
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Does trapped O2 form in the bulk of LiNiO2 during charging?†
Mikkel
Juelsholt‡
 a,
Jun
Chen‡
a,
Miguel A.
Pérez-Osorio
ab,
Gregory J.
Rees
ab,
Sofia
De Sousa Coutinho
ab,
Helen E.
Maynard-Casely
c,
Jue
Liu
d,
Michelle
Everett
d,
Stefano
Agrestini
e,
Mirian
Garcia-Fernandez
e,
Ke-Jin
Zhou
e,
Robert A.
House
*ab and
Peter G.
Bruce
*abf
a,
Jun
Chen‡
a,
Miguel A.
Pérez-Osorio
ab,
Gregory J.
Rees
ab,
Sofia
De Sousa Coutinho
ab,
Helen E.
Maynard-Casely
c,
Jue
Liu
d,
Michelle
Everett
d,
Stefano
Agrestini
e,
Mirian
Garcia-Fernandez
e,
Ke-Jin
Zhou
e,
Robert A.
House
*ab and
Peter G.
Bruce
*abf
aDepartment of Materials, University of Oxford, Oxford, UK. E-mail: robert.house@materials.ox.ac.uk; peter.bruce@materials.ox.ac.uk
bFaraday Institution, Didcot, UK
cAustralian Nuclear Science and Technology Organisation, Kirrawee, New South Wales, Australia
dNeutron Scattering Division, Oak Ridge National Laboratory, Oak Ridge, Tennessee, USA
eDiamond Light Source, Harwell, UK
fDepartment of Chemistry, University of Oxford, Oxford, UK
First published on 27th February 2024
Abstract
LiNiO2 remains a critical archetypal material for high energy density Li-ion batteries, forming the basis of Ni-rich cathodes in use today. Nevertheless, there are still uncertainties surrounding the charging mechanism at high states of charge and the potential role of oxygen redox. We show that oxidation of O2− across the 4.2 V vs. Li+/Li plateau forms O2 trapped in the particles and is accompanied by the formation of 8% Ni vacancies on the transition metal sites of previously fully dense transition metal layers. Such Ni vacancy formation on charging activates O-redox by generating non-bonding O 2p orbitals and is necessary to form vacancy clusters to accommodate O2 in the particles. Ni accumulates at and near the surface of the particles on charging, forming a Ni-rich shell approximately 5 nm thick; enhanced by loss of O2 from the surface, the resulting shell composition is Ni2.3+1.75O2. The overall Ni oxidation state of the particles measured by XAS in fluorescence yield mode after charging across the plateau to 4.3 V vs. Li+/Li is approximately +3.8; however, taking account of the shell thickness and the shell Ni oxidation state of +2.3, this indicates a Ni oxidation state in the core closer to +4 for compositions beyond the plateau.
Broader contextLi-ion batteries are a critical part of global efforts to decarbonise our transport systems thanks to the high energy densities they offer. One of the key factors limiting the amount of energy that Li-ion batteries can store is the cathode, typically a lithium transition metal oxide, e.g. LiNi1/3Mn1/3Co1/3O2. Efforts to further improve these materials and reduce their Co contents lead naturally towards more Ni-rich cathodes, the ultimate example of which is LiNiO2. Unfortunately, LiNiO2 suffers from a number of problems at high states of charge which have been linked with oxygen redox. The phenomenon of oxygen redox is well known in so-called Li-rich transition metal oxide cathodes, where on charge, O2− is oxidised to form molecular O2 trapped within the structure. However, whether this same mechanism extends to materials with fully dense transition metal layers such as LiNiO2 has been a topic of recent debate. Here, we show that trapped O2 does form in LiNiO2, accommodated by Ni vacancies that form in transition metal layers on charging. These results represent an important step towards a universal understanding of oxygen redox, which is critical for developing new high-energy density cathodes. |
Introduction
Ni-rich cathodes, such as LiNi0.8Mn0.1Co0.1O2, currently offer the best-in-class energy density in Li-ion batteries. In the drive to remove Co from these cathodes, it is critical to understand the behaviour of the ultimate Ni-rich archetype, LiNiO2.1–5 At high states of charge, LiNiO2 exhibits O-loss, a contraction in interlayer spacing, particle cracking and poor cycling stability.5–12 Substantial efforts have been made to understand these phenomena and the structural transitions that take place when Li is extracted from LiNiO2, but there remains considerable debate over the extent of Ni oxidation and O-redox in LiNiO2, particularly across the voltage plateau at 4.2 V vs. Li+/Li.13–16 Despite reaching a composition close to NiO2 at the end of charge with Ni nominally in the +4 oxidation state, bulk sensitive X-ray absorption spectroscopy (XAS) appears to show incomplete oxidation of Ni3+ to Ni4+ when the edge position is compared to other Ni4+-containing oxides.14,15 Some argue that this reflects the change in covalency of the Ni–O bond with the electron–hole density shifting more towards O than Ni when Ni is highly oxidised,8,17–19 while others argue O oxidation is invoked.13–15,20Recent research into O oxidation in Li-rich cathodes, such as Li1.2Ni0.13Co0.13Mn0.54O2, has indicated that oxidised oxygen takes the form of molecular O2, which is trapped within vacancy clusters in the cathode structure.21–25 However, in the case of stoichiometric materials like LiNiO2, it has been argued that this same mechanism cannot apply due to the lack of transition metal vacancies in the fully dense transition metal layers (in the Li-rich materials the Li in the transition metal layers are removed on charge and the remaining vacancies cluster to accommodate the O2).14,15
Here, we perform a structural and spectroscopic study of LiNiO2 on charging to investigate the O-redox mechanism. We employ a combination of neutron and synchrotron X-ray powder diffraction analysis that takes account of the stacking faults prevalent in these charged materials, showing that 8% Ni vacancies form in the originally fully dense transition metal layer as the material is charged across the voltage plateau at 4.2 V vs. Li+/Li. High resolution resonant inelastic X-ray scattering (RIXS) at the O K-edge confirms the presence of trapped molecular O2, on charging across the 4.2 V vs. Li+/Li plateau, corresponding to approximately 2% of the O in the material. The Ni vacancies result in non-bonding O 2p states on O2− enabling O2− oxidation and 8% Ni vacancies on the transition metal layers is sufficient to form vacancy clusters to accommodate the resulting O2. Chemical analysis shows that the Ni absent from the bulk does not leave the particles (no Ni was detected in the electrolyte or at the anode after charging). STEM images demonstrate the core–shell nature of the charged particles, with a Ni-rich, Ni1.75O2 rocksalt-like shell approximately 5 nm thick and with a Ni oxidation state of +2.3. The overall Ni oxidation state of the particles charged across the plateau to 4.3 V vs. Li+/Li is determined by Ni L-edge fluorescence yield XAS to be +3.8. However, taking into account the thickness of the shell and its Ni oxidation state of +2.3, the core Ni oxidation state is closer to +4.
Experimental
Materials
Uncoated, polycrystalline LiNiO2 powder was obtained from BASF. The particle morphology is shown on the SEM images in ESI† Fig. S1.Electrochemistry
Self-supporting cathode films of LiNiO2 were prepared by grinding the material first with acetylene black and then with polytetrafluoroethylene in an 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 mass ratio in a pestle and mortar, followed by calendaring to a thickness of ∼100 μm. Typical electrodes were 1 cm2 in size and 15 mg in mass, and the cells were galvanostatically cycled at a rate of 10 mA g−1. Cathodes were assembled into coin cells with 140 μl battery grade 1 M LiPF6 in EC:DMC, 50:50 (Merck) used as the electrolyte and Li metal foil as the counter electrode. Cells were disassembled, and the electrodes were rinsed with dry dimethylcarbonate before characterisation. All handling was performed in an MBraun glovebox under inert atmosphere (<1ppm H2O and O2). Electrochemical charge–discharge cycling was carried out using a Maccor Series 4000. For the neutron and X-ray powder diffraction measurements, cathodes with an 8:2 ratio of active material to carbon were used and no polytetrafluoroethylene. The cathode was cycled in a custom-made coin cell using a Biologic SP-300 potentiostat. The electrodes had a total mass of 1 g and 50 ml battery grade 1 M LiPF6 in EC:DMC, 50:50 (Merck) was used as the electrolyte and Li metal foil as the counter electrode.
:1:1 mass ratio in a pestle and mortar, followed by calendaring to a thickness of ∼100 μm. Typical electrodes were 1 cm2 in size and 15 mg in mass, and the cells were galvanostatically cycled at a rate of 10 mA g−1. Cathodes were assembled into coin cells with 140 μl battery grade 1 M LiPF6 in EC:DMC, 50:50 (Merck) used as the electrolyte and Li metal foil as the counter electrode. Cells were disassembled, and the electrodes were rinsed with dry dimethylcarbonate before characterisation. All handling was performed in an MBraun glovebox under inert atmosphere (<1ppm H2O and O2). Electrochemical charge–discharge cycling was carried out using a Maccor Series 4000. For the neutron and X-ray powder diffraction measurements, cathodes with an 8:2 ratio of active material to carbon were used and no polytetrafluoroethylene. The cathode was cycled in a custom-made coin cell using a Biologic SP-300 potentiostat. The electrodes had a total mass of 1 g and 50 ml battery grade 1 M LiPF6 in EC:DMC, 50:50 (Merck) was used as the electrolyte and Li metal foil as the counter electrode.
