
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecycling of polyurethanes: where we are and where we are going
Gabriele
Rossignolo
a,
Giulio
Malucelli
 *b and
Alessandra
Lorenzetti
a
*b and
Alessandra
Lorenzetti
a
aUniversità di Padova, Dept. of Industrial Engineering, Via F. Marzolo 9, 35131 Padova, Italy
bPolitecnico di Torino Dept. of Applied Science and Technology, Viale Teresa Michel 5, 15121 Alessandria, Italy. E-mail: giulio.malucelli@polito.it
First published on 19th December 2023
Abstract
Polyurethanes (PUs) represent a family of useful synthetic polymers (thermoplastic or thermosetting) obtained from diisocyanates and diols/polyols via polycondensation reactions. Within the circular economy concept and also considering the current need for limiting the environmental impact of plastics, several methods have been designed, assessed, and exploited for the recovery at the end-of-life of polyurethanes and for their recycling. Indeed, the processing of polyurethane wastes can be significantly beneficial not only from an ecological but also from an economic point of view. At present, feedstock (namely, glycolysis) and mechanical recycling are the two most important strategies to recover and recycle polyurethanes; notwithstanding, “biological recycling”, an approach that exploits the biological degradation of the polymer, is gaining interest. This review aims to elucidate the recycling processes of both thermoplastic and thermosetting polyurethanes, providing the reader with some perspectives about their possible future developments.
Introduction
Versatility is undoubtedly one of the main characteristics exhibited by polyurethanes (PUs), an important “family” of thermoplastic and thermosetting polymers containing a remarkable number of urethane groups (–HN–COO–), derived from isocyanates and hydroxyl-containing compounds via the reactions presented in Fig. 1a. This reaction is exothermic and occurs spontaneously even at room temperature; nevertheless, one or more catalysts are commonly used to speed up the reaction. The hydroxyl-containing compound could be a diol, polyol, or water, which is used as a chemical blowing agent for producing polyurethane foams. Indeed, isocyanate reacts with water, forming an unstable substituted carbamic acid that decomposes into an amine species together with carbon dioxide, leading to the blowing of the polyurethane polymer (Fig. 1b). The as-obtained primary amines may react again with isocyanate groups giving a substituted urea (Fig. 1c). At room temperature, this reaction is faster than the urethane formation. Active hydrogen atoms of substituted urea compounds or allophanates (formed by the reaction of isocyanates with urethanes, Fig. 1d) may react again with isocyanates yielding a biuret (Fig. 1e). Isocyanates are highly reactive, so they can also react themselves leading to polymerization reactions: dimerization yields symmetric or asymmetric uretodinediones; trimerization leads to the formation of isocyanurate rings that are responsible for the high thermal stability of polyisocyanurate foams.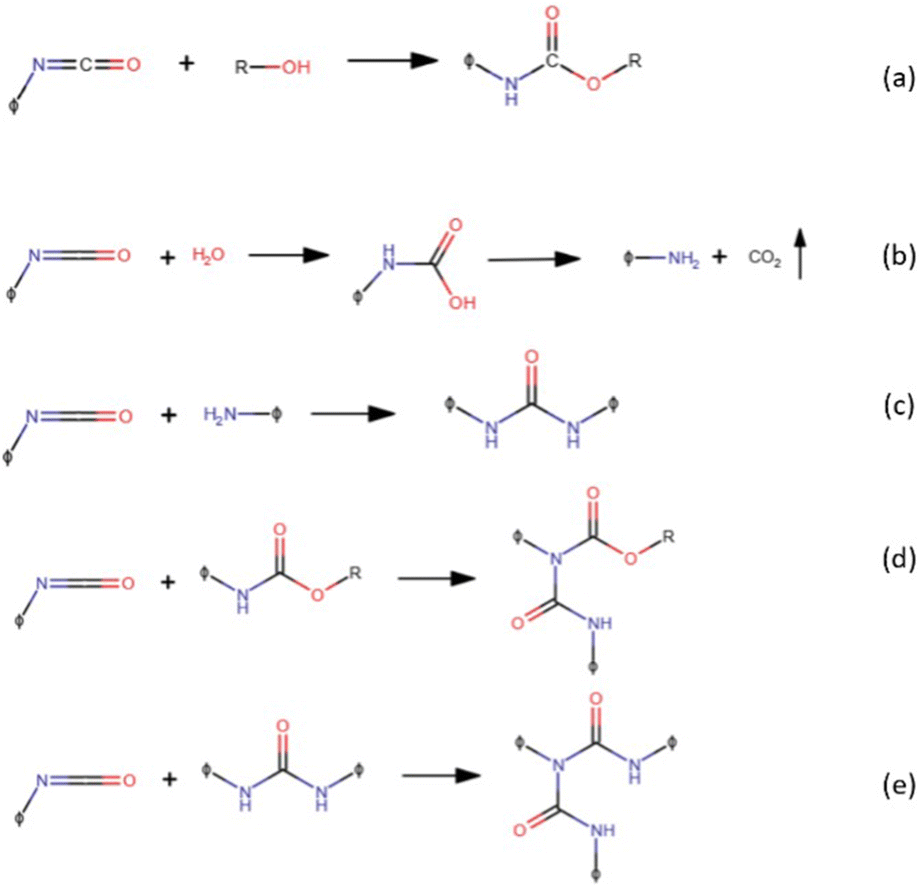 | ||
| Fig. 1 Reaction between isocyanates and hydroxyl-containing compounds (a), water (b), amine (c), urethane (d), and urea (e). | ||
Isocyanates could be aliphatic or aromatic and bifunctional, however, their functionality could be higher than 3. For some specific applications, such as flexible foams, elastomers, and thermoplastic polyurethanes, prepolymeric isocyanate, i.e., an NCO-functional reaction product of aromatic or aliphatic isocyanates and polyols, is commonly used.1 Similarly, a large variety of polyols are offered, but most of the polyols used fall into two major categories: hydroxyl-terminated or amino-terminated polyols. The structure of the polyols has a direct bearing on both processing and finished properties of the polyurethane polymer. In fact, the majority of the linkages found in polyurethanes are derived from the linkages found in the polyols.1 Polyols differ from each other with regard to the molecular weight (up to 6500 Da), hydroxyl number (up to 1000 mg KOH per g) and functionality (up to 8). Polyols with a higher molecular weight and a lower hydroxyl number and functionality are used for elastomers, coatings and flexible foams, while the others are employed for elastoplastics, rigid coatings and rigid foams.1
The wide availability of these co-reactants accounts for the possibility of tailoring the final properties of the obtained PUs, hence manufacturing from thermoplastic to thermosetting polymers, from flexible to semi-rigid and rigid polyurethanes (depending on the ratio between soft segments and hard segments2), which can be employed in different application sectors, including upholstered furniture (as flexible foams), wall and roof insulating rigid foams, thermoplastic footwear and medical devices, commercial refrigerators, adhesives and sealants, and coatings.3 A general classification scheme of PUs and some of their main current industrial applications are presented in Fig. 2 and Table 1. Besides, in EU27 + 3 countries, polyurethanes represent around 7.8% of all the produced plastic materials, which places them in 6th place in the EU27 + 3 plastics demand rank.4
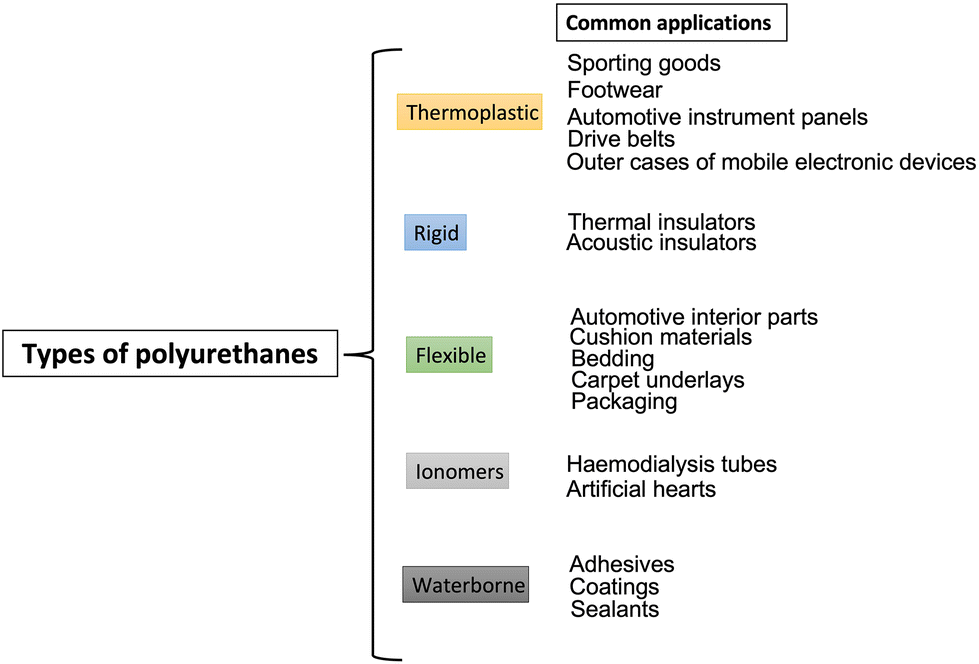 | ||
| Fig. 2 Polyurethanes: classification of general uses. | ||
| Sector | Main applications | Production (%) |
|---|---|---|
| Binders, adhesives, and sealants | Sealants, casting, assembling of wood boards, rubber or elastomeric flooring surfaces | 10 |
| Coatings | Bumpers & side panels of cars | 14 |
| Elastomers | Medical uses | 8 |
| Flexible foams | Mattrasses, seating, cars | 36 |
| Rigid foams | Packaging, appliances, insulation boards | 32 |
The advantage of employing polyurethanes for replacing synthetic plastics (such as polystyrene, polyvinyl chloride, and synthetic rubbers) or natural polymers (i.e., leather) is undeniable, also considering the possibility of using bio-sourced polyols, which reduces the environmental impact of PUs, making these polymer systems greener than the fossil-based counterparts.5 Further, waterborne polyurethane coatings take advantage of the use of water instead of potentially toxic organic solvents.6
Among the most interesting peculiarities, apart from enhanced electrical or adhesive properties, PUs exhibit high durability (higher than polyvinyl chloride) and high resistance toward different solvents/compounds (namely, water, organic solvents, and oils).7 These characteristics justify the very extended lifetimes of several polyurethane-based products, parts and components; however, the extension of their life cycle may have an important environmental impact, due to the accumulation of post-consumer polyurethane garbage, also considering the wearing out over time of such PU-based products as shoes or clothes, or the rapid exchange of some others (like sports equipment, cars, and furniture) for diverse or enhanced counterparts. Therefore, as witnessed by the increased number of publications in peer-reviewed journals on this topic (Fig. 3), the end-of-life recovery and recycling of polyurethanes are currently of extreme importance, in order to avoid landfill confinement (though it is still the most common strategy to exploit, despite the decreasing disposal of landfill spaces8) or incineration (i.e., energy recovery9), due to the toxicity of some products originating during the combustion process. Further, the recovery and recycling strategies of polyurethanes are motivated by the significant depletion of world reserves of such fossil fuels as crude oil, i.e., the raw material, on which the chemistry of polyurethanes is substantially based.
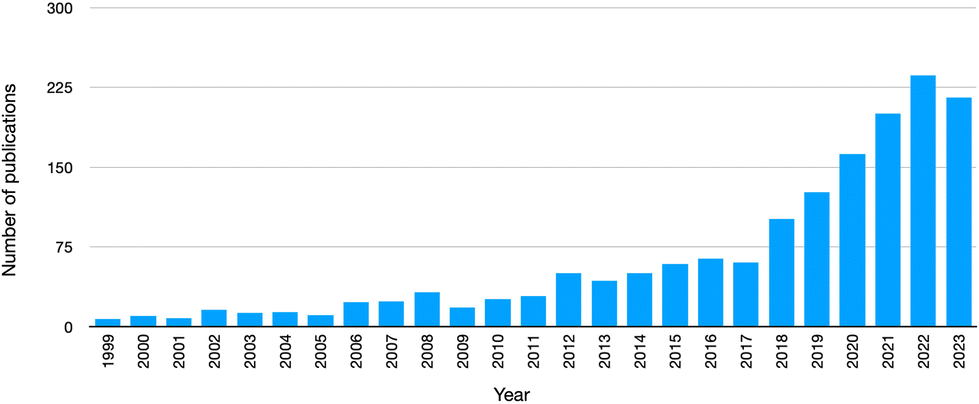 | ||
| Fig. 3 Number of publications (from 1999 to 2023) in peer-reviewed journals, dealing with “recycling AND polyurethanes” (data collected from the Web of Science™ database, accessed on Oct 27, 2023). | ||
In general, the recycling processes can be classified as follows:10
• Primary recycling: the recovered plastic is used in products with performance characteristics that are equivalent to those made using virgin plastics. This comprises closed-loop mechanical recycling.
• Secondary recycling: the recovered plastic is used in products that have less demanding performance requirements than the original application. This comprises open-loop mechanical recycling as well as physical recycling.
• Tertiary recycling: the waste plastic is used as the feedstock in a process that generates chemicals (i.e., the monomers and/or other compounds) and fuel. For this reason, it is also called feedstock recycling and comprises both chemical and thermochemical recycling. Microbial degradation still falls under this category.
• Quaternary recycling: energy is recovered from waste plastics by incineration.
These recycling strategies have been applied to polyurethanes, and they are presented in Fig. 4. Each one shows several peculiarities that not always are very well known by both the academic and industrial communities, especially by the people who are not specifically familiar with this research topic. A comparison among them has been reported by Yang et al.;11 and detailed explanations will be given in each specific section in the following.
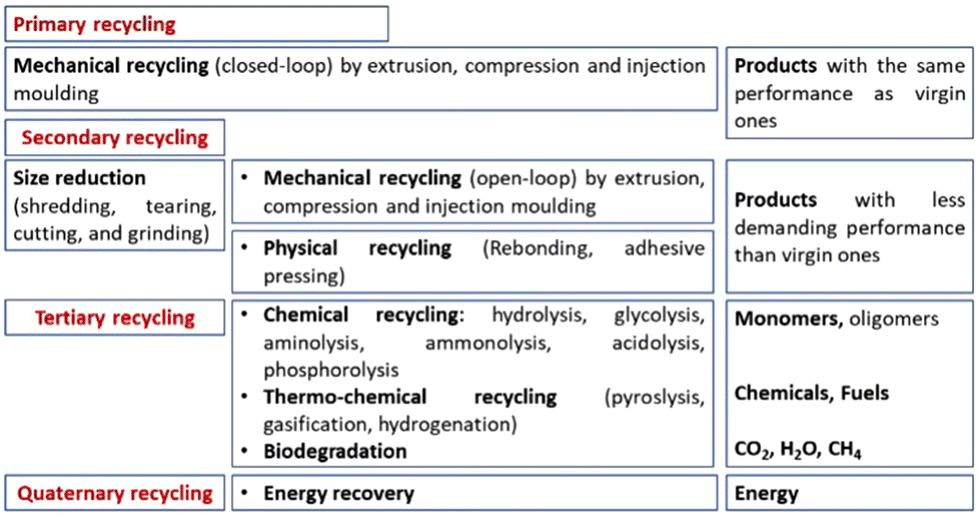 | ||
| Fig. 4 Possible treatment and recycling processes for polyurethane wastes. | ||
At present, the main recovery and recycling routes for polyurethanes, which are the most preferred on a commercial/industrial scale, include mechanical, physical and feedstock recycling that is based on degradation and/or depolymerization reaction. For this latter, hydrolysis or glycolysis represent the recycling strategy that has been mainly exploited: in fact, it allows for the recovery of polyols that can directly be re-used for the manufacturing of new polyurethane systems (i.e., regenerated polyurethanes), replacing up to 50% of virgin polyols with the recovered counterparts in polyurethane foam technology. The three bonds most susceptible to hydrolytic degradation are the ester, urea, and urethane. The ester reverts to the precursor acid and alcohol; this precursor acid further catalyzes ester hydrolysis, and thus the reaction becomes autocatalytic. Because of the autocatalysis of ester hydrolysis, this is the most prevalent hydrolytic degradation reaction. The urea bond can hydrolyze to form a carbamic acid and an amine. The carbamic acid normally is unstable and typically undergoes further reactions. The urethane linkage, although somewhat less susceptible, may undergo hydrolysis to yield a carbamic acid and the precursor alcohol.1
In terms of thermo-chemical process, what is relevant is the thermal stability of the bonds within the polymeric chain. As explained before, there are several chemical groups in a polyurethane chain. The thermal degradation rate and onset are highly dependent on the specific group considered. The onset of allophanate dissociation is around 100–120 °C, while the dissociation temperature of the biuret linkage takes place around 115–125 °C. These reactions are dissociations and somewhat reversible. They revert to the urethane or urea, from which they were formed. The aromatic-based urethane bond begins its thermal dissociation around 180 °C, i.e., before the thermal dissociation of the urea linkage (occurring at about 160–200 °C). The urethane can dissociate into the isocyanate and polyol from which it was formed. This reaction can also lead to the formation of amines and olefins and generates CO2, which is lost as a gas.1 The isocyanurate group is very stable and its decomposition starts around 300 °C.12,13
While the ester is the weakest link in hydrolysis, now it is the ether that is the weakest link in thermooxidation. The order of stability of polyethers to thermooxidation is as follows: polytetra methylene ether glycol is more stable than poly(ethylene oxide) glycols that are, in turn, more stable than poly(propylene oxide) glycols.1
In this context, the present review aims to provide the reader with an overview of the recycling processes of both thermoplastic and thermosetting polyurethanes, suggesting some perspectives about their possible future developments, also considering the strong current need for sustainability and low environmental impact of the designed recycling strategies.
Primary and secondary recycling of polyurethanes
Undoubtedly, primary and secondary (i.e., mechanical and physical) recycling represents the easiest and most basic strategy to recycle PUs, apart from landfill confinement. It is cost-effective and the investment for process equipment is quite low, though many times the performance of the recycled products is decreased, thus reducing the market potential with a low economic benefit.In these recycling processes, the materials have to undergo several processing steps first that are needed for reducing the original particle size to an appropriate level for being reprocessed in secondary manufacturing events. To this aim, polyurethane waste products (comprising old recycled components or production wastes – such as scrap parts and trimmings –) are converted into powders, pellets, or flakes, on the basis of the specific type of polymer that is recycled. Several methods can be successfully employed for fragmenting polyurethanes, namely: shredding, tearing, cutting, and grinding. It is worth noting that the average size of the fragmented particles is strictly related to the fragmentation technique: as an example, two-roll milling can be exploited for obtaining polyurethane particles having an average size below 100–125 μm, which can be employed as a filler in newly processed polyurethanes. Conversely, polyurethane powders having a larger particle size, between 125 and 250 μm, can be obtained by knife cutting. Further, pellet mills can be exploited for preparing PU pellets.
The following steps depend on the nature of polyurethanes, since it shall be kept in mind that the recycling processes are quite different for thermoplastic or thermosetting polyurethanes. Since, by definition, thermoplastic polyurethanes (TPUs) can be melted, they can be mechanically recycled using the common method employed for thermoplastic polymers in general, i.e., by extruding the recycled materials in the form of flakes or pellets, in the presence of some virgin polymers and/or compatibilizers.14–17
Conversely, thermosetting PUs, at least highly crosslinked PUs such as foams, cannot be recycled in such way, but what is generally called “physical recycling” is the eligible method. Physical recycling implies the direct reuse polyurethane wastes, still in form of powders, flakes or small particles without chemical treatment,11 and thus it can be considered among the secondary recycling methods. The PU particles are mixed with another component that will create a continuous matrix in which the particles are embedded. Generally, the use of heat and pressure is required to obtain the final recycled material. Physical recycling method is simple and convenient, with low cost, but there are still certain technical limitations. The performance of the physically recovered products is usually poor, so these are suitable for non-demanding applications only, thus limiting the market potential.11 The different mechanical and physical recycling processes are described in more detail below.
Mechanical recycling by extrusion, compression and injection moulding
Thermoplastic polyurethanes (TPUs) can be reprocessed by means of extrusion, hot compression, or injection moulding, without the use of any binder and obtaining, at the same time, up to 100% recycled material. As mentioned before, the first step in the mechanical recycling is the fragmentation of PU wastes.A general problem of mechanical recycling is related to the degradation of the polymer properties after each recycling cycle, because of the degradation caused by reprocessing (thermo-mechanical degradation) that sums to the degradation during lifetime (thermo-oxidative one).18 In each processing cycle, the material can undergo irreversible thermal and mechanical degradation, which leads to physical and chemical changes and a detriment of the final properties.19 Therefore, the material cannot be subjected to extensive reprocessing cycles,17,20 and the number of reprocessing cycles is generally limited to two or three.17 Sometimes, oligomers have been proposed as chain extenders or recycling aids which, when reacted in simple extrusion or injection moulding equipment with virgin, post-industrial recycle, or post-consumer recycled polyurethanes, effectively revert molecular weight degradation even at very small use levels.20
The possibility of PU waste melting (and bonding in the melted state) is strongly affected by the degree of cross-linking, which increases in the sequence: elastomers < flexible foams < rigid foams. Thus, some reaction injection moulding (RIM) and reinforced RIM (RRIM) polyurethanes, which are strictly speaking thermoset polymers, thanks to their very low crosslinking degree can be still mechanically recycled.21 For example, polyurethane scraps (such as car bumpers at the end of their life; very often the PU wastes primarily derive from RIM polyurethanes) are reground into fine particles that are subsequently compressed at high temperatures (about 180 °C) and pressures (around 350 bar). In this way, it is possible to fabricate such rigid components as motor and pump covers; besides, the incorporation of glass fibers allows for obtaining strengthened parts for door panels and dashboard panels (containing about 6 wt% of reground PU and 15 wt% glass fibers).22 Further, it is possible to use polyurethane particles as fillers for other resins (as an example for polyester resins), taking advantage of the toughening effect exerted by the PU particles compared to mineral fillers. Finally, this technique exhibits two main drawbacks: the difficulty of managing the presence of dyes and pigments in the polyurethane wastes and the limits in the obtainment of finely ground particles.23
When finely ground polyurethane particles cannot be obtained, it is possible to exploit the so-called structure reaction injection moulding (SRIM), employing up to 30 wt% of coarsely ground polymer scrap as an inner layer in a sandwiched assembly made of two glass fiber layers, which, in turn, are covered by a two-component PU resin (Fig. 5).
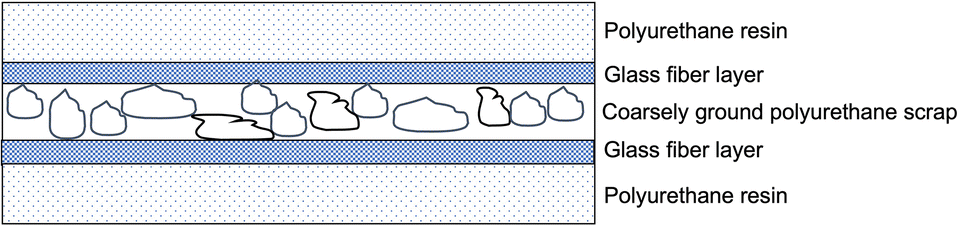 | ||
| Fig. 5 Recycling of PU scraps through structure reaction injection moulding (SRIM). | ||
As assessed by Bayer,24 it is also possible to exploit the injection moulding process to recycle relatively low crosslinked polyurethanes: for this purpose, polyurethane granules (size between 250 and 1000 μm) were employed for producing automotive parts through injection moulding carried out at around 180 °C and high shear compression (beyond 350 bar).
Further, Hulme and Goodhead25 demonstrated the suitability of dual injection moulding processes for recycling and reusing thermoplastic and thermosetting polyurethane wastes and scraps: this technique is based on the concurrent injection moulding into a single mold of a skin (made of virgin polyurethanes) and a core (made of recycled polyurethanes), as shown in Fig. 6. Its main advantages refer to the possibility of obtaining parts and components with enhanced mechanical features and good aesthetical properties (with improved surface finish); besides, it is possible to reprocess colored polyurethanes (as materials for the core).
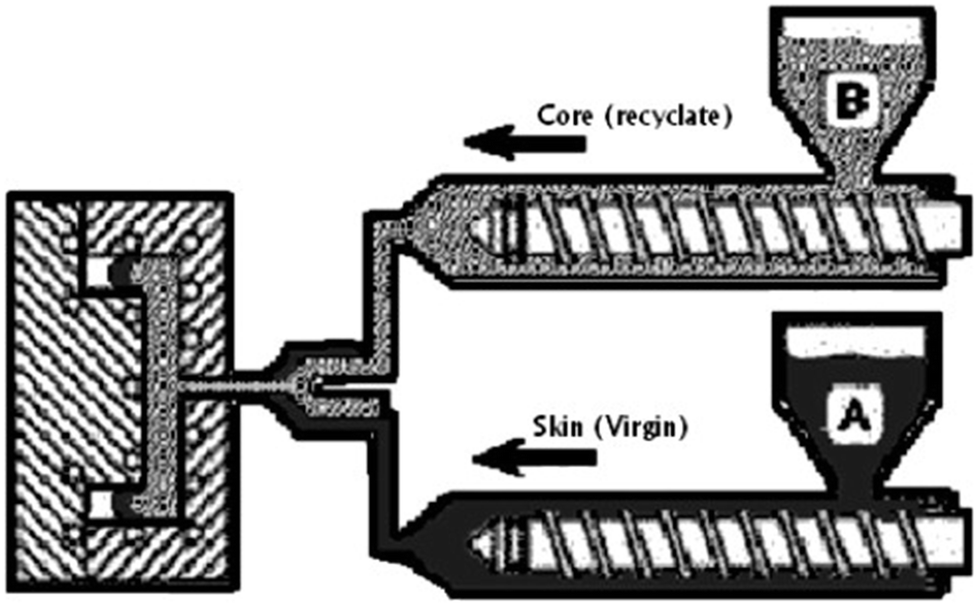 | ||
| Fig. 6 Scheme of the recycling and reuse of polyurethanes by dual injection moulding: two injection machines concurrently inject recycled (B) and virgin (A) polyurethanes; the former is employed for the core of the injection molded part, while the latter, which exhibits good aesthetical properties, is used as an external layer (skin). Reproduced with permission from reference.26 Copyright 2007 Elsevier. | ||
Rebonding re-processing (physical recycling) with adhesives
Polyurethane flakes or granules (hence particles showing an average size of a few mm) are coated with an adhesive at 5–10 wt% loading (such as poly(phenylenemethylene isocyanate) or 4,4′-diphenylmethane diisocyanates), as shown in Fig. 7. Fig. 8 shows the schematic of the rebonding of a flexible PU foam, while Fig. 9 displays the distribution of the foam flakes within the adhesive.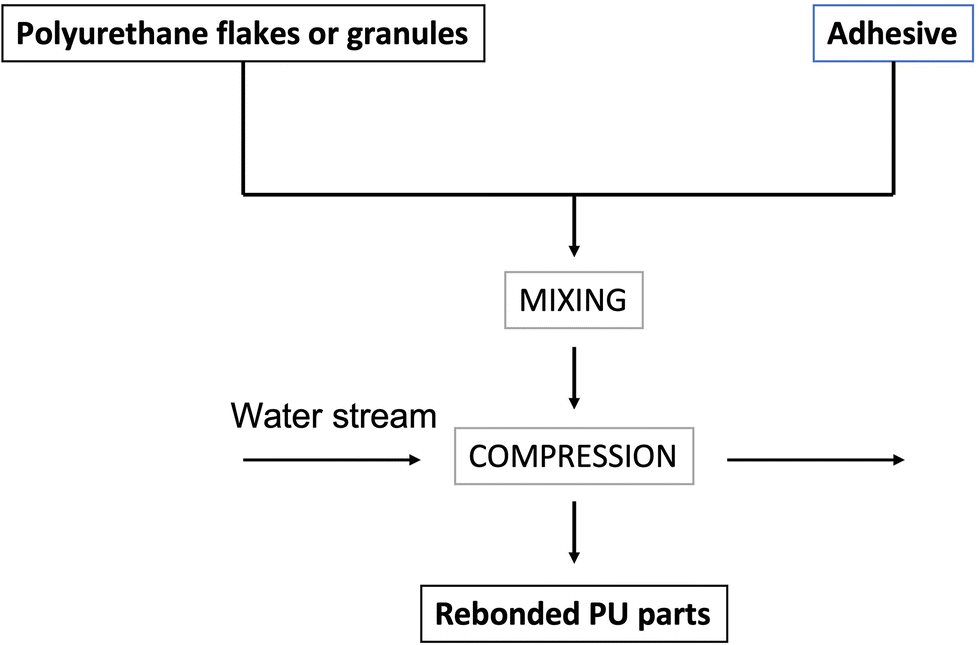 | ||
| Fig. 7 Scheme of the rebonding process for polyurethane flakes or granules. | ||
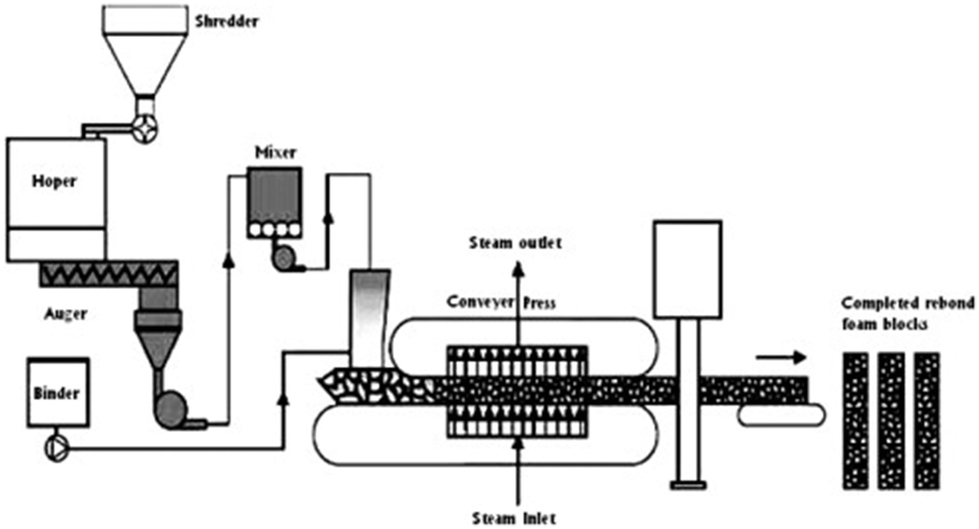 | ||
| Fig. 8 Schematic of flexible foam re-bonding. Reproduced with permission from ref. 27. Copyright 2007 Elsevier. | ||
 | ||
| Fig. 9 Rebonded foam: homogeneous distribution of the flake-binder mixture. Reproduced with permission from ref. 27. Copyright 2007 Elsevier. | ||
Then, a hot water stream (usually at high temperatures, beyond 100 °C) or steam melts the polyurethane that subsequently undergoes compression (usually between 30 and 200 bar). The steam reacts with the prepolymer or binder, which leads to the formation of rebonded PU product. Usually, the squeezed foam flakes are fully saturated with steam, which permits maximum contact with the binder; generally, several minutes are required to saturate compressed PU foams with steam. The rate of reaction can be controlled using catalysts.22 The binder is a crucial component in the production of rebonded PU foams.
This process allows for obtaining contoured parts that can be employed as covers for tires, athletic mats, car linings, and carpet underlays, among other uses.27 Besides, it is possible to manufacture PU building panels exhibiting outstanding insulation features and high resistance to moisture, although characterized by quite a high density and, therefore, with some limits for market exploitation.27
In a recent application of the rebonding strategy, Salino and Catai demonstrated the possibility of using polyurethane foam residues and recycled plasterboard sheet cores for acoustic applications.28 In particular, they found that the recycled materials exhibited high porosity (about 47%) and interesting sound absorption coefficients over a wide range of frequencies (from 250 to 2000 Hz).
Chemical (tertiary) recycling of polyurethanes
Under suitable conditions, it is possible to make the polymerization of polyurethanes partially reversible: generally speaking, gradual depolymerization may occur when the polymer reacts with organic compounds bearing active hydrogen atoms, able to attack the polar groups of the polymer main chain. The most liable chemical groups, involved in the depolymerization process, have been reported in the “introduction” section. In general, depolymerization can be based on the cleavage/exchange of urethane bonds or depolymerization based on the cleavage/exchange of chemical bonds of the precursors used for PU production, such as the ester groups of the polyol.29 This review will primarily focus on depolymerization reactions based on depolymerization involving urethane or urea groups.The possibility of recovering monomers and oligomers from the polymer is strictly correlated with the adopted experimental conditions, namely: selected reactive organic compounds, temperature, pH, pressure, and atmosphere, as well as the type(s) of employed catalysts. After purification and distillation processes carried out on the attained depolymerized products, it is possible to recover monomers with high purity (namely, amines and polyols), which can be exploited for synthesizing not only polyurethanes but also other polymers (such as polyamides, polyesters, and polyureas). Based on the selected degradation pathway, it is possible to recover products with different functionalities and physicochemical features. The most performed processes for the chemical recycling of polyurethanes include hydrolysis, glycolysis, aminolysis, ammonolysis, acidolysis and phosphorolysis, which will be described in the following. It is noteworthy to mention that, compared to mechanical recycling, chemical recycling processes allow for the recovery of higher-value products; besides, although closed-loop recycling is possible, at the same time, these processes are more complicated than the mechanical ones. Further, selected polyurethane wastes are required and higher energy consumption is needed. Hence, the majority of these processes have not achieved the industrial and commercial scale. A full review of the chemolysis process that has gone beyond the limits of the laboratories, in pilot-plant or industrial scale, has been reported by Simón et al.;30 more attention is paid to glycolysis since it is by far the most implemented on a larger scale. In general, chemical recycling is applied to thermoset PUs, while for the thermoplastic ones, as well as for PUs with low crosslinking degrees (e.g., RIM and RRIM), the primary and secondary recycling methods explained before are the preferred option.
Hydrolysis
Hydrolysis can be considered the first chemical technique developed for recycling foamed polyurethane wastes (both as post-consumer wastes and as production scraps). It refers to the reaction of the polymer with water, either in the liquid state or as vapor (steam),31 which gives rise to the formation of different products comprising carbon dioxide, polyols, and amine intermediates (see Scheme 1).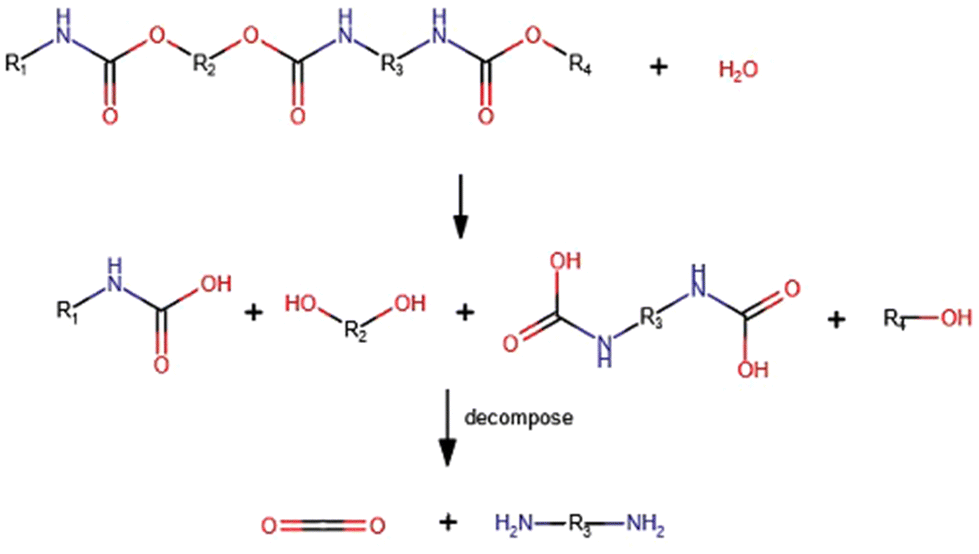 | ||
| Scheme 1 Hydrolysis of polyurethanes. | ||
It should be noted that hydrolysis is almost always a side reaction to chemical recycling of polyurethanes, since any PU foam contains absorbed water (between 5 wt% in viscoelastic foams and 0.5 wt% in aged rigid foams). Water will promote side reactions giving undesired products, especially primary aromatic diamines, such as 4,4′-diaminodiphenylmehane (MDA) or 2,4- and 2,6-tolylene diamines, which are carcinogenic substances.32 This point is discussed in more detail in the “glycolysis” section.
Hydrolysis shows drawbacks, as it is carried out under high operating conditions, i.e., high pressures (often between 15 and 50 atm) and temperatures (even 300–400 °C). It is worth noting that the polyol yield is strictly related to the severity of the adopted conditions. In this context, in a pioneering work, Campbell and Meluch33 demonstrated the suitability of the hydrolysis reaction, carried out between 232 and 316 °C and at atmospheric pressure, for recovering about 20 wt% of polyols from a polyurethane foam, which was then reused for the synthesis of new polyurethane foams. Further, Gerlock and co-workers31 studied the hydrolysis of a poly(ether-urethane) foam between 190 and 230 °C: the reaction yielded high-quality polyols, CO2, and toluene-diamines.
Although it has been reported several times in the scientific literature that hydrolysis can be performed using bases (such as sodium hydroxide) or acids (like hydrochloric acid), basic conditions seem to provide a more efficient process with higher conversions with respect to the acidic reaction medium. Further, acid catalysis combined with increasing hydrolysis temperatures was found to account for enhanced efficiency.34
Then, sodium hydroxide was employed in combination with diaminotoluene, and their catalytic effects on the hydrolysis of post-use polyurethane foams were thoroughly investigated by Dai and co-workers.35 In particular, the concurrent use of the two co-catalysts accounted for the decrease in both depolymerization activation energy and reaction temperature; however, the energy required for the overall process was very high and there were some difficulties in the purification of the resulting polyols.
In a further research effort, Nikje and co-workers36 proposed the use of mixtures of binary (i.e., water and glycerol) or ternary (i.e., water, sorbitol, and glycerol) solutions for chemical recycling polyurethanes, either using a thermal treatment carried out at atmospheric pressure and 160–180 °C, or through microwave irradiation.37 This way it was possible to exploit the synergistic effect provided by the combined action of hydrolysis and glycolysis (in the so-called “hydroglycolysis”), employing milder conditions (temperatures around 200 °C and lithium hydroxide as a catalyst) with respect to hydrolysis only. The obtained decomposition products were employed for synthesizing new polyurethane foams.
Further, Goto38 demonstrated the possibility of carrying out hydrolysis depolymerization reactions on polyurethanes by using sub- or super-critical water.
Actually, hydrolysis processes have only been scaled up to a pilot plant but, in no case, they have achieved the commercial scale due to the high energy input required for the reactor.39 Moreover, the recent use of super and/or subcritical fluids (e.g., CO2 and water) proposed in the literature40 does not seem to be able to overcome this limitation.
Moreover, at the end of the hydrolysis process, as well as hydroglycolysis, it is necessary to completely separate the amines from polyols, if the amines are to be used to produce new isocyanates. The recovered polyols can be used in formulations for making a PU foam, preferably in admixtures with virgin polyethers.32 This separation step is quite complex since it requires solvent extraction, and this hinders the suitability of such a recycling method for industrial applications. Therefore, although the energy requirement of hydroglycolysis is lower than that of hydrolysis, the former is also not applied on an industrial scale for its separation and purification cost.
Glycolysis
Reacting polyurethanes (mainly rigid bulk materials, rigid and flexible foams) with diols at temperatures usually beyond 200 °C and under atmospheric pressure allows for depolymerizing these materials and recovering polyols or glycolysates (i.e., oligomers embedding urethane groups and showing higher molecular weights compared to polyols), which, in turn, can be exploited for producing new polyurethanes. The reaction, called glycolysis (a general scheme is presented in Fig. 10) usually takes place with the glycol, in the presence of a catalyst (such as acetate inorganic salts, hydroxides of alkaline metals, amines, metallo-organic compounds – stannous octoate, dibutyltin dilaurate). It is very important to finely tune the reaction temperature, as the activity of the catalysts is not enough when the reaction is carried out below 180 °C. Conversely, when the glycolysis temperature exceeds 220 °C, undesired side reactions may occur, which lead to the formation of amines rather than polyols or glycolysates.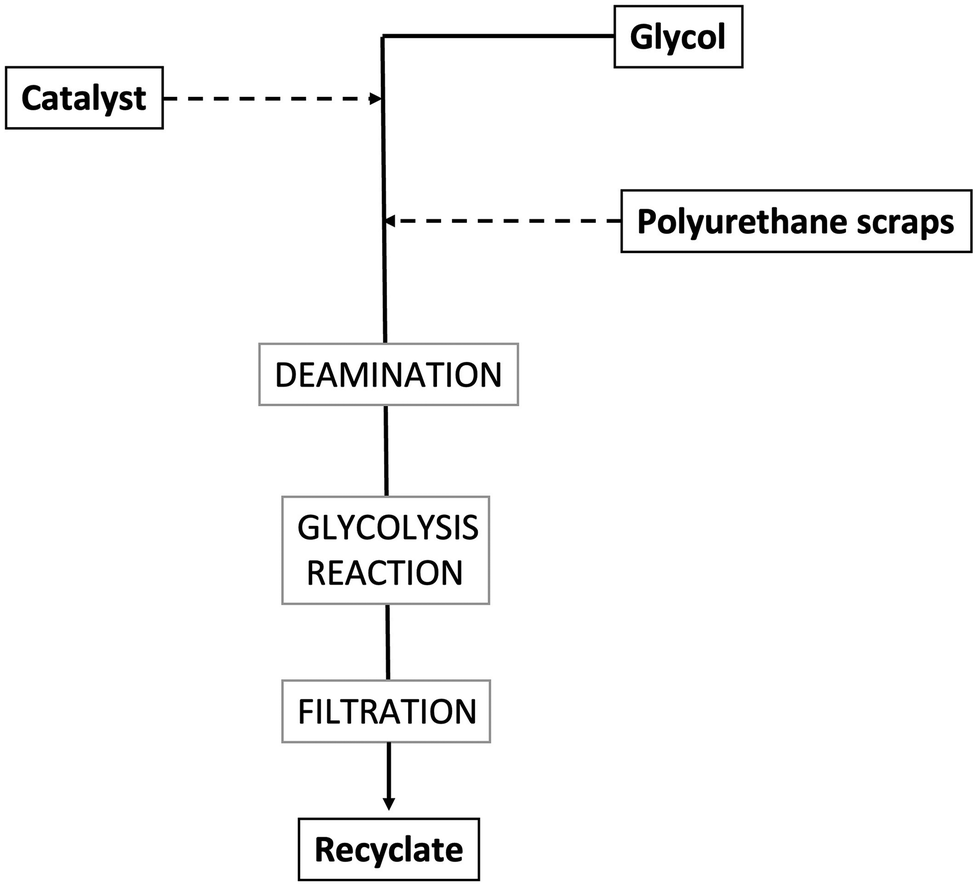 | ||
| Fig. 10 Scheme of the glycolysis process for polyurethane wastes. | ||
After completion, the glycolysis products are cooled down, filtered, and possibly mixed with virgin polyols to reformulate polyurethanes. The overall process requires about 8 h. Several parameters may affect the reaction yields, as well as the quality of the obtained recyclates, namely, temperature and reaction time, mass/molar ratio of polyurethane scraps to glycols, possible stirring of the glycolysis reactor, type and concentration of the employed catalyst, glycol type and size of the polyurethane scraps.41,42
Glycolysis involves two main reactions, namely, the break of biuret and allophanate groups, and the transesterification of urea and urethane bonds. Further, when poly(ester-urethane) scraps are processed, esterification of the polyol-rich moieties (i.e., of the soft segments of the polymers) may occur. Besides, it is very important to limit the presence of amines in the recyclates, as amine groups are more reactive than OH functionalities with isocyanates, leading to faster crosslinking phenomena in the new polyurethanes obtained from the recyclates. Moreover, aromatic amines, formed by PUs comprising aromatic isocyanates (i.e., the largest part of PU) are carcinogenic and thus their content must be limited below 1000 ppm to avoid the need for labelling according to classification and labelling of chemical (CLP) regulation. Indeed, if the concentration of aromatic amines in a polyol exceeds this limit, the polyol can still be used but must be labelled, handled and transported accordingly, increasing the degree of complexity for the managing of the supply chain of recycled polyols for PU production.43 Aromatic amines are formed through the transesterification of urea groups or by the side reaction of urethane with water that is always absorbed in PU wastes or by thermal degradation of urethane because of the high temperature required by the glycolysis process.
It is worth underlining that it is possible to carry out glycolysis reactions without any catalyst, but with much longer reaction times; however, there is a need for higher temperatures, which may account for degradation phenomena involving the processed materials.
Concerning the effect of the mass/molar ratio of polyurethane scraps to glycols, the scientific literature clearly demonstrates that it is possible to perform the glycolysis process with a mass excess of either glycols44 or polyurethane scraps.45Table 2 presents some examples of glycolysis processes performed with a mass excess of polyurethane scraps: the main advantage derived from the glycolysis with the mass excess of polyurethanes refers to the quite high homogeneity of the obtained products (single-phase), which do not need the purification and distillation of glycol excess, as for the process operating with a mass excess of glycol (split-phase). Moreover, the higher the PU/glycol ratio, the lower the investment cost for equipment because of the smaller reactor volume required for the same amount of scraps to be recycled.
| Type of polyurethane scrap | Glycol or diol employed | PU![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :glycol mass ratio :glycol mass ratio |
Catalyst | Reaction temperature (°C) | Ref. |
|---|---|---|---|---|---|
| Flexible PU foam | Diethyleneglycol | 1:0.5 |
Potassium acetate | 215–225 | 57 |
| Flexible poly(ether-urethane) foam | Diethyleneglycol | 1:0.9 |
Stannous octoate | 179 | 58 |
| Flexible PU foam | Ethyleneglycol | 2:1; 5:1; 8:1, 10:1; 20:1 |
Potassium acetate | 195 | 59 |
| Flexible poly(ether-urethane) foam | 1,4-Butanediol | 2:1; 5:1; 8:1; 10:1 |
Potassium acetate | 210–230 | 60 |
| Flexible poly(ether-urethane) foam | Poly(ethylene glycol) | 2:1 |
Potassium acetate | 210–220 | 45 |
| Flexible PU foam | 1,6-Hexanediol | 2:1; 4:1; 6:1; 8:1 |
Potassium acetate | 230–245 | 50 |
| Flexible PU foam | 1,3-Propanediol, 1,4-butanediol, 1,5-pentanediol | 10:1 |
Potassium acetate | 190–250 | 61 |
| PU elastomer | Ethyleneglycol | 2:1; 5:1 |
Potassium acetate | 170–180 | 62 |
| Rigid PU foam | Diethylene glycol, ethyleneglycol, propyleneglycol | 2:1 |
Sodium acetate | 190 | 53 |
Table 3 presents some examples of glycolysis processes carried out with a mass excess of glycols: this condition is adopted when the main target is the recovery of polyols that, after purification and distillation, can be exploited for synthesizing new polyurethane systems.46
| Type of polyurethane scrap | Glycol or diol employed | PU:glycol mass ratio |
Catalyst(s) | Reaction temperature (°C) | Ref. |
|---|---|---|---|---|---|
| Flexible poly(ether-urethane) foam | Diethyleneglycol | 1:1.5 |
K, Ca and Sn(II) octoates | 189 | 63 |
| Flexible poly(ether-urethane) foam | Diethyleneglycol | 1:1.5 |
Diethanolamine, Ti butoxide, K and Ca octoate | 189 | 47 |
| Flexible poly(ether-urethane) foam | Diethyleneglycol | 1:1.5 |
Sn(II) octoate | 190 | 44 |
| Flexible poly(ether-urethane) foam | Diethyleneglycol | 1:1.3; 1:1.5; 1:2 |
Potassium octoate | 190 | 48 |
| Flexible poly(ether-urethane) foam | Ethyleneglycol, diethyleneglycol, propyleneglycol, dipropileneglycol | 1:1.5 |
Diethanolamine | 189 | 49 |
| Flexible PU foam | Ethyleneglycol | 1:2 |
Potassium acetate | 195 | 59 |
| Flexible poly(ether-urethane) foam | Poly(ethylene glycol) | 1:2 |
Potassium acetate | 210–220 | 45 |
| Poly(ether-urethane) elastomer | Ethyleneglycol/diethyleneglycol | 1:2 |
Li Acetate | 170 | 64 |
| Poly(ether-urethane-urea) fibers | Diethyleneglycol | 1:3 |
Dibutyltin dilaurate | 210 | 65 |
| Rigid PU foam | Diethylene glycol, | 1:1, 1:2, 1:3 |
Potassium acetate | 220 | 54 |
| Polyisocyanurate | Dipropyleneglycol | 1:3, 2:3, 9:11 |
Potassium acetate, titanium(IV)-n-butoxide | 200–230 | 66 |
In general, the formation of one or separated phases depends, not only on the PU/glycol ratio, but also on the type of PU waste. The glycolysis of rigid foams generally provides a single homogeneous phase of high viscosity and high hydroxyl number, which can be reused, often mixed with virgin polyols, to make PU rigid foams again. Conversely, this product cannot be used to prepare flexible foams because of the too high hydroxyl number that is due to the high functionality and short molecular chain of these kinds of glycolysates. In case of flexible PU foams, because of their higher urea content, generally a split-phase product is obtained (Fig. 11): the upper phase contains the recovered original polyols, to be eventually further purified by means of distillation, while the bottom phase consists of the glycol excess, together with carbamates and aromatic amines (depending on the type of processed polyurethanes).
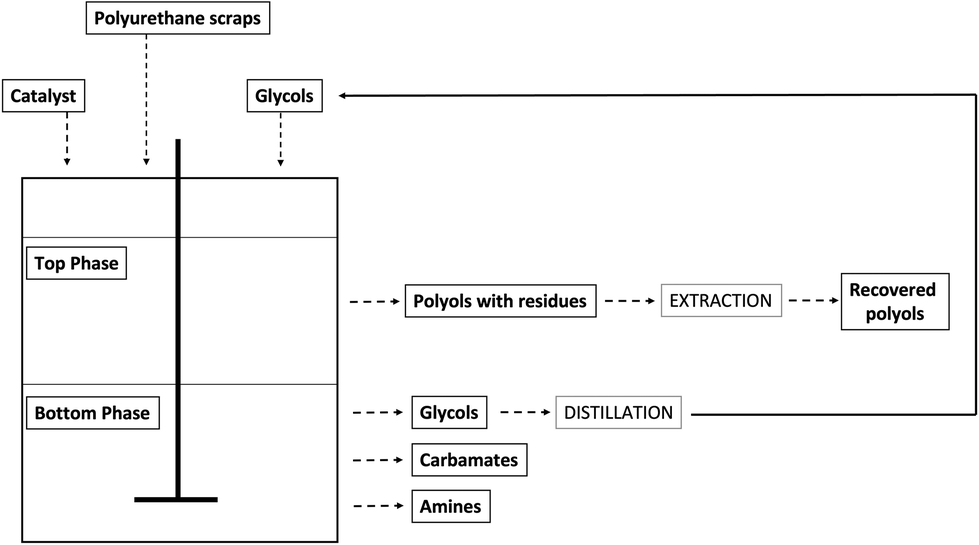 | ||
| Fig. 11 Split-phase glycolysis. | ||
The main advantage of split-phase glycolysis with respect to single phase is related to the high quality of the recovered polyol;47–49 however, split-phase glycolysis processes present a great drawback relying on the huge molar excess of glycol required that causes an important increase in the operation costs, hence making almost impossible the implantation of industrial glycolysis plants.30 Although split-phase situation is more common for flexible foams, the effective final product separation still depends on the PU/glycol ratio. For example, Datta et al.50,51 reported that the single-phase product is obtained from an elastic PU foam using hexamethylene glycol (HDO) when the foam/HDO weight ratio is up to 4/1; for higher ratios (6/1, 8/1, and 10/1), the reaction runs as ‘split-phase’ glycolysis.
Therefore, there is no general rule to define the usable weight ratio of glycol to PU.
