
Ductile adhesive elastomers with force-triggered ultra-high adhesion strength†
Xiao
Zhao
 a,
Zoriana
Demchuk
a,
Jia
Tian
b,
Jiancheng
Luo
a,
Bingrui
Li
c,
Ke
Cao
a,
Alexei P.
Sokolov
ad,
Diana
Hun
*e,
Tomonori
Saito
*ac and
Peng-Fei
Cao
*b
a,
Zoriana
Demchuk
a,
Jia
Tian
b,
Jiancheng
Luo
a,
Bingrui
Li
c,
Ke
Cao
a,
Alexei P.
Sokolov
ad,
Diana
Hun
*e,
Tomonori
Saito
*ac and
Peng-Fei
Cao
*b
aChemical Sciences Division, Oak Ridge National Laboratory, Oak Ridge, TN 37830, USA. E-mail: saitot@ornl.gov
bState Key Laboratory of Organic–Inorganic Composites, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: caopf@buct.edu.cn
cThe Bredesen Center for Interdisciplinary Research and Graduate Education, University of Tennessee, Knoxville, TN 37996, USA
dDepartment of Chemistry, University of Tennessee, Knoxville, TN 37996, USA
eBuildings and Transportation Science Division, Oak Ridge National Laboratory, Oak Ridge, TN 37830, USA. E-mail: hunde@ornl.gov
First published on 6th December 2023
Abstract
Elastomers play a vital role in many forthcoming advanced technologies in which their adhesive properties determine materials’ interface performance. Despite great success in improving the adhesive properties of elastomers, permanent adhesives tend to stick to the surfaces prematurely or result in poor contact depending on the installation method. Thus, elastomers with on-demand adhesion that is not limited to being triggered by UV light or heat, which may not be practical for scenarios that do not allow an additional external source, provide a solution to various challenges in conventional adhesive elastomers. Herein, we report a novel, ready-to-use, ultra high-strength, ductile adhesive elastomer with an on-demand adhesion feature that can be easily triggered by a compression force. The precursor is mainly composed of a capsule-separated, two-component curing system. After a force-trigger and curing process, the ductile adhesive elastomer exhibits a peel strength and a lap shear strength of 1.2 × 104 N m−1 and 7.8 × 103 kPa, respectively, which exceed the reported values for advanced ductile adhesive elastomers. The ultra-high adhesion force is attributed to the excellent surface contact of the liquid-like precursor and to the high elastic modulus of the cured elastomer that is reinforced by a two-phase design. Incorporation of such on-demand adhesion into an elastomer enables a controlled delay between installation and curing so that these can take place under their individual ideal conditions, effectively reducing the energy cost, preventing failures, and improving installation processes.
New conceptsIn this manuscript, we pioneer the incorporation of the unique “on-demand” adhesion feature into functional ductile elastomers, which is completely different from regular pressure-sensitive adhesives. The adhesion feature is easily triggered by mechanical force when needed, and such a feature can effectively reduce the energy cost, prevent damage and improve convenience during handling. After a force trigger and curing process, it exhibits a peel strength and a lap shear strength of 1.2 × 104 N m−1 and 7.8 × 103 kPa, respectively, which top the values reported for advanced functional adhesive elastomers. The rheological properties of the adhesive elastomer after curing reveal that the excellent ability to adapt to rough surfaces and the reinforced elastomer modulus contribute to the ultra-high adhesion strength. The unique “on-demand” adhesion of functional elastomers will shed light on fabricating functional polymeric materials with different applications. |
1. Introduction
Elastomers are rooted in every aspect of our daily life as they are used in consumer and industrial products like sealants, vibration dampers, tires, footwear, toys, furniture, packaging, adhesives, additives, etc. They play a vital role in forthcoming advanced technologies, including but not limited to flexible displays, wearable devices, soft robotics, invasive surgery, building construction, solid-state batteries, and artificial human skin.1–9 Chemical or physical crosslinking of natural rubber, polyurethane, polybutadiene, neoprene, co-polyester, or silicone-based polymers, allow ductile elastomers to possess properties like good extensibility, decent elastic modulus, remarkable recoverability (low mechanical loss), and excellent chemical resistance.10–15 Aside from these fundamental properties, advanced ductile elastomers can have other special features such as self-healing, chemical stability, dielectric response, thermal or magnetic sensitivity, and high optical transparency. These features are typically accomplished by the introduction of single or multiple dynamic bonds, the addition of functional additives, formation of micro-phase separations, manipulation of molecular topology, or engineering into special geometry or composites.16–33 For example, a graphene-containing liquid crystalline elastomer enabled photo-actuation by absorbing near-infrared light and triggering nematic-to-isotropic phase transition, achieving high actuation force and reversible flexibility.22 The interpenetration of the conductive PEDOT:PSS and zwitterionic poly(HEAA-co-SBAA) networks allowed the fabrication of a fully polymeric strain sensor with high mechanical recovery and strong surface adhesion.34 A super stretchable (≈2260%) and recyclable polyurethane elastomer was recently achieved by introducing dynamic π–π motifs and phosphorus-containing moieties.15 An ionic conducting elastomer with autonomous self-healability at 5 °C also serves as an efficient protecting layer for the stable cycling of lithium-metal batteries at relatively low temperatures.35With significantly growing demands of functional ductile elastomers in advanced technologies, their adhesive properties play a critical role in determining their lifetime and reliability in the designated fields.36–45 The incorporation of intermolecular interactions is an important strategy to improve the adhesive properties of elastomers. For example, Chen et al. and Tan et al. independently demonstrated that the incorporation of a small number of hydrogen-bonding 2-ureido-4[1H]-pyrimidinone (UPy) groups enhanced the adhesion force of the elastic materials to organic and inorganic substrates for finger strain sensors and soft robot actuators.46,47 By using the monomer of 4-acryloyl morpholine that intrinsically possesses hydrogen-bonding interactions, a self-adhesive ionic elastomer was synthesized that exhibited a peel strength of 28 N m−1 on a paper substrate and allowed the assembly of stretchable motion energy harvester.48 The addition of 15 mol% dopamine increased the adhesive strength of urethane elastomers from 30 to 71 kPa when used as bio-interfacial electrodes.27 A series of self-healable, adhesive elastomers were also demonstrated to show ultra-high adhesion force, even on dusty surfaces (3488 N m−1), by simply mixing the self-healing polymer and commercially available curable elastomers.25 Aside from physical interactions, adjusting the surface geometries in the micro-scale also facilitates the adhesion of elastomers to various substrates. For example, the adhesion force of the poly(dimethyl siloxane) (PDMS) elastomer to the human skin was improved by 10 times due to the enhanced van der Waals force via the formation of micrometer-sized wrinkles on the elastomer, which was rendered by the addition of 0.004 wt% ethoxylated poly(ethyleneimine).49 By controlling the interfacial interaction-induced surface wrinkling between poly(ι-lactide) and PDMS, a single-component micro-wrinkled PDMS elastomer was shown to have tunable adhesion capabilities.50 Although the enhancement of adhesion force has been demonstrated for functional ductile elastomers, few have the flexibility of the on-demand control when full-adhesion occurs. Therefore, they often require extra protection and labor before and during the installation onto the target substrate and lack convenience during handling. Hence, developing a force-triggered on-demand feature is a more practical, one-step solution for scenarios such as flexible devices or prefab construction that will significantly benefit from having a controlled delay between installation and adhesion.
Microencapsulation technology, which originated from the pursuit of a protective envelope that can control the exchange of materials such as a living cell, has been widely adopted in different applications such as extrinsic self-healing,51–53 food protection,54 drug delivery,55 wound management,56,57 photovoltaic protection,58 thermal sensitive adhesives,59etc. In general, the active ingredients encapsulated in the micro shells are released continuously at a slow rate or completely upon shell rupture. The latter could be triggered by physical stimuli, such as macroscopic fracture,51,60,61 temperature,59,62 ultra-sound,63 and compression.64–66 Among the above, the compression force is the most convenient trigger, as no thermal or acoustic equipment is required. Also, it allows a universal breakage of microcapsules that triggers adhesion in comparison to the crack or fracture that only triggers local healing.