Neutron and X-ray powder diffraction
To obtain sufficient neutron power diffraction data, access was obtained from two sources. Time of flight neutron powder diffraction data were obtained from the NOMAD instrument at the Neutron Spallation Source, Oak Ridge National Laboratory.26 Constant wavelength neutron powder diffraction was measured from the Echidna powder diffractometer27 at the Australian Center for Neutron Scattering with a wavelength of 1.622 Å. X-ray powder diffraction and total scattering measurements were obtained from XPDF at Diamond Light Source with a wavelength of 0.1616 Å. Instrumental contributions to the peak shapes were obtained through the refinement of Si and LaB6 standards. In all cases, the samples were sealed under an inert atmosphere within vanadium cans (neutrons) and borosilicate capillaries (X-rays). All measurements were performed at room temperature.The Rietveld refinements were performed in Topas academic. The background was described by using a combination of a 9th degree Chebyshev polynomial and a scaled scattering pattern of the pure carbon that was used to prepare the electrodes. The X-ray and neutron powder diffraction were refined simultaneously by refining all structural parameters to all detector banks. Each dataset had its individual background and zero error refined. The X-ray and constant wavelength neutron data were modelled using a Thompson-Cox-Hasting pseudo-Voigt profile. The time-of-flight neutron diffraction data were modelled using the instrumental profile of NOMAD implemented in the Topas input file provided by the beamline. The anisotropic peak broadening was modelled using a set of 4 spherical harmonic functions implemented in Topas in the spherical_harmonics_hkl macro.
The pair distribution functions were obtained using PDFgetX328 in xPDFsuite29 and the Pair Distribution Functions were analysed using PDFgui30 The Rietveld refinements were performed in Topas academic.31
STEM
Samples were dispersed in dry dimethylcarbonate (DMC) and transferred onto holey carbon Cu TEM grids by drop-casting inside an argon-filled glovebox and transferred to the microscope using a vacuum transfer holder. ADF-STEM characterisations were obtained using JEOL ARM-200F at the accelerating voltage of 200 kV. The emission current was 5 μA and beam current was 35 pA. A dwell time of 5 μs was used for imaging with a pixel size of 0.006 nm px−1. The inner and outer collecting angles for imaging were 73 and 271 mrad, respectively, with convergence semi-angle of 22.5 mrad.OEMS
Operando electrochemical mass spectrometry (OEMS) was carried out to detect the evolved gases during galvanostatic charging/discharging of LiNiO2. The setup consists of a quadrupole mass spectrometer (Thermo Fischer) and mass-flow controllers (Bronkhorst). A 2-electrode Swagelok electrochemical cell with gas inlet and outlet ports was used for the operando measurements, using LiNiO2 cathode, Li metal anode, Whatman® glass fibre separator, and battery grade 1 M LiPF6 in PC (propylene carbonate) electrolyte. The cells were assembled inside an Ar-filled glovebox. High-purity Ar gas at a flow rate of 1 mL min−1 was used as the carrier gas. 1 cm2 electrodes were used for OEMS with an active cathode material mass loading of 15 mg and cycled at a rate of 10 mA g−1. The OEMS was calibrated for O2, CO2 and Ar using standards of known analyte gas content. The carrier gas was Zero Grade Ar (BOC) and the calibration standard was 1011 ppm CO2 and 1.01% O2 in Ar base gas (BUSE Scientific). Calibration of the instrument was performed using a dedicated method in the instrument software (GasWorks) which compares the ion flux of analyte gases detected in the instrument with their known concentrations in the calibration standard. Before a current was applied, the cell was flushed using Zero Grade Ar carrier gas until the concentration of Ar measured by the mass spec. was above 99.95%. The baseline CO2 and O2 concentrations were then measured for 2 hours before charging the cell.Spectroscopy
Ni L-edge and O K-edge X-ray absorption spectra and high-resolution O K-edge RIXS were collected at beamline I21 Diamond Light Source.32 Samples were transferred to the spectrometer using a vacuum-transfer suitcase to avoid air exposure and were pumped down to ultra-high vacuum (UHV) overnight. All measurements were made at a temperature of 20 K to suppress any possible beam damage. RIXS line scans were recorded at the resonance energy for molecular O2 at 15 different sample locations and averaged together. The acquisition time was 30 seconds for individual RIXS spectra and 1 minute for XAS spectra. To compare the O2 signal intensity, the areas under the vibrational peaks, excluding the elastic line were integrated between 0.1–2 eV.ICP
The concentration of Ni in the electrolyte and at the anode was determined through the analysis of inductively coupled plasma-optical emission spectroscopy (ICP-OES) using a Perkin-Elmer Optima 8000 ICP-OES. To prepare the ICP-OES measurements, the cycled coin cells were disassembled and the cathode film and separator in contact with the cathode were discarded. Every other cell part was then left to soak in a diluted aqua reagia solution (Diluted 10 times, giving a 1.2 M concentration of HCl and 1.5 M HNO3) for 24 hours. The sample solutions were then transferred to the instrument. The ICP-OES was calibrated using a series of diluted standards (200 ppm, 100 ppm, 50 ppm, 10 ppm and 1 ppm) made from an ICP multi-element standard solution IV stock solution obtained from Sigma–Aldrich.NMR
Magic angle spinning (MAS) 1H nuclear magnetic resonance (NMR) was carried out at 9.45 T (ν0 = 400.19 MHz) using a 1.9 mm double air bearing probe (νR = 32 kHz) on a Bruker Avance HD spectrometer. A 15-second recycle delay and 512 transients were employed for each experiment using the same rotor.DFT
Calculations were carried out using the Quantum Espresso (QE) package.33 As exchange–correlation functional, we employed the generalised gradient approximation (GGA) via the Perdew, Burke and Ernzerhof (PBE) functional.34 The interaction between core and valence electrons was described by using optimised norm-conserving Vanderbilt pseudopotentials (ONCVSP).35,36 Hubbard corrections to the 3d states of Ni, with U = 6 eV, were included in the calculations (PBE + U), via the simplified rotational-invariant formulation of Cococcioni and de Gironcoli. van der Waals (vdW) corrections were also applied, using the Tkatchenko–Scheffler dispersion correction method. The wavefunctions and charge density were represented via plane-wave basis sets with energy cutoffs of 120 and 480 Ry, respectively. The energy threshold was set to 1 × 10−8 eV. A 2 × 2 × 2 Monkhorst–Pack k-point grids was employed. Structural models were optimised until forces on the atoms were less than 0.02 eV Å−1 and cell stress was less than 0.5 Kbar. To obtain a model for the charged LiNiO2, a model of the pristine material with composition Li0.96Ni1.04O2 was first constructed from a 3 × 3 × 1 supercell of LiNiO2 in the R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group, containing 26 Li atoms, 28 Ni atoms and 54 O atoms. The lithium was then completed removed and the structure relaxed.
m space group, containing 26 Li atoms, 28 Ni atoms and 54 O atoms. The lithium was then completed removed and the structure relaxed.
Results and discussion
Structure of LiNiO2 over the first cycle
To investigate the Ni occupancies in the transition metal and Li layers on the first cycle (Fig. 1), a structural investigation was performed using a combination of powder X-ray and neutron diffraction (PXRD and PND). For the study, pristine, undoped and uncoated LiNiO2 was obtained from BASF and handled without exposure to air, see Methods. O K-edge XAS data collected in electron and fluorescence yield mode, ESI† Fig. S2, confirm the absence of surface carbonate species which can be observed when LiNiO2 is exposed to air.37 The refinement of PXRD and PND data reveals that the pristine material is slightly Li deficient with a 4.4% Ni occupancy of the Li 3b site, resulting in the final composition [Li0.956Ni0.044]NiO2. The refinements of the pristine material are shown in ESI† Fig. S3 and Table S1.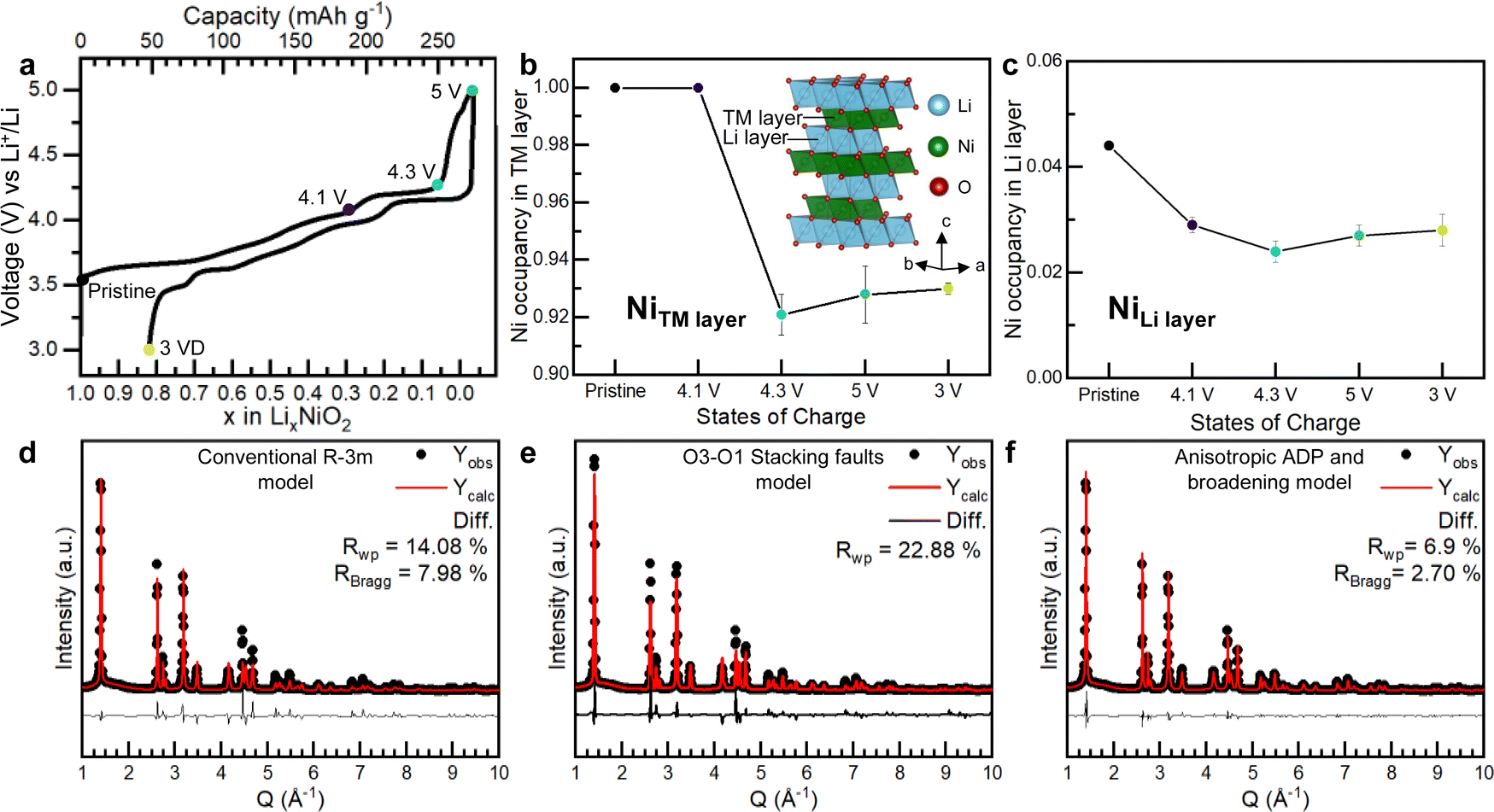 | ||
| Fig. 1 Electrochemistry and Ni site occupancies for LiNiO2 on the first cycle. (a) First cycle load curve for LiNiO2 (b) and (c) refined site occupancies for Ni in the 3a site (transition metal layer) and 3b site (Li layer) of LiNiO2, respectively, at different states of charge. The line is a guide to the eye. Inset in (b) shows the structure of LiNiO2. (d)–(f) Refinement of the synchrotron PXRD for LiNiO2 charged to 4.3 V vs. Li+/Li using the conventional Rm LiNiO2 structure (d), the O3–O1 stacking fault model developed by Croguennec et al.38 refined in Faults39 (e) and Rm LiNiO2 structure, using an anisotropic ADP and peak broadening model (f). | ||
LiNiO2 undergoes a series of well-studied phase transitions upon delithation (H1 → M1 → H2 → H3), between hexagonal phases, H, which differ slightly in their lattice parameters and a monoclinic phase, M.5,38,40–45 Upon reaching the onset of the voltage plateau at 4.1 V vs. Li+/Li, LiNiO2 has transitioned to the H2 phase with an expanded unit cell along the crystallographic c axis, ESI† Fig. S4 and Table S2. At 4.1 V vs. Li+/Li the transition metal layer remains fully dense, Fig. 1b, but the amount of Ni in the Li layer has slightly decreased to 2.9%, Fig. 1c; PXRD and PND refinements at 4.1 V vs. Li+/Li are shown in ESI† Fig. S4 and Table S2. When charging across the voltage plateau and up to 4.3 V vs. Li+/Li, the c lattice parameter contracts, ESI† Table S3, in line with previous reports.5,7,9,10,40,44–46 The simple Rm model of the H3 structure at 4.3 V vs. Li+/Li cannot describe the measured diffraction pattern as seen in Fig. 1d. The discrepancy between the model and the data could stem from the formation of O1 stacking faults as previously reported.38,39,47 At 4.3 V vs. Li+/Li and above, there is little to no Li in the structure, and since we are primarily interested in the Ni behaviour we focused on the PXRD data here as this is most sensitive to the Ni.
O1-type stacking faults in delithiated LiNiO2 have previously been modelled by introducing O1 layers of a well-defined interlayer spacing and a displacement vector into the O3 structure using DIFFaX and Faults software.38,39 However, as shown in Fig. 1e, this model does not adequately describe the diffraction pattern at 4.3 V vs. Li+/Li. As shown in Fig. 1d and e and in ESI† Fig. S5 both the regular Rm model and the O1 stacking fault model cannot describe the intensity and the shape of the Bragg peaks. Instead, we find that using anisotropic atomic displacement parameters (ADP) and taking into account the anisotropic peak broadening by using a set of spherical harmonics substantially improves the refinement, as shown in Fig. 1f and ESI† Fig. S5. This implies that the interlayer distances are more variable than can be accounted for by a binary stacking model.
Using this model, which accurately describes the hkl-dependent peak broadening, the Ni occupancy in the 3a sites in the transition layer was refined. The refinements, which are summarised in Fig. 1f, ESI† Fig. S4 and S5 and Tables S3 and S4, show that the Ni occupancy decreases to around 92–93% in both the 4.3 V vs. Li+/Li and 5 V vs. Li+/Li samples, Fig. 1b. Moreover, joint Rietveld refinements of neutron and X-ray scattering data of the discharged LiNiO2, which does not contain stacking faults, also show there are 8% vacancies in the transition metal layer (ESI† Fig. S7 and Table S5). These results show that the transition metal layer does not remain fully dense on charging across the voltage plateau and that the vacant Ni sites are not repopulated with Ni during the discharge process.
To investigate if the Ni migrates to the Li layer, the Ni occupancy in the 3b sites in the Li layer was refined. As shown in Fig. 1c, the amount of Ni in the Li layers remains constant at about 2–3% across the voltage plateau and on discharge. Refinements were also performed by introducing Ni into tetrahedral interstices in the structure, but this did not improve the fit, ESI† Fig. S8. The refinements, therefore, show that on charging up to and across the 4.2 V vs. Li+/Li plateau, 8% of the Ni is lost from the bulk, corresponding to a composition change from [Li0.956Ni0.044]NiO2 in the pristine material to [Li0Ni0.028]Ni0.926O2 at 5 V vs. Li+/Li and that these Ni occupancies remain on discharge to 3 V vs. Li+/Li.
To determine the destination of the Ni lost from the bulk, the electrolyte along with the solution after dissolving the anode were analysed by Inductively coupled plasma optical emission spectrometry (ICP-OES). The ICP-OES results are presented in ESI† Table S6 and show that only negligible amounts of Ni (147 ppm, which corresponds to 0.3 mmol of Ni loss per mole of cathode) leave the cathode material on charging. These results suggest that the Ni from the bulk remains in the particles and likely accumulates in the near-surface region of the particles, resulting in a core–shell structure.
Local structure of LiNiO2 on charging and surface shell composition
Densification at the cathode particle surface to form a rocksalt-like layer at high degrees of delithiation is a well-studied phenomenon in LiNiO2. This is generally related to O-loss which reduces the O content at the surface and triggers Ni migration inward to sites in the Li layer.5,24,48–53 To quantify O-loss in our sample, operando electrochemical mass spectrometry (OEMS) was performed for LiNiO2 over the first cycle, ESI† Fig. S9 and S10. While no direct O2 evolution was observed throughout the charging/discharging, some CO2 is released, starting at the onset of the voltage plateau. The CO2 arises from an attack of oxidised oxygen species from the cathode lattice on the carbonate solvents in the electrolyte and can be used to quantify the amount of oxygen released from the surface, as previously demonstrated.8,21,54–60 The quantity of CO2 released during charge up to 4.7 V vs. Li+/Li is 0.89 mmol g−1(LNO)−1, indicating that up to 0.04 moles of O are lost per mole of LiNiO2, see Methods.To examine the growth and thickness of the densified layer with state of charge, annular dark field scanning transmission electron microscopy (ADF-STEM) was performed. The morphology of the material is composed of the typical primary/secondary particle agglomerates, see SEM in ESI† Fig. S1. These were sonicated to release the primary particles for STEM analysis. The images are shown in Fig. 2a–l. The high-resolution images in Fig. 2e–h, show the evolution of the surface structure. The layered structure at the surface of the pristine material, Fig. 2e, gives way to the beginning of a cation-mixed, rocksalt-like surface at 4.1 V vs. Li+/Li, evident from the appearance of scattering intensity from the Li layers due to the presence of Ni Fig. 2f. The emergence of this rocksalt-like surface at 4.1 V vs. Li+/Li is in line with the observed small amount of O-loss at the onset of the voltage plateau, ESI† Fig. S8. To determine the thickness of this surface layer, the variations in scattering intensity along three atom rows, i, ii and iii, from the surface to the bulk, Fig. 2e–h, were examined. The intensity of the three lines was averaged and is shown in Fig. 2m–p. The intensities are dominated by the scattering from Ni and show where there are regions of high and low Ni content. The more intense peaks arise from Ni in transition metal layers, and the weaker peaks from Ni in the Li layers. The ratios of the intensity maxima of the high and low-intensity peaks are plotted as the red line in Fig. 2n to p. On moving from bulk to surface, the variation in scattering intensity diminishes in accord with moving from a layered to a rocksalt-like distribution of Ni ions. The transition from layered to rocksalt-like structure does not occur abruptly but has a small transition region. However, the region where the rocksalt dominates appears initially with a thickness of around 1 nm at 4.1 V vs. Li+/Li, Fig. 2n, and grows to around 5 nm by 4.7 V vs. Li+/Li, Fig. 2p.
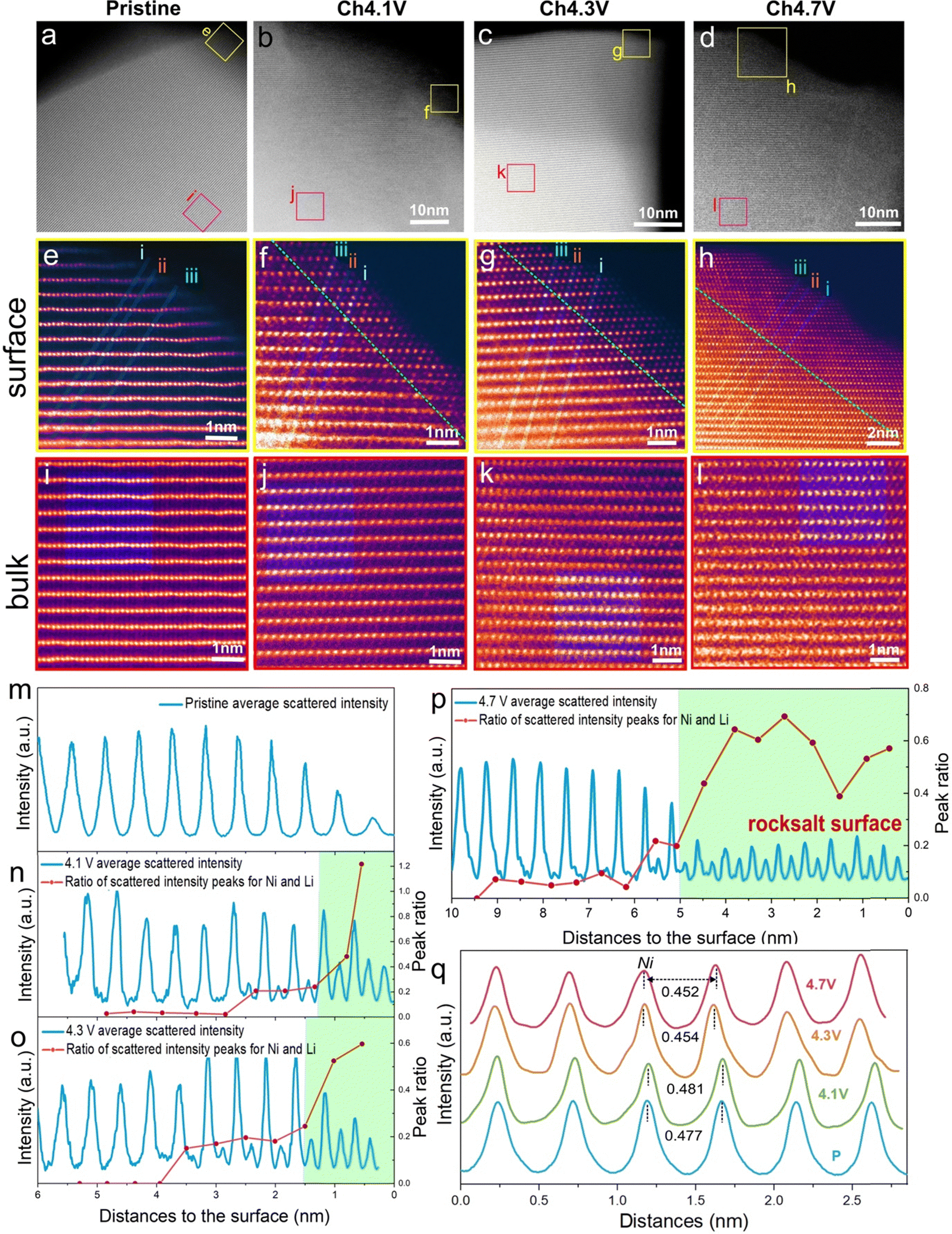 | ||
| Fig. 2 Surface and bulk ADF-STEM images for LiNiO2 on the first charge. (a)–(d) Wide field of view ADF-STEM images for LiNiO2 at increasing states of charge on the first cycle with the surface (yellow boxes) and bulk (red boxes) regions highlighted and examined in closer detail (e)–(h) and (i)–(l), respectively. (e)–(h) ADF-STEM images from the LiNiO2 surface at each state of charge. A shell (surface layer) with a mixing of Ni between the transition and Li layers grows in thickness on charge from 4.1 V vs. Li+/Li. The shell is bound by the green dotted line. The thickness of the evolving shell and therefore position of the green dotted line is established from the data in Fig. 2m to p. (i)–(l) ADF-STEM images of the LiNiO2 bulk regions taken about 50 nm away from the surface at each state of charge showing preservation of the layered structure. (m)–(p) Variation in scattering intensity from the surface to the bulk was obtained by averaging the three numbered line scans in (e)–(h). The ratios of the intensity maxima of the NiLi peak (Ni in the lithium metal layer) to the NiNi peak (Ni in the transition metal layer) are plotted as the red line in the figures. It shows the transition from a rocksalt-like structure (where the Ni occupancies in the Li and transition metal layers are closer) to a layered structure (where they are different). (q) Within the bulk of the particles, Fig. 2(i) to (l), scattering intensities are integrated along the transition metal and Li layers, respectively, within the green-shaded regions, then plotted across the layers. They confirm the layered structure is retained in the bulk on charging. | ||
Considering the images taken from the bulk of the particles (Fig. 2i–l), the layered ordering is maintained throughout the charge, Fig. 2i–l. Intensity analysis was carried out within the blue-shaded regions in Fig. 2i–l. Intensities were integrated along the transition and Li layers, respectively and then plotted across the layers, Fig. 2q. They confirm the retention of the layered structure in the bulk. Between 4.1 V vs. Li+/Li and 4.3 V vs. Li+/Li, the interlayer spacing contracts slightly in agreement with the diffraction results.
Given an average primary particle size of 250 nm as seen in STEM and SEM, ESI† Fig. S1, and a shell thickness of 5 nm, as observed by STEM, Fig. 2h and p, and assuming spherical particles, consistent with the approximately primary particle morphology, seen in ESI† Fig. S1, the rocksalt shell occupies approximately 12% of the volume of the cathode. The 0.04 moles of surface O-loss results in an increase of 0.02 moles of Ni in the densified layer. This, in addition to the 0.08 moles of Ni from the bulk to the surface, yields a shell composition of Ni1.75O2 and, therefore an overall Ni oxidation state of +2.3.
Ni versus O oxidation in LiNiO2
To probe the extent of Ni oxidation in LiNiO2 on charging, Ni L-edge XAS was performed in bulk sensitive fluorescence yield (FY) mode (approximately 50–100 nm penetration depth, i.e. depth of the primary particles). The obtained spectra for LiNiO2 at different states of charge over the first cycle are displayed in Fig. 3a. The pristine spectrum closely resembles other measurements taken on LiNiO2 indicating an initial oxidation state of close to Ni3+.13 On charging, the edge shape changes are consistent with Ni oxidising towards +4. Ni oxidation continues across the voltage plateau at 4.215 V; as seen in the reduction in intensity of the peak at 853 eV, Fig. 3a. Beyond 4.3 V vs. Li+/Li, the L-edge spectra show little further change. It is important to note that the plateau is associated with a two-phase reaction, and the data suggests that the Ni oxidation state is higher in the composition at 4.3 V vs. Li+/Li than in the composition at 4.1 V vs. Li+/Li at the beginning of the plateau. It does not imply a continuous evolution of Ni oxidation state as would be the case for a solid solution. The fact that the spectra for the 4.3 V vs. Li+/Li and 5.0 V vs. Li+/Li samples still exhibit the 853 eV shoulder peak is consistent with an overall Ni oxidation state being less than +4. Comparing the data with the Ni L-edge spectrum of a Ni4+ reference material, Li0.8Ni0.13Co0.13Mn0.54O2, indicates that the overall Ni oxidation state for the particles charged to 4.3 V vs. Li+/Li and beyond is approximately +3.8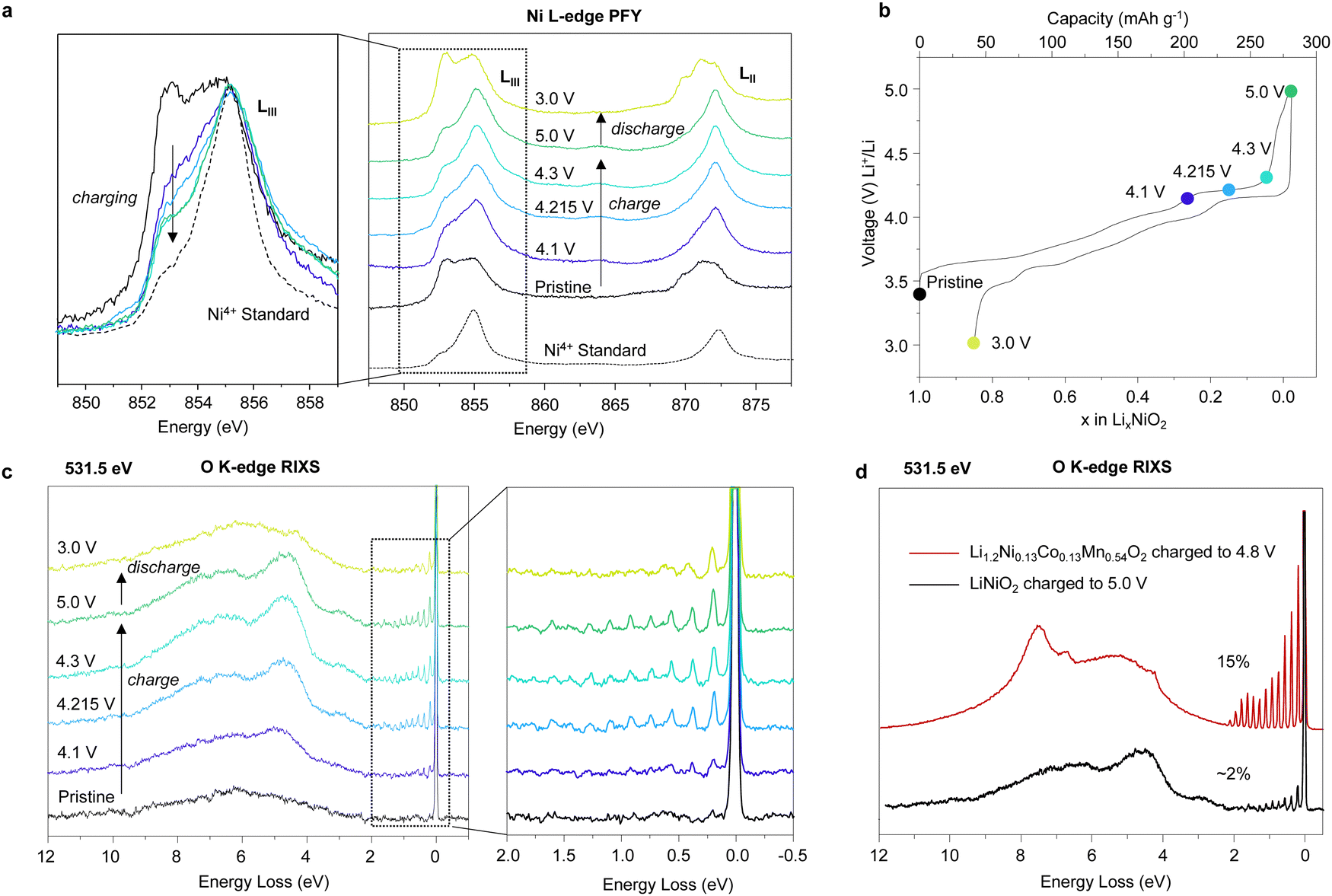 | ||
| Fig. 3 Spectroscopy of LiNiO2 on the first cycle. (a) Ni L-edge X-ray absorption spectra for LiNiO2 at different states of charge over the first cycle, colours correspond to points in (c). Li1.2Ni0.13Co0.13Mn0.54O2 charged to a lithium content of 0.8 is used as a standard reference for Ni4+. Ni LIII-edge spectra show continuous peak intensity reduction at 853 eV, indicating oxidation of Ni across the voltage plateau between 4.1 V vs. Li+/Li and 4.3 V vs. Li+/Li. (b) First cycle load curve for LiNiO2. (c) High-resolution O K-edge RIXS for LiNiO2 measured at the resonance energy for molecular O2. The vibrational peaks between 0 eV and 2 eV energy loss indicate the formation of trapped molecular O2 across the voltage plateau. (d) Comparison of the signal intensity of the vibrational peaks arising from O2 between LiNiO2 charged to 5 V vs. Li+/Li and Li1.2Ni0.13Co0.13Mn0.54O2 are consistent with about 2% of the O in LiNiO2 in the form of molecular O2. The amount of O2 in Li1.2Ni0.13Co0.13Mn0.54O2 is approximately 15%. | ||
While fluorescence yield XAS probes to a depth of 50–100 nm, the signal inevitably contains a contribution from the surface. Electron yield data with a probe depth of around 10–20 nm, ESI† Fig. S11, confirm that the surface Ni is more reduced than that obtained from fluorescence mode, as indicated by a sharp, distinct peak at 853 eV consistent with the 5 nm thick Ni2.3+ shell discussed above. Given that the fluorescence mode contains a contribution of Ni2.3+ from the shell at the surface and that the Ni L-edge fluorescence yield indicates an overall oxidation state of Ni3.8+ the results from the two modes suggest the Ni in the core is closer to +4.
At the end of charge, all of the 0.96 mol of Li have been removed from LiNiO2 (Li0.96Ni0.04[Ni]O2). Given the shell is +2.3, the bulk is closer to +4 and the overall particle Ni oxidation state is approximately +3.8, which is consistent with the XAS spectrum at 5.0 V vs. Li+/Li, approximately 0.16 mol of electrons must arise from O oxidation. Our quantitative OEMS have already shown that surface O-loss accounts for 0.08 mol of lithium extraction (0.04 mol of O lost). We can, therefore, assign the remaining 0.08 mol to bulk oxygen oxidation.
To investigate the bulk O-redox mechanism, O K-edge RIXS measurements were obtained. Previous RIXS measurements on Li-rich cathodes have revealed that O oxidation results in the formation of O2 molecules trapped within the cathode particles.21–23,25,61–63 The RIXS measurements for LiNiO2 (Fig. 3c and d), also show the same vibrational features at an excitation energy of 531.5 eV as those previously attributed to O2, Fig. 3c. The features begin to appear at the onset of the voltage plateau and increase in intensity up to the end of charge. On discharge to 3 V vs. Li+/Li, only a small quantity of O2 remains, indicating an almost complete reduction of O2 back to O2−.
By comparing the area under the signal to RIXS spectra obtained under the same measurement conditions for charged Li1.2Ni0.13Co0.13Mn0.54O2, where the amount of O2 is known, it is possible to estimate approximately how much O2 is present in the LiNiO2 charged to 5 V vs. Li+/Li, see the Methods section and ESI† Fig. S12. The results displayed in Fig. 3d suggest that 2% of the O in LiNiO2 charged to 5 V vs. Li+/Li is in the form of trapped molecular O2, Fig. 3d, corresponding to 0.08 moles of Li extraction (20 mA h g−1), in good accord with the amount of O-oxidation implied from the overall Ni oxidation state. This amount of O2 could feasibly be accommodated by the 8% Ni vacancies in the transition metal layer observed in the charged cathode, if each O2 molecule is trapped within a cluster of four vacancies, as proposed previously.21,23,25,62,64,65
In previous studies of lithium rich materials such as Li[Li0.2Ni0.13Co0.13Mn0.54]O2, we have shown using density functional theory (DFT) how vacancy clusters form in the transition metal layers through metal migration, permitting the formation of trapped O2.21,64 Total energy calculations show that structures containing trapped O2 are substantially lower in energy than other configurations of the charged structure by as much as 410 meV f.u.−1, indicating a thermodynamic driving force behind trapped O2 formation. To demonstrate that LiNiO2 behaves in a consistent manner with our previously investigated systems, we computed the energy difference for charged LiNiO2 before and after the formation of vacancy clusters and trapped O2. The calculations (ESI† Fig. S14 and Table S7) show that the structure with O2 is 205 meV f.u.−1 lower in energy than the structure without, in line with our earlier work.
Close inspection of the relaxed structural model with trapped O2 molecules shows an O–O bond length of 1.2 Å, close to that of free O2, indicating there is little evidence for a strong coordination interaction with the neighbouring Ni. This accords with our previous modelling for Mn-based transition metal oxides, and experimental vibrational RIXS data for the 4d and 5d-TM systems.66
Discussion
Unlike Li-rich layered oxides, Li+ extraction from stoichiometric Ni-rich cathodes could in principle be charge compensated by transition metal oxidation alone. It has further been argued that, due to the lack of cation vacancies in the transition metal layers and the absence of non-bonding O 2p orbitals, O-redox, oxidation of O2− ions, as observed in Li-rich cathodes does not apply to the Ni-rich systems.14,15Our results reveal that the structural instability in the bulk of LiNiO2 is more extensive than previously thought. On oxidation of [Li0.956Ni0.044]NiO2, Li+ extraction is charge compensated by oxidation of Ni +2.96 to +3.63 at the onset of the 4.2 V vs. Li+/Li plateau. On charging across and beyond the plateau, O2− ions are oxidised, resulting in oxygen loss at the surface and the growth of a Ni-rich surface layer reaching approximately 5 nm thick. In the bulk, O2− oxidation forms O2 trapped in the particles. This is enabled by the loss of Ni from the transition metal layers forming 8% Ni vacancies. Ni vacancies on the transition metal sites have two consequences: they generate non-bonding O 2p orbitals activating O2− oxidation and they enable Ni reorganisation and vacancy clustering necessary to accommodate the O2 molecules, explaining how it is possible to form trapped O2 in stoichiometric compounds that do not initially have Li+ or vacancies in the transition metal layers. The Ni no longer present in the bulk is not lost from the particles, instead forming part of a Ni-rich shell with a composition of Ni1.75O2.23,61,62 The overall Ni oxidation state of the particles as determined by Ni L edge XAS in fluorescence yield, i.e. bulk mode, and the oxygen RIXS data show respectively that Ni and O2− are oxidised on charing across the 4.2 V vs. Li+/Li plateau. However, the plateau is associated with a two-phase reaction and all we can conclude is that the phase at the charged end of the plateau at 4.3 V vs. Li+/Li contains Ni in a higher oxidation state than before the plateau and that O-redox has occurred. It does not imply that O-redox commences before Ni is oxidised to +4. The bulk fluorescence yield mode gives the overall Ni oxidation state for the charged particles as +3.8. However, it includes the shell, which has a Ni oxidation state of +2.3 and grows across the plateau. The implication is that the Ni oxidation state in the core of the core–shell particles is very close to +4 from 4.3 V vs. Li+/Li onwards. Therefore, we do not see evidence of O-redox commencing before Ni is oxidised to +4.
O K-edge RIXS data were recently reported by Piper and co-workers15 on LiNi0.98W0.02O2 showing a series of sharp peaks in the emission spectrum between 0–2 eV energy loss at the top of charge.15 While the authors acknowledged that these features were identical to those attributed to molecular O2 in Li-rich cathodes, they argued that since they could not observe vacant Ni sites in the transition metal layer of the charged cathode, there was no possibility of vacancy cluster formation to accommodate O2 in the Ni-rich materials. Instead, Morris and co-workers14 proposed that O–O dimers form by a water-assisted decomposition of oxidised lattice O and they are not intrinsic to the O-redox mechanism.14 According to their model, H2O and OH groups should be present in the empty Li layer. However, 1H magic angle spinning (MAS) NMR data collected on our LiNiO2 sample charged to 5 V vs. Li+/Li do not detect protons, ESI† Fig. S15. In contrast, our results identifying the presence of Ni vacancies in the transition metal layer, suggest that the trapped O2 can arise through a similar process of O2− oxidation to that observed in the Li-rich layered cathodes. The results are summarised in the schematic in Fig. 4.
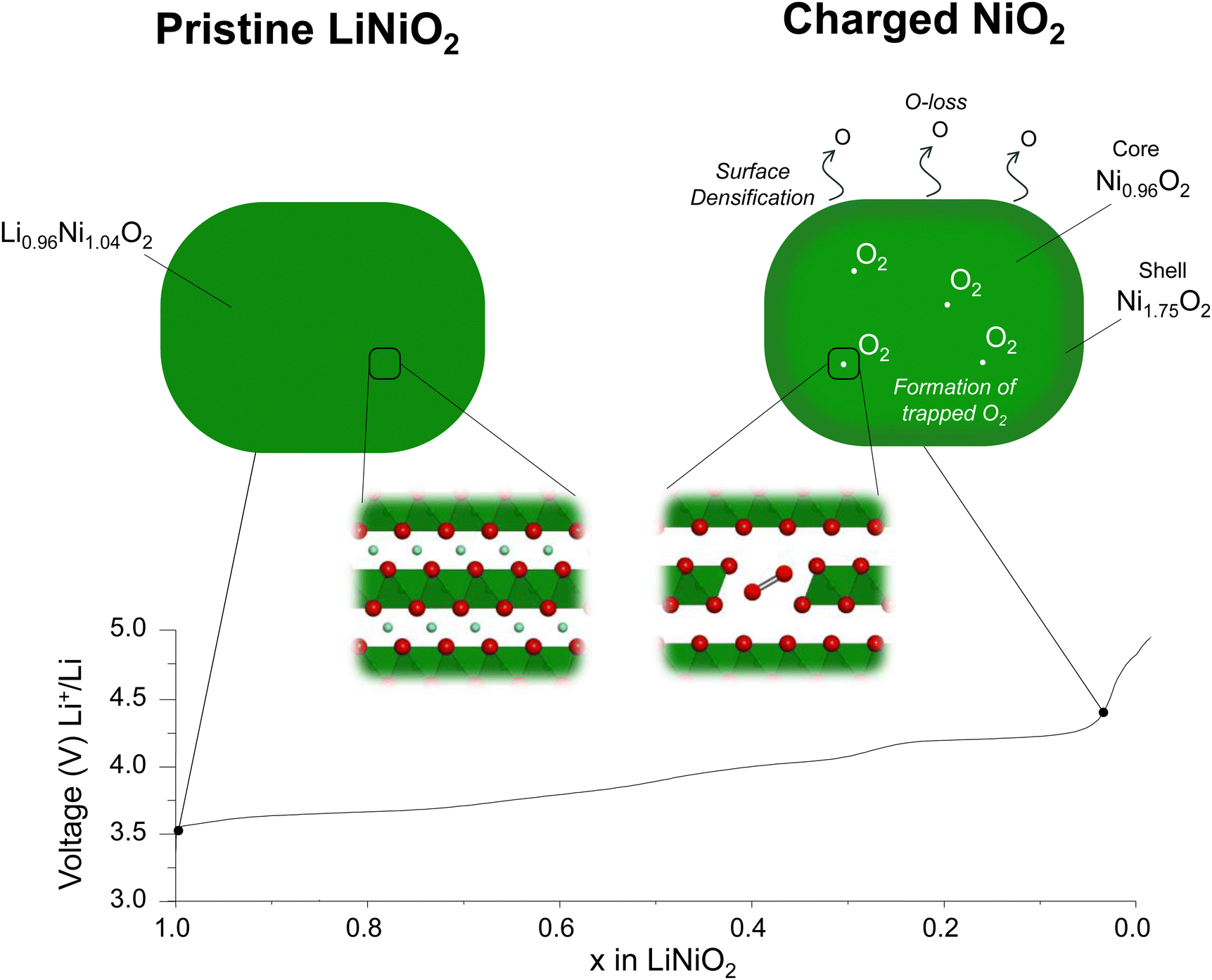 | ||
| Fig. 4 Schematic of charging mechanisms in LiNiO2. Ni vacancies are formed in the transition metal layer on charging across the 4.2 V vs. Li+/Li plateau, generating non-bonded O 2p states that activate O2− oxidation and enabling Ni rearrangement into vacancy clusters that can accommodate trapped O2 molecules. | ||
Conclusion
A combined powder neutron and X-ray diffraction study, taking account of random stacking faults, revealed that charging LiNiO2 across the plateau at 4.2 V vs. Li+/Li induces the formation of Ni vacancies in the previously fully dense transition metal layers in the bulk of the cathode. These vacant sites have two consequences: they activate the oxidation of O2− by forming non-bonding O 2p orbitals and the observed 8% of Ni vacancies are sufficient to form vacancy clusters able to accommodate trapped molecular O2 formed on O2− oxidation which we observe by high-resolution RIXS, similar to the mechanism of O-redox observed in Li-rich cathodes. Beyond the 4.2 V vs. Li+/Li plateau, the particles exhibit a core–shell morphology with a Ni1.75O2 shell and a Ni oxidation state of +2.3, while the core has a Ni oxidation state close to +4. Together, the results reveal that although charging is predominantly associated with Ni oxidation, the bulk structure of delithiated LiNiO2 is liable to changes on charging to accommodate trapped molecular O2. Our study shows the mechanism of O-redox involving the formation of trapped O2 in the bulk is general to both Li-rich materials and LiNiO2 despite the latter possessing fully dense transition metal layers in the pristine state.Author contributions
Mikkel Juelsholt, Jun Chen, Robert A. House, and Peter G. Bruce conceived the project and wrote the manuscript with contributions from all authors. Mikkel Juelsholt and Miguel A. Pérez-Osorio performed the structural analysis of the diffraction data in close collaboration with Helen E. Maynard-Casely, Jue Liu and Michelle Everett. Jun Chen did the STEM measurements and performed their analysis together with Rob A House and Mikkel Juelsholt. Sofia De Sousa Coutinho and Mikkel Juelsholt measured and analysed the ICP. Gregory J. Rees performed and analysed the MAS NMR experiments. Robert A. House, working closely with Stefano Agrestini, Mirian Garcia-Fernandez and Ke-Jin Zhou conducted, processed and interpreted the RIXS and soft XAS measurements. Robert A. House and Peter G. Bruce supervised the project. Miguel A. Pérez-Osorio performed the DFT calculations.Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
We are indebted to the Engineering and Physical Sciences Research Council (EPSRC), the Henry Royce Institute for Advanced Materials (EP/R00661X/1, EP/S019367/1, EP/R010145/1, EP/L019469/1) and the Faraday Institution (FIRG007, FIRG008, FIRG016) for financial support. We acknowledge Diamond Light Source for time on Beamlines I15-1 XPDF and I21 under Proposals MM27764-1 and MM29028-1. We thank the Spallation Neutron Source, a Department of Energy Office of Science User Facility operated by the Oak Ridge National Laboratory, for providing neutron powder diffraction measurements. We acknowledge the support of the Australian Centre for Neutron Scattering, ANSTO and the Australian Government through the National Collaborative Research Infrastructure Strategy in supporting the neutron research infrastructure used in this work via ACNS proposal MI13571. This project was supported by the Royal Academy of Engineering under the Research Fellowship scheme. We also acknowledge the resources provided by the Cambridge Tier-2 system operated by the University of Cambridge Research Computing Service (https://www.hpc.cam.ac.uk) funded by EPSRC Tier-2 capital grant EP/P020259/1, via the BATTSurface, NextCATHODE and SOLEL projects. For the purpose of Open Access, the author has applied a CC BY public copyright licence to any Author Accepted Manuscript version arising from this submission.References
- S.-T. Myung, F. Maglia, K.-J. Park, C. S. Yoon, P. Lamp, S.-J. Kim and Y.-K. Sun, ACS Energy Lett., 2017, 2, 196–223 CrossRef CAS
.
- F. Schipper, E. M. Erickson, C. Erk, J.-Y. Shin, F. F. Chesneau and D. Aurbach, J. Electrochem. Soc., 2017, 164, A6220 CrossRef CAS
- J. Xu, F. Lin, M. M. Doeff and W. Tong, J. Mater. Chem. A, 2017, 5, 874–901 RSC
- S. G. Booth, A. J. Nedoma, N. N. Anthonisamy, P. J. Baker, R. Boston, H. Bronstein, S. J. Clarke, E. J. Cussen, V. Daramalla, M. De Volder, S. E. Dutton, V. Falkowski, N. A. Fleck, H. S. Geddes, N. Gollapally, A. L. Goodwin, J. M. Griffin, A. R. Haworth, M. A. Hayward, S. Hull, B. J. Inkson, B. J. Johnston, Z. Lu, J. L. MacManus-Driscoll, X. Martínez De Irujo Labalde, I. McClelland, K. McCombie, B. Murdock, D. Nayak, S. Park, G. E. Pérez, C. J. Pickard, L. F. J. Piper, H. Y. Playford, S. Price, D. O. Scanlon, J. C. Stallard, N. Tapia-Ruiz, A. R. West, L. Wheatcroft, M. Wilson, L. Zhang, X. Zhi, B. Zhu and S. A. Cussen, APL Mater., 2021, 9, 109201 CrossRef CAS
- M. Bianchini, M. Roca-Ayats, P. Hartmann, T. Brezesinski and J. Janek, Angew. Chem.,Int. Ed., 2019, 58, 10434–10458 CrossRef CAS PubMed
- C. S. Yoon, D. W. Jun, S. T. Myung and Y. K. Sun, ACS Energy Lett., 2017, 2, 1150–1155 CrossRef CAS
- L. de Biasi, A. Schiele, M. Roca-Ayats, G. Garcia, T. Brezesinski, P. Hartmann and J. Janek, ChemSusChem, 2019, 12, 2240–2250 CrossRef CAS
- F. Kong, C. Liang, L. Wang, Y. Zheng, S. Perananthan, R. C. Longo, J. P. Ferraris, M. Kim and K. Cho, Adv. Energy Mater., 2019, 9, 1802586 CrossRef
- C. Xu, P. J. Reeves, Q. Jacquet and C. P. Grey, Adv. Energy Mater., 2021, 11, 2003404 CrossRef CAS
- H. Li, N. Zhang, J. Li and J. R. Dahn, J. Electrochem. Soc., 2018, 165, A2985 CrossRef CAS
- E. Levi, M. D. Levi, G. Salitra, D. Aurbach, R. Oesten, U. Heider and L. Heider, Solid State Ionics, 1999, 126, 97–108 CrossRef CAS
- H. Nguyen, R. Silverstein, A. Zaveri, W. Cui, P. Kurzhals, S. Sicolo, M. Bianchini, K. Seidel and R. J. Clément, Adv. Funct. Mater., 2023, 2306168 CrossRef
- N. Li, S. Sallis, J. K. Papp, J. Wei, B. D. McCloskey, W. Yang and W. Tong, ACS Energy Lett., 2019, 4, 2836–2842 CrossRef CAS
- A. R. Genreith-Schriever, H. Banerjee, A. S. Menon, E. N. Bassey, L. F. J. Piper, C.
P. Grey and A. J. Morris, Joule, 2023, 7, 1623–1640 CrossRef CAS
- A. S. Menon, B. J. Johnston, S. G. Booth, L. Zhang, K. Kress, B. E. Murdock, G. Paez Fajardo, N. N. Anthonisamy, N. Tapia-Ruiz, S. Agrestini, M. Garcia-Fernandez, K. Zhou, P. K. Thakur, T. L. Lee, A. J. Nedoma, S. A. Cussen and L. F. J. Piper, PRX Energy, 2023, 2, 13005 CrossRef
- P. Kurzhals, F. Riewald, M. Bianchini, H. Sommer, H. A. Gasteiger and J. Janek, J. Electrochem. Soc., 2021, 168, 110518 CrossRef CAS
- Dm. M. Korotin, D. Novoselov and V. I. Anisimov, Phys Rev B, 2019, 99, 45106 CrossRef CAS
- K. Foyevtsova, I. Elfimov, J. Rottler and G. A. Sawatzky, Phys Rev B, 2019, 100, 165104 CrossRef CAS
- D. A. Kitchaev, J. Vinckeviciute and A. Van Der Ven, J. Am. Chem. Soc., 2021, 143, 1908–1916 CrossRef CAS PubMed
- N. Li, S. Sallis, J. K. Papp, B. D. McCloskey, W. Yang and W. Tong, Nano Energy, 2020, 78, 105365 CrossRef CAS
- R. A. House, J. J. Marie, M. A. Pérez-Osorio, G. J. Rees, E. Boivin and P. G. Bruce, Nat. Energy, 2021, 6, 781–789 CrossRef CAS
- R. Sharpe, R. A. House, M. J. Clarke, D. Förstermann, J.-J. Marie, G. Cibin, K.-J. Zhou, H. Y. Playford, P. G. Bruce and M. S. Islam, J. Am. Chem. Soc., 2020, 142, 21799–21809 CrossRef CAS
- R. A. House, G. J. Rees, K. McColl, J.-J. Marie, M. Garcia-Fernandez, A. Nag, K.-J. Zhou, S. Cassidy, B. J. Morgan, M. Saiful Islam and P. G. Bruce, Nat. Energy, 2023, 8, 351–360 CrossRef CAS
- J. Feng, Z. Chen, W. Zhou and Z. Hao, Mater. Horiz., 2023, 10, 4686–4709 RSC
- R. A. House, H. Y. Playford, R. I. Smith, J. Holter, I. Griffiths, K.-J. Zhou and P. G. Bruce, Energy Environ. Sci., 2022, 15, 376–383 RSC
- J. Neuefeind, M. Feygenson, J. Carruth, R. Hoffmann and K. K. Chipley, Nucl. Instrum. Methods Phys. Res., Sect. B, 2012, 287, 68–75 CrossRef CAS
- M. Avdeev and J. R. Hester, J. Appl. Crystallogr., 2018, 51, 1597–1604 CrossRef CAS
- P. Juhás, T. Davis, C.-L. Farrow and S.-J.-L. Billinge, J. Appl. Crystallogr., 2013, 46, 560–566 CrossRef
- X. Yang, P. Juhás, C. L. Farrow and S. J. L. Billinge, arXiv: Materials Science.
- C. L. Farrow, P. Juhas, J. W. Liu, D. Bryndin, E. S. Božin, J. Bloch, T. Proffen and S. J. L. Billinge, J. Phys.: Condens. Matter, 2007, 19, 335219 CrossRef CAS
- A. A. Coelho, J. Appl. Crystallogr., 2018, 51, 210–218 CrossRef CAS
- K.-J. Zhou, A. Walters, M. Garcia-Fernandez, T. Rice, M. Hand, A. Nag, J. Li, S. Agrestini, P. Garland, H. Wang, S. Alcock, I. Nistea, B. Nutter, N. Rubies, G. Knap, M. Gaughran, F. Yuan, P. Chang, J. Emmins and G. Howell, J. Synchrotron Radiat., 2022, 29, 563–580 CrossRef CAS
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed
- M. Schlipf and F. Gygi, Comput. Phys. Commun., 2015, 196, 36–44 CrossRef CAS
- D. R. Hamann, Phys Rev B: Condens. Matter Mater. Phys., 2013, 88, 85117 CrossRef
- R. J. Green, H. Wadati, T. Z. Regier, A. J. Achkar, C. McMahon, J. P. Clancy, H. A. Dabkowska, B. D. Gaulin, G. A. Sawatzky and D. G. Hawthorn, 2020.
- L. Croguennec, C. Pouillerie, A. N. Mansour and C. Delmas, J. Mater. Chem., 2001, 11, 131–141 RSC
- M. Casas-Cabanas, M. Reynaud, J. Rikarte, P. Horbach and J. Rodriguez-Carvajal, J. Appl. Crystallogr., 2016, 49, 2259–2269 CrossRef CAS
- J. Molenda, P. Wilk and J. Marzec, Solid State Ionics, 2002, 146, 73–79 CrossRef CAS
- L. Croguennec, C. Pouillerie and C. Delmas, Solid State Ionics, 2000, 135, 259–266 CrossRef CAS
- C. Delmas, J. P. Pérès, A. Rougier, A. Demourgues, F. Weill, A. Chadwick, M. Broussely, F. Perton, Ph Biensan and P. Willmann, J. Power Sources, 1997, 68, 120–125 CrossRef CAS
- J. C. Garcia, J. Gabriel, N. H. Paulson, J. Low, M. Stan and H. Iddir, J. Phys. Chem. C, 2021, 125, 27130–27139 CrossRef CAS
- P.-H. Chien, X. Wu, B. Song, Z. Yang, C. K. Waters, M. S. Everett, F. Lin, Z. Du and J. Liu, Batteries Supercaps, 2021, 4, 1701–1707 CrossRef CAS
- W. Li, J. N. Reimers and J. R. Dahn, Solid State Ionics, 1993, 67, 123–130 CrossRef CAS
- E. Bautista Quisbert, F. Fauth, A. M. Abakumov, M. Blangero, M. Guignard and C. Delmas, Small, 2023, 19, 2300616 CrossRef CAS
- K.-Y. Park, Y. Zhu, C. G. Torres-Castanedo, H. J. Jung, N. S. Luu, O. Kahvecioglu, Y. Yoo, J.-W. T. Seo, J. R. Downing, H.-D. Lim, M. J. Bedzyk, C. Wolverton and M. C. Hersam, Adv. Mater., 2022, 34, 2106402 CrossRef CAS
- P. Xiao, T. Shi, W. Huang and G. Ceder, ACS Energy Lett., 2019, 4, 811–818 CrossRef CAS
- P. Mukherjee, N. V. Faenza, N. Pereira, J. Ciston, L. F. J. Piper, G. G. Amatucci and F. Cosandey, Chem. Mater., 2018, 30, 8431–8445 CrossRef CAS
- D. P. Abraham, R. D. Twesten, M. Balasubramanian, J. Kropf, D. Fischer, J. McBreen, I. Petrov and K. Amine, J. Electrochem. Soc., 2003, 150, A1450 CrossRef CAS
- D. P. Abraham, R. D. Twesten, M. Balasubramanian, I. Petrov, J. McBreen and K. Amine, Electrochem. Commun., 2002, 4, 620–625 CrossRef CAS
- F. Lin, I. M. Markus, D. Nordlund, T.-C. Weng, M. D. Asta, H. L. Xin and M. M. Doeff, Nat. Commun., 2014, 5, 3529 CrossRef
- S. Zheng, R. Huang, Y. Makimura, Y. Ukyo, C. A. J. Fisher, T. Hirayama and Y. Ikuhara, J. Electrochem. Soc., 2011, 158, A357 CrossRef CAS
- A. Mikheenkova, S. Mukherjee, M. Hirsbrunner, P. Törnblom, C.-W. Tai, C. U. Segre, Y. Ding, W. Zhang, T. C. Asmara, Y. Wei, T. Schmitt, H. Rensmo, L. Duda and M. Hahlin, ChemRxiv.
- J. Wandt, A. T. S. Freiberg, A. Ogrodnik and H. A. Gasteiger, Mater. Today, 2018, 21, 825–833 CrossRef CAS
- D. H. Seo, J. Lee, A. Urban, R. Malik, S. Kang and G. Ceder, Nat. Chem., 2016, 8, 692–697 CrossRef CAS
- W. M. Dose, W. Li, I. Temprano, C. A. O’Keefe, B. L. Mehdi, M. F. L. De Volder and C. P. Grey, ACS Energy Lett., 2022, 7, 3524–3530 CrossRef CAS PubMed
- B. Strehle, K. Kleiner, R. Jung, F. Chesneau, M. Mendez, H. A. Gasteiger and M. Piana, J. Electrochem. Soc., 2017, 164, A400 CrossRef CAS
- X. Chen, N. Li, E. Kedzie, B. D. McCloskey, H. Tang and W. Tong, J. Electrochem. Soc., 2019, 166, A4136 CrossRef CAS
- R. Jung, M. Metzger, F. Maglia, C. Stinner and H. A. Gasteiger, J. Electrochem. Soc., 2017, 164, A1361–A1377 CrossRef CAS
- R. Sharpe, R. A. House, M. J. Clarke, D. Förstermann, J.-J. Marie, G. Cibin, K.-J. Zhou, H. Y. Playford, P. G. Bruce and M. S. Islam, J. Am. Chem. Soc., 2020, 142, 21799–21809 CrossRef CAS
- R. A. House, U. Maitra, M. A. Pérez-Osorio, J. G. Lozano, L. Jin, J. W. Somerville, L. C. Duda, A. Nag, A. Walters, K.-J. Zhou, M. R. Roberts and P. G. Bruce, Nature, 2020, 577, 502–508 CrossRef CAS PubMed
- K. Chai, J. Zhang, Q. Li, D. Wong, L. Zheng, C. Schulz, M. Bartkowiak, D. Smirnov and X. Liu, Small, 2022, 18, 2201014 CrossRef CAS
- E. Boivin, R. A. House, M. A. Pérez-Osorio, J.-J. Marie, U. Maitra, G. J. Rees and P. G. Bruce, Joule, 2021, 5, 1267–1280 CrossRef CAS
- U. Maitra, R. A. House, J. W. Somerville, N. Tapia-Ruiz, J. G. Lozano, N. Guerrini, R. Hao, K. Luo, L. Jin, M. A. Pérez-Osorio, F. Massel, D. M. Pickup, S. Ramos, X. Lu, D. E. McNally, A. V. Chadwick, F. Giustino, T. Schmitt, L. C. Duda, M. R. Roberts and P. G. Bruce, Nat. Chem., 2018, 10, 288–295 CrossRef CAS
- R. A. House, J.-J. Marie, J. Park, G. J. Rees, S. Agrestini, A. Nag, M. Garcia-Fernandez, K.-J. Zhou and P. G. Bruce, Nat. Commun., 2021, 12, 2975 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ee04354a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |