sokolov, V. N. Chubarrev, V. V. Tarasov and H. B. Schioth, J. Mol. Sci., 2023, 24, 787 CrossRef CAS PubMed.
- K. B. Johnsen, A. Burkhart, F. Melander, P. Joseph Kempen, J. Bruun Vejlebo, P. Siupka, M. S. Nielsen, T. L. Andresen and T. Moos, Sci. Rep., 2017, 7, 10396 CrossRef PubMed.
- G. R. Topal, M. Mészaros, G. Porkoláb, A. Szecskó, T. F. Polgár, L. Siklós, M. A. Deli, S. Veszelka and A. Bozkir, Pharmaceuticals, 2021, 13, 38 CrossRef CAS PubMed.
- Y.-C. Kuo and C.-W. Tsao, Int. J. Nanomed., 2017, 12, 2857–2869 CrossRef CAS PubMed.
- A. Mwema, P. Bottemanne, A. Paquot, B. Ucakar, K. Vanvarenbdrg, M. Alhouayek, G. G. Muccioli and A. des Rieux, Nanomedicine, 2023, 48, 102633 CrossRef CAS PubMed.
- Y. Zhang, F. Schlachetzki and W. M. Pardridge, Mol. Ther., 2003, 7, 11–18 CrossRef CAS PubMed.
- W. M. Padridges, Trends Mol. Med., 2020, 2, 602236 Search PubMed.
- W. M. Padriges, Trends Mol. Med., 2023, 29, 343 CrossRef PubMed.
- G. Christensesn, D. Urimi, L. Lorenzo-Soler, N. Schipper and F. Paquet-Durand, Eur. J. Pharm. Biopharm., 2023, 187, 175 CrossRef PubMed.
- R. C. Ryals, S. Patel, C. Acosta, M. McKinney, M. E. Pennesi and G. Sahay, PLoS One, 2020, 15, e0241006 CrossRef CAS PubMed.
- H. He, J. Yao, Y. Zhang, Y. Chen, K. Wang, R. J. Lee, B. Yu and X. Zhang, Biochem. Biophys. Res. Commun., 2019, 519, 385–390 CrossRef CAS PubMed.
- S. Sadegh Malvajerd, A. Azadi, Z. Izadi, M. Kurd, T. Dara, M. Dibaei, M. Sharif Zadeh, H. Akbari Javar and M. Hamidi, ACS Chem. Neurosci., 2019, 10, 728–739 CrossRef CAS PubMed.
- A. R. Neves, J. F. Queiroz and S. Reis, J. Nanobiotechnol., 2016, 14, 27 CrossRef PubMed.
- G. Rassu, E. Soddu, A. M. Posadino, G. Pintus, B. Sarmento, P. Giunchedi and E. Gavini, Colloids Surf., B, 2017, 152, 296 CrossRef CAS PubMed.
- A. Zamboulis, S. Nanaki, G. Michailidou, I. Koumentakou, M. Lazaridou, N. M. Ainali, E. Xanthopoulou and D. N. Bikiaris, Polymers, 2020, 12, 1–67 CrossRef PubMed.
- K. M. El-Say and H. S. El-Sawy, Int. J. Pharm., 2017, 528, 675–691 CrossRef CAS PubMed.
- H. Cortés, S. Alcalá-Alcalá, I. H. C. aballero-Florán, S. A. Bernal-Chávez, A. Ávalos-Fuentes, M. González-Torres, M. González-Del Carmen, G. Figueroa-González, O. D. Reyes-Hernández, B. Floran, M. L. Del Prado-Audelo and G. Leyva-Gómez, Membranes, 2020, 2, 212 CrossRef PubMed.
- L. Zhang, J. Fan, G. Li, Z. Yin and B. M. Fu, Cardiovasc. Eng. Technol., 2020, 11, 607–620 CrossRef PubMed.
- Y. Aktaş, M. Yemisci, K. Andrieux, R. N. Gürsoy, M. J. Alonso, E. Fernandez-Megia, R. Novoa-Carballal, E. Quiñoá, R. Riguera, M. F. Sargon, H. H. Çelik, A. S. Demir, A. A. Hıncal, T. Dalkara, Y. Çapan and P. Couvreur, Bioconjugate Chem., 2005, 16, 1503–1511 CrossRef PubMed.
