
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Microporous transport layers facilitating low iridium loadings in polymer electrolyte water electrolysis†
Carl Cesar
Weber
 a,
Salvatore
De Angelis‡
a,
Robin
Meinert
a,
Christian
Appel
b,
Mirko
Holler
b,
Manuel
Guizar-Sicairos
bc,
Lorenz
Gubler
a and
Felix N.
Büchi
*a
a,
Salvatore
De Angelis‡
a,
Robin
Meinert
a,
Christian
Appel
b,
Mirko
Holler
b,
Manuel
Guizar-Sicairos
bc,
Lorenz
Gubler
a and
Felix N.
Büchi
*a
aElectrochemistry Laboratory, Paul Scherrer Institut, 5232 Villigen PSI, Switzerland. E-mail: felix.buechi@psi.ch
bPhoton Science Division, Paul Scherrer Institut, 5232 Villigen PSI, Switzerland
cInstitute of Physics (IPHYS), Ecole Polytechnique Fédérale de Lausanne, Rte Cantonale, Lausanne 1015, Switzerland
First published on 2nd February 2024
Abstract
Minimizing the power-specific iridium loading in polymer electrolyte water electrolysis (PEWE) is essential for the commercialization and upscaling of this technology. However, decreasing the iridium loading can severely affect performance and stability. Microporous layers (MPL) can overcome some of these issues by maximizing catalyst utilization and increasing cell efficiency. In this study, we combined advanced synchrotron and lab-based X-ray imaging techniques and electrochemical characterization to improve the PEWE cell performance at low Ir loadings using novel MPLs. For the first time, the 3D nanostructure of the catalyst layer was characterized under dry and wet conditions using ptychographic X-ray laminography. We prepared catalyst layers (CL) at three iridium loadings between 2.5 and 0.1 mgIrcm−2 in two different configurations: depositing either on the membrane or on the Ti-substrate (MPL). The MPL structure and catalyst distribution at its surface were analyzed using X-ray tomographic microscopy. Moreover, we investigated the effect of introducing a thin protective Pt coating on the MPL. The electrochemical performance was characterized for all cell combinations, and an in-depth kinetic analysis revealed information on CL utilization. The MPLs exhibit significant benefits for reducing iridium loadings, allowing performance to be sustained with only modest voltage losses. The challenges in fabricating anodic CLs with reduced catalyst loadings and the advantages of using an MPL in both configurations are discussed. The findings of this study contribute to accomplishing the required targets in terms of power-specific iridium loadings for future PEWE systems.
Broader contextGreen hydrogen plays a pivotal role in industries that are difficult to decarbonize by electrification, such as steel and fertilizer industries, and heavy-duty transport. Polymer electrolyte water electrolysis (PEWE) is a key technology for producing hydrogen from renewable intermittent energy sources, as wind and solar power. However, high OPEX and CAPEX majorly limit their rapid scale-up and commercialization. A major drawback is the need for substantial amounts of scarce and expensive iridium as the catalyst for the oxygen evolution reaction (OER). Although replacing Ir with a non-noble metal catalyst would be ideal, finding an acceptable substitute in terms of activity and stability is challenging and cannot be immediately implemented on a large scale. Therefore, reducing the amount of iridium to the greatest extent possible is the only tangible solution to scale up green hydrogen production capacity in the near future. The anodic porous transport layer (PTL) interface with the catalyst layer contributes significantly to the efficient use of the catalyst. Consequently, optimizing the PTLs surface properties, for example, by integrating a microporous layer (which has been proven to be essential in the fuel cell field), has major potential in helping to achieve the required iridium loading targets. |
1. Introduction
Hydrogen produced from renewable energy via electrolysis is gradually being established as a versatile energy vector for industries that cannot be easily electrified.1 Polymer electrolyte water electrolysis (PEWE) offers excellent prospects in this regard, as it can be operated at high current densities and has rapid dynamic response times. However, the rapid scale-up of installed capacity and disruptions in the supply chain pose challenges for manufacturers.2,3 Therefore, minimizing the amount of noble metals in PEWE is key for achieving the required increase in the capacity installation rate.4–7 In particular, the scarcity and high price of the Ir required as the anodic catalyst are among the most challenging factors. Thus, to achieve the targeted scalability scenarios, it is essential to reduce the power-specific loading in future systems by up to two orders of magnitude (e.g., from 0.6–0.7 gIr kW−1 to 0.01–0.05 gIr kW−1).5,8 This reduction translates into reducing the iridium loading from currently ∼2 to ∼0.4 mgIr cm−2 while increasing power density from 3 to 8 W cm−2 and system efficiencies from 70% to 75%.5The importance of Ir in catalyzing the oxygen evolution reaction (OER) in PEWE has been extensively investigated.9–12 Numerous studies have presented different and innovative strategies for modifying catalysts to reduce Ir loadings, increase stability, or even replace Ir with non-noble metal catalysts.13–18 However, these drastic changes in catalyst design still need to be scaled up and implemented in technically relevant-sized cells and stacks. Other approaches have focused on minimizing Ir content using commercially available catalysts. For instance, Alia et al. showed that the iridium loading can be reduced significantly by optimizing the coating procedure using commercial unsupported IrO2.19 Taie et al. achieved good performance and high Ir specific current (A mgIr−1) with extremely low iridium loading (as low as 0.011 mgIr cm−2) using only commercially available catalysts and cell materials.20
Decreasing the Ir loading presents significant challenges, to a large extent, owing to the reduction in the associated catalyst layer (CL) thickness.21 One way to address this issue is to reduce the packing density of the catalyst using a catalyst support material.22–26 Bernt et al. showed the impact on the overpotentials when reducing iridium loading using a supported commercial benchmark catalyst.21 The benchmark catalyst of Umicore Elyst is composed of 75 wt% IrO2 on TiO2. Heraeus recently developed a new type of supported OER catalyst with reduced iridium weight percentages (10–45 wt%) on a TiO2 support, which allowed for even lower packing densities.25,27,28 However, because TiO2 is a wide-bandgap semiconductor with poor electronic conductivity, the iridium phase must form a percolation network to sustain an electronic pathway. One solution is to replace TiO2 with a conductive material. However, finding a suitable conductive support (such as carbon in fuel cell catalysts) that can withstand the harsh PEWE anodic environments is challenging.26
In this regard, understanding the microstructures of typical CLs is essential for future optimization. The CL is a porous 3D structure with distinct phases and feature sizes ranging from the micro to the nanometer scale. Therefore, precise structural characterization requires advanced imaging techniques. The CL structure using unsupported Ir–RuOx/Ir was characterized by Hegge et al. using FIB-SEM tomography29 and by Lee et al. using X-ray nanotomography.30 However, in both cases, the crucial ionomer structure could not be resolved and was introduced artificially to estimate the transport properties. De Angelis et al. revealed the structure of a CL using a supported catalyst (Umicore) for the first time by ex situ Ptychographic X-ray computed tomography (PXCT).31 With this technique, it was possible to spatially resolve all phases present in the CL, that is, the IrO2, TiO2, ionomer, and pore phases. Moreover, the ionomer not only covers the IrO2/TiO2 particles but also forms a network that acts as a binder between the particles. Additionally, IrO2 forms a core–shell structure around the TiO2 particles with the additional presence of pure IrO2 particles. In general, the network of the IrO2 phase was highly connected, indicating that the conductivity of the support material had a negligible impact on the overall CL conductivity. Nevertheless, thus far, the CL structure, which may change upon contact with liquid water, has only been investigated under dry conditions.32
Other cell components, such as the porous transport layer (PTL), can significantly impact PEWE performance.33–37 In particular, the interface between the PTL and the CL contributes extensively to the efficient use of the CL.38–44 Schuler et al. correlated the PTL properties with the electrochemical performance, showing that the PTL surface properties significantly affect the CL utilization.38,39 Lopata also investigated the contribution of the PTL/CL interface at different Ir loadings.41 In a recent study, Weber et al. investigated the impact of PTL interface properties on CL utilization using model 2D PTLs, demonstrating that only a small area in close proximity to the pore edge is effectively utilized.44,45 This finding is related on the one hand to mass transport effects under the PTL layer and on the other hand to the low in-plane conductivity of the CL in the open pores,46–48 indicating that small PTL pores and spacing between pores (i.e., small PTL particle size) can favor higher catalyst utilization.
Thus, deliberate engineering of the PTL surface to optimize catalyst utilization is crucial, which can be achieved by modifying the surface30,49 or adding finer microporous layers (MPL).32,50,51 Schuler et al. introduced a well-performing hierarchical PTL with a Ti-based sintered MPL, where a significant increase in CL utilization compared to that in a single-layer PTL was determined. Furthermore, the MPL allowed the use of thinner membranes owing to their smoother surface, reducing damage to the catalyst-coated membrane (CCM) upon compression. However, the influence of PGM coating43,52–54 on the MPLs and its effect on CLs with low iridium loadings has not yet been investigated.
Coating the CL onto a membrane (i.e. CCM approach) is the common choice in PEWE. Nevertheless, coating the CL onto the PTL to create a porous transport electrode (PTE) could provide advantages, such as reducing the working steps in the manufacturing process and allowing for a greater variety of membranes.55 In principle, a PTE improves contact between CL and PTL by reducing CL deformation during membrane swelling.56 Nonetheless, producing PTE has proven to be challenging owing to the rough structure of commercial PTLs, which leads to severe losses of the useable catalyst layers.55–59 Bierling et al. emphasized this issue by extensively characterizing the CL distribution in anodic PTEs (using Ti-felt) via X-ray tomographic microscopy (XTM).55 In this context, coating the CL onto an MPL could be highly beneficial, leading to lower catalyst loss in the void spaces.
In this study, we aim to reduce iridium loadings close to the required targets of 0.05 gIr kW−1 by using novel MPLs in combination with commercial catalyst materials. To this end, we characterized the structures of the catalyst and porous transport layers, as well as investigated the advantages of using an MPL at the interface between the PTL and CL. We identified the challenges of reducing the iridium loading by analyzing the nanostructure of the CL using synchrotron-based ptychographic X-ray laminography (PyXL).60,61 This novel technique allowed us to compute the transport properties, including the electronic conductivity, and the changes induced upon hydrating the sample. The limits and properties of lowering the iridium loading from 2.5 to 0.1 mgIr cm−2 are analyzed by detailed electrochemical and electro-kinetic characterization to determine the following: (i) the influence of a Pt-interlayer coating (between PTL and CL) and (ii) advantages of coating the CL on the membrane (CCM) vs. coating on the transport layer (MPE). The MPL cell performance was benchmarked with that of cells using commercial single-layer PTL materials. The electrochemical results were correlated to the structure of the CL, MPL, and the catalyst distribution in the MPE to answer the following questions: (i) Why does the MPL facilitate low Ir loading? (ii) Why does this work only with a Pt coating? (iii) What is the role of the MPL in coating the CL on the Ti substrate? The findings of this study will facilitate the reduction of power-specific iridium loadings in next-generation PEWE systems.
2. Results
2.1. Morphological characterization
We first aimed to understand the nano and microstructures and transport properties of the anodic porous components in the electrolyzer cell, that is, the PTL, MPL, and CL, as well as the interface between the CL and MPL. Their feature sizes are hierarchical, implying that they gradually decrease (from the flow field toward the membrane). As each layer has a distinct size range, different techniques are required to characterize its morphology.Fig. 1a shows a two-dimensional slice obtained from the entire tomogram. The four phases present in the anodic CL (i.e. voids, ionomer, IrO2, and TiO2) are visible in the grayscale image. The phase fractions obtained from the segmented data of the dry and fully wet structures are listed Table 1. We observed minor differences in the phase fractions when compared with previous work performed by PXCT on a similar catalyst. One reason for this discrepancy may be the cryogenic (90 K) or the vacuum conditions used in the previous study, which could change the effective volume of the ionomer.31 Another reason for the discrepancy in the IrO2 phase fraction might be the different batch number of catalysts used (Umicore MA-1376 in this study vs. MA-292 in ref. 31), which may have resulted in a higher inner porosity of the IrO2 phase. Though, when comparing the phase fraction from the dry structure to the expected values obtained from the ink composition and the CL thickness,63 they differ by less than 2%. Fig. 1b displays the 3D-surface rendering of the segmented image of the same structure, with the ionomer network highlighted in blue. We observed the core–shell structure of IrO2 on the TiO2 as well as pure IrO2 particles. The ionomer forms a film around the catalyst particles and creates a network that binds them together. Fig. 1c shows a high-resolution SEM 2D image of the CL. We observed an interaction between the ionomer and the catalyst particles, confirming the ionomer film and network observed in the PyXL data.
 | ||
| Fig. 1 (a) Grey-scale slice of the CL (dry) extracted from the 3D volume obtained by PyXL (b) surface rendering of the volume with the ionomer phase shown in blue (c) SEM images of the CL particles and ionomer layer at two magnifications. | ||
| Phase | Phase fraction by PyXL (RT, dry) (%) | Phase fraction by PyXL (RT, wet) (%) | Phase fraction by PXCT (cryo, dry)31 (%) | Phase fraction from ink (%) |
|---|---|---|---|---|
| a Shown only for completeness, however segmentation of pore/ionomer in the wet sample may contain errors. | ||||
| Pore | 45 ± 1 | 36 ± 4a | 39 | 44 ± 3 |
| Ionomer | 21 ± 1 | 33 ± 5a | 15 | 20 ± 1 |
| TiO2 | 10 ± 0 | 10 ± 1 | 11 | 11 ± 1 |
| IrO2 | 25 ± 2 | 20 ± 1 | 36 | 25 ± 1 |
In Fig. S2 (ESI†), a 2D slice of the raw PyXL data from the fully wetted volume is shown and compared with that of the dry volume. Note that in the wetted volume, the contrast between the pore space (now filled with water) and the ionomer was much lower than that in the dry volume. The loss in contrast makes the differentiation of the pore and ionomer networks challenging for the wet volume. Still, from the phase fractions of the wet volume in Table 1, we determined that the ionomer phase fraction increased significantly, possibly due to water uptake. However, owing to the low contrast and, consequently, large errors in the segmentation, we cannot be certain about the pore and ionomer networks. Nevertheless, the solid phases (IrO2 and TiO2) were clearly distinguishable and could be well segmented. Therefore, we only characterized the solid phases for the wet CL volume.
Fig. 2 shows the pore and particle size distributions of the four phases of the dry CL and the solid phases of the wet CL volume. The pore size distribution shown in Fig. 2a is wide, with large pores above 1 μm. The ionomer thickness varies between 100 and 300 nm, however, as shown in the SEM image in Fig. 2, there might be thin ionomer layers that cannot be resolved. When comparing the IrO2/TiO2 particle size distributions of the dry and wet samples (Fig. 2c and d), we observed that they did not differ significantly. In general, all four size distributions were comparable to previous data from PXCT by De Angelis et al.31 The transport properties in the voids of the dry CL are listed in Table 2 for all three spatial dimensions. The pore tortuosity factor is 1.5–1.7 in all directions, whereas the relative diffusivity is slightly higher in the through-plane direction. The permeability of the CL is approximately three orders of magnitude lower than that of typical PTLs32,38 and comparable to other reports of CLs in the literature.64 Furthermore, the ionic conductivity of the dry CL was estimated based on the ionomer network (assuming a bulk conductivity of 85 mS cm−1 for Nafion65). The ionic conductivity was higher in the in-plane directions than in the through-plane direction, whereas the contrary trend was observed for electronic conductivity, which was estimated from the IrO2 network, assuming a bulk conductivity of 64 S cm−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 31,66 and that the other phases were insulators. The computed electronic conductivities of the dry anodic CL values were in the same range as previously computed31,64 and measured values.32
31,66 and that the other phases were insulators. The computed electronic conductivities of the dry anodic CL values were in the same range as previously computed31,64 and measured values.32
 | ||
| Fig. 2 Pore and particle size distributions of the four phases of the anode CL by PyXL for (a) pore diameter, (b) ionomer thickness, (c) IrO2 particle diameter, and (d) TiO2 particle diameter. For (c) and (d) the data for the wet CL are included. | ||
| Spatial direction | Tortuosity pore [–] | Rel. diffusivity [%] | Abs. permeability [×10−16 m2] | Ionic conductivity [mS cm−1] | Electronic conductivity [S cm−1] |
|---|---|---|---|---|---|
| X (in-plane) | 1.6 | 17 | 8.7 | 5.2 | 2.6 |
| Y (in-plane) | 1.7 | 15 | 6.2 | 4.9 | 2.4 |
| Z (through-plane) | 1.5 | 20 | 10.5 | 2.1 | 3.2 |
This study aimed to characterize the PEWE performance of cells using an anodic CL with reduced Ir loading. Therefore, the PyXL volumes were virtually cropped to the thicknesses of the CL at the respective Ir loadings. The CL thicknesses of the CCMs at loadings between 0.1 and 3.0 mgIr cm−2 were measured from cross-sectional SEM images, as shown in Fig. S4 (ESI†). In Fig. S4d (ESI†), the measured CL thickness is plotted as a function of the loading. A close-to-linear relationship is present with a nonzero y-axis intercept. The iridium loadings used in the electrochemical part of this study are 2.5, 0.5, and 0.1 mgIr cm−2, corresponding to thicknesses of the anodic CL of 9.7, 2.1, and 0.8 μm, respectively. Fig. S4e (ESI†) shows surface renderings of the cropped CLs for each loading. In Table 3, the computed electronic conductivities for the three CL thicknesses are listed for the dry and wet states. For all loadings, the dry conductivity was two to four times higher than the wet conductivity. Furthermore, in all the cases, the through-plane conductivity increased with lower loading. The in-plane conductivity did not change significantly when the thickness was reduced. Only at the extremely low loading of 0.1 mgIr cm−2, the value was noncalculable since no connected in-plane path exists. This observation is true for the y direction in the dry state and for both in-plane directions (x, y) in the wet state. This finding indicates that a continuous particle network was absent in the present volume (10.5 μm × 10.5 μm × CL thickness) at this specific loading. In fact, this is also observed from the top view SEM image of the anodic CL (0.1 mgIr cm−2) shown in Fig. S3a (ESI†), where “holes” were observed in the CL, in which the membrane could be seen. From these findings, we can conclude that at a loading of 0.1 mgIr cm−2 the CL is not connected in a continuous catalyst particle network, even under the assumption that the particles are evenly distributed.
| Loading (mgIr cm−2) | Spatial direction | 2.5 | 0.5 | 0.1 |
|---|---|---|---|---|
| CL thickness (μm/voxels) | 9.7/278 | 2.1/60 | 0.8/23 | |
| Dry electronic conductivity (S cm−1) | X | 2.7 | 3.3 | 1.4 |
| Y | 2.0 | 1.7 | N.A. | |
| Z | 3.4 | 6.0 | 8.4 | |
| Wet electronic conductivity (S cm−1) | X | 0.7 | 1.1 | N.A. |
| Y | 0.6 | 0.5 | N.A. | |
| Z | 1.1 | 3.4 | 6.1 |
 | ||
| Fig. 3 (a), (c) and (e) Top view SEM images and (b), (d) and (f) XTM surface renderings of (a) and (b) MPL + support layer; (b) and (d) MPE with 2.5 mgIr cm−2 loading; and (e) and (f) Ti-felt (Bekaert) with 2.5 mgIr cm−2. | ||
We investigated the effect of coating the CL on the Ti substrate instead of on the membrane (CCM). To the best of our knowledge, this is the first report in the literature of coating an anodic CL onto a Ti-MPL. For this approach we introduce the terminology: microporous electrode (MPE). Examples of an MPE is shown in the top-view SEM in Fig. 3c and the surface rendering in Fig. 3d. When coating a CL on a standard Ti-PTL (Ti-felt from Bekaert), the result is termed as a porous transport electrode (PTE), as introduced in previous studies.55,57–59 A PTE is shown in the SEM image in Fig. 3e, and in the XTM rendering in Fig. 3f.
The top-view SEM images show that part of the catalyst dissipated into the void spaces of the Ti substrates. An MPE and a PTE with high catalyst loading (2.5 mgIr cm−2) were analyzed by XTM. From the cross-sectional XTM slices shown in Fig. 4a and b, we can visually observe how the catalyst layer distributes at the PTL/CL interface. In the grayscale images, the CL is clearly visible as a white line, and in the segmented volumes, it is visualized in blue for the MPE and in red for the PTE. In the MPE, the CL was homogenously distributed close to the surface, forming a continuous layer. In contrast, in the PTE, a significant fraction of the CL disappears in the large voids of the PTLs, leading to isolated (‘stray’) catalyst regions far from the PTL surface. We visualized the CLs in 3D renderings for the MPE in Fig. 4c and for the PTE in Fig. 4d. The CL connected in a continuous network is shown in black, whereas the isolated parts of the CL are shown in red. For the PTE, more than 5% of the CL was isolated (in red) in comparison to the MPE with only 0.3%. Fig. 4e shows the distribution of the catalyst phase across the PTL thickness. We observed that for the MPE, the distribution was relatively narrow, with most of the CL located below 50 μm from the surface. For the PTE, a substantial portion of the CL penetrated far into the PTL bulk. If we assume that the membrane intrusion (for Nafion 115, dry thickness of 127 μm) is limited to roughly 30–40 μm from the PTL/MPL surface,32,38,55 we can expect that in case of the PTE ∼46% of the CL remains inaccessible and therefore not utilized. This effect is alleviated in the MPE because most parts of the CL are close to the MPL surface and only ∼18% of the CL would be unavailable.
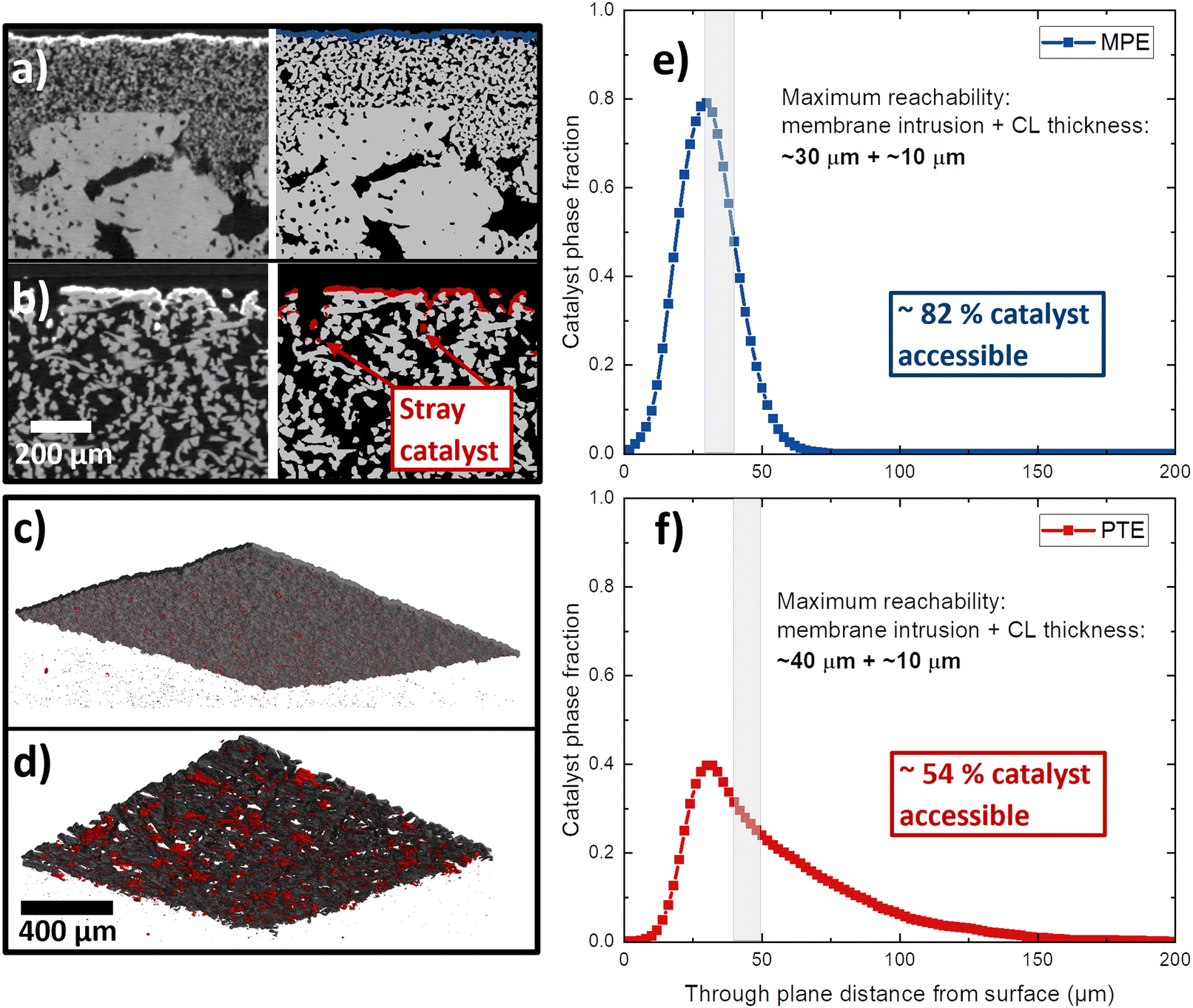 | ||
| Fig. 4 Cross-section XTM slices, as grey-scale and segmented images of (a) an MPE and (b) a PTE (based on Ti-felt) with 2.5 mgIr cm−2 catalyst layers. Three-dimensional visualization of the catalyst layer for (c) the MPE and (c) PTE where the voxels in the CL that are part of the continuous network are shown in black, whereas the isolated voxels are visualized in red. Catalyst layer phase fraction distribution across the thickness of (d) PTE and (e) MPE. | ||
In Fig. 5 the surface maps of the “naked” MPL and PTL as well as for the coated MPE and PTE are shown. The maps are graphical representations of the measured distances from the first contact point. We observed that the MPL exhibited a much lower surface roughness and a more homogenous interface area, whereas the PTL had large valleys or pockets, leading to high roughness. This result is similar to the previously reported MPL/PTL differences.32 On the right side of Fig. 5, we can observe the surface maps of the MPE and PTE. For the MPE, the CL fills most of the open pores making the surface less rough than for the MPL without catalyst. On the PTE surface map, the fiber diameter increased, indicating that the CL was deposited mostly as a shell around the fiber, as shown by Bierling et al.55 Overall, we estimate that for MPEs there is up to ∼28% more catalyst accessible in comparison to the PTE at the same loading. This result may be more pronounced at lower catalyst loadings, where a higher percentage of the total catalyst may be lost in the voids of the PTL. Moreover, Fig. S3 (ESI†) shows a top-view SEM image of an MPE with low iridium (0.1 mgIr cm−2), where only a few catalyst spots are visible on top of the MPL particles.
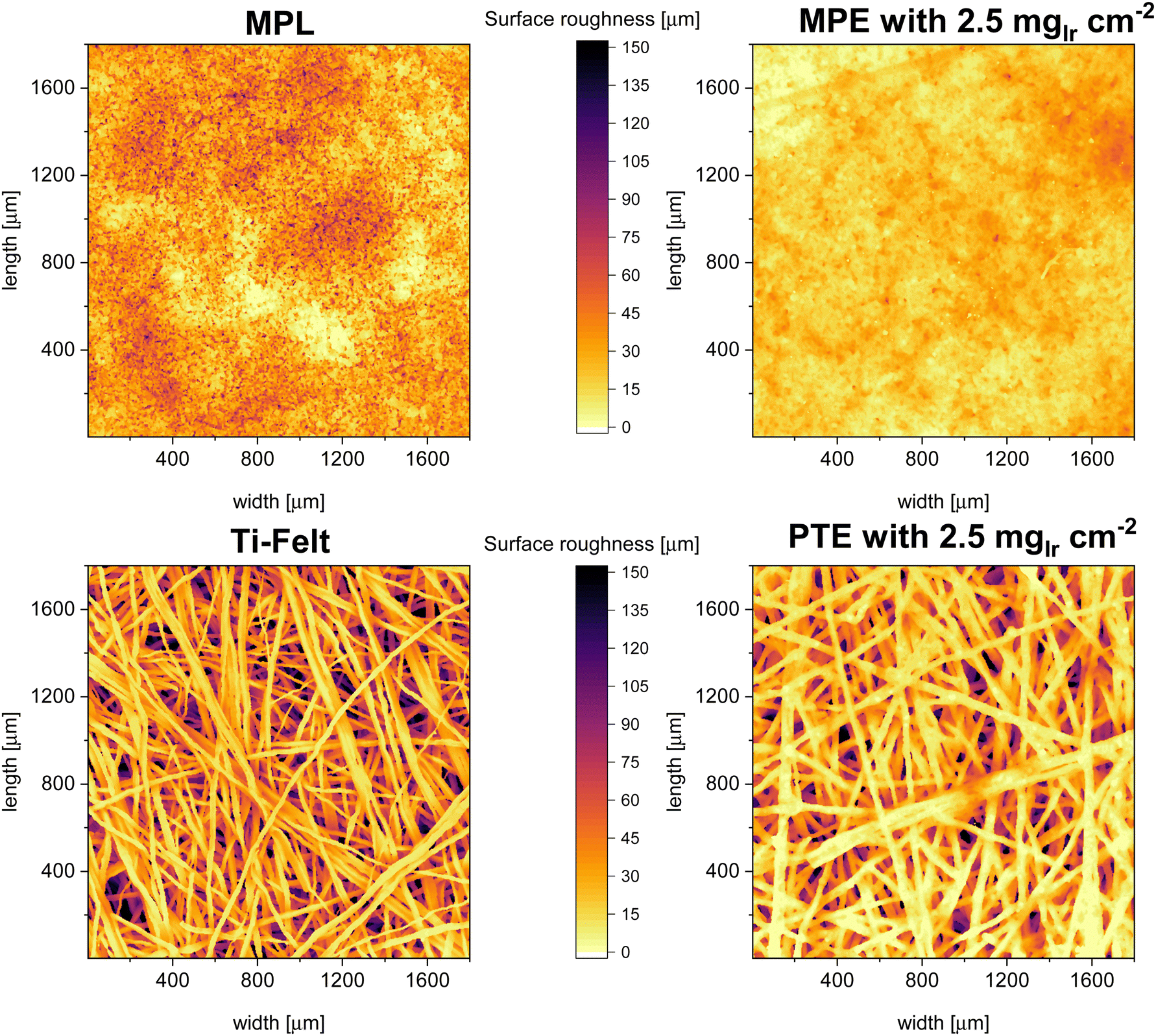 | ||
| Fig. 5 Surface distance maps from the XTM data of the stand-alone MPL and PTL (Ti-felt) as well as the MPE and PTE with a loading of 2.5 mgIr cm−2. | ||
2.2. Electrochemical characterization: towards low Ir loadings using MPLs
In the second part of this study, we focused on the electrochemical characterization of cells at three different Ir loadings: high, medium, and low that is 2.5, 0.5, and 0.1 mgIr cm−2 (±0.02 mgIr cm−2). We compared the three loadings using two different approaches: coating on the membrane (CCM) vs. coating on the MPL (MPE). For each approach (CCM and MPE), we also explored the effect of adding a thin protective Pt coating (∼40 nm, ∼0.085 mgPt cm−2) on the MPL to reduce the interfacial resistances.43 Selected measurements were benchmarked against the respective commercial single-layer materials. Finally, we determined catalyst utilization and mass activity from a kinetic analysis.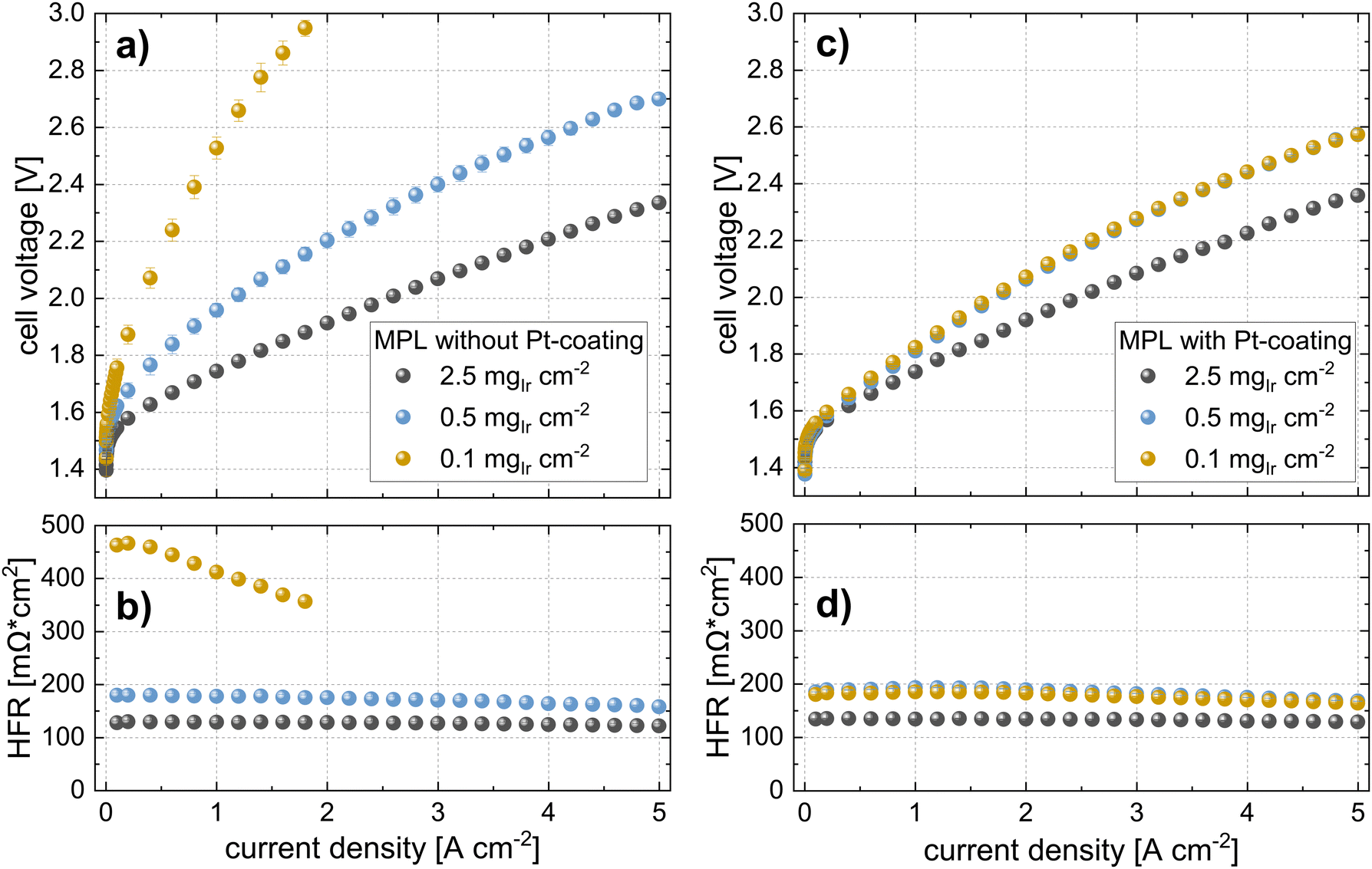 | ||
| Fig. 6 Polarization curves and HFR measurements of cells with a CCM (N115) approach at the different iridium loadings (T = 80 °C, 10 bar balanced pressure) using MPLs (a) and (b) without Pt-coatings and (c) and (d) with Pt-coating (40 nm). | ||
In Fig. 6c, the polarization curves, and in Fig. 6d, the HFR of cells using the same Ir-loadings but with MPLs with a Pt coating (40 nm) are shown. For cells with loadings of 2.5 mgIr cm−2, we did not observe any difference in the performance or HFR when compared to the cells with an MPL and without a Pt coating. In contrast, the cell using 0.5 mgIr cm−2 and, in particular, the cell with 0.1 mgIr cm−2 showed a drastic improvement in performance compared to the cells without Pt-coating on the MPL. Moreover, the cell with 0.1 mgIr cm−2 exhibited similar HFR values in comparison to that of the cell with 0.5 mgIr cm−2, leading to an almost equal performance. From the iR-free cell voltage shown in Fig. S6 (ESI†), the largest contribution to the differences in performance can be attributed to the HFR. The kinetics, which is also affected by the loading and Pt coating, are discussed in a later Section 2.3.
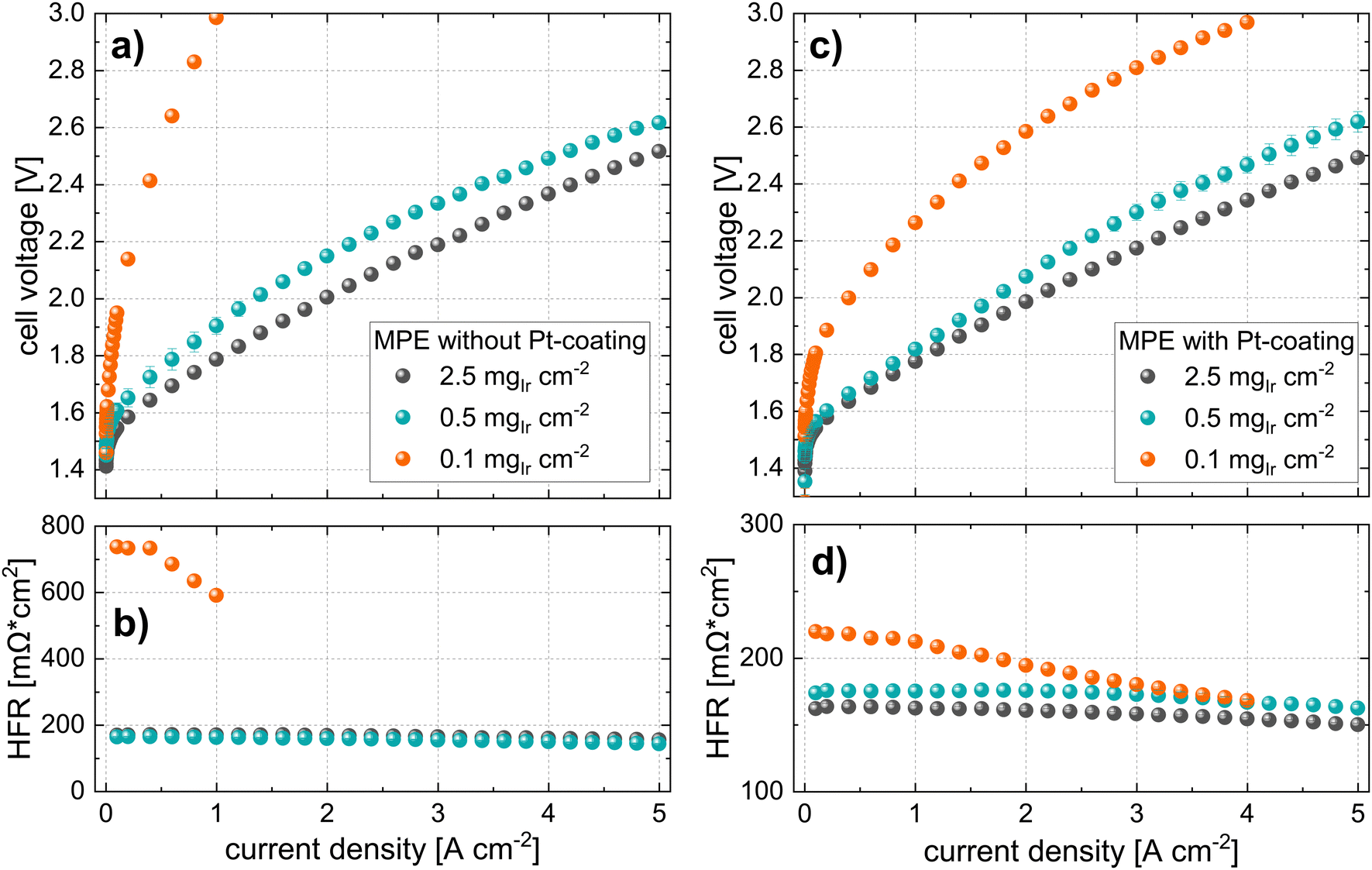 | ||
| Fig. 7 Polarization curves and HFR measurements of cells with an MPE approach at the different iridium loadings (N115, T = 80 °C, 10 bar balanced pressure) using MPLs (a) and (b) without Pt-coatings and (c) and (d) with a Pt-coating (40 nm). | ||
In contrast, the performance for cells using MPE with a loading of 0.5 mgIr cm−2 is good, with only ∼120 mV performance losses at 5 A cm−2 as compared to the high losses observed for the cell using 0.1 mgIr cm−2. Interestingly, when a Pt coating was added at this loading, the performance did not change significantly, which was also observed for the MPE with 2.5 mgIr cm−2, where no performance benefit was present from adding the Pt coating. Furthermore, the high-loading CCMs in Fig. 6 exhibits a higher performance than the MPE with 2.5 mgIr cm−2, which we attribute the loss of catalyst into the open pore space during deposition, even when an MPL was used.
For benchmarking the CCM configuration, polarization curves, and HFR measurements are compared for the lowest loading (0.1 mgIr cm−2). The data are shown in Fig. 8a and b. The cell voltages for the Pt-coated Ti felts were significantly higher (>500 mV at high current densities) than those of the cell with the MPL at the same catalyst loading. The performance difference is clearly given by the higher HFR of the cell using Ti felts (see iR-free cell voltage in Fig. S8, ESI†), as well as by the difference in the kinetic regime, which is subsequently addressed. Our best cell in terms of power-specific iridium loading is the cell using CCM at 0.1 mgIr cm−2 with MPL (Pt-coated) that achieves ∼0.06 gIr kW−1 at 70% cell efficiency (lower heating value) but at moderate current densities of ∼1 A cm−2.
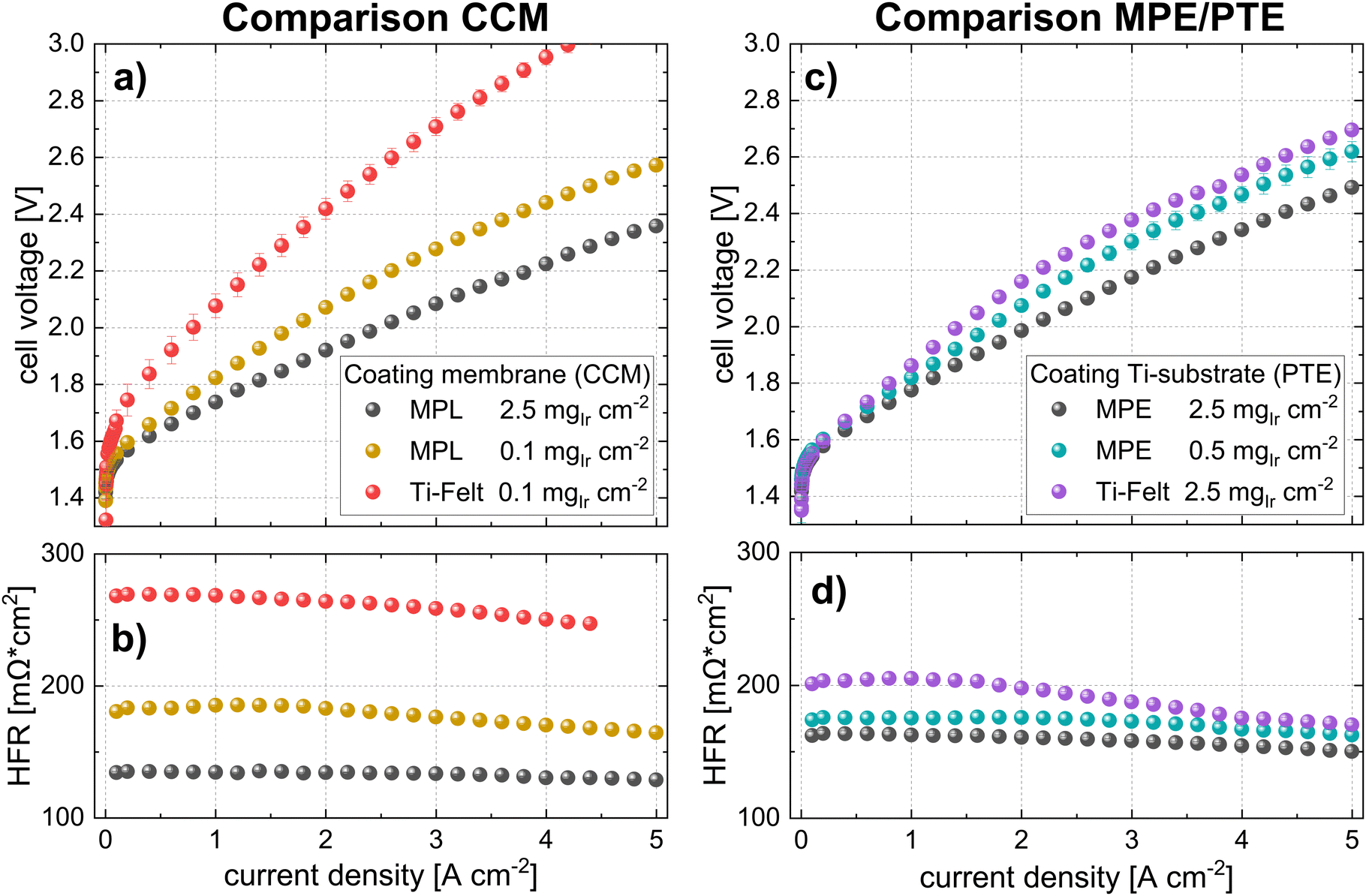 | ||
| Fig. 8 Polarization curves and HFR measurements of selected measurements with MPLs and benchmarks using commercial Ti-felt materials from Bekaert (N115, T = 80 °C, 10 bar balanced pressure) using MPLs (a) and (b) CCM configuration and (c) and (d) MPE/PTE configuration all having an equal Pt-coating of 40 nm. | ||
For comparing the MPE configurations, a cell with a PTE (commercial Bekaert Ti-felt) with a high loading of 2.5 mgIr cm−2, (PTE loading that was characterized in Section 2.1.2) was chosen for benchmarking. The polarization curves in Fig. 8c show that the cell voltage at 5 A cm−2 with a PTE was approximately 200 mV higher than that with an MPE at the same loading. In fact, even at a lower Ir loading (0.5 mgIr cm−2), the cell with the MPE still exhibited a lower voltage than the PTE at high loading (ca. 70 mV@5 A cm−2). This difference majorly originates from the lower HFR, as shown in Fig. 8d and the iR-free plot in Fig. S8 (ESI†). The black points in Fig. 8a and c show the best-performing CCM and MPE configuration with high Ir loading and Pt-coating on the MPL. From this direct comparison, we can deduce that even when using an MPL, the CCM configuration performs better than the MPE approach.
2.3. Kinetic analysis: CL utilization and mass activity
As discussed in the previous sections, part of the performance differences between PTEs and MPEs stem from discrepancies in the HFR, and major differences are also observed in the kinetics. Fig. 9 shows the semi-logarithmic plots of current density vs. iR-free voltage at low current densities (i.e., Tafel plots) for all measurements in the CCM configuration. For the plots of the cells without Pt coating, significant shifts were observed in the location of the Tafel lines and even apparent differences in the Tafel slopes as a function of Ir loading. The Tafel slopes and kinetic parameters are listed in Table 4. On the contrary, the cells with Pt coating in Fig. 9b did not exhibit differences in the slope of the lines and only a minor shift between the 0.5 and 0.1 mgIr cm−2 loadings. In Fig. 9b the comparison with the Ti-felt reference (with Pt-coating at 0.1 mgIr cm−2) is shown. For the commercial Ti-felt, we observed a larger slope and shift in the Tafel line. Notably, the first 10 points at low current densities are missing (between 1 and 10 mA cm−2). For the cell with Ti felt at low loadings, in general, a higher H2 crossover was observed which influenced part of the measurement points in the very low current density range. Hence, these were disregarded from the analysis.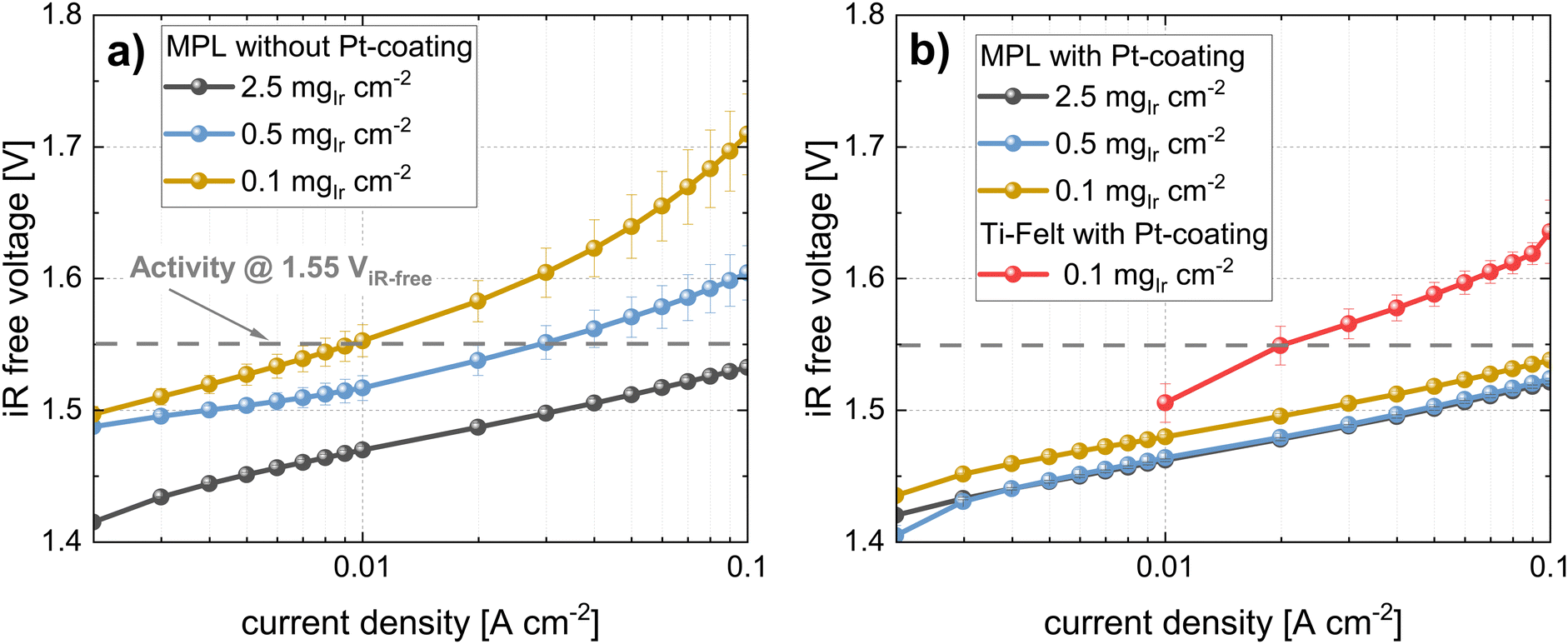 | ||
| Fig. 9 Semi-logarithmic plots of the iR-free cell voltage vs. current density (Tafel-plots) for all loadings in CCM configuration (a) MPLs without Pt-coating (b) MPLs with Pt-coating and comparison with Ti-felt reference with Pt-coating; all measurements with N115, at T = 80 °C and 10 bar balanced pressure. | ||
| CCM | MPL without Pt | MPL with Pt | Ti-felt (Pt) | ||||
|---|---|---|---|---|---|---|---|
| Ir-loading (mgIr cm−2) | 2.5 | 0.5 | 0.1 | 2.5 | 0.5 | 0.1 | 0.1 |
| Tafel slope (mV dec−1) | 62.1 ± 0.2 | 72.7 ± 2.7 | 83.4 ± 1.1 | 56.7 ± 0.5 | 56.9 ± 0.6 | 63.7 ± 1.4 | 106.0 ± 2.5 |
| j 0 (mA cm−2) | 1.32 ± 0.04 | 1.03 ± 0.53 | 1.35 ± 0.24 | 0.78 ± 0.10 | 0.75 ± 0.12 | 1.47 ± 0.42 | 21.2 ± 5.5 |
| j @1.55V (mA cm−2) | 185 | 28.9 | 9.31 | 259 | 232 | 147 | 19.8 |
| Mass activity@1.55 V (A gIr−1) | 74.1 ± 0.6 | 57.7 ± 2.3 | 93.1 ± 19.5 | 104 ± 1 | 464 ± 19 | 1470 ± 300 | 199 ± 42 |
In Table 4 all the kinetic parameters for the cells using the CCM configurations are quantified and summarized. The Tafel slopes were extracted from the measurements using a kinetic analysis tool recently provided by NREL.67 This tool allows for the unbiased and reproducible quantification of Tafel slopes and exchange current densities and extracts the Tafel slope from a different range where the linear Tafel model applies (in our cases somewhere between 1 and 200 mA cm−2 depending on the electrode). For the measurement with cells using MPLs without Pt coating, the Tafel slope values increased with decreasing loading (approximately +10 mV dec−1 per 5 times reduction in Ir-loading). This observation does not apply to the cells with an MPL coated with Pt. In this case, only negligible differences in Tafel slopes were observed between the different loadings, which is in agreement with the Tafel lines in Fig. 9b. For all the cases using MPLs, the exchange current density j0 did not change significantly within the uncertainty. Only the measurement at low loading with the Ti felt exhibited a significantly higher exchange current density. This result might be related to the significantly higher Tafel slope and may not necessarily be reliable because current densities below 10 mA cm−2 could not be evaluated.
To better understand how the available catalyst was utilized, we determined its activity at an iR-free cell voltage of 1.55 V. This voltage was selected based on previous literature.39 From the (extrapolated) Tafel lines, we determined the current density at a given iR-free voltage, which represents the activity. For measurements without Pt coating, the activity dropped drastically from ∼185 to ∼9 mA cm−2 at 1.55 V from the highest to the lowest loading. This decrease represents a reduction in activity by a factor of approximately 18. In comparison, when an MPL with a Pt coating was used, the activity drop was much lower, going from ∼260 to only ∼147 mA cm−2 for the 2.5 to 0.1 mgIr cm−2 range.
Because different loadings were used, we normalized the activity to the Ir loadings:
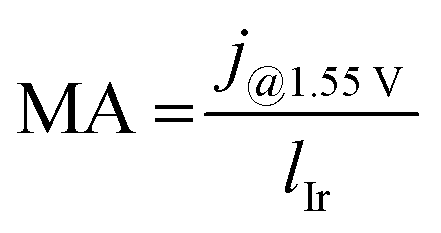 | (1) |
In Fig. 10 the Tafel plots of the cells using an MPE are given. Similarly, as for the CCM approach, the cells with the MPE without Pt coating (see Fig. 10a) exhibited increases in the Tafel slopes and shifts in the Tafel lines with decreasing loading. The shift for the low-loading cases was more pronounced than that for the CCM approach. For the Tafel plots of the cells with Pt coating (Fig. 10b), an improvement in kinetics was observed with higher activities and reduced Tafel slopes, particularly for the lower-loading cells. Fig. 10b shows a comparison with PTE (Ti felt with Pt coating) at a high loading of 2.5 mgIr cm−2. In this case, a small shift in the Tafel line was observed compared to that of the MPE with the same loading.
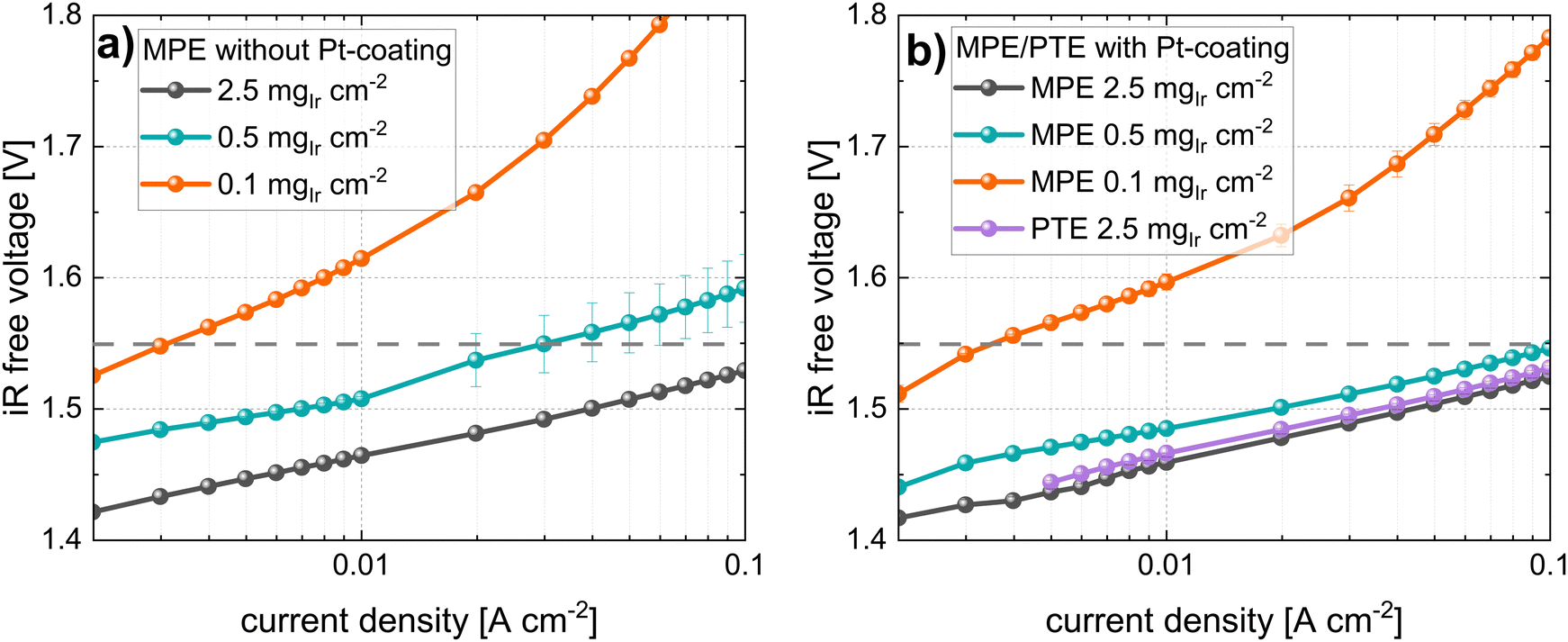 | ||
| Fig. 10 Semi-logarithmic plots of the iR-free cell voltage vs. current density (Tafel-plots) for all loadings in MPE configuration (a) MPLs without Pt-coating, (b) MPLs with Pt-coating and comparison with Ti-felt reference (PTE with Pt-coating); all measurements with N115, at T = 80 °C and 10 bar balanced pressure. | ||
All the kinetic parameters for the cells using MPE/PTEs are listed in Table 5. An apparent increase in the Tafel slope with decreasing Ir loading was again observed for the MPE without the Pt coating. This phenomenon was similar to the increase observed in the CCM configuration, although it had a larger magnitude. Again, this apparent increase in the Tafel slopes disappeared for the cell using the MPE with the Pt coating. The activity (at 1.55 ViR-free) decreased with decreasing loading in all cases, but to a lesser extent with the Pt coating. We observed that the MA decreased in cells with MPE without Pt coating. In contrast, for the MPE with the Pt coating, the MA increased with a reduction in loading. The same observation applied to the CCM approach, although the MAs were significantly lower for the MPE at the same loading. For example, the MA for the CCM with 0.1 mgIr cm−2 (with Pt) was 1470 A gIr−1, whereas that for the respective loading in MPE configuration (with Pt) exhibited only 353 A gIr−1. This effect is clearly more pronounced at low loadings, although the catalyst utilization was consistently higher for the CCM configurations.
| MPE/PTE | MPE without Pt | MPE with Pt | PTE (Pt) | ||||
|---|---|---|---|---|---|---|---|
| Ir-loading (mgIr cm−2) | 2.5 | 0.5 | 0.1 | 2.5 | 0.5 | 0.1 | 2.5 |
| Tafel slope (mV dec−1) | 62.7 ± 0.7 | 85.5 ± 2.1 | 117 ± 0.6 | 63.1 ± 0.1 | 60.1 ± 1.1 | 61.9 ± 0.6 | 72.9 ± 1.0 |
| j 0 (mA cm−2) | 1.78 ± 0.27 | 5.92 ± 1.72 | 5.78 ± 0.34 | 2.25 ± 0.56 | 0.60 ± 0.17 | 1.33 ± 0.19 | 7.21 ± 1.13 |
| j @1.55V (mA cm−2) | 190 | 30.5 | 3.14 | 226 | 113 | 35.3 | 176 |
| Mass activity (A gIr−1) | 76.4 ± 0.6 | 60.9 ± 2.4 | 31.4 ± 6.6 | 90.5 ± 0.7 | 225 ± 9 | 353 ± 74 | 70.4 ± 0.6 |
3. Discussion
The performance and catalyst utilization were dependent on the properties of the PTL/CL interface. In this regard, MPLs exhibited significant benefits for both CCM and MPE configurations, primarily attributed to their smaller pore and particle sizes and, hence, lower surface roughness and more homogenous surface contact. The “MPL boost” is especially vital at low iridium loadings. However, coating the MPL with a PGM, such as Pt, is essential for achieving superior performance and CL utilization for low Ir-loadings in the range of 0.1–0.5 mgIr cm−2. This phenomenon was true in both cases when coating the CL on the membrane and MPL substrate. PGM coatings on PTLs have recently gained attention among researchers. Herein, we show that this coating is also necessary when using MPLs, particularly for low iridium loadings. Furthermore, the primary benefits of Pt-coated MPLs can be narrowed down to two effects: (i) reduction of the HFR and (ii) drastic increase in MA. Therefore, what is the underlying mechanism that explains the advantage of MPLs at low iridium loadings, and why is a PGM coating required?3.1. Why is the MPL facilitating low iridium loadings?
To obtain a clearer picture, the structural characteristics of the CL must be considered. From our morphological analysis, we observed that, at high loadings, a continuous network of the IrO2 phase was formed, rendering large parts of the CL accessible because the in-plane connectivity was preserved. The electronic conductivity was lower in the “wet” state (refer to Section 2.1.1) and this might be even more drastic in the real cell environment.32 However, this network was highly affected by Ir loading. Furthermore, when the CL thickness was reduced (to corresponding loadings in the range of 0.1 mg cm−2) the CL was rather composed of patches of catalyst spots (refer to Section 2.1.1). Therefore, the accessibility of the pore regions at the MPL or PTL/CL interface in terms of electronic paths was highly limited. The smaller the pores and the closer they are spaced, the higher the probability of forming a continuous network between Ti-MPL/PTL particles and the CL, which is exemplified in Fig. 11. Therefore, the small pore and particle sizes of the MPLs are of significant importance, particularly at lower Ir loadings.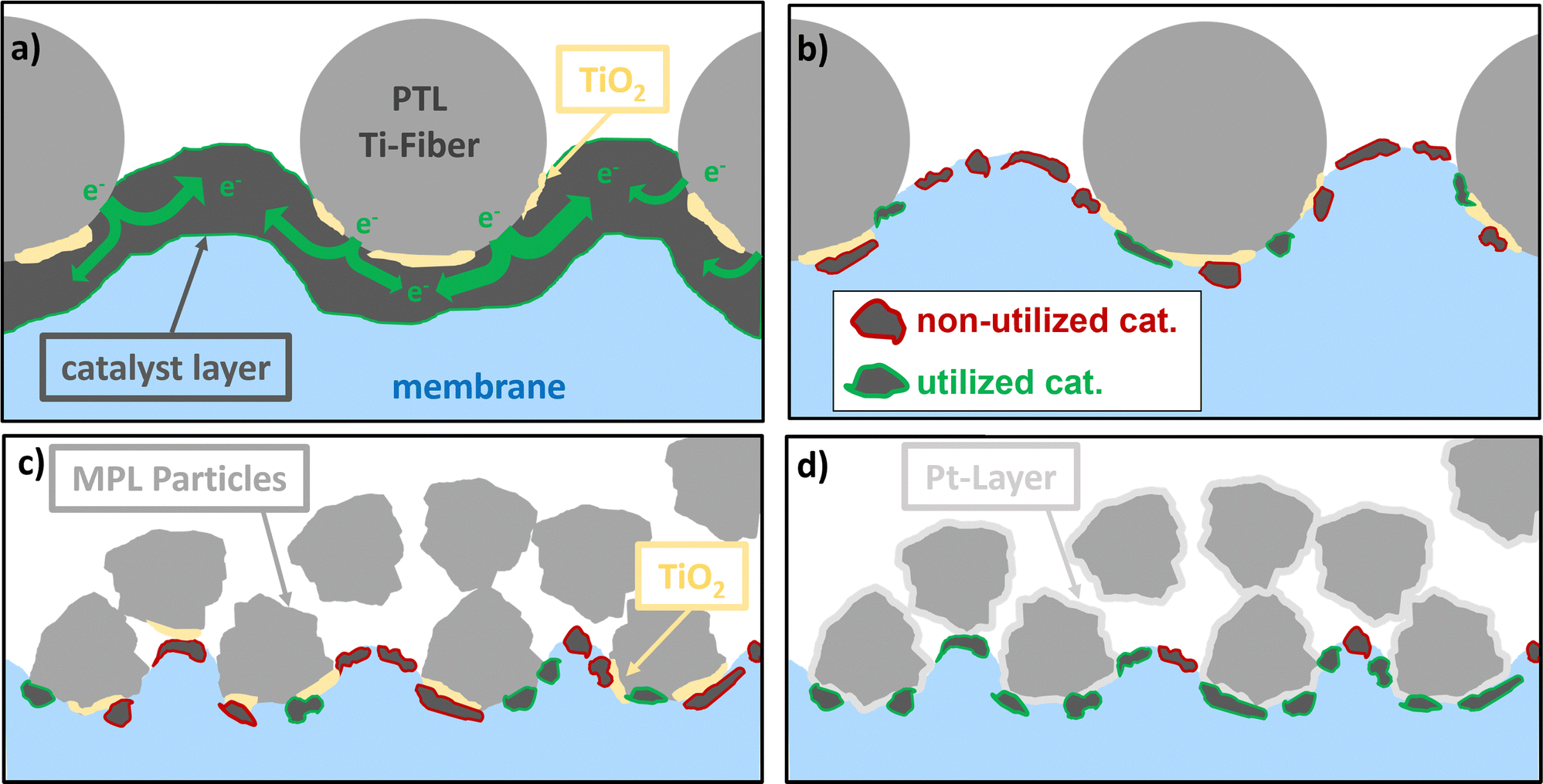 | ||
| Fig. 11 Schematic illustrations showing the influence of MPL on higher utilization of the CL at low iridium loadings in comparison to that of PTLs and the influence of Pt-coatings. (a) PTL with a CCM using high catalyst loading (b) PTL with low catalyst loadings (c) MPL without Pt with low catalyst loading, and (d) MPL with Pt-layer and low catalyst loading leading to better CL utilization. | ||
3.2. Why does it only work with Pt-coating?
A Pt coating is necessary when low Ir loadings are used, which may be attributed to the formation of a (partial) TiOx passivation layer on the MPL/PTL surface. Doo et al. suggested that these issues are associated with upward band bending in the TiOx passivation layers, which leads to a decrease in electronic conductivity.54 Furthermore, the authors pointed out that membrane intrusion through a thin and unconnected CL can affect the interface resistance. Bernt et al. showed that the combination of low electronic conductivity in the CL and poor surface conductivity of the passivation layer (PTL) leads to high resistance and poor performance.25 They also showed that, to achieve a low CL/PTL contact resistance, either a highly conducting catalyst layer or a Pt-coated PTL is required. Our results are in line with this study, showing that a highly loaded CL (good electronic conductivity) exhibits near-equal performance when coupled with either a Pt-coated or non-coated MPL (Fig. 6).Additionally, for low Ir loadings, the CL conductivity was poor owing to the disruption in the electronic percolation network. Therefore, to maintain a satisfactory performance, the addition of a Pt coating is necessary to reduce the contact resistance of the MPL/PTL. However, because several catalyst particles are still inaccessible to the electronic path (d), the resistance of the low iridium loaded catalyst layer is still higher than that of the respective high loadings.
3.3. Role of MPL when coating on the Ti-substrate
A clear improvement in terms of performance and CL utilization was observed when coating the CL on an MPL instead of on a Ti-felt. From the XTM characterization of the MPE/PTE in Section 2.1.2, these benefits are related to the more homogenous catalyst distribution on the MPL. The catalyst is located closer to the MPL surface, leading to higher catalyst utilization, as discussed in Section 2.3. However, cells using MPEs cannot match the performances achieved with equivalently loaded CCM configurations. We hypothesize that even when using an MPL, a portion of the catalyst is lost in the pore space, making it electrochemically unreachable. Although the catalyst is mostly located closer to the MPL surface, membrane intrusion may also be significantly lower when using an MPL because of its small pore size. Therefore, although the accessible catalyst is much higher than that when using PTLs, it is still lower than that for the CCM configurations. This phenomenon might be suppressed if the MPL feature sizes at the CL interface are further reduced, which should be investigated in future studies.It might be possible to improve the quality of MPE/PTE fabrication by using alternative coating techniques, such as slot die coatings68 or doctor blading. These methods involve using inks with higher viscosity, which can reduce the ink's penetration into the MPL/PTL. This, in turn, can increase the accessibility of the catalyst particles, leading to better CL utilization. Further research is needed to explore these alternative coating approaches.
3.4. Low CL utilization in thick electrodes
We have observed that for thicker electrodes with loadings of 2.5 mgIr cm−2, the mass activity is quite low, indicating poor utilization of the CL. This holds for both cases, with and without Pt-coating. It suggests that the in-plane electrical conductivity is not the main cause of the poor CL utilization in thick electrodes. We hypothesize that through-plane ionic conductivity might be a limiting factor. This leads to a potential drop and a corresponding loss in catalyst utilization from the membrane towards the PTL. This indicates an inhomogeneous CL utilization, where higher utilization is expected close to the membrane interface, which drops as the electrode thickness increases. This aligns with the estimations made by Padgett et al.48 for the assumption of a limiting ionic through plane conductivity. Additional studies are required to test this hypothesis, which will be part of future investigations.3.5. Future targets for lowering Ir-loadings
For the future generation of electrolyzers having lower catalyst loadings while maintaining high efficiencies and long-term stability is crucial. This can be accomplished by combining different approaches such as: (i) utilizing advanced multilayer PTLs with MPLs (as presented in this work) (ii) developing next-generation catalyst materials with improved morphologies and better support materials to achieve higher conductivity and CL utilization and (iii) optimizing the coating procedure to produce homogeneously distributed low-loaded electrodes. Future studies should prioritize combining different approaches to maximize CL utilization, which might result in higher-performing and stable electrodes at low catalyst loadings.4. Conclusions
In this study, we investigated the role of the CL morphology and the influence of using novel MPLs in reducing the iridium content in PEWE. To better understand the structure of the CL, we used a novel technique, known as X-ray ptychographic laminography,60 which allowed for the analysis of the CL structure in a relevant field of view in three dimensions on a nanometer scale. Furthermore, for the first time, the morphological changes induced upon contact with liquid water were characterized on this scale. A loss in the in-plane conductivity was observed, which was aggravated upon reducing the CL thickness owing to the loss of connectivity of the conducting IrO2 phase. Therefore, the in-plane distance of the electronic path should be minimized, which renders the use of MPLs beneficial. From characterizing the performance at different iridium loadings (0.1–2.5 mgIr cm−2), we deduced that the MPL allows the use of low-loaded anodic CL with minor setbacks in electrochemical performance. The mass activity increased by a factor of up to 15 when a low Ir loading was used in combination with the MPLs. However, this phenomenon is possible only when a protective Pt coating is used, which can be attributed to the poor electronic conductivity of the low-loaded CL and upward band bending of the TiOx passivation layers.54 Our best cell considering performance and Ir-loading was able to achieve a power-specific iridium loading of 0.06 gIr kW−1 at 70% cell efficiency (lower heating value), which is close to the required targets5 (although only at moderate current densities of ∼1 A cm−2). We believe that further optimization, especially of the CL structure, by adapting the catalyst morphology and fabrication process can achieve the required performance targets.We investigated the possibility of coating a CL onto a Ti-porous substrate (MPE or PTE) and observed an improvement in the performance when the CL was coated onto the MPL instead of the PTL. Furthermore, the MPL allows the production of lower-loaded CL (0.5 mgIr cm−2) with only minor losses in performance. From the XTM characterizations of the MPE/PTE, the CL is much more homogenously distributed on the MPL and closer to the transport layer surface than on the PTE. This phenomenon leads to much better CL utilization and performance, as evidenced by the electrochemical data. However, the performance of the CCM configuration was still superior to that of the MPE, although significant advancement was achieved using an MPL. We believe that this approach has remarkable potential for simplifying the PEWE cell production process and should be investigated in the future.
Overall, we can conclude that MPLs facilitate the use of CL with low Ir loadings in both configurations (CCM and MPE) using only commercially available catalyst materials and without further optimization of the fabrication process. Mass activity can be further improved by engineering the PTL/CL interface properties. Although significant areas for improvement exist in CL fabrication, using MPLs is essential for advancing future PEWE systems toward the required iridium loading targets. Therefore, new fabrication methods to further enhance MPL properties shall be explored in our future studies.
5. Experimental
5.1. Preparation of catalyst layers via ultrasonic spray coating
The spraying procedure in this study was adapted from a prior study.11 Commercial state-of-the-art catalyst materials were employed for the preparation of CLs. On the cathode Pt/C (TEC10E50E, Tanaka Kikinzoku Kogyo) was used as a catalyst. The ink was prepared by dispersing the solid catalyst in ultrapure water, ethanol, and a 5 wt% Nafion dispersion (aliphatic alcohol/water, EW = 1100 g mol−1, DuPont purchased from Sigma-Aldrich), which were added individually in that order. Between each addition, the mixtures were sonicated for 15 min for a total sonication time of 45 min. An ionomer-to-carbon weight ratio of 0.7 was used.As anodic OER catalyst, a 75 wt% IrO2 supported on TiO2 (MA-1376, Umicore) was used. The ink was prepared by dispersing the catalyst powder in a mixture of ultrapure water, 2-proponal and a 5 wt% Nafion dispersion. Each component was added individually in that specific order with a 15 min ultrasonication step in between. The ionomer content was set to 11 wt% of the total electrode weight.63
Nafion membranes (Nafion™, purchased from Quintech, Germany) were mounted in an in-house developed membrane holder with a masked active area of 2 × 2 cm2. An automated programmable benchtop coating system (ExactaCoat, SONO TEC Corporation, USA) was used for the CL preparation. A temperature of 70 °C was chosen for the heating plate. For the cathode, only the membranes were used as substrates (CCM). For the anodic CL, the substrate was either the membrane (CCM), the Ti-porous layer (MPE, PTE), or a thin Kapton foil for the PyXL scans (0.5 cm × 0.5 cm, see next section). Catalyst loading was monitored online by placing a reference check sheet with a defined dry weight in addition to the actual CL. The Pt-loading for the cathodic CL was fixed at 0.50 ± 0.05 mgPt cm−2, which was achieved by 20 spray-coating runs. The loading of the anodic CL was varied between 0.1 and 3 mgIr cm−2 (±0.02 mgIr cm−2) for which 4–120 spray-coating rounds were required. The CCM and MPE/PTEs were dried on a heating plate for 15 min before removal and dried under ambient conditions for at least 12 h before integration into the PEWE cell.
5.2. Synchrotron-based ptychographic X-ray laminography (PyXL)
The CL sample evaluated here was first scanned in a fully hydrated (i.e., wet) state, after which it was dried and scanned again. For wetting, the entire sample was immersed in deionized water for 10 min. Owing to the long scanning times (12–18 h), a sealing to prevent the evaporation of water during the scan was necessary. A second layer of Kapton foil was added to the wet sample on top of the CL. The two Kapton foils were sealed by adding a UV-curing optical adhesive (Norland NOA61, Thorlabs) at the edges of the sample. The glue was irradiated for 5 min with a UV light gun (365 nm) which cured the glue and sealed the two Kapton layers. In a preliminary experiment, we evaluated the leakage in the sample by weighing it twice in a 24-hour interval without any noticeable weight loss. The wetted CL was placed on the sample holder in the LamNI setup and scanned using PyXL (12–18 h scanning time). After completing the scan, the sample was removed from the sample holder. Water droplets were still clearly visible, indicating that the sample did not dry out during scanning.
A dry scan was conducted after the wet scan. For the dry scan, the sealing of the sample was removed with a scalpel without removing from the sample holder, to facilitate finding the identical location. The sample (CL) was then dried under vacuum for 2 h in a desiccator after which it was it was placed in the LamNI setup and scanned in the same field as that of the wetted sample. The dryness of the sample was confirmed by visual inspection under a light microscope. Furthermore, no signs of liquid water were observed on the tomogram.
All images were processed using MATLAB and the open-source software ImageJ. The data were cropped to a volume of 350 × 350 voxels × thickness (voxel size: 34.88 nm) for the analysis. Grayscale images were segmented using Fiji's Trainable Weka Segmentation tool, which is a machine learning tool that leverages a number of manual annotations and trains a classifier to segment the remaining data automatically.76 In the dry state, the four phases (i.e. IrO2, TiO2, ionomer, and void) were successfully segmented. In the wet state, the contrast between the wet ionomer and water (in the void) was insufficient, making the segmentation of these two phases unreliable. All transport parameters were determined from the segmented data using Geodict (Math2Market GmbH, Germany) with parameters similar to those reported in a previous study.38 The electrical conductivity was calculated using Geodict's ConductoDict module77 based on the tortuosity and connectivity of the relevant phase in the respective direction. Here the conductivity of the constituent materials (in our case IrO2 with a bulk conductivity of 64 S cm−1)31,66 is computed by solving one partial differential equation per direction of interest. To estimate ionic conductivity, the bulk conductivity of Nafion of 85 mS cm−1 was assumed.65 The remaining phases were assumed to be either electronic or ionic isolators.
5.3. Laboratory-based X-ray tomographic microscopy (XTM): data acquisition and image processing
X-ray tomography images were acquired using a lab-CT scanner (Phoenix Nanotom M, General Electric, Germany). Cylindrical samples with a diameter of 3 mm, prepared by wire cutting, were mounted perpendicular to the beam. The acquisition parameters were set to 120 kV and 80 μA, and a voxel cube edge length of 2 μm was chosen. We acquired 2000 projections over 360° and 1000 ms exposure times (per projection), resulting in a scan time of approximately 2 h. For all the measured PTLs, a region of interest (ROI) of 1900 × 1900 μm2 × PTL thickness was chosen from the middle of the samples to avoid edge effects. The tomographic grey-scale images were segmented either manually by selecting an appropriate thresholding value or using the trainable Weka segmentation tool from Fiji76 (for MPEs and PTEs). All images were processed using the open-source software ImageJ, transport parameters were estimated using Geodict (Math2Market GmbH, Germany), and surface renderings were prepared using ParaView software.5.4. Electrochemical performance
Two PTFE-coated fiberglass gaskets (FIBERFLON®, 1 × 130 μm and 1 × 260 μm on the cathode and 2 × 60 μm on the anode) were used for gas tightness and electric insulation. The cell included a spring mechanism that maintained a constant CCM compression (2.5 MPa) and was independent of the PTL thickness and clamping pressure (higher for sealing reasons). The cell had gold-coated flow fields with five parallel channels (1 mm depth, 20 mm length, and 2 mm width) separated by 2 mm ribs. A thermostat and thermocouple were located close to the active area in the cell control, and the cell temperature was measured during operation. DI water was recirculated at the anode end with a volume flow of 30 mL min−1 cm−2 through an integrated ion exchange bed to sustain water purity during the performance tests. An in-house developed test bench was used.79 More information related to the cell and the test bench can be found in the ref. 36, 39, 44 and 79.
Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
We thank Umicore© (Germany) for providing the anodic OER catalyst and Dupont© (France) for providing the Kapton foil samples of the required thickness. Furthermore, we would like to thank Dr Zirui Gao (Brookhaven National Laboratory) for assistance and insightful discussions during beamtime. We also extend our gratitude to Martin Amman and Thomas Gloor (PSI) for providing technical support and to Christine Klauser (PSI) for assistance with Pt-sputtering. C. A. has received funding from the EU's Horizon 2020 research and innovation program under the Marie Sklodowska-Curie grant agreement no. 884104 (PSI-FELLOW-III-3i) and from Chalmers initiative for advancement of neutron and X-ray techniques.References
- M. Carmo and D. Stolten, Science and Engineering of Hydrogen-Based Energy Technologies, Elsevier, 2019, pp. 165–199 Search PubMed.
- A. Odenweller, F. Ueckerdt, G. F. Nemet, M. Jensterle and G. Luderer, Nat. Energy, 2022, 7, 854–865 CrossRef.
- Water Electrolyzers and Fuel Cells Supply Chain Deep Dive Assessment | Department of Energy, https://www.energy.gov/eere/fuelcells/water-electrolyzers-and-fuel-cells-supply-chain-deep-dive-assessment, (accessed 26 July 2023).
- J. Kibsgaard and I. Chorkendorff, Nat. Energy, 2019, 4, 430–433 CrossRef.
- C. Minke, M. Suermann, B. Bensmann and R. Hanke-Rauschenbach, Int. J. Hydrogen Energy, 2021, 46, 23581–23590 CrossRef CAS.
- M. Clapp, C. M. Zalitis and M. Ryan, Catal. Today, 2023, 420, 114140 CrossRef CAS.
- C. Mittelsteadt, E. Sorensen and Q. Jia, Energy Fuels, 2023, 37, 12558–12569 CrossRef CAS.
- M. Möckl, M. F. Ernst, M. Kornherr, F. Allebrod, M. Bernt, J. Byrknes, C. Eickes, C. Gebauer, A. Moskovtseva and H. A. Gasteiger, J. Electrochem. Soc., 2022, 169, 064505 CrossRef.
- T. Reier, M. Oezaslan and P. Strasser, ACS Catal., 2012, 2, 1765–1772 CrossRef CAS.
- M. Escudero-Escribano, A. F. Pedersen, E. A. Paoli, R. Frydendal, D. Friebel, P. Malacrida, J. Rossmeisl, I. E. L. Stephens and I. Chorkendorff, J. Phys. Chem. B, 2018, 122, 947–955 CrossRef CAS PubMed.
- T. Schuler, T. Kimura, T. J. Schmidt and F. N. Büchi, Energy Environ. Sci., 2020, 13, 2153 RSC.
- H. N. Nong, L. J. Falling, A. Bergmann, M. Klingenhof, H. P. Tran, C. Spöri, R. Mom, J. Timoshenko, G. Zichittella, A. Knop-Gericke, S. Piccinin, J. Pérez-Ramírez, B. R. Cuenya, R. Schlögl, P. Strasser, D. Teschner and T. E. Jones, Nature, 2020, 587, 408–413 CrossRef CAS PubMed.
- M. Faustini, M. Giraud, D. Jones, J. Rozière, M. Dupont, T. R. Porter, S. Nowak, M. Bahri, O. Ersen, C. Sanchez, C. Boissière, C. Tard and J. Peron, Adv. Energy Mater., 2019, 9, 1802136 CrossRef.
- S. Chatterjee, X. Peng, S. Intikhab, G. Zeng, N. N. Kariuki, D. J. Myers, N. Danilovic and J. Snyder, Adv. Energy Mater., 2021, 2101438 CrossRef CAS.
- Z. Chen, L. Guo, L. Pan, T. Yan, Z. He, Y. Li, C. Shi, Z. Huang, X. Zhang and J. Zou, Adv. Energy Mater., 2022, 12, 2103670 CrossRef CAS.
- K.-R. Yeo, K.-S. Lee, H. Kim, J. Lee and S.-K. Kim, Energy Environ. Sci., 2022, 15, 3449–3461 RSC.
- S. Pan, H. Li, D. Liu, R. Huang, X. Pan, D. Ren, J. Li, M. Shakouri, Q. Zhang, M. Wang, C. Wei, L. Mai, B. Zhang, Y. Zhao, Z. Wang, M. Graetzel and X. Zhang, Nat. Commun., 2022, 13, 2294 CrossRef CAS PubMed.
- J. Torrero, T. Morawietz, D. García Sanchez, D. Galyamin, M. Retuerto, V. Martin-Diaconescu, S. Rojas, J. A. Alonso, A. S. Gago and K. A. Friedrich, Adv. Energy Mater., 2023, 2204169 CrossRef CAS.
- S. M. Alia, S. Stariha and R. L. Borup, J. Electrochem. Soc., 2019, 166, F1164 CrossRef CAS.
- Z. Taie, X. Peng, D. Kulkarni, I. V. Zenyuk, A. Z. Weber, C. Hagen and N. Danilovic, ACS Appl. Mater. Interfaces, 2020, 12, 52701–52712 CrossRef CAS.
- M. Bernt, A. Siebel and H. A. Gasteiger, J. Electrochem. Soc., 2018, 165, F305–F314 CrossRef CAS.
- H. N. Nong, L. Gan, E. Willinger, D. Teschner and P. Strasser, Chem. Sci., 2014, 5, 2955–2963 RSC.
- D. Böhm, M. Beetz, M. Schuster, K. Peters, A. G. Hufnagel, M. Döblinger, B. Böller, T. Bein and D. Fattakhova-Rohlfing, Adv. Funct. Mater., 2020, 30, 1906670 CrossRef.
- A. Hartig-Weiss, M. Miller, H. Beyer, A. Schmitt, A. Siebel, A. T. S. Freiberg, H. A. Gasteiger and H. A. El-Sayed, ACS Appl. Nano Mater., 2020, 3, 2185–2196 CrossRef CAS.
- M. Bernt, C. Schramm, J. Schröter, C. Gebauer, J. Byrknes, C. Eickes and H. A. Gasteiger, J. Electrochem. Soc., 2021, 168, 084513 CrossRef CAS.
- A. S. Pushkarev, I. V. Pushkareva and D. G. Bessarabov, Energy Fuels, 2022, 36, 6613–6625 CrossRef CAS.
- PEM Electrolysis: Iridium Catalysts for Electrodes Heraeus, https://www.heraeus.com/en/hpm/hmp_products_solutions/hydrogensystems/hydrogen_generation/hydrogen_generation.html? websiteCriteria = 0010, (accessed 27 July 2023).
- M. Bernt, A. Hartig-Weiß, M. F. Tovini, H. A. El-Sayed, C. Schramm, J. Schröter, C. Gebauer and H. A. Gasteiger, Chem. Ing. Tech., 2020, 92, 31–39 CrossRef CAS.
- F. Hegge, R. Moroni, P. Trinke, B. Bensmann, R. Hanke-Rauschenbach, S. Thiele and S. Vierrath, J. Power Sources, 2018, 393, 62–66 CrossRef CAS.
- J. K. Lee, T. Schuler, G. Bender, M. Sabharwal, X. Peng, A. Z. Weber and N. Danilovic, Appl. Energy, 2023, 336, 120853 CrossRef CAS.
- S. De Angelis, T. Schuler, M. Sabharwal, M. Holler, M. Guizar-Sicairos, E. Müller and F. N. Büchi, Sci. Rep., 2023, 13, 4280 CrossRef CAS PubMed.
- T. Schuler, J. M. Ciccone, B. Krentscher, F. Marone, C. Peter, T. J. Schmidt and F. N. Büchi, Adv. Energy Mater., 2020, 10, 1903216 CrossRef CAS.
- M. Suermann, K. Takanohashi, A. Lamibrac, T. J. Schmidt and F. N. Büchi, J. Electrochem. Soc., 2017, 164, F973–F980 CrossRef CAS.
- S. Stiber, H. Balzer, A. Wierhake, F. J. Wirkert, J. Roth, U. Rost, M. Brodmann, J. K. Lee, A. Bazylak, W. Waiblinger, A. S. Gago and K. A. Friedrich, Adv. Energy Mater., 2021, 2100630 CrossRef CAS.
- S. De Angelis, T. Schuler, M. A. Charalambous, F. Marone, T. J. Schmidt and F. N. Büchi, J. Mater. Chem. A, 2021, 9, 22102–22113 RSC.
- C. C. Weber, T. Schuler, R. De Bruycker, L. Gubler, F. N. Büchi and S. De Angelis, J. Power Sources Adv., 2022, 15, 100095 CrossRef CAS.
- S. Stiber, N. Sata, T. Morawietz, S. A. Ansar, T. Jahnke, J. K. Lee, A. Bazylak, A. Fallisch, A. S. Gago and K. A. Friedrich, Energy Environ. Sci., 2022, 15, 109–122 RSC.
- T. Schuler, R. De Bruycker, T. J. Schmidt and F. N. Büchi, J. Electrochem. Soc., 2019, 166, F270–F281 CrossRef CAS.
- T. Schuler, T. J. Schmidt and F. N. Büchi, J. Electrochem. Soc., 2019, 166, F555–F565 CrossRef CAS.
- M. Kroschel, A. Bonakdarpour, J. T. H. Kwan, P. Strasser and D. P. Wilkinson, Electrochim. Acta, 2019, 317, 722–736 CrossRef CAS.
- J. Lopata, Z. Kang, J. Young, G. Bender, J. W. Weidner and S. Shimpalee, J. Electrochem. Soc., 2020, 167, 064507 CrossRef CAS.
- X. Peng, P. Satjaritanun, Z. Taie, L. Wiles, A. Keane, C. Capuano, I. V. Zenyuk and N. Danilovic, Adv. Sci., 2021, 2102950 CrossRef CAS.
- Z. Kang, T. Schuler, Y. Chen, M. Wang, F.-Y. Zhang and G. Bender, Electrochim. Acta, 2022, 429, 140942 CrossRef CAS.
- C. C. Weber, J. A. Wrubel, L. Gubler, G. Bender, S. De Angelis and F. N. Büchi, ACS Appl. Mater. Interfaces, 2023, 15, 34750–34763 CrossRef CAS PubMed.
- J. A. Wrubel, Z. Kang, L. Witteman, F.-Y. Zhang, Z. Ma and G. Bender, Int. J. Hydrogen Energy, 2021, 46, 25341–25354 CrossRef CAS.
- Z. Kang, J. Mo, G. Yang, S. T. Retterer, D. A. Cullen, T. J. Toops, J. B. Green Jr, M. M. Mench and F.-Y. Zhang, Energy Environ. Sci., 2017, 10, 166–175 RSC.
- Z. Kang, J. Mo, G. Yang, Y. Li, D. A. Talley, B. Han and F.-Y. Zhang, Electrochim. Acta, 2017, 255, 405–416 CrossRef CAS.
- E. Padgett, G. Bender, A. Haug, K. Lewinski, F. Sun, H. Yu, D. A. Cullen, A. J. Steinbach and S. M. Alia, J. Electrochem. Soc., 2023, 170, 084512 CrossRef.
- M. Suermann, T. Gimpel, L. V. Bühre, W. Schade, B. Bensmann and R. Hanke-Rauschenbach, J. Mater. Chem. A, 2020, 8, 4898–4910 RSC.
- P. Lettenmeier, S. Kolb, N. Sata, A. Fallisch, L. Zielke, S. Thiele, A. S. Gago and K. A. Friedrich, Energy Environ. Sci., 2017, 10, 2521–2533 RSC.
- J. Polonský, R. Kodým, P. Vágner, M. Paidar, B. Bensmann and K. Bouzek, J. Appl. Electrochem., 2017, 47, 1137–1146 CrossRef.
- C. Liu, M. Carmo, G. Bender, A. Everwand, T. Lickert, J. L. Young, T. Smolinka, D. Stolten and W. Lehnert, Electrochem. Commun., 2018, 97, 96–99 CrossRef CAS.
- C. Liu, K. Wippermann, M. Rasinski, Y. Suo, M. Shviro, M. Carmo and W. Lehnert, ACS Appl. Mater. Interfaces, 2021, 13, 16182–16196 CrossRef CAS.
- G. Doo, J. Park, J. Park, J. Heo, J. Jung, D. W. Lee, H. Bae, J. Hyun, E. Oh, J. Kwen, K. M. Kim and H.-T. Kim, ACS Energy Lett., 2023, 8, 2214–2220 CrossRef CAS.
- M. Bierling, D. McLaughlin, B. Mayerhöfer and S. Thiele, Adv. Energy Mater., 2023, 13, 2203636 CrossRef CAS.
- J. K. Lee, G. Anderson, A. W. Tricker, F. Babbe, A. Madan, D. A. Cullen, J. D. Arregui-Mena, N. Danilovic, R. Mukundan, A. Z. Weber and X. Peng, Nat. Commun., 2023, 14, 4592 CrossRef CAS PubMed.
- M. Bühler, P. Holzapfel, D. McLaughlin and S. Thiele, J. Electrochem. Soc., 2019, 166, F1070–F1078 CrossRef.
- M. Bühler, F. Hegge, P. Holzapfel, M. Bierling, M. Suermann, S. Vierrath and S. Thiele, J. Mater. Chem. A, 2019, 7, 26984–26995 RSC.
- E. Leonard, A. D. Shum, N. Danilovic, C. Capuano, K. E. Ayers, L. M. Pant, A. Z. Weber, X. Xiao, D. Y. Parkinson and I. V. Zenyuk, Sustainable Energy Fuels, 2020, 4, 921–931 RSC.
- M. Holler, M. Odstrcil, M. Guizar-Sicairos, M. Lebugle, E. Müller, S. Finizio, G. Tinti, C. David, J. Zusman, W. Unglaub, O. Bunk, J. Raabe, A. F. J. Levi and G. Aeppli, Nat. Electron., 2019, 2, 464–470 CrossRef.
- M. Holler, M. Odstrčil, M. Guizar-Sicairos, M. Lebugle, U. Frommherz, T. Lachat, O. Bunk, J. Raabe and G. Aeppli, J. Synchrotron Radiat., 2020, 27, 730–736 CrossRef CAS PubMed.
- M. Odstrcil, M. Lebugle, T. Lachat, J. Raabe and M. Holler, J. Synchrotron Radiat., 2019, 26, 504–509 CrossRef PubMed.
- M. Bernt and H. A. Gasteiger, J. Electrochem. Soc., 2016, 163, F3179–F3189 CrossRef CAS.
- J. K. Lee, P. Kim, K. Krause, P. Shrestha, M. Balakrishnan, K. Fahy, K. Fatih, N. Shaigan, M. Ge, W.-K. Lee and A. Bazylak, Cell Rep. Phys. Sci., 2023, 4, 101232 CrossRef CAS.
- A. Kusoglu and A. Z. Weber, Chem. Rev., 2017, 117, 987–1104 CrossRef CAS PubMed.
- H. Ohno, S. Nohara, K. Kakinuma, M. Uchida and H. Uchida, Catalysts, 2019, 9, 74 CrossRef.
- A. Dizon, T. Schuler, A. Z. Weber, N. Danilovic and G. Bender, Meet. Abstr., 2022, MA2022-02, 1400 CrossRef.
- M. Stähler, A. Stähler, F. Scheepers, M. Carmo and D. Stolten, Int. J. Hydrogen Energy, 2019, 44, 7053–7058 CrossRef.
- P. Thibault, M. Dierolf, A. Menzel, O. Bunk, C. David and F. Pfeiffer, Science, 2008, 321, 379–382 CrossRef CAS PubMed.
- P. Thibault and M. Guizar-Sicairos, New J. Phys., 2012, 14, 063004 CrossRef.
- K. Wakonig, H.-C. Stadler, M. Odstrčil, E. H. R. Tsai, A. Diaz, M. Holler, I. Usov, J. Raabe, A. Menzel and M. Guizar-Sicairos, J. Appl. Crystallogr., 2020, 53, 574–586 CrossRef CAS PubMed.
- M. Guizar-Sicairos, I. Johnson, A. Diaz, M. Holler, P. Karvinen, H.-C. Stadler, R. Dinapoli, O. Bunk and A. Menzel, Opt. Express, 2014, 22, 14859 CrossRef PubMed.
- M. Odstrčil, M. Holler, J. Raabe and M. Guizar-Sicairos, Opt. Express, 2019, 27, 36637 CrossRef PubMed.
- S. Harasse, W. Yashiro and A. Momose, Opt. Express, 2011, 19, 16560 CrossRef PubMed.
- M. Van Heel and M. Schatz, J. Struct. Biol., 2005, 151, 250–262 CrossRef CAS PubMed.
- I. Arganda-Carreras, V. Kaynig, C. Rueden, K. W. Eliceiri, J. Schindelin, A. Cardona and H. Sebastian Seung, Bioinformatics, 2017, 33, 2424–2426 CrossRef CAS PubMed.
- ConductoDict in GeoDict, https://www.math2market.com/geodict-software/geodict-base-modules/simulation/conductodict.html, (accessed 18 December 2023).
- T. Bystron, M. Vesely, M. Paidar, G. Papakonstantinou, K. Sundmacher, B. Bensmann, R. Hanke-Rauschenbach and K. Bouzek, J. Appl. Electrochem., 2018, 48, 713–723 CrossRef CAS.
- M. Suermann, A. Pătru, T. J. Schmidt and F. N. Büchi, Int. J. Hydrogen Energy, 2017, 42, 12076–12086 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ey00279a |
| ‡ Present address: Technical University of Denmark, 2800 Kgs. Lyngby, Denmark. |
| This journal is © The Royal Society of Chemistry 2024 |