
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntegrated carbon capture and CO production from bicarbonates through bipolar membrane electrolysis†
Hakhyeon
Song‡
 ab,
Carlos A.
Fernández‡
a,
Hyeonuk
Choi‡
c,
Po-Wei
Huang
b,
Jihun
Oh
*c and
Marta C.
Hatzell
*ab
ab,
Carlos A.
Fernández‡
a,
Hyeonuk
Choi‡
c,
Po-Wei
Huang
b,
Jihun
Oh
*c and
Marta C.
Hatzell
*ab
aGeorge W. Woodruff School of Mechanical Engineering, Georgia Institute of Technology, Atlanta, GA 30332, USA. E-mail: marta.hatzell@me.gatech.edu
bSchool of Chemical and Biomolecular Engineering, Georgia Institute of Technology, Atlanta, GA 30332, USA
cDepartment of Materials Science and Engineering, Korea Advanced Institute of Science and Technology (KAIST), 291 Daehak-ro, Yuseong-gu, Daejeon 34141, Republic of Korea. E-mail: jihun.oh@kaist.ac.kr
First published on 16th April 2024
Abstract
The electrochemical CO2 reduction reaction (CO2RR) offers an environmentally friendly method to transform sequestered CO2. While gas-phase electrolysis systems provide high efficiency, gas-phase electrolysis systems face challenges related to carbonate precipitate formation and crossover. As an alternative, liquid-phase (bi)carbonate electrolysis systems based on the use of bipolar membrane (BPM) electrode assemblies have emerged. These systems not only streamline the carbon capture and conversion process but also present economic benefits. However, liquid-phase (bi)carbonate electrolysis cells suffer limited stability and selectivity at relevant operating currents. Here, utilizing a Ni-based single atom catalyst (Ni-SAC) and bicarbonate electrolyte, we demonstrate exceptional CO faradaic efficiency (93%) at a partial current density of −186 mA cm−2 at −3.7 V for over 18 hours with an integrated carbon capture system. Furthermore, we conducted a comparative analysis of various performance metrics between bicarbonate electrolysis and CO2 gas electrolysis. Our results highlight the superior advantages of BPM-based electrolysis in terms of CO2 utilization efficiency, stability, and CO product concentration in the outlet stream. Despite a higher energy demand, BPM-based electrolysis presents a technologically promising alternative to conventional CO2 gas-phase systems. This breakthrough paves the way for efficient direct carbon capture and conversion, offering a promising pathway toward a more sustainable and carbon-neutral future.
Broader contextThe electrochemical CO2 reduction reaction (CO2RR) is a promising process that converts CO2 into useful chemicals and fuels. This process has the potential to mitigate greenhouse gas emissions and combat climate change. Advances in catalyst and reactor technology have significantly improved CO2RR performance, which can achieve industrially relevant current density, faradaic efficiency, and overpotentials. However, an energy-intensive process to supply high-purity CO2 is typically required for efficient CO2RR. Furthermore, most of the CO2 in the reactor remains unreacted and can lead to low carbon efficiency, requiring additional purification and/or separation steps. Our study introduces a groundbreaking approach that bypasses the need for high-purity CO2 by directly electrolyzing carbon capture solutions. By utilizing a Ni singleatom catalyst (Ni-SAC) with a bipolar membrane (BPM)-membrane electrode assembly (MEA), we demonstrate efficient bicarbonate electrolysis. This method achieves exceptional enhancements across various performance metrics for the CO2RR. The integrated carbon capture system in this setup effectively closes the loop, directly converting captured CO2 into valuable products, thereby offering a more sustainable and economically viable approach for carbon management. This research thus marks an important advancement in CO2RR technology, contributing to a more sustainable approach to carbon utilization and signaling a step closer to a carbon-neutral future. |
Introduction
The electrochemical CO2 reduction reaction (CO2RR) holds promise as a sustainable approach for valorizing sequestered CO2.1,2 Over the last two decades, CO2 electrolysis reactor engineering has resulted in several reactor architectures, including gas-phase CO2 anion-exchange membrane (AEM)–membrane electrode assembly (MEA) reactors, cation-exchange membrane (CEM)–MEA reactors, liquid-phase (bi)carbonate bipolar membrane (BPM)–MEA reactors, and even membrane-less microfluidic reactors.3–5 The gas-phase AEM–MEA CO2RR has dominated research and development due to the low mass transfer limitations.6 However, the gas-phase systems suffer from the formation of carbonate.7 This reduces CO2 utilization efficiency to below 50% and stability remains a challenge.7–9 These limitations result in the production of 1.48 tons of CO2 equivalent for every unit of CO produced with a 97 wt% purity, presenting a significant challenge for practical implementation of this approach.10Liquid-phase (bi)carbonate BPM–MEA systems allow for the direct conversion of carbon capture solutions derived from direct air capture (DAC), a technology that directly extracts CO2 from ambient air.11–13 These solutions use an alkaline medium (e.g. KOH) that absorbs and converts gas-phase CO2 into bicarbonates or carbonates depending on the pH. The BPM–MEA has emerged as a favorable alternative to CO2 gas electrolysis systems because of the decreased system complexity. When BPM is placed under “reverse bias” conditions, with the CEM facing a cathode electrode, it can result in a lower local pH in the (bi)carbonate solution.14 This promotes CO2 generation from the (bi)carbonates by shifting the CO2/(bi)carbonate equilibrium condition. The in-situ generated CO2 can then be electrochemically reduced into carbon-based products without requiring a separate CO2 production step. It is important to note that the integration of upstream carbon capture and downstream separation is simpler in the BPM-based configuration compared to AEM-based CO2 gas electrolysis. These advantages not only offer economic benefits but also help overcome the limitations associated with CO2 gas electrolysis systems. However, the acidic conditions at the interface between the catalyst layer and BPM enhance the hydrogen evolution reaction (HER).15 Moreover, if the (bi)carbonate electrolyte is not regenerated during long-term electrolysis, the concentration of (bi)carbonates gradually decreases, while the OH− accumulates in the bulk electrolyte.16 These electrolyte changes can eventually result in reduced stability of the CO2RR performance.
Recently, numerous research groups have conducted comprehensive studies on (bi)carbonate electrolysis in the BPM–MEA systems, including catalyst designs17–20 and optimization of local electrolysis environments.5,11–14,21–23 These treatments can effectively control the mass transfer of reactants, such as bicarbonates/carbonates, in-situ generated CO2, and protons at the catalyst layer, which can enhance CO2RR activity. However, despite various approaches, it is still difficult to achieve >90% FE towards CO in a bicarbonate electrolysis system. Also, to maintain stable production in bicarbonate electrolysis, it is necessary to regularly refresh the bicarbonate concentration. For example, the Berlinguette group demonstrated that a porous Ag electrode can produce stable CO FE over 80 hours by manually refreshing the bicarbonate concentration every 500 seconds.14 Given these challenges, the development of catalysts with high activity under acidic conditions and systems that can efficiently convert OH−, generated by the CO2RR and/or HER, back to reactive carbon solutions becomes critically important.
Here, we evaluate the activity and stability of the bicarbonate-fed BPM–MEA electrolysis system with a Ni single-atom catalyst (Ni-SAC) at the cathode. The Ni-SAC electrode exhibited significant activity in an acidic environment (pH ∼ 2) in a gas-diffusion electrode system (Fig. S1, ESI†). This led to enhanced CO2RR performance, while the HER is suppressed. By continuously introducing gas-phase CO2 into the bicarbonate electrolyte, the system not only regenerated reactive carbons but also operated under near steady-state conditions. After optimizing the Ni-SAC electrode and operating system conditions, we achieved remarkable activity, 93% CO FE (approximately −186 mA cm−2 CO partial current density) at −3.7 V over 18 hours. We compare the performance of the system in terms of FE, cell voltage, energy efficiency (EE), CO2 utilization efficiency, CO2 concentration in the outlet stream, and stability with those of a CO2 gas-phase MEA system. Finally, we conducted a direct comparison of the energy and carbon intensity of bicarbonate electrolysis with CO2 gas electrolysis.