- S. Saleem, B. R. Rachana and R. R. Kannan, ACS Chem. Neurosci., 2022, 13, 2017–2034 CrossRef CAS PubMed.
- T.-M. Don, W.-J. Chang, P.-R. Jheng, Y.-C. Huang and E.-Y. Chuang, Int. J. Biol. Macromol., 2021, 181, 835–846 CrossRef CAS PubMed.
- D. E. Tylawsky, H. Kiguchi, J. Vaynshteyn, J. Gerwin, T. I. Janki Shah, Jacob A. Boyer, D. R. Boué, M. Snuderl, Matthew B. Greenblatt, Y. Shamay, G. Praveen Raju and D. A. Heller, Nat. Mater., 2023, 22, 391–399 CrossRef CAS PubMed.
- D. D. Nguyen, C.-H. Yao, S. J. Lue, C.-J. Yang, Y.-H. Su, C.-C. Huang and J.-Y. Lai, Chem. Eng. J., 2023, 451, 138620 CrossRef CAS.
- K. Kwon, Y. Hwang, J. Jung and G. Tae, Pharmaceutics, 2021, 13, 463 CrossRef CAS PubMed.
- Y. Zhou, F. Zhu, Y. Liu, M. Zheng, Y. Wang, D. Zhang, Y. Anraku, Y. Zou, J. Li, H. Wu, X. Pang, W. Tao, O. Shimoni, A. I. Bush, X. Xue and B. Shi, Sci. Adv., 2020, 6, eabc7031 CrossRef CAS PubMed.
- J. Smith, E. Wood and M. Dornish, Pharm. Res., 2004, 21, 43–49 CrossRef CAS PubMed.
- C. L. Tseng, K. H. Chen, W. Y. Su, Y. H. Lee, C. C. Wu and F. H. Lin, J. Nanomater., 2013, 2013, 1–11 Search PubMed.
- N. Schuerer, E. Stein, A. Inic-Kanada, E. Ghasemian, M. Stojanovic, J. Montanaro, N. Bintner, C. Hohenadl, R. Sachsenhofer and T. Barisani-Asenbauer, J. EuCornea, 2018, 1, 12–18 CrossRef.
- A. M. D. Campos, A. Sánchez and M. J. Alonso, Int. J. Pharm., 2001, 224, 159–168 CrossRef PubMed.
- K. Shao, R. Huang, J. Li, L. Han, L. Ye, J. Lou and C. Jiang, J. Controlled Release, 2010, 147, 118–126 CrossRef CAS PubMed.
- Y. Yu, Z. Pang, W. Lu, Q. Yin, H. Gao and X. Jiang, Pharm. Res., 2012, 29, 83–96 CrossRef CAS PubMed.
- S. Marrache and S. Dhar, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9445–9450 CrossRef CAS PubMed.
- A. Joseph, G. M. Simo, T. Gao, N. Alhindi, N. Xu, D. J. Graham, L. J. Gamble and E. Nance, Biomaterials, 2021, 277, 121086 CrossRef CAS PubMed.
- J. Kreuter, D. Shamenkov, V. Petrov, P. Ramge, K. Cychutek, C. Koch-Brandt and R. Alyautdin, J. Drug Targeting, 2002, 10, 317–325 CrossRef CAS PubMed.
- J. Kreuter, J. Microencapsul., 2013, 30, 49–54 CrossRef CAS PubMed.
- C.-H. Tsai, P.-Y. Wang, I.-C. Lin, H. Huang, G.-S. Liu and C.-L. Tseng, Int. J. Mol. Sci., 2018, 19, 2830 CrossRef PubMed.
- H.-N. P. Voigt, N. Kockentiedt, S. Hintz, W. Tomas and J. Sabel BA, Eur. J. Pharm. Biopharm., 2014, 87, 19–29 CrossRef PubMed.
- Q. You, T. Hopf, W. Hintz, S. Rannabauer, N. Voigt, v Wachem, P. Henrich-Noack and B. A. Sabel, J. Drug Targeting, 2019, 27, 338–346 CrossRef CAS PubMed.
- Q. You, M. Sokolov, L. Grigartzik, W. Hintz, B. G. M. v Wachem, P. Henrich-Noack and B. A. Sabel, Mol. Pharmaceutics, 2019, 16, 5068–5075 CrossRef CAS PubMed.
- H. Hillaireau and P. Couvreur, Cell. Mol. Life Sci., 2009, 66, 2873–2896 CrossRef CAS PubMed.
- I. Cabezón, G. Manich, R. Martín-Venegas, A. Camins, C. Pelegrí and J. Vilaplana, Mol. Pharmaceutics, 2015, 12, 4137–4145 CrossRef PubMed.
- K. Hou, J. Zhao, H. Wang, B. Li, K. Li, X. Shi, K. Wan, J. Ai, J. Lv, D. Wang, Q. Huang, H. Wang, Q. Cao, S. Liu and Z. Tang, Nat. Commun., 2020, 11, 4790 CrossRef CAS PubMed.
- K. J. Mintz, G. Mercado, Y. Zhou, Y. Ji, S. D. Hettiarachchi, P. Y. Liyanage, R. R. Pandey, C. C. Chusuei, J. Dallman and R. M. Leblanc, Colloids Surf., B, 2019, 176, 488–493 CrossRef CAS PubMed.
- A. Barras, F. Sauvage, I. de Hoon, K. Braeckmans, D. Hua, G. Buvat, J. C. Fraire, C. Lethien, J. Sebag, M. Harrington, A. Abderrahmani, R. Boukherroub, S. De Smedt and S. Szunerits, Nanoscale Horiz., 2021, 6, 449–461 RSC.
- I. deHoon, A. Barras, T. Swebocki, B. Vanmeerhaeghe, B. Bogaert, C. Muntean, A. Abderrahmani, R. Boukherroub, S. De Smedt, F. Sauvage and S. Szunerits, ACS Appl. Mater. Interfaces, 2023, 15, 3 CrossRef PubMed.
- S. Kim, B. L. Gates, M. Chang, K. E. Pinkerton, L. V. Winkle, C. J. Murphy, B. C. Leonard, P. Demokritou and S. M. Thomasy, NanoImpact, 2021, 24, 1–10 CrossRef PubMed.
- N. P. Mortensen, M. M. Caffaro, P. R. Patel, M. J. Uddin, S. Aravamudhan, S. J. Sumner and T. R. Fennell, NanoImpact, 2020, 17, 100212 CrossRef PubMed.
- M. Mohammadpour, H. Hashemi, M. Jabbarvand and E. Delrish, Cornea, 2014, 33, 738–743 CrossRef PubMed.
- Y.-J. Chan, P.-L. Liao, C.-H. Tsai, Y.-W. Cheng, F.-L. Lin, J.-D. Ho, C.-Y. Chen and C.-H. Li, Part. Fibre Toxicol., 2021, 18, 1–6 CrossRef PubMed.
- S. N. Tabatabaei, M. S. Tabatabaei, H. Girouard and S. Martel, Int. J. Hyperthermia, 2016, 32, 657–665 CrossRef CAS PubMed.
- K. Nekoueian, M. Amiri, M. Sillanpää, F. Marken, R. Boukherroub and S. Szunerits, Chem. Soc. Rev., 2019, 48, 4281–4316 RSC.
- Y. Zhou, K. J. Mintz, S. K. Sharma and R. M. Leblanc, Langmuirs, 2019, 35, 9115–9132 CrossRef CAS PubMed.
- P. Liyanage, Y. Zhou, A. Alyoubi, A. Bashammakh, M. El-Shahawi, R. M. Graham and R. Leblanc, Nanoscale, 2020, 12, 7927 RSC.
- Y. Zhou, P. Y. Liyanage, D. Devadoss, L. R. Rios Guevara, L. Ling Cheng, R. M. Graham, H. S. Chand, A. O. Al-Youbi, A. S. Bashammakh, M. El-Shahawi and R. M. Leblanc, Nanoscale, 2019, 11, 22387 RSC.
