
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Opening pathways for the conversion of woody biomass into sustainable aviation fuel via catalytic fast pyrolysis and hydrotreating†
Michael B.
Griffin
 *a,
Kristiina
Iisa
a,
Abhijit
Dutta
a,
Xiaolin
Chen
a,
Cody J.
Wrasman
a,
Calvin
Mukarakate
a,
Matthew M.
Yung
a,
Mark R.
Nimlos
a,
Luke
Tuxworth
b,
Xavier
Baucherel
b,
Steven M.
Rowland
a and
Susan E.
Habas
a
*a,
Kristiina
Iisa
a,
Abhijit
Dutta
a,
Xiaolin
Chen
a,
Cody J.
Wrasman
a,
Calvin
Mukarakate
a,
Matthew M.
Yung
a,
Mark R.
Nimlos
a,
Luke
Tuxworth
b,
Xavier
Baucherel
b,
Steven M.
Rowland
a and
Susan E.
Habas
a
aNational Renewable Energy Laboratory, Golden, CO 80401, USA. E-mail: Michael.Griffin@nrel.gov
bJohnson Matthey Technology Center, PO Box 1, Belasis Avenue, Billingham, Cleveland TS23 1 LB, UK
First published on 9th August 2024
Abstract
Meeting aggressive decarbonization targets set by the International Civil Aviation Organization (ICAO) will require the rapid development of technologies to produce sustainable aviation fuel (SAF). Catalytic fast pyrolysis (CFP) can support these efforts by opening pathways for the conversion of woody biomass into an upgraded biogenic oil that can be further processed to SAF and other fuels. However, the absence of end-to-end experimental data for the process leads to uncertainty in the yield, product quality, costs, and sustainability of the pathway. The research presented here serves to address these needs through a series of integrated experimental campaigns in which real biomass feedstocks are converted to a final SAF product using large bench-scale continuous reactor systems. For these campaigns, the degree of catalytic upgrading during CFP was varied to produce CFP-oils with oxygen contents of 17 and 20 wt% on a dry basis. The CFP-oils were then hydrotreated and distilled into gasoline, diesel, and SAF fractions. Detailed yield and compositional data were obtained for each step of the process to inform technoeconomic and lifecycle analyses, and the fuel properties of the SAF fraction were evaluated to provide first-of-its-kind insight into the quality of the final product. This research reveals opportunities to optimize process carbon efficiency by tuning the degree of catalytic upgrading during the CFP step and highlights routes to produce a high-quality cycloalkane-rich SAF with 85–92% reduction in greenhouse gas emissions compared to fossil-based pathways.
Introduction
As part of an aggressive effort to decarbonize air travel, many countries, organizations, and companies have announced net-zero greenhouse gas (GHG) emission targets,1,2 and meeting these goals will require the rapid development of technologies to produce sustainable aviation fuel (SAF). Currently, over 95% of the SAF used in flight has been produced by converting fats, oils, and greases (FOG) into hydrotreated esters and fatty acids (HEFA).3 However, the supply of FOG is limited to approximately 7 million dry tons per year, and additional non-food sourced biogenic oils are needed to enable further growth in this sector.3,4 Woody biomass is a widely available feedstock that can be converted to biogenic oils through fast pyrolysis. The U.S. Department of Energy has estimated that a combined resource potential of 133 million dry tons of forest resources and woody wastes can be produced sustainably in the United States annually.5 The conversion of woody biomass into bio-oil via fast pyrolysis is a commercially proven process. However, the bio-oil generated from fast pyrolysis has several undesirable qualities stemming from its high oxygen content that make it unsuitable for direct down-stream hydrotreating.6–8 These issues can be mitigated through catalytic fast pyrolysis (CFP) to produce a stabilized bio-oil with improved properties that can be tailored for hydroprocessing en route to a final transportation fuel blendstock.9–11A variety of reactor configurations have been explored for CFP including in situ and ex situ approaches.12,13In situ CFP is advantaged in terms of the comparatively low capital requirements since pyrolysis and catalytic upgrading occur in a single unit.14 Alternatively, ex situ CFP, where pyrolysis vapors are catalytically upgraded in a second reactor, offers greater flexibility since each unit operation can be optimized independently. Likewise, ex situ CFP reduces catalyst exposure to inorganic element-containing materials that are present in the pyrolysis reactor, which has the potential to prolong catalyst lifetime and increase process durability.15–19Ex situ CFP can be performed using several different classes of catalysts including zeolites which are commonly operated in inert environments17,20–25 as well as metal-acid bifunctional catalysts which are commonly utilized with co-fed hydrogen.26–31 In all cases, the CFP-oil that is produced exhibits lower oxygen content, reduced acidity, and improved stability compared to non-catalytic fast pyrolysis bio-oil. Importantly, the catalytically upgraded CFP-oil can be hydrotreated using a single-stage system to produce hydrocarbon transportation fuel blendstocks,13,26,29 whereas hydrotreating of non-catalytic fast pyrolysis oil typically requires a two-stage hydrotreating approach, which increases complexity, capital costs and operating expenses.8
Previous CFP research has spanned fundamental laboratory-scale studies to pilot-scale demonstrations. Researchers from the U. S. Department of Agriculture have demonstrated CFP using a variety of biomass feedstock and waste streams including oak,32 eucalyptus,33 switchgrass with polyethylene,34 and barley distillery byproducts.35 This team has also developed advanced process designs to improve performance by recycling product gas36 and enable modular on-farm or in-forest operations.37 Likewise, Dayton and coworkers have developed and demonstrated CFP technologies under both inert and reactive conditions.38–40 Notably, a parametric study was completed at a nominal one ton per day feed rate using a λ-Al2O3 catalyst and pine feedstock. The results revealed the potential to achieve yields of 40–50 gallons per dry ton (12–18% carbon efficiency) with oxygen contents ranging from 21–31 wt%.41 Pilot-scale CFP campaigns have also been performed at VTT in Finland. These experiments were performed at 20 kg h−1 and utilized a HZSM-5 catalyst under inert conditions to generate CFP-oils with an oxygen content of 21.5 wt% and carbon efficiency of 24%.18 Pioneering research from the Brown group at Iowa State University explored alternative process configurations such as autothermal pyrolysis, in which a limited amount of air is introduced to overcome heat transfer bottlenecks through partial oxidation of pyrolysis vapours.42,43 Autothermal experiments with corn stover indicate that the carbon yields to char and aqueous bio-oil light ends decreased by 18.5 and 4.7%, respectively, whereas the desired organic-rich heavy fraction was largely preserved.44 A summary of prior work, including important contributions from many other researchers can be found in recent reviews.45–47
In contrast to thermal pyrolysis, large-scale commercialization of CFP has yet to be fully realized. A notable attempt by KiOR was halted after it failed to meet production targets due to lower-than-expected conversion yields, and the company ceased operations in 2014.48 More recently, Annellotech has reported process yields of 22–24 wt% during six months of continuous operation of their TCat-8 facility in Texas. The Allellotech Bio-TCat™ technology, which was originally developed in the Huber group at the University of Massachusetts-Amherst, is a fluid catalytic process for the conversion of non-food biomass such as loblolly pine into benzene, toluene, and xylene.49 Likewise, the Dutch company BioBTX is developing a process to generate sustainable aromatics from plastic waste and biomass using an ex situ CFP process with a zeolite catalyst. Notably, in 2024 BioBTX secured over €80 million to launch it's first commercial-scale plant in the Netherlands.50 The increased interest in development circular materials and biogenic intermediates for refinery integration is expected to support further growth in this technology area.
Despite the promise of these emerging technologies, there are several research questions that remain unaddressed. Among these, the absence of integrated process data has precluded the ability to understand foundational relationships between the CFP and down-stream processing steps. Consequently, the optimal degree of catalytic upgrading for each unit operation remains an open question, and additional insight is needed to maximize the carbon efficiency of the end-to-end process. Integrated process data is also required for robust technoeconomic and lifecycle analyses to enable benchmarking of fuel selling price and sustainability metrics. Additionally, the composition and fuel properties of the SAF fraction have not been reported, and the resulting uncertainty around the quality of the final product creates a barrier for scale-up and deployment. The research presented herein serves to address these needs through a series of integrated experimental campaigns in which the degree of catalytic upgrading during ex situ CFP was varied to produce two CFP-oils with oxygen contents of 17 and 20 wt% on a dry basis. Each CFP-oil was subsequently hydrotreated at temperatures ranging from 300–385 °C and distilled into gasoline, diesel, and SAF fractions. Detailed yield and compositional data were obtained from each step of the process to inform technoeconomic and lifecycle analysis (TEA/LCA), and the SAF fraction was evaluated to provide first-of-its-kind insight into the fuel properties of the final product.
Results and discussion
Catalytic fast pyrolysis
Biomass fast pyrolysis was conducted in a 2 inch fluidized bed reactor at 500 °C. Compositional analysis of the feedstock, which consisted of 50% clean pine and 50% forest residues, is provided in Table S1.† The pyrolysis vapors were catalytically upgraded at 500 °C in a down-stream bubbling fluidized bed reactor using a ZSM-5 catalyst with an alumina binder and a silica to alumina ratio of 30 (Johnson Matthey). For these experiments, the degree of catalytic upgrading was varied by changing the biomass to catalyst mass ratio from 1.7 to 2.5 to produce two CFP-oils with oxygen contents of 17 and 20 wt% on a dry basis, respectively. These oils will be referred to as ‘17-O’ and ‘20-O’ CFP-oils throughout the manuscript. Select results from the CFP experiments are given in Table 1.| Mass yields, wt% dry biomass basis | 17-O | 20-O |
| Oil | 14.1 ± 0.3 | 17.8 ± 0.9 |
| Condensables | 5.0 ± 0.3 | 5.6 ± 0.3 |
| Gases | 28.9 ± 1.2 | 28.7 ± 0.9 |
| Aqueous | 28.9 ± 0.2 | 27.3 ± 0.7 |
| Char | 12.9 ± 0.2 | 13.1 ± 0.3 |
| Coke | 9.3 ± 0.3 | 7.8 ± 0.3 |
| Water vapor | 1.3 ± 0.2 | 1.2 ± 0.2 |
| Total | 100.4 ± 1.2 | 101.5 ± 1.0 |
| Carbon yields, gC gC −1 in biomass, % | 17-O | 20-O |
| Oil | 21.2 ± 0.8 | 25.7 ± 1.5 |
| Condensables | 7.4 ± 0.5 | 7.9 ± 0.5 |
| Gases | 25.8 ± 0.6 | 25.1 ± 0.6 |
| Aqueous | 3.4 ± 0.1 | 5.8 ± 0.2 |
| Char | 19.8 ± 0.3 | 20.1 ± 0.2 |
| Coke | 15.3 ± 0.5 | 13.1 ± 0.1 |
| Total | 92.8 ± 1.2 | 98.0 ± 2.3 |
| CFP-oil properties | 17-O | 20-O |
| C, wt% dry basis | 76.3 ± 1.2 | 73.6 ± 0.7 |
| H, wt% dry basis | 6.9 ± 0.2 | 6.9 ± 0.0 |
| N, wt% dry basis | 0.1 ± 0.0 | 0.1 ± 0.1 |
| O, wt% dry basis | 16.8 ± 1.3 | 19.5 ± 0.6 |
| H2O, wt% | 3.8 ± 0.2 | 4.8 ± 1.0 |
| S, ppm dry basis | 32 | 32 |
| Carboxylic acid number, mg KOH per g | 19 | 29 |
| Density, g ml−1 | 1.13 | 1.16 |
| Viscosity, @ 40 °C, mPas | 94 | 106 |
The water content of the CFP-oils ranged from 3.8 to 4.8 wt% for the 17-O and 20-O CFP oils, respectively. These values are considerably lower than the 18–21 wt% water content that has been reported for non-catalytic fast pyrolysis of pine.22,51 The comparatively low water content for CFP-oil is associated with the formation of a separate aqueous phase, whereas the organic and aqueous fractions are typically contained within a single phase for non-catalytic pyrolysis. The separation of the aqueous phase during CFP also serves to increases the energy density of CFP-oil compared to non-catalytic fast pyrolysis oils. In these experiments, the aqueous phase contained 3.4% and 5.8% of the biomass carbon for the 17-O and 20-O experiments, respectively. The lower concentration of carbon in the 17-O aqueous phase is consistent with the expected reduction in molecular solubility of organic compounds in water as the degree of deoxygenation is increased.22
An inverse relationship was observed between CFP-oil oxygen content and carbon yield, as has been reported previously.12,17,18,22,23 The reduction in oil carbon yield is primarily associated with the formation of coke on the catalyst and generation of carbon-containing light gases during the upgrading process, which is consistent with the higher coke yields (15.3 vs. 13.1%) and gas yields (26.0 vs. 25.3%) observed during production of the 17-O vs. 20-O CFP-oils. The carbon yield of compounds within the gas phase product stream is shown in Table S2.† The dominant gaseous products were carbon monoxide and carbon dioxide, and a combination of C1–C4 hydrocarbons were also detected. Condensable products refer to volatile compounds such as acetaldehyde, acetone, C5+ hydrocarbons that were detected in the exit gas. Notably, as much as 50% of this category was comprised of fuel-range compounds that could potentially be recovered using an optimized commercial-scale condensation system. These compounds are not included in the CFP-oil yield calculations reported in Table 1 and throughout the manuscript.
A carboxylic acid number of 29 mg KOH per g was measured for the 20-O CFP-oil, which was higher than the 19 mg KOH per g value measured for the 17-O CFP-oil but considerably lower than value of 76 mg KOH per g that has been reported for non-catalytic fast pyrolysis of pine.22 The higher acidity of the 20-O CFP-oil is further supported by compositional analysis provided in Table S3,† which revealed a 2-fold increase in the concentration of acids in the GC-MS detectable fraction of the 20-O vs. 17-O CFP-oil. The sulfur content of both oils was 32 ppm, and slight increases in density and viscosity were observed for the 20-O CFP-oil compared to the 17-O CFP-oil.
The total concentration of GC-MS detectable compounds increased as the oxygen content of the CFP-oil decreased, as shown in Fig. 1a. This effect is attributed to the cracking of high molecular weight oligomers during the upgrading process, resulting in a reduction in the average molecular weight of the oil and an increase in the fraction of the products that can be effectively volatized at the GC inlet. This analysis is further supported by GPC data provided in Fig. S1,† which confirms the lower molecular weight of the 17-O CFP-oil compared to the 20-O CFP-oil. The concentration of GC-MS detectable oxygenates in the 17-O CFP-oil was lower than in the 20-O CFP-oil (Fig. 1b), with the largest reductions observed for sugars and carbonyls. Previous research has identified reactive carbonyls as a primary species contributing to bio-oil instability.52,53 Catalytic upgrading during CFP plays an important role in reducing the concentration of these compounds and mitigates risks associated with the storage, transportation, and down-stream processing of CFP-oils. The concentration of phenolic compounds was slightly higher for the 17-O CFP-oil, which is attributed to the cracking of lignin-derived oligomers into smaller molecules that can be detected by GC-MS. The concentration of aromatic hydrocarbons in the 17-O CFP-oil was higher than in the 20-O CFP-oil, with the largest increases observed for benzene, toluene, xylene, and naphthalenes (Fig. 1c). The production of aromatic hydrocarbons during CFP is attributed to the conversion of oxygenated pyrolysis vapors within the ZSM-5 pore structure through a series of cracking, deoxygenation, oligomerization, cyclization, aromatization, isomerization, and polymerization reactions. Previous research has shown that fresh ZSM-5 exhibits high selectivity aromatic hydrocarbons, whereas oxygenated phenols, cresols and primary vapors become the dominant products as the catalyst is deactivated by coke.17,54
 | ||
| Fig. 1 (a) CFP-oil composition measured by GC-MS-Polyarc-FID, and (b) speciation of oxygenated products and (c) aromatic products by compound class. | ||
To better understand CFP catalyst deactivation rates and regeneration, five reaction-regeneration cycles were performed using a fixed bed CFP system equipped horizontal molecular beam mass spectrometer for online analysis.17 During each cycle, the CFP catalyst was exposed to biomass pyrolysis vapors at 500 °C until a cumulative biomass to catalyst ratio of 3 had been reached. The catalyst was then regenerated in dilute oxygen at 550 °C until the CO2 signal from coke combustion was no longer observed. The performance of the fresh and regenerated catalysts was compared based on selectivity to deoxygenated hydrocarbon and unreacted pyrolysis vapors, as described previously.17Fig. 2a and b show that the selectivity to deoxygenated hydrocarbons decreased and the selectivity to unreacted pyrolysis vapors increased as the catalyst deactivated over the course of an experiment. Notably, the original activity was restored after regeneration, and the rate of deactivation remained consistent over the five consecutive cycles. These data are consistent with previous experiments that suggest deactivation of zeolite catalysts due to coke deposition during CFP is highly reversable.55 These results are further supported by characterization of the catalyst before CFP, following CFP, and after regeneration. As given in Table S4,† measurements using N2 physisorption show that the catalyst surface area changes from 340 m2 g−1 in the fresh catalyst to 85 m2 g−1 in the post-CFP catalyst and then back to 336 m2 g−1 in the regenerated catalyst. Similarly, ammonia temperature programmed desorption shows that the acid site density of the catalyst changes from 765 μmol NH3 per g in the fresh catalyst to 516 μmol NH3 per g in the post-CFP catalyst and then to 722 μmol NH3 per g after regeneration. Collectively, these data suggest that the catalytic activity of the ZSM-5 catalyst can be recovered following oxidative treatment to remove accumulated coke. However, it is important to note that other deactivation mechanisms such as attrition, dealumination, and poisoning due to inorganic biomass contaminants are not addressed in these experiments and may contribute to irreversible catalyst deactivation over longer time periods, as described previously.16,56,57
 | ||
| Fig. 2 Selectivity to (a) deoxygenated hydrocarbons and (b) unreacted pyrolysis vapors observed during five consecutive reaction-regeneration cycles using a fixed bed CFP reactor equipped with a molecular beam mass spectrometer for online analysis. | ||
Hydrotreating
Hydrotreating experiments were performed using a continuous trickle-bed reactor system with a NiMo/Al2O3 catalyst provided by Johnson Matthey. The catalyst was sulfided in situ, and experiments were performed at 125 bar and a WHSV of 0.1 h−1. Results from experiments performed at 385 °C are provided in Table 2.| CFP-oil | 17-O | 20-O |
|---|---|---|
| Oil carbon yield, gC gC−1 CFP-oil | 91.1 ± 1.2% | 92% |
| Aqueous carbon yield, gC gC−1 CFP-oil | 0.12 ± 0.02% | 0.2% |
| Gas carbon yield, gC gC−1 CFP-oil | 4.1 ± 0.1% | 5% |
| Carbon balance | 95.3 + 1.3% | 97% |
| Oil oxygen content, wt% | 0.020 ± 0.005 | 0.04 |
Oil H![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C ratio, mol/mol :C ratio, mol/mol |
1.80 ± 0.03 | 1.76 |
| Oil density, g cm−3 | 0.839 ± 0.006 | 0.828 |
| Hydrogen consumption, g H2 per g CFP-oil | 0.073 ± 0.007 | 0.08 |
Generally, the hydrotreating results were similar for both CFP-oils. The oxygen content was effectively reduced to ≤0.04 wt%, and >90% of the carbon in the CFP-oil was retained in the liquid organic product. Between 4–5% of the carbon in the CFP-oil was converted into C1–C5 hydrocarbons, as shown in Table S5.† Although it was not directly evaluated in this study, these gas phase byproducts create an opportunity for additional energy recovery and may provide a pathway for the production of renewable propane, as is commonly practiced during hydrotreating of waste lipids and vegetable oils.58
The composition of the hydrotreated products determined by GC-vacuum ultraviolet spectroscopy (VUV) analysis is shown in Fig. 3a. Cycloalkanes account for 71–72 wt% of the product, aromatic hydrocarbons for 18–23 wt% of the product, linear alkanes for 6–10 wt% of the product, and oxygenates for <0.3 wt% of the product. The low oxygen content of these oils highlights the ability to produce a high-quality hydrocarbon fuel blendstock via single stage hydrotreating at 385 °C. To probe the impact of hydrotreating temperature, a series of experiments was performed with the 17-O CFP-oil in which the temperature of the isothermal zone was varied between 300 and 385 °C. These data, presented in Fig. 3b, reveal that the composition of the hydrotreated product is highly sensitive to temperature. Selectivity to cycloalkanes decreased from 71 wt% at 385 °C to 41 wt% at 350 °C and 14 wt% at 300 °C. Selectivity to aromatic hydrocarbons exhibited a concomitant increase, and breakthrough of undesired oxygenated phenolics was also observed at 300 °C. These data indicate that the hydrotreating temperature is a key process variable and suggests values of 350 °C or above are preferred to promote deoxygenation and hydrogenation at the pressure and space velocities investigated herein. Notably, the ability to shift the selectivity between aromatic and saturated products by varying the hydrotreating temperature provides a practical pathway to tailor the product compositions based on the process needs and hydrogen availability.
 | ||
| Fig. 3 (a) The composition in wt% determined by GC-VUV analysis of the 17-O and 20-O CFP-oils after hydrotreating at 385 °C, (b) the composition in wt% of the 17-O CFP-oil after hydrotreating at 300, 350, and 385 °C. The error bars represent ±σ/2 for replicate samples collected over a 48 h time period. The sample for 20 wt% O CFP-oil was collected as a single sample and no error bars are included. | ||
A consistent pressure drop in the range of 18–22 kPa was observed over the course of the hydrotreating experiments, as shown in Fig. S2.† Importantly, these data confirm the absence of major process upsets due to plugging or coking of the catalyst bed, as has been previously reported during hydrotreating of non-catalytic pyrolysis oils.59 However, it is important to note that the hydrotreating experiment reported herein were only performed for 72 h per condition, and additional research is needed to better understand long-term catalyst and process durability.
Distillation and fuel property analysis
The hydrotreated samples were distilled to obtain fuel-range cuts using a micro spinning band distillation column, as shown in Table 3. The combined yield of fuel range products was 93 wt% for both CFP-oils that were hydrotreated at 385 °C. The SAF cut comprised 46 wt% and 50 wt% of the product for the 20-O and 17-O CFP-oils, respectively. In both cases the gasoline fraction comprised 26 wt% of the product, and 17–21 wt% of the product boiled in the heavy diesel range. The results suggest a slightly higher fraction of high-boiling compounds in the hydrotreated 20-O than in the 17-O CFP-oil, consistent with the molecular weight distributions of the CFP-oils determined by GC-MS and GPC, above.| CFP-oil and hydrotreating temperature | 20-O 385 °C | 17-O 385 °C | 17-O 350 °C | 17-O 300 °C |
|---|---|---|---|---|
| Gasoline, wt% | 26 | 26 | 17 | 13 |
| SAF, wt% | 46 | 50 | 53 | 45 |
| Diesel, wt% | 21 | 17 | 21 | 15 |
| Residue >330 °C, wt% | 3 | 4 | 6 | 25 |
| Losses, wt% | 4 | 3 | 3 | 2 |
| Total fuel yield, wt% | 93 | 93 | 91 | 73 |
The 17-O CFP-oil that was hydrotreated at 350 °C exhibited a slight increase in SAF and diesel yields with lower gasoline production compared to the same experiment performed at 385 °C. These results are attributed to a reduction in thermal cracking and a corresponding increase in the average molecular weight of the product. These results are consistent with simulated distillation data showing a shift in the boiling point distribution to higher temperatures for CFP-oils that were hydrotreated at 350 °C vs. 385 °C (Fig. S3†). A similar effect likely contributes to the comparatively high residue fraction observed after hydrotreating at 300 °C, which reduced the selectivity to fuel range products from 93 wt% to 73 wt%.
The composition of the SAF fraction measured by GC-MS-Polyarc-FID is provided in Fig. 4. These data reveal 67–73% selectivity to cycloalkanes for the 17-O and 20-O CFP-oils that were hydrotreated at 385 °C. Cycloalkanes are a major component of conventional jet aviation fuels, constituting approximately 30% of Jet-A3. These compounds typically have density, flash point, freeze point, and specific energy properties that exceed conventional fuel specifications. However, emerging SAF production pathways (e.g., Fischer Tropsch, alcohol-to-jet, hydroprocessing of esters and fatty acids) favor the production of linear alkanes.3 The compositional data suggest that CFP could complement other SAF production routes by opening a pathway for the generation of SAF-range cycloalkanes. It has been reported that these molecules may also have desirable seal swelling properties and could potentially reduce the requirement for aromatics in existing jet turbine engines.3,60,61 Reducing the hydrotreating temperature had a considerable impact on the selectivity to cycloalkanes, which was reduced to 35% and 10% for hydrotreating temperatures of 350 and 300 °C, respectively. The reduction in cycloalkanes was accompanied by a concomitant increase in selectivity to aromatics, and oxygenates accounted for 38% of the identified product composition at 300 °C.
 | ||
| Fig. 4 (a) The SAF fraction selectivity for 17-O and 20-O CFP-oils hydrotreated at temperatures from 300–385 °C. Selectivity data are based on GC-MS results in which 55–70 wt% of the total material was identified and quantified. | ||
Properties of the SAF fractions are compiled in Table 4, and the data are compared to target values based on ASTM D4054 (Standard Practice for Evaluation of New Aviation Turbine Fuels and Fuel Additives) and ASTM D1655 (Standard Specifications for Aviation Turbine Fuels). The density, flash point, freeze point, surface tension, volatility, and oxygen content were all within the target ranges for CFP-oils that were hydrotreated at 385 °C. The lower heating value (LHV) for these samples was slightly below the target value. However, the minor deviation was limited to 0.1–0.3 MJ kg−1 and could likely be addressed by optimizing the distillation process to exclude some of the higher boiling point compounds from the SAF fraction. Moreover, the SAF product from CFP is expected to be blended to provide a full range of linear alkanes, cycloalkanes, and aromatics in the final fuel. In this context, it is likely that the CFP SAF could be utilized as produced. The quality of the SAF decreased as the hydrotreating temperature was reduced. The deviation between the target and measured LHV grew to 1.0 MJ kg−1 for experiments performed at 350 °C and 4.7 MJ kg−1 for experiments performed at 300 °C. This trend is attributed to the observed shifts in hydrotreating selectivity from primarily cycloalkanes at higher temperature to less energy-dense aromatics and to oxygenates at lower temperature. Additionally, the flash point of the 350 °C SAF and the density, surface tension, and oxygen content of the 300 °C SAF did not meet the target values. These data further highlight the importance of the hydrotreating temperature on the product quality. Towards meeting jet fuel property specifications, compositional analysis combined with experimental fuel property data are being used to improve predictability of key properties using literature-based correlations, with the goal of integrating those predictions within our TEA models.62
| CFP-oil and hydrotreating temperature | Target values | 20-O 385 °C | 17-O 385 °C | 17-O 350 °C | 17-O 300 °C |
|---|---|---|---|---|---|
| a Based on ASTM D4054 specifications. b Based on ASTM D1655 specifications. c Based on typical ranges reported per ASTM D4054. d Based on ASTM D4050 specifications that C + H must be >99.5 wt%. | |||||
| Density, g mL−1 @ 15 °C | 0.730–0.880a | 0.843 | 0.854 | 0.878 | 0.952 |
| Flash point, °C | >38a | 41.5 | 41.5 | 36.5 | 49.5 |
| Freeze point, °C | <−40a | <−80 | <−80 | <−80 | −48 |
| Lower heating value, MJ kg−1 | >42.8b | 42.7 | 42.5 | 41.8 | 38.1 |
| Surface tension, mN m−1 @ 22 °C | 25–29c | 27.4 | 28.1 | 28.6 | 30.9 |
| D86 Simdist T10, °C | 150–205a | 162 | 162 | 156 | 163 |
| D86 Simdist T50, °C | 165–229a | 186 | 187 | 198 | 203 |
| D86 Simdist T90, °C | 190–262a | 225 | 227 | 246 | 237 |
| D86 Simdist FBP, °C | <300a | 250 | 253 | 271 | 260 |
| Oxygen content, wt% | <0.5d | <0.001 | <0.001 | 0.04 | 3.79 |
End-to-end carbon efficiency
Data from the integrated CFP and hydrotreating experiments can be utilized to inform the optimal degree of catalytic upgrading that should occur during each step to maximize the carbon efficiency of the end-to-end process. This concept is illustrated in Fig. 5, which shows the biomass-to-fuel carbon efficiency for 17-O and 20-O CFP-oils that were hydrotreated at 385 °C. These data reveal an opportunity to increase the total fuel yield from 18% to 22% by shifting the oxygen content of the CFP-oil intermediate from 17 to 20 wt%. This improvement in process-level efficiency is primarily driven by an increase in CFP carbon yield from 21% (17-O) to 26% (20-O), as outlined in Table 1. | ||
| Fig. 5 The integrated biomass-to-fuel carbon efficiency for 17-O and 20-O CFP-oils that were hydrotreated at 385 °C. Notably, this conservative analysis does not include condensable fuel range molecules that were detected in the CFP effluent gas and could potentially be recovered using an optimized commercial-scale condensation system. | ||
A Sankey plot of carbon efficiency for the integrated process shown in Fig. 6 highlights opportunities for further yield improvements. Specific strategies include catalyst and process development to reduce coking and promote coupling via aldol condensation and ketonization of reactive carbonyls in the <C5 condensable fraction. In addition to improving fuel yields, the effective utilization of byproduct streams is a critical step towards the development of a robust and economically viable pathway. Among these byproduct streams, the cumulative production of char and CFP-gases account for 45–46% of the biomass carbon. Identifying scalable strategies to effectively utilize these materials within the context of an integrated process represents a priority for future work.
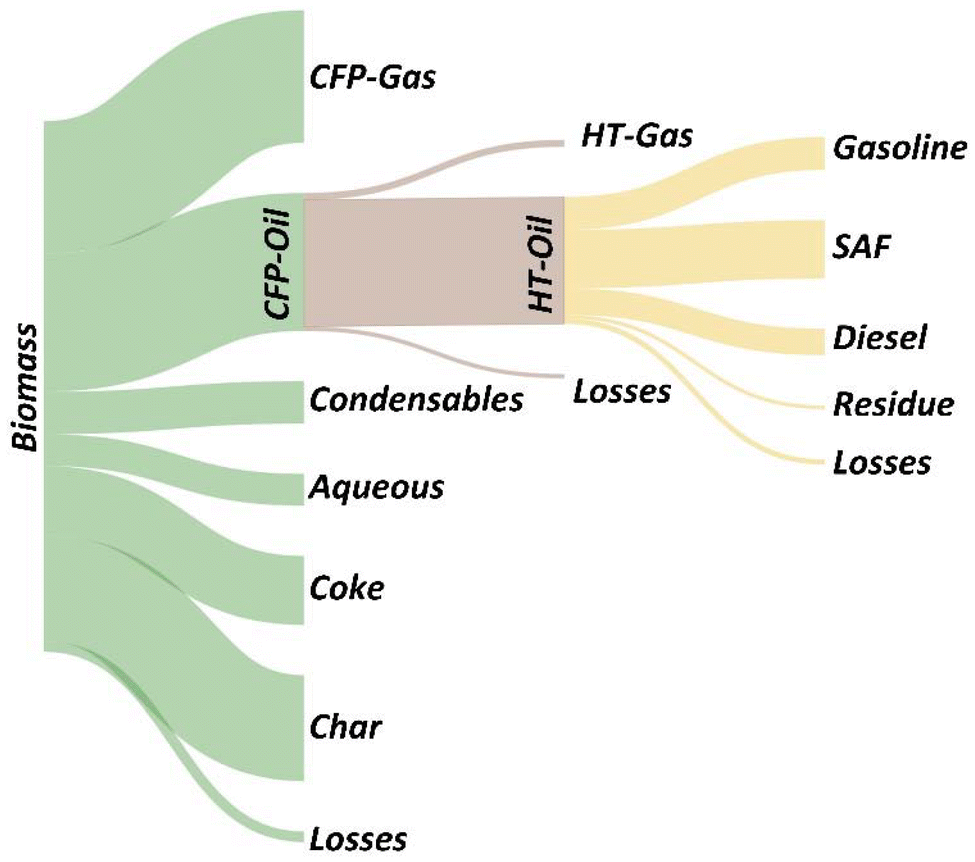 | ||
| Fig. 6 A Sankey plot showing the carbon efficiency for the integrated CFP and hydrotreating process. Yield data are from the 20-O CFP-oil hydrotreated at 385 °C. Image made with SankeyMATIC. | ||
Techno-economic analysis (TEA) and greenhouse gas (GHG) emissions
The minimum fuel selling price (MFSP) for the 17-O and 20-O CFP-oils hydrotreated at 385 °C were estimated using modelling methods that have been previously reported.63 The scaled-up conceptual design was updated based on the process conditions and yields reported herein. The cost breakdowns for the base cases of the 17-O and 20-O models are shown in Fig. 7. As expected, overall carbon efficiency towards liquid fuel products improves the economics of the process. To account for uncertainties in the model, we report the ±10% range values of the MFSP as $6.0–7.4 and $5.5–6.8 per gallon gasoline equivalent (GGE) of fuel. GGE is defined as 116090 Btu per gallon (32.3 MJ L−1) on a lower heating value basis. Total Capital Investment (TCI) estimated using our assumptions were $711 million and $726 million for the 17-O and 20-O cases, respectively, in 2016 US dollars. Further details of capital cost breakdown and tabular cost contributions are included in the ESI.† They are based on the aforementioned assumptions and no credits or subsidies are included in these estimates.
 | ||
| Fig. 7 Cost contributions for the (a) 17-O and (b) 20-O modelled base cases. Costs are in 2016 US dollars. | ||
A GREET 202264 based LCA tool was used to estimate the GHG emissions for the 17-O and 20-O cases, as shown in Fig. 8. GHG reductions over petroleum-based hydrocarbon fuels, represented by petroleum jet fuel (88.7 gCO2e per MJ),65 were estimated at 92% and 85% for the 17-O and 20-O cases. These calculations assume that the hydrogen demand for hydrotreating is met by reforming of pyrolysis off-gases,63,66 and previous reports have shown that the use of fossil-derived hydrogen would lead to increased GHG emissions.67 Feedstock production, transportation, and handling were the largest contributors to the overall GHG emissions.68 Conversely, emissions for the fuel production steps were net negative due to the generation of excess process heat. The LCA model estimates the impact of excess heat by converting it into renewable electricity. However, it should be noted that for overall sustainability and economic benefits, the production of liquid fuels should be maximized over electricity generation, and alternative uses of excess utilities are being explored separately.69,76 The modelled fuel yields for the 17-O and 20-O cases were 41 and 47 GGE per dry metric ton of biomass respectively, while quantities of exported electricity were 12.5 kW h per GGE (0.37 kW h/kW h) and 9.2 kW h per GGE (0.27 kW h/kW h) of liquid fuel produced. These LCA results are consistent with previous studies that report net lifecycle GHG emissions of 17–21 gCO2 per MJ (78–81% reduction) for the production of renewable diesel and gasoline via ex situ CFP using a Pt/TiO2 catalyst under hydrogen-rich conditions.26,70 Similar values ranging from 9.2–10.3 gCO2 per MJ have been reported for conceptual in situ and ex situ CFP using zeolite catalysts.63 A recent report highlighted opportunities to achieve values as low as 3.9 gCO2 per MJ by leveraging existing infrastructure for refinery co-hydroprocessing as well as the generation of high value chemical co-products.67 Notably, this report also revealed the considerable impact of utilizing a low-carbon hydrogen source such as reformed CFP off gasses towards achieving meaningful GHG emission reductions. Additional information regarding TEA and LCA is provided in Tables S6–S12.†
 | ||
| Fig. 8 GHG emissions contributions and credits for the (a) 17-O and (b) 20-O modeled base cases. The modeled biomass feedstock for the CFP process was 50% clean pine/50% forest residues. All cost and GHG emission estimates use a lower heating value basis; petroleum jet fuel was used as the representative hydrocarbon fuel for comparison purposes. Reductions in GHG emissions for RJF (renewable jet fuel, representing all modeled hydrocarbon fuel products) over PJF (petroleum jet fuel) are shown for the 17-O and 22-O cases. Calculated net GHG emissions were 7 and 13.3 gCO2e per MJ for the 17-O and 20-O cases, respectively. PJF GHG emissions are 88.7 gCO2e per MJ. The values of individual contributions shown on these charts are included in the ESI.† | ||
Conclusions
This research represents an important step forward for SAF production from biomass via CFP and hydrotreating. Integrated experimental campaigns provide detailed insight into the yields and product composition for each step of the process and reveal opportunities to increase carbon efficiency by tailoring the degree of catalytic upgrading during CFP. A first-of-its-kind assessment of SAF composition showed >75% selectivity to cycloalkanes, fuel property testing indicated good alignment with ATSM D4054 guidelines, and lifecycle assessment confirmed 85–92% reduction in greenhouse gas emissions compared to fossil-based pathways. Ongoing research is focused on CFP catalyst development to further improve carbon yield by reducing coking and promoting coupling of <C5 oxygenates into fuel-range molecules. Additional research is needed to effectively valorize other byproduct streams (e.g., light gases, char) to achieve a robust and economically viable process.Methods
Catalytic fast pyrolysis
CFP experiments were conducted in a 2-inch fluidized bed reactor (2FBR) system using pine feedstocks with an ash content of 1.0 wt% and a nominal 0.5 mm particle size. Biomass was screw-fed into a 2′′ inner-diameter fluidized bed reactor and nitrogen was used as the fluidizing gas. Vapors were separated from char in a cyclone and then fed into a secondary bubbling bed reactor where they were upgraded using a ZSM-5 catalyst with an alumina binder and a silica-to-alumina ratio of 30 (Johnson Matthey). Fresh catalyst was continuously fed and spent catalyst was continuously removed from the bubbling bed reactor to enable steady-state operation. A hot gas filter (HGF) was used prior to collection of bio-oils to remove any remaining solid material. Products were collected using a fractional condensation unit consisting of (1) a vessel cooled by an ethylene glycol water mixture, (2) an electrostatic precipitator (ESP), (3) two catch pots in dry-ice traps, (4) an iced coalescing filter, and (5) a third catch pot in a dry-ice trap. Oil and aqueous phase yields were determined gravimetrically from the mass increase in the condensation train. Light gas compositions in the exit gases were measured by NDIR analyzers (CO, CO2, and CH4) and by a micro-GC (N2, H2, CO, CO2, and C1–C4 hydrocarbons). The composition of light condensable compounds in the exit gas (C5+) was quantified by an online GC-MS-Polyarc-FID, and the gas flow was measured using a dry gas meter. During all experiments the biomass feed rate was held at a constant 300 g h−1. The biomass:catalyst feed ratio was adjusted by changing the catalyst feed rate. All other operating conditions were maintained. Pyrolysis temp: 500 °C, cyclone: 450 °C, HGF: 450 °C, and VPU: 500 °C. Total nitrogen feed was set to 16.3 slm. System oxidation was conducted between each run.
The liquid phases were analyzed for CHN in a Leco Analyzer, for S by inductively coupled plasma-optical emission spectroscopy (ICP-OES), and for water by Karl-Fisher titration.71 Carboxylic acid number was determined by a modified ASTM D7544 method.72 Chemical composition was analyzed by gas chromatography-mass spectrometry equipped with a Polyarc converter and a flame ionization detector (GC-MS-Polyarc-FID).73 Relative standard deviations for compounds in a representative calibration mixture injected multiple times across an analysis set are ≤5%. Method accuracy determined using calibration standard verification varies depending on the analyte but is typically <5%.
CFP catalyst deactivation and regeneration was evaluated using a fixed bed molecular beam mass spectrometer (MBMS) reactor described previously.17 In this system, a 250 mg bed of catalyst crushed to between 300 and 500 μm was exposed to pulses of pyrolysis vapors generated by pyrolyzing 25 mg pine biomass in quartz boats at 500 °C. 30 successive boats were pyrolyzed to achieve a biomass to catalyst ratio of 3. Pyrolysis vapors were passed over the bed in a stream of 400 mL min−1 He and 5 mL min−1 Ar. The reactor effluent was diluted with 3000 mL min−1 He prior to sampling by the 250 μm MBMS orifice where it was expanded adiabatically in a chamber held at approximately 100 mTorr. This adiabatic expansion cooled incoming gases and stopped further chemical reactions. The cooled gas was skimmed into a molecular beam and ionized via an electron impact ionization source operated at 22.5 eV. The resulting ions were measured using a quadrupole mass spectrometer scanning an m/z range of 10 to 510 every second. Flow through the reactor was normalized between experiments based on the signal of the Ar tracer gas.
Hydrotreating
Hydrotreating experiments were performed using a continuous trickle-bed reactor system with a NiMo/Al2O3 catalyst provided by Johnson Matthey. All experiments were performed at 125 bar and a WHSV of 0.1 h−1. The bed had a temperature profile with an initial transition zone starting at 150 °C followed by an isothermal zone. The temperature of the isothermal zone was varied between 300 and 385 °C. The catalyst was presulfided in situ at 125 bar. During the presulfidation process, the catalyst was heated to 150 °C at a rate of 2.5 °C min−1 in 150 mL min−1 of H2 gas containing 60 ppm of H2S. At this point 35 wt% di-tert-butyl disulfide/65 wt% decane was introduced at a rate of 0.05 ml min−1. The temperature was held at 150 °C for 2 h, increased to 385 °C at 1.5 °C min−1, and held for 4 h. During an experiment, CFP-oil was introduced at 2.5 ml h−1 and the H2/H2S gas mixture was introduced at 175 sml min−1. Liquid products were collected between 24 and 72 h under steady state operation at each condition. Liquids were collected using two alternating vessels that were chilled to 5 °C in the cold-water bath. Residual liquid (<1 mL) in the exit gas flow was chilled by a secondary condenser (−5 °C). The gas flow rate at the exit of the reactor was measured using a Coriolis flow meter, and the effluent gas composition was measured by a micro-GC.The hydrotreated product was analyzed for CHNS at Huffman-Hazen Laboratories in Golden, CO. Oxygen content was measured using an Elementar VL Cube with ALS Autosampler, and water by Karl-Fischer titration. Simulated distillation was performed according to ASTM D2887. Hydrocarbon group type composition was analyzed by gas chromatography with vacuum ultraviolet detection (GC-VUV) according to modified ASTM D8071, and composition by GC-MS-Polyarc-FID.
Distillation and fuel property analysis
Combined liquid samples were distilled to obtain fuel-range cuts using a BR Instruments Micro Spinning Band Distillation column. The hydrotreated product was combined with ca. 5 g of alumina boiling chips into a 250 ml round-bottomed boiling flask with a custom thermowell. The material was then heated and the low-temperature fraction collected from approximately 30 °C to 100 °C. The pot was then cooled to room temperature, about 2 g of fresh alumina boiling chips added, and the collection flask replaced with a fresh collection flask. The system was then subjected to vacuum and operated at 50 Torr. The pot was then heated, and fractions collected for the atmospheric equivalent vapor temperature (VaeT) of 100–115 °C, 115–130 °C, 130–145 °C, 145–245 °C, 245–260 °C, 260–300 °C, and 300–330 °C. All collected fractions were prepared and analyzed via ASTM D2887 (Simulated Distillation). The simulated distillation results were used to identify which cuts could be combined to produce a final SAF blendstock. Candidate SAF samples were evaluated for many of the Tier 1 properties outlined in ASTM D4054. Density at 15 °C was measured using ASTM D4052 (Mettler Toledo DM40), flashpoint by ASTM D6450 (Eralytics Eraflash), heating value by ASTM D240 (IKA C2000), freezing point by D5972 (Phase Technology 70Xi), surface tension by ASTM D1331 using a Whilhelmy plate (Nano Science Sigma 700), and simulated distillation by ASTM D2887 (Agilent 7890).Catalyst characterization
Nitrogen physisorption data were collected at −196 °C using a Quadrasorb instrument. The samples were pretreated under vacuum for 6 h at 300 °C. Surface area was determined using the Brunauer–Emmett–Teller (BET) method.NH3 TPD was performed using an Altamira AMI-300 instrument using pre- and post-reaction catalysts. Prior to analysis, each material was pretreated at 550 °C in He for 2 hours. The reactor was cooled to 120 °C and the catalyst bed was saturated with NH3 for 2 h. He was used to purge the system of non-adsorbed NH3 before NH3 desorption from 120 °C to 500 °C was performed. Desorbed molecules were tracked using a thermal conductivity detector.
TEA and GHG emissions estimation methods
A simplified process flow diagram outlining a conceptual CFP and hydrotreating process is shown in Fig. 9. The TEA generally follows the design and methods outlined in a comprehensive design report prepared in 2015,63 and the financial assumptions are consistent with those reported previously in 2021.66 Additional design updates from the 2015 design report in the current conceptual process include: (1) updated reaction conditions informed by the experimental results reported in this manuscript, (2) CFP operations are at closer to-atmospheric pressure, and that resulted in increases in reactor volumes, thus increasing the capital costs for related equipment, (3) no hydrogen is introduced during CFP, (4) no hydrocracking reactor is included for heavier-than-diesel fuel products, assuming that the heavier hydrocarbons will be routed appropriately through further refining processes, (5) fuel separations, still using two distillation columns as in the previous design, provide a different sets of boiling range cuts that include gasoline, SAF, and the heavier than SAF cut (that includes all heavies including diesel-range product). The MFSP is calculated based on the lower heating value of the entire fuel pool, i.e., gasoline, SAF, and heavies, GHG emissions were estimated using Argonne National Laboratory's “GREET-Based Interactive Life-Cycle Assessment of BETO Biofuel Pathways” tool.74 The approach is similar to previously documented Supply Chain Sustainability Analysis reports,68 in this case using a 50% blend of clean pine with 50% forest residues, as documented in an Idaho National Laboratory report.75 The lifecycle inventory (inputs and outputs) for the 17-O and 20-O base case process models were uploaded to the ANL tool website to calculate the GHG emissions reductions over petroleum jet fuel for the 17-O and 20-O cases respectively.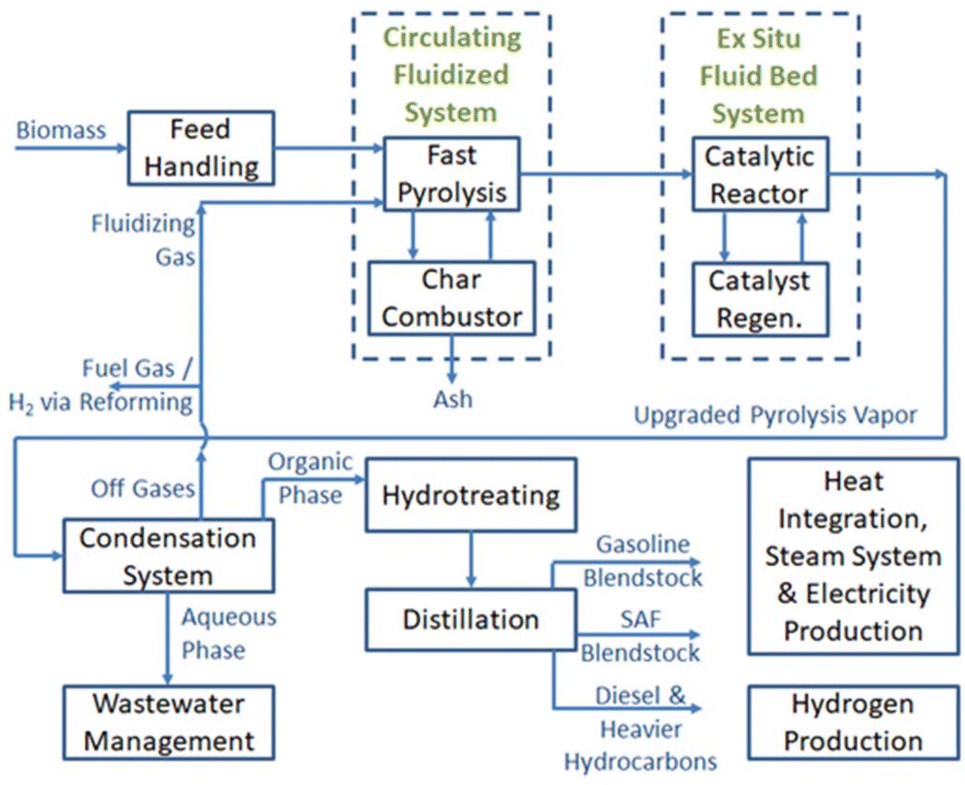 | ||
| Fig. 9 A simplified process flow diagram outlining the conceptual commercial process utilized for technoeconomic and lifecycle assessment. | ||
Data availability
Additional data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was authored by the National Renewable Energy Laboratory, operated by Alliance for Sustainable Energy, LLC, for the U.S. Department of Energy (DOE) under contract no. DE-AC36-08GO28308. The authors gratefully acknowledge funding for this research, provided by the U.S. Department of Energy Office of Energy Efficiency and Renewable Energy Bioenergy Technologies Office. This work was performed in collaboration with the Chemical Catalysis for Bioenergy Consortium (ChemCatBio), a member of the Energy Materials Network. The views expressed in the article do not necessarily represent the views of the U.S. Department of Energy or the United States Government. The U.S. Government retains and the publisher, by accepting the article for publication, acknowledges that the U.S. Government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this work, or allow others to do so, for U.S. Government purposes.We thank Damon Hartley from the Idaho National Laboratory for previously published feedstock cost modelling. For GHG emissions estimates we thank Longwen Ou and Hao Cai from Argonne National Laboratory for the development of the Biofuels Pathways Life-Cycle Assessment tool used in this work, and Eric Tan from NREL for previous guidance and inputs.
References
- N. Höhne, M. J. Gidden, M. den Elzen, F. Hans, C. Fyson, A. Geiges, M. L. Jeffery, S. Gonzales-Zuñiga, S. Mooldijk, W. Hare and J. Rogelj, Wave of net zero emission targets opens window to meeting the Paris Agreement, Nat. Clim. Change, 2021, 11(10), 820–822 CrossRef.
- C. Bergero, G. Gosnell, D. Gielen, S. Kang, M. Bazilian and S. J. Davis, Pathways to net-zero emissions from aviation, Nat. Sustainability, 2023, 6(4), 404–414 CrossRef.
- J. Holladay, Z. Abdullah and J. Heyne, Sustainable Aviation Fuel: Review of Technical Pathways, Department of Energy Energy Efficiency and Renewable Energy, 2020 Search PubMed.
- M. Philbrick, B. Hoffman and R. Nieves, Biofuels and Bioproducts from Wet and Gaseous Waste Streams: Challenges and Opportunities, 2017, DOE/EE-1472 Search PubMed.
- M. H. Langholtz, B. J. Stokes and L. M. Eaton, 2016 Billion-Ton Report: Advancing Domestic Resources for a Thriving Bioeconomy, USDOE Office of Energy Efficiency and Renewable Energy (EERE), Washington, D.C., 2016, DOE/EE-14407439 Search PubMed.
- S. Czernik and A. V. Bridgwater, Overview of Applications of Biomass Fast Pyrolysis Oil, Energy Fuels, 2004, 18(2), 590–598 CrossRef CAS.
- D. A. Ruddy, J. A. Schaidle, J. R. Ferrell III, J. Wang, L. Moens and J. E. Hensley, Recent advances in heterogeneous catalysts for bio-oil upgrading via “ex situ catalytic fast pyrolysis”: catalyst development through the study of model compounds, Green Chem., 2014, 16(2), 454–490 RSC.
- Y. Han, M. Gholizadeh, C.-C. Tran, S. Kaliaguine, C.-Z. Li, M. Olarte and M. Garcia-Perez, Hydrotreatment of pyrolysis bio-oil: A review, Fuel Process. Technol., 2019, 195, 106140 CrossRef CAS.
- R. French and S. Czernik, Catalytic pyrolysis of biomass for biofuels production, Fuel Process. Technol., 2010, 91(1), 25–32 CrossRef CAS.
- C. J. Wrasman, A. N. Wilson, O. D. Mante, K. Iisa, A. Dutta, M. S. Talmadge, D. C. Dayton, S. Uppili, M. J. Watson, X. Xu, M. B. Griffin, C. Mukarakate, J. A. Schaidle and M. R. Nimlos, Catalytic pyrolysis as a platform technology for supporting the circular carbon economy, Nat. Catal., 2023, 6(7), 563–573 CrossRef.
- E. F. Iliopoulou, K. S. Triantafyllidis and A. A. Lappas, Overview of catalytic upgrading of biomass pyrolysis vapors toward the production of fuels and high-value chemicals, Wiley Interdiscip Rev Energy Environ, 2019, 8(1), e322 Search PubMed.
- K. Iisa, R. J. French, K. A. Orton, M. M. Yung, D. K. Johnson, J. ten Dam, M. J. Watson and M. R. Nimlos, In Situ and ex Situ Catalytic Pyrolysis of Pine in a Bench-Scale Fluidized Bed Reactor System, Energy Fuels, 2016, 30(3), 2144–2157 CrossRef CAS.
- K. Iisa, C. Mukarakate, R. J. French, F. A. Agblevor, D. M. Santosa, H. Wang, A. N. Wilson, E. Christensen, M. B. Griffin and J. A. Schaidle, From biomass to fuel blendstocks via catalytic fast pyrolysis and hydrotreating: an evaluation of carbon efficiency and fuel properties for three pathways, Energy Fuels, 2023, 37(24), 19653–19663 CrossRef CAS PubMed.
- D. M. Santosa, C. Zhu, F. A. Agblevor, B. Maddi, B. Q. Roberts, I. V. Kutnyakov, S.-J. Lee and H. Wang, In Situ Catalytic Fast Pyrolysis Using Red Mud Catalyst: Impact of Catalytic Fast Pyrolysis Temperature and Biomass Feedstocks, ACS Sustainable Chem. Eng., 2020, 8(13), 5156–5164 CrossRef CAS.
- K. Wang, J. Zhang, B. H. Shanks and R. C. Brown, The deleterious effect of inorganic salts on hydrocarbon yields from catalytic pyrolysis of lignocellulosic biomass and its mitigation, Appl. Energy, 2015, 148, 115–120 CrossRef CAS.
- A. Eschenbacher, P. A. Jensen, U. B. Henriksen, J. Ahrenfeldt, C. Li, J.Ø Duus, U. V. Mentzel and A. D. Jensen, Impact of ZSM-5 Deactivation on Bio-Oil Quality during Upgrading of Straw Derived Pyrolysis Vapors, Energy Fuels, 2019, 33(1), 397–412 CrossRef CAS.
- C. Mukarakate, X. Zhang, A. R. Stanton, D. J. Robichaud, P. N. Ciesielski, K. Malhotra, B. S. Donohoe, E. Gjersing, R. J. Evans, D. S. Heroux, R. Richards, K. Iisa and M. R. Nimlos, Real-time monitoring of the deactivation of HZSM-5 during upgrading of pine pyrolysis vapors, Green Chem., 2014, 16(3), 1444–1461 RSC.
- V. Paasikallio, C. Lindfors, E. Kuoppala, Y. Solantausta, A. Oasmaa, J. Lehto and J. Lehtonen, Product quality and catalyst deactivation in a four day catalytic fast pyrolysis production run, Green Chem., 2014, 16(7), 3549–3559 RSC.
- F. Lin, Y. Lu, K. A. Unocic, S. E. Habas, M. B. Griffin, J. A. Schaidle, H. M. Meyer III, Y. Wang and H. Wang, Deactivation by Potassium Accumulation on a Pt/TiO2 Bifunctional Catalyst for Biomass Catalytic Fast Pyrolysis, ACS Catal., 2022, 12(1), 465–480 CrossRef CAS.
- K. Iisa, R. J. French, K. A. Orton, S. Budhi, C. Mukarakate, A. R. Stanton, M. M. Yung and M. R. Nimlos, Catalytic Pyrolysis of Pine Over HZSM-5 with Different Binders, Top. Catal., 2016, 59(1), 94–108 CrossRef CAS.
- E. Taarning, C. M. Osmundsen, X. Yang, B. Voss, S. I. Andersen and C. H. Christensen, Zeolite-catalyzed biomass conversion to fuels and chemicals, Energy Environ. Sci., 2011, 4(3), 793–804 RSC.
- K. Iisa, R. J. French, K. A. Orton, A. Dutta and J. A. Schaidle, Production of low-oxygen bio-oil via ex situ catalytic fast pyrolysis and hydrotreating, Fuel, 2017, 207, 413–422 CrossRef CAS.
- V. Paasikallio, K. Kalogiannis, A. Lappas, J. Lehto and J. Lehtonen, Catalytic Fast Pyrolysis: Influencing Bio-Oil Quality with the Catalyst-to-Biomass Ratio, Energy Technol., 2017, 5(1), 94–103 CrossRef CAS.
- J. Liang, G. Shan and Y. Sun, Catalytic fast pyrolysis of lignocellulosic biomass: Critical role of zeolite catalysts, Renewable Sustainable Energy Rev., 2021, 139, 110707 CrossRef CAS.
- B. Luna-Murillo, M. Pala, A. L. Paioni, M. Baldus, F. Ronsse, W. Prins, P. C. A. Bruijnincx and B. M. Weckhuysen, Catalytic Fast Pyrolysis of Biomass: Catalyst Characterization Reveals the Feed-Dependent Deactivation of a Technical ZSM-5-Based Catalyst, ACS Sustainable Chem. Eng., 2021, 9(1), 291–304 CrossRef CAS.
- M. B. Griffin, K. Iisa, H. Wang, A. Dutta, K. A. Orton, R. J. French, D. M. Santosa, N. Wilson, E. Christensen, C. Nash, K. M. Van Allsburg, F. G. Baddour, D. A. Ruddy, E. C. D. Tan, H. Cai, C. Mukarakate and J. A. Schaidle, Driving towards cost-competitive biofuels through catalytic fast pyrolysis by rethinking catalyst selection and reactor configuration, Energy Environ. Sci., 2018, 11(10), 2904–2918 RSC.
- M. Saidi, F. Samimi, D. Karimipourfard, T. Nimmanwudipong, B. C. Gates and M. R. Rahimpour, Upgrading of lignin-derived bio-oils by catalytic hydrodeoxygenation, Energy Environ. Sci., 2014, 7(1), 103–129 RSC.
- M. B. Griffin, G. A. Ferguson, D. A. Ruddy, M. J. Biddy, G. T. Beckham and J. A. Schaidle, Role of the Support and Reaction Conditions on the Vapor-Phase Deoxygenation of m-Cresol over Pt/C and Pt/TiO2 Catalysts, ACS Catal., 2016, 6(4), 2715–2727 CrossRef CAS.
- M. M. Yung, C. Mukarakate, K. Iisa, A. N. Wilson, M. R. Nimlos, S. E. Habas, A. Dutta, K. A. Unocic, J. A. Schaidle and M. B. Griffin, Advancements and challenges in the production of low-carbon fuels via catalytic fast pyrolysis of biomass through refinery integration and co-product generation, Green Chem., 2023, 25(17), 6809–6822 RSC.
- C. Mukarakate, K. Iisa, S. E. Habas, K. A. Orton, M. Xu, C. Nash, Q. Wu, R. M. Happs, R. J. French, A. Kumar, E. M. Miller, M. R. Nimlos and J. A. Schaidle, Accelerating catalyst development for biofuel production through multiscale catalytic fast pyrolysis of biomass over Mo2C, Chem. Catal., 2022, 2(7), 1819–1831 CrossRef CAS.
- R. J. French, K. Iisa, K. A. Orton, M. B. Griffin, E. Christensen, S. Black, K. Brown, S. E. Palmer, J. A. Schaidle, C. Mukarakate and T. D. Foust, Optimizing Process Conditions during Catalytic Fast Pyrolysis of Pine with Pt/TiO2—Improving the Viability of a Multiple-Fixed-Bed Configuration, ACS Sustainable Chem. Eng., 2021, 9(3), 1235–1245 CrossRef CAS.
- C. A. Mullen, A. A. Boateng, D. J. Mihalcik and N. M. Goldberg, Catalytic Fast Pyrolysis of White Oak Wood in a Bubbling Fluidized Bed, Energy Fuels, 2011, 25(11), 5444–5451 CrossRef CAS.
- C. A. Mullen, P. C. Tarves, L. M. Raymundo, E. L. Schultz, A. A. Boateng and J. O. Trierweiler, Fluidized Bed Catalytic Pyrolysis of Eucalyptus over HZSM-5: Effect of Acid Density and Gallium Modification on Catalyst Deactivation, Energy Fuels, 2018, 32(2), 1771–1778 CrossRef CAS.
- C. A. Mullen, C. Dorado and A. A. Boateng, Catalytic co-pyrolysis of switchgrass and polyethylene over HZSM-5: Catalyst deactivation and coke formation, J. Anal. Appl. Pyrolysis, 2018, 129, 195–203 CrossRef CAS.
- C. A. Mullen, A. A. Boateng, K. B. Hicks, N. M. Goldberg and R. A. Moreau, Analysis and Comparison of Bio-Oil Produced by Fast Pyrolysis from Three Barley Biomass/Byproduct Streams, Energy Fuels, 2010, 24(1), 699–706 CrossRef CAS.
- C. A. Mullen, A. A. Boateng and N. M. Goldberg, Production of Deoxygenated Biomass Fast Pyrolysis Oils via Product Gas Recycling, Energy Fuels, 2013, 27(7), 3867–3874 CrossRef CAS.
- A. A. Boateng, M. A. Schaffer, C. A. Mullen and N. M. Goldberg, Mobile demonstration unit for fast- and catalytic pyrolysis: The combustion reduction integrated pyrolysis system (CRIPS), J. Anal. Appl. Pyrolysis, 2019, 137, 185–194 CrossRef CAS.
- K. Wang, D. C. Dayton, J. E. Peters and O. D. Mante, Reactive catalytic fast pyrolysis of biomass to produce high-quality bio-crude, Green Chem., 2017, 19(14), 3243–3251 RSC.
- D. C. Dayton, O. D. Mante, J. Weiner, C. Komnaris, S. Verdier and J. Gabrielsen, Integrated Reactive Catalytic Fast Pyrolysis: Biocrude Production, Upgrading, and Coprocessing, Energy Fuels, 2022, 36(16), 9147–9157 CrossRef CAS.
- D. C. Dayton, J. R. Carpenter, A. Kataria, J. E. Peters, D. Barbee, O. D. Mante and R. Gupta, Design and operation of a pilot-scale catalytic biomass pyrolysis unit, Green Chem., 2015, 17(9), 4680–4689 RSC.
- D. C. Dayton, O. D. Mante and J. Weiner, Effect of Temperature on the Pilot-Scale Catalytic Pyrolysis of Loblolly Pine, Energy Fuels, 2021, 35(16), 13181–13190 CrossRef CAS.
- J. P. Polin, C. A. Peterson, L. E. Whitmer, R. G. Smith and R. C. Brown, Process intensification of biomass fast pyrolysis through autothermal operation of a fluidized bed reactor, Appl. Energy, 2019, 249, 276–285 CrossRef CAS.
- R. C. Brown, Heterodoxy in Fast Pyrolysis of Biomass, Energy Fuels, 2021, 35(2), 987–1010 CrossRef CAS.
- J. P. Polin, H. D. Carr, L. E. Whitmer, R. G. Smith and R. C. Brown, Conventional and autothermal pyrolysis of corn stover: Overcoming the processing challenges of high-ash agricultural residues, J. Anal. Appl. Pyrolysis, 2019, 143, 104679 CrossRef CAS.
- G. Yildiz and W. Prins, Perspectives of Biomass Catalytic Fast Pyrolysis for Co-refining: Review and Correlation of Literature Data from Continuously Operated Setups, Energy Fuels, 2023, 37(2), 805–832 CrossRef CAS.
- A. Eschenbacher, P. Fennell and A. D. Jensen, A Review of Recent Research on Catalytic Biomass Pyrolysis and Low-Pressure Hydropyrolysis, Energy Fuels, 2021, 35(22), 18333–18369 CrossRef CAS.
- T. K. Dada, M. Sheehan, S. Murugavelh and E. Antunes, A review on catalytic pyrolysis for high-quality bio-oil production from biomass, Biomass Convers. Biorefin., 2023, 13(4), 2595–2614 CrossRef CAS.
- K. Fehrenbacher, A Biofuel Dream Gone Bad, Fortune, 2015, https://fortune.com/longform/kior-vinod-khosla-clean-tech/ Search PubMed.
- D. Sudolsky, Bio-TCat™ technology viability confirmed during extensive Anellotech pilot plant campaign, 2019 Search PubMed.
- T. Vries, BioBTX secures over € 80 million to launch world's first renewable aromatics plant, 2024 Search PubMed.
- D. Howe, T. Westover, D. Carpenter, D. Santosa, R. Emerson, S. Deutch, A. Starace, I. Kutnyakov and C. Lukins, Field-to-Fuel Performance Testing of Lignocellulosic Feedstocks: An Integrated Study of the Fast Pyrolysis–Hydrotreating Pathway, Energy Fuels, 2015, 29(5), 3188–3197 CrossRef CAS.
- H. Wang, Y. Liu, L. Zhang, R. Gunawan, Z. Wang and C.-Z. Li, Conversion of carbonyl compounds in bio-oil during the acid/base-catalysed reactive distillation at high pressure, Fuel, 2021, 304, 121492 CrossRef CAS.
- H. Hu, Y. Luo, J. Zou, S. Zhang, D. Yellezuome, M. M. Rahman, Y. Li, C. Li and J. Cai, Exploring aging kinetic mechanisms of bio-oil from biomass pyrolysis based on change in carbonyl content, Renewable Energy, 2022, 199, 782–790 CrossRef CAS.
- C. Engtrakul, C. Mukarakate, A. K. Starace, K. A. Magrini, A. K. Rogers and M. M. Yung, Effect of ZSM-5 acidity on aromatic product selectivity during upgrading of pine pyrolysis vapors, Catal. Today, 2016, 269, 175–181 CrossRef CAS.
- M. M. Yung, A. K. Starace, M. B. Griffin, J. D. Wells, R. E. Patalano, K. R. Smith and J. A. Schaidle, Restoring ZSM-5 performance for catalytic fast pyrolysis of biomass: Effect of regeneration temperature, Catal. Today, 2019, 323, 76–85 CrossRef CAS.
- I. Kariim, H. Swai and T. Kivevele, Bio-Oil Upgrading over ZSM-5 Catalyst: A Review of Catalyst Performance and Deactivation, Int. J. Energy Res., 2023, 2023(1), 4776962 Search PubMed.
- E. Heracleous, E. Pachatouridou, A. M. Hernández-Giménez, H. Hernando, T. Fakin, A. L. Paioni, M. Baldus, D. P. Serrano, P. C. A. Bruijnincx, B. M. Weckhuysen and A. A. Lappas, Characterization of deactivated and regenerated zeolite ZSM-5-based catalyst extrudates used in catalytic pyrolysis of biomass, J. Catal., 2019, 380, 108–122 CrossRef CAS.
- R. M. Baldwin, M. R. Nimlos and Y. Zhang, Techno-Economic, Feasibility, and Life Cycle Analysis of Renewable Propane, National Renewable Energy Lab, 2022 Search PubMed.
- D. C. Elliott, H. Wang, M. Rover, L. Whitmer, R. Smith and R. Brown, Hydrocarbon Liquid Production via Catalytic Hydroprocessing of Phenolic Oils Fractionated from Fast Pyrolysis of Red Oak and Corn Stover, ACS Sustainable Chem. Eng., 2015, 3(5), 892–902 CrossRef CAS.
- A. Landera, R. P. Bambha, N. Hao, S. P. Desai, C. M. Moore, A. D. Sutton and A. George, Building Structure-Property Relationships of Cycloalkanes in Support of Their Use in Sustainable Aviation Fuels, Front. Energy Res., 2022, 9, 771697 CrossRef.
- Z. Yang, Z. Xu, M. Feng, J. R. Cort, R. Gieleciak, J. Heyne and B. Yang, Lignin-based jet fuel and its blending effect with conventional jet fuel, Fuel, 2022, 321, 124040 CrossRef CAS.
- S. Watanasiri, E. Paulechka, K. Iisa, E. Christensen, C. Muzny and A. Dutta, Prediction of sustainable aviation fuel properties for liquid hydrocarbons from hydrotreating biomass catalytic fast pyrolysis derived organic intermediates, Sustainable Energy Fuels, 2023, 7(10), 2413–2427 RSC.
- A. Dutta, A. Sahir, E. C. D. Tan, D. Humbird, L. Snowden-Swan, P. A. Meyer, J. Ross, D. Sexton and J. Lukas, Process Design and Economics for the Conversion of Lignocellulosic Biomass to Hydrocarbon Fuels. Thermochemical Research Pathways with In Situ and Ex Situ Upgrading of Fast Pyrolysis Vapors, National Renewable Energy Laboratory and Pacific Northwest National Laboratory, 2015, NREL/TP-5100-62455, PNNL-23823 Search PubMed.
- M. Wang, A. Elgowainy, U. Lee, K. H. Baek, A. Bafana, P. T. Benavides, A. Burnham, H. Cai, V. Cappello, P. Chen, Y. Gan, U. R. Gracida-Alvarez, T. R. Hawkins, R. K. Iyer, J. C. Kelly, T. Kim, S. Kumar, H. Kwon, K. Lee, X. Liu, Z. Lu, F. H. Masum, C. Ng, L. Ou, K. Reddi, N. Siddique, P. Sun, P. Vyawahare, H. Xu and G. G. Zaimes, Summary of expansions and updates in GREET 2022, Argonne National Laboratory, Lemont, IL, 2022 Search PubMed.
- L. Jing, H. M. El-Houjeiri, J.-C. Monfort, J. Littlefield, A. Al-Qahtani, Y. Dixit, R. L. Speth, A. R. Brandt, M. S. Masnadi, H. L. MacLean, W. Peltier, D. Gordon and J. A. Bergerson, Understanding variability in petroleum jet fuel life cycle greenhouse gas emissions to inform aviation decarbonization, Nat. Commun., 2022, 13(1), 7853 CrossRef CAS PubMed.
- A. Dutta, C. Mukarakate, K. Iisa, H. Wang, M. Talmadge, D. Santosa, K. Harris, F. Baddour, D. Hartely, H. Cai, L. Ou, J. Schaidle and M. Griffin, Ex Situ Catalytic Fast Pyrolysis of Lignocellulosic Biomass to Hydrocarbon Fuels: 2020 State of Technology, 2021, NREL/TP-5100-80291 Search PubMed.
- A. Dutta, H. Cai, M. S. Talmadge, C. Mukarakate, K. Iisa, H. Wang, D. M. Santosa, L. Ou, D. S. Hartley, A. N. Wilson, J. A. Schaidle and M. B. Griffin, Model quantification of the effect of coproducts and refinery co-hydrotreating on the economics and greenhouse gas emissions of a conceptual biomass catalytic fast pyrolysis process, Chem. Eng. J., 2023, 451, 138485 CrossRef CAS.
- H. Cai, L. Ou, M. Wang, R. Davis, A. Dutta, K. Harris, M. Wiatrowski, E. Tan, A. Bartling, B. Klein, D. Hartley, Y. Lin, M. Roni, D. N. Thompson, L. Snowden-Swan and Y. Zhu, Supply Chain Sustainability Analysis of Renewable Hydrocarbon Fuels via Indirect Liquefaction, Ex Situ Catalytic Fast Pyrolysis, Hydrothermal Liquefaction, Combined Algal Processing, and Biochemical Conversion: Update of the 2020 State-of-Technology Cases, Argonne National Laboratory, Lemont, IL, 2021, ANL/ESD-21/1 Rev. 1 Search PubMed.
- A. Dutta, Process Considerations for the Production of Hydrogen via Steam Reforming of Oxygenated Gases from Biomass Pyrolysis and Other Conversion Processes, Adv. Sustainable Syst., 2023, 2300241 CrossRef.
- H. Cai, L. Ou, M. Wang, R. Davis, A. Dutta, K. Harris and M. Wiatrowski, et al., Supply Chain Sustainability Analysis of Renewable Hydrocarbon Fuels via Indirect Liquefaction, Ex Situ Catalytic Fast Pyrolysis, Hydrothermal Liquefaction, Combined Algal Processing, and Biochemical Conversion: Update of the 2020 State-of-Technology Cases, Argonne National Laboratory, 2021 Search PubMed.
- J. Miscall, E. D. Miscall, S. Deutch, R. Jackson and J. R. Ferrell III, Determination of Water Content in Bio-Oils by Volumetric Karl Fischer Titration Laboratory Analytical Procedure (LAP), National Renewable Energy Laboratory, 2021, issue date: October 7, 2021 Search PubMed.
- E. Christensen, J. Ferrell, M. V. Olarte, A. B. Padmaperuma and T. Lemmon, Acid Number Determination of Pyrolysis Bio-oils using Potentiometric Titration Laboratory Analytical Procedure, National Renewable Energy Laboratory, 2016, NREL/TP-5100-65890 Search PubMed.
- E. Christensen, J. Ferrell, M. V. Olarte and A. B. Padmaperuma, Quantification of Semi-Volatile Oxygenated Components of Pyrolysis Bio-Oil by Gas Chromatography/Mass Spectrometry (GC/MS) Laboratory Analytical Procedure (LAP), National Renewbale Energy Laboratory, 2016 Search PubMed, issue date: March 2, 2016.
- L. Ou, M. Sheely and H. Cai, GREET-Based Interactive Life-Cycle Assessment of Biofuel Pathways, 2022, ANL-22/50 177527 Search PubMed.
- D. Hartley, D. Thompson and H. Cai, Woody Feedstocks 2020 State of Technology Report, INL/EXT-20-59976, 2021.
- A. Dutta, M. S. Talmadge, E. C. D. Tan and J. A. Schaidle, Assessment of location and energy utility options for the implementation of pyrolytic biocrude production, Sustainable Energy Fuels, 2023, 7, 4955–4966 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc03333g |
| This journal is © The Royal Society of Chemistry 2024 |