Air-to-CO: catalyst, electrochemical reactors, and carbon capture systems
The Ni-SAC was synthesized using a facile pyrolysis method (refer to the Method section and Fig. S2, ESI†). The Ni-SAC is generally composed of N-coordinated Ni atomic active sites, supported on carbon materials (Fig. 1a). Our Ni-SAC also showed the atomically dispersed Ni atoms on the carbon black, which is proved by aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC-HAADF-STEM), as shown in Fig. 1b. Additionally, energy dispersive X-ray spectroscopy (EDS) images indicate a well-distributed presence of Ni and N elements across the entire surface of the Ni-SAC (Fig. S3, ESI†). However, as a well-known limitation of pyrolysis-based synthesis, the Ni-SAC possess some Ni nanoparticles that are enveloped in a thick carbon shell over 30 nm (Fig. S4, ESI†). The Ni nanoparticles are inevitably formed due to the aggregation of Ni ions during the high-temperature calcination step. Moreover, powder X-ray diffraction (XRD) patterns exhibited three peaks at 44.4°, 51.7°, and 76.3°, corresponding to the (111), (200), and (220) planes of Ni crystallites (Fig. S5, ESI†). Fourier-transform of the extended X-ray absorption fine structure (EXAFS) spectrum revealed peaks at 1.45 Å and 2.10 Å, attributed to Ni–N and Ni–Ni bonds, indicating the coexistence of atomic Ni and Ni nanoparticles in the Ni-SAC (Fig. S6, ESI†). Bulk-scale analyses, including XRD and EXAFS, indicated the formation of Ni nanoparticles in the Ni-SAC during the high-temperature calcination step, necessary for carbon support graphitization. However, surface-scale analysis using X-ray photoelectron spectroscopy (XPS) detected the only atomic Ni, devoid of metallic Ni, Ni(OH)2 and oxidized Ni, due to its shallow sampling depth (around 5 nm) (Fig. S7a, ESI†). Moreover, the N 1s spectrum displayed strong Ni–N bonding peaks, suggesting predominant coordination of N with Ni, which corresponds to the above EXAFS results (Fig. S7b, ESI†). Additionally, to confirm the origin of N in Ni–N, the Ni-SAC was synthesized using a nitrate- or chloride-based nickel precursor. The XPS results, particularly the Ni 2p and N 1s spectra of the chloride-based Ni-SAC, demonstrated a spectrum remarkably similar to that of the nitrate-based Ni-SAC. These findings indicate that the nitrogen atoms primarily derive from the nitrogen-containing organic ligand rather than from the nitrate in the metal precursor (Fig. S7c and d, ESI†). Furthermore, to gain a comprehensive understanding of the structure of Ni-SACs, we quantified the amount of Ni nanoparticles (NPs) and Ni-based single atoms within the Ni-SACs through inductively coupled plasma-mass spectrometry (ICP-MS) and EDS. Utilizing ICP-MS, we determined the total amount of Ni in the Ni-SACs to be 6.32 wt% (Table S3, ESI†). To further elucidate the proportion of atomic Ni in Ni-SAC, EDS analysis was performed specifically at the sites of atomic Ni, excluding areas containing Ni NPs (Fig. S3, ESI†). Based on EDS analysis, the amount of Ni-based single atoms within the Ni-SACs was confirmed to be 1.99 wt%. Although ICP-MS and EDS have different resolutions, which may introduce some challenges in direct comparison, it has been established that Ni-based single atoms constitute 1.99 wt% of the Ni-SACs, while the Ni NPs account for 4.33 wt%. Based on the above analysis results, the synthesized Ni-SAC consists of abundant atomic Ni active sites with some carbon-shelled Ni nanoparticles atop the graphitic carbon support.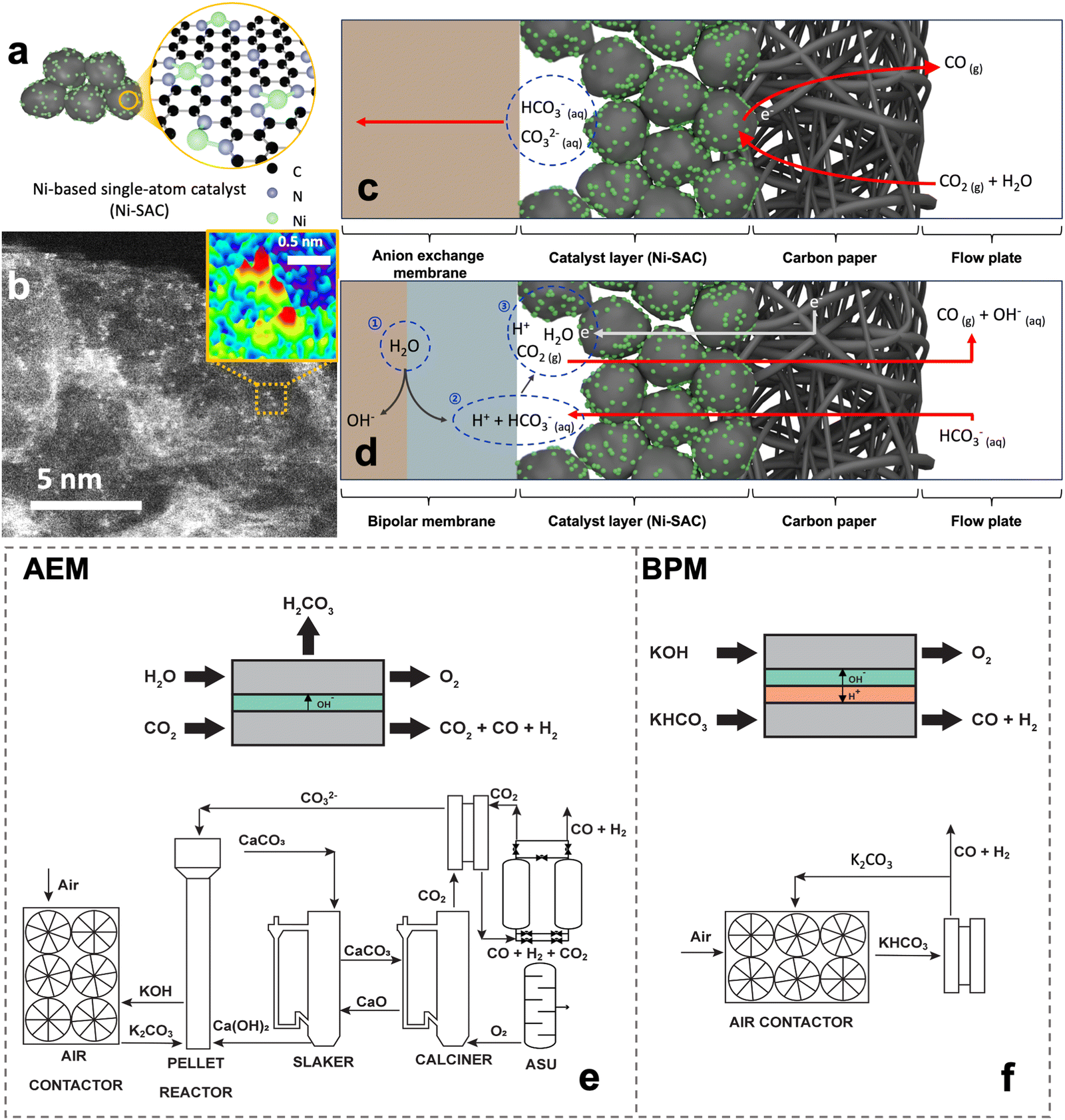 | ||
| Fig. 1 (a) Schematic of a Ni single atom catalyst (Ni-SAC). (b) STEM images of the Ni-SAC. The red area in the inset of (b) represents the presence of Ni atoms. A general schematic of electrochemical CO2 reduction in (c) the CO2 gas-fed anion exchange membrane (AEM)– and (d) bicarbonate-fed bipolar membrane (BPM)–MEA systems. The designed and sized systems for integrated direct air capture and conversion in the (e) AEM–MEA and (f) BPM–MEA. | ||
Gas-phase AEM–MEA-based CO2RR systems have been predominantly used in the lab and industry to convert gaseous CO2 into CO due to the high FE and current density. In these systems (Fig. 1c), humidified CO2 enters the reactor and is reduced to CO while releasing hydroxide ions. The gaseous CO2, hydroxide ions, and water molecules interact to form (bi)carbonate ions. It is estimated that nearly 30% of the gaseous CO2 that enters the reactor is converted into (bi)carbonate ions and transported through the AEM from the cathode to the anode.24,25 From the remaining CO2, only 20% is converted and 50% leaves the cathode as unreacted CO2. As such, for every mole of CO2 that enters the cathode, 0.5 moles of unreacted CO2 exit the cathode, 0.2 moles of CO product exit the cathode, and 0.3 moles of CO2 is lost through carbonate crossover through the anode. A gas-phase AEM–MEA-based CO2RR system consists of an air contactor in which the air comes in contact with aqueous KOH and turns into carbonates. Then, the carbonate solution is transformed into CaCO3 in the pellet reactor through ion exchange between Ca(OH)2 pellets and K2CO3. CO2 is then desorbed from CaCO3 in the calciner, resulting in CO2 and CaO. Finally, CaO is regenerated to form Ca(OH)2 in the slaker. The calciner requires natural gas input to achieve the high temperatures necessary to desorb the CO2. The CO2 lost to carbonates is recycled and fed back into the pellet reactor, slaker, and calciners to regenerate it back to CO2. The unreacted CO2 is separated from the product stream using a pressure swing adsorption system and recycled into the cathode inlet. The component size and energy expenditure for this system are outlined in Tables S1 and S2, ESI.† The gas-phase AEM–MEA-based CO2RR system has two system-related challenges. Firstly, the CO2 that is lost to bicarbonate must be regenerated using a pellet reactor, slaker, and calciner (Fig. 1e). Additionally, the unreacted gaseous CO2 must be separated from the gaseous product stream and recycled. This presents additional capital and energy costs. The CO2RR electrolyzer in this system produces 0.32 MtCO per year. The integrated DAC facility has a capacity of 1.27 MtCO2 per year. From the total capacity, only 0.21 MtCO2 per year is captured from the atmosphere. The remaining 1.06 MtCO2 per year comes from the carbonate that is regenerated (0.77 MtCO2 per year) and from the natural gas used to power the calciner to regenerate the carbonate (0.29 MtCO2 per year).
Alternatives to gas-phase AEM–MEA-based CO2RR systems are BPM-based bicarbonate systems (Fig. 1d). In these systems, a concentrated aqueous bicarbonate solution enters the cathode. During electrolysis, complex reactions occur between the BPM and the catalyst layer, including CO2 recovery and conversion (Fig. 1d): (1) water dissociates into protons (H+) and hydroxide (OH−) ions in the BPM, with the protons flowing to the cathode side. (2) The proton sources react with HCO3−, generating CO2 between the catalyst layer and BPM. (3) Ni-SAC enables the conversion of the in situ generated CO2 into CO. The formed CO and unreacted CO2 gases pass through the Ni-SAC electrode layer. Any unreacted CO2 is absorbed back into the bicarbonate solution and regenerated in the air contactor. As such, we have designed a system that integrates DAC with the electrochemical conversion of bicarbonate to CO in a BPM electrolyzer (Fig. 1f). This system can produce one mole of CO for every mole of captured CO2 from the atmosphere because it requires no external source of heat and as such has no direct CO2 emissions. Additionally, the DAC unit integrated with this system only requires an air contactor as the electrolyzer operates directly with bicarbonate, which lowers the capital cost and energy expenditure compared to the DAC integrated with AEM–MEA-based CO2RR systems, such as those outlined in Fig. 1e. The separation is also simpler as the gaseous product must be separated from a liquid electrolyte and not from another gas.
Impact of the Ni-SAC electrode on bicarbonate electrolysis
We fabricated a Ni-SAC on carbon paper using a spray-coating method. By spraying the catalyst ink onto hydrophilic carbon paper (AvCarb MGL 190), we achieved the desired loading mass (1, 2, and 3 mg cm−2). Scanning electron microscopy (SEM) and EDS mapping revealed a uniform and well-distributed deposition of the Ni-SAC electrode as shown in Fig. 2a–c and Fig. S8, ESI.† It is noteworthy that the electrochemical surface area (ECSA) of the Ni-SAC electrodes is significantly higher (>100x) when compared to that of the carbon paper substrate. As the loading of the Ni-SAC electrodes increases from 1 to 3 mg cm−2, the roughness factor decreases from 637 to 430 (Fig. S9 and S10 and Table S4, ESI†). We conducted electrochemical measurements of the Ni-SAC electrodes in a 3 M KHCO3 solution (Fig. S11, ESI† and Fig. 2d, e). The catalyst loading has a significant impact on the control of CO and H2 distribution (Fig. 2d). At a current density of −100 mA cm−2, the FE for CO exceeds 90% for catalyst loadings of 1 and 2 mg cm−2, but this decreased to 80% when the catalyst loading is increased further to 3 mg cm−2. CO FE also gradually decreased with an increase in applied current density from −100 mA cm−2 to −400 mA cm−2. Notably, the Ni-SAC electrode with a catalyst loading of 2 mg cm−2 shows higher activity towards CO, achieving approximately 50% FE at −400 mA cm−2. The trend of catalyst activity for different loadings within a wider range of current densities (25 to 400 mA cm−2) is depicted in Fig. S12, ESI.