- E. S. Seven, Y. B. Sven, Y. Zhou, S. Poudel-Sharla, J. J. Diaz-Rucco, E. K. Cilingir, G. S. Mitchell, J. D. Van Dyken and R. M. Leblanc, Nanoscale Adv., 2021, 3, 3942–3953 RSC.
- E. S. Seven, Y. Zhou, Y. B. Seven, G. S. Mitchell and R. M. Leblanc, FASEB J., 2019, 33, 785.788–785.788 Search PubMed.
- S. Lu, S. Guo, P. Xu, X. Li, Y. Zhao, G. Gu and M. Xue, Int. J. Nanomed., 2016, 11, 6325–6336 CrossRef CAS PubMed.
- S. Li, Z. Peng, J. Dallman, J. Baker, A. O. Othman, P. L. Blackwelder and R. M. Leblanc, Colloids Surf., B, 2016, 145, 251 CrossRef CAS PubMed.
- S. Xiao, D. Zhou, P. Luan, B. Gu, L. Feng, S. Fan, W. Liao, W. Fang, L. Yang, E. Tao, R. Guo and J. Liu, Biomaterials, 2016, 106, 98–110 CrossRef CAS PubMed.
- M. Kumar, G. Sharma, R. Kumar, B. Singh, O. P. Katare and K. Raza, ACS Biomater. Sci. Eng., 2018, 4, 2134–2142 CrossRef CAS PubMed.
- E. B. Souto, J. Dias-Ferreira, A. López-Machado, M. Ettcheto, A. Cano, A. Camins Espuny, M. Espina, M. L. Luisa Garcia and E. Sánchez-López, Pharmaceutics, 2019, 11, 460 CrossRef CAS PubMed.
- H.-J. Jian, R.-S. Wu, T. Y. Lin, Y.-J. Li, H. J. Lin, S. G. Harroun, J.-Y. Lai and C. C. Huang, ACS Nano, 2017, 11, 6703–6716 CrossRef CAS PubMed.
- A. Shoval, A. Markus, Z. Zhou, X. Liu, R. Cazelles, I. Willner and Y. Mandel, Small, 2019, 15, 1902776 CrossRef PubMed.
- L. Ayers, R. Pink, D. R. F. Carter and R. Nieuwland, J. Extracell. Vesicles, 2019, 8, 1593755 CrossRef CAS PubMed.
- V. V. T. Nguyen, K. W. Witwer, M. C. Verhaar, D. Strunk and B. W. M. van Balkom, J. Extracell. Vesicles, 2020, 10, e12033 CrossRef PubMed.
- A. G. Yates, R. C. Pink, U. Erdbrugger, P. R. Siljander, E. R. Dellar, P. Pantazi, N. Akbar, W. R. Cooke, M. Vatish, E. Dias-Neto, D. C. Anthony and Y. Couch, J. Extracell. Vesicles, 2022, 11, e1215 Search PubMed.
- A. G. Yates, R. C. Pink, U. Erdbrugger, P. R. Siljander, E. R. Dellar, P. Pantazi, N. Akbar, W. R. Cooke, M. Vatish, E. Dias-Neto, D. C. Anthony and Y. Couch, J. Extracell. Vesicles, 2022, 11, e12190 CrossRef CAS PubMed.
- G. Raposo and P. D. Stahl, Nat. Rev. Mol. Cell Biol., 2019, 20, 509–510 CrossRef CAS PubMed.
- G. Raposo and W. Stoorvogel, J. Cell Biol., 2013, 200, 373–383 CrossRef CAS PubMed.
- M. Tkach and C. Théry, Cell, 2016, 164, 1226–1232 CrossRef CAS PubMed.
- S. E. L. Andaloussi, I. Mäger, X. Breakefield and M. J. A. Wood, Nat. Rev. Drug Discovery, 2013, 12, 347–357 CrossRef PubMed.
- V. Agrahari, V. Agrahari, P. A. Burnouf, C. H. Chew and T. Burnouf, Trends Biotechnol., 2019, 37, 707–729 CrossRef CAS PubMed.
- J. Johnson, Y. W. Wu, C. Blyth, G. Lichtfuss, H. Goubran and T. Burnouf, Trends Biotechnol., 2021, 39, 598–612 CrossRef CAS PubMed.
- V. Gratpain, A. Mwema, Y. Labrak, G. G. Muccioli, V. van Pesch and A. des Rieux, Adv. Drug Delivery Rev., 2021, 174, 535–552 CrossRef CAS PubMed.
- S. Herman, I. Fishel and D. Offen, Stem Cells, 2021, 39, 1589–1600 CrossRef PubMed.
- R. L. Webb, E. E. Kaiser, S. L. Scoville, T. A. Thompson, S. Fatima, C. Pandya, K. Sriram, R. L. Swetenburg, K. Vaibhav and A. S. Arbab, Transl. Stroke Res., 2018, 9, 530–539 CrossRef CAS PubMed.
- T. R. Doeppner, J. Herz, A. Görgens, J. Schlechter, A.-K. Ludwig, S. Radtke, K. de Miroschedji, P. A. Horn, B. Giebel and D. M. Hermann, Stem Cells Transl. Med., 2015, 4, 1131–1143 CrossRef CAS PubMed.
- D. R. Ophelders, T. G. Wolfs, R. K. Jellema, A. Zwanenburg, P. Andriessen, T. Delhaas, A.-K. Ludwig, S. Radtke, V. Peters and L. Janssen, Stem Cells Transl. Med., 2016, 5, 754–763 CrossRef CAS PubMed.
- K. Drommelschmidt, M. Serdar, I. Bendix, J. Herz, F. Bertling, S. Prager, M. Keller, A.-K. Ludwig, V. Duhan and S. Radtke, Brain, Behav., Immun., 2017, 60, 220–232 CrossRef CAS PubMed.
- Y. Cai, M.-M. Zhang, M. Wang, Z.-H. Jiang and Z.-G. Tan, J. Neuropathol. Exp. Neurol., 2022, 81, 522–534 CrossRef CAS PubMed.
- A. Nyam-Erdene, O. Nebie, L. Delila, L. Buee, D. Devos, S. Y. Chou, D. Blum and T. Burnouf, ACS Biomater. Sci. Eng., 2021, 7, 5823–5835 CrossRef CAS PubMed.
- O. Nebie, L. Buee, D. Blum and T. Burnouf, Cell. Mol. Life Sci., 2022, 79, 379 CrossRef CAS PubMed.
- O. Nebie, K. Carvalho, L. Barro, L. Delila, E. Faivre, T. Y. Renn, M. L. Chou, Y. W. Wu, A. Nyam-Erdene, S. Y. Chou, L. Buee, C. J. Hu, C. W. Peng, D. Devos, D. Blum and T. Burnouf, Brain, 2021, 144, 3142–3158 CrossRef PubMed.
- W. A. Banks, P. Sharma, K. M. Bullock, K. M. Hansen, N. Ludwig and T. L. Whiteside, Int. J. Mol. Sci., 2020, 21, 4407 CrossRef CAS PubMed.
- A. Chatterjee and R. Singh, Front Cell Dev. Biol., 2023, 11, 1059141 CrossRef PubMed.
- R. Widyaningrum, Y.-W. Wu, L. Delila, D.-Y. Lee, T.-J. wang and T. Burnouf, Platelets, 2022, 33, 1237–1250 CrossRef CAS PubMed.
- R. Widyaningrum, T. Burnouf, O. Nebie, L. Delila and T. J. Wang, Biomed. Pharmacother., 2021, 142, 112046 CrossRef CAS PubMed.
- A. M. Mehta, A. M. Sonabend and J. N. Bruce, Neurotherapeutics, 2017, 14, 358–371 CrossRef CAS PubMed.
- A. J. J. Atkinson, Transl. Clin. Pharmacol., 2017, 25, 117–124 CrossRef PubMed.
- G. Paul, O. Zachrisson, A. Varrone, P. Almqvist, M. Jerling, G. Lind, S. Rehncrona, B. Linderoth, H. Bjartmarz, L. L. Shafer, R. Coffey, M. Svensson, K. Jansson Mercer, A. Forsberg, C. Halldin, P. Svenningsson, H. Widner, J. Frisén, S. Pålhagen and A. Haegerstrand, J. Clin. Invest., 2015, 125, 1339–1346 CrossRef PubMed.
- C. Moreau, A.-S. Rolland, E. Pioli, Q. Qin Li, P. Odou, C. Barthelemy, D. Lannoy, A. Demailly, N. Cart, V. Deramecourt, F. Auger, G. Kuchcinski, C. Laloux, L. Defebvre, R. Bordet, J. Duce, J. C. Devedjian, E. Bezard, M. Fisichella and D. Devos, Neurobiol. Dis., 2020, 139, 104846 CrossRef CAS PubMed.
- C. Laloux, F. Gouel, C. Lachaud, K. Timmerman, B. Do Van, A. Jonneaux, M. Petrault, G. Garcon, N. Rouaix, C. Moreau, R. Bordet, J. A. Duce, J. C. Devedjian and D. Devos, Neurobiol. Dis., 2017, 103, 24–31 CrossRef CAS PubMed.
- N. Zhang, F. Yan, X. Liang, M. Wu, Y. Shen, M. Chen, Y. Xu, G. Zou, P. Jiang, C. Tang, H. Zheng and Z. Dai, Theranostics, 2018, 8, 2264–2277 CrossRef CAS PubMed.
- F. Sauvage, J. C. Fraire, K. Remaut, J. Sebag, K. Peynshaert, M. Harrington, F. Van de Velde, R. Xiong, M.-J. Tassignon, T. Brans, K. Braeckmans and S. De Smedt, ACS Nano, 2019, 13, 8401–8416 CrossRef CAS PubMed.
- N. L. Stone, T. J. England and S. E. O’Sullivan, Front. Cell. Neurosci., 2019, 13, 230 CrossRef CAS PubMed.
- B. Pavan, A. Dalpiaz, N. Ciliberti, C. Biondi, S. Manfredini and S. Vertuani, Molecules, 2008, 13, 1035–1365 CrossRef CAS PubMed.
- G. Botti, A. Dalpiaz and B. Pavan, Pharmaceutics, 2021, 13, 1144 CrossRef CAS PubMed.
- M. Marco Herrera-Barrera, R. C. Ryals, M. Gautam, J. Antony, M. Landry, T. Korzun, M. Gupta, C. Acosta, J. Stoddard, R. Reynaga, W. Tschetter, N. Jacomino, O. Taratula, C. Sun, A. K. Lauer, M. Neuringer and G. Sahay, Sci. Adv., 2023, 9, eadd4623 CrossRef PubMed.
- C. Hoffmann, S. Evcüman, F. Neumaier, B. D. Zlatopolskiy, S. Humpert, D. Bier, M. Holschbach, A. Schulze, H. Endepols and B. Neumaier, ACS Chem. Neurosci., 2021, 12, 3335–3346 CrossRef CAS PubMed.
- A. Gaudin, M. Yemisci, H. Eroglu, S. Lepetre-Mouelhi, O. Faruk Turkoglu, B. Dönmez-Demir, S. Caban, M. F. Sargon, S. Garcia-Argote, G. Pieters, O. Loreau, B. Rousseau, O. Tagit, N. Hildebrandt, Y. Le Dantec, J. Mougin, S. Valetti, H. Chacun, V. Nicolas, D. Desmaële, K. Andrieux, Y. Capan, T. Dalkara and P. Couvreur, Nat. Nanotechnol., 2014, 9, 1054–1062 CrossRef CAS PubMed.
- A. Burgess, K. Shah, O. Hough and K. Hynynen, Exp. Rev. Neurother., 2015, 15, 477–491 CrossRef CAS PubMed.
- S. Ohta, E. Kikuchi, A. Ishijima, T. Azuma, I. Sakuma and T. Ito, Sci. Rep., 2020, 10, 18220 CrossRef CAS PubMed.
- N. Dhas and T. Mehta, J. Drug Delivery Sci. Technol., 2021, 61, 102242 CrossRef CAS.
- K. Pyrć, A. Milewska, E. B. Duran, P. Botwina, A. Dabrowska, M. Jedrysik, M. Benedyk, R. Lopes, A. Arenas-Pinto, M. Badr, R. Mellor, T. L. Kalber, D. Fernandez-Reyes, A. G. Schätzlein and I. F. Uchegbu, Sci. Rep., 2021, 11, 20012 CrossRef PubMed.
- I. Schlachet and A. Sosnik, ACS Appl. Mater. Interfaces, 2019, 11, 21360–21371 CrossRef CAS PubMed.
- E. Gallardo-Toledo, A. Tapia-Arellano, F. Celis, T. Sinai, M. Campos, M. J. Kogan and A. C. Sintov, Int. J. Pharm., 2020, 119957 CrossRef CAS PubMed.
- B. Gabold, F. Adams, S. Brameyer, K. Jung, C. L. Ried, T. Merdan and O. M. Merkel, Drug Delivery Transl. Res., 2023, 13, 822–838 CrossRef CAS PubMed.
- M. L. Formica, D. A. Real, M. L. Picchio, E. Catlin, R. F. Donnelly and A. J. Paredes, App. Mater. Today, 2022, 29, 101631 CrossRef.
- B. Wilson, B. N. Mohamed Alobaid, K. M. Geetha and J. L. Jenita, J. Drug Delivery Sci. Technol., 2021, 61, 102176 CrossRef CAS.
- S. Herman, I. Fishel and D. Offen, Stem Cells, 2021, 39, 1589–1600 CrossRef PubMed.
- M. Losurdo, M. Pedrazzoli, C. D’Agostino, C. A. Elia, F. Massenzio, E. Lonati, M. Mauri, L. Rizzi, L. Molteni and E. Bresciani, Stem Cells Transl. Med., 2020, 9, 1068–1084 CrossRef CAS PubMed.
- X. Zhou, X. Deng, M. Liu, M. He, W. Long, Z. Xu, K. Zhang, T. Liu, K.-F. So and Q.-L. Fu, J. Controlled Release, 2023, 357, 1–19 CrossRef CAS PubMed.
- Y. Guo, D. Hu, L. Lian, L. Zhao, M. Li, H. Bao and S. Xu, Stem Cell Rev. Rep., 2023, 19, 285–308 CrossRef PubMed.
- R. Wang, X. Wang, Y. Zhang, H. Zhao, J. Cui, J. Li and L. Di, J. Controlled Release, 2022, 341, 844–868 CrossRef CAS PubMed.
- W. Neuhaus, Handb. Exp. Pharmacol., 2021, 265, 75–110 CAS.
- C. Eilenberger, M. Rothbauer, F. Selinger, A. Gerhartl, C. Jordan, M. Harasek, B. Schädl, J. Grillari, J. Weghuber, W. Neuhaus, S. Küpcü and P. Ertl, Adv. Sci., 2021, 8, e2004856 CrossRef PubMed.
- L. Koenig, A. P. Ramme, D. Faust, M. Mayer, T. Flötke, A. Gerhartl, A. Brachner, W. Neuhaus, A. Appelt-Menzel, M. Metzger, U. Marx and E. M. Dehne, Cells, 2022, 11, 3295 CrossRef CAS PubMed.
- N. Choublier, M. Taghi, M. C. Menet, M. Le Gall, J. Bruce, P. Chafey, F. Guillonneau, A. Moreau, C. Denizot, Y. Parmentier, S. Nakib, D. Borderie, H. Bouzinba-Segard, P.-O. Couraud, S. Bourdoulous and X. Declèves, Fluids Barries CNS, 2022, 19, 41 CrossRef CAS PubMed.
- J. J. Jamieson, P. C. Searson and S. Gerecht, J. Biol. Eng., 2017, 11, 37 CrossRef PubMed.
- A. R. Santa-Maria, M. Heymans, F. R. Walter, M. Culot, F. Gosselet, M. A. Deli and W. Neuhaus, Handb. Exp. Pharmacol., 2022, 273, 187–204 CAS.
- J. P. Vigh, A. Kincses, B. Ozgür, F. R. Walter, A. R. Santa-Maria, S. Valkai, M. Vastag, W. Neuhaus, B. Brodin, A. Dér and M. A. Deli, Micromachines, 2021, 12, 685 CrossRef PubMed.
- A. Appelt-Menzel, S. Oerter, S. Mathew, U. Haferkamp, C. Hartmann, M. Jung, W. Neuhaus and O. C. Pless, Curr. Protoc. Stem Cell Biol., 2020, 1, e122 CrossRef PubMed.
- A. Appelt-Menzel, A. Cubukova, K. Günther, F. Edenhofer, J. Piontek, G. Krause, T. Stüber, H. Walles, W. Neuhaus and M. Metzger, Stem Cell Rep., 2017, 8, 894–906 CrossRef CAS PubMed.
- S. I. Ahn, Y. J. Sei, H. J. Park, J. Kim, Y. Ryu, J. J. Choi, H. J. Sung, T. J. MacDonald, A. I. Levey and Y. T. Kim, Nat. Commun., 2020, 11, 175 CrossRef CAS PubMed.
- M. Hornof, E. Toropainen and A. Urtti, Biopharmaceutics, 2005, 60, 207–225 CAS.
- Y. Tao, C. Li, A. Yao, Y. Qu, L. Qin, Z. Xiong, J. Zhang and W. Wang, Drug Delivery, 2019, 26, 78–88 CrossRef CAS PubMed.
- R. Catane and L. Wislicki, Harefuah, 1974, 87, 511–512 CAS.
- D. W. Whiteman, D. A. Rosen and R. M. Pinkerton, Am. J. Ophthalmol., 1980, 89, 851–853 CrossRef CAS PubMed.
- R. T. Wallace, G. C. Brown, W. Benson and A. Sivalingham, Am. J. Ophthalmol., 1992, 114, 158–160 CrossRef CAS PubMed.
- J. H. Zeiter, D. M. Corder, M. P. Madion and J. G. McHenry, Am. J. Ophthalmol., 1992, 114, 780–781 CrossRef CAS PubMed.
- R. S. Khan, K. Dine, H. Wessel, L. Brown and K. S. Shindler, J. Neuroophthalmol., 2019, 39, 191–199 CrossRef PubMed.
- S. Craft, L. D. Baker, T. J. Montine, S. Minoshima, G. S. Watson, A. Claxton, M. Arbuckle, M. Callaghan, E. Tsai, S. R. Plymate, P. S. Green, J. Leverenz, D. Cross and B. Gerton, Arch. Neurol., 2012, 69, 29–38 CrossRef PubMed.
- S. M. de la Monte, Aging Health, 2012, 8, 61–64 CrossRef CAS PubMed.
- L. K. Mischley, K. E. Conley, E. G. Shankland, T. J. Kavanagh, M. E. Rosenfeld, J. E. Duda, C. C. White, T. K. Wilbur, P. U. De La Torre and J. M. Padowski, NPJ Parkinsons Dis., 2016, 2, 16002 CrossRef CAS PubMed.
- L. K. Mischley, R. C. Lau, E. G. Shankland, T. K. Wilbur and J. M. Padowski, J. Parkinson's Dis., 2017, 7, 289–299 CAS.
- L. K. Mischley, J. B. Leverenz, R. C. Lau, N. L. Polissar, M. B. Neradilek, A. Samii and L. J. Standish, Mov. Disord, 2015, 30, 1696–1701 CrossRef CAS PubMed.
- L. K. Mischley, M. F. Vespignani and J. S. Finnell, J. Altern. Complement. Med., 2013, 19, 459–463 CrossRef PubMed.
- S. Capsoni, S. Marinelli, M. Ceci, D. Vignone, G. Amato, F. Malerba, F. Paoletti, G. Meli, A. Viegi, F. Pavone and A. Cattaneo, PLoS One, 2012, 7, e37555 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |