
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn ultrafast Na-ion battery chemistry through coupling sustainable organic electrodes with modulated aqueous electrolytes†
Yunpei
Zhu
a,
Xianrong
Guo
b,
Simil
Thomas
c,
Jian
Yin
a,
Youyou
Yuan
b,
Zhengnan
Tian
a,
George T.
Harrison
d,
Stefaan
De Wolf
 d,
Osman M.
Bakr
e,
Omar F.
Mohammed
c and
Husam N.
Alshareef
*a
d,
Osman M.
Bakr
e,
Omar F.
Mohammed
c and
Husam N.
Alshareef
*a
aMaterials Science and Engineering, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia. E-mail: husam.alshareef@kaust.edu.sa
bCore Labs, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia
cAdvanced Membranes and Porous Materials Center, Division of Physical Sciences and Engineering, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia
dKAUST Solar Center, Physical Sciences and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia
eKAUST Catalysis Center, Division of Physical Sciences and Engineering, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia
First published on 5th March 2024
Abstract
Rechargeable Na-ion batteries for grid-scale applications require key battery materials that couple high performance with sustainability. However, there are limited choices of electrodes and electrolytes to achieve this goal. Here we demonstrate that a sustainable organic electrode can deliver unprecedented electrochemical performance through coupling with a rationally designed hybrid electrolyte based on cost-efficient NaClO4, water, and weakly solvating additives (e.g., acetamide). We discover a new Na+ storage mechanism involving simultaneous structural and morphological changes of the organic electrode in the hybrid electrolyte. The unique combination of the organic electrode and hybrid electrolyte can facilitate unprecedented rate performance (>1000C), competitive and even superior to state-of-the-art fast-charging battery chemistries. We identify that the unique electrolyte chemistry enabled by the weakly solvating solvent can significantly promote electron transfer kinetics through the reorganization process, which was proven to be one key enabler for the high rate performance. The electrode and electrolyte chemistries elucidated in this work can propel the development of fast-charging battery technologies based on sustainable and low-cost materials.
Broader contextSustainable batteries represent one key battery technology to decarbonize current energy structures. Aqueous Na-ion batteries will play a critical role in large-scale applications because of the earth abundance and recyclability of the key battery materials. However, compared to other battery chemistries, the study of the fundamental kinetics (i.e., charge rate) for aqueous Na-ion batteries remains at the early stage. Herein, in this study, we illustrate the fundamental interplay between the engineered aqueous electrolytes and electrodes, which typically dominates the kinetics. We design a new aqueous electrolyte featuring weakly solvating amides, which apparently promotes the Na+ storage mechanism of the electrode in a manner of initializing structural changes and promoting charge transfer kinetics. The resultant unprecedented rate capability is found to be related to the facilitated solvation sheath reorganization. This work unlocks a new design strategy to develop high-performance aqueous batteries. |
Introduction
The rapidly growing global market of energy storage calls for rechargeable batteries that can integrate electricity generated from renewable energy into the grid. This large-scale energy storage requires battery components (e.g., electrode materials, electrolytes) to be sustainable, inexpensive, earth-abundant, and environmentally friendly.1 In this regard, aqueous Na-ion batteries (ANIBs) represent a promising energy storage technology because of their intrinsic safety and cost advantages brought by aqueous electrolytes and inexhaustible sodium resources.2–4 In reverse, the electrochemical performance (e.g., power density, cycling stability) of ANIBs is determined by the battery components, especially electrode materials and electrolytes. The search for fundamental solutions that can achieve battery component sustainability and improved battery performance simultaneously has re-emerged as a field of technological and scientific interest for rechargeable ANIBs.Similar to the working mechanism of the state-of-the-art Li-ion batteries, the electrodes of ANIBs undergo reversible Na+ uptake and removal to enable the charge and discharge processes.5 However, compared to Li+, the larger size and heavier molar mass of Na+ deliver two profound issues upon reversibly hosting Na+ in the electrodes, including: (1) serious volume changes of the crystallographic structures lead to accelerated electrode pulverization and performance degradation; and (2) the larger ionic size of Na+ can hinder its solid-state mobility in the electrodes, which proposes a barrier to improve the rate performance. Beyond classical strategies like developing inorganic electrodes with open frameworks to alleviate the two issues,6–8 organic compounds feature intrinsic structural and chemical advantages to facilitate the solid-state diffusion of large Na+.9,10 Structurally, organic compounds are consisted of periodically arranged molecular units that bear redox-active functional groups,9,11,12 wherein the weak interaction (e.g., van der Waals, hydrogen bonding) between molecular units endow organic compounds with intrinsic structural flexibility. This is markedly different from the robust frameworks of inorganic electrodes that are formed by strong ionic or covalent bonds, which necessitates sluggish kinetics (large volume change or even phase transformation) to accommodate large Na+.5–8 Chemically, the redox-active functional groups (e.g., C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O ↔ C–O−) in organic compounds ensure fast redox kinetics since the bond rearrangement in typical battery chemistries is avoided during charge/discharge.13,14 Hypothetically, the structural flexibility and rapid redox chemistry of organic compounds can enable facile Na+ diffusion, even potentially at a rate that is typically unexpected in inorganic electrodes. However, in practice, the rate performance (typically <200C, 1C means that a battery can be fully charged/discharged in one hour) of organic compounds towards Na+ storage, either in organic electrolytes or in aqueous electrolytes, is comparable to or even worse than inorganic electrodes.2,3,5,9,10,15–17 The origin of this discrepancy is still unclear.
O ↔ C–O−) in organic compounds ensure fast redox kinetics since the bond rearrangement in typical battery chemistries is avoided during charge/discharge.13,14 Hypothetically, the structural flexibility and rapid redox chemistry of organic compounds can enable facile Na+ diffusion, even potentially at a rate that is typically unexpected in inorganic electrodes. However, in practice, the rate performance (typically <200C, 1C means that a battery can be fully charged/discharged in one hour) of organic compounds towards Na+ storage, either in organic electrolytes or in aqueous electrolytes, is comparable to or even worse than inorganic electrodes.2,3,5,9,10,15–17 The origin of this discrepancy is still unclear.
Electrolyte design shows equal significance in improving battery performance, especially battery kinetics and cycling stability. Since the electrolyte is the only component that is in contact with other components in a battery, designing better electrolytes signifies tuning and balancing a variety of properties, ranging from bulk (e.g., ion transport, extended liquid structure) to interfacial structure (i.e., ion desolvation, parasitic reaction suppression).18 Water featuring a high dielectric constant and high dipole moment has been proven to be an excellent solvent. Nevertheless, the abundance of free water molecules, featuring percolating hydrogen bond networks, is responsible for undesirable parasitic reactions (e.g., water decomposition) in conventional aqueous electrolytes. Recently, several concepts have been proposed to efficiently suppress the activity of water, including superconcentrated electrolytes,19–22 organic additives,23–25 and nanoconfinement by polymers or porous hosts.26,27 The philosophy behind these breakthrough methodologies is similar, namely, breaking the ubiquitous hydrogen bonding networks formed by water molecules to limit the activity of water from the thermodynamics point of view. For these newly developed electrolytes, their high viscosity and insufficient ionic conductivity are unfavorable for high-power applications as a result of slow ionic transport and sluggish (de)solvation kinetics at the electrode–electrolyte interface. Additionally, economic and environmental concerns (e.g., expensive and toxic F-containing salts) of these electrolytes impede their large-scale energy storage applications.
In this study, we report a strategy to achieve an unprecedented rate performance of aqueous Na-ion batteries through a two-pronged approach: realizing fast redox kinetics of an organic electrode that features remarkable structural flexibility and a non-diffusion controlled enolization redox chemistry (CO ↔ C–O−); and improving bulk ion transport and Na+ desolvation at the electrode–electrolyte interface through designing a modified aqueous electrolyte composed of weakly solvated molecules. We employed both concepts through coupling an organic electrode, perylenetetracarboxylic dianhydride (PTCDA), with an amide-modified aqueous electrolytes. Further, we identified reversible structural transformation of PTCDA accompanied by spontaneous nanostructuring upon Na+ uptake/extraction, which is uniquely enabled by the modified aqueous electrolyte. This mechanism was proven to take account for the ultrahigh rate performance of Na+ storage, comparable to and even superior to state-of-the-art electrodes storing smaller charge carriers like H+ or Li+.28–32
Results and discussion
Rationale for materials selection
PTCDA was chosen as a model organic electrode to illustrate the Na+ storage electrochemistry for three reasons, including: (1) materials sustainability. PTCDA is composed of earth-abundant (C, H, and O) and is a low-cost organic dye molecule with enormous commercial availability; (2) open and flexible crystalline structure (Fig. 1a and b). The crystalline structure of PTCDA shows alternating layers of hexagonally packed molecular units (space group P21/c), wherein the molecular unit consists of a perylene core to which two anhydride groups are attached, one at either side. The open and flexible van der Waals structure of PTCDA is intrinsically superior in storing large Na+; and (3) electrochemistry. The carbonyl groups of PTCDA are known to be redox-active (CO ↔ C–O−) towards hosting cations (e.g., Na+, Mg2+),33 while four carbonyl groups in one PTCDA molecular unit ensures a high theoretical capacity (e.g., 138 mA h g−1 for 2e− reaction). Up to now, compared to inorganic electrodes that have been deeply and widely studied, attention has seldom been paid to the redox mechanisms (e.g., crystal structures, morphology evolution) of organic electrodes.9–13,33
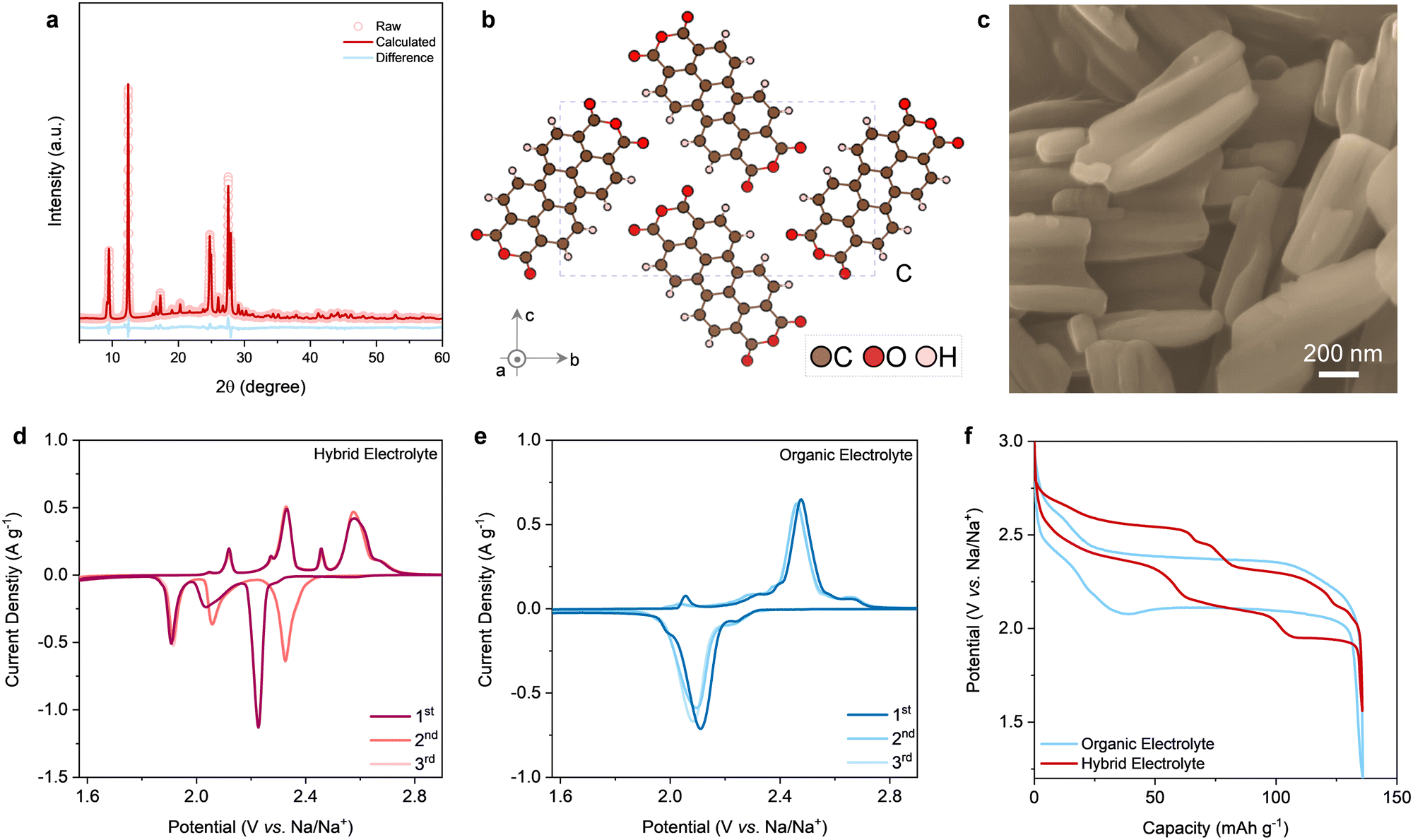 | ||
Fig. 1 Characterizations of PTCDA. (a) XRD pattern with the Rietveld refinement, (b) refined crystal structure viewed along a axis and (c) SEM image showing nanoparticle morphology. The initial three cycles of CV curves of the PTCDA electrode in the hybrid electrolyte with nNaClO4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :nacetamide:nwater = 1:4:4 (d) and organic electrolyte (e) (scan rate: 0.2 mV s−1). (f) Comparison of GCD curves in two different electrolytes recorded at 1C. :nacetamide:nwater = 1:4:4 (d) and organic electrolyte (e) (scan rate: 0.2 mV s−1). (f) Comparison of GCD curves in two different electrolytes recorded at 1C. | ||
PTCDA with carbonyl motifs is an n-type organic electrode that typically shows an average working potential of −0.6 to −0.7 V (versus standard hydrogen electrode) for storing cations.13,33 This electrochemistry feature makes PTCDA a promising anode candidate for aqueous Na-ion batteries. Noticeably, such a low working potential of the anode is normally accompanied by unwanted side reactions, especially water decomposition at the anode in conventional aqueous electrolytes. Following the state-of-the-art strategies in suppressing water activity,19–27 we presume that introducing a co-solvent featuring a wide electrochemical stability window into aqueous electrolytes is an alternative way to achieve the same goal. Application of the co-solvent practice to aqueous electrolytes, similar to the co-solvent strategy in organic Li/Na batteries,34,35 has to be subject to several new constraints. First, the co-solvent should be non-toxic, non-flammable, and cost-efficient. Second, the co-solvent is favored with good miscibility with water in a wide range of electrolyte compositions, thus enabling a high degree of freedom in tuning the hydrogen bonding networks between water molecules. Third and most significantly, the co-solvent should have a simple and small molecular structure and coordinate weakly with Na+, both of which together can deliver favorable bulk properties (e.g., low viscosity, high ionic conductivity) and interfacial behaviors (e.g., low energy barrier for Na+ desolvation). The aforementioned factors rule out most organic solvents like carbonates, ethers, and sulfones as they are either flammable/toxic/expensive or form strong coordination with metal cations, resulting in serious economic and environmental concerns and poor rate performance.
Upon the screening process, we selected acetamide as a logical co-solvent that meets the requirements discussed above. More specifically, acetamide shows extremely high solubility in water (2000 g Lwater−1), while its small molecular structure, high dielectric constant and decent polarity can ensure high ionic conductivity. The amine and carbonyl groups of acetamide have a high tendency towards hydration with water and coordination with Na+. The hydration effect can disturb the extensive hydrogen bonding networks between water molecules, as confirmed by Fourier-transform infrared spectroscopy (FT-IR) (Fig. S1, ESI†) and differential scanning calorimetry (DSC) (Fig. S2, ESI†). The weak coordination allows for facile Na+ transport and low desolvation energy barriers to achieve fast Na+ storage kinetics. A transparent, liquid-state electrolyte can be formed by mixing acetamide, water, and NaClO4 (Fig. S3, ESI†). Noticeably, NaClO4 was utilized because of its cost efficiency, sustainability, and non-toxicity, making it a decent candidate for large-scale energy storage. Herein, the electrolyte exhibits well-balanced bulk properties at the molar ratio of nNaClO4:nAcatamide:nH2O = 1:4:4 (Fig. S4, ESI†), including low viscosity (11.4 mPa s), high ionic conductivity (28.6 mS cm−1, approximately three times organic Na-ion battery electrolytes36), and low water content (14.9 wt%). For simplicity, this electrolyte is labelled as the hybrid electrolyte in the following.
Electrochemistry of organic electrodes towards Na+ storage
Fundamentally, the electrochemical performance of an electrode, especially classical inorganic electrodes, strongly depends on the electrolytes. One performance difference between organic and aqueous electrolytes is the rate performance, wherein a higher rate performance can be generally achieved due to the higher ion transport rate (conductivity) and more facile desolvation in aqueous electrolytes. Another mechanism difference is the stored cation species. Protons are oftentimes reversibly co-hosted by inorganic electrodes in aqueous electrolytes.4,37 To date, compared to inorganic electrodes, the electrochemical energy storage mechanism of organic electrodes has been much underdeveloped either in organic or in aqueous electrolytes. The systematic illustrations of the redox reaction mechanism of organic electrodes can bring new opportunities to design better organic electrodes and compatible electrolytes.Herein, the electrochemical behavior of PTCDA was evaluated in two different electrolyte systems, i.e., the classical organic electrolyte (NaClO4 dissolved in ethylene carbonate and diethyl carbonate) and the hybrid electrolyte. A classical three-electrode set-up was used to evaluate the electrochemical performance of PTCDA in different electrolytes (Fig. S5, ESI†). All the electrode potentials versus the reference electrode were converted to standard Na/Na+ potentials for comparison purposes. Proton co-intercalation mechanism in the hybrid electrolyte has been excluded based on the ex situ X-ray photoelectron spectroscopy (XPS) (Fig. S6, ESI†). Na+ has been confirmed to be the only cation that is reversibly uptaken/extracted during charge/discharge in both electrolytes. This can be rationalized by the suppressed water activity of the hybrid electrolyte (Fig. S7, ESI†).
Two important features of PTCDA can be observed when working in the two different electrolytes, as indicated by the cyclic voltammetry (CV) curves. One remarkable feature is the discrepancy in the CV plots between the first cycle and the following cycles in the hybrid electrolyte (Fig. 1d), but not in the organic electrolyte (Fig. 1e). One profound irreversible peak upon cathodic Na+ uptake can be seen during the first cycle when operating PTCDA in the hybrid electrolyte. Additionally, a couple of pairs of redox peaks can be identified in the hybrid electrolyte, whereas only one pair of dominant redox peaks is seen in the organic electrolyte. Such a difference in electrochemical behavior is more visually obvious in the corresponding galvanostatic charge–discharge (GCD) curves (Fig. 1f), wherein several well-defined plateaus appear when discharging/charging PTCDA in the hybrid electrolyte, in contrast to the relatively straightforward charge/discharge profiles in the organic electrolyte.38 The overpotentials of the GCD curves are reasonably related to the low electronic conductivity of the PTCDA electrode and pronounced structural changes of PTCDA during (de)sodiation, as will be discussed in the next section. Apparently, the electrochemical reaction of PTCDA during Na+ storage in the case of hybrid electrolyte follows a reaction mechanism that is hitherto unknown, especially distinct reaction pathways between the initial and subsequent sodiation/desodiation processes.
Analysis of Na+ storage mechanism
In the above two electrolytes, PTCDA delivers nearly the same capacity that approaches theoretical capacity enabled by 2e− reaction at 1C rate (Fig. 1f), an indication of reversible enolization redox chemistry involving two carbonyl groups in one PTCDA molecule towards electrochemical Na+ storage. Considering the large single crystals and structural flexibility of PTCDA (Fig. 1a–c), we hypothesize that the inconsistent electrochemical behaviors are associated with different structural transformation processes of PTCDA in the two electrolytes. We first performed in situ X-ray diffraction (XRD) to illustrate the origin of voltage profile evolution between cycles in different conditions (Fig. S8, ESI†).According to the in situ XRD patterns of PTCDA working in the organic electrolyte (Fig. 2a), PTCDA maintains its monoclinic structure during the entire sodiation/desodiation process. Closer examination indicates the lattice expansion upon sodiation and reversible lattice shrinkage during desodiation. The reversible structural changes are reminiscent of the Na+ intercalation chemistry for classical inorganic electrodes.5,7,8 As expected, the in situ XRD patterns of the PTCDA electrode reveal completely different structural evolutions on charge/discharge in the hybrid electrolyte (Fig. 2b). The foremost difference is the complicated yet reversible diffraction evolutions upon sodiation/desodiation, in accordance with the multiple pairs of redox peaks in the voltage profiles (Fig. 1d).
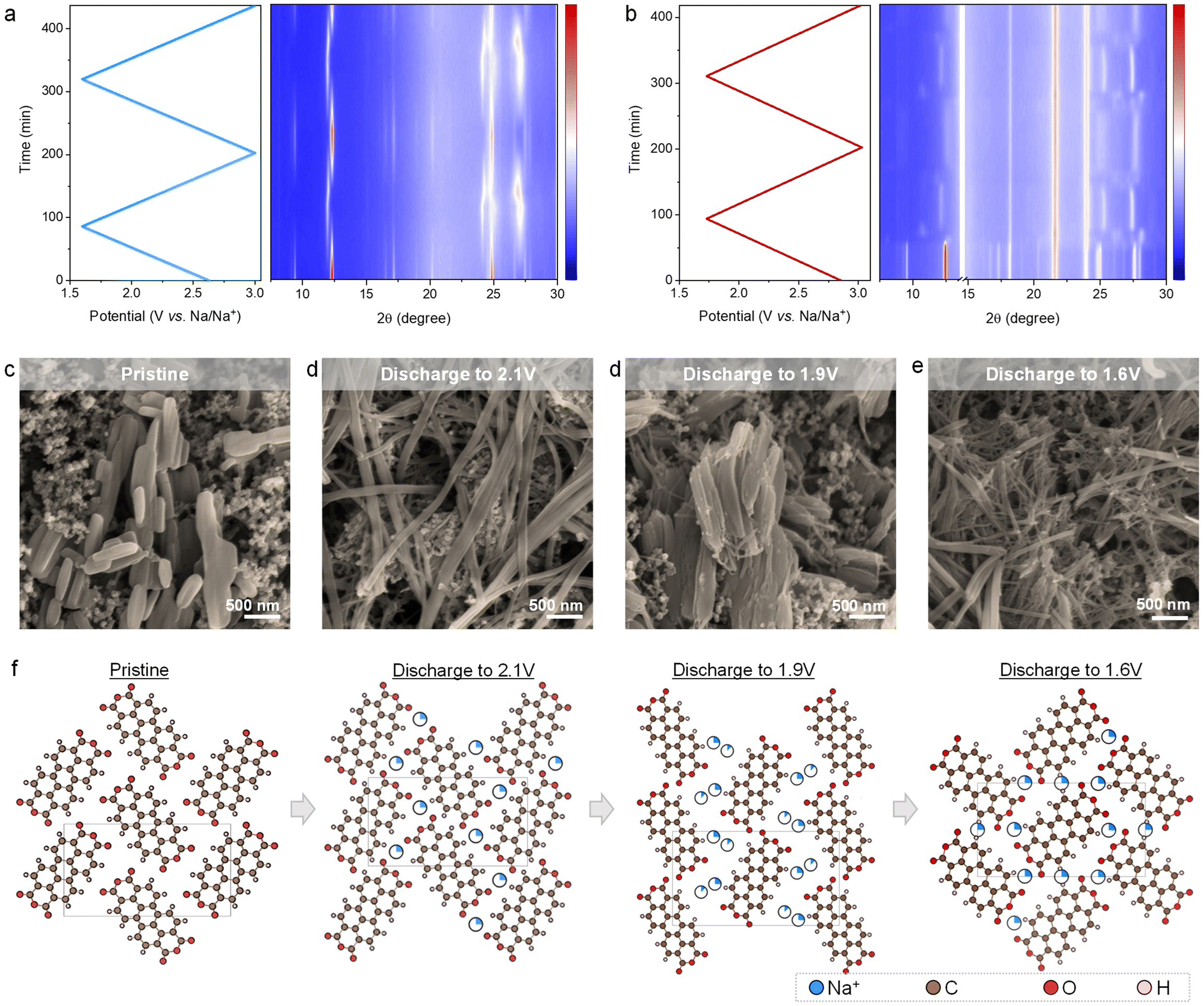 | ||
| Fig. 2 Structural and morphological evolutions of the PTCDA electrodes upon Na+ storage. Operando XRD patterns of the PTCDA electrode in the organic electrolyte (a) and hybrid electrolyte with nNaClO4:nacetamide:nwater = 1:4:4 (b). (c–e) Ex situ SEM images and (f) the corresponding fitted crystalline structures of the PTCDA electrodes upon discharging (Na+ intercalation) to different potentials in the hybrid electrolyte at nNaClO4:nacetamide:nwater = 1:4:4. The intercalated Na+ is shown in the way of occupancy. Occupancy is defined as the faction of Na+ present at the site. | ||
The other intriguing structural transformation is the sharp loss of diffraction intensity upon sweeping the potential across the irreversible cathodic peak in the discharge curve of the first cycle (Fig. 1d). This is indicative of losing crystallization, whereas this process is irreversible, as confirmed by the weak diffractions in the following sodiation/desodiation steps. To uncover the underlying reasons, we conducted ex-situ scanning electron microscopy (SEM) to characterize the morphology of the PTCDA electrode (Fig. 2d). The SEM images show a remarkable morphology change from the pristine nanorods to nanofibers with largely increased length-to-width ratios. According to previous studies, drastic morphological changes of electrode materials (e.g., metal oxides, sulfur) can induce significant voltage hysteresis,9,39–41 This is a kinetically limited process that is accompanied by large polarization in the voltage curves. Accordingly, compared to the stable and reversible potential profiles afterward, we attribute the irreversible cathodic peak in the first cycle to the significant textual transformation during sodiation. Upon further Na+ uptake, the PTCDA nanoparticles experience a process of continuous morphological changes, whereas the nanoparticles become much smaller in length and/or width compared to the pristine PTCDA (Fig. 2d). Once fully desodiated, the electrode can not recover the original particulate morphology (Fig. S9, ESI†), as confirmed by the much weakened diffraction intensity after the first cycle in the in situ XRD patterns (Fig. 2b). This noticeable morphological change is repeatable cycle by cycle after the first cycle, in line with our observations of reproducible in situ XRD patterns during each sodiation/desodiation cycle and stable voltage profiles in the following cycles. In sharp contrast, we did not observe significant morphological evolutions of PTCDA nanoparticles in the organic electrolyte (Fig. S10, ESI†). This result coincides well with the strong diffractions in the corresponding in situ XRD patterns during cycling (Fig. 2a).
We selectively fitted the diffraction patterns of the PTCDA electrode at various charge/discharge states to accurately illustrate how the structural evolutions relate to the morphological changes. The pristine PTCDA features a β-phase crystal structure that is attributed to the monoclinic space group of P21/c (unit-cell volume: 777.29 Å3), while the monoclinic space group was maintained upon Na+ uptake/removal when testing PTCDA in the organic and hybrid electrolytes. In the organic electrolyte, Na+ tends to occupy the 4e site to achieve a high capacity approaching the theoretical 2-Na+ storage capacity (Fig. S11, ESI†). The unit-cell volume of PTCDA shows insignificant changes during (de)sodiation (Table S1, ESI†).
However, the structural evolution of PTCDA followed a different mechanism when storing Na+ in the hybrid electrolyte (Fig. 2f and Fig. S12, ESI†). Structural fitting results reveal that Na-ions occupy the 4e site (i.e., the basal parts of the PTCDA molecules) until storing 1.5 Na-ions per unit cell. Upon discharging to 2.1 V (storing 1 Na-ion per unit cell), the lattice parameters and unit-cell volume were insignificantly changed. On discharging to 1.9 V (storing 1.5 Na-ions per unit cell), the unit-cell volume (972.19 Å3) was increased by 25%, along with the elongation of the unit cell along the b-axis and c-axis. This is due to the steric limitation as the 1.5 Na-ions still occupy the 4e sites (occupancy: 0.375). We notice that discharging to 1.6 V (storing 2 Na-ions per unit cell) leads to the contraction of the unit cell (unit-cell volume: 805 Å3). The reason behind this is besides 4e site (occupancy: 0.25), Na-ions start occupying the hole parts of 2d site (occupancy: 0.25), thereby resulting in a decreased steric effect. The intercalation of two positively charged Na-ions at two different sites can attenuate the carbonyl/carbonyl repulsion and become coordinated by two different carbonyl groups. Reversely, upon charging, Na-ions at the 2d site were deintercalated first, followed by the removal of those at the 4a site (Fig. S12, ESI†). This indicates the high structural reversibility of PTCDA in the hybrid electrolyte, which is essential for efficient Na+ storage in PTCDA over multiple cycles.
Based on the above investigation, we conclude that PTCDA undergoes completely different crystalline structural and morphological evolutions when working in the hybrid electrolyte, while the crystalline structure transformations and morphological changes are intimately correlated. During the first sodiation process, we hypothesize that a large energy barrier is required for the phase transformation from the pristine monoclinic phase to the phase storing 1.5 Na per PTCDA unit (Na1.5-PTCDA) in the second sodiation step, as the direct nucleation of Na1.5-PTCDA that involves the entire slab shift is kinetically sluggish and difficult. On downsizing the nanoparticles, the kinetic barrier for the reversible phase transformation in the hybrid electrolyte can be efficiently decreased.9,39–41 Apparently, compared to the organic electrolyte, the hybrid electrolyte appears to be more efficient in facilitating morphological evolutions and reversible phase transformation simultaneously.
Electrochemical and spectroscopic analysis of the electrolytes
An electrolyte is a complicated system that involves intermolecular interactions among cations, anions, and solvent molecules (e.g., solvation sheath structure of Na+). Quantifying the influence of these interactions on the electrochemical behavior of an electrode is not straightforward. In this regard, chemical potential, μNa+, turns out to be an appropriate macroscopic descriptor. Indeed, μNa+ defines the molar Gibbs free energy change upon adding an infinitesimal amount of Na+ into the electrolyte system.42 Since Na+ coordinates with solvent molecules and/or anions, its solvation sheath structure (coordination environment) plays an indispensable role in tuning μNa+, and hence, the equilibrium potential of the electrode. To confirm this, we tested the equilibrium potential shift based on electromotive force measurements on the Na3−xV2(PO4)3 (x ∼ 2) in different Na-ion battery electrolytes (Fig. 3a). The details of this experimental design is given in Fig. S13 and S14 (ESI†). For reference, the equilibrium potential shift expected in ideal solutions (assuming a Na+ activity coefficient of 1, i.e., irrespective of the Na+ concentrations) is depicted in Fig. 3a. For the pure aqueous electrolytes, the equilibrium potential shifts upwards with the increase of Na+ molality. It is noteworthy that the upward potential shift is markedly larger than that expected in ideal solutions, which suggests an increased Na+ activity coefficient.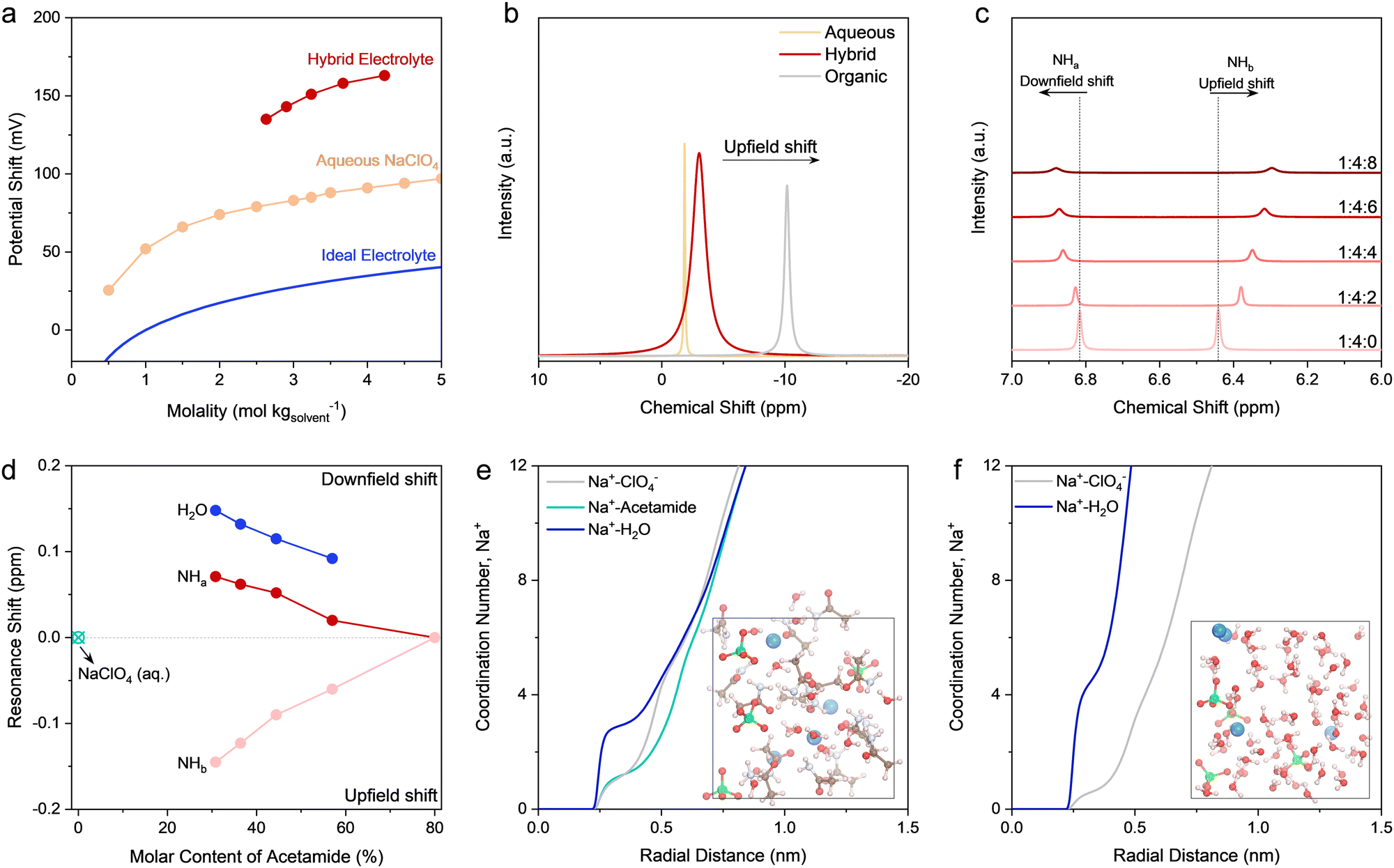 | ||
| Fig. 3 Physicochemical characterization of the electrolytes. (a) Equilibrium potential shift of Na+ intercalation reaction as a function of Na+ molality for different electrolytes, while the potential shift of the Na3−xV2(PO4)3 electrode in different electrolytes was recorded versus the classical Ag/AgCl in the cell setup shown in the ESI.† The electrolytes include the hybrid electrolytes and typical aqueous NaClO4 electrolytes with a series of concentrations. For the hybrid electrolytes, the resultant concentration comes down when introducing higher molar ratio of water like 4.23 m for the composition of nNaClO4:nacetamide:nwater = 1:4:8 and 3.24 m for nNaClO4:nacetamide:nwater = 1:4:4. The potential shift of the ideal electrolytes, calculated based on the Nernst equation, is included for comparison, in which the activity coefficient of Na+ is supposed to be 1. (b) 23Na NMR spectra of the organic electrolyte, hybrid electrolyte (nNaClO4:nacetamide:nwater = 1:4:4), and aqueous NaClO4 electrolyte (3.24 m). (c) 1H (–NH2) NMR spectra of the hybrid electrolytes with a series of compositions, shown as nNaClO4:nacetamide:nwater in panel (c). The two resonance signals are labeled as –NHa and –NHb. (d) Summary of the relative resonance shifts of different protons, i.e., 1H (H2O), 1H (–NHa), and 1H (–NHb). The peak shifts of amide protons were calculated taking the control electrolyte with nNaClO4:nacetamide:nwater = 1:4:0 as the reference, whereas the peak shifts of 1H (H2O) were determined using the aqueous NaClO4 (3.24 m) as the reference. Radial distribution function (RDF) plots of the hybrid electrolyte at nNaClO4:nacetamide:nwater = 1:4:4 (e) and 3.24 m aqueous NaClO4 electrolyte (f), which were calculated based on ab initio MD. Insets show the corresponding snapshots of equilibrium trajectories for these two electrolytes. | ||
When introducing acetamide to make the hybrid electrolytes, the upward shift of equilibrium potential undergoes an even larger extent in comparison with that of pure aqueous electrolytes. From the thermodynamics point of view, the control over the molar ratio of acetamide to water can efficiently modulate the solvation structure of Na+, which provides a straightforward strategy to illustrate electrolyte chemistry in detail. Closer observation indicates that the decrease of water content in the hybrid electrolytes causes the upward shift in potential. These results signify the different solvation sheath structures of Na+ upon incorporating acetamide, whereas water plays a pivotal role. To verify this assumption, we conducted liquid-state nuclear magnetic resonance (NMR) spectroscopy to investigate the chemical environment of core elements in the electrolytes (Fig. S15, ESI†). Fig. 3b is the comparison of 23Na NMR spectra for three representative electrolytes. Compared to the aqueous electrolyte, the 23Na resonance peak for the hybrid electrolyte shows a pronounced upfield shift. This indicates a strong shielding effect introduced by acetamide (Fig. S16, ESI†), while acetamide starts modifying the solvation sheath of Na+ in the hybrid electrolyte, as further confirmed by molecular dynamics (MD) simulations discussed later.
The changes in the chemical environment for the hybrid electrolytes can be further revealed by the NMR spectra for amine protons of acetamide (Fig. 3c). Compared to the pristine acetamide (Fig. S17, ESI†), one notable characteristic of the hybrid electrolytes is the appearance of two resonance signals of amine protons. This indicates two different chemical environments for the acetamide molecules in the hybrid electrolytes. Another key characteristic is the different shift directions of these two resonance peaks: upfield shift for the resonance at ∼6.45 ppm and downfield shift for the resonance at ∼6.82 ppm. The downfield and upfield resonance shifts reveal decreased and increased electron density of the nuclei, respectively.43 Accordingly, we attribute the resonance at ∼6.82 ppm to amine protons of acetamide interacting with water molecules through hydrogen bonding and the resonance at ∼6.45 ppm to amine protons that interact with water molecules in the solvation sheath of Na+. This conclusion can be endorsed by the downfield shift of water protons (Fig. S15, ESI†) and the relative amount of proton resonance shift (Fig. 3d) upon decreasing the molar content of acetamide in the hybrid electrolytes. Conversely, when decreasing the molar content of acetamide, the resonance at ∼6.45 ppm shows an upfield shift yet a similar extent of relative peak shift compared to that of water protons. However, less pronounced peak shifts are observed for the peaks around 6.8 ppm. Therefore, we hypothesize that water can be partially freed from the solvation sheath by acetamide replacement, while the hydrogen bonding networks of water molecules can be substantially disrupted by acetamide. The most pronounced upfield shift occurs in the organic electrolyte, which can be attributed to the strong shielding effect caused by anions in the solvation sheath of Na+. The increased line width for the hybrid electrolyte entails improved interaction and complexation among Na+, ClO4−, H2O, and acetamide, whereas the fast exchange between Na+ and molecules/anions in the solvation shell can be preserved.44
Ab initio MD simulations were further performed to provide theoretical insights into the electrolytes. The solvation structures of different electrolytes are in good agreement with experimental results. MD simulations exhibit that ClO4− anions dominate the solvation sheath of Na+ in the organic electrolyte (Fig. S18, ESI†), matching well with the NMR spectroscopic results (Fig. S15, ESI†). Similarly, the first solvation shell of Na+ features ClO4− coordination in the aqueous electrolytes to form contact ionic pairs (CIPs) (Fig. 3e), while a free water network formed by uncoordinated water molecules exists, which can lead to side reactions and fast capacity degradation at the anode (Fig. S19, ESI†). Differently, in the hybrid electrolyte, the entrance of acetamide into the first solvation sheath of Na+ displaces water in the solvation shell (Fig. 3f). Acetamide bears an electron-donating functional group of –NH2 to induce a strong dipole moment of acetamide (3.76 Debye vs. 1.85 Debye for water). Therefore, an increased affinity between acetamide and Na+ (increased ion–dipole interaction) can be initiated, which in turn promotes the Na+–O (acetamide) association to displace water molecules in the solvation sheath,45 as further confirmed by the NMR and FT-IR results (Fig. S1 and S15, ESI†). As expected, the –NH2 and –CO groups in acetamide can assist its penetration into the hydrogen-bond network of water molecules, which disrupts the intermolecular network in free water clusters and thereby suppresses the water activity thermodynamically.18–21,23 This is a critical enabler for the high-efficiency PTCDA anode working at low redox potentials.
Mechanism for morphological and phase evolutions
As illustrated by electrochemistry, electron microscopy, and in situ XRD experiments, two noticeable changes can be identified in the organic PTCDA electrode when storing Na+ in different electrolytes: morphology and phase transformation. In the following experiment, PTCDA was tested in a water-free electrolyte made of NaClO4 and acetamide (Fig. S20, ESI†). Similar voltage profiles to the case of organic electrolyte are observed, indicating a similar Na+ storage mechanism in these two electrolytes. Upon introducing a minor amount of water into this water-free electrolyte (Fig. S20, ESI†), we observe nearly identical voltage profile evolution between the first and following sodiation processes to that of the hybrid electrolyte.This indicates the critical role of water in enabling the morphological phase evolutions of the organic PTCDA electrode. This conclusion can be further reinforced by the large discrepancy between the initial and subsequent cycles in the voltage plots when operating PTCDA in an aqueous NaClO4 electrolyte (Fig. S19, ESI†).
According to the spectroscopic analysis and MD simulations, the solvation sheath of Na+ in the hybrid electrolyte features a significant contribution of water molecules. During sodiation, Na+ with this unique hydrated solvation structure approaches the electrode/electrolyte interface to create a water-enriched Helmholtz layer, as proven by previous theoretical simulations.46 This indicates a certain extent of hydration of the surface region of the PTCDA electrode in the hybrid electrolyte (Fig. S21 and S22, ESI†).12,32 Given the much higher solubility of PTCDA in water than that in organic solvent,9 we hypothesize that slight dissolution of negatively charged PTCDA units upon uptaking Na+ in the water-containing interfacial region is one key enabler for the morphological changes (Fig. S21–S24 and Supplementary Note 1, ESI†). On the other hand, different from other organic electrodes with covalent frameworks (e.g., polymers), monomer PTCDA particulates are constructed by aromatic organic units featuring weak interactions (e.g., π–π, hydrogen bond) among them. This endows the molecular units with sufficient flexibility for morphological evolutions especially when introducing strong Na−O ionic bonds between the molecular units on sodiation.
The second question would be the mechanism of different crystalline structure transformations of PTCDA in organic and hybrid electrolytes. We hypothesized the crucial role of water in initiating structural evolutions. The electrode reaction of PTCDA in the hybrid electrolytes (or generally in the presence of water, as analyzed above) can be ascribed to a heterogeneous liquid (soluble Nax-PTCDA)–solid (insoluble PTCDA) reaction. This reaction resembles that of sulfur in storing alkaline metal ions in organic electrolytes.40,47 Given the structural flexibility of monomer PTCDA, a different storage mechanism is thermodynamically feasible considering the high activity of Na+ in the hybrid electrolyte. It is reasonable to conclude that the weak solvation of water and acetamide for solvated Na+ can facilitate Na+ desolvation and insertion into the structure of PTCDA, which in turn reduces the kinetic barrier for the unit-cell volume change. Similarly, a new conversion reaction mechanism of sulfur cathodes was recently found in aqueous electrolytes compared to that in organic electrolytes.48
The different mechanisms of Na+ storage in PTCDA can be further revealed by different Na+ solid diffusion kinetics in these two electrolytes. As revealed by galvanostatic intermittent titration technique (GITT) plots (Fig. S25, ESI†), the voltage response of PTCDA in the hybrid electrolyte shows remarkably decreased overpotentials during (de)sodiation compared to that of organic electrolyte. This signifies faster Na+ diffusion in PTCDA when working in the hybrid electrolyte, wherein the diffusion coefficient is about one order of magnitude higher than that enabled by organic electrolyte.
Ultrahigh rate performance and mechanism
The understanding of the sodiation mechanism of PTCDA motivated us to further evaluate its rate performance in the hybrid electrolyte, especially considering the facile solid-state diffusion of Na+ and the unique solvation environment of the hybrid electrolyte. On the basis of the GCD profiles recorded in a three-electrode setup (Fig. 4a), there are limited polarization and profile distortion even at a high rate of 1000C, delivering a capacity of 118 mA h g−1 (85.5% capacity retention of the theoretical capacity). Such high rate performance is competitive to and even superior to state-of-the-art battery electrodes for smaller cations (e.g., H+, Li+) storage (Fig. S26, ESI†).28–32 On the other hand, PTCDA exhibits considerably poorer rate performance when working in the organic electrolyte (Fig. 4b). As expected, inferior rate performance in the organic electrolyte is associated with lower Na+ diffusion rate (or ionic conductivity) and sluggish solid-state diffusion. Dissociating Na+ from the strong solvation of CIPs in the organic electrolyte can further create kinetic barriers for Na+ storage.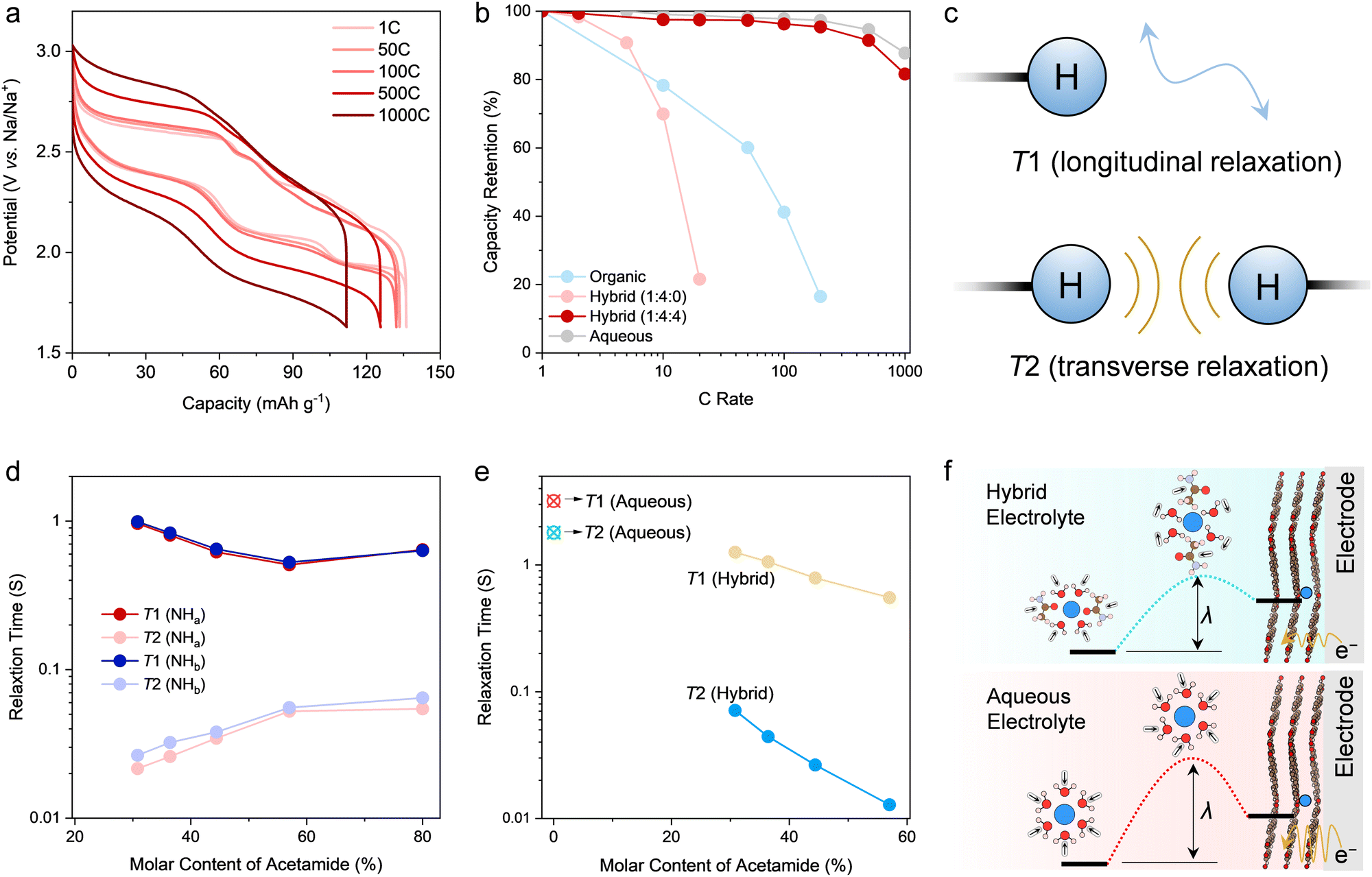 | ||
| Fig. 4 Rate performance and mechanism analysis. (a) Galvanostatic charge–discharge plots of the PTCDA electrodes in the hybrid electrolyte with nNaClO4:nacetamide:nwater = 1:4:4. (b) Comparison of rate performance of the PTCDA electrode in different electrolytes. (c) Scheme of the T1 relaxation (nuclei tumbling) and T2 relaxation (spin–spin interactions). Herein, the protons of the solvents were taken as the probes to uncover roles of acetamide and water in the electrolytes. Comparison of the T1 and T2 values for the amide protons (d), i.e., 1H (–NH2), and water protons (e), 1H (H2O). Aqueous NaClO4 electrolyte (3.24 m) was included in panel (e) for comparison. The molar content of acetamide for the x axis of panels (d) and (e) was calculated based on the nNaClO4:nacetamide:nwater for the hybrid electrolytes, for example, 80% for nNaClO4:nacetamide:nwater = 1:4:0 and 44.4% for nNaClO4:nacetamide:nwater = 1:4:4. In this way, the relationship between relaxation times and the electrolyte compositions can be clearly demonstrated. (f) Scheme of the concerted Na+ and electron transfer limited by the solvation shell reorganization, while the hybrid electrolyte delivers lower energy barrier for solvation sheath reorganization and faster electron transfer kinetics to facilitate the high rate performance. | ||
It is noteworthy that PTCDA working in the hybrid electrolyte delivers a competitive rate performance to that of the other control electrolyte of aqueous NaClO4 (Fig. 4b), which is the same molality as the hybrid electrolyte. However, the aqueous electrolyte shows higher ionic conductivity (128 mS cm−1). Accordingly, the solvation environment of Na+ is the key to understanding the mechanism of how acetamide enhances charge transfer for Na+ storage. Comparing the hybrid electrolyte with the aqueous electrolyte, MD simulations and spectroscopic results uncover that the role of acetamide is to displace water molecules from the solvation shell of Na+. This is ascribed to the higher affinity between acetamide and Na+, as initiated by the strong dipole moment of acetamide, to facilitate Na+–O (acetamide) association. This in turn delivers increased binding strength between acetamide and Na+, revealing more stable solvation sheaths for the hybrid electrolyte. However, solvates in the hybrid electrolyte featuring higher association strength than that in aqueous electrolyte support faster (de)sodiation. For an electrode-bound electrochemical reaction, the electron transfer can be approximated as non-adiabatic. This process involves ionic intermediates featuring a reorganized solvation sheath according to the Marcus theory.42,49 The corresponding reorganization energy (λ) denotes the energy required to reorganize the solvation sheath, herein Na+, to facilitate electron transfer. Accordingly, the species in the solvation sheath of Na+ show an impact through their contribution to λ. Compared with smaller molecules, larger molecules in the solvation shells tend to show lower λ.42 Since large acetamide appears in the solvation sheath of Na+, as proven by our spectroscopic tests and ab initio MD simulations, it is reasonable to infer that the reduced λ for the hybrid electrolyte can facilitate the desolvation and electron transfer to enable high-rate performance during Na+ storage (Fig. 4f).
The influence of solvents on electron transfer is larger than simply through their energetic contribution to solvent reorganization upon charge transfer. The dynamics of solvent reorganization can be represented by the longitudinal relaxation time (T1) of the solvent.42,50 Analyzing T1 is thus expected to deliver insights on how acetamide impacts the dynamics of solvent reorganization to enable the unprecedented high rate performance. Herein, we conducted inversion recovery technique based on 1H NMR to measure the T1 values for different electrolytes (Fig. S27–S29; see Experimental details; see calculation details in Supplementary Note 2, ESI†).51 Hydrogen nuclei, especially –NH2 of acetamide and –OH of water, were chosen as the probes to study relaxation behaviors of solvents (acetamide and H2O) because of protons’ intrinsic high sensitivity and spectrum resolution, distinctive chemical shifts for different species (Fig. 4c), and their direct indication with chemical environment (solvation and interaction with water through hydrogen bond). T1 is related to the size of a solvent molecule containing hydrogen nuclei and in particular the tumbling rate of the molecule (typically known as molecular motion). According to the Marcus theory microscopic model,42,52 heterogeneous electrochemical reaction rate constant (k0) is proportional to T1−θ, wherein θ is a fraction between 0 and 1 (Supplementary Note 3, ESI†). Based on our 1H NMR longitudinal relaxation experiments, T1 for 1H (H2O) is four times decreased for the hybrid electrolyte compared to that of aqueous electrolyte (Fig. 4d). This corresponds to approximately four times increase of heterogeneous reaction rate upon solvent motions or reorganizations in the hybrid electrolyte.
Indeed, it is reasonable to find clues about the decreased T1 of 1H (H2O) from the chemical environment of the hybrid electrolyte, as previously depicted by MD simulations and spectroscopic measurements. Asymmetric acetamide with a high dipole moment can displace water in the solvation sheath of Na+ and to break percolating hydrogen bonding networks between water molecules. This creates more polarizable environments especially around Na+ and induces increased intermolecular dipole–dipole interaction to decrease T1 for both acetamide and water (Fig. S30–S32, ESI†).53 This phenomenon can be further evidenced by the paradoxical trends of transverse or spin–spin relaxation time (Fig. 4d and e), i.e., T2, for 1H (–NH2) and 1H (H2O) upon increasing the molar ratio of water in the hybrid electrolytes. Specifically, when decreasing the molar ratio of water to acetamide, T2 values for 1H (H2O) are shortened (Fig. 4e), following the same trend as that of T1 for 1H (H2O). This can be attributed to the increased dipolar interactions among solvent molecules and reduced tumbling rates of water, which can be further revealed by the increased viscosity and decreased ionic conductivity of the electrolytes on decreasing water content (Supplementary Fig. S4, ESI†). Nonetheless, T2 values for 1H (–NH2) decrease along with the increase of T1 values for 1H (–NH2) when changing solvent ratios (Fig. 4d). The rationale is the less frequent spin–spin interaction between acetamide molecules and water molecules as free acetamide and water molecules move rapidly (decreased viscosity) when water content in the hybrid electrolyte is increased. T1 reveals the dynamic feature of solvent molecules (herein water and acetamide), while T1 is intimately related to the electrolyte chemistry. Accordingly, the modulation of solvation structures can efficiently regulate the dynamic behaviors (e.g., orientation) of solvent molecules to influence the interfacial electrochemistry. To summarize, the more polarizable and less compact solvation shells, as induced by acetamide, can facilitate the kinetics of solvent reorganization through coherent motions of acetamide and water in the first solvation shells,54,55 which consequently promotes electron transfer for the unprecedented rate capability of PTCDA in the hybrid electrolyte (Fig. 4f).
Another factor also needs attention in this process. It is the downsizing effect of PTCDA in the hybrid electrolyte to minimize the kinetic barrier of structural evolution to ensure fast solid-state Na+ diffusion. Herein, we can conclude that it is the combination of the unique redox chemistry of PTCDA and the hybrid electrolyte design that makes the unprecedented rate performance achievable.
High-performance aqueous full Na-ion batteries
The cycle life has been one obstacle for organic monomer electrodes in aqueous electrolytes, which show rapid capacity degradation due to the dissolution of organic electrodes and continuous loss of active electrode materials during cycling. This issue can be efficiently suppressed in the hybrid electrolyte because of limited free water. The PTCDA electrode shows decent cycling stability in the hybrid electrolyte, though a capacity attenuation of 38% still occurs after 1350 cycles at a rate of 100C (Fig. 5a). To further control the dissolution issue, a thin and lightweight membrane of two-dimensional (2D) Ti3C2Tx (Tx represent negatively charged surface functional groups like –O and –OH) MXene was introduced between the PTCDA electrode and the separator to trap the soluble intermediate. Ti3C2Tx MXene turns out to be an excellent interlayer to cope with the instability issue of organic electrodes without coming at the cost of the rate capability significantly (Fig. 5b and c). Firstly, the negatively charged surface caused by the functional groups on the MXene surface can act as ion-hopping sites of positively charged carriers,56 namely, Na+ in this study, and impede the transport of negatively charged species (e.g., PTCDAx−, x ≤ 2) as a result of electrostatic repulsions (Fig. S33, ESI†). Furthermore, 2D MXene nanosheets (Fig. S34, ESI†) can act as physical barriers. Last but not least, Ti3C2Tx MXene features metallic electrical conductivity, while its 2D morphology with large sufficient interlayer spacing can facilitate fast Na+ transport (Fig. S35 and S36 and Supplementary Notes 4 and 5, ESI†). After introducing the Ti3C2Tx MXene interlayer (Fig. S35, ESI†), the capacity retention of the PTCDA electrode reaches 70% after 20000 cycles (Fig. 5a).
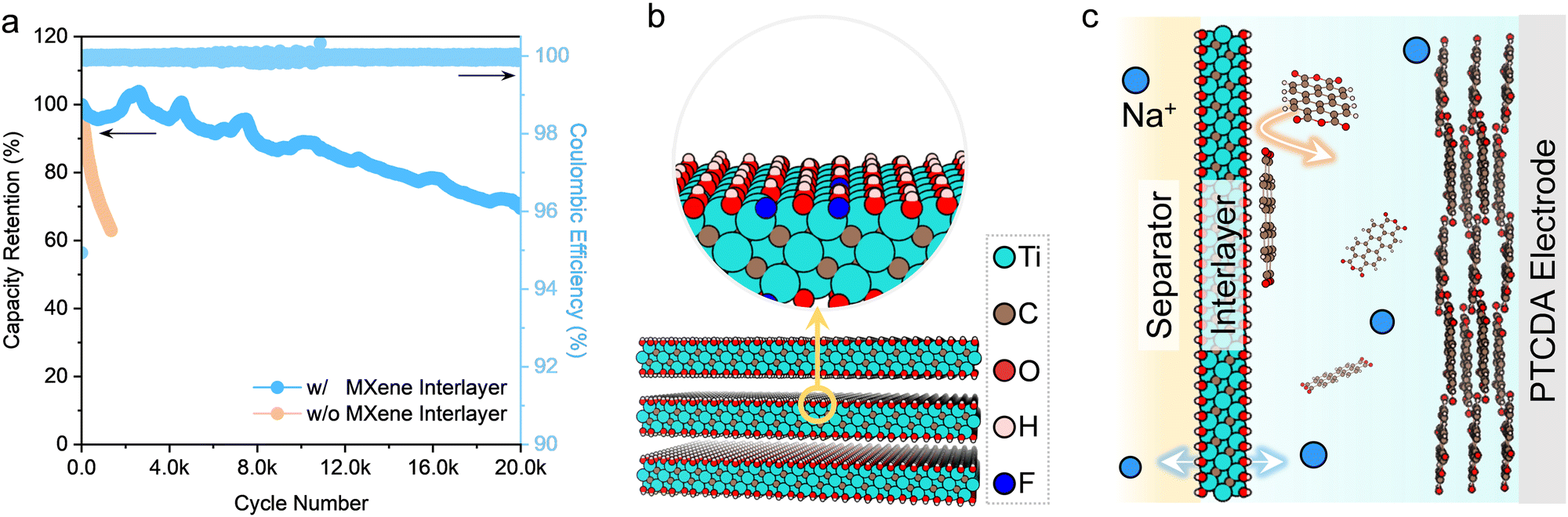 | ||
| Fig. 5 Cycling stability tests. Cycling stability of the PTCDA electrode in the hybrid electrolyte (nNaClO4:nacetamide:nwater = 1:4:4) with and without the MXene interlayer (current rate: 100C). Schemes of the 2D MXene nanosheets with negatively charged surfaces (b) and a cell configuration with the MXene interlayer to mitigate the dissolution of PTCDA during operation (c). | ||
To further illustrate the concept of aqueous Na-ion batteries, we assembled a full battery with a PTCDA anode, a Prussian blue analogue cathode (Fig. S38, ESI†), a Ti3C2Tx MXene interlayer between separator and anode, and the hybrid electrolyte. The full battery delivers a high rate performance (up to 80C) and a promising cycling performance (capacity retention of 70% after 4000 cycles) (Fig. S39, ESI†).
Conclusions
In this work, we have achieved the mechanistic understanding of charge storage in a classical organic electrode of PTCDA, in different electrolyte systems, including organic, aqueous, and hybrid electrolytes. We discovered a redox mechanism associated with simultaneous morphological and structural changes when operating PTCDA in the hybrid electrolyte, whereas water was confirmed to play a crucial role. Moreover, we introduced the relaxation time of the solvent that can be used as the descriptor, which can be employed to evaluate the reorganization of solvent to enable Na+ desolvation and intercalation and the coupled electron transfer kinetics. A small and polar molecule of acetamide was demonstrated as an efficient additive to modulate the solvation sheath of Na+ and electrolyte chemistry, while acetamide can efficiently facilitate the Na+ desolvation and electron transfer to unlock the ultrahigh rate performance. Our design principles of electrolytes and the methodology of evaluating solvent reorganization processes are generally applicable to many other metal-ion batteries. Thermodynamically and kinetically, solvent reorganization and interfacial electron transfer can be modulated by controlling the molecular structures and compositions, e.g., creating less compact structure and introducing donor atoms, to further improve the fast charging capability of the key battery technologies. This is urgently needed for stationary energy storage of renewable energy and transport electrification.Author contributions
Y. Z. and H. N. A. conceived the idea and conceptualized the work. Y. Z. conducted the materials synthesis, electrochemical tests, in situ XRD experiments and materials/electrolyte characterizations. X. G. performed the NMR experiments. S. T. performed the simulations. J. Y. and Z. T. conducted electron microscopy and contact angle experiments. Y. Y. performed the fittings of XRD patterns. G. T. H. performed the XPS experiments and analysis. Y. Z. wrote the paper, and all authors contributed to the manuscript preparation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research conducted in this work was supported by King Abdullah University of Science and Technology (KAUST).Notes and references
- T. M. Gür, Energy Environ. Sci., 2018, 11, 2696 RSC.
- H. Kim, J. Hong, K.-Y. Park, H. Kim, S.-W. Kim and K. Kang, Chem. Rev., 2014, 114, 11788 CrossRef CAS PubMed.
- D. Chao, W. Zhou, F. Xie, C. Ye, H. Li, M. Jaroniec and S.-Z. Qiao, Sci. Adv., 2020, 6, eaba4098 CrossRef CAS PubMed.
- Y. Liang and Y. Yao, Nat. Rev. Mater., 2023, 8, 109 CrossRef.
- R. Usiskin, Y. Lu, J. Popovic, M. Law, P. Balaya, Y.-S. Hu and J. Maier, Nat. Rev. Mater., 2021, 6, 1020 CrossRef CAS.
- Y. Li, Q. Zhou, S. Weng, F. Ding, X. Qi, J. Lu, Y. Li, X. Zhang, X. Rong, Y. Lu, X. Wang, R. Xiao, H. Li, X. Huang, L. Chen and Y.-S. Hu, Nat. Energy, 2022, 7, 511 CrossRef CAS.
- H.-W. Lee, R. Y. Wang, M. Pasta, S. W. Lee, N. Liu and Y. Cui, Nat. Commun., 2014, 5, 5280 CrossRef CAS PubMed.
- X. Li, Y. Wang, D. Wu, L. Liu, S.-H. Bo and G. Ceder, Chem. Mater., 2016, 28, 6575 CrossRef CAS.
- M. Lee, J. Hong, J. Lopez, Y. Sun, D. Feng, K. Lim, W. C. Chueh, M. F. Toney, Y. Cui and Z. Bao, Nat. Energy, 2017, 2, 861 CrossRef CAS.
- S. Wu, W. Wang, M. Li, L. Cao, F. Lyu, M. Yang, Z. Wang, Y. Shi, B. Nan, S. Yu, Z. Sun, Y. Liu and Z. Lu, Nat. Commun., 2016, 7, 13318 CrossRef CAS PubMed.
- W. Walker, S. Grugeon, O. Mentre, S. Laruelle, J.-M. Tarascon and F. Wudl, J. Am. Chem. Soc., 2010, 132, 6517 CrossRef CAS PubMed.
- I. A. Rodríguez-Pérez, Y. Yuan, C. Bommier, X. Wang, L. Ma, D. P. Leonard, M. M. Lerner, R. G. Carter, T. Wu, P. A. Greaney, J. Lu and X. Ji, J. Am. Chem. Soc., 2017, 139, 13031 CrossRef PubMed.
- Y. Lu and J. Chen, Nat. Rev. Chem., 2020, 4, 127 CrossRef CAS PubMed.
- K. Qin, J. Huang, K. Holguina and C. Luo, Energy Environ. Sci., 2020, 13, 3950 RSC.
- Y. Hu, Q. Yu, W. Tang, M. Cheng, X. Wang, S. Liu, J. Gao, M. Wang, M. Xiong, J. Hu, C. Liu, T. Zou and C. Fan, Energy Storage Mater., 2021, 41, 738 CrossRef.
- Y. J. Kim, W. Wu, S.-E. Chun, J. F. Whitacre and C. J. Bettinger, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 20912 CrossRef CAS PubMed.
- Y. Xu, M. Zhou and Y. Lei, Mater. Today, 2018, 21, 60 CrossRef CAS.
- Y. S. Meng, V. Srinivasan and K. Xu, Science, 2022, 378, eabq3750 CrossRef CAS PubMed.
- L. Suo, O. Borogin, T. Gao, M. Olguin, J. Ho, X. Fan, C. Luo, C. Wang and K. Xu, Science, 2015, 350, 938 CrossRef CAS PubMed.
- Y. Yamada, J. Wang, S. Ko, W. Watanabe and A. Yamada, Nat. Energy, 2019, 4, 269 CrossRef CAS.
- J. Zheng, G. Tan, P. Shan, T. Liu, J. Hu, Y. Feng, L. Yang, M. Zhang, Z. Chen, Y. Lin, J. Lu, J. C. Neuefeind, Y. Ren, K. Amine, L.-W. Wang, K. Xu and F. Pan, Chem, 2018, 4, 2872 CAS.
- S. Sayah, A. Ghosh, M. Baazizi, R. Amine, M. Dahbi, Y. Amine, F. Ghamouss and K. Amine, Nano Energy, 2022, 98, 107336 CrossRef CAS.
- J. Xie, Z. Liang and Y.-C. Lu, Nat. Mater., 2020, 19, 1006 CrossRef CAS PubMed.
- F. Ming, Y. Zhu, G. Huang, A.-H. Emwas, H. Liang, Y. Cui and H. N. Alshareef, J. Am. Chem. Soc., 2022, 144, 7160 CrossRef CAS PubMed.
- D. Han, C. Cui, K. Zhang, Z. Wang, J. Gao, Y. Guo, Z. Zhang, S. Wu, L. Yin, Z. Weng, F. Kang and Q.-H. Yang, Nat. Sustainability, 2022, 5, 205 CrossRef.
- X. He, B. Yan, X. Zhang, Z. Liu, D. Bresser, J. Wang, R. Wang, X. Cao, Y. Su, H. Jia, C. P. Grey, H. Frielinghaus, D. G. Truhlar, M. Winter, J. Li and E. Paillard, Nat. Commun., 2018, 9, 5320 CrossRef CAS PubMed.
- Y. Song, P. Ruan, C. Mao, Y. Chang, L. Wang, L. Dai, P. Zhou, B. Lu, J. Zhou and Z. He, Nano-Micro Lett., 2022, 14, 218 CrossRef CAS PubMed.
- X. Wu, J. J. Hong, W. Shin, L. Ma, T. Liu, X. Bi, Y. Yuan, Y. Qi, T. W. Surta, W. Huang, J. Neuefeind, T. Wu, P. A. Greaney, J. Lu and X. Ji, Nat. Energy, 2019, 4, 123 CrossRef CAS.
- R. Emanuelsson, M. Sterby, M. Strømme and M. Sjödin, J. Am. Chem. Soc., 2017, 139, 4828 CrossRef CAS PubMed.
- S. W. Lee, N. Yabuuchi, B. M. Gallant, S. Chen, B.-S. Kim, P. T. Hammond and Y. Shao-Horn, Nat. Nanotechnol., 2010, 5, 531 CrossRef CAS PubMed.
- B. Kang and G. Ceder, Nature, 2009, 458, 190 CrossRef CAS PubMed.
- J. Zheng, Y. Hou, Y. Duan, X. Song, Y. Wei, T. Liu, J. Hu, H. Guo, Z. Zhuo, L. Liu, Z. Chang, X. Wang, D. Zherebetskyy, Y. Fang, Y. Lin, K. Xu, L.-W. Wang, Y. Wu and F. Pan, Nano Lett., 2015, 15, 6102 CrossRef CAS PubMed.
- J. Kim, Y. Kim, J. Yoo, G. Kwon, Y. Ko and K. Kang, Nat. Rev. Mater., 2023, 8, 54 CrossRef.
- Y. Jin, P. M. L. Le, P. Gao, Y. Xu, B. Xiao, M. H. Engelhard, X. Cao, T. D. Vo, J. Hu, L. Zhong, B. E. Matthews, R. Yi, C. Wang, X. Li, J. Liu and J.-G. Zhang, Nat. Energy, 2022, 7, 718 CrossRef CAS.
- S. Ko, T. Obukata, T. Shimada, N. Takenaka, M. Nakayama, A. Yamada and Y. Yamada, Nat. Energy, 2022, 7, 1217 CrossRef CAS.
- D. Monti, E. Jónsson, A. Boschin, M. R. Palacín, A. Ponrouch and P. Johansson, Phys. Chem. Chem. Phys., 2020, 22, 22768 RSC.
- R. Demir-Cakan, M. R. Palacin and L. Croguennec, J. Mater. Chem. A, 2019, 7, 20519 RSC.
- W. Luo, M. Allen, V. Raju and X. Ji, Adv. Energy Mater., 2014, 4, 1400554 CrossRef.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, Nature, 2020, 407, 496 CrossRef PubMed.
- Q. Pang, A. Shyamsunder, B. Narayanan, C. Y. Kwok, L. A. Curtiss and L. F. Nazar, Nat. Energy, 2018, 3, 783 CrossRef CAS.
- C. Wang, Y. Fang, Y. Xu, L. Liang, M. Zhou, H. Zhao and Y. Lei, Adv. Funct. Mater., 2016, 26, 1777 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, 2nd edn, John Wiley & Sons, New Jersey, 2001 Search PubMed.
- M. S. Kim, Z. Zhang, P. E. Rudnicki, Z. Yu, J. Wang, H. Wang, S. T. Oyakhire, Y. Chen, S. C. Kim, W. Zhang, D. T. Boyle, X. Kong, R. Xu, Z. Huang, W. Huang, S. F. Bent, L.-W. Wang, J. Qin, Z. Bao and Y. Cui, Nat. Mater., 2022, 21, 445 CrossRef CAS PubMed.
- E. G. Bloor and R. G. Kidd, Can. J. Chem., 1968, 46, 3425 CrossRef CAS.
- S. Hou, X. Ji, K. Gaskell, P.-F. Wang, L. Wang, J. Xu, R. Sun, O. Borodin and C. Wang, Science, 2021, 374, 172 CrossRef CAS PubMed.
- Y.-H. Wang, S. Zheng, W.-M. Yang, R.-Y. Zhou, Q.-F. He, P. Radjenovic, J.-C. Dong, S. Li, J. Zheng, Z.-L. Yang, G. Attard, F. Pan, Z.-Q. Tian and J.-F. Li, Nature, 2021, 600, 81 CrossRef CAS PubMed.
- J. Conder, R. Bouchet, S. Trabesinger, C. Marino, L. Gubler and C. Villevieille, Nat. Energy, 2017, 2, 17069 CrossRef CAS.
- C. Yang, L. Suo, O. Borodin, F. Wang, W. Sun, T. Gao, X. Fan, S. Hou, Z. Ma, K. Amine, K. Xu and C. Wang, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6197 CrossRef CAS PubMed.
- A. Chandra, A theoretical study of outer sphere electron transfer reactions in electrolyte solutions, J. Chem. Phys., 1999, 110, 1569 CrossRef CAS.
- M. Kozik, N. Sutin and J. R. Winkler, Coord. Chem. Rev., 1990, 97, 23 CrossRef CAS.
- R. S. Macomber, A Complete Introduction to Modern NMR Spectroscopy, John Wiley & Sons, New Jersey, 1997 Search PubMed.
- W. R. Fawcett and M. Opallo, J. Electroanal. Chem., 1993, 349, 273 CrossRef CAS.
- J. Karjalainen, H. Henschel, M. J. Nissi, M. T. Nieminen and M. Hanni, J. Phys. Chem. B, 2022, 126, 2538 CrossRef CAS PubMed.
- E. Biasin, Z. W. Fox, A. Andersen, K. Ledbetter, K. S. Kjær, R. Alonso-Mori, J. M. Carlstad, M. Chollet, J. D. Gaynor, J. M. Glownia, K. Hong, T. Kroll, J. H. Lee, C. Liekhus-Schmaltz, M. Reinhard, D. Sokaras, Y. Zhang, G. Doumy, A. M. March, S. H. Southworth, S. Mukamel, K. J. Gaffney, R. W. Schoenlein, N. Govind, A. A. Cordones and M. Khalil, Nat. Chem., 2021, 13, 343 CrossRef CAS PubMed.
- P. A. Zagrebin, R. Buchner, R. R. Nazmutdinov and G. A. Tsirlina, J. Phys. Chem. B, 2010, 114, 311 CrossRef CAS PubMed.
- B. Anasori, M. R. Lukatskaya and Y. Gogotsi, Nat. Rev. Mater., 2017, 2, 16098 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of experiments, characterizations, and simulations. See DOI: https://doi.org/10.1039/d4ee00367e |
| This journal is © The Royal Society of Chemistry 2024 |