Herein, we demonstrate a novel design for an on-demand, ultra-high strength ductile adhesive elastomer, whose adhesion is triggered by compression force. This triggering method is more robust than the moisture or light-triggered curing, since such factors are normally difficult to avoid under ambient conditions.67 In general, we selected a urea-based elastomer that is fabricated by methylene diphenyl diisocyanate (MDI)-prepolymer and polypropylene glycol (PPG)-diamine as resin and curing agents, respectively. The on-demand feature is achieved through a compression force to break the encapsulated resin agent and trigger the curing reaction, as illustrated in Fig. 1. Notably, our design is different from the two/multi-component self-healing,51,60,61 of which our adhesive elastomer is homogenously cured with mechanical performance and interfacial binding significantly improved, whereas the microencapsulation-based self-healing aims at repeatedly restoring the local damage. Also, this adhesive elastomer is different from the conventional pressure-sensitive adhesives, as the latter involves merely physical tacking and typically low adhesion on rough surfaces as will be discussed later. Here, the combination of the intrinsic adaptability on the receiving substrate, i.e., micro-scale geometry adaption, and the reinforced mechanical strength enabled the developed ductile adhesive elastomer to achieve an ultra-high adhesion force with peel strength and adhesion strength exceeding magnitudes of 104 N m−1 and 103 kPa, respectively. To the best of our knowledge, the adhesion force of such elastomers outperforms that of the currently reported ductile adhesive elastomers. With such ultra-high adhesion force and on-demand adhesion that can be conveniently applied by human force, it is anticipated that such ductile adhesive elastomer can be used in a large variety of applications.
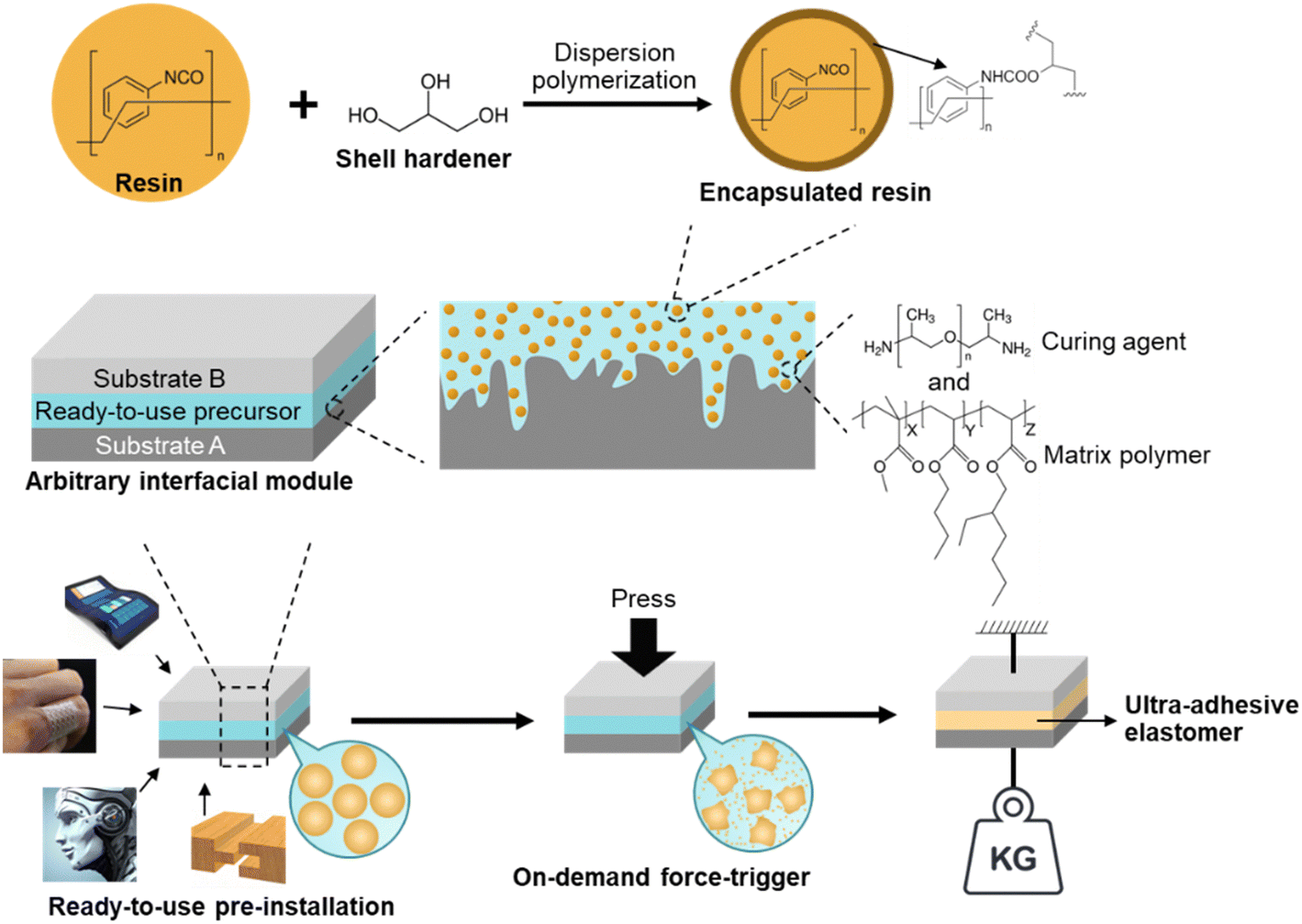 | ||
| Fig. 1 Illustration of the on-demand ultra-high adhesion formation of the ready-to-use precursor and its preparation process. The resin, curing agent, and matrix polymer are MDI-prepolymer, PPG-diamine, and acrylates copolymer, respectively. | ||
2. Results and discussion
The ready-to-use elastomer precursor is composed of an encapsulated MDI-prepolymer and PPG-diamine, as reactive and curing agents, respectively, along with a matrix polymer of methyl methacrylate-butyl acrylate-ethylhexyl acrylate random copolymer poly(MMA-r-BA-r-EHA), as depicted in Fig. 1. The reactive and curing agents were selected based on pre-screens of curing rate from various two-component elastomer systems, between a reactive agent, including bisphenol A diglycidyl ether, unsaturation-modified epoxy, hexamethylene diisocyanate (HDI) prepolymer, and MDI-prepolymer, and a curing agent, including polyethyleneimine (PEI), PPG-triamine, PPG-diamine, and PPG-di-OH.68–70 A suitable curing rate allows adequate time for the substrates that will be adhered to each other to be adjusted while they did not require too long waiting time (<2 hours). The pre-screen tests (ESI,† Table S1) showed that neither diol- nor PEI-type curing agent cured at a reasonable rate. Additional PPG-triamine is required to slow down the PEI-type curing agent, which adds to the complexity. Regarding the reactive agents, the epoxy-based agents cured relatively slow with diamine/triamine, and HDI-prepolymer cured too fast with the diamine in which the additional diol is required for a suitable curing rate. Therefore, the combination of the MDI-prepolymer and PPG-diamine was the most suitable among them, considering their curing rate as well as the raw materials availability and the ability to be scaled up. Both the MDI-prepolymer and PPG-diamine contain reactive terminal groups with sufficient chain length to attain flexibility for the obtained elastomer without harming its cohesive strength.71–73 The adhesive random copolymer, i.e., poly(methyl methacrylate-r-butyl acrylate-r-2-ethylhexyl acrylate) was utilized as a polymer matrix, which improves the longevity and interfacial adhesion for the elastomers, especially before the force-triggered curing.The microencapsulation process is established,52,53 and the encapsulated MDI-prepolymer was synthesized using a single-step dispersion polymerization between MDI-prepolymer and glycerol, as illustrated in Fig. 1. Specifically, gum arabic, dodecyl trimethylammonium bromide (DTAB), and glycerol were used as the surfactant, segregation aid, and shell extender with the synthesis protocol demonstrated in Scheme S1 (ESI†). The well-dispersed MDI-prepolymer droplets were partially crosslinked by glycerol to form the shell of the microcapsules,68 sealing the unreacted MDI-prepolymer inside. It is noteworthy that the addition of a cationic surfactant, i.e., DTAB, introduces coulombic repulsion on the surface of the microcapsules that effectively prevents microcapsule aggregation (ESI,† Fig. S1), which increases the robustness of microcapsules during handling. The characteristic chemical structure of the shell was demonstrated by FT-IR as shown in Fig. 2A. The peaks at 1235 and 1310 cm−1 correspond to the urethane ester bond formed between isocyanate groups from MDI-prepolymer and hydroxyl units in glycerol.74,75 Moreover, a significant peak at 2250 cm−1 that corresponds to the isocyanate group was observed for both broken microcapsules and MDI-prepolymers,76 indicating the presence of a significant amount of reactive isocyanate groups inside the microcapsule. The DSC result (ESI,† Fig. S2) of the MDI-prepolymer-containing microcapsule also confirmed the presence of an isocyanate group. The diameter of the microcapsule was tunable from 10 to 900 μm by changing the mixing speed of the agitator during dispersion. The plot of microcapsule diameters vs. agitating speeds is provided in Fig. 2B, along with the representative optical microscope images for samples prepared at 300 and 1000 rpm. Tuning the size of microcapsules can potentially affect two important factors: (1) the force required to break the microcapsules and trigger the curing reaction; (2) the dispersion of released reactive agents in the polymer matrix. The scanning electron microscopy (SEM) images (ESI,† Fig. S3) clearly showed that the microcapsules can be broken by sufficient external force.
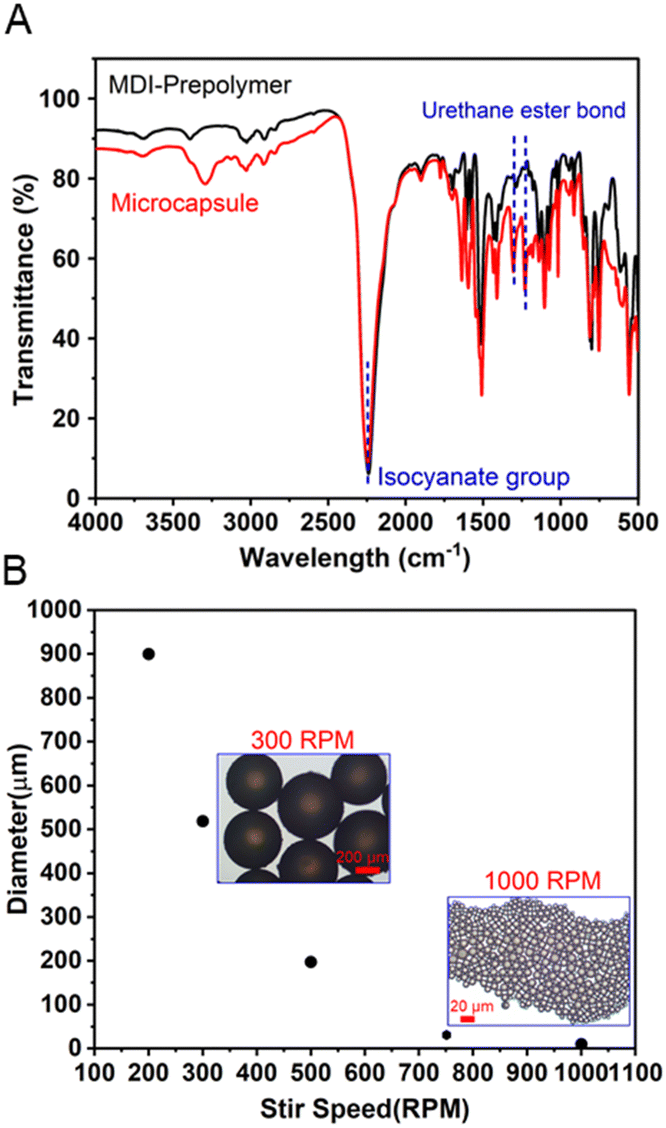 | ||
| Fig. 2 (A) FT-IR spectra of MDI-prepolymer reagent and encapsulated MDI-prepolymer. (B) Diameter of the synthesized microcapsules vs. the agitation speed during emulsion polymerization, along with visualizations of the microcapsules by an optical microscope. | ||
The matrix polymer was synthesized by free-radical random copolymerization of methyl methacrylate (MMA), butyl acrylate (BA), and ethylhexyl acrylate (EHA).77 The random copolymer with the feed ratio of MMA of 35 mol% was selected due to its suitable glass transition temperature (Tg), which optimized the adhesion and self-healability; the BA to 2-EHA feed ratio was 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by weight. The chemical structure of the synthesized matrix polymer was confirmed by 1H NMR (Fig. 3A) and FT-IR spectra (ESI,† Fig. S4), where the composition of the MMA unit was 29 mol%, as calculated by integrating the corresponding 1H NMR peaks, consistent with the feed ratio. The molecular weight (Mn) of the copolymer was 23.6 kDa as measured by GPC, and the relatively broad polydispersity (PDI = 2.62) is typical for such random radical copolymerization. The low Tg (−33 °C using DSC, ESI† Fig. S5) manifested the rubbery nature of such copolymers over a wide temperature range. The dynamic mechanical analysis (DMA) result (ESI,† Fig. S6) clearly demonstrated that the modulus of the matrix polymer was around 2–5 MPa at room temperature, which is important for good interfacial adhesion. The tensile test exhibited excellent extensibility of such matrix polymer with elongation before breaks over 1500%. Having low Tg, high flexibility and adhesion also contributes to the self-healing capability. The self-healability was examined by mechanically cutting the cured adhesive into two parts and subsequently arranging them so that they were in contact with each other. After a 24 hour healing process under ambient conditions, the matrix polymer could fully recover its mechanical properties as shown in Fig. 3B.
:1 by weight. The chemical structure of the synthesized matrix polymer was confirmed by 1H NMR (Fig. 3A) and FT-IR spectra (ESI,† Fig. S4), where the composition of the MMA unit was 29 mol%, as calculated by integrating the corresponding 1H NMR peaks, consistent with the feed ratio. The molecular weight (Mn) of the copolymer was 23.6 kDa as measured by GPC, and the relatively broad polydispersity (PDI = 2.62) is typical for such random radical copolymerization. The low Tg (−33 °C using DSC, ESI† Fig. S5) manifested the rubbery nature of such copolymers over a wide temperature range. The dynamic mechanical analysis (DMA) result (ESI,† Fig. S6) clearly demonstrated that the modulus of the matrix polymer was around 2–5 MPa at room temperature, which is important for good interfacial adhesion. The tensile test exhibited excellent extensibility of such matrix polymer with elongation before breaks over 1500%. Having low Tg, high flexibility and adhesion also contributes to the self-healing capability. The self-healability was examined by mechanically cutting the cured adhesive into two parts and subsequently arranging them so that they were in contact with each other. After a 24 hour healing process under ambient conditions, the matrix polymer could fully recover its mechanical properties as shown in Fig. 3B.
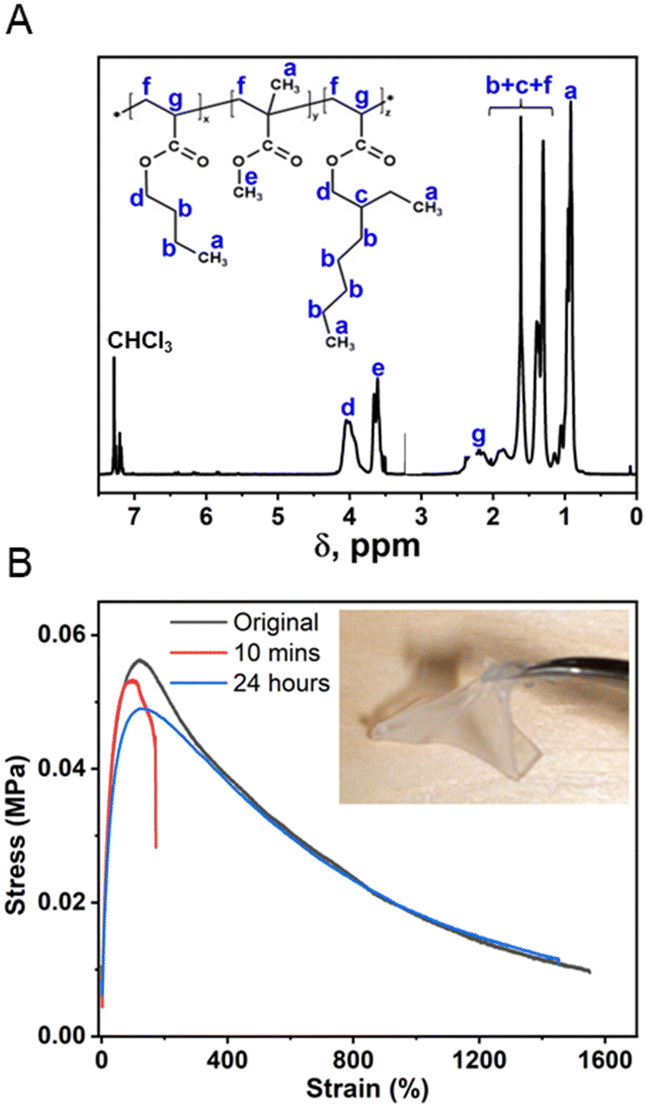 | ||
| Fig. 3 (A) 1H-NMR spectrum of the matrix polymer with characteristic chemical shift peaks marked on the chemical structure. (B) Tensile results of the cut matrix polymer that self-healed for 10 min and 24 h in comparison to the original uncut material. | ||
The ready-to-use precursor was further formulated using the synthesized microcapsules with the encapsulated MDI-prepolymer, PPG-diamine, and matrix polymer. Specifically, we chose the microcapsules that were synthesized under 300 rpm for the following results. The initial formulation of the elastomer precursor contained 60 wt% microcapsules, 30 wt% PPG-diamine, and 10 wt% matrix polymer, which afforded the cured elastomer with adhesion force in the order of magnitude of 103 N m−1 (ESI,† Fig. S7). To optimize the composition ratios for the maximal reaction extent, the isocyanate content in the microcapsules with a diameter of 520 μm was calculated through titration using bromophenol and an excess quantity of amines (see Scheme S2, ESI† for the titration protocol). Based on the calculated result (isocyanate content 20.9 wt%), we optimized the composition of the ready-to-use precursor to be 50 wt% encapsulated MDI-prepolymer, 40 wt% PPG-diamine, and 10 wt% matrix polymer, so the unreacted excess agents were minimized. Such formulation was utilized for further studies.
To better control the force during the pre-installation process and on-demand curing, we loaded the precursor on to a customized shallow groove-shaped container. The curing reaction was then triggered by applying a 500 N compression force for 30 seconds on a 10 cm2 flat surface, and such a short dwell time was sufficient to allow a homogeneous force distribution against the stress relaxation of the precursor. The 500 N compression force was selected based on the preliminary screening of the breaking state of microcapsules and the resultant peel strength (ESI,† Table S2, and Fig. S8) against different compression forces. A significant increase (500%) in peel strength was observed for 500 N, indicating sufficient breakage of microcapsules. Notably, this does not necessarily mean that all microcapsules were broken by such compression force, because there might be a small portion of smaller microcapsules requiring larger compression force. However, the amount of such a small portion was inadequate to cause significant macroscopic defects that influence the cohesive strength of the elastomer. Such a hypothesis was indirectly proven by similar tests for microcapsules synthesized at 200 rpm (Table S3 and Fig. S9, ESI†), where the critical inflection region became less steep due to the broader distribution of microcapsule size. On the other hand, the microcapsules synthesized at 200 rpm exhibit inferior adhesion performance compared to those at 300 rpm. The advantage of microcapsules synthesized at 200 rpm is that a lower trigger force, 150 N, was sufficient to trigger the curing. Obviously, the triggering force could be further tuned by varying the microcapsule sizes and adjusting the precursor formulation, whereas this fine-tuning confronted the challenge as the capsule thickness needed to be proportionally scaled against its size. After compression, the sample was cured for three days for complete cure at 25 °C in a temperature-controlled chamber prior to being processed for the tensile test (Fig. 4A). Herein, the tensile test was performed at a tensile rate of 1 mm s−1 with the result plotted in Fig. 4B. The cured elastomer illustrated a maximum tensile stress of 1.75 MPa and an elongation at breakage of 223%, a decent mechanical performance in comparison with advanced ductile adhesive elastomers.48,49,78–81
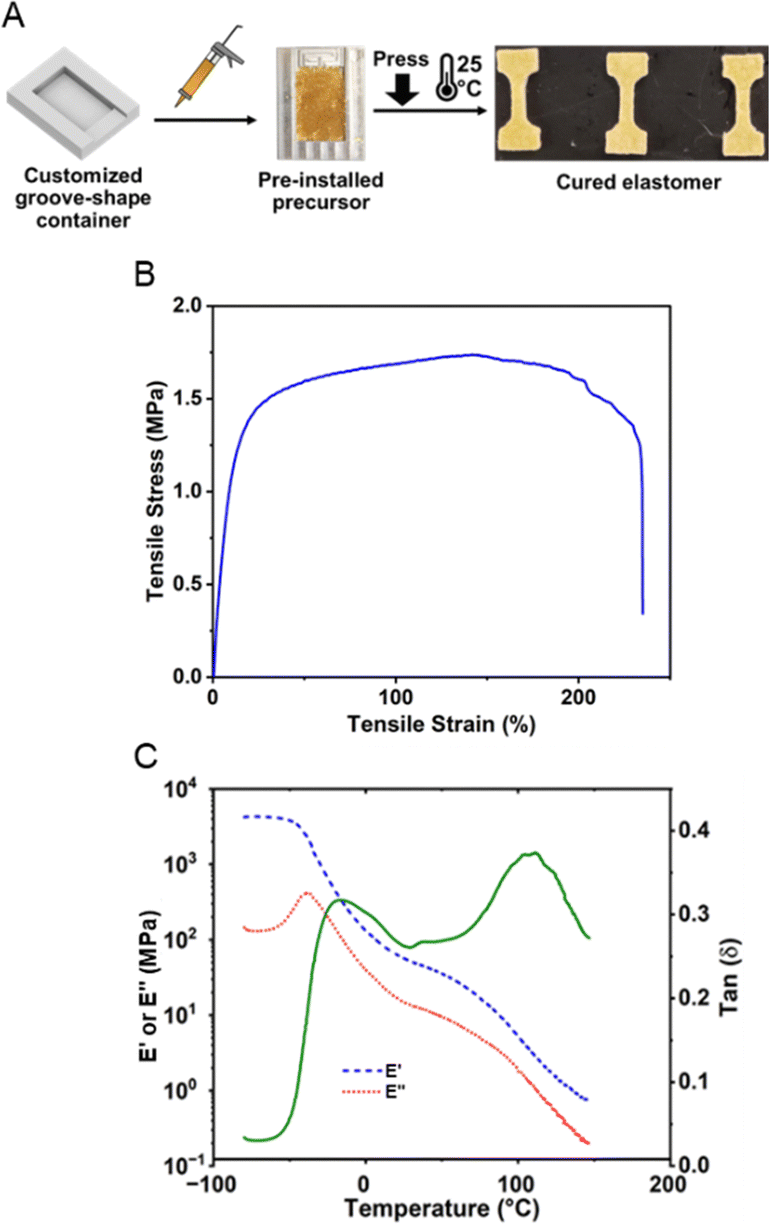 | ||
| Fig. 4 (A) Protocols to cure the pre-installed precursor and preparation of samples for tensile tests. (B) Tensile result of the ductile adhesive elastomer that was cured at 25 °C for three days. (C) Dynamic modulus and phase angle of the cured ductile adhesive elastomer by temperature sweep using DMA. | ||
The viscoelasticity of the cured elastomer was further investigated by temperature sweep using DMA as shown in Fig. 4C. At temperatures below −35 °C, a typical glassy modulus of around 4.3 GPa was observed. As the temperature increases, the result of the phase angle clearly indicates a broad transition from −25 °C to 0 °C, due to the devitrification of the cured ductile polyurea elastomer network. Herein, longer polyether chains were selected between crosslinks to avoid high rigidity, and therefore the Tg of the network was relatively low compared to rigid polyurea materials.82 From 0 to 75 °C, a temperature-dependent elastic region was demonstrated for cured elastomers, mainly due to the strongly temperature-dependent mechanical strength of the matrix polymer. The characteristic elastic modulus is around 40 MPa, which is at least 10 times higher than the typical rubber-state modulus.83–87 The high elastic modulus intrinsically enhances the cohesive strength of the adhesive elastomer.88–91 Such enhancement was presumably caused by the mechanical reinforcement of the highly crosslinked microcapsule shells phase-separated in the elastomer matrix.31,92–94 The existence of phase separation was indicated in the tan (δ) spectrum (Fig. 4C) as a high-temperature transition peak at 110 °C, and such temperature agreed with the Tg of crosslinked microcapsules (ESI,† Fig. S2).
As mentioned earlier, the novel design achieves the on-demand, ultra-high adhesion strength. The on-demand feature is important especially when the ductile adhesive elastomeric materials need a controlled delay between installation and adhesion, which often requires exterior protection and lacks flexibility of the installation. Some previous research (even current commercial products) have demonstrated the moisture- or light-triggered adhesion, although such triggering factors were easily disturbed by ambient humidity or light.67 Conversely, the mechanical-force trigger can be easily controlled under ambient conditions and readily applied by end users. Herein, the on-demand feature is easily achieved by applying a uniform compression force on the pre-installed ready-to-use precursor. Such a strategy is different from the well-reported pressure-sensitive adhesives, although the latter also has an on-demand feature. The regular pressure-sensitive adhesives involve no chemical changes during installation, and the external force triggers a viscoelastic change to tack to the substrate. Therefore, its crosslinking extent and matrix rigidity are limited, as the peel strength is around 103 N m−1.95–97 Moreover, the thickness of pressure-sensitive adhesives is often less than 100 μm, to retain the shear resistance,98–100 which limits its performance on various rough surfaces. In contrast, our force-triggering strategy involves a chemical curing process that provides at least 10 times higher strength to adhere to the substrate and maintain cohesion, as shown below. Moreover, the flexibility of the ready-to-use precursors and unlimited choices of thickness facilitate versatile adaption with various surface conditions.
The adhesion force of the ductile adhesive elastomer was studied by the peel and lap shear strength to benchmark the reported ductile adhesive elastomers. Here, we used customized test kits that contain an embedded groove, similar to the container illustrated in Fig. 4A, and the testing methods comply with ASTM C794 and C961 standards. The test results of peel strength and lap shear on both aluminum and wood substrates, along with pictures of the testing setup, are shown in Fig. 5A and B. After curing at room temperature, the peel strength (tensile rate of 50 mm min−1) was 1.2 × 104 N m−1 (294 N per inch) and 7.5 × 103 N m−1 (192 N per inch) for aluminum and wood substrates, respectively. The lap shear strength (tensile rate of 12.7 mm min−1) was 2.1 × 103 and 7.8 × 103 kPa for aluminium and wood substrates, respectively. To the best of our knowledge, in terms of adhesion force, our on-demand ductile adhesive elastomer tops the reported advanced ductile adhesive elastomers as illustrated by a summary of adhesion performance vs. materials shown in Fig. 6.
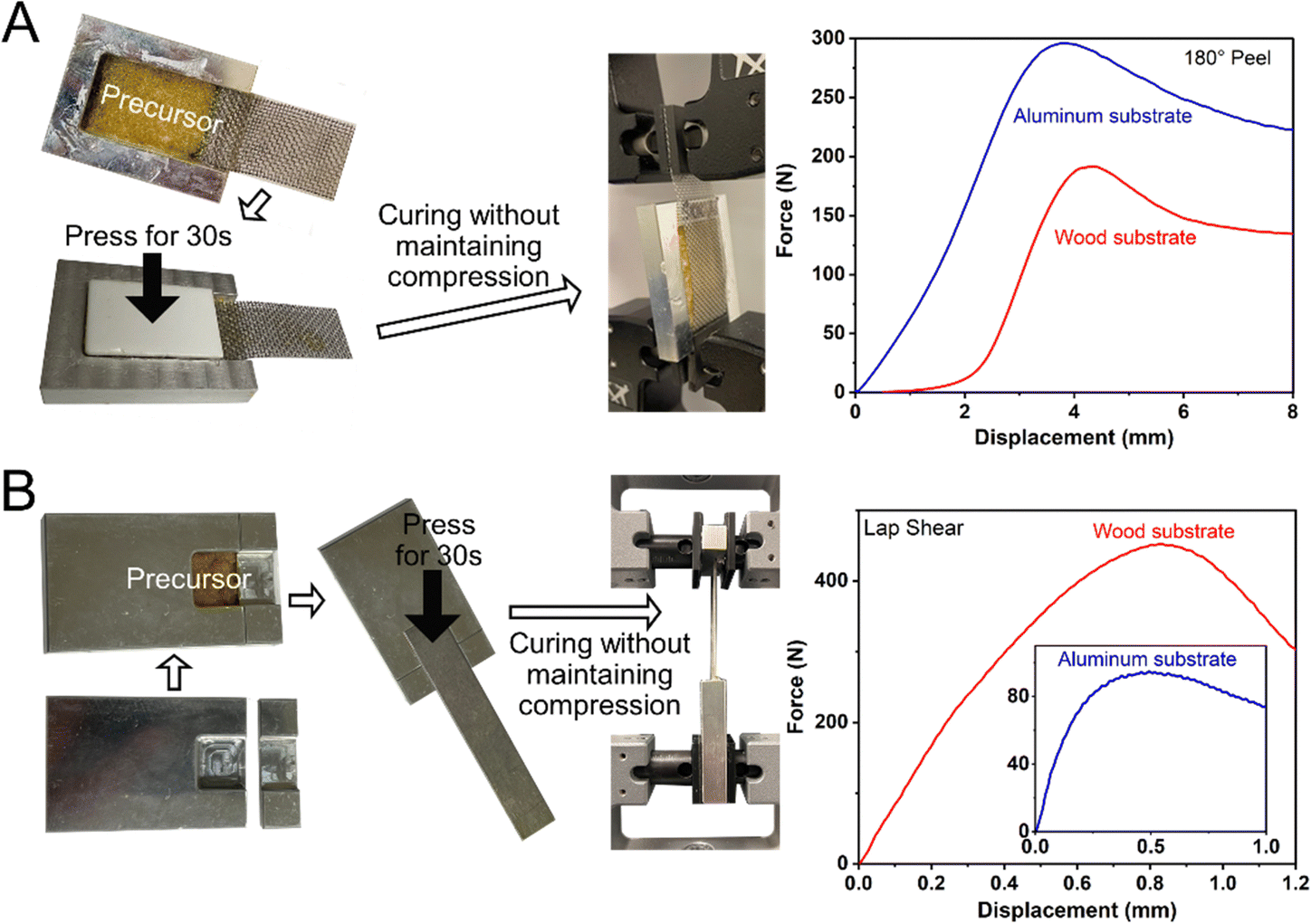 | ||
| Fig. 5 (A) Peeling result of the cured ductile adhesive elastomer using a 180° peel setup. (B) Tensile result of the cured ductile adhesive elastomer using a lap shear setup. | ||
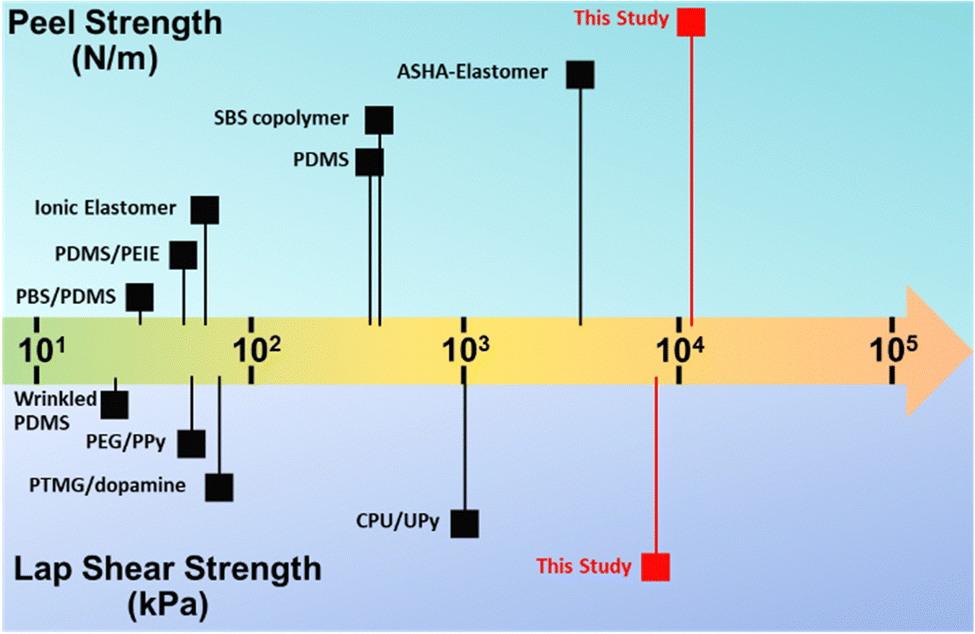 | ||
| Fig. 6 Summary of adhesion performance of currently reported advanced ductile adhesive elastomers, including the reported chemistries: ASHA-elastomer,25 SBS copolymer,39 PDMS,21 ionic elastomer,48 PDMS/PEIE,49 PBS/PDMS,64 CPU/UPy,47 PTMG/dopamine,27 PEG/PPy,46 and wrinkled PDMS.50 | ||
This ultra-high adhesion force provides a robust strategy to be utilized for applications on various substrate surfaces, and it can be explained by the following two reasons. First, the high mechanical robustness of the cured elastomer increases the cohesion strength. As measured by DMA, it showed an elastic modulus at least 10 times higher than regular rubbers, which provided reinforced cohesion during the peel test. Second, the ready-to-use precursor was pre-installed as a relatively liquid-like state that allowed the elastomer to adapt well to the texture roughness of the interface, significantly increasing the contact area. To examine the adhesion contribution from the matrix polymer, we also applied the peel test on the matrix polymer using the same abovementioned method. As shown in Fig. S10 (ESI†), the peel strength of the matrix polymer was only 1.8 × 103 N m−1 with clear cohesion failure observed (vs. 2.1 × 104 N m−1 for our ductile adhesive elastomer). These results confirm the advantage of such a unique design that has a soft nature for efficient contact during installation and is mechanically robust after a force-triggered curing reaction.
The on-demand adhesion feature has great potential for prefab construction,101–103 as the precursor could be easily pre-installed at the prefab plant and triggered at the construction site when components are assembled, and hence effectively reducing assembly time and cost and improving installation quality and performance. To demonstrate the potential application, we prepared an example of a prefab tongue and grove joint that is commonly used with wood sheathings or boards with our elastomer precursor (Fig. 7A). The joint was compressed for 30 seconds and cured under ambient conditions. Our high-resolution Instron tensile machine could barely pull the 5 cm-wide joints apart, as it exceeded the maximum load of 1000 N (Fig. 7A). The ultra-high adhesion force is further illustrated in Fig. 7B as the cured ductile adhesive elastomer could withstand the weight of 4 jugs of water, roughly 80 kg.
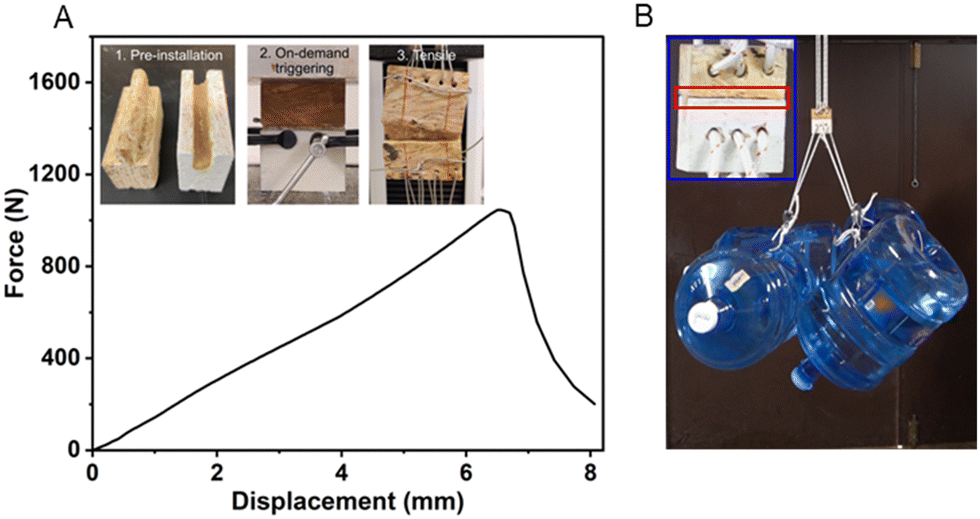 | ||
| Fig. 7 (A) Tensile result of the bench-scale prefab joints connected by our cured elastomer, along with the protocol of setting up the bench-scale prefab joint. (B) Illustration of the ultra-high adhesion performance of our ductile adhesive elastomer using the bench-scale prefab joints. | ||
3. Conclusions
In summary, we report a novel design of ductile adhesive elastomers whose ultra-high adhesion strength can be easily triggered by an external force. The ready-to-use precursor was easily prepared with the encapsulated MDI-prepolymer as a reactive agent, PPG-diamine as the curing agent, and polyacrylates as a matrix polymer. The encapsulated MDI-prepolymer was synthesized by partially crosslinking the MDI-prepolymer and the diameter of the microcapsules was adjustable. The polyacrylate matrix polymer improved the adhesion of the precursor and provided the advantage of self-healability. The on-demand adhesion feature was achieved by pre-installing the precursor onto a surface and the feature was easily triggered by compression force. After curing, our ductile adhesive elastomer exhibited a peel strength and a lap shear strength of 1.2 × 104 N m−1 and 7.8 × 103 kPa, respectively, which tops the reported adhesion force for advanced ductile elastomers. This ultra-high adhesion force is mainly due to the excellent surface contact of the liquid-like precursor and the high elastic modulus of the cured elastomer intrinsically reinforced by a two-phase design. We further demonstrated that the on-demand adhesion can be effectively used to seal joints between prefab components that are used in construction, as a prefab joint can withstand a force of at least 1000 N in a 5 cm-wide joint. Our study sheds light on the on-demand adhesion features of advanced elastomer materials and provides predominant adhesion and flexibility over regular pressure-sensitive adhesives or ductile adhesive elastomers.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was funded by the Advanced Building Construction Initiative under the Building Technologies Office of the U.S. Department of Energy (DOE), under contract no. DE-FOA-0002099. The National Natural Science Foundation of China (grant no. 52373275) supported contributions to the writing and editing of the manuscript, as well as a few experiments. This manuscript was authored by UT-Battelle, LLC under contract no. DE-AC05-00OR22725 with the U.S. Department of Energy. The United States Government retains the publisher by accepting the article for publication, acknowledges that the United States Government retains a non-exclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this manuscript, or allow others to do so, for United States Government purposes. The Department of Energy will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (https://energy.gov/downloads/doe-public-access-plan).References
- G. Demirci, M. J. Niedźwiedź, N. Kantor-Malujdy and M. E. Fray, Polymers, 2022, 14(9), 1822 CrossRef CAS PubMed.
- C. Ma, M.-G. Ma, C. Si, X.-X. Ji and P. Wan, Adv. Funct. Mater., 2021, 31(22), 2009524 CrossRef CAS.
- Y. Li, J. Li, W. Li and H. Du, Smart Mater. Struct., 2014, 23(12), 123001 CrossRef.
- C. S. Luo, P. Wan, H. Yang, S. A. A. Shah and X. Chen, Adv. Funct. Mater., 2017, 27(23), 1606339 CrossRef.
- Y. Guo, L. Liu, Y. Liu and J. Leng, Adv. Intell. Syst., 2021, 3(10), 2000282 CrossRef.
- S. Gao, M. Zhang, C. Gainaru, A. P. Sokolov, H. Yang and P.-F. Cao, Matter, 2022, 5(8), 2457–2460 CrossRef CAS.
- Q. Zhang, S. Niu, L. Wang, J. Lopez, S. Chen, Y. Cai, R. Du, Y. Liu, J.-C. Lai, L. Liu, C.-H. Li, X. Yan, C. Liu, J. B.-H. Tok, X. Jia and Z. Bao, Adv. Mater., 2018, 30(33), 1801435 CrossRef PubMed.
- B. T. White and T. E. Long, Macromol. Rapid Commun., 2019, 40(1), 1800521 CrossRef PubMed.
- D. Zhang, Y. Tang, X. Gong, Y. Chang and J. Zheng, SmartMat, 2023, e1160 CrossRef.
- K. Parvathi, M. A. Al-Maghrabi, M. Subburaj and M. T. Ramesan, Polym. Compos., 2021, 42(9), 4586–4595 CrossRef CAS.
- C.-Y. Shi, Q. Zhang, C.-Y. Yu, S.-J. Rao, S. Yang, H. Tian and D.-H. Qu, Adv. Mater., 2020, 32(23), 2000345 CrossRef CAS PubMed.
- P. K. Behera, S. Mohanty and V. K. Gupta, Polym. Chem., 2021, 12(11), 1598–1621 RSC.
- J. Huang, Y. Cai, C. Xue, J. Ge, H. Zhao and S.-H. Yu, Nano Res., 2021, 14(10), 3636–3642 CrossRef CAS.
- T. E. Long and J. Scheirs, Modern Polyesters: Chemistry and Technology of Polyesters and Copolyesters, John Wiley & Sons, 2005 Search PubMed.
- Y. Xue, J. Lin, T. Wan, Y. Luo, Z. Ma, Y. Zhou, B. T. Tuten, M. Zhang, X. Tao and P. Song, Adv. Sci., 2023, 2207268 CrossRef CAS PubMed.
- Y. Lai, X. Kuang, P. Zhu, M. Huang, X. Dong and D. Wang, Adv. Mater., 2018, 30(38), 1802556 CrossRef PubMed.
- X. Zhou, L. Wang, Z. Wei, G. Weng and J. He, Adv. Funct. Mater., 2019, 29(34), 1903543 CrossRef.
- J. H. Koo, D. C. Kim, H. J. Shim, T.-H. Kim and D.-H. Kim, Adv. Funct. Mater., 2018, 28(35), 1801834 CrossRef.
- L.-J. Yin, Y. Zhao, J. Zhu, M. Yang, H. Zhao, J.-Y. Pei, S.-L. Zhong and Z.-M. Dang, Nat. Commun., 2021, 12(1), 4517 CrossRef CAS PubMed.
- E. Siéfert, E. Reyssat, J. Bico and B. Roman, Nat. Mater., 2019, 18(1), 24–28 CrossRef PubMed.
- A. M. Hubbard, W. Cui, Y. Huang, R. Takahashi, M. D. Dickey, J. Genzer, D. R. King and J. P. Gong, Matter, 2019, 1(3), 674–689 CrossRef CAS.
- Y. Yang, W. Zhan, R. Peng, C. He, X. Pang, D. Shi, T. Jiang and Z. Lin, Adv. Mater., 2015, 27(41), 6376–6381 CrossRef CAS PubMed.
- S. Liu, S. Wang, S. Xuan, S. Zhang, X. Fan, H. Jiang, P. Song and X. Gong, ACS Appl. Mater. Interfaces, 2020, 12(13), 15675–15685 CrossRef CAS PubMed.
- J. Kang, D. Son, G.-J. N. Wang, Y. Liu, J. Lopez, Y. Kim, J. Y. Oh, T. Katsumata, J. Mun, Y. Lee, L. Jin, J. B.-H. Tok and Z. Bao, Adv. Mater., 2018, 30(13), 1706846 CrossRef PubMed.
- Z. Zhang, N. Ghezawi, B. Li, S. Ge, S. Zhao, T. Saito, D. Hun and P.-F. Cao, Adv. Funct. Mater., 2021, 31(4), 2006298 CrossRef CAS.
- J. Luo, Z. Demchuk, X. Zhao, T. Saito, M. Tian, A. P. Sokolov and P.-F. Cao, Matter, 2022, 5(5), 1391–1422 CrossRef CAS.
- Z. Xu, L. Chen, L. Lu, R. Du, W. Ma, Y. Cai, X. An, H. Wu, Q. Luo, Q. Xu, Q. Zhang and X. Jia, Adv. Funct. Mater., 2021, 31(1), 2006432 CrossRef CAS.
- R. Du, Z. Xu, C. Zhu, Y. Jiang, H. Yan, H.-C. Wu, O. Vardoulis, Y. Cai, X. Zhu, Z. Bao, Q. Zhang and X. Jia, Adv. Funct. Mater., 2020, 30(7), 1907139 CrossRef CAS.
- Y. Chen, Z. Tang, X. Zhang, Y. Liu, S. Wu and B. Guo, ACS Appl. Mater. Interfaces, 2018, 10(28), 24224–24231 CrossRef CAS PubMed.
- Y. Chen, Z. Tang, Y. Liu, S. Wu and B. Guo, Macromolecules, 2019, 52(10), 3805–3812 CrossRef CAS.
- P. Wei and E. B. Pentzer, Matter, 2022, 5(8), 2479–2481 CrossRef CAS.
- P. Song and H. Wang, Adv. Mater., 2020, 32(18), 1901244 CrossRef CAS PubMed.
- Q. Ma, S. Liao, Y. Ma, Y. Chu and Y. Wang, Adv. Mater., 2021, 33(36), 2102096 CrossRef CAS PubMed.
- D. Zhang, Y. Tang, Y. Zhang, F. Yang, Y. Liu, X. Wang, J. Yang, X. Gong and J. Zheng, J. Mater. Chem. A, 2020, 8(39), 20474–20485 RSC.
- F. Sun, Z. Li, S. Gao, Y. He, J. Luo, X. Zhao, D. Yang, T. Gao, H. Yang and P.-F. Cao, ACS Appl. Mater. Interfaces, 2022, 14(22), 26014–26023 CrossRef CAS PubMed.
- X. Chu, R. Wang, H. Zhao, M. Kuang, J. Yan, B. Wang, H. Ma, M. Cui and X. Zhang, ACS Appl. Mater. Interfaces, 2022, 14(14), 16631–16640 CrossRef CAS PubMed.
- K. Guk, G. Han, J. Lim, K. Jeong, T. Kang, E.-K. Lim and J. Jung, Nanomaterials, 2019, 9(6), 813 CrossRef CAS PubMed.
- Y. Fang and J. Xia, ACS Appl. Mater. Interfaces, 2022, 14(33), 38398–38408 CrossRef CAS PubMed.
- N. Sato, A. Murata, T. Fujie and S. Takeoka, Soft Matter, 2016, 12(45), 9202–9209 RSC.
- L. Meng, J. He and C. Pan, Materials, 2022, 15(7), 2548 CrossRef CAS PubMed.
- A. B. Croll, N. Hosseini and M. D. Bartlett, Adv. Mater. Technol., 2019, 4(8), 1900193 CrossRef.
- R. Coulson, C. J. Stabile, K. T. Turner and C. Majidi, Soft Rob., 2022, 9(2), 189–200 CrossRef PubMed.
- H. Iwasaki, F. Lefevre, D. D. Damian, E. Iwase and S. Miyashita, IEEE Rob. Autom. Lett., 2020, 5(2), 2015–2022 Search PubMed.
- Z. Ma, G. Bao and J. Li, Adv. Mater., 2021, 33(24), 2007663 CrossRef CAS PubMed.
- M. Runciman, A. Darzi and G. P. Mylonas, Soft Robot., 2019, 6(4), 423–443 CrossRef PubMed.
- J. Chen, J. Liu, T. Thundat and H. Zeng, ACS Appl. Mater. Interfaces, 2019, 11(20), 18720–18729 CrossRef CAS PubMed.
- M. W. M. Tan, G. Thangavel and P. S. Lee, Adv. Funct. Mater., 2021, 31(34), 2103097 CrossRef CAS.
- L. Wang, Y. Wang, S. Yang, X. Tao, Y. Zi and W. A. Daoud, Nano Energy, 2022, 91, 106611 CrossRef CAS.
- S. H. Jeong, S. Zhang, K. Hjort, J. Hilborn and Z. Wu, Adv. Mater., 2016, 28(28), 5830–5836 CrossRef CAS PubMed.
- C.-H. Lin, C.-Y. Huang, J.-Y. Ho and H.-Y. Hsueh, ACS Appl. Mater. Interfaces, 2020, 12(19), 22365–22377 CrossRef CAS PubMed.
- B. J. Blaiszik, S. L. B. Kramer, S. C. Olugebefola, J. S. Moore, N. R. Sottos and S. R. White, Annu. Rev. Mater. Res., 2010, 40(1), 179–211 CrossRef CAS.
- D. Y. Zhu, M. Z. Rong and M. Q. Zhang, Prog. Polym. Sci., 2015, 49–50, 175–220 CrossRef CAS.
- K. Wazarkar, D. Patil, A. Rane, D. Balgude, M. Kathalewar and A. Sabnis, RSC Adv., 2016, 6(108), 106964–106979 RSC.
- V. Nedovic, A. Kalusevic, V. Manojlovic, S. Levic and B. Bugarski, Procedia Food Sci., 2011, 1, 1806–1815 CrossRef CAS.
- C. J. Martínez Rivas, M. Tarhini, W. Badri, K. Miladi, H. Greige-Gerges, Q. A. Nazari, S. A. Galindo Rodríguez, R. Á. Román, H. Fessi and A. Elaissari, Int. J. Pharm., 2017, 532(1), 66–81 CrossRef PubMed.
- M. Villiou, J. I. Paez and A. del Campo, ACS Appl. Mater. Interfaces, 2020, 12(34), 37862–37872 CrossRef CAS PubMed.
- W. Sliwka, Angew. Chem., Int. Ed. Engl., 1975, 14(8), 539–550 CrossRef CAS PubMed.
- Q. Lu, Z. Yang, X. Meng, Y. Yue, M. A. Ahmad, W. Zhang, S. Zhang, Y. Zhang, Z. Liu and W. Chen, Adv. Funct. Mater., 2021, 31(23), 2100151 CrossRef CAS.
- S. M. Jee, C.-H. Ahn, J. H. Park, T. A. Kim and M. Park, Composites, Part B, 2020, 202, 108438 CrossRef CAS.
- M. M. Caruso, B. J. Blaiszik, S. R. White, N. R. Sottos and J. S. Moore, Adv. Funct. Mater., 2008, 18(13), 1898–1904 CrossRef CAS.
- S. H. Cho, H. M. Andersson, S. R. White, N. R. Sottos and P. V. Braun, Adv. Mater., 2006, 18(8), 997–1000 CrossRef CAS.
- M. J. Shin, Y. J. Shin, S. W. Hwang and J. S. Shin, J. Appl. Polym. Sci., 2013, 129(3), 1036–1044 CrossRef CAS.
- Y. Zhang, J. Yu, H. N. Bomba, Y. Zhu and Z. Gu, Chem. Rev., 2016, 116(19), 12536–12563 CrossRef CAS PubMed.
- J. Giro-Paloma, C. Barreneche, M. Martínez, B. Šumiga, A. I. Fernández and L. F. Cabeza, Renewable Energy, 2016, 85, 732–739 CrossRef CAS.
- H. Zhao, X. Fei, L. Cao, B. Zhang and X. Liu, Materials, 2019, 12(3), 393 CrossRef CAS PubMed.
- A. Ghaemi, A. Philipp, A. Bauer, K. Last, A. Fery and S. Gekle, Chem. Eng. Sci., 2016, 142, 236–243 CrossRef CAS.
- A. P. Esser-Kahn, S. A. Odom, N. R. Sottos, S. R. White and J. S. Moore, Macromolecules, 2011, 44(14), 5539–5553 CrossRef CAS.
- J. Yang, M. W. Keller, J. S. Moore, S. R. White and N. R. Sottos, Macromolecules, 2008, 41(24), 9650–9655 CrossRef CAS.
- P.-F. Cao, B. Li, T. Hong, K. Xing, D. N. Voylov, S. Cheng, P. Yin, A. Kisliuk, S. M. Mahurin, A. P. Sokolov and T. Saito, ACS Appl. Mater. Interfaces, 2017, 9(31), 26483–26491 CrossRef CAS PubMed.
- T. Yin, M. Z. Rong, M. Q. Zhang and G. C. Yang, Compos. Sci. Technol., 2007, 67(2), 201–212 CrossRef CAS.
- J. S. Nakka, K. M. B. Jansen and L. J. Ernst, J. Polym. Res., 2011, 18(6), 1879–1888 CrossRef CAS.
- G. Yang, S.-Y. Fu and J.-P. Yang, Polymer, 2007, 48(1), 302–310 CrossRef CAS.
- E. Urbaczewski-Espuche, J. Galy, J.-F. Gerard, J.-P. Pascault and H. Sautereau, Polym. Eng. Sci., 1991, 31(22), 1572–1580 CrossRef CAS.
- S. J. McCarthy, G. F. Meijs, N. Mitchell, P. A. Gunatillake, G. Heath, A. Brandwood and K. Schindhelm, Biomaterials, 1997, 18(21), 1387–1409 CrossRef CAS PubMed.
- M. Strankowski, D. Włodarczyk, Ł. Piszczyk and J. Strankowska, J. Spectrosc., 2016, 2016, 7520741 Search PubMed.
- Z. Wen, M. K. McBride, X. Zhang, X. Han, A. M. Martinez, R. Shao, C. Zhu, R. Visvanathan, N. A. Clark, Y. Wang, K. Yang and C. N. Bowman, Macromolecules, 2018, 51(15), 5812–5819 CrossRef CAS.
- V. A. Gabriel and M. A. Dubé, Macromol. React. Eng., 2019, 13(2), 1800057 CrossRef.
- M. Tang, P. Zheng, K. Wang, Y. Qin, Y. Jiang, Y. Cheng, Z. Li and L. Wu, J. Mater. Chem. A, 2019, 7(48), 27278–27288 RSC.
- J. Li, A. Celiz, J. Yang, Q. Yang, I. Wamala, W. Whyte, B. Seo, N. Vasilyev, J. Vlassak and Z. Suo, Science, 2017, 357(6349), 378–381 CrossRef CAS PubMed.
- D. F. S. Saldanha, C. Canto, L. F. M. da Silva, R. J. C. Carbas, F. J. P. Chaves, K. Nomura and T. Ueda, Int. J. Adhes. Adhes., 2013, 47, 91–98 CrossRef CAS.
- L. Wang, G. Gao, Y. Zhou, T. Xu, J. Chen, R. Wang, R. Zhang and J. Fu, ACS Appl. Mater. Interfaces, 2019, 11(3), 3506–3515 CrossRef CAS PubMed.
- J. S. Santana, E. S. Cardoso, E. R. Triboni and M. J. Politi, Polymers, 2021, 13(24), 4393 CrossRef CAS PubMed.
- J. D. Ferry, Viscoelastic Properties of Polymers, John Wiley & Sons, 1980 Search PubMed.
- T. Hwang, Z. Frank, J. Neubauer and K. J. Kim, Sci. Rep., 2019, 9(1), 9658 CrossRef PubMed.
- M. Hao, L. Li, S. Wang, F. Sun, Y. Bai, Z. Cao, C. Qu and T. Zhang, Microsyst. Nanoeng., 2019, 5(1), 9 CrossRef PubMed.
- T. Noguchi, M. Endo, K. Niihara, H. Jinnai and A. Isogai, Compos. Sci. Technol., 2020, 188, 108005 CrossRef CAS.
- B. M. Boyle, O. Heinz, G. M. Miyake and Y. Ding, Macromolecules, 2019, 52(9), 3426–3434 CrossRef CAS PubMed.
- B. D. B. Tiu, P. Delparastan, M. R. Ney, M. Gerst and P. B. Messersmith, Angew. Chem., Int. Ed., 2020, 59(38), 16616–16624 CrossRef CAS PubMed.
- A. Zosel, J. Adhes., 1989, 30(1–4), 135–149 CrossRef CAS.
- T. L. Gordon and M. E. Fakley, Int. J. Adhes. Adhes., 2003, 23(2), 95–100 CrossRef CAS.
- F. P. M. Mercx, A. Benzina, A. D. van Langeveld and P. J. Lemstra, J. Mater. Sci., 1993, 28(3), 753–759 CrossRef CAS.
- Z. Zhang, J. Luo, S. Zhao, S. Ge, J.-M. Y. Carrillo, J. K. Keum, C. Do, S. Cheng, Y. Wang, A. P. Sokolov and P.-F. Cao, Matter, 2022, 5(1), 237–252 CrossRef CAS.
- Y. Zheng, R. Kiyama, T. Matsuda, K. Cui, X. Li, W. Cui, Y. Guo, T. Nakajima, T. Kurokawa and J. P. Gong, Chem. Mater., 2021, 33(9), 3321–3334 CrossRef.
- Y. Zhuo, Z. Xia, Y. Qi, T. Sumigawa, J. Wu, P. Šesták, Y. Lu, V. Håkonsen, T. Li, F. Wang, W. Chen, S. Xiao, R. Long, T. Kitamura, L. Li, J. He and Z. Zhang, Adv. Mater., 2021, 33(23), 2008523 CrossRef CAS PubMed.
- S. Sun, M. Li and A. Liu, Int. J. Adhes. Adhes., 2013, 41, 98–106 CrossRef CAS.
- S. Mapari, S. Mestry and S. T. Mhaske, Polym. Bull., 2021, 78(7), 4075–4108 CrossRef CAS.
- R. Jovanović and M. A. Dubé, J. Macromol. Sci., Part C, 2004, 44(1), 1–51 CrossRef.
- B. T. Poh and H. K. Kwo, Polym.-Plast. Technol. Eng., 2007, 46(10), 1021–1024 CrossRef CAS.
- B. T. Poh and H. K. Kwo, J. Appl. Polym. Sci., 2007, 105(2), 680–684 CrossRef CAS.
- I. Khan and B. T. Poh, J. Polym. Environ., 2011, 19(3), 793–811 CrossRef CAS.
- S. G. Naoum, Int. J. Prod. Perform. Manage., 2016, 65(3), 401–421 CrossRef.
- K. M. A. El-Abidi and F. E. M. Ghazali, Appl. Mech. Mater., 2015, 802, 668–675 Search PubMed.
- F. E. Boafo, J.-H. Kim and J.-T. Kim, Sustainability, 2016, 8(6), 558 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3mh01280h |
| This journal is © The Royal Society of Chemistry 2024 |