
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegulating electrode/electrolyte interfacial chemistry enables 4.6 V ultra-stable fast charging of commercial LiCoO2†
Anping
Zhang
af,
Zhihong
Bi
af,
Gongrui
Wang
a,
Shihao
Liao
a,
Pratteek
Das
a,
Hu
Lin
a,
Mingrun
Li
a,
Yan
Yu
 *c,
Xinliang
Feng
*bd,
Xinhe
Bao
a and
Zhong-Shuai
Wu
*ae
*c,
Xinliang
Feng
*bd,
Xinhe
Bao
a and
Zhong-Shuai
Wu
*ae
aState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 457 Zhongshan Road, Dalian 116023, China. E-mail: wuzs@dicp.ac.cn
bCenter for Advancing Electronics Dresden (cfaed), Faculty of Chemistry and Food Chemistry, Technische Universität Dresden, Dresden 01062, Germany. E-mail: xinliang.feng@tu-dresden.de
cHefei National Research Center for Physical Sciences at the Microscale, Department of Materials Science and Engineering, CAS Key Laboratory of Materials for Energy Conversion, University of Science and Technology of China, Hefei 230026, Anhui, China. E-mail: yanyumse@ustc.edu.cn
dMax Planck Institute of Microstructure Physics, Halle (Saale) 06120, Germany
eDalian National Laboratory for Clean Energy, Chinese Academy of Sciences, 457 Zhongshan Road, Dalian 116023, China
fUniversity of Chinese Academy of Sciences, 19 A Yuquan Road, Shijingshan District, Beijing 100049, China
First published on 7th March 2024
Abstract
The difficulty of achieving fast-charging high-voltage lithium-ion batteries arises from severely unstable electrode–electrolyte interfaces with sluggish kinetics. Here we overcome this challenge by developing a “cocktail electrolyte” enabling commercial LiCoO2 with ultra-stable fast-charging in a wide-temperature range. Unlike commercial carbonate electrolytes, our electrolyte synergistically contributes to fast ion transport and robust electrode/electrolyte interphases, which suppresses interfacial side reactions, accelerates interfacial reaction kinetics on the cathode side, and prevents Li-dendrites on anodes even at extremely high-rates (3C and 5C). Consequently, the Li||LiCoO2 coin cell displays ultra-high stability both at a fast-charging rate (5C, 73.2% retention after 1000 cycles) and under extreme conditions (−20 and 45 °C), far beyond the state-of-the-art electrolytes. Moreover, we show the practical and general applicability of our electrolyte through the stable operation of a graphite||LiCoO2 pouch cell (72.1% retention after 2000 cycles) and other advanced high-Ni or Co-free cathodes. This work proposes deep insights and a practical strategy for high-energy-density and fast-charging batteries.
Broader contextHigh-voltage lithium cobaltate (LiCoO2, LCO)-based lithium-ion batteries (LIBs) have always been the primary power source for portable electronics due to their high energy density. The increasing demand for commercial applications drives the need for LIBs with higher energy density, longer cycle life, and faster charging capability. However, the severely unstable electrode/electrolyte interfaces with sluggish kinetics in commercial carbonate electrolytes have become one of the major challenges limiting high-voltage fast-charging LIBs. In the present work, we demonstrate that our fluorinated “cocktail electrolyte” highly stabilizes the electrode/electrolyte interfaces on both the cathode and the anode. The synergistic cooperation of multi-component additives was validated to promote the enrichment of inorganic components (LiF and Li3PO4) with low resistance and high ion conductivity in electrode/electrolyte interfaces, thus suppressing interfacial side reactions and accelerating interfacial reaction kinetics. As a result, the Li||LCO coin cell in our cocktail electrolyte displays ultra-high stability both at a fast-charging rate of 5C over 1000 cycles and under extreme conditions (−20 and 45 °C). Moreover, our graphite||LCO pouch cell over 3800 cycles and other advanced high-Ni or Co-free cathodes demonstrate the practical and general applicability of our electrolyte. |
Introduction
Since their inception, lithium-ion batteries (LIBs) consisting of a LiCoO2 (LCO) cathode and graphite anode, have been the major power source for portable electronics.1,2 It is well known that LCO has been the best yet primary cathode material of LIBs for powering 3C electronic products due to its high output voltage and high theoretical capacity of 274 mA h g−1, which is equal to an energy density of 1070 W h kg−1. To meet future demands in 3C industry electronics of computer, communication, and consumer electronics, LIBs must achieve higher energy density, longer cycling life, faster charging capability, and wider operating temperature range.3,4 However, limited by the stable voltage window (4.4 V vs. Li+/Li) of traditional carbonate electrolytes, commercial LCO cathodes can only provide 62% (170 mA h g−1) of the theoretical capacity and volumetric/gravimetric energy density (2812 W h L−1/669.6 W h kg−1 at 4.4 V).5–7 Moreover, the applications of LCO batteries in the fields of fast-charging and wide-temperature range are still far from satisfactory.5Elevating the working cut-off voltage and charging rate is the most straightforward and efficient strategy to obtain fast-charging LIBs with high energy density.8,9 When the charge cut-off voltage is increased to 4.6 V, 80% of the total Li+ is extracted from the LCO cathode (Li1−xCoO2, x ≈ 0.8) and the specific capacity increases to 220 mA h g−1. However, the increases of the voltage and charging rate will cause the continuous dissolution of transition metals, irreversible release of reactive oxygen species, and aggravation of side reactions at electrode/electrolyte interfaces.3,10,11 There have been some reports that the insertion of Li+ into cathodes is strongly impeded by interfacial kinetics when LiPF6 salt is used, which largely depends on the choice of electrolytes.11 However, the traditional carbonate electrolytes, such as LiPF6/ethylene carbonate (EC)/diethyl carbonate (DEC), will decompose and induce the generation of gases such as COx when the voltage is higher than 4.4 V.7 These by-products, together with high-valence transition metals, will further catalyze the rapid decomposition of LiPF6 to produce corrosive HF, leading to the continuous formation of a high internal resistance cathode–electrolyte interphase (CEI) layer.7,12 Although the ionic conductivities of current liquid electrolytes (mS cm−1 level) have reached the requirement of fast-charging LIBs, the instability of the electrode/electrolyte interphase is the major bottleneck for electrolyte depletion, lithium dendrite growth, and restricted interfacial charge transfer kinetics under fast charging.13
Thus far, researchers have verified that customized high-voltage electrolytes and additives can promote the in situ formation of dense CEI layers, which can significantly mitigate cathode surface degradation and electrolyte decomposition, eventually providing high-energy-density LIBs.14–19 For instance, the inorganic-rich CEI layers derived in electrolytes can serve as robust and protective films to improve the stability of the high-voltage cathodes.12,20–23 However, the advanced electrolytes reported so far still cannot well coordinate the high energy density and fast-charging performance. With the increase of energy density, the sluggish interfacial kinetics vastly limits the simultaneous realization of the above two indicators, which is rarely discussed in currently reported high-voltage electrolytes.11,24 Thus, the development of high-voltage and fast-charging electrolytes is a key factor in achieving fast-charging batteries with high energy density, in which the coinstantaneous implementation of fast interfacial kinetics at the cathode and anode is crucial for fast-charging LIBs. However, so far, the issues related to fast-charging on the cathode side have received far less attention than those at anodes, especially the cathode/electrolyte interface, which will have negative impacts on the development of high-energy and high-power batteries.
Herein, we demonstrate a “cocktail electrolyte” based on the synergistic cooperation of multi-component additives and lithium salt, which demonstrates various merits toward high-voltage (4.6 V) fast-charging LIBs. (i) LiPO2F2 is preferentially oxidized on the cathode surface to generate a robust and fast ion transport CEI layer, thus inhibiting the interfacial side reactions and enhancing cathode interfacial kinetics. (ii) Difluoroethylene carbonate (DFEC), fluoroethylene carbonate (FEC) and LiPO2F2 synergistically promote the lithium dendrite-free anode through film formation and passivation under fast charging. (iii) Appropriate amounts of LiPO2F2 can effectively inhibit the decomposition of LiPF6 according to the “same-ion effect”, thereby improving the long-term cyclability of batteries. The “cocktail electrolyte” enables ultra-stable high-voltage (4.6 V) fast-charging of commercial LCO in Li||LCO and practical graphite||LCO cells, even over a wide temperature range (−20–45 °C). The capacity retention of the Li||LCO cell is as high as 73.2% at 5C over 1000 cycles, among the best fast-charging cyclability up to date. More importantly, the pouch-type cells of the graphite||LCO battery can maintain 72.1% capacity retention over 2000 cycles, demonstrative of practical applicability. Extendedly, the “cocktail electrolyte” also exhibits unexpected generalization in other high-voltage Ni-rich and Co-free cathodes.
Results and discussion
Design principle and properties of the “cocktail electrolyte”
Compared with traditional carbonate electrolytes, fluorinated solvents and additives can effectively improve the antioxidant activity, low-temperature performance, flame retardancy, and wettability of the electrolyte.12,25,26 As shown in Fig. 1a, methyl 2,2,2-trifluoroethyl carbonate (FEMC) is chosen as a fluorinated solvent to endow the high-voltage LCO with stable operation and it exhibits considerable passivation ability for the cathode surface.27 DFEC displays excellent solid electrolyte interphase (SEI) formation ability on the anode benefiting from the lowest unoccupied molecular orbital (LUMO) energy among the solvents of the fluorinated electrolyte.28 Compared with DFEC and FEMC, FEC can effectively eliminate the detrimental decomposition of FEMC and Li+ solvated products on the surface of lithium metal due to its stronger solvation ability.29,30 Thus, the synergistic effect of FEC and DFEC ensures the excellent passivation of the anode, which will be discussed in detail later. To further strengthen the robustness of the CEI layer on the high-voltage cathode and the SEI layer on the anode, LiPO2F2 with high HOMO (highest occupied molecular orbital) and low LUMO energy levels is screened out. As a typical film-forming additive, LiPO2F2 can form a thin and dense interfacial film (Fig. 1a and b), which exhibits high ionic conductivity and low impedance.22,31,32 Moreover, LiPO2F2 can suppress the detrimental decomposition of LiPF6 through the “same-ion effect” between LiPO2F2 and LiPF6, thereby reducing the production of the corrosive species (HF).20,22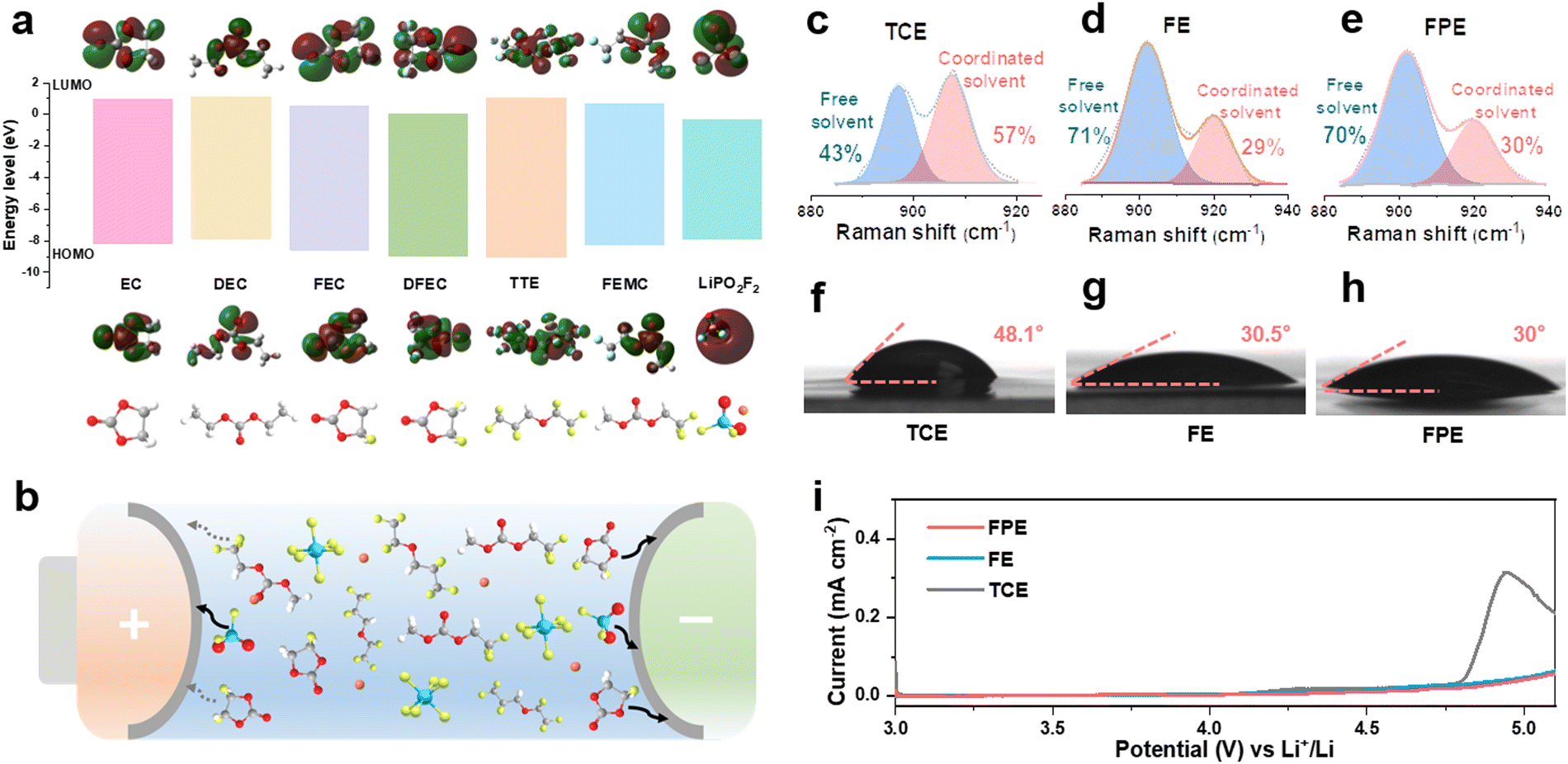 | ||
| Fig. 1 Design principle and properties of fluorinated solvents and functional film-forming additives in high voltage FPE electrolyte. (a) LUMO/HOMO energy levels of different solvents and additives in electrolytes. (b) Schematic diagram of the role of each component in stabilizing the interface of the LCO cathode and anode. (c)–(e) Fitted Raman spectra of (c) TCE, (d) FE and (e) FPE. (f)–(h) Contact angles of (f) TCE, (g) FE, and (h) FPE with the separator. (i) LSV curves of Li||LCO cells in these three electrolytes. | ||
Given that fluorination could weaken the interaction between solvents and Li+, the solvation structures of electrolytes are analyzed.33 As displayed in Fig. S1, ESI,† there are fewer negative charges around O atoms in FEC, DFEC, and FEMC than in EC and DEC, illustrating that their bindings to Li+ ions are relatively weaker, as further confirmed by the Raman spectra (Fig. 1c–e).34–36 In the commercial carbonate electrolyte (1 M LiPF6 in EC/DEC, denoted as TCE), the proportion of Li+-solvent interaction is as high as 57%, while it is only 29% in the designed fluorinated electrolyte (1 M LiPF6 in FEMC/DFEC/FEC/TTE (1,1,2,2-tetrafluoroethyl-2,2,3,3-tetrafluoropropylether), denoted as FE) according to the fitted Raman spectra. The presence of slight LiPO2F2 additives in the electrolyte (1 M LiPF6 in FEMC/DFEC/FEC/TTE + 0.02 M LiPO2F2, denoted as FPE) has no negative effect on the solvation structure. The weak coordination between Li+ and fluoride solvents plays a key role in accelerating Li+ desolvation, reducing the interface resistance, and improving the fast-charging and cycling performance, especially at low temperatures.
Fluorinated reagents, particularly the TTE component (commonly utilized as the diluent), ensure good wettability on the separators and electrodes.12 As shown in Fig. 1f–h and Fig. S2, ESI,† the interfacial contact between the electrolyte and the separator is markedly meliorative in FE and FPE, as the contact angle decreases from 48.1° (TCE) to 30.5° (FE) and 30° (FPE), respectively. The high wettability ensures that Li ions can quickly and evenly pass through the separator, particularly at high rates, which is beneficial to eliminate local polarization, reduce internal resistance, and inhibit the growth of lithium dendrites.37 Although fluorination slightly decreases the ionic conductivity of the electrolyte, the value of 5.77 mS cm−1 still makes FPE an ideal choice for a fast-charging electrolyte (Fig. S3a, ESI†). To verify the theoretical calculation results of molecular orbitals, linear sweep voltammetry (LSV) was used with the Li||Al (coated with Carbon ECP600JD and poly(vinylidene fluoride)) configuration at 0.1 mV s−1. As demonstrated in Fig. 1i, the onset of oxidation in FPE is above 5 V, while the current in TCE increases sharply at around 4.6 V. The excellent oxidation resistance is crucial for stably operating LCO cathodes at high voltage.
Electrochemical performance of 4.6 V LCO
Electrochemical performances of Li||LCO cells in the FPE, TCE, and FE electrolytes were firstly evaluated at a high charge cut-off voltage of 4.6 V. Floating currents of batteries under the constant potential of 4.6 V with time reveals that the static leakage current of the battery in FPE is much lower than that in TCE (Fig. S3b, ESI†), indicating that the side reactions between the cathode and the organic electrolyte are greatly suppressed.38 The selected charge–discharge curves of LCO cathodes in FPE, TCE, and FE are displayed in Fig. 2a and b and Fig. S4, ESI.† Compared with that in TCE and FE, the polarization degree of the LCO cathode in FPE is much smaller and the plateau voltage capacity decay decreases little at high voltage. Furthermore, it is displayed that the initial coulombic efficiency is greatly improved from 79.8% in TCE to 90.5% in FPE (Table S1, ESI†). Moreover, with an optimal content of LiPO2F2 additive (at 0.02 M, Fig. 2c and Fig. S5, ESI†), the LCO cathode achieves remarkable cyclability with 84.1% capacity retention after 500 cycles at 1C.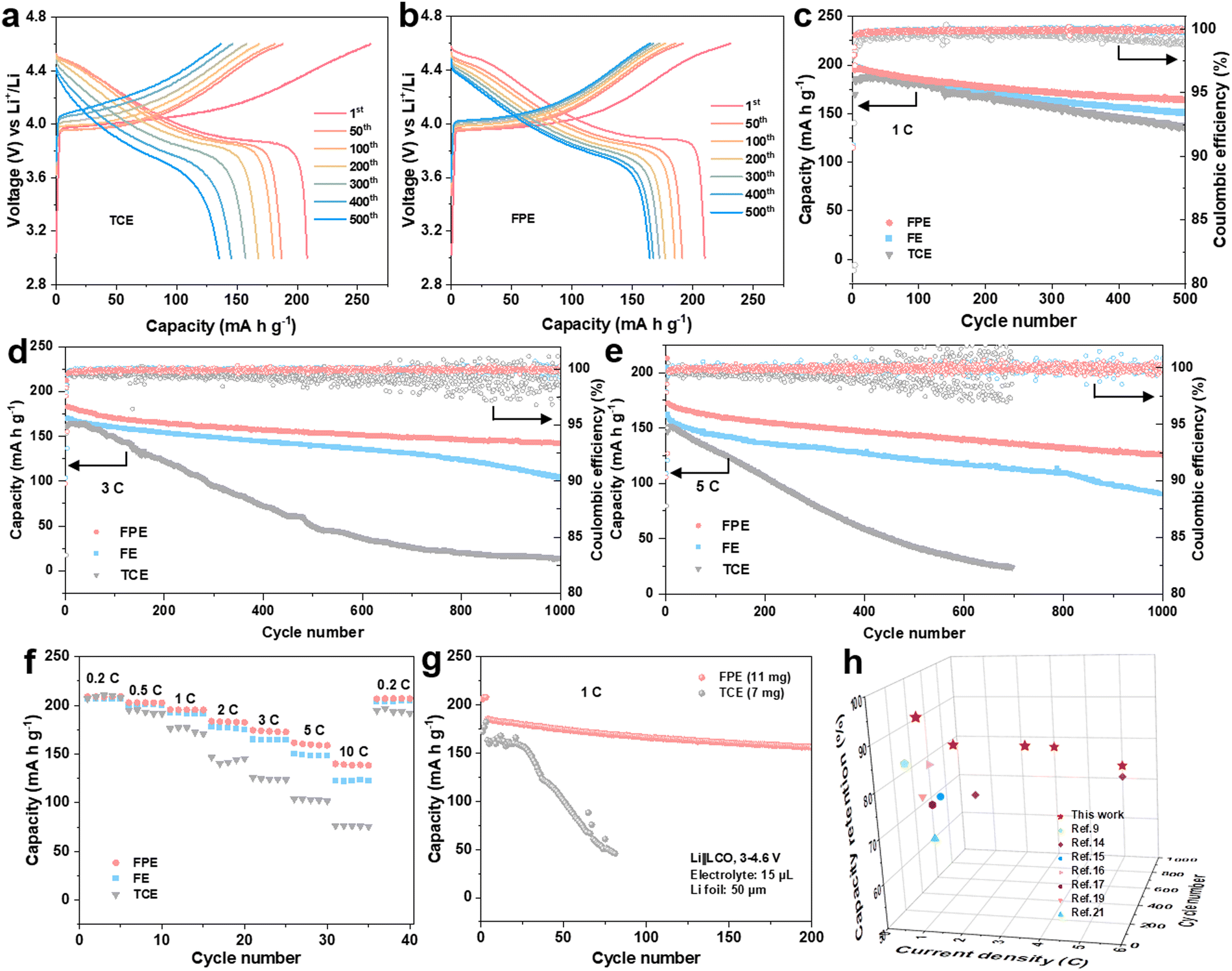 | ||
| Fig. 2 Electrochemical performance of a 4.6 V LCO cathode at 3–4.6 V (vs. Li+/Li) in FPE. (a) and (b) Charge–discharge voltage profiles of Li||LCO cell at 1C (1C = 274 mA g−1) in (a) TCE and (b) FPE. (c)–(e) Cycling stabilities at (c) 1C, (d) 3C fast charging/discharging, and (e) 5C superfast charging/discharging performance of Li||LCO cells in three electrolytes. (f) Rate capability of Li||LCO cells in three electrolytes. (g) High-voltage Li||LCO full cells in practical conditions (thick cathode of 10 mg cm−2, thin Li anode of 50 μm, and lean electrolyte of 15 μL) in TCE and FPE. (h) Comparison of cycling stability and fast-charging performance of 4.6 V LCO cathodes with reported LCO works in advanced electrolytes. | ||
Besides the improved cyclability, the robust electrode/electrolyte interface film, together with synergistic effects among components in FPE, also plays a vital role in enhancing the fast-charging performance of 4.6 V LCO. As illustrated in Fig. 2d and Fig. S6, ESI,† the capacity retention in FPE is as high as 77.6% and exhibits a reversible capacity of 142 mA h g−1 after 1000 cycles at 3C, which is much higher than that in TCE (only 14 mA h g−1 after 1000 cycles with 8.9% capacity retention). Moreover, LCO in FPE exhibits superfast charging performance at 5C (Fig. 2e), in which capacity retention reaches up to 73.2%. Median charge/discharge voltages presented in Fig. S7, ESI,† confirm a more stable voltage platform and lower polarization achieved in FPE than in TCE.
Moreover, the LCO cathode offers superior rate performance in FPE, and high average capacities of 160 and 140 mA h g−1 can be delivered even at ultrahigh rates of 5C and 10C, respectively (Fig. 2f). In sharp contrast, LCO in TCE displays a worse rate capability with capacities of only 103 and 76 mA h g−1 at 5C and 10C, respectively. Furthermore, the Li||LCO cells were also assembled, with a thick cathode (∼10 mg cm−2), thin Li (50 μm), and a lean electrolyte (15 μL) (Fig. 2g). The cell in FPE achieves superior cycling performance under such harsh conditions, preserving a high capacity of 156 mA h g−1 at the 200th cycle with 84.3% capacity retention, much higher than that in TCE (28.5% after 80 cycles). It should be emphasized that the fast-charging capability and cycling stability of commercial LCO working in our proposed FPE are amongst the best-reported electrolyte systems for 4.6 V LCO (Fig. 2h and Table S2, ESI†).
Interfacial kinetics and wide-temperature performance
To examine the electrochemical behavior of the LCO cathode in the FPE, TCE, and FE, the selected differential capacity (dQ/dV) and cyclic voltammetry (CV) curves of corresponding Li||LCO cells are compared (Fig. 3a and b and Fig. S8, S9, ESI†). The redox peaks in FPE are still clearly visible after cycling compared to the obvious decay in FE and TCE, demonstrating that the structure and interface of LCO are well encapsulated and maintained, which is attributed to the robust and dense CEI layer formed, promoting stable reaction kinetics of lithium insertion/extraction.39 The kinetics of Li+ transport in LCO was examined through electrochemical impedance spectroscopy (EIS) after 500 cycles at open circuit voltage (Fig. 3c). It is calculated that the CEI resistance (RCEI) and charge transfer resistance (Rct) in FPE (5.8 and 5.1 Ω) are lower than those in FE (9.7 and 7.3Ω) and TCE (7.9 and 15.2 Ω), implying the significantly improved interfacial charge-transfer capability of the CEI layer derived from the film-forming additive LiPO2F2 in FPE.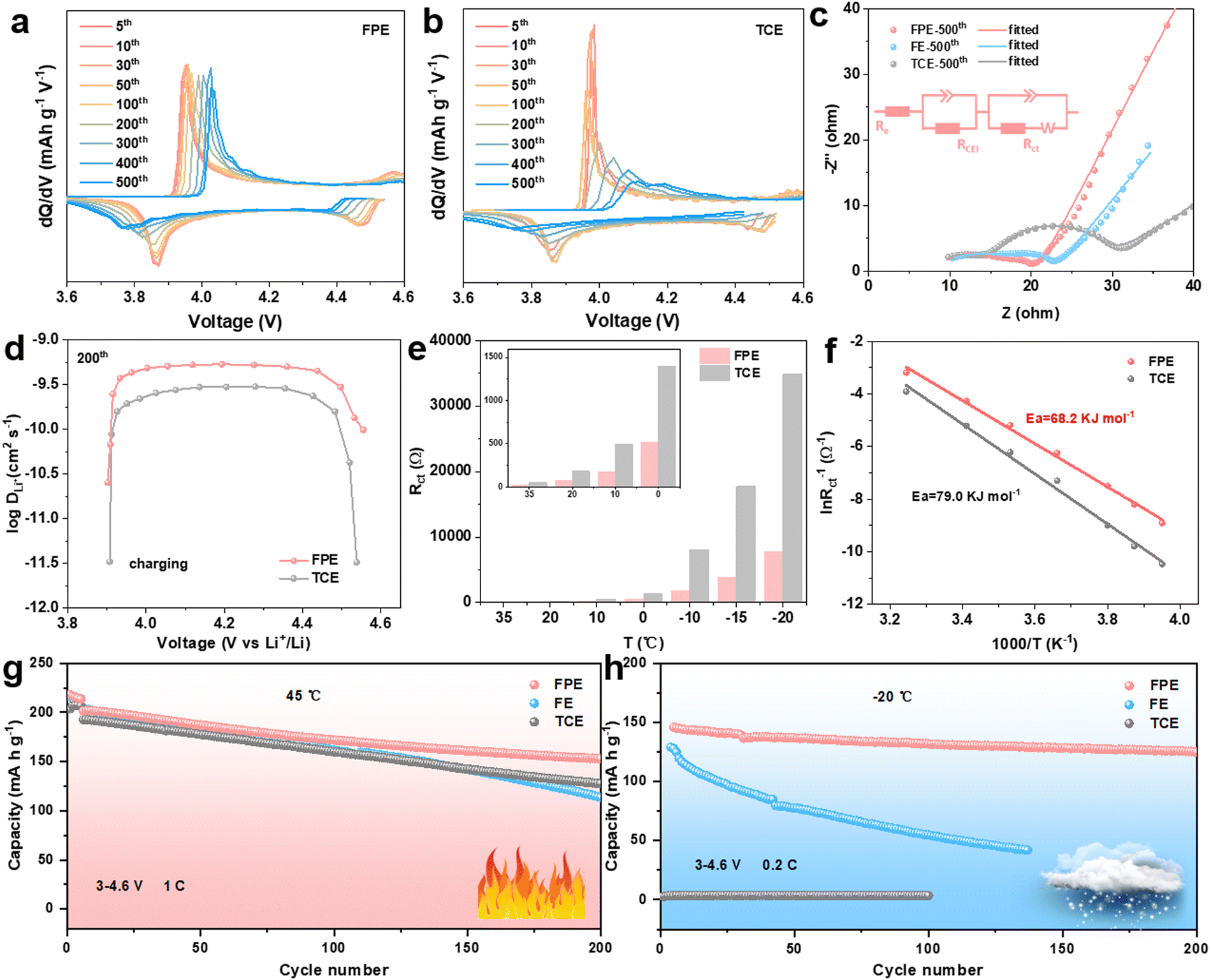 | ||
| Fig. 3 Interfacial kinetics near the 4.6 V LCO cathode/electrolyte interface and wide-temperature electrochemical performance. (a) and (b) The dQ/dV curves of Li||LCO cells at the selected cycles in (a) FPE and (b) TCE at 3–4.6 V (vs. Li+/Li). (c) EIS spectra of Li||LCO cells cycled in different electrolytes after 500 cycles. (d) The calculated lithium-ion diffusion coefficients according to the GITT during the 200th charging process in two electrolytes. (e) The Rct values and (f) Arrhenius plots for Rct of LCO in two electrolytes at different temperatures at open circuit voltage. (g) and (h) Cycling stabilities of Li||LCO cells in the voltage range of 3–4.6 V (vs. Li+/Li) at (g) 45 °C at 1C and (h) −20 °C at 0.2C. | ||
The galvanostatic intermittent titration technique (GITT) and CV measurements were performed to gain further insight into the remarkable fast-charging kinetic behavior of 4.6 V LCO (Fig. 3d and Fig. S10, S11, ESI†).18 It is worth noting that the lithium-ion diffusion coefficient (DLi+) value of LCO in FPE at the 6th cycle calculated from the GITT is larger than that in TCE, and then the DLi+ value is well-maintained in FPE (about 5 × 10−10 cm2 s−1) at the 200th cycle, while it decreases dramatically in TCE (about 2 × 10−10 cm2 s−1). The results calculated by CV are consistent with those of the GITT. Consequently, the LCO cathode in FPE exhibits much higher Li+ diffusion coefficients than that in TCE, which confirms that the CEI layer formed in FPE is beneficial for alleviating the external surface structural transition of the LCO cathode, thereby maintaining the excellent diffusion kinetics and enabling more reversible and faster Li+ insertion/extraction during cycling.10
In view of the dramatic changes in the interfacial chemistry of CEI films near the LCO cathode, it is crucial to elucidate the role of different electrolyte-derived CEI in interfacial kinetics.40 Temperature-dependent EIS measurements were carried out from −20 °C to 35 °C (Fig. 3e and Fig. S12, ESI†). Arrhenius formula was utilized to fit and analyse Rct, and the activation energy of the charge transfer step of the LCO cathode in FPE (68.2 kJ mol−1) is about 10.8 kJ mol−1 lower than that in TCE (79.0 kJ mol−1) (Fig. 3f and Fig. S13, ESI†). The rapid interface kinetics not only facilitates room-temperature fast-charging performance, but is also critical for low-temperature operation.20,41–44 As shown in Fig. 3h and Fig. S14, ESI,† the Li||LCO cell in TCE could hardly operate at −20 °C, mainly due to the solidification of the liquid electrolyte according to the sharp exothermic peaks during cooling (Fig. S15, ESI†), resulting in the blockage of the transmission of Li+ in the electrolyte. Fluorination can lower the freezing point of solvents and accelerate Li+ desolvation, thus ensuring the operation of cells in FPE and FE at low temperatures. Benefitting from the synergy between multiple components in the “cocktail electrolyte”, the cell with faster interfacial kinetics (Fig. S16, ESI†) displays much higher cycling stability in FPE (90.4%) than that in FE (42.8%) after 100 cycles, and the capacity retention in FPE is as high as 85.6% (equal to 125 mA h g−1) after 200 cycles. In addition, the LCO in FPE displays greatly improved cyclability at a high temperature of 45 °C, offering a capacity retention of up to 76% (Fig. 3g).
Structure and phase investigations
The structural evolution and reversibility of the LCO cathode during the lithiation/de-lithiation process in FPE, TCE, and FE were examined to uncover the underlying mechanisms. As revealed from ex situ X-ray powder diffraction (XRD) tests (Fig. 4a and h), similar shifts occur at the (003) peak in both FPE and TCE. Specifically, slight initial shifts to low angles (phase transition from H2 to M1 at 3.9 V, M1 to H3 at 4.1 V) and then abrupt shifts to high angles during charging (phase transition from O3 to H1–3 at 4.5 V), and vice versa upon discharging.45 It is worth noting that the (003) peak is able to return to the original position after discharging to 3 V in FPE but could not do so in TCE. The different behavior of the (003) peak in these two electrolytes during cycling further illustrates that the FPE-derived CEI can effectively mitigate the electrochemically induced mechanical degradation and thus maintain the structural integrity of the cathode during cycling at high voltage (Fig. S17, ESI†).46,47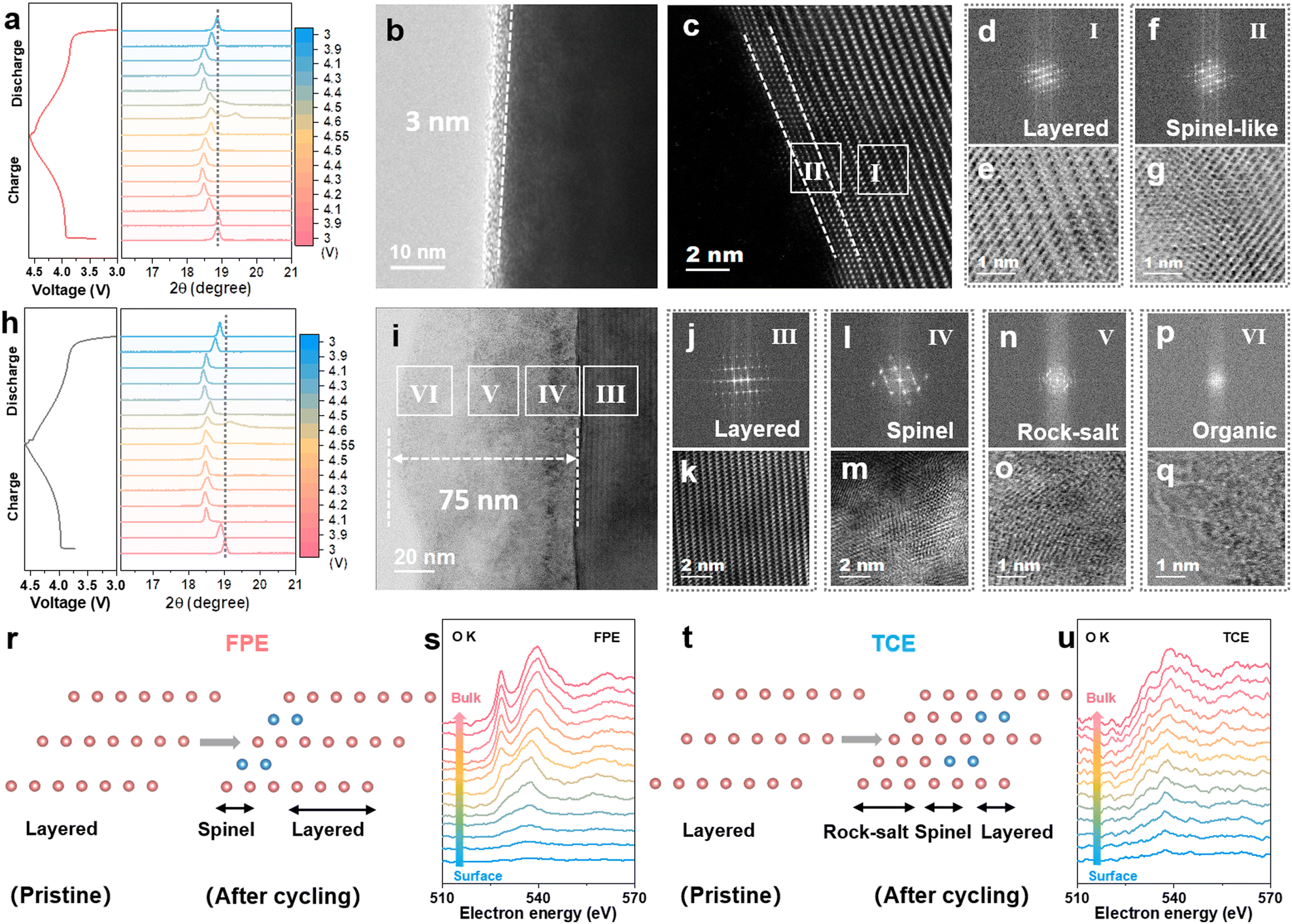 | ||
| Fig. 4 Characterization of the CEI layer and surface evolution of the 4.6 V LCO cathode during the charge/discharge processes. (a) Selected region of the (003) plane from the ex situ XRD patterns of LCO electrode cycled in FPE during the 4th charging–discharging process at 0.2C. (b) HRTEM and (c) high angle annular dark field scanning transmission electron microscope images of 4.6 V LCO cathode particles after 500 cycles in FPE. (d) The fast Fourier transform (FFT) and (e) inverse fast Fourier transform (IFFT) patterns of selected regions marked (I). (f) The FFT and (g) IFFT patterns of selected regions marked (II). (h) Selected region of the (003) plane from the ex situ XRD patterns of LCO electrode cycled in TCE during the 4th charging–discharging process at 0.2C. (i) HRTEM image of 4.6 V LCO cathode particles after 500 cycles in TCE. (j) The FFT and (k) IFFT patterns of selected regions marked (III). (l) The FFT and (m) IFFT patterns of selected regions marked (IV). (n) The FFT and (o) IFFT patterns of selected regions marked (V). (p) The FFT and (q) IFFT patterns of selected regions marked (VI). (r) Schematic diagram of atomic configuration evolution and (s) EELS spectra of O K-edge from the surface to the inner bulk of the cycled LCO cathodes in FPE. (t) Schematic diagram of atomic configuration evolution and (u) EELS spectra of O K-edge from the surface to the inner bulk of the cycled LCO cathodes in TCE. | ||
Afterwards, the electrodes post 500 cycles were characterized through high-resolution transmission electron microscopy (HRTEM). Clearly, compared with the discontinuous CEI on the surface of LCO after cycling in FE (Fig. S18, ESI†), it is observed that a thin and uniform CEI layer (about 3 nm) is formed, derived from the film-forming additive LiPO2F2 in FPE (Fig. 4b), while a thick and heterogeneous CEI and reconstruction layer with a thickness of 75 nm is observed in TCE due to the decomposition of the carbonate electrolyte at high voltage (Fig. 4i). Although the spinel phase (region II) appeared on the near-surface lattice of cycled LCO in FPE, its thickness is less than 2 nm and it did not convert further into the rock-salt phase (Fig. 4c–g and r). There is a significant difference in TCE, in which there is a severe spinel phase (region IV, Fig. 4l and m), rock-salt phase (region V, Fig. 4n and o), and disordered area (region VI, Fig. 4p and q). Large numbers of voids in the cycled LCO in TCE reflect the grievous transition metal loss and oxygen release (Fig. S19, ESI†).48,49
Electron energy loss spectroscopy (EELS) was utilized to further understand the mechanism of cathode/electrolyte interphase decay and phase transition of the high-voltage LCO cathode.50 A series of O K- and Co L-edge peaks are obtained from the surface to the bulk phase of different LCOs (10 nm away from the surface) at an interval of 1 nm per scan (Fig. 4s, u and Fig. S20, ESI†). The O K-edge is associated with the valence states of the Co according to the amount of oxygen loss.32 In contrast to the clearly visible O K-edge peaks of the LCO cathode cycled in FPE, the nearly disappearing O K-edge peaks at 528 eV in TCE are attributed to the phase transition from the layered to the rock-salt/spinel structure. Coincidentally, the intensities of Co L-edge peaks in FPE are higher than that in TCE, especially at the surface region. It is further demonstrated that the dense CEI layer derived in FPE can effectively isolate the electrode from the electrolyte and protect the LCO cathode surface from corrosion by HF, thereby inhibiting the irreversible phase transition caused by oxygen loss and transition metal ion valence decline at high voltage.50,51
More morphological and structural details of the cycled LCO cathode were determined by scanning electron microscopy (SEM). As displayed in Fig. S21, ESI,† the surface of the LCO cathode is smooth and crack-free even after 500 cycles in FPE (3C, 3–4.6 V), while the cracks arising from the mechanical stress during the fast-charging process are visible in FE and TCE.18,52 Furthermore, Raman spectra show the deep structural difference of 4.6 V LCO (Fig. S22, ESI†), in which two characteristic peaks at 488 and 596.9 cm−1 are assigned to the variation of O–Co–O bending (Eg) and Co–O stretching (A1g) of LCO, respectively.45,53 With the increase in the number of cycles, the Eg and A1g peaks of LCO in TCE exhibit serious attenuation, while the two peaks in FPE maintain satisfactory reversibility, demonstrating that the surface oxygen loss was significantly suppressed.
Fast charging will aggravate polarization and lead to the precipitation of Li-dendrites on the anode surface, resulting in short-circuiting and safety hazards. Li||Cu cells were assembled to gain insight into the compatibility of electrolytes with the anode (Fig. S23, ESI†). The cell cycle in FPE displays highly reversible Li plating/stripping kinetics with more stable coulombic efficiencies and lower polarization than that in TCE, which is attributed to the robust and low-resistance SEI film formed on the Li metal anode in FPE.15,38 The synergistic effect of DFEC and FEC in FPE for the passivation of the anode can be verified by the smooth and Li dendrite-free anode morphology after cycling in Li||Cu cells (Fig. S24, ESI†). Furthermore, the cycled Li||LCO cells were disassembled (Fig. S25 and S26, ESI†), and the smooth Li anode surface and clean diaphragm indicate that the FPE-derived SEI effectively suppresses the over-growth of lithium dendrites at high current densities, improving the safety and reliability of LIBs.43,54,55
Interfacial chemistry
To identify the interfacial chemistry, the components of CEI layers formed on the LCO after 500 cycles in Li||LCO cells were systematically evaluated. The F signals in CEI layers on the surface of LCO were characterized by XPS depth profiling with continuous Ar-ion sputtering from the CEI surface to the bulk. A strong LiF peak (686 eV) is observed, and the signal persisted during the whole sputtering process (Fig. 5a), demonstrating the abundant and stable LiF composition of the thin CEI layer in FPE. While in TCE and FE, the inorganic component LiF is much lower than organic fluorides (CFx and LixPFyOz) generated by the decomposition of LiPF6 and carbonate solvents (Fig. 5b and Fig. S27, S28, ESI†).12,27,56 In P 2p XPS spectra (Fig. 5c and f), two peaks at 135.8 eV and 134.3 eV are assigned to LixPFyOz and Li3PO4, respectively, which are the decomposition products of LiPF6 and LiPO2F2 (Fig. S29, ESI†).20,32,51 Notably, LiPO2F2 can effectively suppress the consumption and decomposition of LiPF6 according to “same-ion effect”.20,22 Moreover, the SEI characterization of the Li anode reveals a LiF- and Li3PO4-rich layer after only 4 cycles in FPE (Fig. S30, ESI†), facilitating the formation of kinetically matched and fast interfacial interphases at both the cathode and the anode in 4.6 V LCO batteries.22,57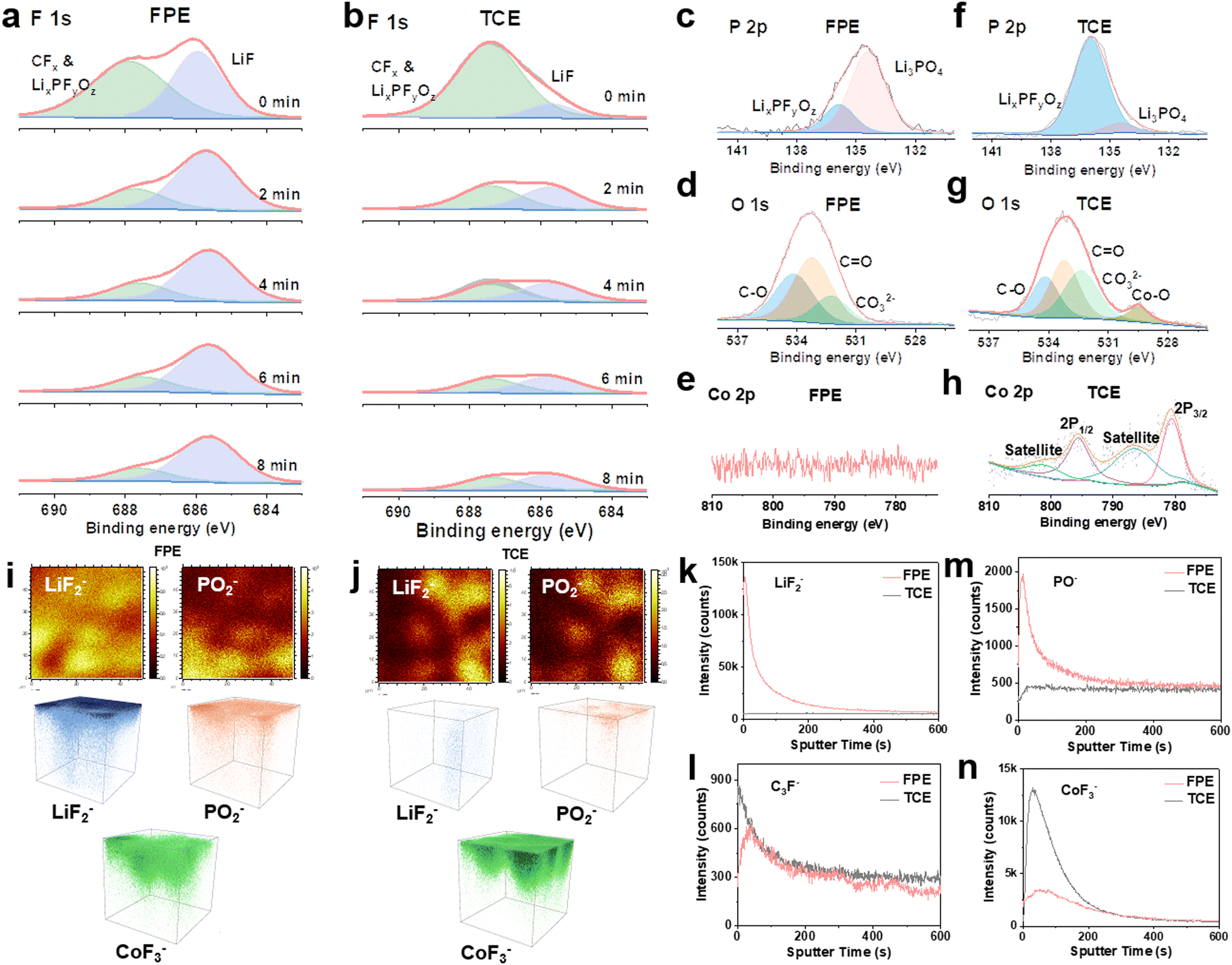 | ||
| Fig. 5 Interfacial chemistry between the 4.6 V LCO cathode and the electrolyte. (a) and (b) XPS depth profiles of F 1s of CEI layers formed on the LCO surface in (a) FPE and (b) TCE after 500 cycles. (c) P 2p, (d) O 1s, and (e) Co 2p XPS spectra of CEI layers formed on the LCO surface in FPE after 500 cycles. (f) P 2p, (g) O 1s, and (h) Co 2p XPS spectra of CEI layers formed on the LCO surface in TCE after 500 cycles. (i) and (j) TOF-SIMS 2D and 3D mapping images of several representative secondary ion fragments obtained from LCO cathodes after cycling in (i) FPE and (j) TCE. (k)–(n) TOF-SIMS depth profiles of (k) LiF2−, (l) C3F−, (m) PO−, and (n) CoF3− species in the CEI layer from the LCO cathode after cycling in two electrolytes. | ||
In addition, the high peak of CO32− (∼532 eV) in O 1s XPS spectra in TCE (Fig. 5d and g) is the result of the combination of reoxidation of alkyl lithium carbonate and poor hydrolysis stability, leading to the impedance increase of batteries.27,57 The obvious signal of metal oxide (Co–O) is observed in TCE (Fig. 5h), which is attributed to the attack on the LCO cathode by HF and POF3 produced by the decomposition of LiFP6 and carbonate solvents, resulting in Co dissolution and subsequent attachment to the electrode surface.31 In contrast, there is almost no Co–O signal in FPE (Fig. 5e), indicating that the robust CEI film can effectively stabilize the cathode structure.
To further explore the components and distribution of the CEI films formed after 500 cycles, time of flight secondary ion mass spectrometry (TOF-SIMS) was employed. Apparently, LiF2−/F− (representing LiF) and PO−/PO2− (representing Li3PO4) are enriched in the thin CEI layer on the LCO cathode in FPE, consistent with the XPS results. This can be further confirmed by TOF-SIMS 2D and 3D mapping images (Fig. 5i–k, m and Fig. S31, ESI†).31,58,59 The organic species C3F− mainly comes from the oxidative decomposition of carbonate solvents, and the lower intensity in FPE confirms that the oxidative decomposition of electrolyte solvents is suppressed (Fig. 5l and Fig. S32, ESI†). Through the significantly reduced CoF3− signal in FPE (Fig. 5n and Fig. S32, ESI†), it is reflected that Co dissolution is inhibited, which demonstrates that the passivated LCO electrode interface by the dense CEI layer could be against acid leaching.60,61
Working mechanism, practical applications and generalization
Given the above experimental and theoretical results, we generalize the comprehensive relationships between the CEI morphologies and components, interface side reactions, high-voltage cyclability and fast-charging capability (Fig. 6a). On the one hand, a thin, dense and fast ion transport CEI is derived from FPE, which inhibits the escape of Co/O elements and interface side reactions, and accelerates interfacial reaction kinetics. On the other hand, the “cocktail electrolyte” also enables the lithium dendrite-free anodes even at a fast-charging rate of 5C. Benefitting from the synergy between multiple components in the “cocktail electrolyte”, the LCO battery exhibits fast-charging capability and outstanding long-term stability even at harsh temperatures.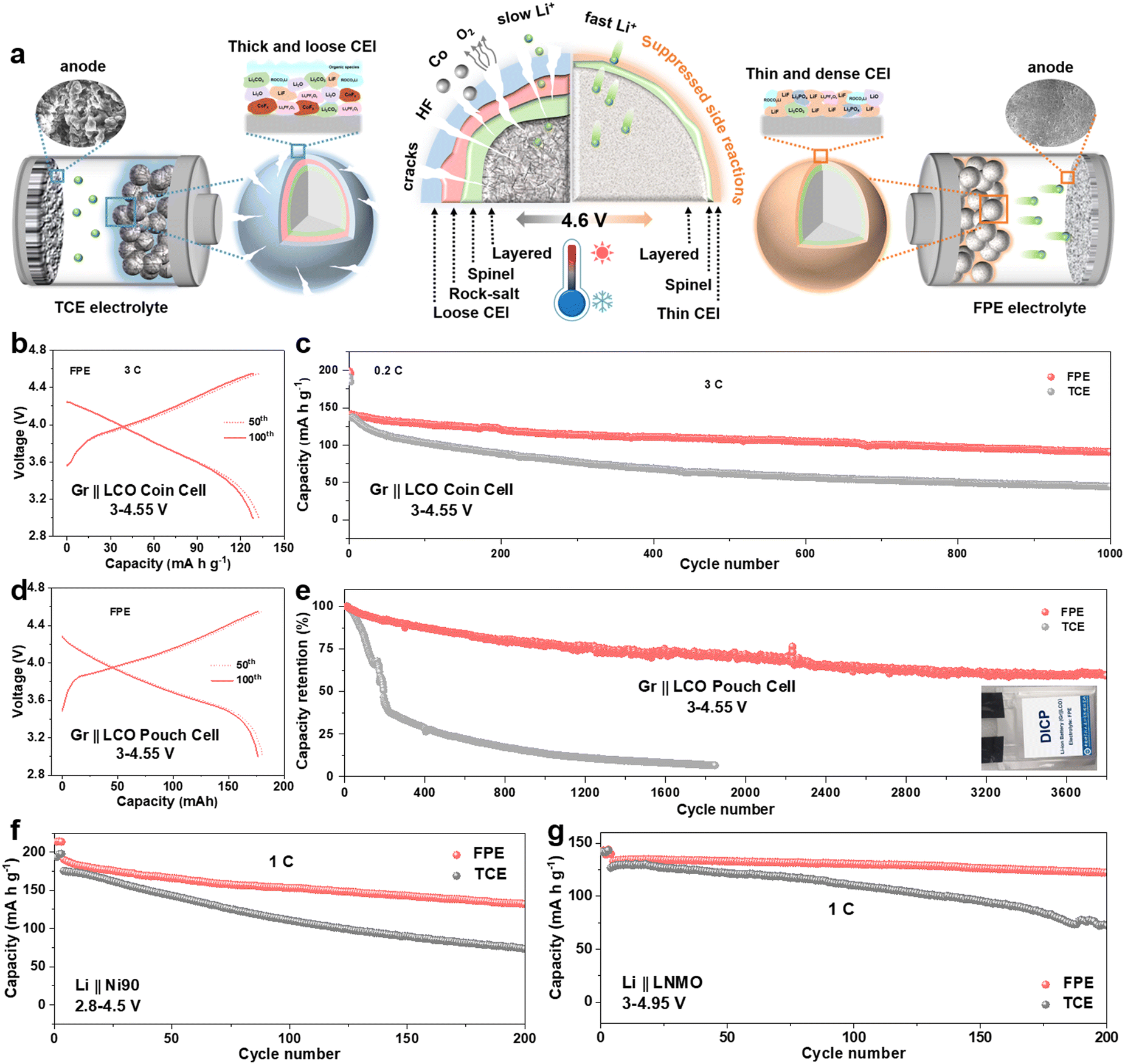 | ||
| Fig. 6 Working mechanism of the performance enhancement, practical application and versatilities of FPE. (a) Schematic illustration of the mechanism on the stabilized 4.6 V LCO batteries. (b) Charge–discharge voltage profiles and (c) long-term cycling performance of Gr||LCO coin cells obtained at 3C at 3–4.55 V (equivalent to 4.6 V vs. Li+/Li). (d) Charge–discharge voltage profiles and (e) long-term cyclability of Gr||LCO pouch cells measured at 290 mA at 3–4.55 V. (f) Cycling performance of Li||Ni90 cells at 1C obtained at 2.8–4.5 V. (g) Cycling performance of the Li||LNMO cell measured at 1C at 3–4.95 V. | ||
Coupling our LCO cathode with a commercial graphite (Gr) anode, we verified the application of FPE in Gr||LCO full cells. As presented in Fig. 6b and c, the Gr||LCO coin cells in FPE displayed excellent fast-charging performance with 64.5% capacity retention after 1000 cycles at 3C, much higher than that in TCE (32.8%, Fig. S33–S36, ESI†). Furthermore, Gr||LCO pouch type cells were cycled in FPE (Fig. 6d and e), which delivers an ultra-high capacity retention rate of 85.5% with a capacity of 158 mA h after 500 cycles (vs. 24.8% in TCE) and 72.1% after 2000 cycles, and long-term cyclability over 3800 cycles (Fig. S37, ESI†), demonstrative of the scalable industrial application potential.
Besides 4.6 V LCO, we further confirm the generalization of our “cocktail electrolyte” using other advanced cathodes, such as Ni-rich (LiNi0.8Mn0.1Co0.1O2 (NCM811) and LiNi0.9Mn0.05Co0.05O2 (Ni90)) and Co-free LiNi0.5Mn1.5O4 (LNMO) cathodes (Fig. 6f, g and Fig. S38–S42, ESI†). The capacity retentions of Li||Ni90 and Li||NCM811 reach up to 70% and 82%, respectively, which are much higher than those in TCE (only 42% and 67%, respectively). Furthermore, the Co-free LNMO cathode also exhibits ultra-high cycling stability and rate capability in FPE even at 4.9 V and 4.95 V, corresponding to capacity retention rates of 93% and 92% after 200 cycles, respectively.
Conclusions
In summary, we report an additive-containing “cocktail electrolyte” paired with a 4.6 V LCO cathode that exhibits ultra-stable fast-charging cycling stability, even at harsh temperatures and in practical applications. The proposed electrolyte effectively stabilizes the LCO cathode surface, thus suppressing the irreversible phase transition, dissolution of transition metals and side reactions. Sufficient surface characterization studies demonstrate that our electrolyte promotes more robust, lower resistance and higher ion conductivity inorganic components (LiF and Li3PO4) in the CEI than traditional commercial carbonate electrolytes. In addition, the tailored “cocktail electrolyte” is also compatible with anodes, which vastly eliminates the polarization at fast charging and low temperatures, realizing lithium dendrite-free anodes. Even at a high current density of 5C, the capacity retention of the LCO battery is as high as 73.2% after 1000 cycles at 4.6 V. Moreover, the ultra-long cyclability of both Gr||LCO coin cells and pouch type cells and unexpected generalization in high-voltage Ni-rich and Co-free cathodes further demonstrate the practicality of FPE. Therefore, our strategy presents a general electrolyte design principle via synergistic utilization of additives and is expected to be a leap in the pursuit of practically feasible high-energy-density LCO batteries with fast charging ability, long-term life, and wide temperature operation.Author contributions
Z.-S. Wu, X. H. Bao, Y. Yu and X. L. Feng conceived the experiments and supervised this project. A. P. Zhang performed the preparation, characterization and performance measurement of electrodes, electrolytes and coin cells, and the theoretical calculation. Z. H. Bi and H. Lin performed the preparation and characterization of coin cells. S. H. Liao performed the fabrication and performance testing of the pouch cells. M. R. Li conducted STEM-EELS characterization. A. P. Zhang, G. R. Wang, P. Das, Y. Yu, X. L. Feng, X. H. Bao and Z.-S. Wu wrote the manuscript. All the authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 22125903), the National Key R&D Program of China (Grants 2022YFA1504100, 2023YFB4005204), Dalian Innovation Support Plan for High Level Talents (2019RT09), the Dalian National Laboratory For Clean Energy (DNL), CAS, DNL Cooperation Fund, CAS (DNL202016, DNL202019), and the Exploratory Research Project of Yanchang Petroleum International Limited and DICP (yc-hw-2022ky-01). We acknowledge the Vacuum Interconnected Nanotech Workstation (NANO-X) for conducting the FIB, XPS-depth and TOF-SIMS experiments. The authors thank Prof. Yi Cui, Tong Liu, Xuefei Weng, and Rong Huang in NANO-X (Suzhou Institute of Nanotechnology and Nano-bionics, Chinese Academy of Sciences) for kindly conducting FIB, XPS-depth and TOF-SIMS characterization studies.Notes and references
- J. B. Goodenough and Y. Kim, Chem. Mater., 2009, 22, 587–603 CrossRef.
- H. Li, Joule, 2019, 3, 911–914 CAS.
- M. Weiss, R. Ruess, J. Kasnatscheew, Y. Levartovsky, N. R. Levy, P. Minnmann, L. Stolz, T. Waldmann, M. Wohlfahrt-Mehrens, D. Aurbach, M. Winter, Y. Ein-Eli and J. Janek, Adv. Energy Mater., 2021, 11, 2101126 CrossRef CAS.
- T. Islam, M. Li, A. Blanton, K. A. Pitton, K. R. Rao, S. Bayat, K. M. Wiaderek, M. A. Weret, S. C. Roy, R. Feng, D. Li, R. Alam, J. Nie, O. Oketola, A. Pramanik, B. S. Guiton, C. Risko, I. Belharouak, R. Amin and S. M. Islam, ACS Energy Lett., 2024, 9, 1–9 CrossRef CAS.
- Y. Lyu, X. Wu, K. Wang, Z. Feng, T. Cheng, Y. Liu, M. Wang, R. Chen, L. Xu, J. Zhou, Y. Lu and B. Guo, Adv. Energy Mater., 2021, 11, 2000982 CAS.
- K. Wang, J. Wan, Y. Xiang, J. Zhu, Q. Leng, M. Wang, L. Xu and Y. Yang, J. Power Sources, 2020, 460, 228062 CrossRef CAS.
- X. Fan and C. Wang, Chem. Soc. Rev., 2021, 50, 10486–10566 RSC.
- J.-N. Zhang, Q. Li, C. Ouyang, X. Yu, M. Ge, X. Huang, E. Hu, C. Ma, S. Li, R. Xiao, W. Yang, Y. Chu, Y. Liu, H. Yu, X.-Q. Yang, X. Huang, L. Chen and H. Li, Nat. Energy, 2019, 4, 594–603 CrossRef CAS.
- W. Xue, R. Gao, Z. Shi, X. Xiao, W. Zhang, Y. Zhang, Y. G. Zhu, I. Waluyo, Y. Li, M. R. Hill, Z. Zhu, S. Li, O. Kuznetsov, Y. Zhang, W.-K. Lee, A. Hunt, A. Harutyunyan, Y. Shao-Horn, J. A. Johnson and J. Li, Energy Environ. Sci., 2021, 14, 6030–6040 RSC.
- X. Yang, C. Wang, P. Yan, T. Jiao, J. Hao, Y. Jiang, F. Ren, W. Zhang, J. Zheng, Y. Cheng, X. Wang, W. Yang, J. Zhu, S. Pan, M. Lin, L. Zeng, Z. Gong, J. Li and Y. Yang, Adv. Energy Mater., 2022, 12, 2200197 CrossRef CAS.
- B. Wen, Z. Deng, P.-C. Tsai, Z. W. Lebens-Higgins, L. F. J. Piper, S. P. Ong and Y.-M. Chiang, Nat. Energy, 2020, 5, 578–586 CrossRef CAS.
- L. Xia, H. Miao, C. Zhang, G. Z. Chen and J. Yuan, Energy Storage Mater., 2021, 38, 542–570 CrossRef.
- J. Xu, J. Zhang, T. P. Pollard, Q. Li, S. Tan, S. Hou, H. Wan, F. Chen, H. He, E. Hu, K. Xu, X. Q. Yang, O. Borodin and C. Wang, Nature, 2023, 614, 694–700 CrossRef CAS PubMed.
- A. Fu, J. Lin, Z. Zhang, C. Xu, Y. Zou, C. Liu, P. Yan, D.-Y. Wu, Y. Yang and J. Zheng, ACS Energy Lett., 2022, 7, 1364–1373 CrossRef CAS.
- X. Yang, M. Lin, G. Zheng, J. Wu, X. Wang, F. Ren, W. Zhang, Y. Liao, W. Zhao, Z. Zhang, N. Xu, W. Yang and Y. Yang, Adv. Funct. Mater., 2020, 30, 2004664 CrossRef CAS.
- Y. Zou, J. Zhang, J. Lin, D.-Y. Wu, Y. Yang and J. Zheng, J. Power Sources, 2022, 524, 231049 CrossRef CAS.
- Y. Qin, K. Xu, Q. Wang, M. Ge, T. Cheng, M. Liu, H. Cheng, Y. Hu, C. Shen, D. Wang, Y. Liu and B. Guo, Nano Energy, 2022, 96, 107082 CrossRef CAS.
- X. Wen, M. Chen, X. Zhou, S. Chen, H. Huang, J. Chen, D. Ruan, W. Xiang, G. Zhang and W. Li, J. Phys. Chem. C, 2021, 126, 282–295 CrossRef.
- J. Zhang, P. F. Wang, P. Bai, H. Wan, S. Liu, S. Hou, X. Pu, J. Xia, W. Zhang, Z. Wang, B. Nan, X. Zhang, J. Xu and C. Wang, Adv. Mater., 2022, 34, 2108353 CrossRef CAS PubMed.
- S. Tan, Z. Shadike, J. Li, X. Wang, Y. Yang, R. Lin, A. Cresce, J. Hu, A. Hunt, I. Waluyo, L. Ma, F. Monaco, P. Cloetens, J. Xiao, Y. Liu, X.-Q. Yang, K. Xu and E. Hu, Nat. Energy, 2022, 7, 484–494 CrossRef CAS.
- P. Bai, X. Ji, J. Zhang, W. Zhang, S. Hou, H. Su, M. Li, T. Deng, L. Cao, S. Liu, X. He, Y. Xu and C. Wang, Angew. Chem., Int. Ed., 2022, 61, e202202731 CrossRef CAS PubMed.
- W. Deng, W. Dai, X. Zhou, Q. Han, W. Fang, N. Dong, B. He and Z. Liu, ACS Energy Lett., 2020, 6, 115–123 CrossRef.
- X. Fan, L. Chen, O. Borodin, X. Ji, J. Chen, S. Hou, T. Deng, J. Zheng, C. Yang, S. C. Liou, K. Amine, K. Xu and C. Wang, Nat. Nanotechnol., 2018, 13, 715–722 CrossRef CAS PubMed.
- S. Tu, B. Zhang, Y. Zhang, Z. Chen, X. Wang, R. Zhan, Y. Ou, W. Wang, X. Liu, X. Duan, L. Wang and Y. Sun, Nat. Energy, 2023, 8, 1365–1374 CrossRef CAS.
- X. Fan, X. Ji, L. Chen, J. Chen, T. Deng, F. Han, J. Yue, N. Piao, R. Wang, X. Zhou, X. Xiao, L. Chen and C. Wang, Nat. Energy, 2019, 4, 882–890 CrossRef CAS.
- T. Deng, X. Fan, L. Cao, J. Chen, S. Hou, X. Ji, L. Chen, S. Li, X. Zhou, E. Hu, D. Su, X.-Q. Yang and C. Wang, Joule, 2019, 3, 2550–2564 CrossRef CAS.
- C. Luo, Q. Liu, X. Wang, Y. Tian, Z. Liu, F. Kang and B. Li, Nano Energy, 2023, 109, 108323 CrossRef CAS.
- D. Aurbach, E. Markevich and G. Salitra, J. Am. Chem. Soc., 2021, 143, 21161–21176 CrossRef CAS PubMed.
- C.-C. Su, M. He, M. Cai, J. Shi, R. Amine, N. D. Rago, J. Guo, T. Rojas, A. T. Ngo and K. Amine, Nano Energy, 2022, 92, 106720 CrossRef CAS.
- Y. Chen, W. Zhao, Q. Zhang, G. Yang, J. Zheng, W. Tang, Q. Xu, C. Lai, J. Yang and C. Peng, Adv. Funct. Mater., 2020, 30, 2000396 CrossRef CAS.
- X. Cui, J. Zhang, J. Wang, P. Wang, J. Sun, H. Dong, D. Zhao, C. Li, S. Wen and S. Li, ACS Appl. Mater. Interfaces, 2021, 13, 59580–59590 CrossRef CAS PubMed.
- S. Kim, S. O. Park, M.-Y. Lee, J.-A. Lee, I. Kristanto, T. K. Lee, D. Hwang, J. Kim, T.-U. Wi, H.-W. Lee, S. K. Kwak and N.-S. Choi, Energy Storage Mater., 2022, 45, 1–13 CrossRef.
- Z. Piao, R. Gao, Y. Liu, G. Zhou and H.-M. Cheng, Adv. Mater., 2023, 35, 2206009 CrossRef CAS PubMed.
- T. Liu, X. Han, Z. Zhang, Z. Chen, P. Wang, P. Han, N. Ding and G. Cui, J. Power Sources, 2019, 437, 226942 CrossRef CAS.
- Z. Cao, M. Hashinokuchi, T. Doi and M. Inaba, J. Electrochem. Soc., 2019, 166, A82 CrossRef CAS.
- J.-Y. Hwang, H. M. Kim and Y.-K. Sun, J. Electrochem. Soc., 2018, 165, A5006 CrossRef CAS.
- P. Xiao, Y. Zhao, Z. Piao, B. Li, G. Zhou and H.-M. Cheng, Energy Environ. Sci., 2022, 15, 2435–2444 RSC.
- X. Zhang, L. Zou, Z. Cui, H. Jia, M. H. Engelhard, B. E. Matthews, X. Cao, Q. Xie, C. Wang, A. Manthiram, J.-G. Zhang and W. Xu, Mater. Today, 2021, 44, 15–24 CrossRef CAS.
- Y. Liao, Z. Wang, X. Dai, H. Chen, F. Wu, J. Li, Y. Mai and S. Li, J. Phys. Chem. C, 2022, 126, 16627–16635 CrossRef CAS.
- L. Deng, K. Goh, F.-D. Yu, Y. Xia, Y.-S. Jiang, W. Ke, Y. Han, L.-F. Que, J. Zhou and Z.-B. Wang, Energy Storage Mater., 2022, 44, 82–92 CrossRef.
- K. Li, J. Zhang, D. Lin, D. W. Wang, B. Li, W. Lv, S. Sun, Y. B. He, F. Kang, Q. H. Yang, L. Zhou and T. Y. Zhang, Nat. Commun., 2019, 10, 725 CrossRef CAS PubMed.
- W. Zhang, X. Sun, Y. Tang, H. Xia, Y. Zeng, L. Qiao, Z. Zhu, Z. Lv, Y. Zhang, X. Ge, S. Xi, Z. Wang, Y. Du and X. Chen, J. Am. Chem. Soc., 2019, 141, 14038–14042 CrossRef CAS PubMed.
- W. Y. Huang, Q. Zhao, M. J. Zhang, S. Y. Xu, H. Y. Xue, C. Zhu, J. J. Fang, W. G. Zhao, G. X. Ren, R. Z. Qin, Q. H. Zhao, H. B. Chen and F. Pan, Adv. Energy Mater., 2022, 12, 2200813 CrossRef CAS.
- J. Liu, B. Yuan, N. He, L. Dong, D. Chen, S. Zhong, Y. Ji, J. Han, C. Yang, Y. Liu and W. He, Energy Environ. Sci., 2023, 16, 1024–1034 RSC.
- Y. Wang, Q. Zhang, Z.-C. Xue, L. Yang, J. Wang, F. Meng, Q. Li, H. Pan, J.-N. Zhang, Z. Jiang, W. Yang, X. Yu, L. Gu and H. Li, Adv. Energy Mater., 2020, 10, 2001413 CrossRef CAS.
- L. Ni, S. Zhang, A. Di, W. Deng, G. Zou, H. Hou and X. Ji, Adv. Energy Mater., 2022, 12, 2201510 CrossRef CAS.
- H. Liu, M. Wolfman, K. Karki, Y. S. Yu, E. A. Stach, J. Cabana, K. W. Chapman and P. J. Chupas, Nano Lett., 2017, 17, 3452–3457 CrossRef CAS PubMed.
- X. Zhang, H. Jia, L. Zou, Y. Xu, L. Mu, Z. Yang, M. H. Engelhard, J.-M. Kim, J. Hu, B. E. Matthews, C. Niu, C. Wang, H. Xin, F. Lin and W. Xu, ACS Energy Lett., 2021, 6, 1324–1332 CrossRef CAS.
- F. Wu, J. Dong, L. Chen, L. Bao, N. Li, D. Cao, Y. Lu, R. Xue, N. Liu, L. Wei, Z. Wang, S. Chen and Y. Su, Energy Storage Mater., 2021, 41, 495–504 CrossRef.
- F. Lin, D. Nordlund, I. M. Markus, T.-C. Weng, H. L. Xin and M. M. Doeff, Energy Environ. Sci., 2014, 7, 3077 RSC.
- Y. Zou, A. Fu, J. Zhang, T. Jiao, Y. Yang and J. Zheng, ACS Sustainable Chem. Eng., 2021, 9, 15042–15052 CrossRef CAS.
- Q. Gan, N. Qin, Z. Wang, Z. Li, Y. Zhu, Y. Li, S. Gu, H. Yuan, W. Luo, L. Lu, Z. Xu and Z. Lu, ACS Appl. Energy Mater., 2020, 3, 7445–7455 CrossRef CAS.
- H.-H. Ryu, B. Namkoong, J.-H. Kim, I. Belharouak, C. S. Yoon and Y.-K. Sun, ACS Energy Lett., 2021, 6, 2726–2734 CrossRef CAS.
- E. R. Logan and J. R. Dahn, Trends Chem., 2020, 2, 354–366 CrossRef CAS.
- S. S. Zhang, InfoMat, 2020, 3, 125–130 CrossRef.
- D. Wu, J. He, J. Liu, M. Wu, S. Qi, H. Wang, J. Huang, F. Li, D. Tang and J. Ma, Adv. Energy Mater., 2022, 12, 2200337 CrossRef CAS.
- Z. Jiang, Z. Zeng, H. Zhang, L. Yang, W. Hu, X. Liang, J. Feng, C. Yu, S. Cheng and J. Xie, iScience, 2022, 25, 103490 CrossRef CAS PubMed.
- E. M. Erickson, W. Li, A. Dolocan and A. Manthiram, ACS Appl. Mater. Interfaces, 2020, 12, 16451–16461 CrossRef CAS PubMed.
- W. Li, A. Dolocan, P. Oh, H. Celio, S. Park, J. Cho and A. Manthiram, Nat. Commun., 2017, 8, 14589 CrossRef PubMed.
- J. Li, W. Li, Y. You and A. Manthiram, Adv. Energy Mater., 2018, 8, 1801957 CrossRef.
- Z. Zhu, H. Wang, Y. Li, R. Gao, X. Xiao, Q. Yu, C. Wang, I. Waluyo, J. Ding, A. Hunt and J. Li, Adv. Mater., 2020, 32, e2005182 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee00676c |
| This journal is © The Royal Society of Chemistry 2024 |