† Moreover, we confirmed that the morphology and surface states of the Ni-SAC remain largely unchanged after electrolysis (Fig. S13, ESI†). The CO FE of the Ni-SAC electrode surpasses that of commercial Ag on the same carbon paper by over threefold in the current density range of −25 to −200 mA cm−2 (Fig. S14, ESI†). This difference highlights the exceptional intrinsic activity of the Ni-SAC electrode for the CO2RR. Previous studies have also highlighted the distinctions in intrinsic activity between Ni-SAC and Ag catalysts, attributing the difference to the unique properties of the Ni-SAC. Particularly, the Ni-SAC possesses narrow d states and larger adsorbate dipoles, which facilitate charge transfer to the CO2 molecule, thereby promoting the easy formation of chemically adsorbed CO2δ− species.26,27 In contrast, the adsorption of CO2*, which tends to be the rate-limiting step on Ag catalysts, is not a significant barrier for the Ni-SAC. Instead, the Ni-SAC faces limitations primarily due to the proton–electron transfer reaction necessary for the formation of COOH* species.26 Furthermore, the Ni-SAC demonstrates a lower activation energy for the CO2RR compared to Ag, underscoring its superior intrinsic capability for CO production. These observations align with our results, indicating that the CO2RR performance of our Ni-SAC surpasses that of Ag catalysts.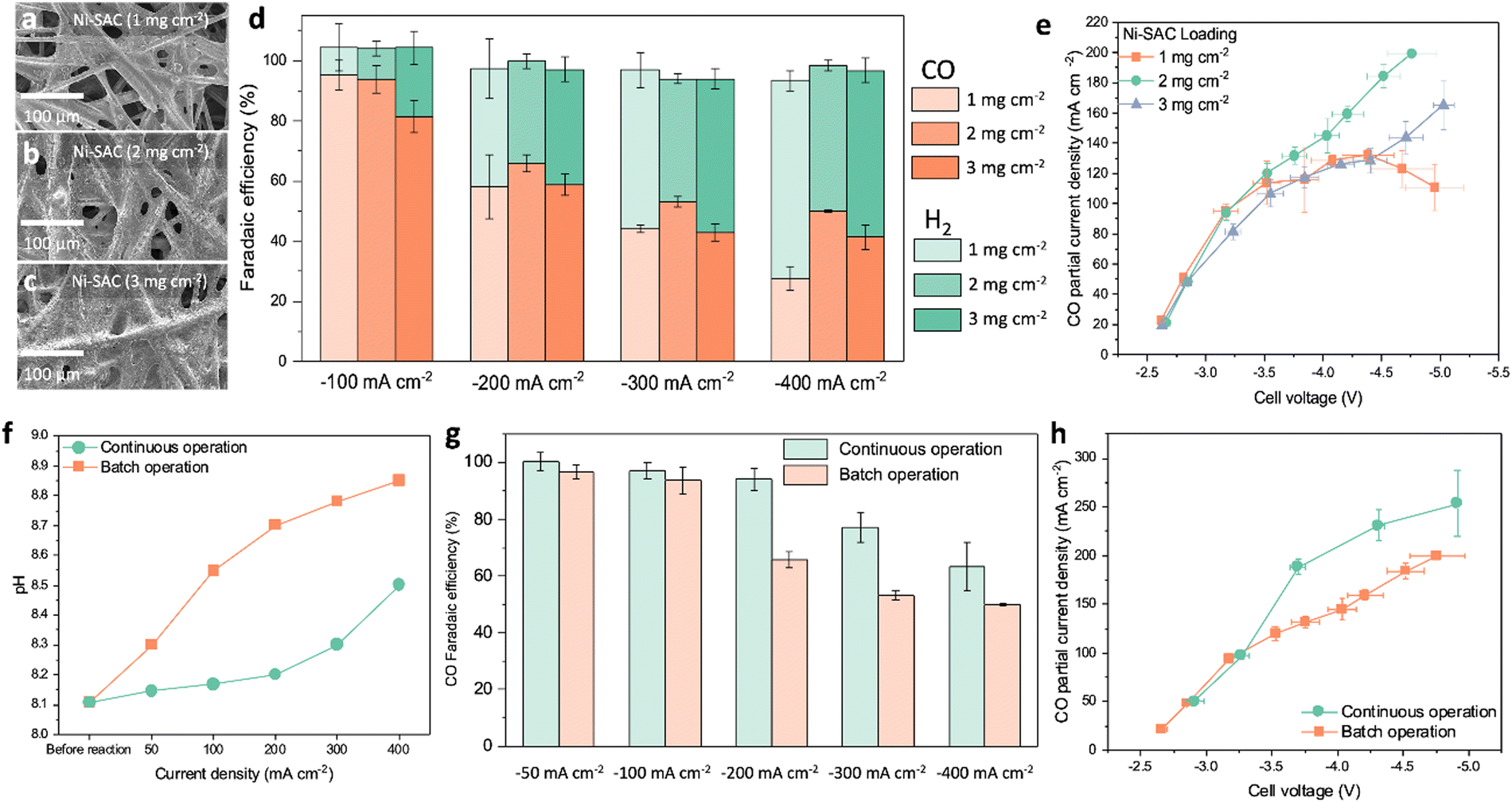 | ||
| Fig. 2 SEM images of the Ni-SAC electrode on carbon paper with different loadings of (a) 1, (b) 2, and (c) 3 mg cm−2. (d) Faradaic efficiencies as a function of applied current densities (−100 to −400 mA cm−2) for 1, 2, and 3 mg cm−2 Ni-SAC in a 3 M KHCO3 solution, with an electrolysis time of 30 minutes. (e) Partial current densities of CO at different Ni-SAC loadings (1, 2, and 3 mg cm−2). (f) The bulk pH of a 3 M KHCO3 solution before and after a 30 min electrolysis process with continuous and batch operation. (g) Faradaic efficiencies for CO with continuous and batch operation as a function of applied current density (−50 to −400 mA cm−2). (h) Partial current densities towards CO with continuous and batch operation. Measurements were performed in 3 M KHCO3 over 30 min, with a CO2 flow of 200 sccm for the CO2 flow system. Error bars represent standard deviations from three repeated measurements. | ||
The Ni-SAC electrode with a loading of 2 mg cm−2 achieved a steady enhancement in CO current density, reaching −200 mA cm−2 at −4.6 V (Fig. 2e). In contrast, other catalyst loadings (1 and 3 mg cm−2) exhibit lower CO current densities in the −3.5 to −5.0 V range. For example, the Ni-SAC with 1 mg cm−2 loading has similar CO current densities to 2 mg cm−2 from −2.6 V to −3.5 V. However, the CO partial current densities remain constant at −100 mA cm−2 to −120 mA cm−2 above −3.5 V. Similarly, the Ni-SAC with 3 mg cm−2 loading shows comparable current densities up to −2.8 V, but the CO partial current densities gradually decline with increasing cell voltages. This trend is attributed to changes in mass transfer and reactant permeability due to evolving electrode structure.17 As catalyst loading increases, the electrode pores decrease, causing the Ni-SAC electrode to transition from a three-dimensional electrode to a more planar structure (Fig. 2a–c). The ECSA also showed a decline with increased catalyst loading, confirming this transition (Fig. S10 and Table S4, ESI†). Consequently, in the low voltage range of −2.8 to −3.5 V, where mass transfer limitations have less impact, the 1 and 2 mg cm−2 electrodes with relatively high surface area can enhance CO reaction rates when compared to the 3 mg cm−2 electrode. However, in the high cell voltage regions of −3.5 to −5.0 V, where mass transfer becomes more critical, the 2 mg cm−2 electrode, with its balanced porosity and active sites, facilitates higher CO current densities. This suggests that maintaining an optimal level of catalyst loading and structure is key to achieving high efficiency in different voltage ranges.
Bicarbonate electrolysis with integrated carbon capture: HCO3− regeneration by introduction of CO2 in a bicarbonate solution
Even with a continuous circulation of highly concentrated bicarbonates to the electrode, CO2RR activity gradually diminishes due to the accumulation of OH− in the electrolyte, a phenomenon inherent of the nature of the recycle system (batch operation). At higher bulk pH, CO32− is the predominate form of CO2 as per: OH− + HCO3− → CO32− + H2O. As the pH of (bi)carbonate solutions increases, the amount of in situ generated CO2 decreases, leading to reduced activity of the CO2RR. Moreover, we confirmed that the 3 M KHCO3 produces two orders of magnitude more in situ CO2 compared to the 1.5 M K2CO3 solution (Fig. S15, ESI†). Bicarbonate and carbonate electrolytes also result in different local pH values (3 M KHCO3: ∼6 and 1.5 M K2CO3: ∼10) at the interfaces between a catalyst layer and membrane.21,28 This pH environment in a bicarbonate solution allows for the generation of an adequate amount of CO2 through the equilibrium relationship between CO2 and HCO3−, which is dependent on the pH conditions. The high CO2 concentration facilitates enhanced CO2RR activity, resulting in significant differences in CO2RR activity between 3 M KHCO3 and 1.5 M K2CO3 as shown in Fig. S16 (ESI†).We demonstrate bicarbonate electrolysis with integrated carbon capture (BE-ICC) by introducing CO2 in the bicarbonate solution (continuous operation) (Fig. S17 and S18, ESI†). Rather than directly using CO2 gas as a reactant, in prevailing CO2 electrolysis methods, here CO2 primarily serves to replenish HCO3− concentrations. This approach ensures consistent HCO3− concentrations throughout the reactor and maintains the overall bulk pH as shown in Fig. 2f. Instead of forming carbonates and accumulating OH−, the CO2 flow during bicarbonate electrolysis regenerates HCO3− by reacting with OH−: CO2 + OH− ↔ HCO3− (Fig. S17, ESI†). Accordingly, the BE-ICC system leads to a nearly steady-state electrolysis environment like single-pass continuous flow conditions. Impressively, by translating batch to continuous operation, electrolysis at −200 mA cm−2 leads to a minimal increase in pH, indicating the ability of the regeneration to sustain bicarbonate concentrations up to −200 mA cm−2 (Fig. 2f). However, there is a pH rise, likely due to a higher generation of OH− ions surpassing the conversion of HCO3− ions under ambient conditions. Consistent with pH results, the BE-ICC system shows >90% CO FE up to −200 mA cm−2, whereas the CO2RR performance of bicarbonate electrolysis with batch operation gradually decreases after −100 mA cm−2 (Fig. 2g). Remarkably, the BE-ICC shows superior CO2RR current densities, reaching as high as −253 mA cm−2 at −4.9 V (Fig. 2h and Table S5, ESI†). To the best of our knowledge, this activity is the highest for the (bi)carbonate electrolysis (Table S5, ESI†). We carried out control experiments with the HCO3− regeneration system using an AEM instead of a BPM to examine the impact of dissolved CO2 by CO2 flow in a bicarbonate solution (Fig. S19, ESI†). Since the AEM–MEA system does not produce in situ generated CO2 from a bicarbonate solution, the CO2RR is solely dependent on dissolved CO2. The AEM–MEA can produce 16% CO FE (∼32 mA cm−2) at −200 mA cm−2, suggesting that the majority of the CO2RR proceeds with in situ generated CO2 gas at the three-phase interfaces (CO2 gas/electrolyte/catalyst) like a gas-diffusion electrode system.29 Interestingly, recent research showed over 70% CH4 FE in a CO2 flow bicarbonate (0.3 M KHCO3)-fed system over Cu mesh, suggesting that dissolved CO2 plays a major role in the CO2RR.30 This contrasting trend is attributed to different CO2 concentration requirements for CO and CH4 activation. While CH4 production decreases with high CO2 concentrations, CO production is directly proportional to CO2 concentrations.31 A 0.3 M KHCO3 concentration, which is tenfold more dilute than ours, can contribute to limited in situ generated CO2 concentration. The environment in limited CO2 could induce CH4 activation due to the accelerating protonation of CO intermediates.32 Furthermore, we have examined the impact of impurities such as NO3−, NO2−, and SO42− within the bicarbonate electrolysis system (Fig. S20, ESI†).13 Impressively, the Ni-SAC catalyst in the BE-ICC system sustained a high level of activity for CO production, achieving 96% CO FE in the presence of NO3− and SO42− impurities. However, the addition of NO2− led to a slight decrease in the CO2RR (11–24%), which appears to promote an increase in ammonia production. Thus, precise modulation of CO2 and bicarbonate concentrations, along with vigilant management of impurities, is imperative, tailored to the target CO2RR products.
Comparison of performance metrics with conventional CO2 gas electrolysis
For the CO2 gas electrolysis, we utilized the same Ni-SAC with a catalyst loading of 2 mg cm−2. The CO2 gas-fed AEM–MEA system shows over 90% CO FE within the current density range from −25 to −300 mA cm−2 (Fig. 3a). This system requires lower cell voltages (−3.2 V) when compared to the bicarbonate electrolysis (−3.7 V) at −200 mA cm−2. This is because of the different reactants (bicarbonates vs. CO2 gas) as well as additional overpotentials associated with water dissociation in a BPM. Over a period of 4 hours, the FE towards CO of the gas-fed system shows a gradual decrease, going from approximately 100% to 73.9% (Fig. 3b). It is crucial to note that after 4 hours of electrolysis, the formation of carbonates in the flow plate completely obstructs the transfer of CO2, and CO production declines to 0% FE (Fig. S21, ESI†). Similarly, bicarbonate electrolysis without CO2 addition also shows a gradual decrease in the CO2RR over time, resulting in a reduction of CO FE by half within two hours (Fig. 3b). The decrease results from rapid reduction of in situ generated CO2 from HCO3− (Fig. S22, ESI†). The BE-ICC system ensures a stable and continuous production of CO with >90% FE over 18 hours (Fig. 3b). Beyond this duration, a slight decrease in CO FE to just below 90% after 20 hours is observed (Fig. S23, ESI†). This can be attributed to the partial detachment of the Ni-SAC catalyst during electrolysis (Fig. S24a, ESI†). Despite this detachment, the intrinsic material properties of the catalyst remained unchanged (Fig. S24b and c, ESI†). Furthermore, refreshing the electrolyte did not improve the CO FE, indicating that the observed degradation in stability is not a result of operational conditions within the BE-ICC system (Fig. S23, ESI†). Thus, the overall performance and stability of the bicarbonate electrolysis process are supported over extended periods by the efficient recovery of HCO3− and steady-state conditions, enhanced by continuous CO2 flow.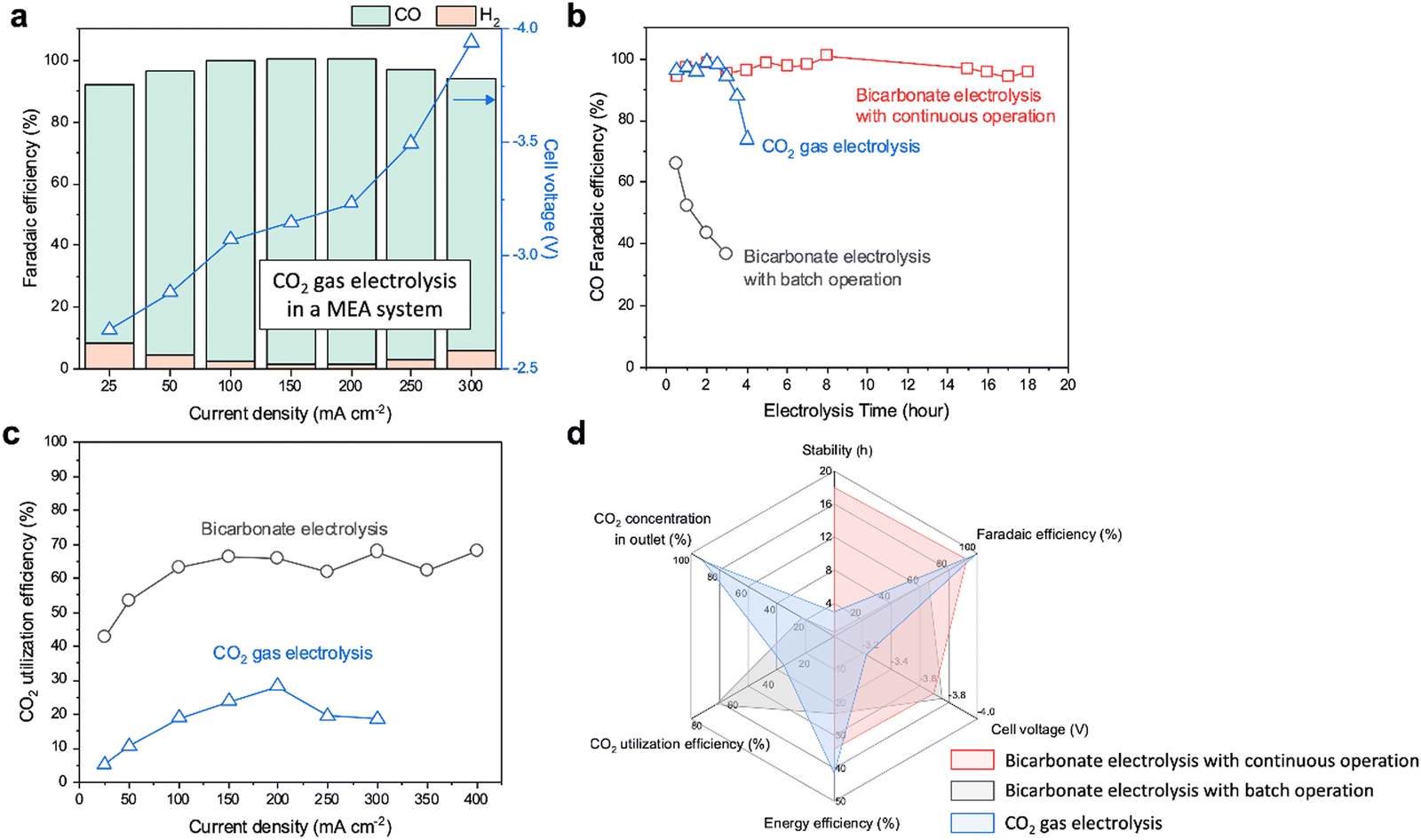 | ||
| Fig. 3 Comparison of performance metrics for CO2 gas electrolysis and bicarbonate electrolysis with batch and continuous operation. (a) Faradaic efficiencies and cell voltages in an AEM–MEA system as a function of applied current densities (−25 to −300 mA cm−2), using Ni-SAC (2 mg cm−2). The system uses humidified CO2 at the cathode and 0.1 M KHCO3 at the anode, respectively. (b) Stability comparison between CO2 gas electrolysis and bicarbonate electrolysis with batch and continuous operation at −200 mA cm−2. (c) CO2 utilization efficiencies of bicarbonate and CO2 gas electrolysis as a function of applied current densities. (d) A radar chart illustrating various performance metrics such as the stability, faradaic efficiency, cell voltage, energy efficiency, CO2 utilization efficiency, CO2 concentration in the outlet stream for the CO2 gas electrolysis and bicarbonate electrolysis with batch and continuous operation at −200 mA cm−2. | ||
In alkaline gas-phase systems with an AEM, the formation and crossover of carbonates significantly reduce the carbon utilization efficiency (theoretically maximum CO2 utilization efficiency is 50% for CO production).8 Also, the gas-fed systems generally require a substantial flow of CO2 to ensure adequate CO2-rich environments. Despite demonstrating high CO2RR performance in the CO2 gas electrolysis system, our AEM–MEA system exhibited a CO2 utilization efficiency of less than 30% within the current density range of −25 to −300 mA cm−2 (Fig. 3c). In contrast, bicarbonate electrolysis accomplishes a significantly higher CO2 utilization. Not only does it exceed 60%, but it also maintains a steady rate of 60–70% for current densities beyond −100 mA cm−2. This can be attributed to the equilibrium between CO2 generation and utilization as well as the prevention of (bi)carbonate crossover through the BPM.33 As the applied current density increases, the BPM accelerates water dissociation, resulting in increased H+ flux.34 This allows for generation of appropriate CO2 concentrations corresponding to the varying applied current densities.
We further evaluate and compare several key performance metrics of bicarbonate electrolysis with batch and continuous operation, as well as CO2 gas electrolysis (Fig. 3d). These metrics provide insights into the stability, FE, cell voltage, CO2 utilization efficiency, EE, and product gas concentration in the outlet stream at a current density of −200 mA cm−2. In the gas-fed electrolysis, the Ni-SAC electrode exhibits exemplary performance, achieving an FE of over 90%, a cell voltage of −3.2 V, and an EE of 41% at −200 mA cm−2. Nevertheless, the system's limitations include reduced stability of 4 hours and a CO2 utilization efficiency of 28%. Notably, besides the suboptimal CO2 utilization, a significant 91% of the CO2 does not engage in the reaction and is subsequently released through the outlet. On the other hand, bicarbonate electrolysis with batch and/or continuous operation overcomes the challenges associated with CO2 electrolysis, showing improved stability (18 hours) or higher CO2 utilization efficiency (60–70%) than CO2 gas electrolysis. Furthermore, in the bicarbonate electrolysis system, the CO2 concentration at the outlet is measured as 22%. When compared to the CO2 gas system, such improved carbon efficiency translates to greater economic viability. Considering the various performance metrics evaluated, the bicarbonate electrolysis system emerges as a promising solution to address the limitations of CO2 gas-phase systems for CO2 capture and conversion. However, it is important to note that these performance metrics are limited to the reactor level. To ensure a successful transition from concept to practical application, in-depth analysis is essential to assess not only the scalability and stability but also the economic feasibility of the system in real-world scenarios, taking into account factors such as CO2 capture, conversion, and separation. It is worth mentioning that the carbon utilization efficiency of the BE-ICC system theoretically reaches 100%, provided the CO2 is continuously recirculated in the bicarbonate feed. In contrast, CO2 flow systems would face carbon losses and requirement of additional energy for separation and regeneration.
Comparison of energy expenditure and carbon abatement with conventional CO2 gas electrolysis
The integration of DAC with bicarbonate-to-CO conversion through a BPM electrolyzer has the potential to enhance the energy efficiency and the CO2 abatement potential compared to coupling DAC with CO2 gas-phase AEM–MEA systems. Thus, we analyze the energy and emissions associated with electrochemical CO production from DAC using both gas-phase and (bi)carbonate-based systems.The energy required to produce one metric ton of CO from air depends on the electrochemical reactor's cell voltage and the CO FE. For a gas-phase AEM–MEA-based CO2RR system (Fig. 4a), the energy required to produce a metric ton of CO is broken down for each component in the system (Tables S1 and S2, ESI†). Experimental results for CO2 gas electrolysis show an FE of over 80% and cell voltages around 3 V at relevant current densities. With a cell voltage of 3 V and an FE of over 80%, the energy required for this system ranges between 8 MW h and 10 MW h per tonne of CO produced. As a reference, electrified reverse water gas shift requires around 7.2 MW h per tonne of CO produced.35
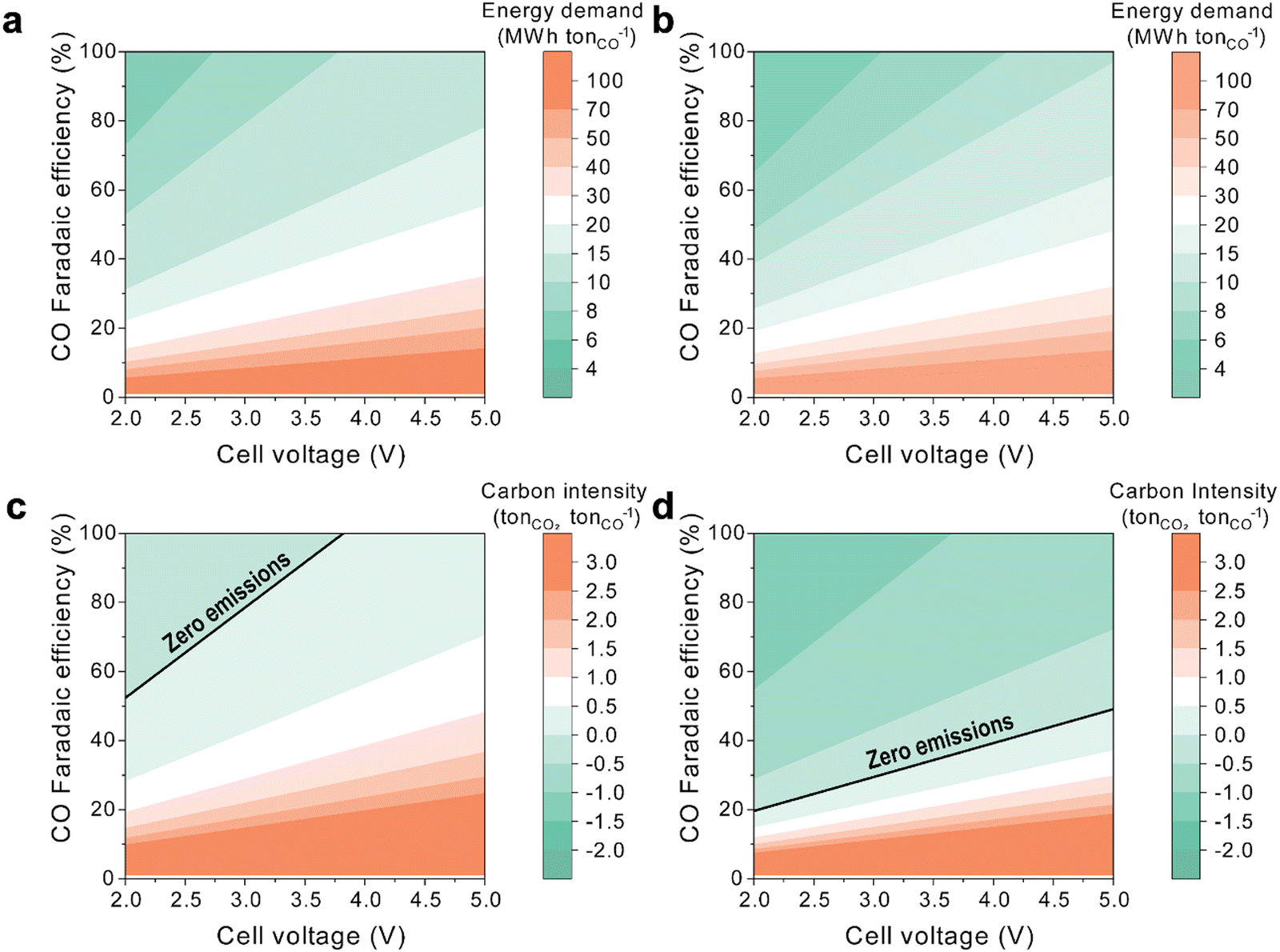 | ||
| Fig. 4 Energy requirement for (a) a gas-phase AEM–MEA-based CO2RR system and for (b) a liquid-phase BPM-based CO2RR system. Carbon dioxide emissions abatement by (c) a gas-phase AEM–MEA-based CO2RR system and (d) a liquid-phase BPM-based CO2RR system. | ||
In contrast, the energy requirement for a BPM-based CO2RR system (Fig. 4b) is slightly lower for the same cell voltage and FE due to the lower energy demand from the DAC system, as this system works with carbonate, which eliminates the need for a pellet reactor, slaker, and calciner (Fig. 1f). For the same voltage (3 V), a BPM-based CO2RR system has an energy requirement between 6 MWh and 8 MWh per tonne of CO produced. However, BPM reactors typically operate at higher voltages than gas-phase reactors due to the additional water dissociation overpotential from the bipolar membrane. Thus, we anticipate at least a 1 V increase in the overall cell potential. For a similar BPM-based CO2RR system operating at 4 V, the energy requirement ranges from 8 MWh to 10 MWh per tonne of CO produced, which is similar to a gas-phase AEM–MEA-based CO2RR system operating at 3 V. The increased energy needed to power the BPM is offset by the decrease in energy from the DAC system.
The CO2RR has the potential to capture and transform CO2 directly from air. As such, we compare the CO2 capture potential per unit of CO produced, contrasting the outcomes of coupling DAC with both gas-phase and carbonate-based electrochemical systems. The gas-phase AEM–MEA-based CO2RR system demonstrates positive CO2 emissions across all FEs when cell voltages exceed 4 V or when FE remains below 50% (Fig. 4c). In essence, AEM–MEA-based systems are capable of capturing a maximum of 0.5 metric tonnes of CO2 per metric tonne of CO produced. Such limitations are attributed to emissions stemming from the calciner process and inefficiencies linked to regenerating the carbonate generated and transported through the AEM back to CO2 that is recycled and used as a feedstock in the cathode.
In contrast, a BPM-based CO2RR system has the potential to capture more CO2 than the AEM-based system (Fig. 4d). The advantage comes from utilizing carbonate as a feedstock to the electrochemical reactor instead of CO2, which eliminates emissions associated with the conversion of carbonate to CO2 within the calciner, where natural gas serves as the heat source. Thus, a BPM-based CO2RR system can achieve negative emissions at all cell voltages surveyed and with FE above 20%. For a system operating at 4 V and 80% FE, a BPM-based system is able to capture between 0.5 and 1 tonne of CO2 per tonne of CO produced. If the voltage is further reduced to 3 V, the system can capture over 1 tonne of CO2 per tonne of CO produced.
Conclusions
In our comprehensive study, we have demonstrated the exceptional capability of a Ni-SAC based BPM–MEA system to directly convert a carbon capture medium to CO, thereby highlighting its potential to transform captured CO2 into valuable products. Our approach, ranging from materials synthesis to system integration, ensures that the bicarbonate electrolysis system rivals alternative reactors for CO2 valorization. The BE-ICC system demonstrates a high CO2RR performance, thus providing a robust solution to the challenges faced by bicarbonate electrolysis. However, despite the superior performance of the BPM-based system, further enhancements in functionality and improvements in energy efficiency are required for practical application. These findings underscore the necessity for further innovation in system optimization and catalyst development, as well as policy support, to ensure that bicarbonate electrolysis becomes a competitive and sustainable approach for CO2 utilization in the future.Author contributions
H. S., C. A. F., and M. C. H. initiated the project and conceptualized the research idea. H. S. designed and performed the experiments. H. C. synthesized and characterized catalysts. H. S., H. C., and P. H. performed the electrochemical measurements. H. S., C. A. F., and H. C. prepared the manuscript. J. O. and M. C. H. supervised the project. All authors collaborated in discussions and contributed to the manuscript's writing and review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded through the ECS and the Toyota Research Institute North America – Toyota Young Investigator Fellowship. J. O. acknowledges the support of the National Research Foundation of Korea (NRF) grant funded by the Ministry of Science and ICT (MSIT), Republic of Korea (NRF-2020M3H7A1096388, NRF-2021R1A2C3007280, and NRF-2021R1A5A1084921).References
- A. Gawel, T. Jaster, D. Siegmund, J. Holzmann, H. Lohmann, E. Klemm and U.-P. Apfel, iScience, 2022, 25, 104011 CrossRef CAS PubMed.
- I. E. Stephens, K. Chan, A. Bagger, S. W. Boettcher, J. Bonin, E. Boutin, A. K. Buckley, R. Buonsanti, E. R. Cave and X. Chang, J. Phys-Energy, 2022, 4, 042003 CrossRef CAS.
- S. C. Perry, P.-K. Leung, L. Wang and C. Ponce de León, Curr. Opin. Electrochem., 2020, 20, 88–98 CrossRef CAS.
- Z. Zhang, X. Huang, Z. Chen, J. Zhu, B. Endrődi, C. Janáky and D. Deng, Angew. Chem., Int. Ed., 2023, 62, e202302789 CrossRef CAS PubMed.
- T. Li, E. W. Lees, M. Goldman, D. A. Salvatore, D. M. Weekes and C. P. Berlinguette, Joule, 2019, 3, 1487–1497 CrossRef CAS.
- J. T. Song, H. Song, B. Kim and J. Oh, Catalysts, 2019, 9, 224 CrossRef.
- Y. Xu, J. P. Edwards, S. Liu, R. K. Miao, J. E. Huang, C. M. Gabardo, C. P. O’Brien, J. Li, E. H. Sargent and D. Sinton, ACS Energy Lett., 2021, 6, 809–815 CrossRef CAS.
- J. E. Huang, F. Li, A. Ozden, A. Sedighian Rasouli, F. P. García de Arquer, S. Liu, S. Zhang, M. Luo, X. Wang, Y. Lum, Y. Xu, K. Bertens, R. K. Miao, C.-T. Dinh, D. Sinton and E. H. Sargent, Science, 2021, 372, 1074–1078 CrossRef CAS PubMed.
- G. O. Larrazábal, P. Strøm-Hansen, J. P. Heli, K. Zeiter, K. T. Therkildsen, I. Chorkendorff and B. Seger, ACS Appl. Mater. Interfaces, 2019, 11, 41281–41288 CrossRef PubMed.
- J. Kim, T. H. Ha, J. Kim, G. H. Jeong, S. O. Kim, W. Chung, K. Roh, J. H. Lee and J. Oh, Appl. Catal., B, 2023, 339, 123160 CrossRef CAS.
- Y. C. Xiao, C. M. Gabardo, S. Liu, G. Lee, Y. Zhao, C. P. O’Brien, R. K. Miao, Y. Xu, J. P. Edwards, M. Fan, J. E. Huang, J. Li, P. Papangelakis, T. Alkayyali, A. Sedighian Rasouli, J. Zhang, E. H. Sargent and D. Sinton, EES Catal., 2023, 1, 54–61 RSC.
- Y. C. Li, G. Lee, T. Yuan, Y. Wang, D.-H. Nam, Z. Wang, F. P. García de Arquer, Y. Lum, C.-T. Dinh, O. Voznyy and E. H. Sargent, ACS Energy Lett., 2019, 4, 1427–1431 CrossRef CAS.
- D. J. D. Pimlott, A. Jewlal, B. A. W. Mowbray and C. P. Berlinguette, ACS Energy Lett., 2023, 8, 1779–1784 CrossRef CAS.
- Z. Zhang, E. W. Lees, F. Habibzadeh, D. A. Salvatore, S. Ren, G. L. Simpson, D. G. Wheeler, A. Liu and C. P. Berlinguette, Energy Environ. Sci., 2022, 15, 705–713 RSC.
- Z. Yan, J. L. Hitt, Z. Zeng, M. A. Hickner and T. E. Mallouk, Nat. Chem., 2021, 13, 33–40 CrossRef CAS PubMed.
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442–1453 RSC.
- Y. Kim, E. W. Lees and C. P. Berlinguette, ACS Energy Lett., 2022, 7, 2382–2387 CrossRef CAS.
- J. Lee, H. Liu and W. Li, ChemSusChem, 2022, 15, e202201329 CrossRef CAS PubMed.
- E. W. Lees, M. Goldman, A. G. Fink, D. J. Dvorak, D. A. Salvatore, Z. Zhang, N. W. X. Loo and C. P. Berlinguette, ACS Energy Lett., 2020, 5, 2165–2173 CrossRef CAS.
- H. Song, C. A. Fernández, A. Venkataraman, V. D. Brandão, S. S. Dhingra, S. S. Arora, S. S. Bhargava, C. M. Villa, C. Sievers, S. Nair and M. C. Hatzell, ACS Appl. Energy Mater., 2024, 7(3), 1224–1233 CrossRef CAS.
- G. Lee, A. S. Rasouli, B.-H. Lee, J. Zhang, D. H. Won, Y. C. Xiao, J. P. Edwards, M. G. Lee, E. D. Jung, F. Arabyarmohammadi, H. Liu, I. Grigioni, J. Abed, T. Alkayyali, S. Liu, K. Xie, R. K. Miao, S. Park, R. Dorakhan, Y. Zhao, C. P. O’Brien, Z. Chen, D. Sinton and E. Sargent, Joule, 2023, 7, 1277–1288 CrossRef CAS.
- Z. Zhang, E. W. Lees, S. Ren, B. A. W. Mowbray, A. Huang and C. P. Berlinguette, ACS Central Sci., 2022, 8, 749–755 CrossRef CAS PubMed.
- Z. Zhang, L. Melo, R. P. Jansonius, F. Habibzadeh, E. R. Grant and C. P. Berlinguette, ACS Energy Lett., 2020, 5, 3101–3107 CrossRef CAS.
- Y. C. Li, Z. Yan, J. Hitt, R. Wycisk, P. N. Pintauro and T. E. Mallouk, Adv. Sustain. Syst., 2018, 2, 1700187 CrossRef.
- L.-C. Weng, A. T. Bell and A. Z. Weber, Energy Environ. Sci., 2019, 12, 1950–1968 RSC.
- S. Vijay, W. Ju, S. Brückner, S.-C. Tsang, P. Strasser and K. Chan, Nat. Catal., 2021, 4, 1024–1031 CrossRef CAS.
- H. B. Yang, S.-F. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H.-Y. Wang, W. Cai, R. Chen, J. Gao, X. Yang, W. Chen, Y. Huang, H. M. Chen, C. M. Li, T. Zhang and B. Liu, Nat. Energy, 2018, 3, 140–147 CrossRef CAS.
- R. Kas, K. Yang, G. P. Yewale, A. Crow, T. Burdyny and W. A. Smith, Ind. Eng. Chem. Res., 2022, 61, 10461–10473 CrossRef CAS.
- F. P. García de Arquer, C.-T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D.-H. Nam, C. Gabardo, A. Seifitokaldani, X. Wang, Y. C. Li, F. Li, J. Edwards, L. J. Richter, S. J. Thorpe, D. Sinton and E. H. Sargent, Science, 2020, 367, 661–666 CrossRef PubMed.
- C. A. Obasanjo, G. Gao, J. Crane, V. Golovanova, F. P. García de Arquer and C.-T. Dinh, Nat. Commun., 2023, 14, 3176 CrossRef CAS PubMed.
- M. Moradzaman, C. S. Martínez and G. Mul, Sustain. Energy Fuels, 2020, 4, 5195–5202 RSC.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- H.-M. Chang and I. V. Zenyuk, Commun. Chem., 2023, 6, 2 CrossRef CAS PubMed.
- S. Z. Oener, M. J. Foster and S. W. Boettcher, Science, 2020, 369, 1099–1103 CrossRef CAS PubMed.
- L. Zheng, M. Ambrosetti, A. Beretta, G. Groppi and E. Tronconi, Chem. Eng. J., 2023, 466, 143154 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee00048j |
| ‡ These authors contributed equally: Hakhyeon Song, Carlos A. Fernández, Hyeonuk Choi. |
| This journal is © The Royal Society of Chemistry 2024 |