
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAntimicrobial peptide-conjugated phage-mimicking nanoparticles exhibit potent bactericidal action against Streptococcus pyogenes in murine wound infection models†
Johanna
Olesk
a,
Deborah
Donahue
 b,
Jessica
Ross
c,
Conor
Sheehan
d,
Zach
Bennett
a,
Kevin
Armknecht
e,
Carlie
Kudary
f,
Juliane
Hopf
f,
Victoria A.
Ploplis
b,
Francis J.
Castellino
b,
Shaun W.
Lee
c and
Prakash D.
Nallathamby
*af
b,
Jessica
Ross
c,
Conor
Sheehan
d,
Zach
Bennett
a,
Kevin
Armknecht
e,
Carlie
Kudary
f,
Juliane
Hopf
f,
Victoria A.
Ploplis
b,
Francis J.
Castellino
b,
Shaun W.
Lee
c and
Prakash D.
Nallathamby
*af
aDepartment of Aerospace and Mechanical Engineering, University of Notre Dame, Notre Dame, Indiana, USA. E-mail: pnallath@nd.edu; Tel: +1 574 631 7868
bW. M. Keck Center for Transgene Research, University of Notre Dame, Notre Dame, Indiana, USA
cDepartment of Biological Sciences, University of Notre Dame, Notre Dame, Indiana, USA
dDepartment of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, Indiana, USA
eDepartment of Pre-Professional Studies, University of Notre Dame, Notre Dame, Indiana, USA
fBerthiaume Institute for Precision Health, University of Notre Dame, Notre Dame, Indiana, USA
First published on 10th January 2024
Abstract
Streptococcus pyogenes is a causative agent for strep throat, impetigo, and more invasive diseases. The main reason for the treatment failure of streptococcal infections is increased antibiotic resistance. In recent years, infectious diseases caused by pyogenic streptococci resistant to multiple antibiotics have been rising with a significant impact on public health and the veterinary industry. The development of antibiotic resistance and the resulting emergence of multidrug-resistant bacteria have become primary threats to the public health system, commonly leading to nosocomial infections. Many researchers have turned their focus to developing alternative classes of antibacterial agent based on various nanomaterials. We have developed an antibiotic-free nanoparticle system inspired by naturally occurring bacteriophages to fight antibiotic-resistant bacteria. Our phage-mimicking nanoparticles (PhaNPs) display structural mimicry of protein-turret distribution on the head structure of bacteriophages. By mimicking phages, we can take advantage of their evolutionary constant shape and high antibacterial activity while avoiding the immune reactions of the human body experienced by biologically derived phages. We describe the synthesis of hierarchically arranged core–shell nanoparticles, with a silica core conjugated with silver-coated gold nanospheres to which we have chemisorbed the synthetic antimicrobial peptide Syn-71 on the PhaNPs surface, and increased the rapidity of the antibacterial activity of the nanoparticles (PhaNP@Syn71). The antibacterial effect of the PhaNP@Syn71 was tested in vitro and in vivo in mouse wound infection models. In vitro, results showed a dose-dependent complete inhibition of bacterial growth (>99.99%). Cytocompatibility testing on HaCaT human skin keratinocytes showed minimal cytotoxicity of PhaNP@Syn71, being comparable to the vehicle cytotoxicity levels even at higher concentrations, thus proving that our design is biocompatible with human cells. There was a minimum cutoff dosage above which there was no evolution of resistance after prolonged exposure to sub-MIC dosages of PhaNP@Syn71. Application of PhaNP@Syn71 to a mouse wound infection model exhibited high biocompatibility in vivo while showing immediate stabilization of the wound size, and infection free wound healing. Our results suggest the robust utility of antimicrobial peptide-conjugated phage-mimicking nanoparticles as a highly effective antibacterial system that can combat bacterial infections consistently while avoiding the emergence of resistant bacterial strains.
1 Introduction
Streptococcus pyogenes, otherwise known as a β-hemolytic Gram+ Group A Streptococcus (GAS), is currently one of the significant problems in the global healthcare system, especially in hospital-acquired infections.1–3 It causes a wide range of infections, varying from mild pathologies such as pharyngotonsillitis to severe invasive infections such as necrotizing fasciitis and sepsis,4–6 with one-fifth of the invasive infections being severe bloodstream infections.7S. pyogenes mainly infects school-aged children and young adults. However, they have the potential to infect people of all ages. In the United States alone, 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–24000 people suffer from a severe GAS disease, with almost 2000 dying directly due to S. pyogenes infections,4 making it a top 10 cause of death globally. The prevalence of S. pyogenes infections in the USA has almost doubled, increasing from 3.4 to 4.3 per 100000 people in 2012 to 7.26 per 100000 people in 2017,8 mainly due to changes in virulence,9 or the development of antibiotic resistance in S. pyogenes or through co-infection with other antibiotic-resistant bacteria such as Staphylococcus aureus.10,11
000–24000 people suffer from a severe GAS disease, with almost 2000 dying directly due to S. pyogenes infections,4 making it a top 10 cause of death globally. The prevalence of S. pyogenes infections in the USA has almost doubled, increasing from 3.4 to 4.3 per 100000 people in 2012 to 7.26 per 100000 people in 2017,8 mainly due to changes in virulence,9 or the development of antibiotic resistance in S. pyogenes or through co-infection with other antibiotic-resistant bacteria such as Staphylococcus aureus.10,11
Antibiotics have been the primary method of treating bacterial infections, including S. pyogenes infections. For decades, the gold standard of treatment of S. pyogenes infections has been penicillin.12 Due to widespread use of antibiotics,10 and the last new class of antibiotics being introduced in 2003,13 bacteria have developed antibiotic resistance from chronic exposure to low levels of antibiotics over long periods, allowing the bacteria to adapt and build survival mechanisms against antibiotics. Antibiotic resistance is an increasingly growing global health concern. Like other bacteria, S. pyogenes can also develop antibiotic resistance, making the infections more challenging to treat as the treatment becomes ineffective.14–16 Although S. pyogenes has remained relatively susceptible to β-lactam antibiotics, the resistance has increased radically in recent years.15 It has been reported that in approximately 35% of pharyngotonsillitis patients, the eradication of S. pyogenes fails due to bacterial antibiotic resistance.16 Therefore, several studies have focused their research on developing alternative methods against antibiotic-resistant bacteria.17 There are various approaches that could provide the potential to treat antibiotic-resistant bacterial infections, such as antimicrobial peptides, phage therapy, or nanoparticle-based systems.
The development of antibacterial nanoparticles has become one of the most researched areas to fight antibiotic-resistant infections.18–22 An advantage of nanoparticles is that they can be designed to be target-specific, reducing toxicity to human cells and tissues. Metal nanoparticles have been extensively studied and are among the most promising antibacterial nanoparticles, showing strong antibacterial effect via various mechanisms of action,23 including the generation of reactive oxygen species (ROS), the release of cations, bacterial membrane binding, and disruption due to their positive charge, and biomolecular damage by interacting with biomolecules, such as lipopolysaccharides.24,25 Heavy metal nanoparticles, such as silver nanoparticles, generate ROS with high affinity to thiol (–SH) carriers such as cysteine.25,26 This affinity breaks the disulfide (–S–S–) bridges of the proteins and disrupts enzymatic functions, leading to cell death. The antibacterial properties of silver have been a widely researched area due to its long history of usage in treating infections,27 with silver nanoparticles being the most effective against bacteria.28 Silver nanoparticles have unique physicochemical properties, such as size distribution, agglomeration, surface chemistry, and ion release, that influence their biological activity.29 Silver nanoparticle have the ability to adhere to bacterial cell walls and membranes, penetrate the cells and bind with bacterial biomolecules, causing their disruption and intracellular damage.29–31 More importantly, silver nanoparticles generate ROS and free radicals that trigger the oxidative stress of the bacterial cell and interfere with bacterial signal transduction pathways.30 However, silver-generated ROS have been shown to exhibit significant toxic effects on human cells despite their counterbalancing by the activation of the eukaryotic antioxidant network, which severely limits silver nanoparticle use for treating infections.32,33
More recent studies have focused on the synergistic antibacterial effect exhibited by the nanoparticles and antimicrobial peptides (AMPs) to enhance the overall antibacterial effect.17,34–36 All these studies focused on electrostatic immobilization of the AMPs on silver nanoparticles, unlike ours, which has the AMP chemisorbed on gold-silver nano turrets on a silica core. AMPs are an important novel source of antibacterial compounds as an alternative to conventional antibiotics. They are usually positively charged short peptides, existing as part of the innate immune system of humans, animals, and plants. AMPs exhibit a broad antibacterial spectrum with complex mechanisms induced by the direct interactions of the peptides with bacterial membranes, leading to membrane disruption or entry into the bacterial cell and interactions with intracellular components.8,37 Acting through various mechanisms reduces the possibility of bacteria developing resistance towards AMPs compared to antibiotics.38 To make AMPs more biocompatible and increase their effectiveness and stability, researchers have created synthetic AMPs and developed combinations of nanoparticles and AMPs to induce synergistic effects.39
Here, we discuss the significant antibacterial activity and high biocompatibility of modularly assembled phage-mimicking nanoparticles (PhaNPs) capped with a rationally designed, cysteine-modified, antimicrobial peptide (Syn-71) (Fig. 1). Our nanoparticle design was based on the structure of tailless bacteriophages, taking advantage of assembling antibacterial components hierarchically in order to maximize the desired antibacterial effects while minimizing the toxicity of the particles to human cells. PhaNPs are composed of a silica core with silver-coated gold nanospheres distributed on the silica surface to mimic the protein turret on the surface of bacteriophages.40 The combination of hierarchically arranged materials allows the PhaNPs to inhibit bacterial growth and kill pathogenic bacteria. It was shown that this hierarchical design is highly bacteriostatic (99.9% retardation in Staphylococcus aureus USA300, Pseudomonas aeruginosa FRD1, Enterococcus faecalis, and Corynebacterium striatum) while showing little toxicity to mammalian cells. We have made the PhaNPs rapidly bactericidal by chemisorbing synthetic antimicrobial Syn71 peptide on to the gold-silver nanodots on the PhaNPs. Syn71 peptide is highly bactericidal to the human pathogen S. pyogenes, but its therapeutic window is limited by its toxicity to human cells at doses higher than 8 μM.40,41 By incorporating the peptide onto the overall PhaNPs design, we hypothesized that we would be able to increase the rapidity of bactericidal activity of our nanoparticles towards S. pyogenes while keeping the cytotoxicity of the nanoparticles towards mammalian cells minimal. The antibacterial effect of the PhaNP@Syn71 was tested in vitro and in vivo against S. pyogenes. In vitro results displayed dose-dependent bactericidal activity (up to >99.99%). Cytocompatibility testing of PhaNP@Syn71 on HaCaT human skin keratinocytes showed significantly low cytotoxicity, the toxicity levels being comparable to the vehicle level even at the highest concentration tested (up to 64 μM), demonstrating the biocompatibility of our designed nanoparticle system with human cells. In vitro evolution of resistance studies indicated a cutoff sub-MIC dosage above which there was no evolved resistance of S. pyogenes to PhaNP@Syn71 after repeated exposure of ∼1000 generations of S. pyogenes to PhaNP@Syn71. In vivo studies on a mouse wound infection model exhibited immediate wound size stabilization compared to wound flaring in controls while showing no toxicity to the surrounding mammalian cells and mouse organs. Our results suggest that PhaNP@Syn71 has opened the door for a new class of antibacterial nanocarriers that can sterically separate and orient antimicrobial peptides to be a highly effective antibacterial system to combat bacterial infections without antibiotics and prevent the emergence of antibiotic resistance.
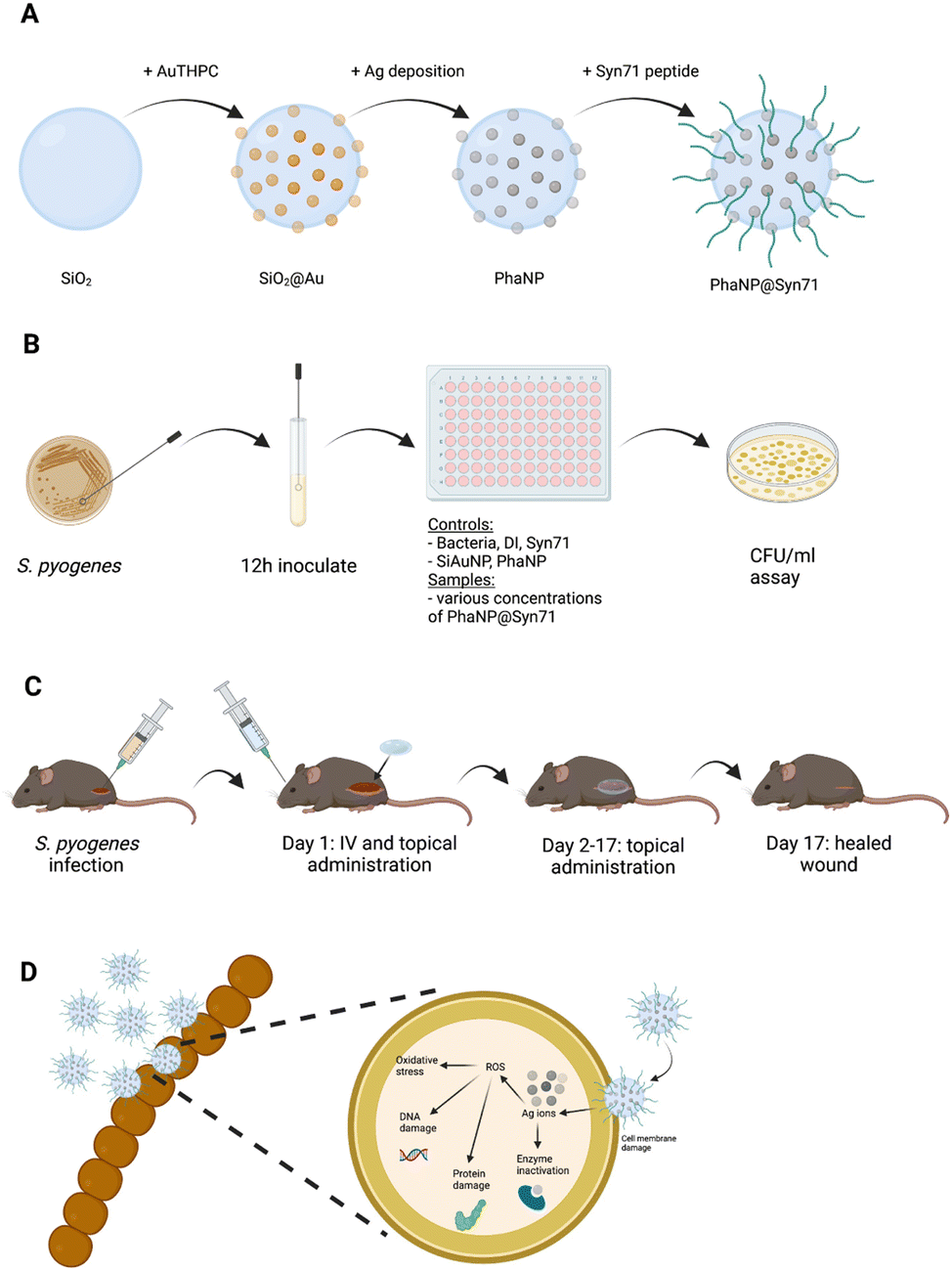 | ||
| Fig. 1 The overall experimental workflow. (A) A visual representation of the modular assembly of PhaNP@Syn71 synthesis workflow, (B) in vitro bacteria growth inhibition testing workflow, (C) in vivo mouse wound infection model experimental workflow, (D) antibacterial mechanisms of PhaNP@Syn71 is through the synergistic effect of PhaNPs and Syn71. Bacterial membrane penetration by Syn71 peptides on PhaNP@Syn71 enhances the period of contact of PhaNPs with the bacteria, disrupting the bacterial integrity. Illustrations are not to scale. | ||
2 Materials and methods
2.1 Materials
Streptococcus pyogenes strain AP53 Covs+; LB broth (Miller; Sigma-Aldrich); agar (Sigma-Aldrich); 200 proof ethanol (VWR); 20% v/v ammonium hydroxide (NH4OH; BDH); tetraethyl orthosilicate (TEOS; Sigma Aldrich, 99% GC grade); 3-aminopropyltriethoxysilane (APTES; Sigma Aldrich, 99%); sodium hydroxide (NaOH; Sigma Aldrich, >97%); sodium citrate dihydrate (C6H5Na3O7·2H2O; Sigma Aldrich, >99%); tetrakis(hydroxymethyl)phosphonium chloride (80% purity) (THPC; Sigma Aldrich, 80% in water); gold chloride (anhydrous; Sigma Aldrich); silver nitrate (AgNO3; Sigma Aldrich, 99% ACS grade); hydroquinone (Sigma Aldrich, >99% reagent grade); deionized water (DI water); 4% buffered paraformaldehyde (VWR), phosphate-buffered saline (PBS) without calcium and magnesium (1X; Cytiva); rhodamine B isothiocyanate (Sigma-Aldrich); AFdye488-Maleimide (Fluoroprobes), Cys-Syn71 peptide (CAGTEKIFQRLKKTIQEGKKIAKRWW, GenScript).2.2 Nanoparticle synthesis
:2 and stirred overnight in the dark with a smooth vortex, yielding SiO2@Au core–shell nanoparticles. The solution was centrifuged at 9000 RCF for 15 min at room temperature. The pellet was washed with 10 ml ethanol, sonicated (pulse for 20 s at 40% amplitude), and rinsed with 30 ml ethanol by centrifugation.

|
2.3 Nanoparticle characterization
000×. A drop of resuspended PhaNPs sample was placed onto an aluminum SEM stub and dried under a vacuum. An EDXS map was created to scan over a site of PhaNPs on the stub, with a count rate of 993 cps.
:1 hydrochloric acid:nitric acid), followed by the addition of 9.5 ml DI water to a final concentration of 5% Aqua Regia. Si, Au, and Ag emission spectra were collected at 251.611 nm, 267.595 nm, and 328.068 nm, respectively. Emission spectra for yttrium (Y), an internal standard, were collected at 371.029 nm.
2.3.5.1. Dynamic light scattering (DLS). The size distributions of the plain and peptide-bearing nanoparticles were measured using a Malvern Zetasizer Nano-ZS (Model ZEN3600) instrument by performing DLS. Each DLS sample was prepared by diluting 100 μl of the nanoparticle stock solution with 900 μl of 1X phosphate-buffered saline (PBS). The resulting dilutions were carefully pipetted into 1 ml polystyrene cuvettes (Malvern part number DTS0012) to prevent bubble formation. The cuvettes were loaded into the Zetasizer one at a time by orienting the arrow onto the cuvette to the arrow on the instrument and closing the device's lid. The Malvern Zetasizer software was used to take the size measurements. The program uses the absorption and refractive index of the nanoparticle's silica core material (Ri = 1.459, A = 0.035) and the refractive index, dielectric constant, and viscosity of the PBS dispersant ((Ri)PBS = 1.330, K = 79, μ = 0.8882 cP) as well as its internal measurements to determine the size distribution of the nanoparticles. The instrument reports three size distributions that correspond to the three trials the instrument automatically performs when run. The software determines the sizes reported for each trial by averaging eleven different measurements taken during each trial.
2.3.5.2. Electrophoretic light scattering (ELS). The zeta potential values of the plain and peptide-bearing nanoparticles were also found with the Malvern Zetasizer Nano-ZS (Model ZEN3600) instrument by performing ELS. Each ELS sample was prepared by diluting 100 μl of the nanoparticle stock solution with 900 μl of 1X phosphate-buffered saline (PBS), as was done for the DLS samples. These dilutions were carefully pipetted into folded capillary zeta cells (Malvern part number DTS1070) to prevent bubble formation. These cells were loaded into the warmed Zetasizer one at a time by matching the arrow on the cell to the arrow on the instrument and closing the device's lid. The Malvern Zetasizer Nano-ZS software was used to take the zeta potential measurements. The program uses the absorption and refractive index of the silica core material ((Ri)Si = 1.459, A = 0.035) and the refractive index, dielectric constant, and viscosity of the PBS dispersant ((Ri)PBS = 1.330, K = 79, μ = 0.8882 cP) as well as its internal measurements to determine the zeta potential. The instrument reports three zeta potential measurements that correspond to the three trials the instrument automatically performs when run. The software displays the average zeta potential found over the 100 measurements the device takes during each trial.
2.4 Cytocompatibility testing
To test the cytocompatibility of the PhaNP@Syn71, HaCaT human epithelial keratinocytes were used. The cells were cultured and maintained in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) at 37 °C with 5% CO2 in 100 mm culture dishes. First, HaCaT cells were grown to 75–80% confluency in 24-well tissue culture plates supplemented with DMEM. Before adding the samples (PhaNP@Syn71, PhaNPs, Syn71), DMEM was aspirated from the wells, and the cells were washed with 1X PBS. The PBS was aspirated, and the sample solutions in DMEM were added to the wells with washed HaCaT cells. The cells were incubated at 37 °C with 5% CO2 for 16 h, after which DMEM was aspirated, and cells were washed with 1X PBS to remove free nanoparticles. To determine the cytotoxicity of various concentrations of PhaNP@Syn71 as well as PhaNPs and Syn71 peptide alone, ethidium homodimer cell death assay30 was used. 4 μM ethidium homodimer in 1X PBS was added to HaCaT cells and incubated at 37 °C for 30 min. The plate reader was set to 528 nm excitation and 617 nm emission with a cutoff value of 590 nm to determine the level of fluorescence. To determine the percentage of dead cells, all cells were permeabilized by the addition of 0.1% (w/v) saponin to each well after the initial reading, and incubated at room temperature for 20 min, followed by shaking the plate on an orbital shaker. The plate reader was used in the same settings as before to measure the fluorescence. Percent membrane permeabilization was calculated by dividing the fluorescence values with intact cells by the fluorescence values after cell disruption by saponification. Each treatment condition was performed in triplicate, with the average cell viability (as a percent of the control) plus the standard deviation of all conditions plotted together for comparison.000 HaCaT cells were added to each well. Corresponding volumes of free Syn71 peptide (8 μM), SiO2AuNPs, and PhaNPs, volumes corresponding to the volume of PhaNP@Syn71 containing 4 μM or 8 μM Syn71 were added. Enough DMEM media was added to reach the final volume of 300 μl per well. The cells were allowed to attach to the well bottom for 26 h at 37 °C, followed by creating the scratch by using a sterile pipette tip to scrape the cell monolayer in a straight line. The chamber slide was live imaged under the microscope at 37 °C for 24 h.
2.5 Biocidal activity against planktonic bacteria
The antibacterial effect of various concentrations of PhaNP@Syn71 together with SiO2@Au nanoparticles, non-peptide PhaNPs and Syn71 peptide alone was screened against S. pyogenes. Growth curve measurements and CFU per ml assays were used to evaluate the overall antibacterial effect and are described below. | (1) |
2.6 Bacteria imaging
2.6.2.1 Maleimide AF488 staining for cysteine residues on fixed S. pyogenes. Streptococcus pyogenes was grown in LB media for 16 hours. The bacteria were pelleted and fixed in 4% buffered para-formaldehyde for 1 hour. Bacteria were then pelleted by centrifugation and resuspended in 1X PBS. The OD600 nm was adjusted to 0.05 using 1X PBS for the fixed S. pyogenes bacteria. Then Af488-maleimide dye (5 mg ml−1) in DMSO was diluted 100-fold by direct addition to the OD600 nm = 0.05 bacteria solution. The solution was mixed by shaking in the fridge for 12 hours. The bacteria were rinsed thrice by centrifugation (4500 rpm on benchtop centrifuge for 5 minutes) and then resuspended in 1X PBS. An aliquot of 10 μL of the samples was dropped on a glass slide and imaged using dark-field microscopy, epi-fluorescence microscopy, and a 40X objective.
2.6.2.2 Red fluorescent silica@Au NPs staining of fixed S. pyogenes. Streptococcus pyogenes was grown in LB media for 16 hours. The bacteria were pelleted and fixed in 4% buffered para-formaldehyde for 1 hour. Bacteria were then pelleted by centrifugation and resuspended in 1X PBS. The OD600 nm was adjusted to 0.05 using 1x PBS for the fixed S. pyogenes bacteria. Then 1% v/v red fluorescent silica@Au NPs was diluted 5-fold by direct addition to the OD600 nm = 0.05 bacteria solution. The solution was mixed by shaking in the fridge for 12 hours. The bacteria were rinsed thrice by centrifugation (4500 rpm on benchtop centrifuge for 5 minutes) and then resuspended in 1X PBS. An aliquot of 10 μL of the samples were dropped on a glass slide and imaged using dark-field microscopy, epi-fluorescence microscopy, and a 40X objective.
2.7 In vivo wound healing studies
Male wild type (C57bl/6) mice (age 6–10 weeks) were prepared with NAIR on the right flank. An aliquot of 100 μl of Streptococcus pyogenes strain AP53S+ was injected SubQ with a concentration of 1–3 × 108 CFU per mouse under isoflurane anesthesia. After the wound was established, on day 2, daily measurements were measured and a 100 μl dose of either vehicle, peptide, or nanoparticles with peptide was given topically. The topical dose w.r.t peptide was 150 μM, suspended in 20% w/v Pluronic F-127. After 15 days, the mice were sacrificed. In the second experiment, all the previous steps were followed. The intravenous (IV) dose w.r.t peptide was 100 μM, suspended in 1X PBS. A retro-orbital injection of 100 μl was done with either vehicle, peptide or nanoparticles on day 2 when topical treatment was begun. Wound sizes were recorded daily. The mice were allowed to fully heal before anesthetizing with a rodent cocktail (9 parts ketamine (100 mg ml−1) + 9 parts xylazine (20 mg ml−1) + 3 parts acepromazine (10 mg ml−1) + 79 parts saline), (body weight × 10) − 50 μl = μL per mouse and collection of blood and tissues. All studies were approved by the Institutional Animal Care and Use Committee at the University of Notre Dame and were conducted in accordance with the guidelines of the U.S. Public Health Service Policy for Humane Care and Use of Laboratory Animals. All efforts were made to minimize the suffering of the mice. The wound size data were normalized for the analysis based on the size on day 1. The normalized wound sizes were then averaged for each day, and standard deviations were calculated. After the mice were sacrificed, they were dissected, and tissues and organs were collected and fixed in 4% buffered para-formaldehyde. The histology samples were prepared for H & E staining and Gram-staining. Stained samples were imaged under Swift Binocular Compound Microscope SW350B with SWIFT 1.3 Megapixel Digital Camera for Microscope Eyepiece Mount Windows/Mac to record the images.:3 nitric acid:hydrochloric acid) was added to begin digestion. A glass rod was used to break down the sample into small pieces. The samples were allowed to digest for 1–7 days. Samples were diluted to 5% Aqua Regia with purified DI water. Samples were mixed by vortexing and then filtered through a 1.0 μm Nylon syringe filter before testing. The samples were run on a Perkins Elmer Avio 200 using a 5-point standard curve created from Si, Ag, and Au standards (LabChem). The instrument was blanked using a 5% Aqua Regia and purified DI water solution. The concentrations of elements in each sample were related to the weight of digested samples.
3 Results and discussion
3.1 Nanoparticle characterization
Our research group has confirmed that the structure of our synthesized nanoparticles resembles naturally occurring bacteriophages, with smaller-sized nanoparticles displaying closer mimicry with bacteriophages.40 The nanoparticles mimic bacteriophages from the family of Microviridae with 88% similarity. Microviridae are tailless bacteriophages, sized 25–27 nm. To visualize the PhaNPs, TEM analysis was carried out (Fig. 2A and B), revealing that the diameter of SiO2 nanoparticle core was 20.5 nm ± 2.1 nm (Fig. 2A). Analysis of PhaNP@Syn71 showed a nanoparticle diameter of 20.2 ± 2.4 nm (Fig. 2B), verifying that further synthesis processes (addition of gold nanospheres, silver, and Syn71 peptide) do not alter the base diameter of the nanoparticle silica core. In addition, TEM image analysis of PhaNPs and PhaNP@Syn71 demonstrated the presence of gold NPs on the silica core. EDXS compositional analysis of the PhaNPs (Fig. 2C) revealed the presence of Si, Au and Ag, in agreement with the synthesis process, with ICP-OES measurements (ESI A Table S1†) further confirming the presence of Au on SiO2AuNPs, and Au and Ag on PhaNPs. The concentration of Si was 3.829 mg ml−1 and 12.068 mg ml−1 for SiO2AuNPs and PhaNPs, respectively. The concentration of Au on SiO2AuNPs was 0.124 mg ml−1, while the Au and Ag concentrations of PhaNPs were 0.054 mg ml−1 and 3.924 mg ml−1, respectively. This is consistent with the synthesis process, showing similar gold content of SiO2AuNPs and PhaNPs, with the addition of Ag on PhaNPs.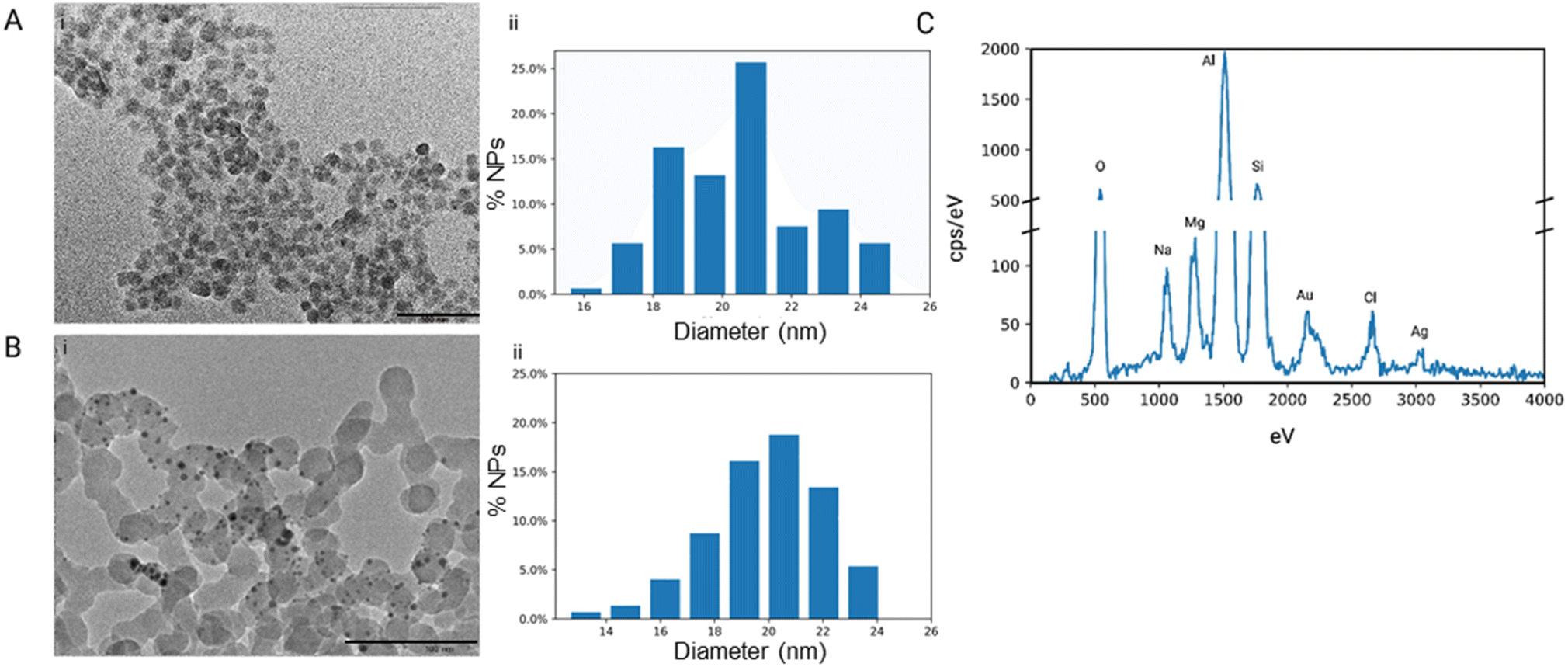 | ||
| Fig. 2 Nanoparticle characterization. (A) (i) TEM image of silica NPs before surface modifications (scale bar = 100 nm) and (ii) the size distribution of silica NPs showing an average diameter of 20.5 nm ± 2.1 nm. (B) (i) TEM image of PhaNPs used for Syn71 conjugation (scale bar = 100 nm) and (ii) the size distribution of PhaNPs showing an average diameter of 20.2 nm ± 2.4 nm. (C) The energy-dispersive X-ray spectrum (EDXS) of PhaNPs confirmed their elemental composition. Distinct energy peaks for Si (Kα = 1.740 keV), O (Kα = 0.525 keV), Au (Mα = 2.123 keV, Lα = 9.713 keV) and Ag (Kα = 22.163 keV, Lα = 2.983 keV) were detected. Al peak is present due to SEM stub material; Mg, Na and Cl peaks are due to the solvent used (PBS buffer). | ||
PhaNPs were functionalized with the cysteine terminated Syn71 peptide, chosen from syn-larvacin library created by Ross et al.41 Syn-larvacin-71 is a synthetic, unmodified peptide derived from the active region of the enterocin AS-48-like bacteriocin homologue, ribosomally produced by Paenibacillus larvae. It was identified using a homology-based search of the truncated enterocin AS-48 previously identified to exhibit antibacterial properties. The 25-mer larvacin AS-48 peptide scaffold was used to create a novel, synthetic peptide library. Rational design techniques hypothesized to increase antibacterial activity were employed on the larvacin peptide scaffold. Briefly, in a stepwise fashion, lysine and tryptophan substitutions were made for aliphatic and short-chain amino acids, hypothesized to increase lipid membrane affinity and penetration, respectively. Through the inversion of hydrophobic amino acids within the helical wheel, amphipathicity was increased, and hypothesized to bind to and disrupt the lipid membrane. In total, 95 peptide variants were designed and deemed the syn-larvacin library. To identify potent peptide variants against S. pyogenes, minimal inhibitory concentration (MIC) assays were performed using the syn-larvacin scaffold peptide (Table 1). Following the identification of the MIC, peptide variants were screened at half the concentration to identify peptide variants with increased antibacterial activity. After identifying optimized peptide variants, MIC assays, and minimal bactericidal concentration assays were performed to evaluate potency. The 25-mer Syn-laravacin-71 was robustly immobilized on PhaNPs by adding a cysteine residue at the N-terminal, and we designated the 26-mer cysteine-modified peptide as Syn71.
DLS analysis was conducted on PhaNPs without the Syn71 peptide and PhaNP@Syn71 peptide-modified nanoparticles. The results showed an average hydrodynamic diameter of the nanoparticles to be 781.7 ± 498.5 nm, indicating that it is possible that the PhaNPs clump in PBS solvent (ESI B Table S2†). PhaNP@Syn71 had an average diameter of 2582 ± 641.2 nm, showing a further increase in hydrodynamic radius compared to PhaNPs. The zeta sizer indicated that the ionic strength of the solvent and the surface modification of nanoparticles play a key role in the hydrodynamic radius of colloidal particles in solution.52–54 These results indicate the actual behavior of the nanoparticles in the PBS solution. The zeta potential results were −16.0 mV for PhaNPs with a conductivity of 17.8 mS cm−1, while the zeta potential for PhaNP@Syn71 was −8.54 mV with a conductivity of 16.8 mS cm−1, indicating a decrease in surface potential when the PhaNPs are modified with Syn71 peptide. Both zeta potential and DLS analyses confirmed the Syn71 peptide addition to the PhaNPs surface, showing the change in zeta potential and the size, which has been demonstrated in previously published works to indicate successful peptide addition to the nanoparticles.36
LC-MS analysis on the supernatant of PhaNP@Syn71 revealed that there is no residual Syn71 peptide present in the supernatant (ESI C Fig. S2†), indicating that >99.9% of the peptide added during the synthesis process had been absorbed on the PhaNPs. Change in zeta potential when comparing PhaNPs to PhaNP@Syn71 further verified the chemisorption of Syn71 onto the PhaNPs surface.
3.2 Biocompatibility with human skin cell line
Various concentrations of PhaNP@Syn71 (vehicle, 0.25 μM, 0.5 μM, 1 μM, 2 μM, 4 μM, 8 μM, 16 μM, 32 μM and 64 μM) were tested on HaCaT human skin keratinocytes (Fig. 3A) and compared against PhaNPs and free Syn71 peptide. The results showed that the PhaNP@Syn71 had significantly lower cytotoxicity than the free peptide, as well as slightly lower toxicity than PhaNPs, displaying almost no change in cytotoxicity from the vehicle up to relatively high concentrations. Syn71 peptide alone showed significantly increased cytotoxicity above 8 μM. In addition, the SEM imaging confirmed that PhaNP@Syn71 attaches to the HaCaT cells and does not lyse them (Fig. 3B).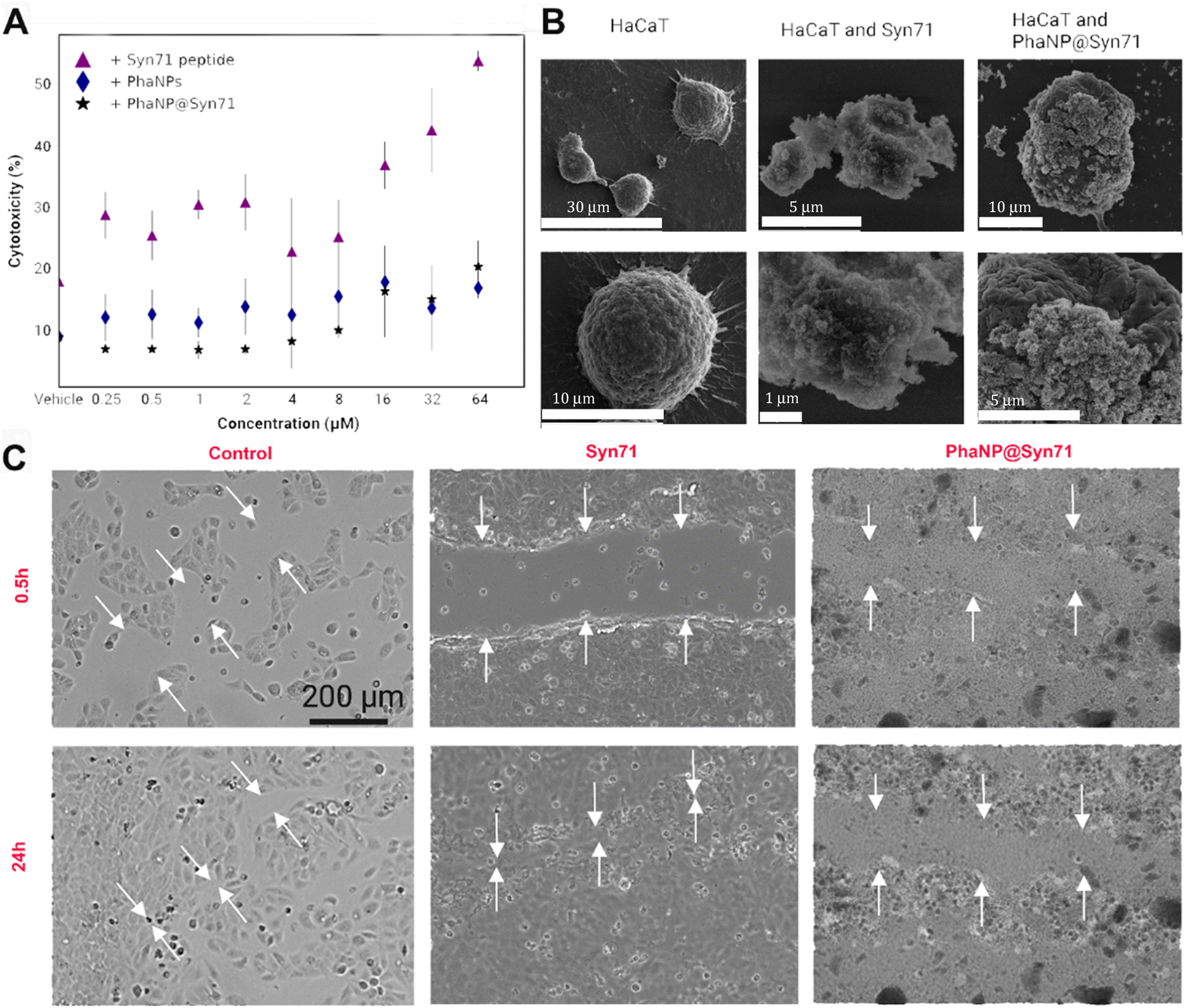 | ||
| Fig. 3 (A) Cytocompatibility of PhaNP@Syn71 on the HaCaT human keratinocyte cell line. PhaNP@Syn71 were cytocompatible at all concentrations tested similar to vehicle only control. Syn71 peptide was cytocompatible to 8 μM only. The average of four replicates for each sample was computed together with standard deviations. (B) SEM images of HaCaT cells incubated with PhaNP@Syn71. This indicated that the HaCaT cells integrity was not hampered by Syn71 or PhaNP@Syn71 at 8 μM concentration w.r.t Syn71. (C) In vitro scratch closure assay on HaCaT cells showed no cell migration in the presence of PhaNP@Syn71, while Syn71 peptide only presence, or no treatment group control allowed cell migration and scratch closure. This is because the PhaNP@Syn71 had a net negative charge, which will allow them to coat the positively charged tissue culture treated gap, as a consequence of which the surface charge of the gap becomes negative and repels cells migration. The free peptides are positively charged and do not hinder cell migration. 200 μm scale bar is applicable to all optical images. | ||
Silica-based materials are highly compatible with human cells55,56, with amorphous silica nanoparticles with APTES molecules similar to our designed core nanoparticles have been demonstrated to be biocompatible with human cells.57 Ag nanoparticles are also considered relatively biocompatible compared to other metallic nanoparticles. Therefore, they have been recommended as a good alternative for antibiotics to eradicate bacterial infections with minimal toxicity to surrounding human cells.58 Ag nanoparticle cytotoxicity is size-dependent, surface-dependent, and dose-dependent, with high doses of total Ag concentration being toxic.59 Our design chose 5 nm Au/Ag nanospheres, and the arrangement of the nanospheres on the silica core ensures they are not presented as <5 nm, reducing the probability of the nanoparticles permeabilizing mammalian cells.40 According to Environmental Protection Agency (EPA), the chronic oral Reference Dose (RfD) is 250 μg top 750 μg per person per day.60 However, our PhaNP@Syn71 exhibits the maximum Ag content of 171.36 μg per ml, leaving it under the EPA-established safety limits.
Several works have shown that the Minimum Inhibitory Concentration (MIC) of free antibacterial peptides is relatively high for inhibiting the growth of pathogenic bacteria, the MICs being higher than the MIC to affect human cells.61,62 Therefore, integration of the antibacterial peptides with nanoparticles to decrease their MIC, has been pursued in studies showing that in some cases peptide integration into nanoparticles results in smaller MICs than that of the free peptide, although that wasn't the stated goal of our study.63,64 A few studies have also indicated that the amino acid sequence plays a key role in the cytocompatibility and biocompatibility of peptide coated NPs.65,66
Syn71 peptide exhibits dose-dependent toxicity to human cells at >8 μM and mild hemolytic activity at 8 μM. Our designed PhaNP@Syn71 showed significantly decreased cytotoxicity toward mammalian cells compared to free peptides at the same concentration. At relatively high concentrations (64 μM), PhaNP@Syn71 cytotoxicity was at 20%, while the free peptide displayed a cytotoxicity of over 50%. Very low cytotoxicity was displayed at concentrations up to 8 μM (10%), as the free peptide showed cytotoxicity higher than that of the highest concentration of PhaNP@Syn71, confirming the success of lowering the overall cytotoxicity with the developed nanoparticle-peptide constructs.
3.3 Biocidal activity of PhaNP@Syn71 against planktonic S. pyogenes
Our designed nanoparticles take advantage of the Ag antibacterial effect coupled with the antibacterial effect of the antimicrobial Syn71 peptide. By alloying Ag onto Au nanospheres, we can minimize the leaching of Ag ions, leading to lower off-target toxicity.40 Silver and antimicrobial peptide containing PhaNP@Syn71 have successfully shown to inhibit the bacteria growth in vitro (Fig. 4A) at concentrations between 4 and 50 μM. In addition, free Syn71 peptide was tested for concentrations 8 μM and 4 μM. The comparison of free Syn71 concentrations with corresponding PhaNP@Syn71 concentrations shows that at both concentrations designed PhaNP@Syn71 has higher antibacterial activity than the free peptide. Maximum 8 μM Syn71 peptide was used since the concentrations above are highly toxic to mammalian cells. Here, we have shown that the S. pyogenes growth is almost fully inhibited by concentrations of 25 μM to 50 μM. PhaNP@Syn71 concentration of 8–10 μM also showed a significant halt on bacterial growth after initial growth. The lowest concentration of 4 μM PhaNP@Syn71 inhibited the bacteria growth up to 50%, showing an immediate constant inhibition from the beginning. The minimum inhibitory concentration of PhaNP@Syn71was determined to be 8.62 μM with concentration in respect to Syn71 loaded on NPs. Colony Forming Units per ml (CFU per ml) further confirmed that at 50 μM concentration of PhaNP@Syn71 kills the S. pyogenes (Table 2). Although other concentrations show some live bacteria still present post-treatment, the live bacteria decreased from 3.17 × 107 in the control group to 3.93 × 106, with the lowest number of live bacteria being present in the sample treated with 10 μM PhaNP@Syn71. In the growth curve, 25 μM PhaNP@Syn71 exhibits similar high inhibition as 50 μM PhaNP@Syn71, however, the CFU per ml assay shows live bacteria present post-treatment, meaning that at concentrations lower than 50 μM, the nanoparticles do not completely eradicate viable bacteria. These results are relevant in demonstrating that our designed nanoparticle-peptide constructs inhibit S. pyogenes growth at very low concentrations compared to a free peptide. The PhaNP@Syn71 are also completely bactericidal at >25 μM concentration while being cytocompatible to human cells, a concentration at which free Syn71 peptides cannot be utilized due to their high cytotoxicity to human cells beyond 8 μM. For the PhaNP@Syn71, the MIC was 8.62 μM while the cysteine modified Syn71 alone had an MIC of 8 μM.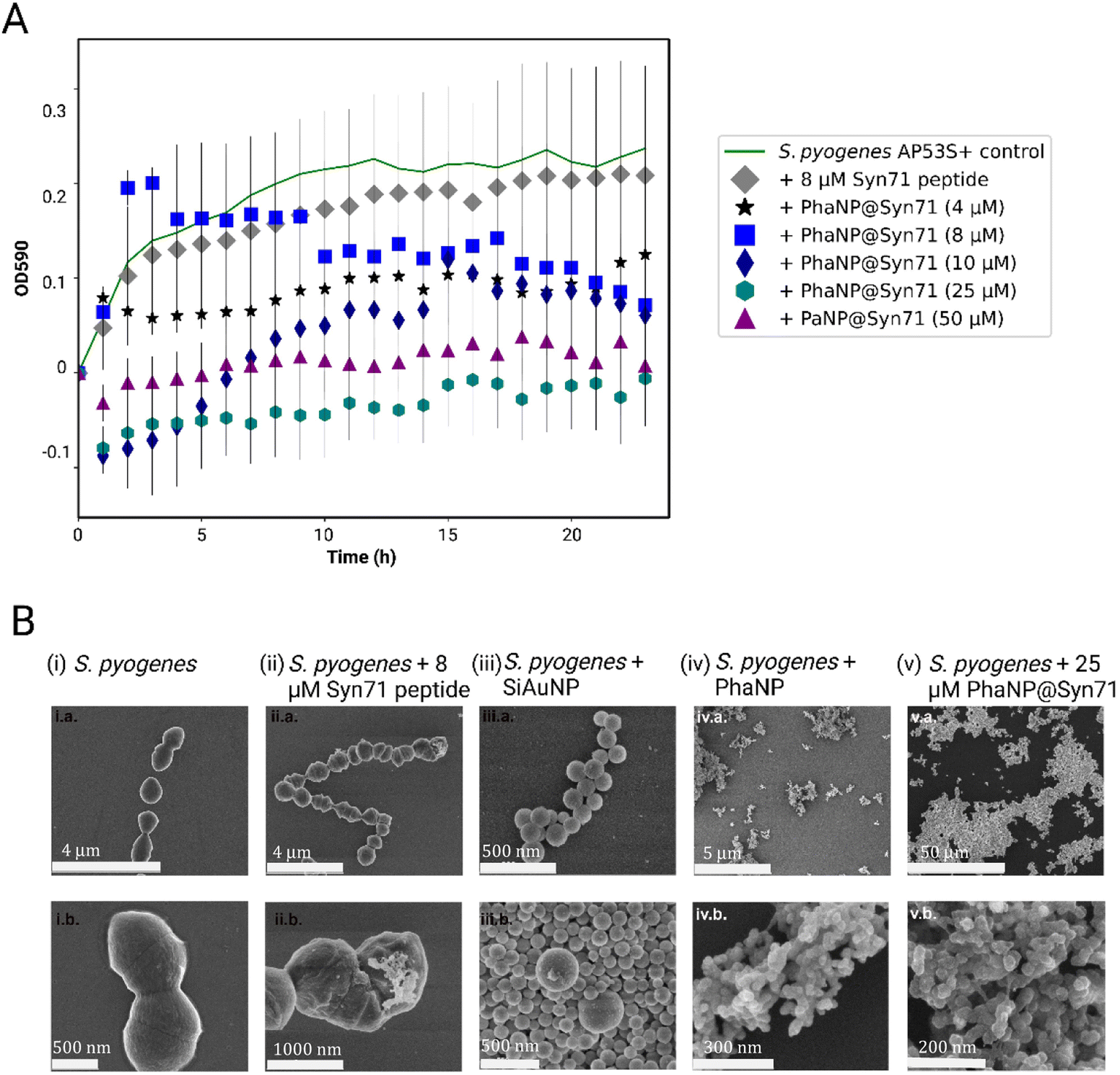 | ||
| Fig. 4 In vitro antibacterial activity of PhaNP@Syn71. (A) S. pyogenes growth curve in the presence of various concentrations of PhaNP@Syn71. The average of four replicates for each sample was computed together with standard deviations. The 4 μM and 8 μM Syn71 peptide concentrations inhibited bacterial growth. The PhaNP@Syn71 with 4 μM Syn71 showed 50% inhibition of bacterial growth immediately. Consistent bacterial inhibition occurred at 10 μM Syn71 on PhaNP@Syn71. >25 μM of Syn71 on PhaNP@Syn71 led to 100% bactericidal activity. (B) SEM images of S. pyogenes incubated with PhaNP@Syn71 compared to controls. (i) S. pyogenes formed classic chain structures. (ii) Syn71 peptide clusters latched onto bacterial structures but bacterial structure stayed intact. (iii) SiO2@AuNP were found bound to bacterial membranes in large numbers and interestingly prevented bacterial chain formation. (iv) The introduction of silver dramatically altered the outcome. PhaNPs resulted in no intact bacteria. (v) PhaNP@Syn71 resulted in no intact bacteria and large rafts of nanoparticles. | ||
| ID | CFU per ml |
|---|---|
| S. pyogenes | 3.17 × 107 ± 5.69 × 106 |
| S. pyogenes + SiO2AuNP | 8.93 × 106 ± 9.45 × 105 |
| S. pyogenes + PhaNPs | 5.13 × 106 ± 5.13 × 105 |
| S. pyogenes + PhaNP@Syn71 (50 μM) | 0 |
| S. pyogenes + PhaNP@Syn71 (25 μM) | 5.13 × 106 ± 1.53 × 105 |
| S. pyogenes + PhaNP@Syn71 (10 μM) | 3.93 × 106 ± 1.36 × 106 |
| S. pyogenes + PhaNP@Syn71 (8 μM) | 4.53 × 106 ± 4.04 × 105 |
| S. pyogenes + PhaNP@Syn71 (4 μM) | 4.27 × 106 ± 2.31 × 105 |
SEM imaging was used to visualize the attachment of nanoparticles onto the bacterial cell membrane as well as the further internalization of the nanoparticles inside the bacteria cells (Fig. 4B). S. pyogenes bacteria form chain-like structures in their natural environment. The analysis of the SEM images confirmed the round coccus shape and formation of chain-like structures of untreated S. pyogenes (Fig. 4B(i)). It has been previously shown that enterocin AS-48 and AS-48-like antimicrobial peptides act via their interaction with and disruption of S. pyogenes membrane.41,67 In this case, the Syn71 peptide formed microclusters on the bacteria and punctured the bacterial cell wall, confirming the previously studied effect of the peptide on the bacteria (Fig. 4B(ii)). PhaNPs with Au but without Ag and peptide showed binding to the bacteria surface, this way disrupting the bacterial chain formation (Fig. 4B(iii)). In contrast, PhaNPs with Ag but without the peptide exhibited significant bacterial lysis due to antibacterial properties of Ag (Fig. 4B(iv)).2 PhaNP@Syn71 nanoparticles exhibited rapid lysis of bacteria, leading to complete destruction of the bacterial structure with no viable bacteria present in the sample, and only PhaNP@Syn71 were observed on SEM images (Fig. 4B(v)), showing that our designed nanoparticles can eradicate the S. pyogenes from a given site.
Additionally, maleimide dyes were utilized to selectively block the thiol on bacteria to test to see if thiol residues were the primary points of adhesion of PhaNPs (sans peptide and silver) to bacteria (ESI E Fig. S4†). We synthesized fluorescent PhaNPs(Ag(-)peptide(-)) by volume-labelling the silica core with rhodamine using techniques previously developed in our laboratory to track the PhaNPs interaction with bacteria utilizing fluorescence microscopy.68–72 The blocking of thiol residues on the bacterial surface did not hinder the ability of PhaNPs to disrupt the chain forming ability of the bacteria. The results (ESI E Fig. S4†) indicated that PhaNPs(Ag(-)peptide(-)) can bind to bacteria by targeting surface thiols, reducing disulfide bridges to thiols, and bind to amine rich peptide sequences used by the bacteria for forming chain like structures.73–75
3.4 In vivo testing on mouse wound infection healing model
The antibacterial effect of PhaNP@Syn71 was tested in vivo on male wild type (C57bl/6) mouse (n = 3–4) using a Streptococcus pyogenes wound healing model which was approved by the University of Notre Dame's Institutional Animal Care and Use Committee. To introduce the infection, the S. pyogenes strain AP53S+ was injected into the right flank of the mouse subcutaneously (SubQ). On day 2 after the infection, a wound had established, and treatment was begun with two treatment variants: topical application only or a combination of intravenous (IV) injection and topical application.Topical-only treatment of the infected wounds with PhaNP@Syn71 indicated clear stabilization of the wound size (ESI F Fig. S5†), while the vehicle-only and Syn71 peptide-only treatment exhibited an initial increase in the wound size, indicating irritation of the wound. By day 9 the wound had decreased to half of the initial size and fully healed by the end of the treatment course (day 17) with PhaNP@Syn71 topical only treatment. The number of mice with wounds decreased from the initial four to three by day 11 (ESI F Fig. S5†). By day 15, it further decreased to two mice with wounds and by the end of the treatment period (day 17), half of the infected mice had been healed. At the end of the treatment period (day 17), the wound treated with PhaNP@Syn71 was fully healed, while the control and Syn71 peptide-only treatments still showed the presence of a wound (ESI F Fig. S5†).
The combination of the one-time IV injection on day 2 and further daily topical treatment was carried out using vehicle only for both IV and topical (VV), peptide only for both (PP), peptide only for IV in combination with PhaNP@Syn71 topical (NP), and PhaNP@Syn71 only for both IV and topical (NN). The combination of Syn71 peptide IV injections with PhaNP@Syn71 topical treatment resulted in an initial small increase in the wound size, similar to the vehicle-only and Syn71 peptide-only treatments, while PhaNP@Syn71 IV injection combined with PhaNP@Syn71 daily topical treatment of the infected wounds confirmed the initial stabilization of the wound size (Fig. 5A). The latter treatment method resulted in the wound size decreasing to half of the initial wound size by day 8, and wounds being fully healed by day 17. The number of mice with wounds decreased from the initial four to three by day 15 (Fig. 5B). By day 17 there were no mice with wounds present, indicating all mice were healed.
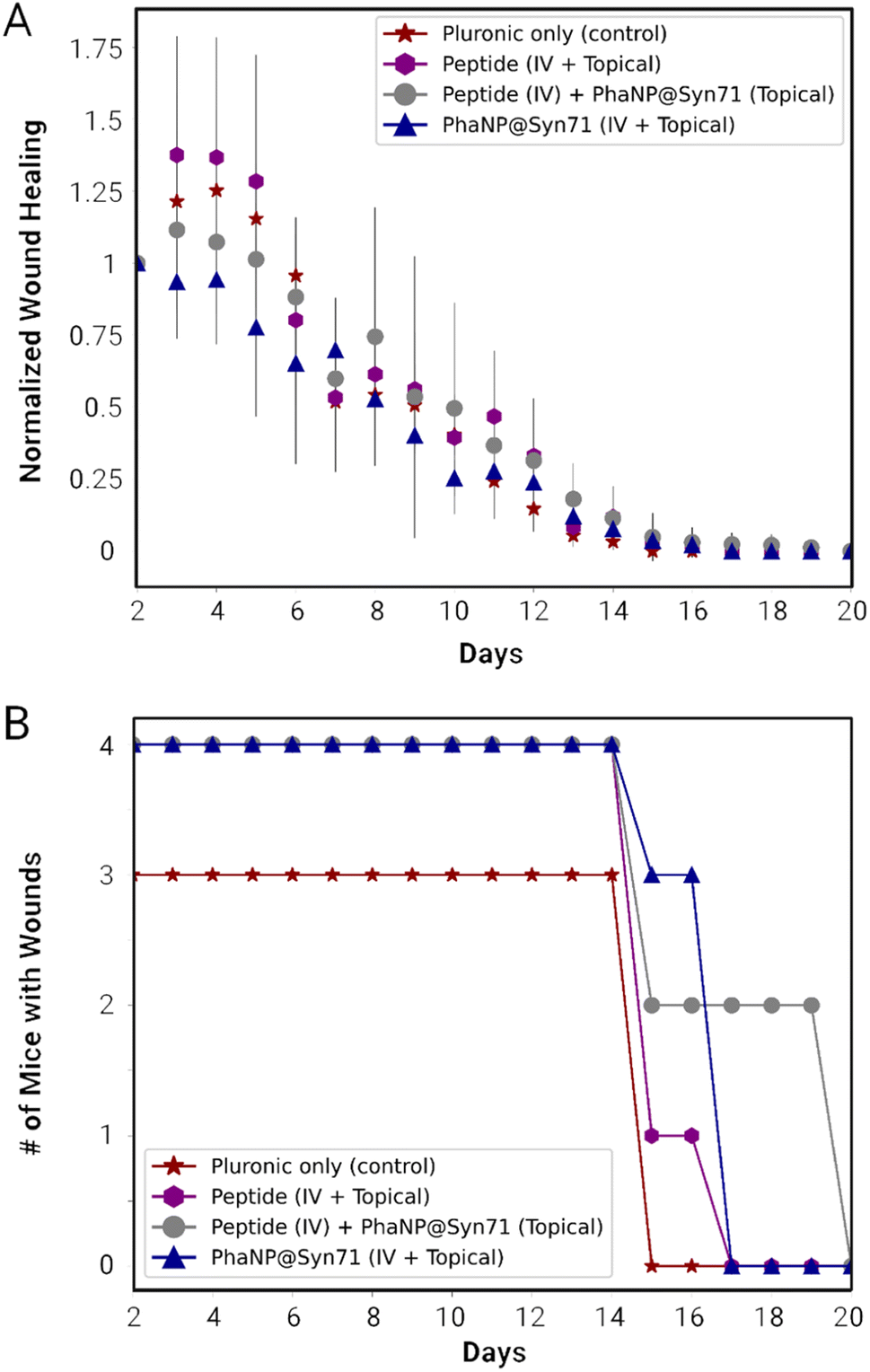 | ||
| Fig. 5 In vivo mouse wound model experiments with a combinatory treatment of one intravenous (IV) injection (100 μM w.r.t Syn71) followed by a daily topical dose of PhaNP@Syn71 (150 μM w.r.t Syn71). (A) Normalized wound healing size in mice treated with PhaNP@Syn71 vs. controls. (B) Number of mice with wounds throughout the treatment. The PhaNP@Syn71 did not seem to increase the rate of wound healing in comparison to the controls, but it immediately stabilized the wound size and prevented it from increasing in size from the first dosage application. Abbreviation's key: pluronic control (VV = IV + topical), peptide control (PP = IV + topical), peptide IV + PhaNP@Syn71 topical (NP) and PhaNP@Syn71 (NN = IV + topical). | ||
Hematoxylin and Eosin (H & E) staining of the dissected organs and skin tissue (Fig. 6A) was carried out by a pathologist to score post-treatment inflammation. There was no significant abnormal inflammation noted. Inflammation was present on skin tissue with the wound for all treatment groups. However, this is a natural occurrence due to the presence of macrophages clearing the bacteria and debris while also releasing cytokines necessary for wound healing and tissue remodeling. In mice treated with Syn71 peptide only IV injection and topical application, and the combination of Syn71 peptide IV injection and PhaNP@Syn71, there was some inflammation noted in the lungs. No additional inflammation of organs was observed in mice treated with the combination of IV injection and topical application using PhaNP@Syn71 treatment. SEM analysis of the skin tissue was carried out (ESI F Fig. S7†) confirming the histology results. Some inflammation was observed, consistent with the histology analysis, since the infected wound disrupted the tissues and created an immune response leading to inflammation. However, there were also many hair follicles present, indicating tissue regeneration.
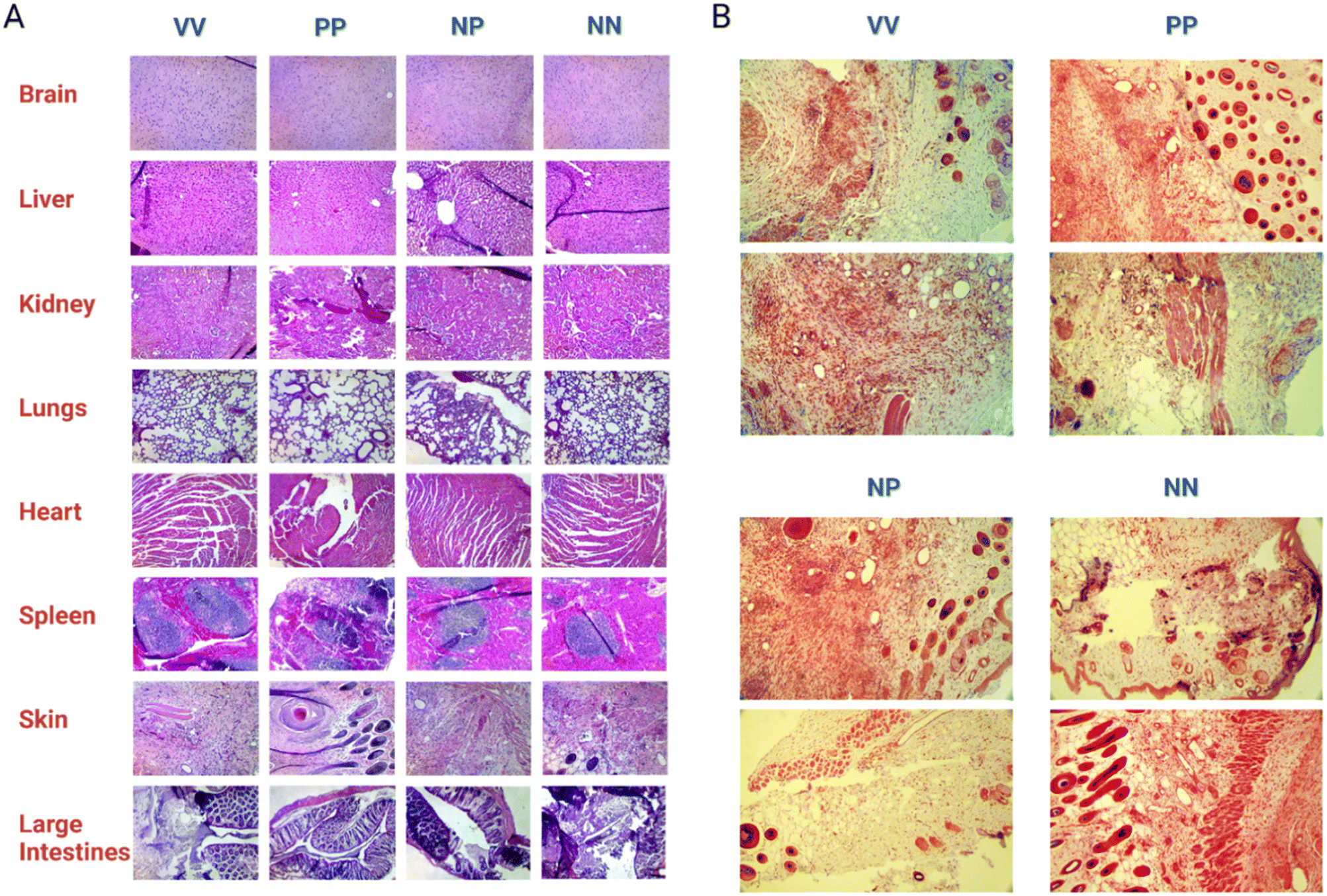 | ||
| Fig. 6 Histology samples of mice organs and skin tissue. (A) H & E staining on mouse organs and skin tissue was analyzed and scored for inflammation by a board-certified pathologist. The results showed no damaging inflammation of the organs in any of the mice. Skin tissue showed signs of inflammation from the wound infection. Abbreviations: VV – vehicle (IV + topical), PP – peptide (IV + topical), NP – peptide (IV) + PhaNP@Syn71 (topical), NN – PhaNP@Syn71 (IV + topical). (B) Serial staining of histology sections of skins by H & E and Gram staining did not reveal any bacterial colonies in the subcutaneous skin sections at the endpoint. Hair growth in the wound indicated wound healing. Histology sections were imaged at 10× magnification. | ||
Although there was mild inflammation in the skin tissue due to the wound infection, the histology samples of mice treated with PhaNP@Syn71 did not show increased inflammation of any of the organs, proving that our nanoparticle design is biocompatible, and does not result in the accumulation of nanoparticles in organs. The Gram-staining of the skin tissue wounds (Fig. 6B) indicated no bacteria present in the wound after the end of the treatment period (day 17). Therefore, PhaNP@Syn71 fully eradicated the bacteria cells within the treatment period. In vivo results indicate that PhaNP@Syn71 topical treatment, as well as the combination of the IV injection and topical treatment, immediately stabilizes the infected wound, while the control and Syn71 peptide-only treatment led to a progression in wound size before the animal's natural immunity stabilizes the wound. This proves the PhaNP@Syn71 design is highly effective in immediately stopping wound growth and in clearing infection.
ICP-OES analysis was carried out on dissected organs and tissues to quantify the elements from nanoparticles (i.e., Si, Au and Ag) present in the organs post-treatment, indicating how much nanoparticles were retained in different organs and tissues (see ESI G Tables S3–S6†). The results were given in mg of elements per 1 g of organ/tissue. In any of the treatment options (VV, PP, NP and NN), there was no Ag present, which would indicate Ag being cleared out or being below the limits of detection of the analytical instrument (5 μg L−1 or 5 ppb). It was observed that there is a higher amount of Si present in the lungs, spleen, and fecal pellet post-treatment. It has been shown previously that silica nanoparticles mainly accumulate in the spleen, liver, and lungs due to endocytosis by macrophages.76 They get trapped in the lungs due to passive entrapment in the endothelium opening of the liver and spleen or in pulmonary capillaries since the reticuloendothelial system internalizes silica nanoparticles after sequestration in the liver and spleen.77 The accumulation of Si in fecal pellets indicates that the clearance of the nanoparticles is via the GI tract due to their degradation in the liver and spleen.78 The Au particles have mainly accumulated in the lungs, spleen and heart. However, as noted before there was no inflammation of any organs due to the PhaNP@Syn71. It was noted that there is a higher accumulation of Si and Au in the NP treatment group but these are classified as GRAS compounds (Generally Regarded as Safe) by the FDA due to their approved use in pharmaceuticals as fillers.79
4 Conclusions
In this study, we have successfully demonstrated that our modular design of phage-mimicking nanoparticles (PhaNPs) modified with antimicrobial peptide Syn71 (PhaNP@Syn71) has high antibacterial activity while exhibiting a low cytotoxic effect on mammalian cells. The biocompatibility tests on HaCaT cells exhibited significantly lower cytotoxicity to the cells (at least 8-fold) than that of the corresponding concentrations of free Syn71 peptide. In addition, PhaNP@Syn71 obtained stably low cytotoxicity up to higher concentrations, with the highest concentration (64 μM) of the PhaNP@Syn71 exhibiting cytotoxicity no higher than the vehicle-only control. Regarding the antibacterial effect of PhaNP@Syn71, they significantly inhibit the growth of Streptococcus pyogenes at concentrations as low as 10 μM and completely kill the bacteria at concentrations >25 μM. S. pyogenes does not achieve its maximum growth potential at any of the PhaNP@Syn71 concentrations tested (4 μM to 64 μM). At equivalent concentrations, PhaNP@Syn71 displayed stronger antibacterial activity than that of its corresponding concentrations of the free Syn71 peptide. SEM analysis to study the mechanisms of action for the antibacterial effect of PhaNP@Syn71 revealed that the PhaNP@Syn71 had attached to S. pyogenes cell wall, lysing the cell and therefore inhibiting its growth and killing it since no bacterial cells were observed on SEM images when incubated with PhaNP@Syn71.PhaNP@Syn71 showed a significant ability to improve the infection treatment outcome in the in vivo mouse wound healing model, and immediately stabilized the S. pyogenes induced wound size from day 1 through the treatment period of 15–17 days. Further histological analysis displayed mild inflammation in skin tissues, while no inflammation was observed in any of the organs, indicating that the PhaNP@Syn71 design is biocompatible. While analyzing the post-treatment accumulation of the nanoparticles, it was shown that the nanoparticles tend to accumulate in the lungs, spleen and large intestines. Therefore, these data show that the post-treatment clearance of the PhaNP@Syn71occurs primarily via the GI tract.
The MIC of PhaNP@Syn71 to S. pyogenes was 8.62 μM, which was slightly higher than the MIC of cysteine-modified Syn71 peptide alone, which was 8 μM. But the cytocompatibility of Syn71 on PhaNP@Syn71 was approximately an order of magnitude higher than the Syn71 peptide alone. The increase range of cytocompatible doses allowed us to test previously untestable concentrations of Syn71 for bactericidal activity without cause for concern of increasing toxicity to human cells. The evolution of resistance study also indicated the absence of resistance after more than 1000 generations of S. pyogenes were exposed to 0.35XMIC (sub-MIC) of PhaNP@Syn71 which is highly significant for the long-term use of PhaNP@Syn71 as an alternate to antibiotics. The therapeutic dose used to treat wound infection was 20X MIC of PhaNP@Syn1 to S. pyogenes.
In conclusion, the results of this study demonstrate that the PhaNP@Syn71 design exhibits high antibacterial activity towards S. pyogenes infections while exhibiting higher biocompatibility with mammalian cells than the free Syn71 peptide, without promoting emergence of resistant bacterial strains when utilized at the appropriate concentration range. This study therefore, lays a foundation for a novel antibiotic-free nanoparticle system to clear bacterial infections with low side-effects, through multiple doses without promoting the emergence of resistant strains of bacteria. This study has demonstrated that PhaNP@Syn71 design will be a key component in the fight against the emergence and spread of antimicrobial resistant strains of bacteria.
Author contributions
Conceptualization: Nallathamby, P. D., Lee, S., Castellino, F., Ploplis, V., Donahue, D.; data curation: Olesk, J., Donahue, D., Ross, J., Hopf, J., Sheehan, C., Armknecht, K., Bennett, Z., and Nallathamby, P. D.; formal analysis: Olesk, J., Donahue, D., Ross, J., Bennett, Z., and Nallathamby, P. D.; funding acquisition: Nallathamby, P. D., Ploplis, V., Castellino, F., and Lee, S.; investigation: Olesk, J., Donahue, D., Ross, J., Kudary, C., Hopf, J., Sheehan, C., Armknecht, K., and Bennett, Z.; methodology: Hopf, J., Donahue, D., Ross, J., and Nallathamby, P. D.,; project administration: Nallathamby, P. D.; resources: Lee, S., Ploplis, V., Castellino, F., and Nallathamby, P. D.; supervision: Nallathamby, P. D., and Kudary, C.; validation: Nallathamby, P. D.; visualization: Bennett, Z., Olesk, J., Donahue, D., and Nallathamby, P. D.; writing – original draft: Olesk, J., and Nallathamby, P. D.; writing – review & editing: Donahue, D., Ross, J., Lee, S., Ploplis, V., Castellino, F., and Nallathamby, P. D.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Nallathamby lab would like to acknowledge the financial and material support of the Notre Dame Berthiaume Institute for Precision Health (BIPH), and the Notre Dame Centre for Nano Science and Technology's (NDNano) undergraduate summer research fellowships (NURF). We thank the staff of the Materials Characterization Facility, ASEND facility, CEST-ND, Freimann Life Sciences Center, technician at the Berthiaume Institute for Precision Health, and the Notre Dame Integrated Imaging Facility for analytical services. ICP-OES measurements were performed on a PerkinElmer Optima 8000 in the Center for Environmental Science and Technology at the University of Notre Dame. Conor Sheehan, Kevin Armknecht and Zachary Bennett were undergraduate research associates. Conor Sheehan (NURF'22) and Kevin Armknecht (NURF'23) had NDNano undergraduate summer research fellowships. Johanna Olesk was funded by the Notre Dame (ND) Energy Materials Science and Engineering Fellowship. The research was supported by a Project Development Team grant from the Indiana Clinical and Translational Sciences Institute (CTSI-PDT: 373037-31005-FY19CTSIK), Institutional Research Grants from the American Cancer Society (ACS IRG-17-182-04, ACS IRG-17-182-04-2) and seed grants from the Notre Dame Berthiaume Institute for Precision Health (372333-31025) and (372717-43310-FY17RFP).References
- S. S. Ahmed, K. E. Diebold, J. M. Brandvold, S. S. Ewaidah, S. Black, A. Ogundimu, Z. Li, N. D. Stone and C. A. Van Beneden, The role of wound care in 2 group A streptococcal outbreaks in a Chicago skilled nursing facility 2015–2016, Open Forum Infect. Dis., 2018, 5, ofy145 CrossRef PubMed.
- M. Deutscher, S. Schillie, C. Gould, J. Baumbach, M. Mueller, C. Avery and C. A. Van Beneden, Investigation of a group A streptococcal outbreak among residents of a long-term acute care hospital, Clin. Infect. Dis., 2011, 52(8), 988–994 CrossRef.
- S. A. Nanduri, B. J. Metcalf, M. A. Arwady, C. Edens, M. A. Lavin, J. Morgan, W. Clegg, A. Beron, J. P. Albertson and R. Link-Gelles, et al., Prolonged and large outbreak of invasive group A Streptococcus disease within a nursing home: repeated intrafacility transmission of a single strain, Clin. Microbiol. Infect., 2019, 25(2), 248.e1–248.e7 CrossRef PubMed.
- K. Fay, J. Onukwube, S. Chochua, W. Schaffner, P. Cieslak, R. Lynfield, A. Muse, C. Smelser, L. H. Harrison and M. Farley, et al., Patterns of antibiotic nonsusceptibility among invasive group A Streptococcus infections—United States, 2006–2017, Clin. Infect. Dis., 2021, 73(11), 1957–1964 CrossRef PubMed.
- J. Ibrahim, J. A. Eisen, G. Jospin, D. A. Coil, G. Khazen and S. Tokajian, Genome analysis of Streptococcus pyogenes associated with pharyngitis and skin infections, PLoS One, 2016, 11(12), e0168177 CrossRef.
- S. Kanwal and P. Vaitla, Streptococcus Pyogenes, StatPearls, 2023 Search PubMed.
- M. K. Siggins, N. N. Lynskey, L. E. Lamb, L. A. Johnson, K. K. Huse, M. Pearson, S. Banerji, C. E. Turner, K. Woollard and D. G. Jackson, et al., Extracellular bacterial lymphatic metastasis drives Streptococcus pyogenes systemic infection, Nat. Commun., 2020, 11(1), 1–12 CrossRef PubMed.
- Y. Li, J. Rivers, S. Mathis, Z. Li, S. Velusamy, S. A. Nanduri, C. A. Van Beneden, P. Snippes-Vagnone, R. Lynfield and L. McGee, et al., Genomic surveillance of Streptococcus pyogenes strains causing invasive disease, United States, 2016–2017, Front. Microbiol., 2020, 11, 1547 CrossRef PubMed.
- L. Zhu, R. J. Olsen, W. Nasser, I. Riva Morales and J. M. Musser, Trading capsule for increased cytotoxin production: contribution to virulence of a newly emerged clade of emm89 Streptococcus pyogenes, mBio, 2015, 6(5), e01378 CrossRef PubMed.
- W. C. Albrich, D. L. Monnet and S. Harbarth, Antibiotic selection pressure and resistance in Streptococcus pneumoniae and Streptococcus pyogenes, Emerging Infect. Dis., 2004, 10(3), 514 CrossRef PubMed.
- M. J. Bottery, J. W. Pitchford and V. P. Friman, Ecology and evolution of antimicrobial resistance in bacterial communities, ISME J., 2021, 15(4), 939–948, DOI:10.1038/s41396-020-00832-7.
- A. F. Johnson and C. N. LaRock, Antibiotic treatment, mechanisms for failure, and adjunctive therapies for infections by group A Streptococcus, Front. Microbiol., 2021, 12, 760255 CrossRef.
- P. Makvandi, C.-Y. Wang, E. N. Zare, A. Borzacchiello, L.-N. Niu and F. R. Tay, Metal-based nanomaterials in biomedical applications: antimicrobial activity and cytotoxicity aspects, Adv. Funct. Mater., 2020, 30(22), 1910021 CrossRef.
- J. M. Munita and C. A. Arias, Mechanisms of antibiotic resistance, Microbiol. Spectr., 2016, 4(2), 0016 Search PubMed.
- V. Cattoir, Mechanisms of Streptococcus pyogenes Antibiotic Resistance, Streptococcus pyogenes: Basic Biology to Clinical Manifestations, 2nd edn, 2022, ch. 30 Search PubMed.
- D. Passàli, M. Lauriello, G. C. Passàli, F. M. Passàli and L. Bellussi, Group A streptococcus and its antibiotic resistance, Acta Otorhinolaryngol. Ital., 2007, 27(1), 27 Search PubMed.
- M. Gakiya-Teruya, L. Palomino-Marcelo, S. Pierce, A. M. Angeles-Boza, V. Krishna and J. C. F. Rodriguez-Reyes, Enhanced antimicrobial activity of silver nanoparticles conjugated with synthetic peptide by click chemistry, J. Nanopart. Res., 2020, 22(4), 1–11 CrossRef.
- A. Gupta, R. F. Landis, C.-H. Li, M. Schnurr, R. Das, Y.-W. Lee, M. Yazdani, Y. Liu, A. Kozlova and V. M. Rotello, Engineered polymer nanoparticles with unprecedented antimicrobial effi cacy and therapeutic indices against multidrug-resistant bacteria and biofilms, J. Am. Chem. Soc., 2018, 140(38), 12137–12143 CrossRef.
- J. S. Kim, K. N. Yu, J.-H. Kim, S. J. Park, H. J. Lee, S. H. Kim, Y. K. Park, Y. H. Park and C.-Y. Hwang, et al., Antimicrobial effects of silver nanoparticles, Nanomed. Nanotechnol. Biol. Med., 2007, 3(1), 95–101 CrossRef PubMed.
- J. Li, W. Sun, Z. Yang, G. Gao, H.-H. Ran, K.-F. Xu, Q.-Y. Duan, X. Liu and F.-G. Wu, Rational design of self-assembled cationic porphyrin-based nanoparticl es for efficient photodynamic inactivation of bacteria, ACS Appl. Mater. Interfaces, 2020, 12(49), 54378–54386 CrossRef PubMed.
- C. A. Marangon, V. C. A. Martins, M. H. Ling, C. C. Melo, A. M. G. Plepis, R. L. Meyer and M. Nitschke, Combination of rhamnolipid and chitosan in nanoparticles boosts their antimicrobial efficacy, ACS Appl. Mater. Interfaces, 2020, 12(5), 5488–5499 CrossRef CAS PubMed.
- P. D. Nallathamby, K. J. Lee, T. Desai and X. H. Xu, Study of the multidrug membrane transporter of single living Pseudomonas aeruginosa cells using size-dependent plasmonic nanoparticle optical probes, Biochemistry, 2010, 49(28), 5942–5953, DOI:10.1021/bi100268k.
- S. M. Dizaj, F. Lotfipour, M. Barzegar-Jalali, M. H. Zarrintan and K. Adibkia, Antimicrobial activity of the metals and metal oxide nanoparticles, Mater. Sci. Eng. C, 2014, 44, 278–284 CrossRef CAS PubMed.
- M. A. Ansari, H. M. Khan, A. A. Khan, M. K. Ahmad, A. A. Mahdi, R. Pal and S. S. Cameotra, Interaction of silver nanoparticles with Escherichia coli and their cell envelope biomolecules, J. Basic Microbiol., 2014, 54(9), 905–915 CrossRef CAS PubMed.
- P. Jena, M. Bhattacharya, G. Bhattacharjee, B. Satpati, P. Mukherjee, D. Senapati and R. Srinivasan, Bimetallic gold-silver nanoparticles mediate bacterial killing by disr upting the actin cytoskeleton MreB, Nanoscale, 2020, 12(6), 3731–3749 RSC.
- T. Bruna, F. Maldonado-Bravo, P. Jara and N. Caro, Silver nanoparticles and their antibacterial applications, Int. J. Mol. Sci., 2021, 22(13), 7202 CrossRef PubMed.
- G. Zhao and S. E. Stevens, Multiple parameters for the comprehensive evaluation of the susceptibility of Escherichia coli to the silver ion, BioMetals, 1998, 11, 27–32 CrossRef PubMed.
- A. Ivask, A. ElBadawy, C. Kaweeteerawat, D. Boren, H. Fischer, Z. Ji, C. H. Chang, R. Liu, T. Tolaymat and D. Telesca, et al., Toxicity mechanisms in Escherichia coli vary for silver nanoparticles and differ from ionic silver, ACS Nano, 2014, 8(1), 374–386 CrossRef PubMed.
- A. Salleh, R. Naomi, N. D. Utami, A. W. Mohammad, E. Mahmoudi, N. Mustafa and M. B. Fauzi, The potential of silver nanoparticles for antiviral and antibacterial applications: a mechanism of action, Nanomaterials, 2020, 10(8), 1566 CrossRef PubMed.
- S. S. I. Abdalla, H. Katas, J. Y. Chan, P. Ganasan, F. Azmi and M. F. M. Busra, Antimicrobial activity of multifaceted lactoferrin or graphene oxide functionalized silver nanocomposites biosynthesized using mushroom waste and chitosan, RSC Adv., 2020, 10(9), 4969–4983 RSC.
- O. Choi, C.-P. Yu, G. E. Fernández and Z. Hu, Interactions of nanosilver with Escherichia coli cells in plank tonic and biofilm cultures, Water Res., 2010, 44(20), 6095–6103 CrossRef CAS PubMed.
- P. V. AshaRani, G. Low Kah Mun, M. P. Hande and S. Valiyaveettil, Cytotoxicity and genotoxicity of silver nanoparticles in human cells, ACS Nano, 2009, 3(2), 279–290 CrossRef CAS PubMed.
- P. D. Nallathamby and X. H. Xu, Study of cytotoxic and therapeutic effects of stable and purified silver nanoparticles on tumor cells, Nanoscale, 2010, 2(6), 942–952, 10.1039/c0nr00080a.
- B. Casciaro, M. Moros, S. Rivera-Fernandez, A. Bellelli, J. M. Fuente and M. L. Mangoni, Gold-nanoparticles coated with the antimicrobial peptide esculentin-1a (1-21) NH2 as a reliable strategy for antipseudomonal drugs, Acta Biomater., 2017, 47, 170–181 CrossRef PubMed.
- J. Gao, H. Na, R. Zhong, M. Yuan, J. Guo, L. Zhao, Y. Wang, L. Wang and F. Zhang, One step synthesis of antimicrobial peptide protected silver nanoparticles: the core-shell mutual enhancement of antibacterial activity, Colloids Surf., B, 2020, 186, 110704 CrossRef.
- W. Li, Y. Li, P. Sun, N. Zhang, Y. Zhao, S. Qin and Y. Zhao, Antimicrobial peptide-modified silver nanoparticles for enhancing the antibacterial efficacy, RSC Adv., 2020, 10(64), 38746–38754 RSC.
- S. Srivastava, K. Dashora, K. L. Ameta, N. P. Singh, H. A. El-Enshasy, M. C. Pagano, A. E.-L. Hesham, G. D. Sharma, M. Sharma and A. Bhargava, Cysteine-rich antimicrobial peptides from plants: the future of antimicrobial therapy, Phytother. Res., 2021, 35(1), 256–277 CrossRef PubMed.
- L. Wang, C. Hu and L. Shao, The antimicrobial activity of nanoparticles: present situation and pro spects for the future, Int. J. Nanomed., 2017, 12, 1227 CrossRef PubMed.
- S. Ruden, K. Hilpert, M. Berditsch, P. Wadhwani and A. S. Ulrich, Synergistic interaction between silver nanoparticles and membrane-perm eabilizing antimicrobial peptides, Antimicrob. Agents Chemother., 2009, 53(8), 3538–3540 CrossRef PubMed.
- J. Hopf, M. Waters, V. Kalwajtys, K. E. Carothers, R. K. Roeder, J. D. Shrout, S. W. Lee and P. D. Nallathamby, Phage-mimicking antibacterial core-shell nanoparticles, Nanoscale Adv., 2019, 1(12), 4812–4826 RSC.
- J. N. Ross, F. R. Fields, V. R. Kalwajtys, A. J. Gonzalez, S. O’Connor, A. Zhang, T. E. Moran, D. E. Hammers, K. E. Carothers and S. W. Lee, Synthetic peptide libraries designed from a minimal alpha-helical doma in of AS-48-bacteriocin homologs exhibit potent antibacterial activity, Front. Microbiol., 2020, 11, 589666 CrossRef PubMed.
- E. M. Windels, J. E. Michiels, M. Fauvart, T. Wenseleers, B. Bergh and J. Michiels, Bacterial persistence promotes the evolution of antibiotic resistance by increasing survival and mutation rates, ISME J., 2019, 13(5), 1239–1251 CrossRef PubMed.
- S. T. Gentry, S. J. Fredericks and R. Krchnavek, Controlled particle growth of silver sols through the use of hydroquin one as a selective reducing agent, Langmuir, 2009, 25(5), 2613–2621 CrossRef PubMed.
- T. Huang, P. D. Nallathamby and X. H. Xu, Photostable single-molecule nanoparticle optical biosensors for real-time sensing of single cytokine molecules and their binding reactions, J. Am. Chem. Soc., 2008, 130(50), 17095–17105, DOI:10.1021/ja8068853.
- T. Huang, P. D. Nallathamby, D. Gillet and X. H. Xu, Design and synthesis of single-nanoparticle optical biosensors for imaging and characterization of single receptor molecules on single living cells, Anal. Chem., 2007, 79(20), 7708–7718, DOI:10.1021/ac0709706.
- Y. Xue, X. Li, H. Li and W. Zhang, Quantifying thiol-gold interactions towards the efficient strength control, Nat. Commun., 2014, 5, 4348, DOI:10.1038/ncomms5348.
- Y. Lyu, L. M. Becerril, M. Vanzan, S. Corni, M. Cattelan, G. Granozzi, M. Frasconi, P. Rajak, P. Banerjee and R. Ciancio, et al., The Interaction of Amines with Gold Nanoparticles, Adv. Mater., 2023, e2211624, DOI:10.1002/adma.202211624.
- C.-C. Liang, A. Y. Park and J.-L. Guan, In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro, Nat. Protoc., 2007, 2(2), 329–333 CrossRef CAS PubMed.
- R. Humphries, A. M. Bobenchik, J. A. Hindler and A. N. Schuetz, Overview of Changes to the Clinical and Laboratory Standards Institute Performance Standards for Antimicrobial Susceptibility Testing, M100, 31st Edition, J. Clin. Microbiol., 2021, 59(12), e0021321, DOI:10.1128/JCM.00213-21.
- R. Vinchhi, C. Jena and N. Matange, Adaptive laboratory evolution of antimicrobial resistance in bacteria for genetic and phenotypic analyses, STAR Protoc., 2023, 4(1), 102005, DOI:10.1016/j.xpro.2022.102005.
- M. Lagator, H. Uecker and P. Neve, Adaptation at different points along antibiotic concentration gradients, Biol. Lett., 2021, 17(5), 20200913, DOI:10.1098/rsbl.2020.0913.
- T. L. McGinnity, V. Sokolova, O. Prymak, P. D. Nallathamby, M. Epple and R. K. Roeder, Colloidal stability, cytotoxicity, and cellular uptake of HfO2 nanoparticles, J. Biomed. Mater. Res., Part B, 2021, 109(10), 1407–1417 CrossRef CAS PubMed.
- K. Suttiponparnit, J. Jiang, M. Sahu, S. Suvachittanont, T. Charinpanitkul and P. Biswas, Role of Surface Area, Primary Particle Size, and Crystal Phase on Titanium Dioxide Nanoparticle Dispersion Properties, Nanoscale Res. Lett., 2010, 6(1), 27, DOI:10.1007/s11671-010-9772-1.
- D. S. Nikam, S. V. Jadhav, V. M. Khot, R. S. Ningthoujam, C. K. Hong, S. S. Mali and S. H. Pawar, Colloidal stability of polyethylene glycol functionalized Co0.5Zn0.5Fe2O4 nanoparticles: effect of pH, sample and salt concentration for hyperthermia application, RSC Adv., 2014, 4(25), 12662–12671, 10.1039/C3RA47319H.
- H. Jaganathan and B. Godin, Biocompatibility assessment of Si-based nano-and micro-particles, Adv. Drug Delivery Rev., 2012, 64(15), 1800–1819 CrossRef CAS.
- J. F. Zimmerman, R. Parameswaran, G. Murray, Y. Wang, M. Burke and B. Tian, Cellular uptake and dynamics of unlabeled freestanding silicon nanowires, Sci. Adv., 2016, 2(12), e1601039 CrossRef PubMed.
- J. McCarthy, I. Inkielewicz-Stępniak, J. J. Corbalan and M. W. Radomski, Mechanisms of toxicity of amorphous silica nanoparticles on human lung submucosal cells in vitro: protective effects of fisetin, Chem. Res. Toxicol., 2012, 25(10), 2227–2235 Search PubMed.
- R. Salomoni, P. Léo, A. F. Montemor, B. G. Rinaldi and M. F. A. Rodrigues, Antibacterial effect of silver nanoparticles in Pseudomonas aeruginosa, Nanotechnol. Sci. Appl., 2017, 115–121 Search PubMed.
- W. Liu, Y. Wu, C. Wang, H. C. Li, T. Wang, C. Y. Liao, L. Cui, Q. F. Zhou, B. Yan and G. B. Jiang, Impact of silver nanoparticles on human cells: effect of particle size, Nanotoxicology, 2010, 4(3), 319–330 CrossRef CAS.
- R. Krishnan, V. Arumugam and S. K. Vasaviah, The MIC and MBC of silver nanoparticles against Enterococcus faecalis - a facultative anaerobe, J. Nanomed. Nanotechnol., 2015, 6(3), 285 Search PubMed.
- J. M. Conlon, E. Ahmed and E. Condamine, Antimicrobial properties of brevinin-2-related peptide and its analogs: efficacy against multidrug-resistant Acinetobacter baumannii, Chem. Biol. Drug Des., 2009, 74(5), 488–493 CrossRef CAS.
- J. M. Conlon, E. Ahmed, L. Coquet, T. Jouenne, J. Leprince, H. Vaudry and J. D. King, Peptides with potent cytolytic activity from the skin secretions of th e North American leopard frogs, Lithobates blairi and Lithob ates yavapaiensis, Toxicon, 2009, 53(7–8), 699–705 CrossRef.
- L.-H. Peng, Y.-F. Huang, C.-Z. Zhang, J. Niu, Y. Chen, Y. Chu, Z.-H. Jiang, J.-Q. Gao and Z.-W. Mao, Integration of antimicrobial peptides with gold nanoparticles as uniqu e non-viral vectors for gene delivery to mesenchymal stem cells with a ntibacterial activity, Biomaterials, 2016, 103, 137–149 CrossRef PubMed.
- C. Zannella, S. Shinde, M. Vitiello, A. Falanga, E. Galdiero, A. Fahmi, B. Santella, L. Nucci, R. Gasparro and M. Galdiero, et al., Antibacterial activity of indolicidin-coated silver nanoparticles in oral disease, Appl. Sci., 2020, 10(5), 1837 CrossRef.
- L. M. Browning, K. J. Lee, P. D. Nallathamby, P. K. Cherukuri, T. Huang, S. Warren and X. N. Xu, Single Nanoparticle Plasmonic Spectroscopy for Study of Charge-Dependent Efflux Function of Multidrug ABC Transporters of Single Live Bacillus subtilis Cells, J. Phys. Chem. C. Nanomater. Interfaces, 2016, 120(37), 21007–21016, DOI:10.1021/acs.jpcc.6b03313.
- K. J. Lee, L. M. Browning, P. D. Nallathamby and X. H. Xu, Study of charge-dependent transport and toxicity of peptide-functionalized silver nanoparticles using zebrafish embryos and single nanoparticle plasmonic spectroscopy, Chem. Res. Toxicol., 2013, 26(6), 904–917, DOI:10.1021/tx400087d.
- M. Maqueda, A. Gálvez, M. M. Bueno, M. J. Sanchez-Barrena, C. González, A. Albert, M. Rico and E. Valdivia, Peptide AS-48: prototype of a new class of cyclic bacteriocins, Curr. Protein Pept. Sci., 2004, 5(5), 399–416 CrossRef PubMed.
- P. D. Nallathamby, J. Hopf, L. E. Irimata, T. L. McGinnity and R. K. Roeder, Preparation of fluorescent Au-SiO(2) core-shell nanoparticles and nanorods with tunable silica shell thickness and surface modification for immunotargeting, J. Mater. Chem. B, 2016, 4(32), 5418–5428, 10.1039/c6tb01659f.
- W. Wang, P. D. Nallathamby, C. M. Foster, J. L. Morrell-Falvey, N. P. Mortensen, M. J. Doktycz, B. Gu and S. T. Retterer, Volume labeling with Alexa Fluor dyes and surface functionalization of highly sensitive fluorescent silica (SiO2) nanoparticles, Nanoscale, 2013, 5(21), 10369–10375, 10.1039/c3nr02639f.
- M. Waters, J. Hopf, E. Tam, S. Wallace, J. Chang, Z. Bennett, H. Aquino, R. K. Roeder, P. Helquist and M. S. Stack, et al., Biocompatible, Multi-Mode, Fluorescent, T(2) MRI Contrast Magnetoelectric-Silica Nanoparticles (MagSiNs), for On-Demand Doxorubicin Delivery to Metastatic Cancer Cells, Pharmaceuticals, 2022, 15(10), 1216 CrossRef.
- Y. Zhang, P. D. Nallathamby, G. D. Vigil, A. A. Khan, D. E. Mason, J. D. Boerckel, R. K. Roeder and S. S. Howard, Super-resolution fluorescence microscopy by stepwise optical saturation, Biomed. Opt. Express, 2018, 9(4), 1613–1629, DOI:10.1364/BOE.9.001613.
- X.-H. N. Xu, Y. Song and P. Nallathamby, Probing membrane transport of single live cells using single-molecule detection and single nanoparticle assay, in New Frontiers in Ultrasensitive Bioanalysis, 2007, pp. 41–70 Search PubMed.
- I. M. Frick, M. Morgelin and L. Bjorck, Virulent aggregates of Streptococcus pyogenes are generated by homophilic protein-protein interactions, Mol. Microbiol., 2000, 37(5), 1232–1247, DOI:10.1046/j.1365-2958.2000.02084.x.
- J. Kim, M. Yeom, T. Lee, H. O. Kim, W. Na, A. Kang, J. W. Lim, G. Park, C. Park and D. Song, et al., Porous gold nanoparticles for attenuating infectivity of influenza A virus, J. Nanobiotechnol., 2020, 18(1), 54, DOI:10.1186/s12951-020-00611-8.
- Y. Lyu, L. M. Becerril, M. Vanzan, S. Corni, M. Cattelan, G. Granozzi, M. Frasconi, P. Rajak, P. Banerjee and R. Ciancio, et al., The Interaction of Amines with Gold Nanoparticles, Adv. Mater., 2023, 2211624 CrossRef.
- G. Xie, J. Sun, G. Zhong, L. Shi and D. Zhang, Biodistribution and toxicity of intravenously administered silica nano particles in mice, Arch. Toxicol., 2010, 84, 183–190 CrossRef.
- T. Yu, D. Hubbard, A. Ray and H. Ghandehari, In vivo biodistribution and pharmacokinetics of silica nanopart icles as a function of geometry, porosity and surface characteristics, J. Controlled Release, 2012, 163(1), 46–54 CrossRef.
- R. Kumar, I. Roy, T. Y. Ohulchanskky, L. A. Vathy, E. J. Bergey, M. Sajjad and P. N. Prasad, In vivo biodistribution and clearance studies using multimodal organically modified silica nanoparticles, ACS Nano, 2010, 4(2), 699–708 CrossRef.
- E. A. Steele, C. Breen, E. Campbell and R. Martin, Food regulations and enforcement in the USA, in Reference Module in Food Science, Elsevier, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3na00620d |
| This journal is © The Royal Society of Chemistry 2024 |