
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence“Turn-on” and pinhole-free ultrathin core–shell Au@SiO2 nanoparticle-based metal-enhanced fluorescent (MEF) chemodosimeter for Hg2+†
Ying
Cui‡
ab,
Shanji
Fan‡
c,
Yunran
Zhai
a,
Yingjie
Liu
c,
Junhua
Li
b,
Jiawen
Hu
*a and
Lijia
Wang
 *d
*d
aHunan Key Laboratory of Two-Dimensional Materials, Advanced Catalytic Engineering Research Center of the Ministry of Education, College of Chemistry and Chemical Engineering, Hunan University, Changsha 410082, China. E-mail: jwhu@hnu.edu.cn
bKey Laboratory of Functional Metal-Organic Compounds of Hunan Province, College of Chemistry and Materials Science, Hengyang Normal University, Hengyang, 421001, PR China
cDepartment of Breast and Thyroid Surgery, The First Affiliated Hospital, Hengyang Medical School, University of South China, Hengyang 421000, China
dChildren's Hospital, Zhejiang University School of Medicine, National Clinical Research Center for Child Health, National Children's Regional Medical Center, Hangzhou, Zhejiang 310052, China. E-mail: wanglijia@zju.edu.cn
First published on 2nd April 2024
Abstract
This study reports a metal-enhanced fluorescence chemodosimeter for highly sensitive detection of Hg2+ ions. Silica-coated Au nanoparticles (Au@SiO2 NPs) with a pinhole-free 4–5 nm shell were synthesized and functionalized with a monolayer of turn-on fluorescent probes. Compared to other organic fluorescent probes suffering from poor biocompatibility and detection limits, this design of a monolayer of turn-on fluorescent probes immobilized on the Au@SiO2 NPs with a pinhole-free 4–5 nm shell avoids fluorescence quenching and allows the fluorescent probe within the field of the inner Au NPs to experience metal-enhanced fluorescence. With this design, the chemodosimeter permits fluorescence emission in the presence of Hg2+ ions, because they trigger the ring-opening reaction of the fluorescent probe immobilized on the Au@SiO2 NPs. Additionally, the fluorescent probe is distanced by the thin SiO2 shell from directly attaching to the metallic Au NPs, which not only avoids fluorescence quenching but allows the fluorescent probe within the long-ranged field of the inner Au NPs to experience metal-enhanced fluorescence. As a result, the detection limit for the chemodosimeter can reach up to 5.0 × 10−11 M, nearly two orders of magnitude higher than that achieved for the free fluorescent probe. We also demonstrate the acquisition of images of Hg2+ in HTC116 cells and zebrafish using a simple fluorescence confocal imaging technique. The fluorescence response results for HTC116 cells and zebrafish show that the probes can permeate into cells and organisms. Considering the availability of the many organic fluorescent probes that have been designed, the current designed metal-enhanced fluorescence chemodosimeter holds great potential for fluorescence detection of diverse species and fluorescence imaging.
Introduction
Heavy metal ions in the environment, foods and water have received intensive attention for decades.1 Among them, Hg2+ ions are one of the most toxic ions, which, when accumulated through the food chain, may cause damage to skin, respiratory, and gastrointestinal tissues.2–4 According to the US Environmental Protection Agency (EPA), 10 nM (2 ppb) is the maximum residue limit of Hg2+ allowed in drinkable water.5 To monitor mercury levels in populations occupationally exposed to mercury, numerous methods have been developed, such as atomic absorption spectroscopy (AAS),6,7 atomic emission spectroscopy (AES),8,9 and inductively coupled plasma atomic mass spectrometry (ICP-MS)10,11 methods. Generally, these methods are more precise than other methods but have the drawbacks of requiring expensive equipment and a time-consuming analysis process.12,13In recent years, fluorescence-based colorimetric chemodosimeters for the detection of Hg2+ ions have attracted increasing attention because of their high selectivity, sensitivity, irreversible binding ability, mild reaction conditions (e.g., room temperature), and water solubility.14,15 In 1992, Chae et al. first introduced the term chemodosimeter, which refers to an abiotic molecule that recognizes the analyte and at the same time irreversibly transduces an observable signal.16 Generally, mechanisms for chemodosimeters can be divided into two main modes: the analyte can react with the chemodosimeter or act as a catalyst.17 For example, one fluorescence chemodosimeter relies on the binding of Hg2+ for the desulfurization of a thioamide to amide to produce fluorescence signals.18–20
Because the fluorescence intensity determines the sensitivity of a fluorescent probe, metal-enhanced fluorescence (MEF) has been widely used to enhance the intensity of the fluorescence.21,22 Surface plasmon resonance is the light-induced of collective oscillation of free electrons in a metal, and thus generates a very strong electromagnetic field that can improve the fluorescence intensity.23,24 This phenomenon is called metal-enhanced fluorescence (MEF) and has been reported for various organic fluorophores25,26 and quantum dots27,28 in proximity to gold or silver substrates or other metal nanostructures. It has also been found that metal nanoparticles (NPs) can affect fluorescence intensity. This effect mostly depends on the distance between the metal nanostructures and fluorophores; too-great proximity results in quenching, while separation by several nanometers produces enhancement.29 However, when the distance is too large, the fluorophore will be far from the magnetic field generated by the plasma resonance of the metal nanoparticles, and the fluorescence intensity of the fluorophore will not be affected. Therefore, optimization of fluorophore-metal NP distance is critical for fluorescence enhancement.30 To date, a great deal of work has been performed using metal NPs deposited on glass,31 plastics,32,33 paper34 or other non-metallic plate substrate surfaces via self-assembly,35 to study the MEF effect on photoluminescent materials. Therefore, it is desirable to fabricate uniform and disperse nanocomposite systems to realize the MEF effect in biological,36 nanophase electronic,37 single nanoparticle sensing38,39 and fluorescence imaging.40
Fluorescent chemosensors for Hg2+ have been extensively explored, including cyclen,41 hydroxyquinoline,42 azine,43,44 diazatetrathia crown ethers36 and calixarene.45 However, many of these sensors suffer from limitations in terms of synthetic difficulty, high cost of starting materials, high detection limits or lack of selectivity towards potential competitors such as copper (Cu2+) and silver (Ag+) due to the similarity of their chemical behavior to that of Hg2+.46 In addition, two different modes, namely, the “turn-off” mode47 and “turn-on” mode,48 have been proposed for optical sensors based on fluorescence. Most of the reported fluorescent sensors have been based on the “turn-off” mode, in which false positive results occur due to other quenchers in real samples.49 Compared to the traditional “turn-off” mode, the latter approach based on the “turn-on” fluorescence change (quenching recovery) can not only expand the detection range but also increase stability and reduce environmental interferences.50
In this study, we developed a facile, highly sensitive and selective chemodosimeter for the detection of Hg2+ ions based on MEF. This was accomplished by immobilizing a fluorescent probe on ultrathin SiO2-shell-coated Au NPs51 (Au@SiO2 NPs, where the diameter of the SiO2 shell was 4–5 nm). With this core–shell type configuration, the plasmon of the inner Au NPs produces long-range electromagnetic fields to enhance the fluorescence emission. Additionally, the ultrathin SiO2 shell can avoid fluorescence quenching through separating the fluorophore probe from the metal. Furthermore, the fluorescence can be further enhanced via optimizing the core size. To demonstrate the feasibility of the MEF-based fluorescence platform, we demonstrated its application for the detection of Hg2+ ions. The optimized detection limit achieved was 5.0 × 10−11 M, which was about two orders of magnitude lower than that achieved using the free fluorescent probe (8.5 × 10−9 M) and far lower than the permissible EPA limit for Hg2+ ions in drinkable water (<1.0 × 10−9 M). Additionally, this MEF-based fluorescence platform has high selectivity for Hg2+ over other metal ions. The proposed strategy opens new avenues to fabricate a sustainable chemodosimeter that affords superior performance in the detection of Hg2+ in real environments, such as food samples and even whole cells.
Results and discussion
Scheme 1 shows a schematic illustration of the preparation flow for the fluorophore-labeled Au@SiO2 probe and detection mechanism of Hg2+ ions. In the absence of Hg2+ ions, the fluorophore-labeled Au@SiO2 probe exhibits relatively weak fluorescence emission. In contrast, because of their strong thiophilic affinity, the presence of Hg2+ ions can easily open the spirolactam ring (via desulfurization reaction) of the fluorophore immobilized on the SiO2 shell, thereby turning on fluorescence emission.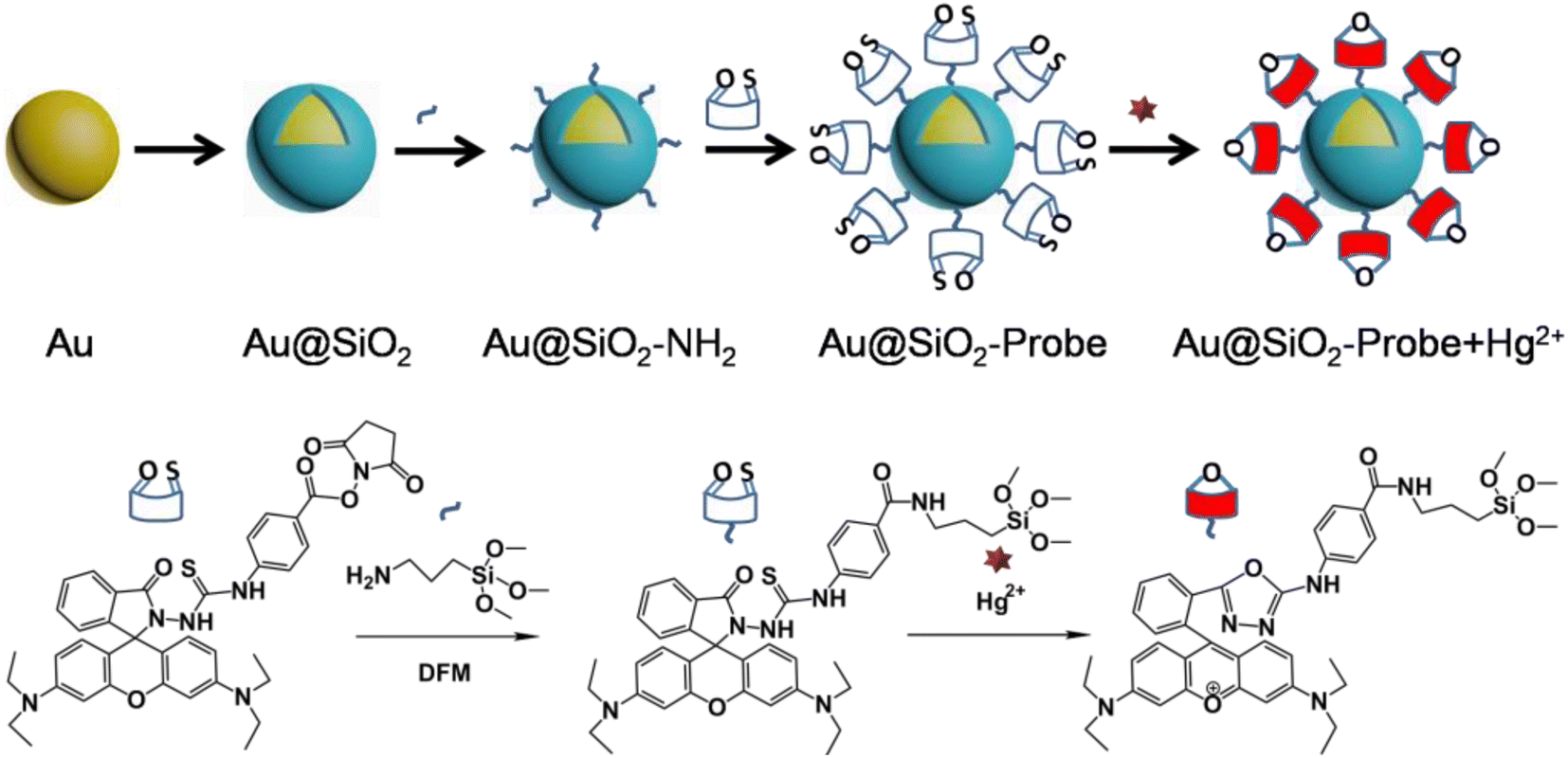 | ||
| Scheme 1 Schematic illustration of the preparation flow for the fluorophore-labeled Au@SiO2 NP probe and its detection mechanism for Hg2+ ions. | ||
Silica coating was performed according to the method established by Li et al.51 By carefully controlling reaction parameters such as pH, reaction time, temperature, and the amount of silica source, this method allows coating of the Au NPs with a controlled-thickness SiO2 shell. Fig. 1 shows the TEM images of the 80 nm Au NPs (a) and the corresponding Au@SiO2 NPs with a 4–5 nm SiO2 shell (b). Upon silica coating, a thin and uniform SiO2 shell of ∼4–5 nm is clearly seen coating each isolated Au NP. Moreover, the Au NPs supported on the GC electrode show typical electrochemical responses of a polycrystalline Au surface (Fig. 1c), with a wide Au oxide peak initiating at about 1.2 V and a corresponding stripping peak for the Au oxide at 0.9 V. On the contrary, the thin SiO2 shell completely restrains the formation of the Au oxide peak during the cationic scan and accordingly strips the peak of the Au oxide in the cyclic voltammetry (CV) curve of the Au@SiO2 NPs (Fig. 1c). Therefore, the SiO2 shell coating is “pinhole-free”, as further confirmed by the lack of any discernable SERS peaks for pyridine molecules when immersing the Au@SiO2 NPs in pyridine solution (for details, see Fig. 1d). The naked pure Au NPs show strong SERS peaks because of their easily available surface, which allows the adsorption of the pyridine molecules to experience SERS enhancement. In contrast, the Au@SiO2 NPs do not show any SERS peaks from pyridine, because the SiO2 shell effectively blocks the pyridine molecules from approaching and adsorbing on the inner Au NPs. This “pinhole-free” SiO2 shell can thus effectively separate the fluorophore from direct contact with the Au metal underneath, thereby avoiding fluorescence quenching. Conversely, the SiO2 shell is very thin, so that the field created by the Au NPs underneath can sufficiently penetrate the shell and enhance the fluorescence of the fluorophore immobilized therein. Moreover, a comparison material, 50 nm Au NPs, and the corresponding Au55 nm@SiO2 NPs were synthesized (for details, see ESI, Fig. S1 and S2†).
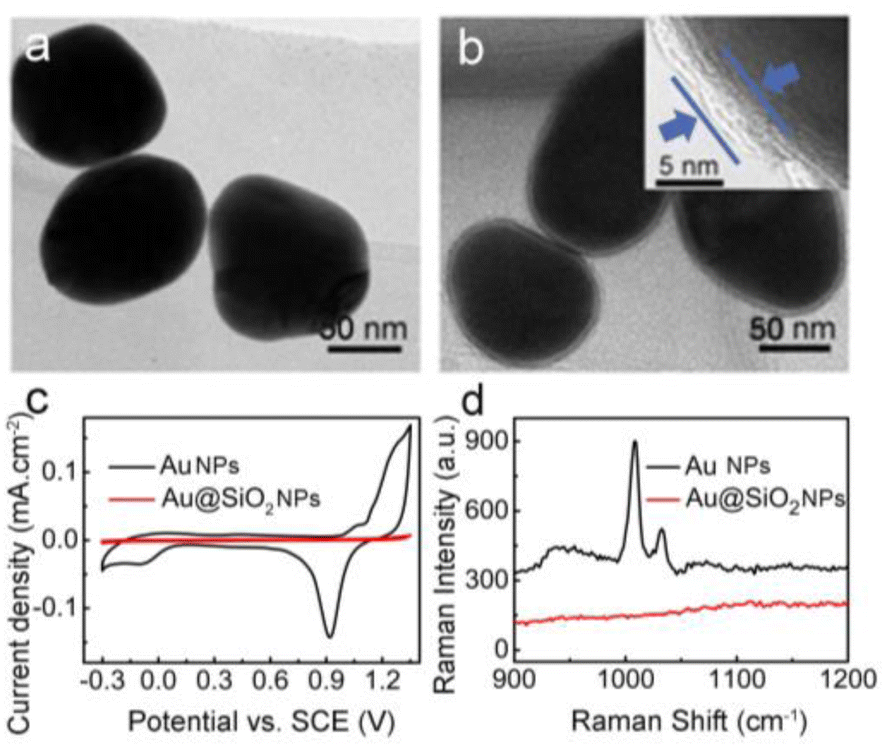 | ||
| Fig. 1 TEM images for the (a) 80 nm Au NPs and (b) corresponding Au@SiO2 NPs with an external 4–5 nm SiO2 shell. Inset is a TEM image of Ag@SiO2 NPs to show the silica shell. (c) Cyclic voltammograms of Au NPs and Au@SiO2 in 0.5 M H2SO4 solution, scan rate: 0.1 V s−1. (d) Raman spectrum of pyridine; 0.001 M pyridine on Au and Au@SiO2 on a Si wafer, respectively. | ||
Fig. 2a shows the UV-vis spectrum of the free fluorescent molecule, which exhibits a strong adsorption peak with a maximum wavelength (λmax) of 563 nm. Upon the addition of Hg2+ ions, the λmax red-shifts to 570 nm. At the same time, as shown in the corresponding fluorescence spectrum in Fig. 2b, the fluorescence intensity of the free fluorescent probe was enhanced after the addition of Hg2+, indicating that the ring-opening reaction occurred in the fluorescent molecule. Interestingly, the fluorescence intensity was dramatically enhanced when Hg2+ was added to the solution of the probe-labeled-Au@SiO2, which is because the excitation region of the probe is consistent with the plasmon of Au@SiO2 (Fig. 2b). The enhancement factor therefore results from the mercury-induced ring-opening reaction. More importantly, MEF, as a leading effect, causes a significant fluorescence enhancement. Because of the much wider resonance, the enhancement effect of the probe-labeled Au@SiO2 NPs is more remarkable than that of the free fluorescent probe. A comparison was made using 55 nm gold NPs, and it was found that the 80 nm NPs have better MEF performance (for details, see ESI, Fig. S3†). The time profile of the fluorescence response of the probes (0.5 μM) in the presence of Hg2+ (1.0 equiv.) in pH 7.4 PBS buffer is displayed in Fig. S4,† showing that the response of probes to sulfite was very quick. In addition, to study the stability of the probes at different pH values, the fluorescence spectra of the response of the probes toward Hg2+ under different pH conditions were evaluated (Fig. S5, see ESI†). These results clearly show that this probe can be used in a broad pH range of 6.0–9.0. This result shows that the probes can be applied to biological systems.
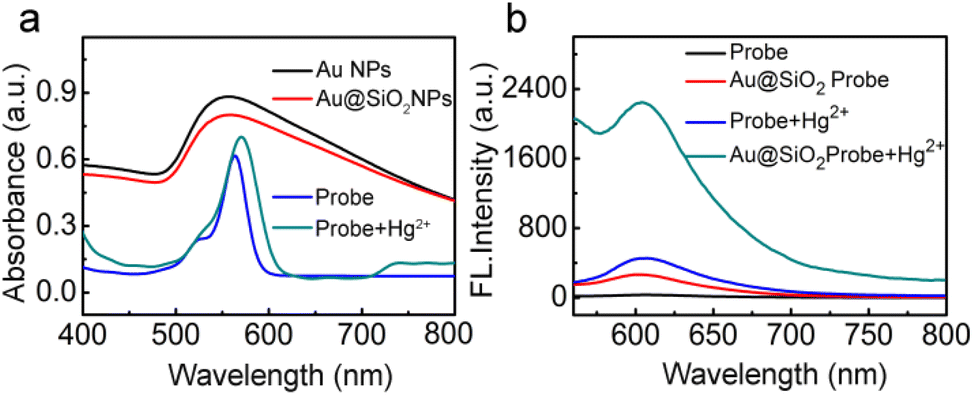 | ||
| Fig. 2 (a) UV-vis spectra for 0.5 μM of the free fluorescent probe, Au@SiO2 NPs, free fluorescent probe and fluorophore-labeled Au@SiO2 probe with the addition of Hg2+. (b) Fluorescence spectra of the free probe, fluorophore-labeled Au@SiO2 probe, and corresponding changes in fluorescence intensity after the addition of Hg2+. The concentration of the probe and Hg2+ are all 0.5 μM. | ||
Fig. 3a and b show the fluorescence spectrum of the free fluorescent probe and fluorophore-labeled Au@SiO2 probe, respectively, in the presence of different amounts of Hg2+ ions. For reliable comparison, the number of fluorescent probes immobilized on the Au@SiO2 NPs was adjusted to the same amount (all achieved 0.5 μM). After that, Hg2+ ions were introduced with increasing molar ratio to the probe-labeled Au@SiO2 NP solutions. With increasing amount of Hg2+ ions, the intensity of the fluorescence initially quickly increased. The fluorescence intensities then became saturated at a molar ratio of 1.8 for the probe-labeled Au@SiO2 NPs and 1.4 for the free fluorescent probe, as shown in Fig. 3b and d, because the mercury-induced ring-opening reactions reached equilibrium. The addition of Hg2+ to the probe-labeled Au@SiO2 solutions causes strong yellow fluorescence, as shown in the inset of Fig. 3c. Consistent with its large fluorescence enhancement ability (Fig. 3a and c), the fluorescence enhancement achieved on the Au@SiO2 probe is more prominent than that achieved for the free fluorescent probes. The number of fluorescent molecules immobilized on the surface of the Au@SiO2 NPs can be estimated, as described in the ESI Fig. S6† section;52 the fluorescence enhancement achieved on the Au@SiO2 NPs was 8.2-fold. With this core–shell type configuration, the fluorescence enhancement arises because the plasmon of the inner Au NPs is resonant with maximum UV-vis absorption peaks for the fluorescent probe. This large fluorescence enhancement makes the Au@SiO2 NPs very attractive for the practical detection of Hg2+ ions, which relies on fluorescence as the signaling pathway. The plots shown in Fig. 3b and d reveal that the fluorescence intensities changed as a function of the Hg2+ concentration, and the insets show the linear plot ranges of fluorescence intensity versus Hg2+ ionic concentration that were extracted from Fig. 3b and d. For the free fluorescent probe (Fig. 3b), the detection limit achieved is 8.5 × 10−9 M, while that achieved for the Au@SiO2 NPs is 5.0 × 10−11 M. The capability of our MEF probe to detect Hg2+ was comparable to that of other materials (Table 1). Apparently, owing to the large fluorescence enhancement, the fluorophore-labeled Au@SiO2 probes show much improved sensitivity toward Hg2+ ions. To study the stability of the probe RS (Rhodamine spirocyclic type fluorescent molecular probe) at different pH values, the fluorescence spectra of the RS response toward Hg2+ under different pH conditions were evaluated (Fig. S5, see ESI†). These results clearly show that this probe can be used in a broad range pH of 6.0–9.0.
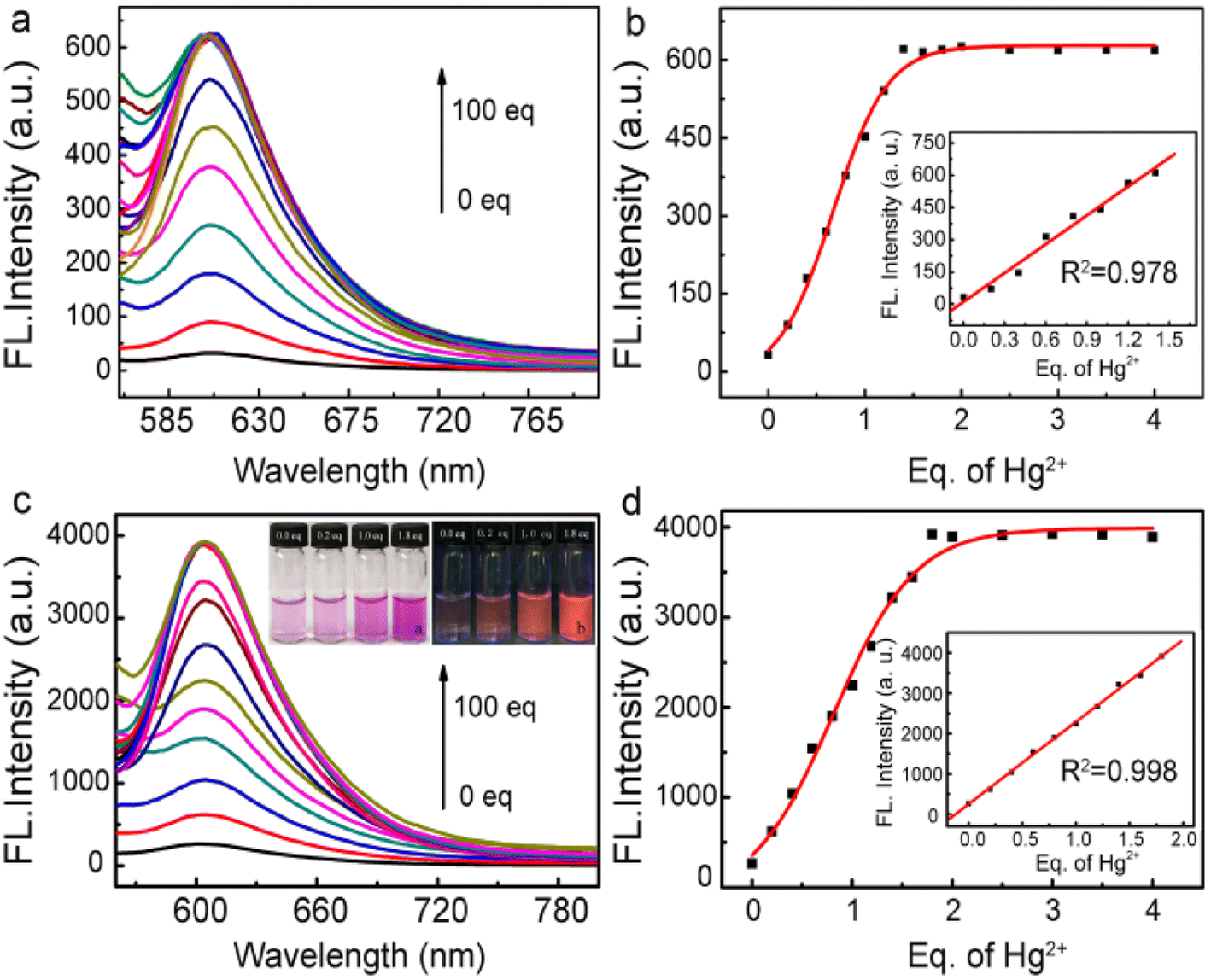 | ||
| Fig. 3 (a) Fluorescence spectra of the free fluorescent probe (0.5 μM) in the presence of different amounts of Hg2+ ions and (b) fluorescence intensity as a function of the amount of Hg2+ ions and corresponding linearity in the inset. (c) Fluorescence spectra for the fluorophore-labeled Au@SiO2 probe with various amounts of Hg2+, and corresponding fluorescence images with various amounts of Hg2+ in the inset. (d) Change in fluorescence intensity as a function of Hg2+ ion concentration and the corresponding linearity in the inset. The final concentrations for both probes were 0.5 μM. Excitation at 560 nm. | ||
| Material | LOD/mol L−1 | Year | Ref. |
|---|---|---|---|
| SBA-15 | 0.6 × 10−6 | 2008 | 53 |
| NaYF4:Yb3+ | 0.6 × 10−10 | 2010 | 54 |
| CdTe QDs | 0.7 × 10−7 | 2012 | 55 |
| Rhodamine derivative | 2.9 × 10−8 | 2013 | 56 |
| Au NP | 5.0 × 10−9 | 2016 | 57 |
| Aptamer-Ag@SiO2 | 3.3 × 10−10 | 2015 | 58 |
| Unsymmetrical quinoline type tolans | 2.0 × 10−10 | 2016 | 59 |
| G-quadruplex DNA | 1.3 × 10−10 | 2017 | 60 |
| Au/N-CQDs | 1.2 × 10−5 | 2018 | 61 |
| Ag@SiO2 NPs | 2.0 × 10−19 | 2022 | 62 |
| Au@SiO2-probe | 5.0 × 10−11 | Now | This work |
To further reveal their resistance against interferent ions, Fig. 4a and b shows fluorescence spectra for the Au@SiO2 probes in the presence of other metal ions, namely, Co2+, Fe3+, Cd2+, Fe2+, K+, Cr2+, Cu2+, Pb2+, Mg2+, Ag+, Ba2+, and Ni2+ ions. Clearly, only the presence of Hg2+ ions can trigger the high fluorescence intensity of the probe, thereby indicating the excellent selectivity of the Au@SiO2 fluorescent probes. In addition, we further examined the fluorescence response of the probe toward Hg2+ ions in the presence of other potentially competing species. These other species only displayed minimum interference (Fig. S7†). This suggests that the Au@SiO2 probe is potentially useful for sensing Hg2+ ions in the presence of other related species in pH 7.4 PBS buffer.
 | ||
| Fig. 4 (a) Fluorescence spectra for the fluorophore-labeled Au@SiO2 probe in the presence of 1.0 equivalent of Hg2+, Co2+, Fe3+, Cd2+, Fe2+, K+, Cr2+, Cu2+, Pb2+, Mg2+, Ag+, Ba2+, and Ni2+. The final concentration of the probe and metal ions are all 0.5 μM. (b) Selectivity of NP sensors plotted in histogram form. | ||
Because of the desirable sensitivity and selectivity of this fluorophore-labeled Au@SiO2 probe toward Hg2+ ions, it shows potential application in real water sample analysis. For demonstration, the proposed fluorescent probe was applied to detect Hg2+ ions in many different kinds of water collected from local places (Xiangjiang River, Peach Lake, and Yuelu Mountain Spring, in Changsha, China). The fluorescence intensity of the solutions was recorded after adding the fluorophore-labeled Au@SiO2 and a spiking concentration of Hg2+ ions to the water samples. To prove the reliability of our sensor, the same detection was carried out using ICP (inductive coupled plasma emission spectrometry), and the results are summarized in Table 2. The results of the two methods show good agreement between expected and found values, which proved that the Au@SiO2 NP fluorescent probes exhibit satisfactory performances in real environmental water samples.
| Sample | Added (equiv.) | Measured | ICP (mol L−1) |
|---|---|---|---|
| a N = no Hg2+ was detected. | |||
| River water | 0 | N | N |
| 1.00 | 1.13 | 5.27 × 10−7 | |
| 1.60 | 1.75 | 8.18 × 10−7 | |
| Lake water | 0 | N | N |
| 1.00 | 1.32 | 5.41 × 10−7 | |
| 1.60 | 1.94 | 8.33 × 10−7 | |
| Spring water | 0 | N | N |
| 1.00 | 0.92 | 4.66 × 10−7 | |
| 1.60 | 1.43 | 7.72 × 10−7 | |
To demonstrate the potential use of the probe and Au@SiO2 probe in bioimaging applications, we tested the cytotoxicity of the Au@SiO2 probe toward HCT116 colorectal cancer cells using the reduction activity of the methylthiazolyltetrazolium (MTT) assay (Fig. S8, see ESI†). The viability of untreated cells was assumed to be 100%. Upon the incubation with 0.5 μM of the probe or Au@SiO2 probe for 24 h, no significant difference in the proliferation of the cells was observed. Specifically, a cell viability of about 80% was observed after 24 h even at a high-dose concentration of 1.8 equiv. Hg2+. These data indicated the satisfactory biocompatibility of the Hg2+ fluorescent probe at all dosages, enabling the probe and Au@SiO2 probe to serve as a potential probe for fluorescence bioimaging.
To extend the application of the probes to more complex metrics, we examined the imaging characteristics of the probes in cultured living cells in vitro (HCT116, human colorectal cancer cells) using fluorescence microscopy (Fig. 5). The cells were incubated with 0.5 μM of the probe and Au@SiO2 probe for 1 h and 6 h at 37 °C, respectively. The cells were then washed with PBS three times and mounted on a microscope stage. As shown in Fig. 5a, the cells display modest intracellular staining after incubation with the probe and subsequent addition of 1.8 equiv. Hg2+. Upon incubation for 1 h, striking turn-on fluorescence is observed inside the HCT116 cells, indicating the formation of the probe + Hg2+ complex, which was in agreement with studies performed in solution. Under same conditions, the Au@SiO2 probe (0.5 μM) was incubated for 6 h and 1.8 equiv. Hg2+ was added and incubated for 1 h, and a notable enhancement was observed (Fig. 5b). In contrast, the Au@SiO2 probe produces a stronger fluorescence imaging effect than the free probe. The fluorescence microscopy analysis strongly suggested that the Au@SiO2 probe could cross the membrane barrier, permeate into the HCT116 cells, and rapidly sense intracellular Hg2+. It is significant to mention that bright field imaging of treated cells did not reveal any gross morphological perturbations, which suggested that the HCT116 cells were viable. These preliminary experimental results demonstrated that the Au@SiO2 probe could be applied for the fluorescence imaging of Hg2+ in biological samples with high resolution.
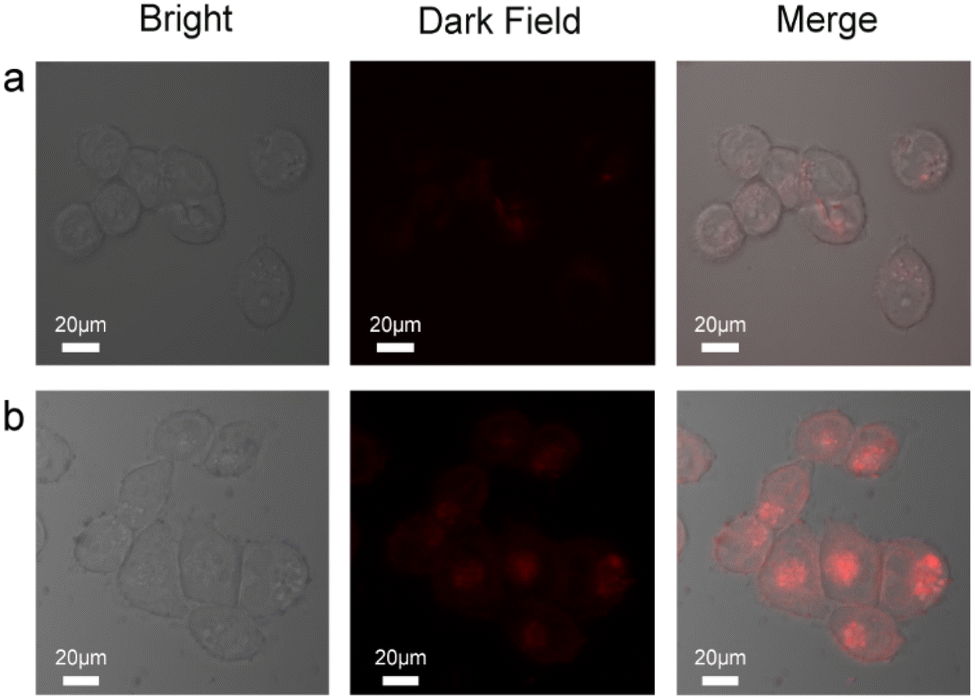 | ||
| Fig. 5 Fluorescence microscopic images of HCT116 cells: (a) after treatment with 0.5 μM probe and the addition of 5 μM Hg2+ (under red light) to probe-treated cells. (b) After treatment with 0.5 μM Au@SiO2 probe and the addition of 5 μM Hg2+ (under red light) to probe-treated cells. | ||
Whole-organism experiments were also carried out to examine whether the probe could be used to image Hg2+ in living organisms. A 3 day-old zebrafish was incubated with 1 μM of probe and Au@SiO2 probe in E3 embryo media for 4 h and 24 h at 28 °C and later incubated in a solution containing 1.8 equiv. Hg2+ for 2 h. Subsequently, the samples were washed with PBS to remove remaining probes. The treated zebrafish is shown in Fig. 6. The zebrafish remained alive throughout imaging experiments. The results of fluorescence microscopy analysis of these specimens showed that Hg2+ in the zebrafish was fluorescently detected by the probes (Table 1). In this case, the Au@SiO2 probe has stronger fluorescence intensity than the free probe. These results indicate that the probes are useful for the study of the toxicity or bioactivity of Hg2+ in living organisms.
 | ||
| Fig. 6 Fluorescence microscopic images of three-day-old zebrafish incubated with probe (a) and Au@SiO2 probe and (b) in the presence of Hg2+. Bright field image (left), fluorescence image (middle), and merged image (right). | ||
Experimental
Materials and methods
Sodium silicate solution (≥27 wt% SiO2), 3-aminopropyltrimethoxysilane (APTS, 97%), and chloroauric acid (≥99.9%) were obtained from Sigma-Aldrich, Alfa Aesar, and Jiuyue Chemical Ltd Co. (Shanghai, China), respectively. Sodium citrate, hydrochloric acid, mercuric nitrate (Hg(NO3)2), phosphate buffer saline (PBS), ethanol, and dimethyl formamide (DMF), all of analytical grade, were purchased from Sinopharm Chemical Reagent Ltd Co. (Shanghai, China). DMEM (Dulbecco's Modified Eagle Medium) were purchased from Sangon Biotech Reagent Ltd Co. (Shanghai, China). Other materials were obtained from native suppliers (Changsha Longhe Chemical and Glass Experimental Products Ltd Co. (Changsha, China)), and all materials were used as received without further purification.Synthesis of Au@SiO2 core–shell NPs
The synthesis of the Au@SiO2 core–shell NPs involves the preparation of the Au NPs and subsequent silica coating.51 In brief, 80 nm (in diameter) Au NPs were synthesized following Frens's method.63 After that, freshly prepared APTS solution (0.63 mL, 1 mM) and as-synthesized 80 nm Au colloids (90 mL) were mixed at room temperature, followed by vigorous stirring for 15 min. To the above mixture was then added activated sodium silicate solution (6.3 mL, 0.54 wt%). Prior to use, the silica solution was activated by adjusting its pH to 10.2–10.3 using 0.5 M HCl solution. After stirring for 3 min, aforementioned mixtures were transferred into a 95 °C water bath and stirred at this temperature for another 1 h to accomplish the silica coating. The final Au@SiO2 NPs were collected by centrifugation at 2500 rpm and stored in water (20 mL).Examining the coating quality of the synthesized Au@SiO2 NPs
The coating quality of the SiO2 shell on the Au nanoparticles (NPs) was examined using cyclic voltammetry (CV) and surface-enhanced Raman scattering (SERS) studies.51 For CV measurements, the working electrode was prepared by drying concentrated dispersions of Au NPs or Au@SiO2 NPs (40 mL, concentrated from 4 mL original colloids) on a glass carbon (GC) electrode (3 mm, in diameter). The CV curves for the Au NPs and Au@SiO2 NPs were measured in 0.5 M H2SO4 solution. For SERS measurements, a drop of the concentrated dispersion of Au@SiO2 NPs was dried on a Si wafer, onto which was spread a drop of 0.001 M pyridine solution; the wafer was then covered with a cover glass to prevent water evaporation.Fluorescence labeling of the Au@SiO2 NPs
The fluorescent probe, 2,5-dioxopyrrolidin-1-yl-4-(3-(3′,6′-bis(diethylamino)-3-oxospiro[isoindoline-1,9′-xanthen]-2-yl)thioureido) benzoate was synthesized according to a literature report.20 Prior to use, the fluorescent probe was dissolved in a mixed solution of PBS and ethanol (v:v, 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) with pH = 7.4. To immobilize the fluorescent probe onto the Au@SiO2 NPs, APTS was used as the linker.64 Briefly, freshly prepared APTS solution (1.12 mL, 1 mM) was added to a dispersion of Au@SiO2 NPs (4 mL) with vigorous stirring and were stirred for 15 min. The residual, unlinked APTS was removed by centrifugation/re-dispersion of the above mixtures in DMF three times, and the collected Au@SiO2 NPs were redispersed in DMF (20 mL). After that, the fluorescent probe solution (4.26 mL, 5 μM) was added to the DMF dispersion of the Au@SiO2 NPs and stirred in the dark and at room temperature for 24 h.65 The fluorescent probe was immobilized onto the surface of the Au@SiO2 NPs by the amidogen of APTS reacting with the ester group of the fluorescent probe. The fluorescent-probe-labeled Au@SiO2 NPs were collected by centrifugation, washed with ethanol and PBS/ethanol (v/v, 7/3) solution twice each, and re-dispersed in PBS/ethanol solution (10 mL).
:3) with pH = 7.4. To immobilize the fluorescent probe onto the Au@SiO2 NPs, APTS was used as the linker.64 Briefly, freshly prepared APTS solution (1.12 mL, 1 mM) was added to a dispersion of Au@SiO2 NPs (4 mL) with vigorous stirring and were stirred for 15 min. The residual, unlinked APTS was removed by centrifugation/re-dispersion of the above mixtures in DMF three times, and the collected Au@SiO2 NPs were redispersed in DMF (20 mL). After that, the fluorescent probe solution (4.26 mL, 5 μM) was added to the DMF dispersion of the Au@SiO2 NPs and stirred in the dark and at room temperature for 24 h.65 The fluorescent probe was immobilized onto the surface of the Au@SiO2 NPs by the amidogen of APTS reacting with the ester group of the fluorescent probe. The fluorescent-probe-labeled Au@SiO2 NPs were collected by centrifugation, washed with ethanol and PBS/ethanol (v/v, 7/3) solution twice each, and re-dispersed in PBS/ethanol solution (10 mL).
Fluorescence detection of Hg2+ ion
To test Hg2+ ions, the probe-labeled Au@SiO2 NP dispersion (254 μL, 0.98 μM) was placed into 2 mL test tubes, followed by the addition of a certain amount of Hg2+ ions (Hg(NO3)2 in PBS/ethanol solution). For all these measurements, the final probe concentration in the solution was 0.5 μM, with the molar ratio of Hg2+ ion to probe varying from 0 to 100.0. After the addition of Hg2+ ions, the solutions were allowed to stand undisturbed at room temperature for 2 h, followed by fluorescence measurement. For comparison, the fluorescence detection of other interferent ions (CoCl2, FeCl3, CdCl2, FeSO4, KCl, Cr(NO3)2, CuCl2, Pb(NO3)2, MgCl2, AgNO3, BaCl2 and Ni(NO3)2 in PBS/ethanol solution) was similarly tested using the fluorescent-probe-labeled Au@SiO2 NPs at a resultant concentration of 0.5 μM for the fluorescent probe and a 1.0 molar ratio of the different ions.Analysis of real water samples
To evaluate the practical potential of the Au@SiO2 NPs fluorescent probe, it was also used for the detection of Hg2+ ions in real water samples. Three real water samples were taken from Xiang River, Peach Lake, and Yuelu Mountain Spring (Changsha, China). Before measurements, the water samples were filtered through a 0.22 μm membrane syringe filter to filter out impurities. Then, 254 μL of the Au@SiO2 NPs fluorescent probe solution was dropped into the water sample. Subsequently, Hg2+ solutions were added to the Au@SiO2 NRs/water system at a final concentration of 0, 100, or 200 nM, and the water samples were then determined using fluorescence and ICP-MS.Cell cultures
The human colorectal cancer cell line HCT116 cells were provided by the Institute of Biochemistry and Cell Biology, Hunan University (Changsha, China). The cells were propagated in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% (v/v) fetal bovine serum, penicillin (100 μg mL−1), and streptomycin (100 μg mL−1). Cells were maintained under a humidified atmosphere of 5% CO2 and at 37 °C in an incubator as mentioned before.Cytotoxicity of the Au@SiO2 probe
The cytotoxic effect of the Au@SiO2 probe was determined using an MTT assay following manufacturer's instructions (Sigma-Aldrich, MO). Human colorectal cancer cell line HCT116 cells, provided by the Institute of Biochemistry and Cell Biology, Hunan University (Changsha, China), were seeded in a 96-well plate (2000 cells per well) with DMEM in an atmosphere of 5% CO2 and 95% air at 37 °C for 24 h. Then, the cells were incubated with DMEM containing various concentrations of the Au@SiO2 probe with different final concentrations (0.5, 1, 3, 5, 10 and 20 μM), while some cells were treated with Hg(NO3)2 (0.9 μM) alone. The cells were then incubated at 37 °C in an atmosphere of 5% CO2 and 95% air at 37 °C for 24 h, followed by MTT assays. An untreated assay with DMEM (n = 3) was also conducted under same conditions.Cell imaging experiments
For cell imaging studies, cells were seeded into a confocal dish and incubated at 37 °C in a CO2 incubator for 2 h. After one day, the cells were washed three times with phosphate buffered saline (pH 7.4) and incubated with 0.5 μM probe or Au@SiO2 probe in DMEM at 37 °C for 1 h and 6 h in a CO2 incubator and later incubated in DMEM with 1.8 equiv. Hg(NO3)2 for 1 h. The cells were again washed thrice with PBS (pH 7.4) to remove the probe or Au@SiO2 probe. Finally, the samples were observed under a TI-E + A1 SI Nikon laser confocal microscope and images were taken.Imaging of zebrafish
Zebrafish were obtained from the School of Life Sciences of Hunan Normal University (Changsha, China). The zebrafish were maintained in E3 embryo media (15 mM NaCl, 0.5 mM KCl, 1.0 mM MgSO4, 1.0 mM CaCl2, 0.15 mM KH2PO4, 0.05 mM Na2HPO4, 0.7 mM NaHCO3, and 5–10% methylene blue, pH 7.5). In fluorescence imaging experiments, three-day-old zebrafish were incubated with 1 μM probe and Au@SiO2 probe in E3 embryo media for 4 h and 24 h at 28 °C and later incubated in a solution containing 1.8 equiv. Hg2+ for 1 h at 28 °C, respectively. Subsequently, the samples were washed with PBS to remove remaining probes and examined using a TI-E + A1 SI Nikon laser confocal microscope.Characterization
UV-vis spectra and transmission electron microscopy (TEM) images were obtained using a UV-1800 spectrophotometer using 1 cm quartz cells (Shimadzu, Japan) and a JEM-2100F microscope (JEOL, Japan), respectively. For TEM characterization, the samples were prepared by drying a drop of dilute particle dispersion on a carbon-coated Cu grid. Fluorescence spectra were measured using a F-4600 fluorescence spectrophotometer (Hitachi, Tokyo, Japan) under the excitation of a 560 nm laser. Confocal laser scanning microscopy images were recorded on a TI-E + A1 SI, Nikon.Conclusions
In conclusion, we have developed a highly sensitive and selective metal-enhanced fluorescence chemodosimeter for the detection of Hg2+ ions, prepared by the immobilization of “turn-on” fluorescent probes on Au@SiO2 NPs. With this configuration, the chemodosimeter Au@SiO2 probe with an 80 nm core can produce an 8.2-fold fluorescence enhancement, while the thin SiO2 shell effectively prevents fluorescence quenching. The optimized detection limit achieved was 5.0 × 10−11 M, which was about two orders of magnitude lower than that achieved using the free fluorescent probe (8.5 × 10−9 M) and far lower than the permissible EPA limit of Hg2+ ions in drinkable water (<10 nM). These results suggest that the fluorophore-labeled Au@SiO2 probe has great potential for practical detection of Hg2+ ions in the environment, food samples, and even whole cells.Author contributions
Ying Cui and Shanji Fan contributed equally to this work. Ying Cui, conceptualization, methodology, investigation, writing – original draft, Shanji Fan, conceptualization, methodology, writing original draft, funding acquisition, review & editing, Yunran Zhai, investigation, Yingjie Liu, investigation, Junhua Li, review & editing, Jiawen Hu, methodology, writing, review & editing, Lijia Wang, methodology, writing, review & editing, funding acquisition, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research work was supported by Hunan Provincial Natural Science Foundation of China (Grant No. 2022JJ40393 and 2023JJ50093), Zhejiang Provincial Natural Science Foundation of China (Grant No. LBY22H180013), Science Foundation of Hengyang Normal University of China (No. 2021QD03), Hengyang Normal University Industrial Supported Project (No. HXKY2023042), Key Laboratory of Functional Metal-Organic Compounds of Hunan Province of Hengyang Normal University (Grant No. 2023HSKFJJ007), and Hunan Provincial Health Commission Research (Grant No. 202204014247). This research work was also supported by Medical and Health Science and Technology Program of Zhejiang Province (Grant No. 2021RC085).Notes and references
- J. O. Nriagu, Nature, 1979, 279, 409–411 CrossRef CAS PubMed.
- G. Cabana and J. B. Rasmussen, Nature, 1994, 372(6503), 255–257 CrossRef CAS.
- T. W. Clarkson, L. Magos and G. J. Myers, N. Engl. J. Med., 2003, 349(18), 1731–1737 CrossRef CAS PubMed.
- A. Renzoni, F. Zino and E. Franchi, Environ. Res., 1998, 77(2), 68–72 CrossRef CAS PubMed.
- U. EPA, National Primary Drinking Water Regulations, Arsenic and Clarifications, 2009 Search PubMed.
- O. Caylak, S. G. Elci, A. Hol, A. Akdogan, U. Divrikli and L. Elci, Food Chem., 2019, 274, 487–493 CrossRef CAS PubMed.
- F. M. Teeny, J. Agric. Food Chem., 1975, 23(4), 668–671 CrossRef CAS PubMed.
- F. X. X. Han, W. D. Patterson, Y. J. Xia, B. B. Sridhar and Y. Su, Water, Air, Soil Pollut., 2006, 170(1–4), 161–171 CrossRef CAS.
- J. Mo, Q. Li, X. Guo, G. Zhang and Z. Wang, Anal. Chem., 2017, 89(19), 10353–10360 CrossRef CAS PubMed.
- V. L. Dressler, F. G. Antes, C. M. Moreira, D. Pozebon and F. A. Duarte, Int. J. Mass Spectrom., 2011, 307(1–3), 149–162 CrossRef CAS.
- C. D. Palmer, M. E. Lewis, C. M. Geraghty Jr, F. Barbosa Jr and P. J. Parsons, Spectrochim. Acta, Part B, 2006, 61(8), 980–990 CrossRef.
- O. T. Butler, W. R. L. Cairns and J. M. Cookc, J. Anal. At. Spectrom., 2017, 32(1), 11–57 RSC.
- Y. Yin, J. Liu and G. Jiang, Chin. Sci. Bull., 2013, 58(2), 150–161 CrossRef CAS.
- Q. Lin, P. Chen, J. Liu, Y. Fu, Y. Zhang and T. Wei, Prog. Chem., 2013, 25(7), 1177–1186 CAS.
- E. M. Nolan and S. J. Lippard, J. Am. Chem. Soc., 2003, 125(47), 14270–14271 Search PubMed.
- M. Y. Chae and A. W. Czarnik, J. Am. Chem. Soc., 1992, 114, 9704–9705 CrossRef CAS.
- Y. Xu, Z. Jiang, Y. Xiao, T. T. Zhang, J. Y. Miao and B. X. Zhao, Anal. Chim. Acta, 2014, 807, 126–134 CrossRef CAS PubMed.
- M. G. Choi, H. G. Im, J. H. Noh, D. H. Ryu and S. K. Chang, Sens. Actuators, B, 2013, 177, 583–588 CrossRef CAS.
- A. K. Manna, J. Mondal, R. Chandra, K. Rout and G. K. Patra, J. Photochem. Photobiol., A, 2018, 356, 477–488 CrossRef CAS.
- Y. K. Yang, K. J. Yook and J. Tae, J. Am. Chem. Soc., 2005, 127(48), 16760–16761 CrossRef CAS PubMed.
- K. H. Drexhage, J. Lumin., 1970, 1–2, 693–701 CrossRef.
- Y. Jeong, Y. M. Kook, K. Lee and W. G. Koh, Biosens. Bioelectron., 2018, 111, 102–116 CrossRef CAS PubMed.
- H. Pei, S. Zhu, M. Yang, R. Kong, Y. Zheng and F. Qu, Biosens. Bioelectron., 2015, 74, 909–914 CrossRef CAS PubMed.
- Z. Zhou, H. Huang, Y. Chen, F. Liu, C. Z. Huang and N. Li, Biosens. Bioelectron., 2014, 52, 367–373 CrossRef CAS PubMed.
- H. Wang, T. Kang, X. Wang and L. Feng, Sens. Actuators, B, 2018, 264(3), 391–397 CrossRef CAS.
- X. Wang, L. Liu, S. Zhu and L. Li, Phys. Status Solidi RRL, 2019, 13(2), 1800521 CrossRef.
- C. Li, Y. Zhu, X. Zhang, X. Yang and C. Li, RSC Adv., 2012, 2(5), 1765–1768 RSC.
- C. Zhai, Z. Chen, S. Lv, Y. Lin, Z. Zhang and D. Pang, Prog. Chem., 2017, 29(8), 814–823 CAS.
- J. R. Bhamore, Z. V. P. Murthy and S. K. Kailasa, J. Mol. Liq., 2019, 280, 18–24 CrossRef CAS.
- A. I. Dragan, E. S. Bishop, J. R. Casas-Finet, R. J. Strouse, J. McGivney, M. A. Schenerman and C. D. Geddes, Plasmonics, 2012, 7, 739–744 CrossRef CAS.
- H. I. Peng, C. M. Strohsahl, K. E. Leach, T. D. Krauss and B. L. Miller, ACS Nano, 2009, 3, 2265–2273 CrossRef CAS PubMed.
- K. Aslan, R. Badugu, J. R. Lakowicz and C. D. Geddes, J. Fluoresc., 2018, 15(2), 99–104 CrossRef PubMed.
- V. Srinivasan, A. U. Andar, Y. Kostov and G. Rao, Plasmonics, 2019, 14(3), 731–736 CrossRef.
- Y. Zhang, K. Aslan, M. J. R. Previte and C. D. Geddes, Dyes Pigm., 2008, 77, 545–549 CrossRef CAS.
- G. Hong, S. M. Tabakman, K. Welsher, H. Wang, X. Wang and H. Dai, J. Am. Chem. Soc., 2010, 132, 15920–15923 CrossRef CAS PubMed.
- L. Jiang, X. Hang, P. Zhang, J. Wang, Y. Wang and L. Ren, Microchem. J., 2019, 148, 285–290 CrossRef CAS.
- A. V. Veglia and A. Guillermo Bracamonte, J. Nanophotonics, 2018, 12(3), 033004 Search PubMed.
- K. Aslan, M. Wu, J. R. Lakowicz and C. D. Geddes, J. Am. Chem. Soc., 2007, 129(6), 1524–1525 CrossRef CAS PubMed.
- I. G. Theodorou, Q. Jiang, L. Malms, X. Xie, R. C. Coombes, E. O. Aboagye, A. E. Porter, M. P. Ryan and F. Xie, Nanoscale, 2018, 10(33), 15854–15864 RSC.
- C. Joyce, S. M. Fothergill and F. Xie, Mater. Today Adv., 2020, 7, 100073 CrossRef.
- Z. Shen, C. Zhang, X. Yu, J. Li, Z. Wang, Z. Zhang and B. Liu, J. Mater. Chem. C, 2018, 6(36), 9636–9641 RSC.
- H. Jiang, D. Tang, Z. J. Li, J. W. Li, H. B. Liu, Q. J. Meng, Q. X. Han and X. A. Liu, Spectrochim. Acta.A., 2020, 243, 118784 CrossRef CAS PubMed.
- A. Caballero, R. Martinez, V. Lloveras, I. Ratera, J. Vidal-Gancedo, K. Wurst, A. Tarraga, P. Molina and J. Veciana, J. Am. Chem. Soc., 2005, 127(45), 15666–15667 CrossRef CAS PubMed.
- V. Narayanan, V. Ganesan, E. Shanmugasundram, S. Durganandini, K. Vellaisamy, H. Amirthalingam and S. Thambusamy, J. Photochem. Photobiol., A, 2023, 445, 115069 CrossRef CAS.
- G. G. Talanova, N. S. Elkarim, V. S. Talanov and R. A. Bartsch, Anal. Chem., 1999, 71, 3106–3109 CrossRef CAS PubMed.
- Z. Cheng, G. Li and M. Liu, J. Hazard. Mater., 2015, 287, 402–411 CrossRef CAS PubMed.
- S. Ishida, D. Kitagawa, S. Kobatake, S. Kim, S. Kurihara and T. Fukaminato, Chem. Commun., 2019, 55, 5681–5684 RSC.
- Y. Pang, Z. Rong, R. Xiao and S. Wang, Sci. Rep., 2015, 5, 11005–11012 CrossRef PubMed.
- Y. P. Li, X. H. Zhu, S. N. Li, Y. C. Jiang, M. C. Hu and Q. G. Zhai, ACS Appl. Mater. Interfaces, 2019, 11(12), 11338–11348 CrossRef CAS PubMed.
- Y. P. Li, X. H. Zhu, S. N. Li, Y. C. Jiang, M. C. Hu and Q. G. Zhai, ACS Appl. Mater. Interfaces, 2019, 11, 11338–11348 CrossRef CAS PubMed.
- J. F. Li, Y. F. Huang, Y. Ding, Z. L. Yang, S. B. Li, X. S. Zhou, F. R. Fan, W. Zhang, Z. Y. Zhou, D. Y. Wu, B. Ren, Z. L. Wang and Z. Q. Tian, Nature, 2010, 464(7287), 392–395 CrossRef CAS PubMed.
- J. Peng, W. Xu, C. L. Teoh, S. Han, B. Kim, A. Samanta, J. C. Er, L. Wang, L. Yuan, X. Liu and Y. T. Chang, J. Am. Chem. Soc., 2015, 137, 2336–2342 CrossRef CAS PubMed.
- J. Wang, H. Li, X. Min, L. Long, W. Ying, G. Ling, J. Zhu and Z. Zou, Appl. Surf. Sci., 2008, 254(17), 5329–5335 CrossRef CAS.
- M. Kumar and P. Zhang, Biosens. Bioelectron., 2010, 25(11), 2431–2435 CrossRef CAS PubMed.
- J. Pei, H. Zhu, X. Wang, H. Zhang and X. Yang, Anal. Chim. Acta, 2012, 757, 63–68 CrossRef CAS PubMed.
- X. Zhou, W. Yan, T. Zhao, Z. Tian and X. Wu, Tetrahedron, 2013, 69(46), 9535–9539 CrossRef CAS.
- J. Li, J. Chen, Y. Chen, Y. Li and C. Yu, Analyst, 2015, 141(1), 346 RSC.
- Y. Pang, Z. Rong, R. Xiao and S. Wang, Sci. Rep., 2015, 5, 9451 Search PubMed.
- J. H. Hong, S. Kurapati, Y. Jo, J. H. Shin and D. G. Cho, Chem. Commun., 2016, 52(71), 10759–10762 RSC.
- X. Zhang, B. Ding, W. U. Hua, J. Wang and H. A. Yang, Anal. Sci., 2017, 33(2), 165–169 CrossRef CAS PubMed.
- A. Meng, Q. Xu, K. Zhao, Z. Li, J. Liang and Q. A. Li, Sens. Actuators, B, 2018, 255(1), 657–665 CrossRef CAS.
- A. Picard-Lafond, D. Larivière and D. Boudreau, ACS Omega, 2022, 7(26), 22944–22955 CrossRef CAS PubMed.
- G. Frens, Nat. Phys. Sci., 1973, 241(105), 20–22 CrossRef CAS.
- R. Bardhan, N. K. Grady and N. J. Halas, Small, 2008, 4, 1716–1722 CrossRef CAS PubMed.
- A. Kaur, M. A. Haghighatbin, C. F. Hogan and E. J. New, Chem. Commun., 2015, 51, 10510–10513 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3na00746d |
| ‡ Co-first authors. |
| This journal is © The Royal Society of Chemistry 2024 |