Not only the weight but also the type of employed glycol greatly influences the glycolysis process. Dipropylene glycol and tetra ethylene glycol dissolved PUF in the shortest time among polypropylene glycols and polyethylene glycols (e.g., DEG), respectively. PUF dissolution time was reduced to one-half for each 10 °C rise in the range of 170–200 °C. Moreover, PUF dissolution time was inversely proportional to the KOH (catalyst) concentration, while the dibutyltindilaurate concentration had less influence on the PUF dissolution time than the KOH concentration. Furthermore, smaller PUF particles dissolved in a shorter time. More specifically, the initial glycolysis conversion of PUF was proportional to the total surface area of PUF particles.52
Moreover, the polyols produced from the reaction with DEG were more viscous than those from propylene (PG) or ethylene (EG) glycols due to the higher molecular weight of DEG and the slower reaction process.53
The glycolysate final composition depends also on the catalyst used because of the different catalysts’ selectivity toward transesterification and/or hydrolysis reactions. The use of potassium acetate (KOAc) is known to increase the amounts of amines in the glycolysis products of PU foams.54 It has been shown43 that aromatic methylenedianiline (MDA) content increases almost linearly with the reaction time and its formation is exponentially related to the KOAc concentration. An optimal KOAc shall be identified as a good trade-off between the reduced MDA concentration and the sufficient depolymerization degree to produce a recycled polyol with suitable (i.e., not too high) viscosity. While a proper solution to reduce the free aromatic amine content for flexible PU foam wastes is available thanks to the implementation of an acidolysis process (as it will be explained later), for rigid PU wastes, the most effective solution to obtain products with a low amine content is adding chemical reagents to the glycolysis system, such as glycidyl ethers, hexamethylenetetramine, ethyl or methyl carbamate, propylene oxide and acetic anhydride or by alkoxylation of the final product.43,55,56
Aminolysis
Aliphatic diamines or even polyamines bearing primary and secondary amine functionalities can be employed for the chemical decomposition of polyurethane scraps through transesterification (i.e., aminolysis). The reaction kinetics is strictly related to the basic character of the nitrogen of the amine groups: in particular, it is possible to perform the reaction under mild conditions (i.e., low temperatures) when highly reactive amines are utilized. The general reaction is displayed in Scheme 2.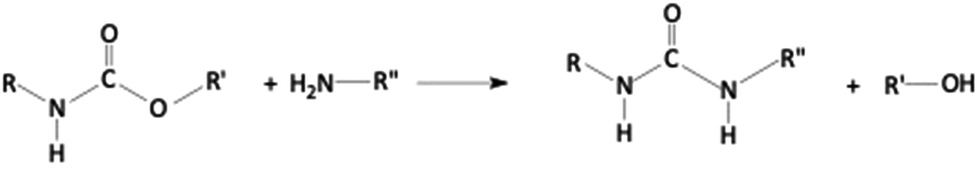 | ||
| Scheme 2 Aminolysis of polyurethanes. | ||
In one of the pioneering works, Xue and co-workers67 investigated the aminolysis of rigid polyurethanes and polyisocyanurate foams carried out with different polyamines (namely, tetraethylenepentamine, triethylenetetramine, and diethylenetriamine); several experimental parameters were considered (i.e., foam/polyamine ratio, the structure of the polyamine, and the reaction temperature). The resulting decomposed products were directly employed as curing agents for epoxy systems, particularly for designing new structural adhesives. The molecular weight of the polyamines turned out to remarkably affect the reaction kinetics: more specifically, low-molecular weight polyamines increased the degradation rates and the total amine number as well. Besides, the viscosity of the resulting decomposed products lowered with the increase in the reaction temperature and with the decrease in the foam/polyamine ratio.
Further, Kanaya and co-workers68 demonstrated the feasibility of using alkanolamines for decomposing flexible polyurethane foams. More specifically, the reaction was carried out at 150 °C without employing any catalyst and obtaining two layered phases: the upper phase was rich in polyether polyol, while the lower consisted of 4,4′-methylene diphenylamine and some derivatives of alkanolamines. Despite its good potential, the aminolysis process is still on the research/lab scale.
Ammonolysis
The chemical decomposition of polyurethane scraps using ammonia or ammonium hydroxide is known as ammonolysis (Scheme 3).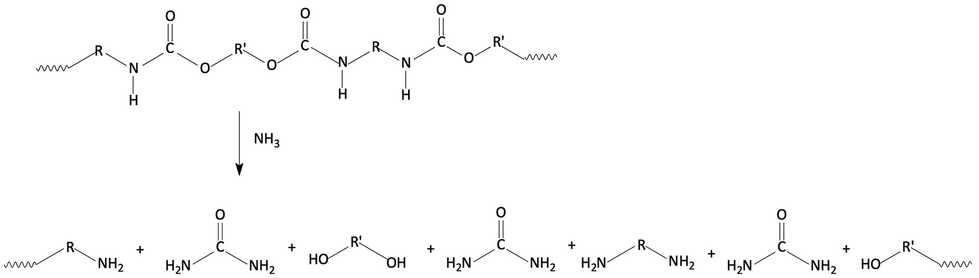 | ||
| Scheme 3 Ammonolysis of polyurethanes. | ||
The reaction, as aminolysis, is a transesterification that originates such decomposition products as polyols (usually suitable for synthesizing rigid polyurethane foams), amines, and urea, due to the higher nucleophilic character of ammonia with respect to H2O. Besides, the reaction kinetics increases with the increase in the ammonia-to-polyurethane mass ratio.
In addition to the “standard” ammonolysis, Lentz and Mormann69 succeeded in developing a particular ammonolysis process (Fig. 12), suitable for the feedstock recycling of rigid polyurethane elastomers and flexible foams, working with supercritical ammonia. The mass ratio of ammonia to polyurethane scrap was fixed at 1/1 and the reaction was performed at 139 °C, and 140 atm, for 2 h.
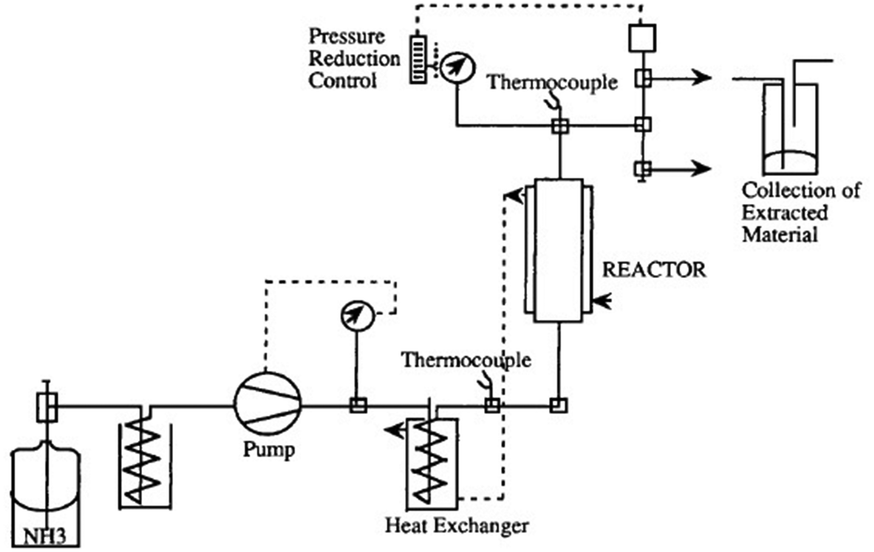 | ||
| Fig. 12 Schematic of the ammonolysis apparatus working with supercritical ammonia. Reprinted with permission from ref. 69. Copyright 1992 Wiley. | ||
Acidolysis
Despite the fact that the first investigation on the acidolysis of PUs dated back to 1992, only recently acidolysis has started to emerge as a promising method for the chemical recycling of PUs. It is a method that converts the PU into a polyalcohol, i.e., a polyol, using acids,70,71 possibly in the presence of a polyol, used as a solvent. Organic or inorganic acids can be successfully exploited for acidolysis recycling of polyurethane scraps: in particular, inorganic acids (such as HCl) give rise to the formation of polyols, CO2, and amine salts. Conversely, the reaction of organic acids (usually dicarboxylic structures) with the urethane groups accounts for the formation of amide groups. This is one of the main advantages of the use of organic acids, i.e., the absence or the very low amount of aromatic amines in the final product. Acidolysis is particularly convenient for obtaining a recycled polyol with a very low hydroxyl value, suitable for flexible PU applications, while, for rigid PU applications, since a high hydroxyl value is needed, glycolysis is the most convenient choice with respect to acidolysis.43Besides, two main types of organic acids can be employed, namely, saturated (such as adipic acid or succinic acid) and unsaturated (like fumaric or maleic acid) dicarboxylic acids. The saturated acids allow for carrying out the chemical decomposition reactions under milder conditions (i.e., low temperatures – around 60 °C – and short reaction times), giving rise to the formation of oligomers functionalized with amines, amides, urea compounds, or –OH groups. Conversely, the use of unsaturated dicarboxylic acids accounts for the formation of oligomers bearing double bonds, suitable for the formulation of adhesives.72 Though organic acids work as reagents and catalysts as well, He et al.73 proposed zinc acetate to speed up the process, without negatively affecting the quality of the recovered polyol.
Gama et al.74 studied the acidolysis of the flexible PU foam by the design of experiment (DOE) method using succinic, phthalic, or adipic dicarboxylic acids (DA). They analyzed the process temperature in the range of 190–200 °C, with the PU/DA ratio between 4 and 5 and reaction time between 4.5 and 5.5 hours. They found that the temperature and PU/DA ratio are the input variables with a higher influence on the hydroxyl number of the recovered polyol, although its variation is quite limited by changing the reaction conditions and it is generally about 50 mg KOH per g, which is suitable for the production of flexible foams.
Phosphorolysis
Using esters of phosphoric or phosphonic acid allows for the decomposition of polyurethane scraps, by exploiting phosphorolysis reactions. Troev and co-workers have exploited this reaction for the chemical decomposition of both flexible foams and microcellular elastomers, as witnessed by their significant contribution to the scientific literature.75–78 Phosphorolysis reactions were performed between 140 and 180 °C, according to the type of polyurethane scrap and P-containing ester; dimethyl phosphonate, diethyl phosphonate, triethyl phosphate, and tris(1-methyl-2-chloroethyl) phosphate were successfully employed. Because of the presence of phosphorus, the recovered products (usually a mixture of P-containing oligo-urethanes) were utilized for synthesizing novel flame-retarded polyurethanes.Thermo-chemical (tertiary) recycling of polyurethanes
Thermo-chemical recycling of polyurethane wastes was carried out to recover chemical products, energy, and fuels, among others. The main strategies that can be successfully exploited are pyrolysis, gasification, and hydrogenation, as described in the following paragraphs.Pyrolysis of polyurethane scraps
Generally speaking, this process refers to the thermal decomposition of long macromolecular chains, with the formation of lower molecular weight products; the reaction is usually performed at high pressures and in inert gas. The main decomposition products comprise gases, oils, and ashes.Specifically referring to polyurethane wastes, their pyrolysis occurs according to a two-step process: the first step, taking place between 100 and 300 °C, refers to the thermal decomposition of polyol-rich moieties (about 50% of the initial mass is lost), while the second step (occurring between 300 and 800 °C) involves the isocyanate parts of the polymer; the final ash content is usually below 3 wt%.79
As reported by Cullis and Hirschler,80 the thermal decomposition of polyurethanes may involve three different possible dissociation pathways of the urethane bond, namely:
- dissociation into olefin and carbamic acid
- dissociation into alcohol and isocyanate
- dissociation of carbamic acid into secondary amines and carbon dioxide.
All these reactions usually take place between 200 and 300 °C.
As observed by Takamoto and co-workers,81 the pyrolysis of reaction injection molded polyurethane scraps carried out beyond 450 °C normally originates 10–45 wt% oils (extremely viscous and prone to solidify over time), 5–25 wt% char and more than 40 wt% gases. Further, the use of either the char produced by the pyrolysis or activated carbon, during a secondary pyrolysis step, accounted for the enhanced amount and quality of the resulting pyrolyzed products, especially of the liquid fraction (oils), which showed lowered viscosity.
Moroi and co-workers82,83 highlighted the possible catalytic effect exerted by metal salts (namely, FeCl3, NaCl, MnCl2, CoCl2, CuCl2, and CrCl3) on the pyrolysis of poly(ester-urethane)s. In particular, copper and chromium salts accounted for the best catalytic effect, favoring the occurrence of thermal decomposition at lower temperatures, but, at the same time, stabilizing the intermediate pyrolysis products. Conversely, all the other metal salts, despite an improvement in the thermal decomposition of the polymer, were not able to stabilize the intermediate degradation products.
Gasification
Gasification is a very exothermic reaction that is performed at high temperatures (ranging from 1200 to 1500 °C) and pressures (20–80 bar), in an oxygen (air) atmosphere; it produces heat, syngas (i.e., synthetic gas, a gaseous mixture containing large amounts of hydrogen and carbon monoxide) and ashes. Further, it really takes a few seconds to achieve very high conversions of fuels (around 98–99%). Fig. 13 shows the general scheme of the gasification process.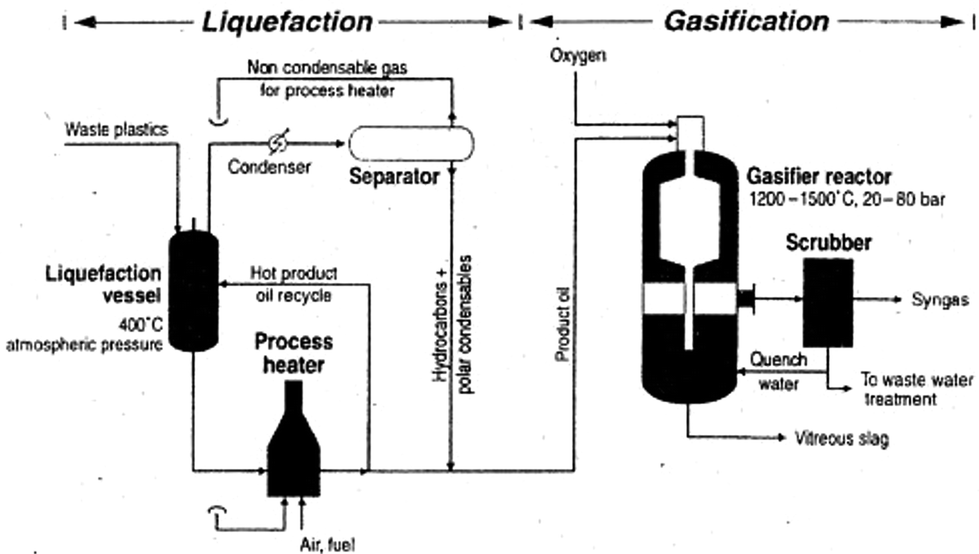 | ||
| Fig. 13 Schematic of the liquefaction/gasification process. Reprinted with permission from ref. 27. Copyright 2007 Elsevier. | ||
The obtained syngas can be employed to synthesize isocyanates and polyetherols, suitable for producing new polyurethanes.84
Hydrogenation
To recover both liquid and gaseous products from polyurethane wastes, it is possible to perform hydrogenation processes, combining heat with high-pressure H2. Therefore, this process is in between pyrolysis (heat treatment) and gasification (treatment with high-pressure gas). Compared to simple pyrolysis, hydrogenation allows for obtaining purer recyclates. Similarly to other depolymerization processes, hydrogenation has not been implemented on a large scale yet.Biodegradation of polyurethanes
It is possible to exploit such living microorganisms as bacteria and fungi to biodegrade polyurethane wastes, including post-consumer materials:29,85–87 because of the interaction with the microorganisms, either under aerobic or anaerobic conditions, the macromolecular chains are shortened, and the molecular weight of the polymer significantly decreases. Further, according to the adopted experimental conditions, it is possible to achieve the full mineralization of the biodegraded polymer. Biodegradation takes place according to three successive steps: break of the macromolecular chains and formation of oligomeric structures, further degradation to low–molecular weight species, and final conversion to CO2 and H2O (under aerobic conditions) and CH4 (under anaerobic conditions). Compared to thermochemical degradation, biodegradation is more environmentally friendly, as it does not need the use of specific impacting reagents and takes place at room temperature (i.e., no heat must be provided to the degrading system). Three different types of biodegradation can be considered, namely, fungal, bacterial, and enzymatic degradation; each of them usually suits better specific types of polyurethanes. As an example, fungal biodegradation is preferred to biodegrade polyether polyurethane foams, as the mycelium of fungi can easily penetrate the foam cells; conversely, bacterial degradation is ideal to biodegrade polyurethane coatings, as bacteria can effortlessly exploit the smooth surface of these coatings for the biofilm formation (Table 4).| Polyurethane | Fungi | Bacteria |
|---|---|---|
| Thermoplastic polyester polyurethane | Penicillium sp.89 | Bacillus sp.90 |
| Alternaria sp.89 | Comamonas acidovorans 91 | |
| Thermoplastic polyether polyurethane | Staphilococcus epidermidis 92 | |
| Polyester polyurethane foam | Alycycliphilus sp.93 | |
| Pseudomonas aeruginosa 94 | ||
| Polyester polyurethane coating | Penicillium crysogenum 95 | Bacillus suptilis 97 |
| Cladosporium sp.96 | Psedomonas putida 98 | |
| Polyether polyurethane foam | Alternaria sp.99 | |
| Cladosporium herbarum 100 |
Besides, it is worth highlighting that biodegradation is affected by several experimental factors including the crosslinking degree of the polyurethane substrate, its crystallinity, and molecular orientation as well. In particular, because of the easier accessibility, the amorphous parts of the polyurethane wastes are generally more prone to biodegrade with respect to their crystalline counterparts.88
As far as enzymatic degradation is considered, many of the employable enzymes belong to the class of hydrolases (such as proteases, elastase, esterases, and ureases, see Table 5) and are suitable for polyurethane coatings and thermoplastic bulk polymers. Further, despite the quite high potential of biodegradation, the research is still at the lab scale, also considering that it usually requires a lot of time and research efforts to obtain reliable data.
| Polyurethane | Enzyme | Ref. |
|---|---|---|
| Thermoplastic polyester polyurethane | Lipase | 101 |
| Esterase | 102 | |
| Protease | 103 | |
| Thermoplastic polyether polyurethane | Esterase | 103 |
| Protease | 104 | |
| Chymotrypsin | 105 | |
| Thermoplastic poly(ester urea) polyurethane | Lipase | 106 |
| Cholesterol esterase | 107 | |
| Thermoplastic poly(ether urea) polyurethane | Elastase | 108 |
| Papain | 109 | |
| Polyester polyurethane coating | Lipase | 110 |
Energy recovery (quaternary recycling) from polyurethanes
Energy recovery (through incineration or combustion processes) is exploited only when polyurethane wastes and scraps are not suitable for any other recycling strategy; besides, it is very appropriate when PU-based products at the end of life are contaminated. The main difference between incineration and combustion is that the former is an incomplete combustion that may result in the production of toxic gases when not properly carried out.Besides, two main advantages can be highlighted: energy recovery allows for reducing the volume of the wastes that are landfill confined by up to 99%; then, the produced energy is comparable to that which originated from coal combustion (the average calorific value of polyurethanes is around 25–30 MJ kg−1) and it is marginally lower than that gathered from fuel combustion.111 Conversely, the process is not feasible when flame-retarded polyurethane wastes or scraps have to be recycled, because of the presence of flame-retardant additives having a high environmental impact. Further, another issue arising from energy recovery refers to the possible generation of toxic and harmful gases, including nitrogen oxides, CO, and HCN.112 This latter problem can successfully be solved by recirculating the exhaust gases that are generated during combustion and further oxidizing the not-fully oxidized products with a secondary airflow.
Generally speaking, three main energy recovery approaches for polyurethane wastes have been considered so far, namely: co-combustion of polyurethanes with municipal solid wastes,111 fluidized bed combustion,113 and two-stage incineration.9
Recent advances in polyurethane recycling
This paragraph aims to describe some recent examples that clearly indicate the direction taken for this research topic during the last years, especially toward the design and exploitation of strategies with low environmental impacts.Zhang and co-workers114 succeeded in isolating a fungal strain (namely, Cladosporium halotolerans) from the deep sea, able to exploit a polyurethane foam (namely Impranil PU) as the only carbon source. In particular, as assessed by FTIR spectroscopy measurements, Cladosporium halotolerans accounted for the degradation of the foam up to 80% after 3 days of incubation at 28 °C (Fig. 14), through the breakage of C–N–H bonds and carbonyl groups. Further, gas chromatography-mass spectrometry measurements highlighted the formation of alkanes and polyols as degradation intermediates, due to the hydrolysis of urethane and ester bonds. Finally, urease and esterase activities were identified in 7 days old cultures utilizing the polyurethane foam as the only carbon source.
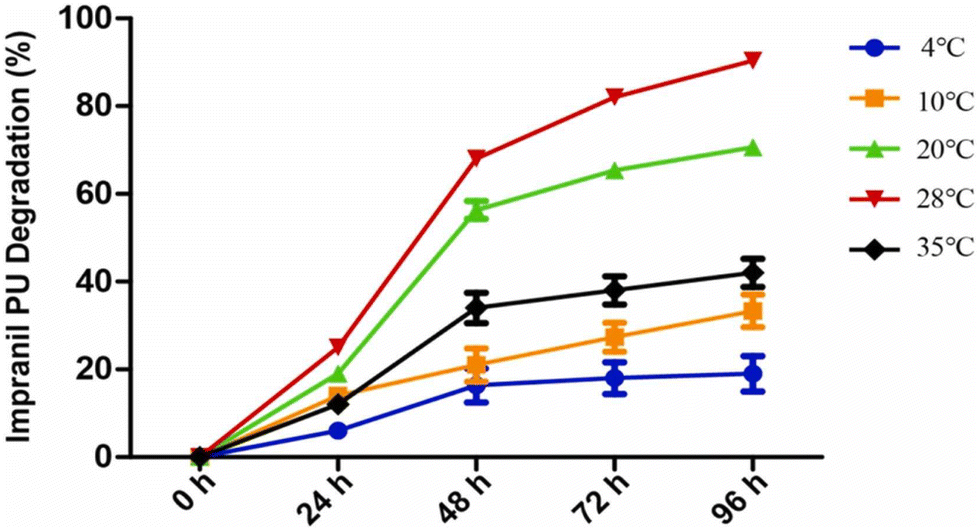 | ||
| Fig. 14 Time course of the degradation of Impranil PUs by Cladosporium halotolerans 6UPA1 growing at different temperatures. Reprinted with permission from ref. 114. Copyright 2022 Elsevier. | ||
Recently, Branson et al.115 demonstrated the suitability and effectiveness of urethanases, discovered from a metagenome library constructed from the soil, for recycling polyether-polyurethane foams through a chemo-enzymatic process. In particular, the urethanase was able not only to hydrolyze low–molecular weight dicarbamates resulting from chemical glycolysis of the foam but also to recover the polyether-polyols derived from chemocatalytic glycolysis.
Jeong and co-workers116 synthesized bio-polyurethane foams by reacting toluene diisocyanate with sucrose and propolis. A foaming rate as high as 1440% was observed for the polyurethane formulation containing 15 wt% of sucrose and 20 wt% of propolis. Besides, the presence of the sugar accounted for a high enzymatic degradability (using invertase), while the incorporation of the propolis was responsible for high antibacterial features, with about 94% bacteria reduction.
Recently, Xu and Hong117 nicely reviewed the potential of thermoplastic biodegradable polyurethanes for tissue repair and regeneration (Fig. 15), highlighting the possibility of synthesizing them starting from such aliphatic diisocyanates as 1,4-butane diisocyanate, 1,6-hexamethylene diisocyanate, and lysine-based diisocyanate, and exploiting their nontoxic biodegradation products (namely, 1,4-butanediamine, 1,6-hexanediamine, and lysine, respectively).
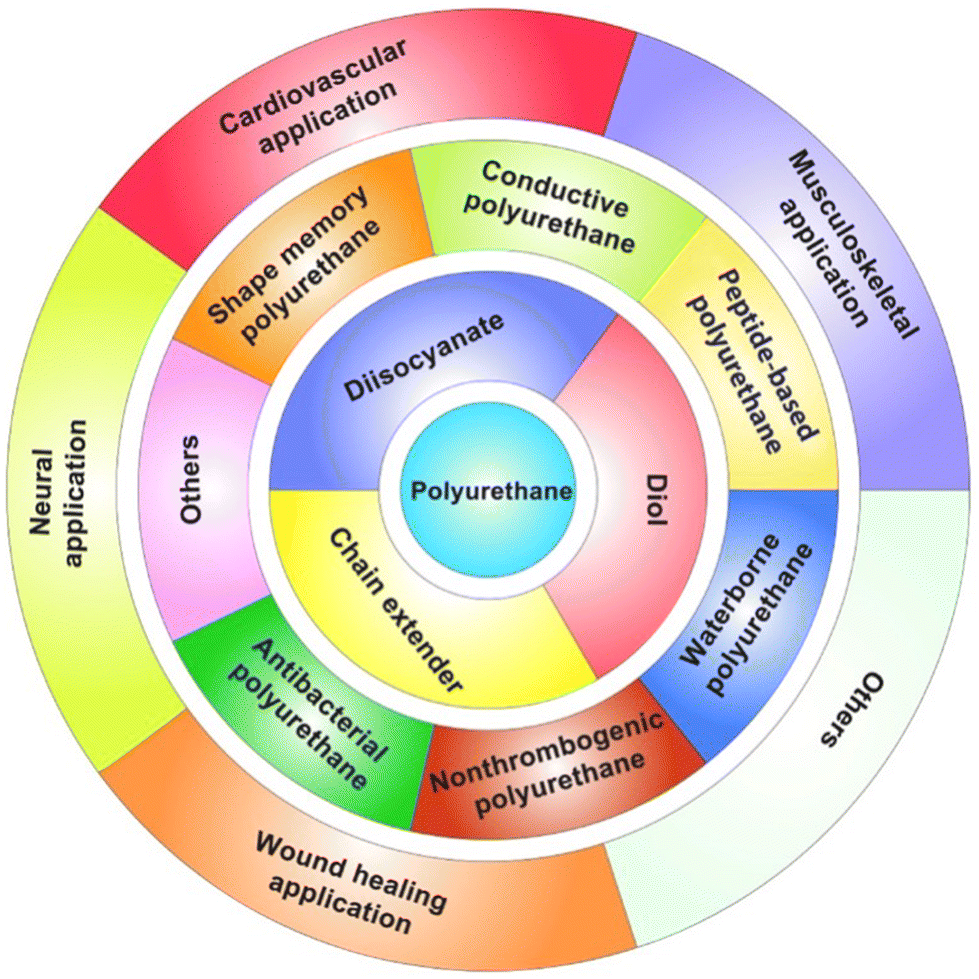 | ||
| Fig. 15 Biodegradable polyurethanes for tissue repair and regeneration. Reprinted from ref. 117 under CC BY-NC-ND 4.0 License. | ||
Pfohl and co-workers118 thoroughly investigated the biodegradation in compost (monitoring the carbon dioxide evolution) of (thermoplastic) polyurethanes differing with regard to their hard-to-soft segment ratios, the type of polymer backbone (aromatic or aliphatic), the presence of specific additives (i.e., hydrolysis stabilizers), and the possible degree of crosslinking. The increase in both the degree of crosslinking and the hard segments’ content turned out to lower the rate and extent of the biodegradation rate (the maximum value achieved was about 72%). Besides, the incorporation of a hydrolysis stabilizer accounted for a decrease in the polyurethane fragmentation, without significantly impacting the conversion of the polyurethane carbon into carbon dioxide.
Espinosa et al.119 demonstrated the possibility of employing a soil bacterium, namely Pseudomonas sp. as an effective strain for biodegrading both a polyurethane-diol solution (i.e., a polyurethane oligomer) and 2,4-diaminotoluene, a very common precursor in the synthesis of polyurethanes and, at the same time, a putative degradation intermediate.
Then, the possibility of designing renewable low viscosity polyester-polyols obtained from sebacic acid, azelaic acid and bio-based 1,3-propanediol, suitable for the synthesis of biodegradable thermoplastic polyurethanes was successfully demonstrated by Rajput and co-workers.120 The resulting polyurethanes showed remarkable biodegradation under compost environmental conditions, with an as high as 57% decrease in molecular weight after 9 weeks of biodegradation, and up to 97% biodegradation over 120 days, as assessed by respirometric analyses.
Very recently, Liu et al.121 isolated 20 strains (namely, 9 fungi and 11 bacteria) and assessed their suitability for biodegrading a poly(1,4-butylene adipate)-based polyurethane. Among the different tested strains, Cladosporium sp. P7 was found to be the most efficient for biodegrading the polymer: in particular, it was found that both ester and urethane bonds were cleaved by this strain, leading to the formation of six metabolites comprising adipic acid, 1,4-butanediol, and 4,4′-methylenedianiline. Besides, after 28 days of cultivation, about 32 and 44% of the polyurethanes were converted into soluble small molecules when employed as the sole carbon source or in a medium with other co-carbon sources, respectively.
Concerning some recent outcomes in the mechanical and chemical recycling of polyurethanes, new strategies based on mechanochemistry have been reported in the literature. Mechanochemistry refers to the phenomenon of chemical reactions of substances caused by the input of mechanical energy (such as crushing in a ball mill or shearing). Guo and co-workers122 performed high-shear grinding on flexible polyurethane foam scraps by means of two-roll milling; the resulting powder was employed for partially replacing polyols in new polyurethane foams. In fact, the high shear grinding accounted for the activation of the powder, because of the appearance of several hydroxyl groups on its surface. In particular, the powder obtained by performing 7 milling cycles carried out at room temperature showed an average size of around 98 μm, and successfully replaced 15 wt% of the polyols in the preparation of the new polyurethane foams, whose microstructure and density were similar to those of the pristine polyurethane scraps. He and co-workers123 proposed a mechano–chemical reaction for recycling rigid polyurethane insulation board, employing a self-made pulverizer. The accumulation of mechanical energy during the process was responsible for the concentration of the impact energy on individual macromolecular chain segments, hence determining the break of their chemical bonds. The crushing parameters (feed amount – 90 g, crushing speed – 500 rpm, and crushing time – 30 min) were optimized; finally, the recovered polyurethane powder (exhibiting high activity of functional groups and reduced the cross-linking density as well) was exploited for producing composite panels with polypropylene.
A further step in mechanochemical recycling is related to the use of covalent adaptable networks (CANs). The polymer materials with reversible CANs are described as vitrimers.124
Under specific stimuli such as light and heat, the breaking, recombination and exchange of dynamic covalent bonds occur, which means that the crosslinked networks are no longer fixed, and the topological structures will change under external forces. Indeed, topological rearrangement reactions occur, which make the thermoset polymers flowable and thus allow for their reprocessing. Vitrimers have mechanical properties similar to common thermoset resins, but they can be remodeled during the bond exchange process, like thermoplastics since they can flow under shear stress. This bond exchange process needs a catalyst that can be inserted during the synthesis of new PU polymers but, to recycle an existing PU waste, the catalyst must be introduced into the PU just before reprocessing, i.e., after reducing the size of the material by crushing and milling, among others. For example, Li et al.125 studied the effects of different pretreatment methods on pulverized PU foams (PUF), after adding a dibutyltin dilaurate (DBTDL) catalyst, on the reprocessing properties of commercial rigid PUF. In particular, they considered direct pretreatment, swelling pretreatment, and ball milling pretreatment; they also tested multiple reprocessing sets by ball milling. The treated powders were placed in a mold and pressed into sheets for 30 min at a pressure of 30 MPa at 170 °C. They showed that, through strong mechanical forces, the ball milling pretreatment method could effectively reduce the size of PUF particles, increase the surface area, and generate new reactive groups via mechanochemical action, which effectively promoted urethane exchange and improved the reprocessing properties of PUF by final mechanical recycling. Bandegi et al.126 proposed a similar approach, by using triazabicyclodecene (TBD) as a catalyst, and found that the permanent crosslinked structure of the PU thermoset foam can be converted into a dynamic network upon vitrimerization. The vitrimerized network retains high mechanical strength and can also be foamed by applying low pressures at high temperatures. Liu et al.127 still used TBD to show that three-dimensional photo-printable resins with tunable material mechanical properties—which are superior to commercial high-performance counterparts—can be formulated with the addition of various network reforming additives, starting from waste PU foams. They thus showed that a more attractive approach with respect to chemical recycling is to partially disintegrate the network to a point that allows reworking. There are two notable benefits with this approach: (1) the reaction conditions can be much milder and (2) the obtained mixture can be fully converted into a network without requiring purification. A more circular, direct and continuous foam-to-foam recycling method using twin screw-extrusion by leveraging the melt processability of PU CANs has been proposed by Kim et al.,128 by using zirconium(IV) acetylacetonate as a replacement for toxic tin catalysts in PU CANs and azodicarbonamide as a chemical blowing agent. Their results indicate that waste PU foams can be directly recycled into a new foam while preserving its structural properties and chemical integrity. Bai and co-workers129 exploited the combination of coordination and dynamic chemistry for facilitating the thermal and chemical recycling of polyurethanes. A general scheme is shown in Fig. 16. In particular, aluminum acetylacetonate was employed as a crosslinker for the synthesis of the polyurethanes, which gave rise to the formation of thermally reversible bonds between isocyanate and acetylacetone groups. This way it was possible to provide the crosslinked polyurethanes with a worthy thermal remoulding capability at temperatures beyond 100 °C and, at the same time, allow for a simple chemical recycling route based on the pH-sensitive Al–acetylacetone coordination, using acetic acid for promoting acid hydrolysis. In fact, the coordination chemistry accounted for the break of the coordination bonds dissociating in aluminum acetylacetonate and the reassociation of the same bonds after the removal of acetic acid through evaporation.
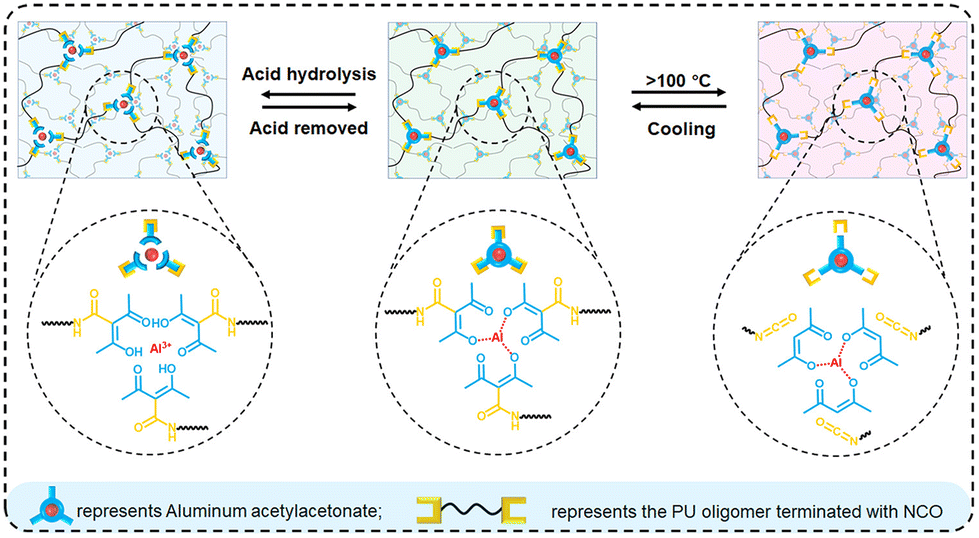 | ||
| Fig. 16 Schematic of the thermal (via dynamic chemistry) and chemical (via coordination) recycling of polyurethanes. Reprinted with permission from ref. 129. Copyright 2022 ACS. | ||
Disulfide as well as thiourethane, imine, urea, and boronic ester can be used to create CAN PUs.124,130
Contrary to mechanical recycling, on which the research has not been focused so much during the last years, chemical recycling has highlighted significant outcomes.
In particular, Donadini and co-workers131 exploited a micro-wave-assisted glycolysis reaction catalyzed by monoethanolamine, stannous octoate, or potassium acetate, for recovering a polyol-like liquid product from rigid polyurethane foam wastes. The use of the microwave reactor accounted for a remarkable decrease in the reaction time (by about 94% with respect to conventional-heated processes) and significant energy saving (by about 45%). Finally, the replacement of virgin polyol with the recovered counterpart allowed for a slight decrease in the thermal conductivity of the resulting re-foamed material, as well as for increasing its compression strength (by around 20%).
A microwave-assisted acidolysis reaction with adipic acid for recycling flexible polyurethane foams was proposed by Grdadolnik and co-workers,132 who demonstrated the feasibility of the process for the recovery of polyether polyols to be employed for the preparation of new polyurethanes. The molar ratio of adipic acid to urethane groups of polyurethane foam was set at 1.1 and the reaction was carried out at different temperatures (namely, 210, 220, and 230 °C) and reaction times (15, 30, and 40 min). The microwave reactor accounted for a significant decrease in the reaction time (below 1 h) compared to the acidolysis with conventional heating (several hours). It is worth noting that, with the increase in the amount of adipic acid, the content of toluendiamine (TDA) in the polyol decreased and the degree of degradation of the urethane groups increased, but at the expense of a higher degree of esterification of the hydroxyl groups of the polyol, resulting in the recovered polyols (RPs) being terminated with carboxyl groups to a greater extent. In other words, none of the reaction conditions employed resulted in the formation of a completely hydroxyl-functionalized polyol.
The results indicated that with the increase in the carboxyl functionality of the RPs in the formulation, the carboxyl-terminated polyol negatively affects the quality and performance of the flexible PUFs.
In a further research effort, Amundarain and co-workers133 exploited the polyols, recovered from post-industrial rigid polyurethane foams by means of glycolysis reactions, for preparing new rigid foams. The recovered counterparts showed remarkably higher values for viscosity, acidity, and average molecular weight and more hydroxyl groups than those of commercial polyols: these findings were ascribed to both the residual ethylene glycol in the polyol after the purification of the glycolysate and in the presence of glycolysis by-products (such as amines and carbamates). Though it was not possible to incorporate more than 15 wt% of the recovered polyols into the formulation of the final polyurethanes, the latter showed enhanced tensile strength, likely due to a higher cross-linking of the foams containing the recycled polyols.
Pursuing this research, the same group134 employed a glycolysis process for the recovery of polyols from rigid polyurethane foams either aromatic or aliphatic. Ethylene glycol or diethylene glycol was employed as a glycolysis reagent; sodium hydroxide, sodium acetate or diethanolamine was utilized as a catalyst. Unlike the glycolysis of the aromatic polyurethane, which achieved high conversions (about 90%) in about 2 h at 198 °C and without any catalyst, the presence of any catalyst was mandatory for the decomposition of the aliphatic polyurethane, leading to higher conversions (around 98%) under the same reaction conditions adopted for the aromatic counterpart. As assessed by gel permeation chromatography, the structure of the recovered polyols was similar to that of the pristine ones employed in the formulation of the initial polyurethanes, hence allowing for a partial replacement of “virgin” polyols with the recovered molecules (up to 15 wt%) in the preparation of new polyurethane systems.
Another interesting approach well matching the circular economy concept has been recently proposed by Sternberg and Pilla,135 who investigated the feasibility of a high-pressure hydrolysis/hydroglycolysis recycling technique, for the recovery of lignin (exhibiting both improved solubility and lower hydroxyl contents) from a lignin-derived non-isocyanate foam and its reuse for the obtainment of second-generation polyurethane foams. The latter showed a mechanical behavior comparable to the foams made of virgin lignin, despite a slight decrease in the compressive strength (126 vs. 133 kPa, for the recycled lignin foams and the virgin lignin–containing counterparts, respectively), ascribed to a faintly lower crosslinking density due to the decreased hydroxyl content of the recycled lignin.
Yuan and co-workers136 synthesized biodegradable and chemically recyclable polyurethanes using bio-based telechelic hydroxyl-terminated poly(γ-butyrolactone) diols as suitable precursors. Changing the molar mass of the diols allowed for obtaining polyurethanes with thermoplastic or elastomeric features. Finally, the authors demonstrated that simple heating of the polyurethanes in bulk at 170 °C for 2 h, in the presence of 4 wt% of stannous octoate as a catalyst, was enough for easily recovering highly pure γ-butyrolactone in about 99% yield.
Gausas et al.137 synthesized an Mn-based complex for catalyzing the hydrogenative depolymerization of both real-life and end-of-life flexible polyurethane foams, using KOH, isopropyl alcohol, and H2. This way, it was possible to deconstruct the foams into polyol and aniline fractions; the process was implemented on a gram-scale with a total of 90% mass recovery.
Gu and co-workers138 employed four types of glycols (namely, butanediol, propylene glycol, diethylene glycol, and monodiethylene glycol) for degrading waste polyurethane elastomers. As an example, Fig. 17 shows the catalytic degradation mechanism of the polyurethane elastomer, using butanediol or diethylene glycol.
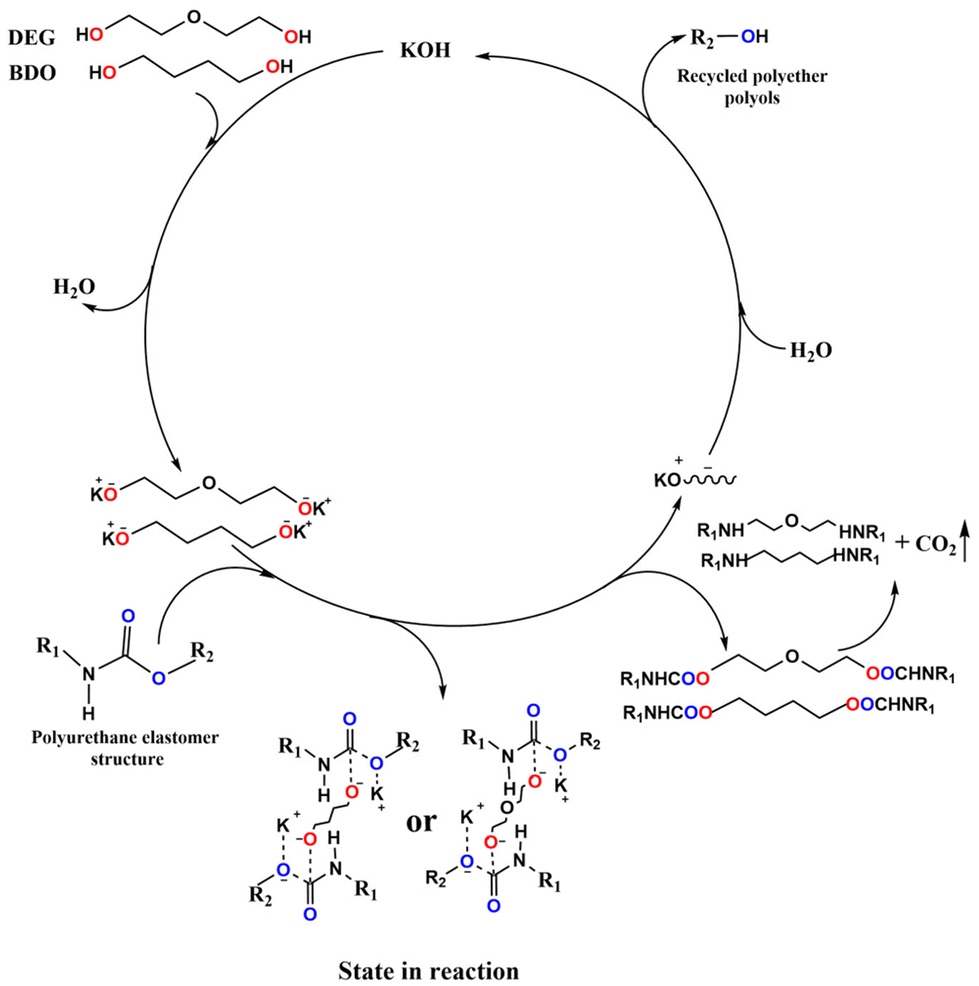 | ||
| Fig. 17 Catalytic degradation mechanism of the polyurethane elastomer. Reprinted from ref. 138 under Creative Commons Attribution 4.0 International License. | ||
The recovered polyols were utilized for the preparation of regenerated polyurethane rigid foams showing physical, mechanical, and thermal features suitable for the envisaged applications. In particular, the foaming time of the regenerated polyurethane rigid foam was usually between 40 and 140 s. The compressive strength ranged from 0.131 to 0.176 MPa and the water absorption from 0.7265 to 1.9923%. Further, the apparent density of the regenerated foams was between 30 and 40 kg m−3, and the thermal conductivity ranged from 0.0151 to 0.0202 W m−1 K−1.
A very recent application of recyclable thermoplastic polyurethanes refers to the design of flexible strain sensors, suitable for detecting little changes in strain and pressure.139 For this purpose, a thermoplastic polyurethane was mixed with graphene (at different loadings, ranging from 1 to 10 wt%) in a Haake internal mixer, working at 185 °C and 60 rpm for 2.5 min; the desired sheet geometry was obtained by subsequent compression moulding. The optimum graphene loading for the envisaged application was 7 wt%: the related nanocomposite sensor exhibited good strain sensitivity, even after three mechanical recycling cycles, as displayed in Fig. 18.
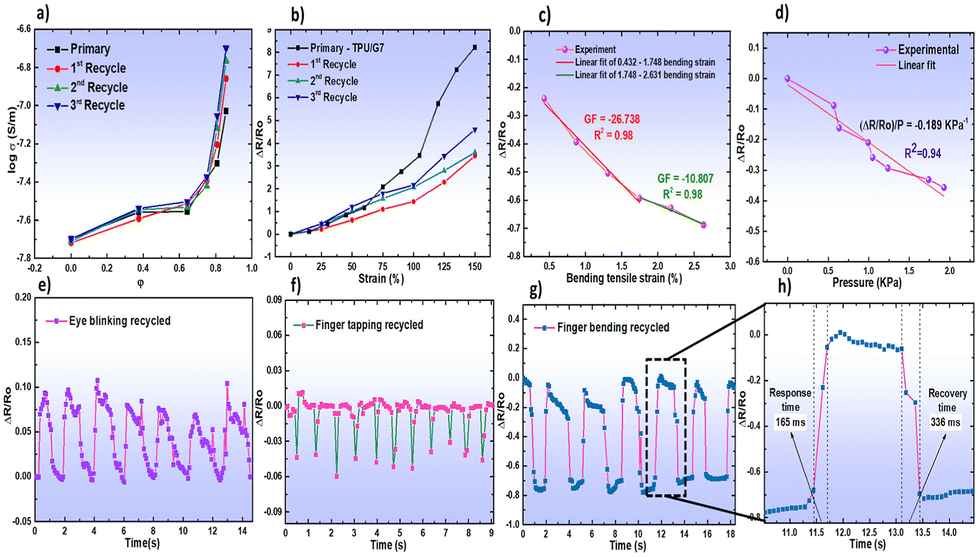 | ||
| Fig. 18 Recyclability of thermoplastic polyurethane (TPU)/graphene nanocomposites: (a) log conductivity of TPU/graphene nanocomposites as a function of graphene content (φ) of each recycle; (b) tensile strain response of the strain sensor containing 7 wt% of graphene (sample coded as TPU/G7) for each recycling cycle; (c) relative resistance changes of the strain sensor as a function of the bending angle for the third recycled nanocomposite; (d) pressure response of the third recycled TPU/G7 nanocomposite. Human motion detection using the sensor made of the three-time recycled TPU/graphene nanocomposite: (e) eye blinking; (f) finger tapping; (g) finger bending. (h) Response and recovery time of the recycled sensor. Reprinted with permission from ref. 139. Copyright 2023 ACS. | ||
The scalability and cost-effectiveness of the chemical recycling process have been and are still very challenging. A potential solution to solve the high operating cost of split-phase glycolysis has been first proposed by Simón et al.,140 who used crude glycerol, i.e., a cheap by-product of biodiesel production, as a glycolyzing agent. It has been demonstrated that crude glycerol provides an upper phase with a lower content of by-products and transesterification agents than those in the case of using the best “common” option (i.e., diethylene glycol (DEG)), as a consequence of its higher dielectric constant. Furthermore, as a result of this latter, a glycolysis bottom phase free of polyol, has been obtained thus increasing the net yield of the glycolysis process.
In a more recent work, a detailed evaluation of the economic feasibility of a split-phase glycolysis process (up to a pilot scale, Fig. 19) for the recycling of any kind of flexible polyurethane foam wastes, utilizing crude glycerol as cleavage agent, has been published by Del Amo and co-workers.141 In particular, a Net Present value of 1464555€, with an Internal Rate of Return of 27.99%, and a Payback Time between 4 and 5 years were estimated, hence confirming the economic viability of the proposed recycling process.
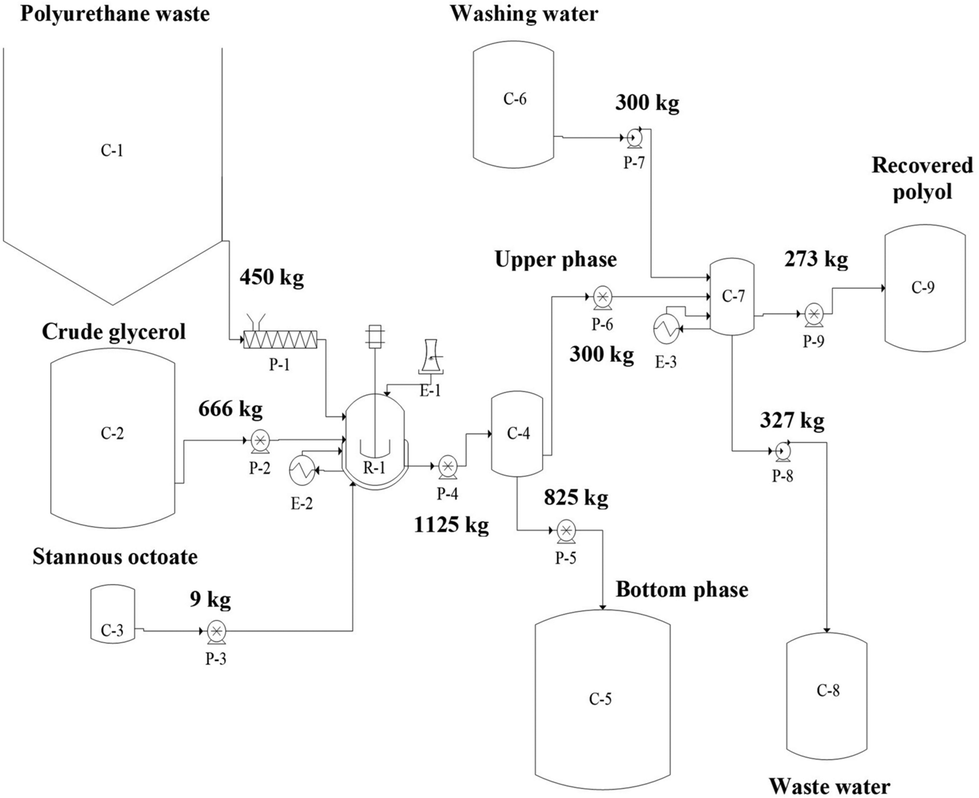 | ||
| Fig. 19 Flow diagram of the split-phase glycolysis plant and mass balance per batch. Legend: C-1 = polyurethane silo; C-2, C-3, C-5, C-6, C-8 and C-9 = storage tanks; C-4 = decanter; C-7 = liquid–liquid extractor; R-1 = glycolysis reactor; E-1 = condenser; E-2 and E-3 = heat exchangers; P-1 = Archimedean screw; P-2, P-3, P-4, P-5, P-6 and P-9 = Gear pumps; P-7 and P-8 = centrifugal pumps. Reprinted from ref. 141 under Creative Commons Attribution 4.0 International License. | ||
Zhang and co-workers142 prepared a series of fluorinated polyurethane rigid foams using fluorine-containing recycled polyols obtained from the degradation of waste polyurethane foams. The glycolysis reaction was carried out at 200 °C for 2 h, in the presence of ethylene glycol, diethylene glycol, and a fluorodiol synthesized on purpose. In particular, the incorporation of 8 wt% of fluorodiol in the waste polyurethane foam accounted for: (i) a significant increase in the compressive strength of the regenerated fluorinated rigid polyurethane foam (+ 43% with respect to that of the non-fluorinated regenerated counterpart), (ii) an enhanced waterproof behavior and (iii) a decreased thermal conductivity (which was measured as low as 0.0227 W m−1 K−1).
A recent trend is also the “upcycling” of the polyurethane waste.143,144 The term ‘upcycling’ was first used to refer to any process that transforms by-products, undesired, unwanted or waste products into new materials of higher values.145
Li et al.146 upcycled TPU wastes into activated carbon through two steps of controllable carbonization and activation and demonstrated its excellent wastewater treatment performance.
Daniel et al.147 showed the synthesis of iron–nitrogen–carbon (Fe–N–C) electrocatalysts for oxygen reduction reactions starting from a mix of waste polyethylene (PE) and polyurethane (PU) by adding FeCl3. In another work,148 a PUR waste foam was converted into N-doped hierarchical porous carbon (NHPC) via an autogenic atmosphere pyrolysis (AAP)-KOH activation approach for electrochemical energy storage applications. The functional upcycling of the PU waste is also extensively reviewed in a recent paper,149 where several strategies for the generation of new high-performance materials, especially high-capacity adsorbents for metal ions or oil–water separation, are proposed.
Another recent advance in the polyurethane field is the development of a non-isocyanate polyurethane (NIPU). These are a fairly new class of polyurethanes, which were initially discovered in 1957 by Dyer and Scott as a method to produce polyurethanes without using moisture-sensitive isocyanates. Because of the recent toxicity issues arisen around the use of isocyanates, they have recently attracted enormous attention. Advances in the synthesis of cyclic carbonates via the chemical insertion of CO2 into epoxy resins have also contributed to push activity in NIPUs.150 Although quite intense work dealing with NIPUs and several reviews have already appeared,151–154 only very few papers deal with the recycling of this new class of materials. Many of these latter rely on the use of dynamic covalent bonds (i.e., use the covalent adaptable network concept) to guarantee the recyclability of such polymers, by heating and pressurizing, both in case of compact and foamed PUs.155–158 More specifically, they combine the recent trends in material synthesis (i.e., NIPU) with recent trends in recycling (i.e., vitrimers). There are very few examples dealing with the chemical recycling of NIPUs. Liu et al.159 developed a series of isoeugenol-based NIPUs from bio-/CO2-derived bis(6-membered cyclic carbonate) cured with amines, and showed that they can completely be degraded into pre-monomers by reacting them with ethanol in 30 wt% NaOH at 90 °C for 4 h, i.e., under quite mild conditions. The pre-monomers can be separated by filtration with a high recovery ratio (90%) and used again, with CO2, to produce the monomers. These recovered monomers were then exploited for preparing a NIPU that showed nearly the same mechanical properties of the virgin one. Despite a fairly limited number of papers on their chemical recycling, it can be expected that NIPUs can solve, at least partly, the “aromatic amine issue” in the chemical recycling process since aromatic amines cannot be formed. It can also be expected that NIPUs can help in closing the material loop, with a good recycling efficiency under relatively mild degradation conditions.
Conclusions
Undoubtedly, the possibility of designing and effectively applying feasible and reliable recycling strategies to such fossil-derived polymers as polyurethanes has significantly reduced the general concern about their end-of-life and their difficult recyclability, while improving their overall image as recyclable materials with an important intrinsic added-value.However, the recycling strategies that have been designed and exploited so far, or are being developed for polyurethane waste streams, have to face the great complexity of these latter, considering the wide range of chemical structures, molecular weights, degree of crystallinity, crosslinking density, and hard-to-soft segment ratios, among other parameters, all of which significantly affect the recycling processes and the properties of the final recycled materials and products. Therefore, it is not possible to identify an ideal, unique, feasible, reliable, and even scalable recycling technology that should be suitable for all polyurethanes, but it is necessary to develop specific recycling approaches that maximize the yields, purity, and exploitability of the resulting recycled products, trying, at the same time, to minimize the environmental impact and the overall energy consumption. As reported in the review, several recycling methods have been designed and successfully applied: among them, the most promising ones, also considering industrial exploitation, are mechanical recycling (through regrinding and compression moulding) and chemical recycling (mainly through glycolysis reactions). It is expected that these two recycling strategies will continue to be exploited in the future, as they are currently quite well-established and consolidated, even on an industrial scale. Conversely, almost all the other recycling strategies are still focused on lab-scale research investigation, despite the significant outcomes achieved up to now; it is, therefore, difficult to foresee their development and exploitation on a larger processing scale. Both regrinding and glycolysis are cost-effective and limit the impact on the environment; at variance, the energy recovery through pyrolysis, gasification, and two-stage combustion, despite the important reduction of the waste volumes that are landfill-confined and the high value of the recovered energy, are still limited by the quite complicate control of the emission of toxic and hazardous products.
Further, although biodegradation can be performed using mild conditions (i.e., at room temperature and without the need for hazardous chemicals), it is still limited by the quite restricted number of suitable microorganisms and enzymes (indeed, the isolation of new microorganisms able to degrade polyurethanes is usually labor-intensive and time-consuming), and also by the biodegradation time that is usually high, and therefore has not expressed its full potential yet. To overcome these issues, the recently proposed metagenomic analysis of microbial population involved in the biodegradation of polyurethanes160,161 could help elucidating the structure of the microbial community, mining the enzymes involved in the polymer degradation and predicting their biodegradation ability in situ.
A further issue that undoubtedly may slow down the progress in polyurethane recycling regards the diversified forms (i.e., foams, bulk materials, and elastomers) and structures of the manufactured polyurethanes, which makes it more difficult to identify the most appropriate recycling strategy. In this context, a selective collection of polyurethane wastes could help a lot.
Further, it is essential to assess the influence of the polyurethane recyclates on the features (thermal and mechanical, among others) of the final products, into which recyclates are incorporated.
Finally, the development of reliable and feasible recycling strategies for polyurethane wastes and scraps demands cost-effectiveness and environmental friendliness: at present, these are still two challenging issues, especially when they are not considered individually. Besides, at present, the recovery and recycling of polyurethane wastes have no structured market; further, the cost and emission factors of the recovery processes have not been consolidated yet.162
However, it is expected that, in the next few years, research on the recycling of end-of-life polyurethanes will make further progress, at least starting from the lab scale, leading to a significant improvement in the recycling processes (also including low environmental impact approaches) and their reliable use, hopefully on an industrial scale. These possible outcomes will certainly contribute to ameliorating the overall management of end-of-life polyurethanes, offering some new perspectives within the circular economy concept, to select which polyurethanes should progressively take part as valuable materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The European Union—NextGenerationEU (National Sustainable Mobility Center CN00000023, Italian Ministry of University and Research Decree n. 1033-17/06/2022, Spoke 11—Innovative Materials & Lightweighting) is gratefully acknowledged. The opinions expressed are those of the author only and should not be considered as representative of the European Union or the European Commission's official position. Neither the European Union nor the European Commission can be held responsible for them.References
- M. Szycher, Szycher's Handbook of Polyurethanes, CRC Press, Boca Raton, 2nd edn, 2017 Search PubMed.
- S. Laurichesse and L. Avérous, Prog. Polym. Sci., 2014, 39, 1266–1290 CrossRef CAS.
- J. O. Akindoyo, M. D. H. Beg, S. Ghazali, M. R. Islam, N. Jeyaratnam and A. R. Yuvaraj, RSC Adv., 2016, 6, 114453–114482 RSC.
- PlasticsEurope Association of Plastics Manufacturers Plastics—The Facts 2021. An analysis of European Plastics Production, Demand and Waste Data. Available online: https://www.plasticseurope.org/en/resources/market-data (accessed on 23 May 2023).
- M. Kuranska, A. Prociak, M. Kirpluks and U. Cabulis, Ind. Crops Prod., 2015, 74, 849–857 CrossRef CAS.
- S. Bhargava, M. Kubota, R. D. Lewis, S. G. Advani, A. K. Prasad and J. M. Deitzel, Prog. Org. Coat., 2015, 79, 75–82 CrossRef CAS.
- D. H. Morton-Jones and J. W. Ellis, Polymer Products, Design, Materials and Processing, Chapman and Hall Ltd, London, UK, 1986. ISBN 9789401083201 Search PubMed.
- J. Datta, P. Kopczynska, D. Simón and J. F. Rodríguez, J. Polym. Environ., 2018, 26, 166–174 CrossRef CAS.
- Y. Rogaume, F. Jabouille, M. Auzanneau and J. C. Goudeau, Proc. of the Fifth Int. Conf. on Technologies and Combustion for a Clean Environment, Lisbon, Portugal, 1999, pp. 345–351.
- A. Merrington, in Applied Plastics Engineering Handbook, ed. M. Kutz, Elsevier, 2011, pp. 177–192 Search PubMed.
- W. Yang, Q. Dong, S. Liu, H. Xie, L. Liu and J. Li, Procedia Environ. Sci., 2012, 16, 167–175 CrossRef CAS.
- K. Ashida, Polyisocyanurate Foams in Polyurethane and Related Foams Chemistry and Technology, CRC Press, Boca Raton, 1st edn, 2007, pp. 101–102 Search PubMed.
- C. Dick, E. Dominguez-Rosado, B. Eling, J. J. Liggat, C. I. Lindsay, S. C. Martin, M. H. Mohammed, G. Seeley and C. E. Snape, Polymer, 2001, 42, 913–923 CrossRef CAS.
- A. Nanni, L. Crosetta, G. La Fauci, F. Biagi, M. Parisi, D. Colombo and M. Colonna, Sustainable Chem. Pharm., 2023, 33, 101059 CrossRef CAS.
- B. Wölfel, A. Seefried, V. Allen, J. Kaschta, C. Holmes and D. W. Schubert, Polymers, 2020, 12, 1–13 CrossRef PubMed.
- V. Skrockiene, K. Žukiene, V. Jankauskaite, A. Baltušnikas and S. Petraitiene, J. Elastomers Plast., 2016, 48, 266–286 CrossRef CAS.
- Y. H. Lee, B. K. Kang, H. Do Kim, H. J. Yoo, J. S. Kim, J. H. Huh, Y. J. Jung and D. J. Lee, Macromol. Res., 2009, 17, 616–622 CrossRef CAS.
- F. P. La, Mantia. Basic Concepts on the Recycling of Homogeneous and Heterogeneous Plastics, in Recycling of PVC and Mixed Plastic Waste, ed. F. P. La Mantia, Chemtec Publishing, Toronto, 1996, pp. 63–76 Search PubMed.
- T. Calvo-Correas, M. Benitez, I. Larraza, L. Ugarte, C. Peña-Rodríguez and A. Eceiza, Polym. Degrad. Stab., 2022, 198, 109880 CrossRef CAS.
- M. Villalobos, A. Awojulu, T. Greeley, G. Turco and G. Deeter, Energy, 2006, 31, 3227–3234 CrossRef CAS.
- J. Datta, E. Głowińska and M. Włoch, Mechanical Recycling via Regrinding, Rebonding, Adhesive Pressing, and Molding, in Recycling of Polyurethane Foams, ed. T. Sabu, A. V. Rane, K. Krishnan, V. K. Abitha and G. T. Martin, William Andrew Publishing, 2018, pp. 57–65 Search PubMed.
- E. Głowinska and J. Datta, Cellulose, 2015, 22, 2471–2481 CrossRef.
- G. J. Bleys, H. A. James and E. F. Cassidy, Method for the Preparation of Polyurethane Elastomers, U.S. Patent6,069,184A, 2000 Search PubMed.
- J. Scheirs, Polymer Recycling, John Wiley & Sons, Chichester, UK, 1998, ch. 10. ISBN: 978-0-471-97054-5 Search PubMed.
- A. J. Hulme and T. C. Goodhead, J. Mater. Process. Technol., 2003, 139, 322–326 CrossRef CAS.
- K. M. Zia, H. N. Bhatti and I. A. Bhatti, React. Funct. Polym., 2007, 67, 675–692 CrossRef CAS.
- J. L. Nafziger, S. B. Lowenkron, C. E. Koehler and B. N. Stevens, Flexible polyurethane rebond foam having improved tear resistance and method for the preparation thereof, U.S. Patent5312888, 1994 Search PubMed.
- R. E. Salino and R. E. Catai, Constr. Build. Mater., 2023, 387, 131201 CrossRef CAS.
- L. Polo Fonseca, A. Duval, E. Luna, M. Ximenis, S. De Meester, L. Avérous and H. Sardon, Curr. Opin. Green Sustainable Chem., 2023, 41, 100802 CrossRef CAS.
- D. Simón, A. M. Borreguero, A. de Lucas and J. F. Rodríguez, Waste Manage., 2018, 76, 147–171 CrossRef PubMed.
- J. L. Gerlock, J. Braslaw, L. R. Mahoney and F. C. Ferris, J. Polym. Sci., Polym. Chem. Ed., 1980, 18, 541–557 CrossRef CAS.
- M. Chanda, Adv. Ind. Eng. Polym. Res., 2021, 4, 133–150 CAS.
- G. A. Campbell and W. C. Meluch, Environ. Sci. Technol., 1976, 10, 182–185 CrossRef CAS.
- Y. Shi, X. Zhan, Q. Zhang and F. Chen, Chem. React. Eng. Technol., 2009, 25, 88–92 Search PubMed.
- Z. Dai, B. Hatano, J. Kadokawa and H. Tagaya, Polym. Degrad. Stab., 2002, 76, 179–184 CrossRef CAS.
- M. M. A. Nikje and F. H. A. Mohammadi, Polimery, 2009, 54, 541–545 CrossRef.
- M. M. A. Nikje, M. Nikrah and F. H. A. Mohammadi, J. Cell. Plast., 2008, 44, 367–380 CrossRef CAS.
- M. Goto, J. Supercrit. Fluids, 2009, 47, 500–507 CrossRef CAS.
- G. Behrendt and B. W. Naber, J. Univ. Chem. Technol. Metall., 2009, 44, 3–23 CAS.
- S. Motokucho, Y. Nakayama, H. Morikawa and H. Nakatani, J. Appl. Polym. Sci., 2018, 135, 14–17 CrossRef.
- J. Datta and M. Rohn, Polimery, 2007, 9, 627–633 CrossRef.
- J. Datta and M. Rohn, Polimery, 2007, 52, 579–582 CrossRef CAS.
- R. Donadini, C. Boaretti, L. Scopel, A. Lorenzetti and M. Modesti, Chem. – Eur. J. DOI:10.1002/chem.202301919.
- D. Simon, A. M. Borreguero, A. de Lucas and J. F. Rodrıguez, Polym. Degrad. Stab., 2014, 109, 115–121 CrossRef CAS.
- H. Benes, J. Rosner, P. Holler, H. Synkova, J. Kotek and Z. Horak, Polym. Adv. Technol., 2007, 18, 149–156 CrossRef CAS.
- J. Datta and M. Włoch, Recycling of Polyurethanes, in Polyurethane Polymers: Blends and Interpenetrating Polymer Networks, ed. S. Thomas, J. Datta, T. Haponiuk and A. Reghunnadhan. Elsevier, Amsterdam, 2017 Search PubMed.
- C. Molero, A. de Lucas and J. F. Rodriguez, Polym. Degrad. Stab., 2006, 91, 894–901 CrossRef CAS.
- C. Molero, A. de Lucas and J. F. Rodrıguez, Polym. Degrad. Stab., 2008, 93, 353–361 CrossRef CAS.
- C. Molero, A. de Lucas and J. F. Rodrıguez, Polym. Degrad. Stab., 2006, 91, 221–228 CrossRef CAS.
- J. Datta and M. Rohn, J. Therm. Anal. Calorim., 2007, 88, 437–440 CrossRef CAS.
- J. Datta, J. Elastomers Plast., 2010, 42, 117–127 CrossRef CAS.
- M. Murai, M. Sanou, T. Fujimoto and F. Baba, J. Cell. Plast., 2003, 39, 15–27 CrossRef CAS.
- J. Y. Lee and D. Kim, J. Appl. Polym. Sci., 2000, 77, 2646–2656 CrossRef CAS.
- C. H. Wu, C. Y. Chang and J. K. Li, Polym. Degrad. Stab., 2002, 75, 413–421 CrossRef CAS.
- M. Gassan, B. Naber, V. Neiss, P. Moeckel and W. Weissflog, Preparation of recyclate polyols, and the use thereof in the preparation of polyurethanes, US Patent5357006, 1994 Search PubMed.
- H. R. Van Der Wal, J. Reinf. Plast. Compos., 1994, 13, 87–96 CrossRef.
- C. H. Wu, C. Y. Chang, C. M. Cheng and H. C. Huang, Polym. Degrad. Stab., 2003, 80, 103–111 CrossRef CAS.
- D. Simon, M. T. Garcıa, A. de Lucas, A. M. Borreguero and J. F. Rodrıguez, Polym. Degrad. Stab., 2013, 98, 144–149 CrossRef CAS.
- J. Datta and S. Pasternak, Polimery, 2005, 50, 352–357 CrossRef CAS.
- J. Datta and K. Pniewska, Polimery, 2008, 53, 27–32 CrossRef CAS.
- J. Datta, J. Therm. Anal. Calorim., 2012, 109, 517–520 CrossRef CAS.
- J. Datta and M. Janicka, Przem. Chem., 2007, 86, 624–626 CAS.
- C. Molero, A. de Lucas, F. Romero and J. F. Rodriguez, J. Mater. Cycles Waste Manage., 2009, 11, 130–132 CrossRef CAS.
- X. Wang, H. Chen, C. Chen and H. Li, Fibers Polym., 2011, 12, 857–863 CrossRef CAS.
- J. Wang, D. Chen and Q. Zhang, Fibers Polym., 2007, 8, 13–18 CrossRef CAS.
- M. Modesti, F. Costantini, E. dal Lago, F. Piovesan, M. Roso, C. Boaretti and A. Lorenzetti, Polym. Degrad. Stab., 2018, 156, 151–160 CrossRef CAS.
- S. Xue, M. Omoto, T. Hidai and Y. Imai, J. Appl. Polym. Sci., 1995, 56, 127–134 CrossRef.
- K. Kanaya and S. Takahashi, J. Appl. Polym. Sci., 1994, 51, 675–682 CrossRef CAS.
- H. Lentz and W. Mormann, Makromol. Chem., Macromol. Symp., 1992, 57, 305–310 CrossRef CAS.
- M. Soltysinski, K. Piszczek, D. Romecki, S. Narozniak, J. Tomaszewska and K. Skórczewska, Polimery/Polymers, 2018, 63, 234–238 CAS.
- N. Gama, B. Godinho, G. Marques, R. Silva, A. Barros-Timmons and A. Ferreira, Chem. Eng. J., 2020, 395, 125102 CrossRef CAS.
- R. Arunima and T. Sabu, Polyurethanes: Structure, Properties, Synthesis, Characterization, and Applications, in Polyurethane Polymers: Blends and Interpenetrating Polymer Networks, ed. T. Sabu, J. Datta, T. Haponiuk and A. Reghunnadhan, Elsevier, Amsterdam, 2017 Search PubMed.
- H. He, H. Su, H. Yu, K. Du, F. Yang, Y. Zhu, M. Ma, Y. Shi, X. Zhang, S. Chen and X. Wang, ACS Sustainable Chem. Eng., 2023, 11, 5515–5523 CrossRef CAS.
- N. Gama, B. Godinho, G. Marques, R. Silva, A. Barros-Timmons and A. Ferreira, Polymer, 2021, 219, 123561 CrossRef CAS.
- K. Troev, A. Tsekova and R. Tsevi, J. Appl. Polym. Sci., 2000, 78, 2565–2573 CrossRef CAS.
- K. Troev, A. Tsekova and R. Tsevi, Polym. Degrad. Stab., 2000, 67, 397–405 CrossRef CAS.
- K. Troev, V. I. Atanasov and R. Tsevi, J. Appl. Polym. Sci., 2000, 76, 886–893 CrossRef CAS.
- K. Troev, G. Grancharov and R. Tsevi, Polym. Degrad. Stab., 2000, 70, 43–48 CrossRef CAS.
- G. Jomaa, P. Goblet, C. Coquelet and V. Morlot, Thermochim. Acta, 2015, 612, 10–18 CrossRef CAS.
- C. F. Cullis and M. M. Hirschler, The Combustion of Organic Polymers, Clarendon Press, United Kingdom, 1981, ISBN: 9780198513513 Search PubMed.
- D. Y. Takamoto and M. A. Petrich, Ind. Eng. Chem. Res., 1994, 33, 3004–3009 CrossRef CAS.
- G. Moroi, C. Ciobanu, E. Costea, N. Blba and I. Palamaru, Thermochim. Acta, 1997, 291, 95–99 CrossRef CAS.
- G. Moroi, J. Anal. Appl. Pyrolysis, 2004, 71, 485–500 CrossRef CAS.
- M. M. Esperanza, A. N. Garcia, R. Font and J. A. Conesa, J. Anal. Appl. Pyrolysis, 1999, 52, 151–166 CrossRef CAS.
- A. A. Shah, F. Hasan, A. Hameed and S. Ahmed, Biotechnol. Adv., 2008, 26, 246–265 CrossRef CAS PubMed.
- P. Bhavsar, M. Bhave and H. K. Webb, World J. Microbiol. Biotechnol., 2023, 39, 1–11 CrossRef PubMed.
- J. Banik, D. Chakraborty, M. Rizwan, A. H. Shaik and M. R. Chandan, Waste Manage. Res., 2023, 41, 1063–1080 CrossRef CAS PubMed.
- G. T. Howard, Int. Biodeterior. Biodegrad., 2002, 49, 245–252 CrossRef CAS.
- A. Magnin, L. Hoornaert, E. Pollet, S. Laurichesse, V. Phalip and L. Avérous, Microb. Biotechnol., 2018, 12, 544–555 CrossRef PubMed.
- A. A. Shah, F. Hasan, J. I. Akhter, A. Hameed and S. Ahmed, Ann. Microbiol., 2008, 58, 381–386 CrossRef CAS.
- Y. Akutsu, T. Nakajima-Kambe, N. Nomura and T. Nakahara, Appl. Environ. Microbiol., 1998, 64, 62–67 CrossRef CAS PubMed.
- B. Jansen, F. Schumacher-Perdreau, G. Peters and G. Pulverer, Zentralbl. Bakteriol., 1991, 276, 36–45 CrossRef CAS PubMed.
- L. F. Pérez-Lara, M. Vargas-Suárez, N. N. Lõpez-Castillo, M. J. Cruz-Gõmez and H. Loza-Tavera, J. Appl. Polym. Sci., 2016, 133, 42992 CrossRef.
- M. J. Kay, L. H. G. Morton and E. L. Prince, Int. Biodeterior., 1991, 27, 205–222 CrossRef CAS.
- J. Álvarez-Barragán, L. Domínguez-Malfavón, M. Vargas-Suárez, R. González-Hernández, G. Aguilar-Osorio and H. Loza-Tavera, Appl. Environ. Microbiol., 2016, 82, 5225–5235 CrossRef PubMed.
- J. R. Crabbe, J. R. Campbell, L. Thompson, S. L. Walz and W. W. Schultz, Int. Biodeterior. Biodegrad., 1994, 33, 103–113 CrossRef.
- A. Nakkabi, M. Sadiki, M. Fahim, N. Ittobane, S. Ibnsouda Koraichi, H. Barkai and S. El Abed, Int. J. Environ. Res., 2015, 9, 157–162 CAS.
- Y. H. Peng, Y. H. Shih, Y. C. Lai, Y. Z. Liu, Y. T. Liu and N. C. Lin, Environ. Sci. Pollut. Res., 2014, 21, 9529–9537 CrossRef CAS PubMed.
- Y. Matsumiya, N. Murata, E. Tanabe, K. Kubota and M. Kubo, J. Appl. Microbiol., 2010, 108, 1946–1953 CAS.
- Z. Filip, Eur. J. Appl. Microbiol. Biotechnol., 1979, 7, 277–280 CrossRef CAS.
- T. Takamoto, H. Shirasaka, H. Uyama and S. Kobayashi, Chem. Lett., 2001, 30, 492–493 CrossRef.
- A. Magnin, E. Pollet, R. Perrin, C. Ullmann, C. Persillon, V. Phalip and L. Avérous, Waste Manage., 2019, 85, 141–150 CrossRef CAS PubMed.
- R. Smith, D. F. Williams and C. Oliver, J. Biomed. Mater. Res., 1987, 21, 1149–1166 CrossRef CAS PubMed.
- C. Ferris, M. V. De Paz, F. Zamora and J. A. Galbis, Polym. Degrad. Stab., 2010, 95, 1480–1487 CrossRef CAS.
- B. D. Ratner, K. W. Gladhill and T. A. Horbett, J. Biomed. Mater. Res., 1988, 22, 509–527 CrossRef CAS PubMed.
- J. Fang, S. H. Ye, V. Shankarraman, Y. Huang, X. Mo and W. R. Wagner, Acta Biomater., 2014, 10, 4639–4649 CrossRef CAS PubMed.
- G. B. Wang, R. S. Labow and J. P. Santerre, J. Biomed. Mater. Res., 1997, 36, 407–417 CrossRef CAS PubMed.
- R. S. Labow, D. J. Erfle and J. P. Santerre, Biomaterials, 1996, 17, 2381–2388 CrossRef CAS PubMed.
- Q. Zhao, R. E. Marchant, J. M. Anderson and A. Hiltner, Polymer, 1987, 28, 2040–2046 CrossRef CAS.
- B. Pilch-Pitera, J. Polym. Environ., 2013, 21, 215–223 CrossRef CAS.
- A. Kemona and M. Piotrowska, Polymers, 2020, 12, 1752 CrossRef CAS PubMed.
- M. Paabo and B. C. Levin, Fire Mater., 1987, 11, 1–29 CrossRef CAS.
- E. Weigand, J. Wagner and G. Waltenberger, AbfallwirtschaftsJournal, 1996, 3, 40–45 Search PubMed.
- K. Zhang, J. Hu, S. Yang, W. Xu, Z. Wang, P. Zhuang, H. P. Grossart and Z. Luo, J. Hazard. Mater., 2022, 437, 129406 CrossRef CAS PubMed.
- Y. Branson, S. Söltl, C. Buchmann, R. Wei, L. Schaffert, C. P. S. Badenhorst, L. Reisky, G. Jäger and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2023, 62, e202216220 CrossRef CAS PubMed.
- J. Jeong, D. Oh and M. Goh, J. Ind. Eng. Chem., 2022, 109, 182–188 CrossRef CAS.
- C. Xu and Y. Hong, Bioact. Mater., 2022, 15, 250–271 CAS.
- P. Pfohl, D. Bahl, M. Rückel, M. Wagner, L. Meyer, P. Bolduan, G. Battagliarin, T. Hüffer, M. Zumstein, T. Hofmann and W. Wohlleben, Environ. Sci. Technol., 2022, 56, 16873–11688 CrossRef CAS PubMed.
- M. J. C. Espinosa, A. C. Blanco, T. Schmidgall, A. K. Atanasoff-Kardjalieff, U. Kappelmeyer, D. Tischler, D. H. Pieper, H. J. Heipieper and C. Eberlein, Front. Microbiol., 2020, 11, 1–9 CrossRef PubMed.
- B. S. Rajput, T. A. P. Hai, N. R. Gunawan, M. Tessman, N. Neelakantan, G. B. Scofield, J. Brizuela, A. A. Samoylov, M. Modi, J. Shepherd, A. Patel, R. S. Pomeroy, N. Pourahmady, S. P. Mayfield and M. D. Burkart, J. Appl. Polym. Sci., 2022, 139, e53062 CrossRef CAS.
- J. Liu, Q. Zeng, H. Lei, K. Xin, A. Xu, R. Wei, D. Li, J. Zhou, W. Dong and M. Jiang, J. Hazard. Mater., 2023, 448, 130776 CrossRef CAS PubMed.
- L. Guo, L. Wang, X. Guo, K. Hao, H. Liu, Y. Xu, G. Liu, S. Guo, L. Bai, D. Ren and F. Liu, Materials, 2022, 15, 6047 CrossRef CAS PubMed.
- P. He, H. Lu, H. Ruan, C. Wang, Q. Zhang, Z. Huang and J. Liu, Polymers, 2022, 14, 3277 CrossRef CAS PubMed.
- S. A. Madbouly, Curr. Opin. Green Sustainable Chem., 2023, 42, 100835 CrossRef CAS.
- J. Li, H. Zhu, D. Fang, X. Huang, C. Zhang and Y. Luo, J. Environ. Chem. Eng., 2023, 11, 110269 CrossRef CAS.
- A. Bandegi, M. Montemayor and I. Manas-Zloczower, Polym. Adv. Technol., 2022, 33, 3750–3758 CrossRef CAS.
- Z. Liu, Z. Fang, N. Zheng, K. Yang, Z. Sun, S. Li, W. Li, J. Wu and T. Xie, Nat. Chem., 2023, 15, 1773–1779 CrossRef CAS PubMed.
- S. Kim, K. Li, A. Alsbaiee, J. P. Brutman and W. R. Dichtel, Adv. Mater., 2023, 2305387 CrossRef CAS PubMed.
- J. Bai, L. Zhang, Z. Shi and X. Jiang, Chem. Mater., 2022, 34, 10172–10180 CrossRef CAS.
- Y. Tao, X. Liang, J. Zhang, I. M. Lei and J. Liu, J. Polym. Sci., 2022, 2233–2253 Search PubMed.
- R. Donadini, C. Boaretti, A. Lorenzetti, M. Roso, D. Penzo, E. Dal Lago and M. Modesti, ACS Omega, 2023, 8, 4655–4666 CrossRef CAS PubMed.
- M. Grdadolnik, A. Drinčić, A. Oreški, O. C. Onder, P. Utroša, D. Pahovnik and E. Žagar, ACS Sustainable Chem. Eng., 2022, 10, 1323–1332 CrossRef CAS PubMed.
- I. Amundarain, R. Miguel-Fernández, A. Asueta, S. García-Fernández and S. Arnaiz, Polymers, 2022, 14, 1157 CrossRef CAS PubMed.
- R. Miguel-Fernández, I. Amundarain, A. Asueta, S. García-Fernández, S. Arnaiz, N. Lardiés Miazza, E. Montón, B. Rodríguez-García and E. Bianca-Benchea, Polymers, 2022, 14, 2936 CrossRef PubMed.
- J. Sternberg and S. Pilla, Nat. Sustainability, 2023, 6, 316–324 CrossRef.
- L. Yuan, W. Zhou, Y. Shen and Z. Li, Polym. Degrad. Stab., 2022, 204, 110116 CrossRef CAS.
- L. Gausas, B. S. Donslund, S. K. Kristensen and T. Skrydstrup, ChemSusChem, 2022, 15, e202101705 CrossRef CAS PubMed.
- X. Gu, X. Wang, X. Guo, S. Liu, Q. Li and Y. Liu, Polymers, 2023, 15, 1445 CrossRef CAS PubMed.
- A. Haridas, S. Sharma, K. Naskar and T. Mondal, ACS Appl. Mater. Interfaces, 2023, 15, 17279–17292 CrossRef PubMed.
- D. Simón, A. M. Borreguero, A. De Lucas and J. F. Rodríguez, Polym. Degrad. Stab., 2015, 121, 126–136 CrossRef.
- J. Del Amo, D. Simón, M. J. Ramos, J. F. Rodríguez, A. De Lucas and A. M. Borreguero, J. Mater. Cycles Waste Manage., 2022, 24, 1059–1071 CrossRef CAS.
- D. S. Zhang, X. H. Gu, S. W. Liu, Y. Liu, Q. Y. Zhou, S. W. Zhu and Y. W. Zhu, Sustainability, 2022, 14, 15685 CrossRef CAS.
- B. Wang, Y. Wang, F. Li, S. Du, J. Zhu and S. Ma, J. Polym. Sci. DOI:10.1002/pol.20230424.
- Z. Liu, Z. Fang, N. Zheng, K. Yang, Z. Sun, S. Li, W. Li, J. Wu and T. Xie, Nat. Chem., 2023, 15, 1773–1779 CrossRef CAS PubMed.
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y. X. Chen, F. A. Leibfarth and H. Sardon, Nature, 2022, 603, 803–814 CrossRef CAS PubMed.
- Z. Li, K. Chen, Z. Chen, W. Li, B. W. Biney, A. Guo and D. Liu, J. Environ. Chem. Eng., 2021, 9, 104704 CrossRef CAS.
- G. Daniel, T. Kosmala, M. C. Dalconi, L. Nodari, D. Badocco, P. Pastore, A. Lorenzetti, G. Granozzi and C. Durante, Electrochim. Acta, 2020, 362, 137200 CrossRef CAS.
- X. Zhou, L. Zhu, Y. Yang, L. Xu, X. Qian, J. Zhou, W. Dong and M. Jiang, Chemosphere, 2022, 300, 134552 CrossRef CAS PubMed.
- O. Guselnikova, O. Semyonov, E. Sviridova, R. Gulyaev, A. Gorbunova, D. Kogolev, A. Trelin, Y. Yamauchi, R. Boukherroub and P. Postnikov, Chem. Soc. Rev., 2023, 52, 4755–4832 RSC.
- A. Gomez-Lopez, F. Elizalde, I. Calvo and H. Sardon, Chem. Commun., 2021, 57, 12254–12265 RSC.
- J. Guan, Y. Song, Y. Lin, X. Yin, M. Zuo, Y. Zhao, X. Tao and Q. Zheng, Ind. Eng. Chem., 2017, 50, 6517–6527 CrossRef.
- M. S. Kathalewar, P. B. Joshi, A. S. Sabnis and V. C. Malshe, RSC Adv., 2013, 3, 4110–4129 RSC.
- M. Włoch and J. Datta, Nonisocyanate Polyurethanes, in Polyurethane Polymers: Blends and Interpenetrating Polymer Networks, ed. T. Sabu, J. Datta, T. Haponiuk and A. Reghunnadhan, Elsevier, Amsterdam, 2017, pp. 169–202 Search PubMed.
- H. Khatoon, S. Iqbal, M. Irfan, A. Darda and N. K. Rawat, Prog. Org. Coat., 2021, 154, 106124 CrossRef CAS.
- P. Miao, J. Liu, M. He, X. Leng and Y. Li, Chem. Eng. J., 2023, 475, 146398 CrossRef CAS.
- S. Hu, X. Chen and J. M. Torkelson, ACS Sustainable Chem. Eng., 2019, 7, 10025–10034 CrossRef CAS.
- F. Monie, B. Grignard and C. Detrembleur, ACS Macro Lett., 2022, 11, 236–242 CrossRef CAS PubMed.
- N. S. Purwanto, Y. Chen, T. Wang and J. M. Torkelson, Polymer, 2023, 272, 125858 CrossRef CAS.
- J. Liu, P. Miao, X. Leng, J. Che, Z. Wei and Y. Li, Macromol. Rapid Commun., 2023, 44, 2300263 CrossRef CAS PubMed.
- J. Purohit, A. Chattopadhyay and B. Teli, Curr. Genomics, 2020, 21, 253–270 CAS.
- X. Jin, J. Dong, X. Guo, M. Ding, R. Bao and Y. Luo, Polym. Int., 2022, 71, 1384–1392 CrossRef CAS.
- M. Ravina, I. Bianco, B. Ruffino, M. Minardi, D. Panepinto and M. Zanetti, J. Cleaner Prod., 2023, 394, 136227 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |