
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ultra-high surface area ionic-liquid-derived carbons that meet both gravimetric and volumetric methane storage targets†
Nawaf
Albeladi
 ab,
L. Scott
Blankenship
a and
Robert
Mokaya
*a
ab,
L. Scott
Blankenship
a and
Robert
Mokaya
*a
aSchool of Chemistry, University of Nottingham, University Park, Nottingham NG7 2RD, UK. E-mail: r.mokaya@nottingham.ac.uk
bTaibah University, Yanbu Al Bahr, 46423, Saudi Arabia
First published on 3rd April 2024
Abstract
The storage of methane, to enable vehicular use, may be achieved in porous solids, but to date, there is no material that meets the gravimetric and volumetric targets (for example those set by the US Department of Energy, DOE) for such use. Here, in an effort to address this challenge, we explore the use of carbonised N-rich crosslinkable imidazolium-based ionic liquid (IL), 1-butyl-3-methylimidazolium tricyanomethanide, ([BMIm][C(CN)3]), as a precursor for porous carbons. On carbonisation, the IL yields carbonaceous matter (IL-C) with the unusual combination of high N content and low O content (i.e., low O/C atomic ratio). Activation of the IL-derived carbonaceous matter (IL-C) with KOH generates activated carbons with a mix of microporosity and mesoporosity, ultra-high surface area of up to ∼4000 m2 g−1, pore volume of up to 3.3 cm3 g−1, and relatively high packing density. The enhanced porosity and comparatively high packing density of the activated carbons is a consequence of the elemental composition of the IL-C precursor. The presence of N, which acts as a porogen, favours generation of carbons with high mesoporosity and high surface area while a low O/C ratio acts in a reverse manner favouring the formation of microporous carbons with high packing density. The overall effect is that the carbons have porosity and packing density that is suited for optimising both the gravimetric and volumetric uptake of methane, which reaches 0.53 g g−1 and 289 cm3 (STP) cm−3, respectively, at 25 °C and 100 bar. The uptake, therefore, surpasses both the gravimetric and volumetric methane storage targets that would enable widespread use for vehicular transport. The IL-derived activated carbons are the first porous materials (carbon or MOF) to meet both gravimetric and volumetric methane storage targets for experimentally determined values.
Broader contextNatural gas, whose main ingredient is methane, is an alternative fuel for vehicular transport due to its lower carbon emission compared to petroleum based fuels. Methane storage for vehicular use may be achieved in porous materials, but to date, there is no material that meets the required gravimetric and volumetric targets for such use. To address this challenge, we explore carbonised N-rich and O-poor (i.e. low O/C ratio) crosslinkable imidazolium-based ionic liquid as precursor for porous carbons. On activation, the carbonised ionic liquid yields activated carbons with surface area and pore volume of up to ∼4000 m2 g−1 and 3.3 cm3 g−1, respectively, along with high packing density. Here we show that the carbons have excellent methane uptake of up to 0.53 g g−1 and 289 cm3 (STP) cm−3, respectively, at 25 °C and 100 bar, which surpasses both gravimetric and volumetric storage targets required for vehicular transport. |
1. Introduction
The continuous emission of greenhouse gases into the atmosphere, largely driven by rapid global development that has relied on the accelerated use of fossil fuels, has raised concerns over the impact on the environment, and on climate and global ecosystems. In particular, the concentration of CO2, the largest contributor to global warming, has reached unprecedented levels in the atmosphere.1 The need for alternative fuels to enable a transition away from conventional petroleum is becoming more urgent in order to meet the mitigation target set by international pledges, such as the Paris Agreement, to reduce atmospheric CO2 levels.2 Efforts are currently focussed on the development of environmentally friendly and low cost technology that will enable both the remediation of environmental damage caused by rising CO2 levels and the transition to cleaner fuels.3–6 A possible alternative fuel is natural gas (NG), which primarily consists of methane. NG has recently received a great deal of interest as an alternative fuel for vehicular transport due to its abundance and lower carbon emissions.6,7 However, the use of NG as fuel for mobile applications faces challenges associated with storage and transportation.5,8–10 Conventionally, NG is stored as compressed natural gas (CNG) in high-pressure vessels (>250 bar), which pose safety concerns. NG may also be stored as liquefied natural gas (LNG) under cryogenic conditions. However, LNG technology requires high compression costs and is therefore impractical for use in vehicles. On the other hand, adsorbed natural gas (ANG) technology, which involves adsorption onto solid materials, is promising and can circumvent the difficulties of CNG and LNG by combining much lower pressures than those needed for CNG along with the possibility of achieving sufficient storage at room temperature, unlike LNG. Successful application of ANG technology relies on finding adsorbent materials that are able to store sufficient amounts of natural gas or methane. In this regard, a variety of porous materials have been investigated, including metal–organic frameworks (MOFs),6,11 zeolites12 and porous carbons.13,14 Porous carbons, and in particularly activated carbons, have gained increasing interest due to several factors, including their relatively low cost, excellent chemical and mechanical stability, and ability to tune their textural properties.15–18The storage of methane in zeolites is limited by their low to moderate surface area. This means that they have modest gravimetric methane storage capacity of up to ca. 10 mmol g−1 (i.e., 0.16 g g−1) at 100 bar and 25 °C.12 On the other hand, due to their much higher surface area and pore volume, MOFs can achieve greater gravimetric methane uptake.6,11 For example, at 23 °C, it has been reported that methane storage in MOFs can reach 25 and 33 mmol g−1 at 65 bar and 100 bar, respectively.11 However, the low packing density of MOFs means that their volumetric uptake does not reach sufficiently high levels.6,11 The packing density of MOFs may be increased if they are compacted, but such compaction or densification reduces porosity, which has deleterious effects on the gravimetric methane storage capacity and consequently does not improve the volumetric uptake.6,11 Porous carbons, on the other hand, can achieve high surface area and, due to their high mechanical stability, can also be compacted without loss of porosity meaning that they retain good gravimetric and volumetric uptake capacity for methane. The ideal porous carbon would need to have both high porosity (surface area and pore volume) and high packing density. Such a combination, while possible for porous carbons, would be difficult to achieve for zeolites and MOFs.
The textural properties of activated carbons are determined by the type and amount of activating agent, the activation temperature, and the nature and elemental composition of the carbonaceous precursor from which they are derived.16 Regarding the impact of the nature of the carbonaceous precursor, we have recently shown that it is possible to tailor the porosity of activated carbons by choice of precursors based on their elemental composition.13,19,20 We have explored the use of pre-mixed precursors (e.g., a mixture of polypyrrole and sawdust) to control, tailor and enhance the porosity of activated carbons.19 More specifically, we have shown that the nature of the carbonaceous precursor, and in particular the elemental composition, can determine susceptibility to activation.13,14,16,20 Knowledge of the link between the elemental composition of a carbon precursor and susceptibility to activation is important as it can enable predicting of the textural parameters of the resulting activated carbon, i.e., proportion and level of microporosity and mesoporosity. We have shown that the O/C atomic ratio of a carbonaceous precursor can play a role in determining susceptibility to activation. Carbonaceous matter with a high O/C ratio are more susceptible to activation and may result in activated carbons with high surface area and greater levels of mesoporosity and wider pores. On the other hand, carbonaceous matter with a low O/C ratio are resistant to so-called over-activation and thus offer more control over textural properties and favour the formation of micropores (i.e., are microporous) to the exclusion of mesopores and mesoporosity. However, such low O/C ratio precursors are only able to generate activated carbons with relatively low surface area due to their high resistance to activation. This means that the gravimetric gas (e.g., methane) uptake of activated carbons derived from such precursors tends to be low since gas storage at moderate to high pressure (35 to 100 bar) is very dependent on the surface area of the adsorbent.13,14
The presence of N in the framework of a carbonaceous precursor also plays a role in determining the textural properties of activated carbons. For example, adding N-containing additives to carbonaceous precursors can tune the porosity of activated carbons towards achievement of high surface area and can be used to tailor the balance between microporosity and mesoporosity.21–24 The N in the precursors acts as an in situ porogen and favours the generation of carbons with high mesoporosity and high surface area but which have low packing density. In a sense, the presence of N can be considered to have an opposite effect to that of low O/C ratio. While the presence of N favours generation of high surface area carbons that possess significant mesoporosity, a low O/C ratio, on the other hand, acts in a reverse manner and favours the formation of microporous carbons with moderate surface area but high packing density. In order to optimise both the gravimetric and volumetric uptake of methane, a suitable carbon material would ideally have high surface area, a mix of mesoporosity and microporosity, and a high packing density. Activated carbons with such a mix of properties are most likely to be obtained from carbonaceous precursors that have a high N content along with a low O/C ratio.
In this work, we show that by choosing a suitable precursor it is possible to prepare activated carbons with a combination of attractive properties that have previously only been achieved via the use of pre-mixed precursors or with added N-containing doping agents. We report on an ionic liquid derived carbonaceous precursor that generates activated carbons with high surface area but without excessive diminution of microporosity or packing density. Ionic liquids (ILs) have recently gained attention as precursors for carbon materials.25–29 ILs are composed entirely of ions, which coupled with their carbon-rich nature, means that they can function as precursors for porous carbons. In this regard, ILs offer the following advantages: (i) because of their structure and high electrostatic interaction between electrical charges, they exhibit negligible vapour pressure and high thermal stability leading to reduced mass loss during carbonisation processes, (ii) homogeneous incorporation of N and C atoms in situ into the carbon materials, which eliminates the need for using doping agents, and (iii) structural diversity that can be modified to meet various requirements by the selection of the cation and anion thus offering additional possibilities for control over structure and properties of carbon materials at the molecular level. We show that carbonisation of ILs can yield carbonaceous matter that possesses both a high N content and a low O/C ratio. We explore the porosity of carbons prepared from the IL-derived carbonaceous matter, and probe the impact of the presence of N on the methane storage capacity and manner of sorption. The expectation, which is proven here, is that such (i.e., high N content and a low O/C ratio) precursors can generate porous carbons with both high porosity and high packing density, a combination that is suitable for optimising both the gravimetric and volumetric storage of methane.
2. Experimental section
2.1 Materials
1-Butyl-3-methylimidazolium tricyanomethanide, ([BMIm][C(CN)3]), (TCI), and potassium hydroxide (KOH), (Sigma-Aldrich) were used as received. Ionic liquid-derived activated carbons (IL-ACs) were prepared in two steps, namely, carbonisation followed by activation. For carbonisation, the required amount of [BMIm][C(CN)3], in an alumina boat, was placed in a horizontal tube furnace and heated, at a heating ramp rate of 10 °C min−1, to 800 °C and held for 1 h under an inert (N2 gas) atmosphere. The furnace was then allowed to cool to room temperature under a flow of N2 gas. The resulting carbonaceous matter was designated as IL-C (IL = ionic liquid, and C = carbonised). The IL-C carbon was activated as follows; IL-C was added to KOH at KOH/IL-C ratio of 2, 4, or 6 in an agate mortar and thoroughly mixed. The mixture was then placed in an alumina boat and heated in a furnace under a flow of N2 gas (100 mL min−1) at ramp rate of 5 °C min−1 to the target temperature (700, 800, or 900 °C) and held for 1 h. The furnace was then allowed to cool to room temperature under a flow of N2 gas. The resulting activated carbons were washed with 1 M HCl by stirring at room temperature, followed by washing with deionised water until the filtrate achieved neutral pH (i.e., pH ∼ 7), and then dried in oven at 120 °C. The activated carbons were designated as IL-ACxT, where x is the KOH/IL-C mass ratio, and T is the activation temperature (in °C). Thus, a carbon activated at a KOH/IL-C ratio of 4 and at 800 °C is designated as IL-AC4800. The packing density of the carbons was determined by pressing a known amount of carbon in a 1.3 cm die at pressure of 7 MPa for 5 min. The packing density was then calculated from the mass and volume of the resulting pellet/disc.2.2 Materials characterisation
Thermogravimetric analysis (TGA) was performed using a TA Instruments SDT Q600 analyser under flowing air or N2 conditions (100 ml min−1) and heating ramp rate of 10 °C min−1. Powder XRD patterns were obtained using a PANalytical X’Pert PRO diffractometer with Cu-Kα radiation (λ = 1.5418 Å) (40 kV, 40 mA), 0.02 step size and 5 s step time between 2° and 80°. Elemental (CHN) analysis was performed on an Exeter Analytical CE-440 Elemental Analyser. Nitrogen sorption (at −196 °C) isotherms and textural properties of the activated carbons were determined using a Micromeritics 3Flex sorptometer. Prior to analysis, the carbons were degassed at 300 °C for 16 h under vacuum. The surface areas were calculated using the Brunauer–Emmett–Teller (BET) method using the nitrogen adsorption data in the relative pressure (P/P0) range between 0.05–0.22. Total pore volume was determined from the amount of nitrogen adsorbed at a relative pressure of 0.99. The micropore surface area and micropore volume were estimated by t-plot analysis. The porosity measures (surface area and pore volume) were also calculated using the non-local density functional theory (NLDFT) model. Furthermore, to ensure consistency of the porosity measures, BET surface identification (BETSI) was also performed for which the minimum number of points in the linear region was 10, with a minimum r2 of 0.995, and Rouquerol criterion maximum error of 20%.30–32 The pore size distribution (PSD) curves were calculated using the SAIEUS software and the 2D-NLDFT heterogeneous surface kernel with a fitting parameter, λ, of 5 for all isotherms.Infra-red spectroscopy (FTIR) was performed using a Bruker ALPHA FTIR Spectrometer. Scanning electron microscopy (SEM) images were obtained using a JEOL 7000F FEG-SEM microscope. Transmission electron microscopy (TEM) images were obtained using a JEOL 2100F instrument operating at 200 kV and equipped with a Gatan Orius CCD for imaging. Prior to analysis, the carbon samples were suspended in distilled water and dispersed onto lacey carbon support films. For X-ray photoelectron spectroscopy (XPS), a Kratos AXIS ULTRA with a monochromated A1 K X-ray source (1486.6 eV) was used at 10 mA emission current and 12 kV anode potential. To avoid surface charging, a charge neutralizer filament was utilised. The analysis chamber pressure was better than 5 × 10−9 Torr. Each sample was analysed in three different areas. XPS spectra were analysed using CasaXPS™ software (version 2.3.22PR1.0), with the Kratos sensitivity factors to derive atomic percentage values from the peak regions. The binding energies were calibrated by setting C-1s to 284.6 eV.
2.3 Methane uptake measurements
Methane uptake was determined using a Hiden Isochema XEMIS Analyser. Sample masses were at least 30 mg, and the mass measurement precision of XEMIS is 0.1 μg with a mass stability of 1 μg. Methane (BOC, purity 99.9995%) uptake isotherms were measured up to 100 bar. The pressure precision for any isotherm point was 0.02% at any point, and the base vacuum pressure was <1 × 10−6 mbar. Equilibrium uptake was determined by identifying the asymptote of rate of change in uptake after a relaxation of 99% (tolerance 2%). Data was sampled at an interval of 1 s. Prior to isotherm measurement, the carbons were degassed in situ at 240 °C (ramp rate 10 °C min−1) for at least 4 h until sample mass was stable.Isosteric enthalpies of adsorption (Qst) were calculated from methane uptake isotherms using the Whittaker method,33 for which a specific python module was created in pyGAPS.34 Information on installation and basic use is available in the ESI.† Owing to the PointIsotherm and ModelIsotherm classes in pyGAPS, calculations require only an isotherm as an input because physical parameters (critical point, triple point, and saturation pressures) are automatically calculated from the identity of the adsorbate and analysis temperature. In the case of a supercritical isotherm, a pseudo-saturation pressure is calculated. As the Whittaker method requires determination of enthalpy of vaporisation of the adsorbate, the module returns the values of Qst only at loadings corresponding to pressures between the critical and saturation pressures. For convenience, the module can also output a plot of Qstvs. loading. For this work, Toth was found to fit the isotherms best so this model was used in the Whittaker Qst calculations.
3. Results and discussion
3.1 Nature and composition of the ionic liquid-derived carbons
The ionic liquid, 1-butyl-3-methylimidazolium tricyanomethanide (Fig. S1, ESI†) was chosen as starting material due to a combination of factors aimed at achieving the desired properties in the carbonisation products. The ionic liquid is rich in N (C12H15N5), and the high electrostatic interaction between the ions leads to negligible vapour pressure and high thermal stability, minimising mass loss during carbonisation. The imidazolium based-cation can induce high surface area in any resulting carbons, while the nitrile group (tricyanomethanide) favours the formation of 3D-connected frameworks via cross-linking during pyrolysis. The yields and elemental composition of the samples are summarized in Table 1. We first noticed that, due to the high thermal stability of the [BMIm][C(CN)3] ionic liquid, the IL-C carbonaceous matter has a graphite-like appearance and texture (Fig. S2, ESI†). This is attributed to the presence of the [C(CN)3] anion, which does not decompose completely at high temperature and has a yield (for IL-C) of ∼30 wt% with respect to the IL. This is comparable to hydrochar yields from biomass following hydrothermal carbonisation.19,20,24 The molar N/C ratio of the IL is 0.42 with N content of 30 wt%, which is consistent with the molecular formula (C12H15N5). After carbonisation, the N content of IL-C remains high at 19.3 wt%, while the C content increases from ca. 62 wt% for the IL to 69 wt% for IL-C, and the O content rises from a nominal value of 2.1 wt to 10.7 wt%. The H content decreases from 6.6 wt for the IL to 0.9 wt for IL-C. It is interesting to note that the O/C ratio of IL-C is, therefore, very low at 0.116, which is one of the lowest reported for any activatable carbonaceous matter.13,14 This means that IL-C achieves the unusual elemental composition of having both a high N content (19.3 wt%) and very low O/C ratio (0.116). We are not aware of any previous study that has deliberately targeted such carbonaceous matter as precursor for activated carbons or conducted a systematic exploration of its activation.| Sample | Yield [wt%] | C [%] | H [%] | N [%] | O [%] | (O/C)a | (N/C)a |
|---|---|---|---|---|---|---|---|
| a Atomic ratio. b Nominal O content of 2.1% obtained as O = 100-C–H–N, which gives O/C ratio of 0.026. | |||||||
| ILb | — | 61.6 | 6.6 | 29.7 | 0.482 | ||
| IL-C | 28 | 69.1 | 0.9 | 19.3 | 10.7 | 0.116 | 0.279 |
| IL-AC2700 | 46 | 72.2 | 0.6 | 6.7 | 20.5 | 0.213 | 0.093 |
| IL-AC2800 | 33 | 87.9 | 0.5 | 1.6 | 10.0 | 0.085 | 0.018 |
| IL-AC2900 | 17 | 90.0 | 0.2 | 0.3 | 9.5 | 0.079 | 0.004 |
| IL-AC4700 | 34 | 83.2 | 0.8 | 2.3 | 13.7 | 0.124 | 0.027 |
| IL-AC4800 | 29 | 93.6 | 0.1 | 0.5 | 5.8 | 0.047 | 0.005 |
| IL-AC4900 | 16 | 92.9 | 0.0 | 0.4 | 6.7 | 0.054 | 0.004 |
| IL-AC6700 | 23 | 86.6 | 0.4 | 2.3 | 10.7 | 0.093 | 0.027 |
| IL-AC6800 | 18 | 90.3 | 0.8 | 0.2 | 8.7 | 0.072 | 0.002 |
| IL-AC6900 | 13 | 96.0 | 0.0 | 0.2 | 3.8 | 0.030 | 0.001 |
In general, the yields of the activated carbons (with respect to the IL-C) depend on the amount of KOH and the activation temperature. For samples activated at 900 °C, the yields vary from 13 to 17%, while at 700 °C the yield is between 23 and 46%. For samples activated at 800 °C the yield is in the range of 18 to 33 wt%. The activated samples show a high C content, which increases from ca. 69 wt% for IL-C to between 72 and 96 wt% for IL-ACxT carbons. The highest C content (96 wt%) was observed for the most severely activated sample (IL-AC6900), while the lowest (72 wt%) was for the least severely activated (IL-AC2700). The N content of the activated carbons decreases as the activation temperature or amount of KOH rises, and ranges from 6.7 wt% for the sample activated under the mildest conditions (IL-AC2700) to 0.2 wt% for the most severely activated carbons (IL-AC6800 and IL-AC6900). These results indicate that a high amount of N is lost during activation, which can be attributed to the fact that N is oxidised and eliminated more readily than C during the activation process, which is the genesis of the N's action as an in situ porogen.35 The H and O content vary less drastically on activation but generally decrease with increasing severity of activation.
The thermal properties of the IL, IL-C, and the IL-ACxT activated carbons were probed using TGA, and the resulting curves are shown in Fig. 1. When the IL was thermally treated to 800 °C with a ramp rate of 10 °C min−1 under an inert atmosphere of (N2 flow of 100 ml min−1), the TGA curve was consistent with the carbonisation yield of ca. 30 wt%. The purity (in terms of being purely carbonaceous with no inorganic components) and thermal stability of IL-C and IL-ACxT carbons were probed by heating under an air flow of 100 ml min−1. The TGA curves in Fig. 1 indicate high thermal stability up to at least 500 °C, followed by almost complete mass loss between 500–750 °C, due to combustion of carbon. The IL-ACs burn off at roughly the same temperature, indicating that they are similar in terms of the nature of their carbon. The activated carbons were burnt off with hardly any residual mass, which indicates their fully carbonaceous nature.
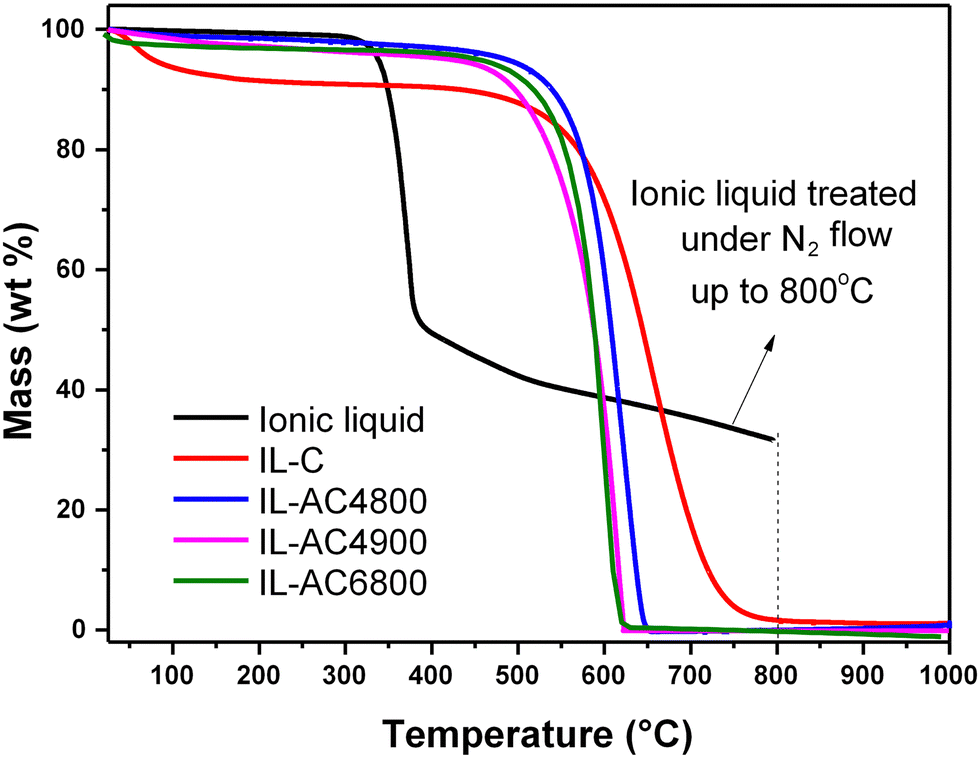 | ||
| Fig. 1 Thermal stability and purity of ionic liquid, IL-C and activated carbons. TGA curves of the ionic liquid (thermally treated under N2 gas), and IL-C and some IL-C derived activated carbons (IL-ACxT), which were thermally treated in air. | ||
Powder X-ray diffraction was used to study the structure and purity (in terms of absence of any crystalline inorganic phases) of IL-C and the activated carbons. The XRD pattern of IL-C (Fig. S3, ESI†) shows a well-developed graphitic stacking peak at 2θ of ∼26°, which is assigned to the (002) diffraction of graphitic carbon. A further weak peak is observed at 2θ of ∼44° which is the (100) diffraction arising from graphene stacks. The intensity of these peaks reduced greatly on activation of IL-C (Fig. S3, ESI†). Carbons activated under milder conditions (e.g., IL-AC2700) still show the peaks, which are, however, much broader. For the other more severely activated carbons, no graphitic peaks were observed, which indicates that the nature of the carbon is amorphous. This is consistent with the expectation that activation should break up any graphitic domains.
The surface structure and morphology of IL-C and IL-AC carbons were explored using scanning electron microscopy (SEM) so as to illustrate the structural transformation following carbonisation and activation. The SEM images of IL-C appear to show a non-porous surface morphology (Fig. S4, ESI†), which agrees with the expected low porosity of the carbonised product. In contrast, the SEM images for IL-AC carbons (Fig. S5 and S6, ESI†) suggest well developed porous structures. Transmission electron microscopy (TEM) was used to further probe the microstructural differences between IL-C and IL-ACs. The TEM images of IL-C reveal a graphitic structure (Fig. S7, ESI†), which agrees well with the XRD pattern (Fig. S3, ESI†). The formation of a graphitic carbon can be attributed to the dynamic trimerization of the nitrile group based-anion during carbonisation.36 On the other hand, the TEM images of IL-ACs (Fig. S8, ESI†) are similar to each other and reveal amorphous carbon structures, which is consistent with the XRD patterns and the expected break up of graphitic domains during activation. Overall, however, the morphology of the IL-AC carbons is similar to that previously reported for activated carbons.15–20
X-ray photoelectron spectroscopy (XPS) was used to further probe the elemental composition of IL-C and the activated carbons and the chemical state of any elements present. The XPS wide scan spectra of IL-C and IL-ACxT carbons are compared in Fig. 2. The spectrum of IL-C revealed a high N content, which is consistent with the elemental composition (Table 1). The content of N reduced after activation and the reduction was greater at higher activation temperature/and or amounts of KOH. The high resolution N 1s-XPS spectra (Fig. S9, ESI†) show that the N has the following bonding: the N environment in IL-C is dominated by pyridine-N (peak at 398.4 eV) and quarternary-graphitic N (peak at 401.0 eV).37 For IL-AC2700 (Fig. S9, ESI†) there are three peaks ascribed to pyridinic N (N-6 at 398.9 eV), quaternary N (N-Q at 401.1 eV) and oxidized N (N-X at 405.3 eV). The intensity of the N-6 peak decreased after activation, which is in agreement with previous reports.38 It is likely that the activation is responsible for generating the oxidised-N group. The XPS spectrum of IL-AC4900, which was prepared under harsh conditions, appeared to show no N content in agreement with the elemental composition (Table 1).
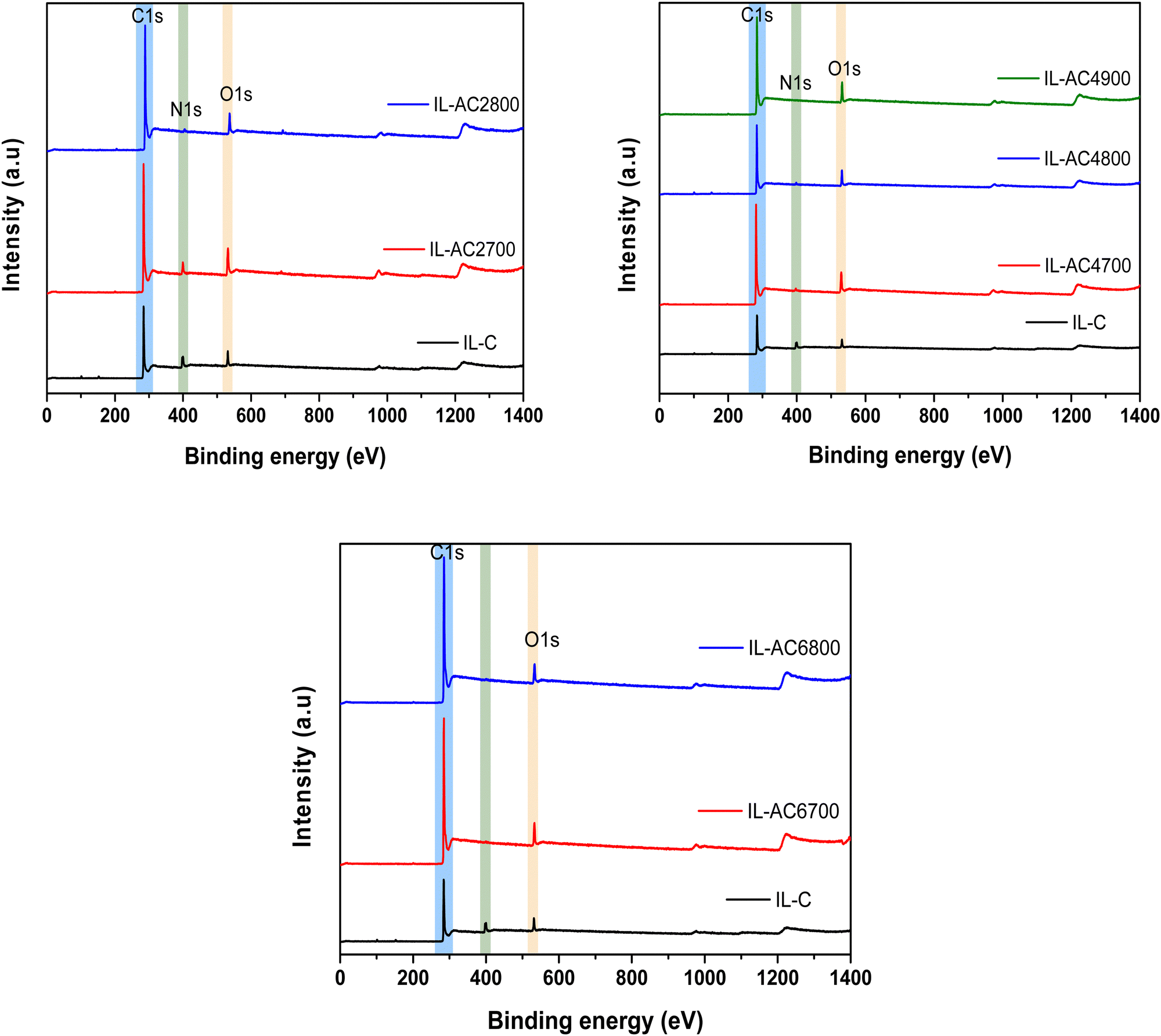 | ||
| Fig. 2 X-ray photoelectron spectroscopy (XPS) of IL-C and activated carbons. Wide scan XPS spectra of carbonised ionic liquid (IL-C) and IL-ACxT activated carbons. | ||
In order to clarify on the mechanism underlying the conversion of the IL to IL-C and finally to the activated carbons, we performed Fourier transform Infra-red spectroscopy (FTIR) of all the respective samples in the transformation chain. The IR spectrum of the 1-butyl-3-methylimidazolium tricyanomethanide ionic liquid (Fig. S10, ESI†) exhibits peaks at 2960 cm−1 and 2870 cm−1, which arise from C–H stretching vibrations of the alkyl chain. The peak at ca. 3100 cm−1 can be attributed to the stretching mode of the aromatic C–H bonds of the imidazolium cation. The strong peak at 2156 cm−1 arises from stretching vibrations of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bonds of the tricyanomethanide moiety. The peaks within the fingerprint region (600–1500 cm−1) can be attributed to the imidazolium ring, i.e., stretching and bending vibrations of the aromatic structure. The peaks at 1570 cm−1 and 1450 cm−1 are due to C
N bonds of the tricyanomethanide moiety. The peaks within the fingerprint region (600–1500 cm−1) can be attributed to the imidazolium ring, i.e., stretching and bending vibrations of the aromatic structure. The peaks at 1570 cm−1 and 1450 cm−1 are due to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CN stretching, while those at 750 cm−1 and 620 cm−1 are due to C–N stretching. Interestingly, the spectra of IL-C (Fig. S10, ESI†) shows some retained features in the fingerprint region due to retention of the imidazolium backbone in the carbonaceous matter. Interestingly, these ‘imidazolium backbone’ features are retained in the IL-ACxT activated carbons (Fig. S11, ESI†). Importantly, however, the spectra of IL-C and the IL-ACxT activated carbons do not show the sharp tricyanomethanide (CN) peak expected at 2156 cm−1. These observations are important in clarifying the mechanism of conversion of the IL to the IL-C carbonaceous matter and finally to the IL-ACxT activated carbons especially with regard to role of the imidazolium backbone and the N in the anionic fragments. The IR spectra are consistent with the assumption that the N in the anionic fragments serves as a source of N within the carbonised IL (IL-C), which is then available to influence the nature of porosity developed in the activated carbons. On the other hand, the imidazolium backbone serves as a main C source for the activated carbons along with any carbon arising from the alkyl chains.
C and CN stretching, while those at 750 cm−1 and 620 cm−1 are due to C–N stretching. Interestingly, the spectra of IL-C (Fig. S10, ESI†) shows some retained features in the fingerprint region due to retention of the imidazolium backbone in the carbonaceous matter. Interestingly, these ‘imidazolium backbone’ features are retained in the IL-ACxT activated carbons (Fig. S11, ESI†). Importantly, however, the spectra of IL-C and the IL-ACxT activated carbons do not show the sharp tricyanomethanide (CN) peak expected at 2156 cm−1. These observations are important in clarifying the mechanism of conversion of the IL to the IL-C carbonaceous matter and finally to the IL-ACxT activated carbons especially with regard to role of the imidazolium backbone and the N in the anionic fragments. The IR spectra are consistent with the assumption that the N in the anionic fragments serves as a source of N within the carbonised IL (IL-C), which is then available to influence the nature of porosity developed in the activated carbons. On the other hand, the imidazolium backbone serves as a main C source for the activated carbons along with any carbon arising from the alkyl chains.
3.2 Porosity of the ionic-liquid derived activated carbons
The IL-C carbon had limited porosity with surface area and pore volume of 40 m2 g−1 and 0.03 cm3 g−1, respectively. The nitrogen sorption isotherm and pore size distribution of IL-C (Fig. S12, ESI†) confirm that the IL-C carbon is very lowly porous with hardly any developed pores. The nitrogen sorption isotherms and corresponding pore size distribution (PSD) curves of the activated carbons are shown in Fig. 3. For IL-AC2T, the isotherm changes from type I (microporous) to type IV (mesoporous) with increase in activation temperature. This change is reflected in the pore structure of the activated carbons by the formation of wider pores as shown by the PSD curves (Fig. 3 and Fig. S13, ESI†), which indicate the presence of micropores and small mesopores. For KOH/IL-C ratio of 4 or 6 (Fig. 3 and Fig. S13, ESI†), predominantly mesoporous carbons with isotherm type IV are produced at all activation temperatures. Thus, unlike IL-AC2T carbons, the IL-AC4T and IL-AC6T samples are predominantly mesoporous but with a significant proportion of microporosity. The adsorbed nitrogen levels, as per the isotherms, are significantly higher than the average for activated carbons.18,39,40 We attribute the enhanced porosity to the presence of N in the IL-C precursor. However, it is also noticeable that despite the enhanced porosity, the isotherms and PSD curves still show that some microporosity is retained even under the harshest activating conditions. We attribute the retention of some microporosity to the mitigating effect of the low O/C ratio of IL-C. The isotherms of IL-AC2700, IL-AC2800, IL-AC4700 and IL-AC6700, which exhibit a relatively more microporous nature, do not show any hysteresis loop. The rest of the samples show type H2 hysteresis loop, which can arise from hierarchical porosity, which is consistent with their higher level of mesoporosity accompanied with a significant proportion of microporosity. The size of the hysteresis loop appears to increase with the level of mesoporosity. Overall, the hysteresis loops are consistent with slit-shaped pores and the nature of connectivity expected for activated carbons.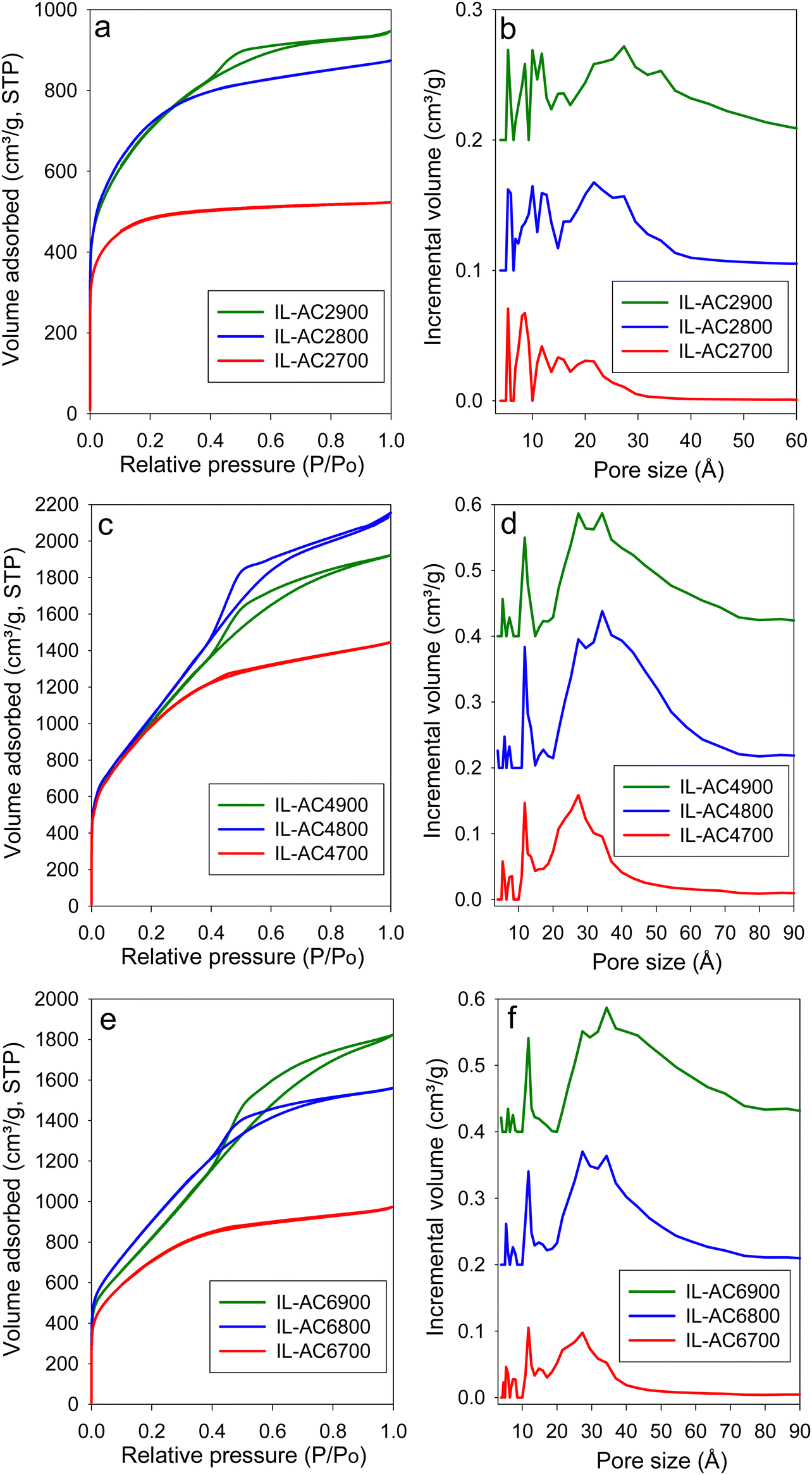 | ||
| Fig. 3 Porosity of IL-ACxT activated carbons. (a), (c) and (e) Nitrogen sorption isotherms of IL-AC2T, IL-AC4T and IL-AC6T carbons, respectively. (b), (d) and (f) Pore size distribution (PSD) curves of IL-AC2T, IL-AC4T and IL-AC6T carbons, respectively. | ||
Table 2 summarises the textural parameters of the activated carbons. The surface area of the activated carbons is in the high to ultra-high range of 1803 to 3995 m2 g−1, depending on the severity of activation. In general, the surface area increases when the activation temperature rises from 700 to 800 °C, followed by a slight reduction at 900 °C, which is in line with the concept of “overactivation”.15 The surface area for IL-AC2T carbons (IL-AC2700, IL-AC2800 and IL-AC2900) is 1803, 2619 and 2534 m2 g−1, respectively, which shows a clear impact of the activation temperature and that 800 °C offers the optimum conditions for maximising the surface area. For IL-AC4T carbons (IL-AC4700, IL-AC4800 and IL-AC4900), the surface area increased to 3662, 3995 and 3818 m2 g−1, respectively. Further increase in amount of KOH (KOH/IL-C ratio of 6) resulted in a decrease in the surface area to 2567, 3394 and 3135 m2 g−1, respectively for IL-AC6700, IL-AC6800 & IL-AC6900 carbons. The carbons have high pore volume that ranges from 1.35 to 3.3 cm3 g−1, except the sample IL-AC2700 (0.81 cm3 g−1). The surface area and pore volume are amongst the highest ever reported for activated carbons from any precursor under any given activation conditions. Indeed, to the best of our knowledge, IL-AC4800 has one of the highest surface areas of any activated carbons reported to date.13,24,39–45 The level of microporosity in the carbons depends on the severity of activation, and generally decreases at higher activation temperature and KOH/IL-C ratio. Thus, whilst IL-AC2T carbons are predominantly microporous (57–79% of surface area and 45–72% of pore volume arising from micropores), IL-AC4T show a mix of micro and mesoporosity, and IL-AC6T are mesoporous in nature. Similar to recently described polypyrrole-derived carbons, IL-C appears to be susceptible to the creation of larger pores and mesoporosity, suggesting that, despite having a low O/C ratio, it is considerably less resistant to activation due to a high N content.18,40,41
| Sample | Surface area (m2 g−1) | Micropore surface areaa (m2 g−1) | Pore volume (cm3 g−1) | Micropore volumeb (cm3 g−1) | Surface area densityc (m2 cm−3) | Packing densityd (g cm−3) | Volumetric surface areae (m2 cm−3) |
|---|---|---|---|---|---|---|---|
| a Values in parenthesis are % of surface area from micropores. b Values in parenthesis are % of pore volume from micropores. c Surface area density is obtained as ratio of total surface area to total pore volume. d The packing density was determined by pressing a known amount of carbon in a 1.3 cm die at pressure of 7 MPa for 5 min. e Volumetric surface area determined as surface area × packing density. | |||||||
| IL-AC2700 | 1803 | 1424 (79) | 0.81 | 0.58 (72) | 2226 | 0.76 | 1370 |
| IL-AC2800 | 2619 | 1853 (71) | 1.35 | 0.80 (60) | 1940 | 0.50 | 1310 |
| IL-AC2900 | 2524 | 1433 (57) | 1.46 | 0.66 (45) | 1729 | 0.39 | 984 |
| IL-AC4700 | 3662 | 1745 (48) | 2.23 | 0.79 (35) | 1642 | 0.49 | 1794 |
| IL-AC4800 | 3995 | 1523 (38) | 3.30 | 0.81 (25) | 1210 | 0.39 | 1558 |
| IL-AC4900 | 3817 | 1172 (31) | 2.97 | 0.64 (22) | 1285 | 0.31 | 1183 |
| IL-AC6700 | 2567 | 1322 (52) | 1.50 | 0.6 (40) | 1711 | 0.56 | 1438 |
| IL-AC6800 | 3394 | 1305 (38) | 2.40 | 0.71 (30) | 1414 | 0.37 | 1256 |
| IL-AC6900 | 3135 | 876 (28) | 2.81 | 0.61 (22) | 1116 | 0.31 | 972 |
Recently, there has been some discussion on the effectiveness of the use of the BET equation in reproducibly calculating the surface area of porous materials.42 A round-robin study involving a large number of research groups found significant variability in the BET surface area calculated from identical nitrogen sorption isotherms.42 To address such variability, the study developed a computational method, namely, BET surface identification (BETSI), that was claimed to more accurately and unambiguously determine the BET surface area of porous materials.42 Therefore, to ensure consistency of our porosity measures, we calculated the surface area of the activated carbons using the BETSI method wherein the minimum number of points in the linear region was 10, with a minimum r2 of 0.995, and Rouquerol criterion maximum error of 20% (Table S1 and Fig. S14–S22, ESI†). For completeness, we also analysed the porosity and calculated the porosity measures (surface area and pore volume) using the non-local density functional theory (NLDFT) model (Fig. S23–S25, ESI†). The comparative porosity data from BET, BETSI and NLDFT analysis (Table S2, ESI†) shows that our BET values are very close to the BETSI derived surface area, which signifies their veracity. Most of our BET surface area values are within 1% of the BETSI surface areas, which validates our porosity calculation protocols. The surface area obtained via the NLDFT method is lower than that from BET and BETSI methods. However, the trends in surface area are similar for all the three models.
Recent reports have shown that there is a link between the ratio of surface area to pore volume ratio, i.e., the surface area density (SAD), and the tendency to generate micropores rather than larger pores.14,19 Microporous materials have high SAD, and the SAD can reduce with the introduction of mesopores. A strong linear correlation has been demonstrated between the O/C of carbonaceous precursors and surface area density of their resulting carbons.14 This means that SAD is a good indicator of the susceptibility or resistance to activation of any carbonaceous precursor.14,19 The SAD of the IL-ACxT carbons (Table 2) covers a wide range from 1116 to 2226 m2 cm−3, and is dependent on the activation conditions (KOH/IL-C ratio and activation temperature). To further clarify on the contrasting effects of the presence of N and low O/C ratio on the activation behavior of IL-C, we compared it with other forms of carbonaceous matter that have been used as starting material for activated carbons (Table S3, ESI†). The carbonaceous matter include air-carbonised carbon from biomass (date seed,14 CNL1 carbon,46 cloves,47 sawdust48), hydrothermally carbonised hydrochar from biomass (starch,49 cellulose hydrochar,49,50 sawdust,19,49 lignin,51 jujun grass,52Camellia Japonica,52 cloves47), hydrochar from fresh cigarette filters,43 hydrochar from smoked cigarette filters,43 raw sawdust,53 hydrochar from cellulose acetate,50 and also carbon nanotube composites,54 and polypyrrole.18 It is interesting to note that IL-C has the lowest O/C ratio amongst the wide range of carbonaceous matter (Table S3, ESI†), which suggests that IL-ACxT carbons should exhibit comparatively high SAD. Thus, we compared (Tables S4 and S5, ESI†) the SAD of selected IL-AC carbons with those of similarly activated (in terms of KOH/precursor ratio and activation temperature) carbons from the other carbonaceous matter (Table S3, ESI†). The expectation is that the IL-AC carbons should have the highest SAD under any given activations conditions because of their low O/C ratio. In general, there is a clear linear relationship between the O/C ratio and SAD as has previously been demonstrated.14 However, the IL-AC samples, both IL-AC4700 and IL-AC4800, do not follow this trend (Fig. S26 and S27, ESI†). Whilst the expectation is that the IL-AC samples should have the highest SAD amongst the suite of carbons, the reality is that their SAD is much lower. This anomalous behavior can be explained by the counter effect of the high N content in IL-C. This behavior reveals the unique nature of the IL-C carbon, which is the genesis for generating ultra-high surface area carbons that still possess significant microporosity. These observations confirm that careful selection of a carbonaceous precursor can allow tailoring of the porosity of activated carbons. In the present case, it is clear that knowledge of the link between N-content and O/C ratio of a precursor and SAD can be a basis for the preparation of carbons with tailored or targeted porosity for specific applications.
The packing density of the IL-AC carbons (Table 2) reflects their micro/mesoporous nature. The packing density reduces as the overall porosity rises but still remains comparatively high. The volumetric surface area (defined as surface area × packing density) of the carbons in Table 2 shows some interesting trends. The volumetric surface area of IL-AC2T carbons is in the range 984 and 1370 m2 cm−3 and for IL-AC4T it is between 1183 and 1794 m2 cm−3 while for IL-AC6T carbons it ranges from 972 to 1438 m2 cm−3. The volumetric surface area of IL-AC2T carbons is limited by their relatively low surface area, while that of IL-AC6T is constrained by low packing density. On the other hand, samples IL-C4700 and IL-AC4800 have the highest volumetric surface area of, respectively, 1794 and 1558 m2 cm−3, due to their high surface area and moderate packing density. Such volumetric surface area is amongst the highest reported for any porous materials.14,19,55–60 Although some MOFs such as NU-1501-Al have been reported to have volumetric surface area as high as 2060 m2 cm−3, it is likely that such values are overestimated due to the fact that they are calculated using the crystallographic density of the MOFs instead of the packing density.60 The performance of porous materials in gas storage, and especially methane uptake, has recently been linked to their volumetric surface area.13,14,47,55 The expectation, therefore, is that the IL-ACxT samples that have high volumetric surface area (e.g. IL-AC4700 and IL-AC4800) would exhibit attractive methane uptake at pressures of 35 to 100 bar on both a gravimetric basis (due to high surface area and pore volume) and volumetric basis (due to enhanced packing density and volumetric surface area).
3.3 Methane storage of ionic-liquid derived activated carbons
To achieve efficient methane storage at medium to high pressures (35–100 bar), porous materials should possess a combination of micropores and mesopores and have high surface area. Given the unique properties of the IL-AC carbons in terms of their porosity and packing density, they are potentially excellent candidates for achieving enhanced methane storage at moderate to high pressures. The methane storage capacity was measured at 25 °C and pressures ranging from 0 to 100 bar. Uptake at 35, 65, and 100 bar was the main focus of this study since these pressures have been used to evaluate the performance of porous materials in many previous reports and therefore provide a convenient basis for comparing the performance of the present carbons to the state-of-the-art materials.6,8,10,13,55,57–62 The methane uptake measurements determined the excess storage capacity. The total methane storage capacity was then calculated from the excess uptake data by taking into account the total pore volume of the activated carbon, along with the density of methane at any given temperature and pressure. The following equation was applied:| θT = θExc + (dCH4 × VT) |
Fig. 4 shows the excess gravimetric methane uptake isotherms of the activated carbons. The excess methane storage capacity at 35, 65, and 100 bar is summarised in Table 3. The excess gravimetric uptake shows the following trends; (i) increases as KOH/IL-C ratio rises from 2 to 4 and then reduces for samples prepared at a ratio of 6, and (ii) increases as the activating temperature rises from 700 °C to 800 °C and then slightly decreases for carbons prepared at 900 °C. For example, at 35 bar, the excess methane uptake of samples IL-AC2700, IL-AC2800 and IL-AC2900 is 8.1, 12.5 and 11.1 mmol g−1, respectively, whereas IL-AC4700, IL-AC4800 and IL-AC4900 have higher uptake of 11.6, 12.7 and 12.5 mmol g−1, respectively. For IL-AC6700, IL-AC6800 and IL-AC6900, uptake was 9.7, 12.1 and 12.3 mmol g−1, respectively. The excess methane uptake of IL-AC4800 (12.7 mmol g−1) at 25 °C and 35 bar is among the best reported for any porous material.6,8,10,13,55,57–62 At higher pressures of 65 and 100 bar, the excess methane uptake ranges from 9.5 to 15.7 mmol g−1 and 10.1 to 17.2 mmol g−1, respectively, with IL-AC4T samples possessing the highest storage capacity. In general, the performance of the IL-AC4T samples is superior to the IL-AC2T or IL-AC6T series, which is in line with the trend in surface area and volumetric surface area (Table 2).
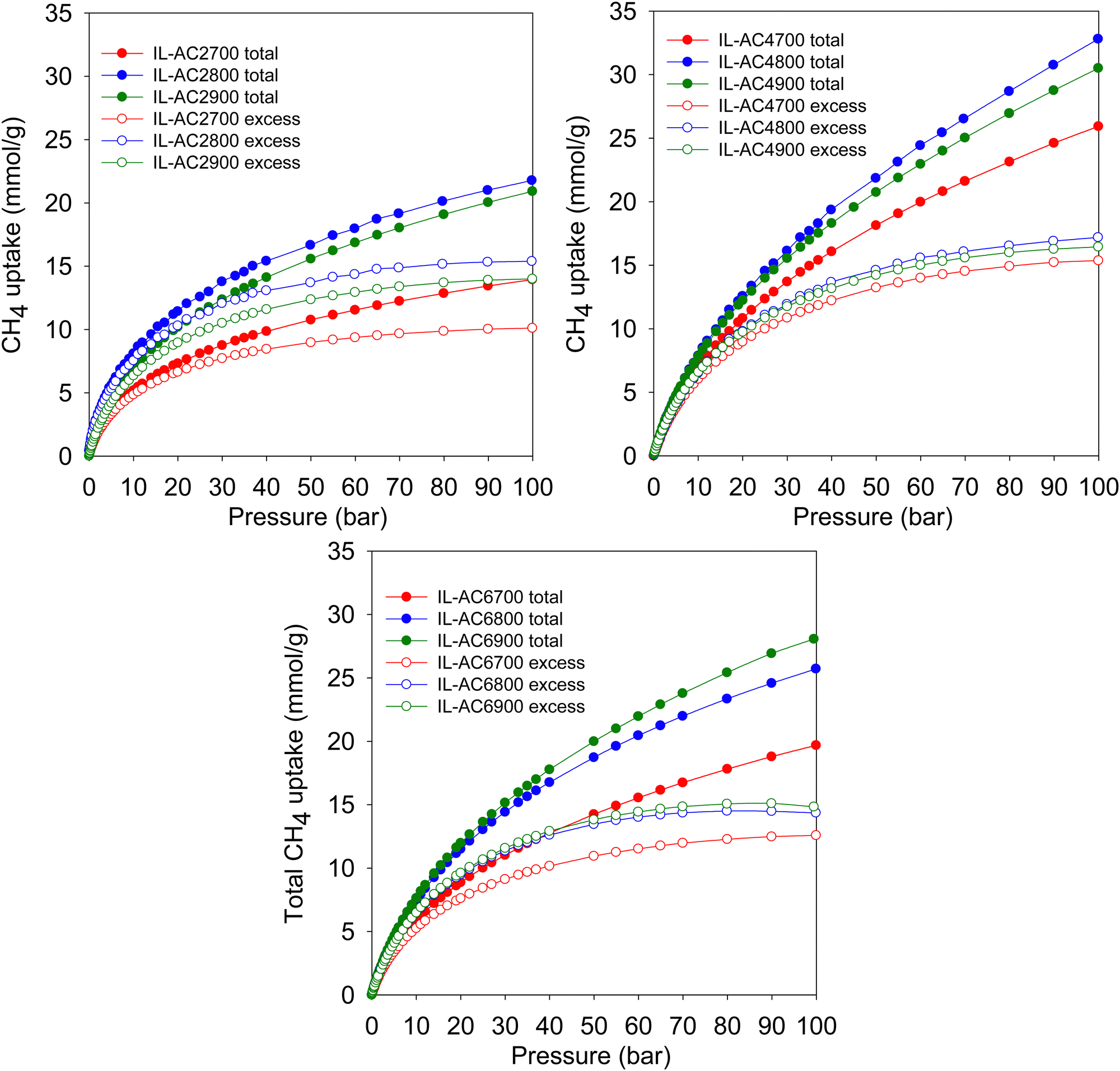 | ||
| Fig. 4 Gravimetric methane uptake isotherms. Excess and total gravimetric methane uptake at 25 °C for IL-ACxT activated carbons. | ||
| Sample | Gravimetric methane uptake | |||||
|---|---|---|---|---|---|---|
| 35 bar | 65 bar | 100 bar | ||||
| (mmol g−1) | (g g−1) | (mmol g−1) | (g g−1) | (mmol g−1) | (g g−1) | |
| IL-AC2700 | 9.3 (8.1) | 0.15 (0.13) | 11.9 (9.5) | 0.19 (0.15) | 13.9 (10.1) | 0.23 (0.16) |
| IL-AC2800 | 14.6 (12.5) | 0.23 (0.20) | 18.8 (14.9) | 0.30 (0.24) | 21.8 (15.4) | 0.35 (0.25) |
| IL-AC2900 | 13.3 (11.1) | 0.21 (0.18) | 17.5 (13.2) | 0.28 (0.21) | 20.9 (14.0) | 0.34 (0.22) |
| IL-AC4700 | 14.9 (11.6) | 0.24 (0.19) | 20.8 (14.3) | 0.33 (0.23) | 25.9 (15.4) | 0.42 (0.25) |
| IL-AC4800 | 17.7 (12.7) | 0.28 (0.20) | 25.4 (15.7) | 0.41 (0.25) | 32.8 (17.2) | 0.53 (0.28) |
| IL-AC4900 | 17.0 (12.5) | 0.27 (0.20) | 24.0 (15.3) | 0.39 (0.25) | 30.5 (16.5) | 0.49 (0.26) |
| IL-AC6700 | 11.9 (9.7) | 0.19 (0.16) | 16.2 (11.8) | 0.26 (0.19) | 19.7 (12.6) | 0.32 (0.20) |
| IL-AC6800 | 15.6 (12.1) | 0.25 (0.19) | 21.2 (14.2) | 0.34 (0.23) | 25.7 (14.4) | 0.41 (0.23) |
| IL-AC6900 | 16.5 (12.3) | 0.26 (0.20) | 22.9 (14.7) | 0.37 (0.24) | 28.1 (14.8) | 0.45 (0.24) |
The total gravimetric methane uptake isotherms are shown in Fig. 4. Table 3 shows a summary of the total gravimetric uptake at 35, 65, and 100 bar. The total methane uptake for IL-AC2T carbons is in the range of 9.3–14.6 mmol g−1 (at 35 bar), 11.9–18.8 mmol g−1 (at 65 bar) and 13.9–21.8 mmol g−1 (at 100 bar), respectively. The total gravimetric methane uptake at 35 bar ranged from 14.9 to 17.7 mmol g−1 for IL-AC4T carbons and from 11.9 to 16.5 mmol g−1 for IL-AC6T samples. The total gravimetric methane uptake by the IL-AC4T carbons is, in particular, higher than that of other reported carbon materials.12,13,63,64 Additionally, the total gravimetric methane uptake of IL-AC4T carbons at 65 bar ranges from 20.8 to 25.4 mmol g−1, while that of IL-AC6T carbons ranges from 16.2 to 22.9 mmol g−1. Total methane uptake for IL-AC4T carbons at 100 bar varies between 25.9 and 32.8 mmol g−1, while for IL-AC6T samples, the range is 19.7 to 28.1 mmol g−1. It is clear that the carbons prepared at KOH/IL-C, ratio 4 have the highest gravimetric uptake, which is in line with their high surface area and pore volume. Indeed, the total gravimetric uptake of IL-AC4800 at 100 bar and 25 °C at 32.8 mmol g−1 is one of the best for any carbon or MOF material.6,8,10,12–14,47,57–59,61–68
The United States Department of Energy (DOE) has set a methane uptake target of 0.5 g g−1 at 25 °C and pressure of 35 to 100 bar. The present IL-AC carbons have impressive gravimetric storage performance for methane storage, as shown in Table 4. In particular, the IL-AC4800 carbon surpasses the DOE gravimetric methane storage target with uptake of 0.53 g g−1 at 100 bar and 25 °C. Such a high gravimetric uptake matches or exceeds that of the best benchmark materials reported to date.6,8,10,12,13,47,57–59,61,62,64–68 Furthermore, other IL-AC samples (Table 4) also have gravimetric uptake that is very close to the DOE target; 0.42 g g−1 (IL-AC4700), 0.49 g g−1 (IL-AC4900), 0.41 g g−1 (IL-AC6800) and 0.45 g g−1 (IL-AC6900). As far as we are aware, these are the highest values ever reported for carbons and are comparable with the top values reported for MOFs.6,8,10,12,13,47,57–59,61,62,64–68 If the high gravimetric uptake of the IL-ACxT activated carbons were accompanied with high volumetric uptake, it would provide unique optimised materials for methane storage.
| Sample | Total volumetric methane uptakea (cm3 (STP) cm−3) | ||
|---|---|---|---|
| 35 bar | 65 bar | 100 bar | |
| a The numbers in parentheses represent the volumetric working capacity (i.e., deliverable methane), which is the difference between the gas uptake at pressure of 5 bar and the specified pressure (35, 65, or 100 bar). | |||
| IL-AC2700 | 159 (100) | 203 (144) | 238 (179) |
| IL-AC2800 | 163 (101) | 211 (149) | 244 (182) |
| IL-AC2900 | 118 (78) | 156 (116) | 187 (147) |
| IL-AC4700 | 165 (119) | 230 (184) | 287 (241) |
| IL-AC4800 | 156 (115) | 223 (182) | 289 (248) |
| IL-AC4900 | 116 (84) | 164 (132) | 209 (177) |
| IL-AC6700 | 151 (105) | 205 (159) | 249 (203) |
| IL-AC6800 | 130 (93) | 176 (139) | 213 (176) |
| IL-AC6900 | 115 (83) | 159 (127) | 195 (163) |
Methane storage performance is often measured in terms of the volumetric uptake. The DOE has set a target of 263 cm3 (STP) cm−3 at 25 °C and moderate pressure (i.e., 35–100 bar), in addition to gravimetric uptake of at least 0.5 g g−1. The gravimetric uptake and the packing density of an adsorbent are the two most important factors in determining volumetric uptake. Fig. 5 displays the excess and total volumetric methane uptake isotherms and Table 4 summarises the total volumetric uptake and the working capacity (i.e., deliverable methane at 5 bar) for storage at 35, 65, and 100 bar. The working capacity is calculated as the difference between adsorption at 5 bar (delivery pressure) and at uptake pressure (35, 65 or 100 bar). For IL-AC2T carbons, the total volumetric uptake (cm3 (STP) cm−3) was in the range of 118–163, 156–211 and 187–244 at 35, 65 and 100 bar, respectively. For IL-AC6T carbons, the uptake ranges between 115 and 151 at 35 bar, 159 and 205 at 65 bar, and 195 and 249 at 100 bar. The volumetric uptake of IL-AC4700, IL-AC4800 and IL-AC4900 carbons is even more attractive; 165, 156 and 116, respectively, at 35 bar, 230, 223 and 164, respectively, at 65 bar, and 287, 289 and 209, respectively, at 100 bar. The IL-AC4T carbons show no saturation at 100 bar in their total volumetric uptake, which suggests that the carbons are capable of storing more methane at higher pressures. This contrasts with MOFs, which tend to show saturation at ca. 80 bar.69,70 The volumetric uptake is amongst the best reported to date for carbon or MOF materials, and is crucially based on experimentally determined packing density.6,8–10,12–14,55,57–60,62–64,69–72 The present carbons also show impressive performance in terms of their working capacity, which is higher than that of most porous materials; the highest deliverable methane for pressure swing of 100 to 5 bar at 25 °C is for IL-AC4700 and IL-AC4800 at 241 and 248 cm3 (STP) cm−3, respectively. More importantly, the present IL-AC carbons, and in particular sample IL-AC4800, are the first porous materials (carbon or MOF) to meet both gravimetric and volumetric storage targets at 25 °C.6,8–10,12–14,47,55,57–64,69–72 At 100 bar, the volumetric uptake capacity of IL-AC4700, IL-AC4800 and IL-AC4900 at, respectively, 287, 289 and 209 cm3 (STP) cm−3, coupled with their high gravimetric uptake of 0.42, 0.53 and 0.49 g g−1, offer impressive storage combinations that are superior to any previous reports for porous carbon or MOF materials.6,8–10,12–14,47,55,57–64,69–72
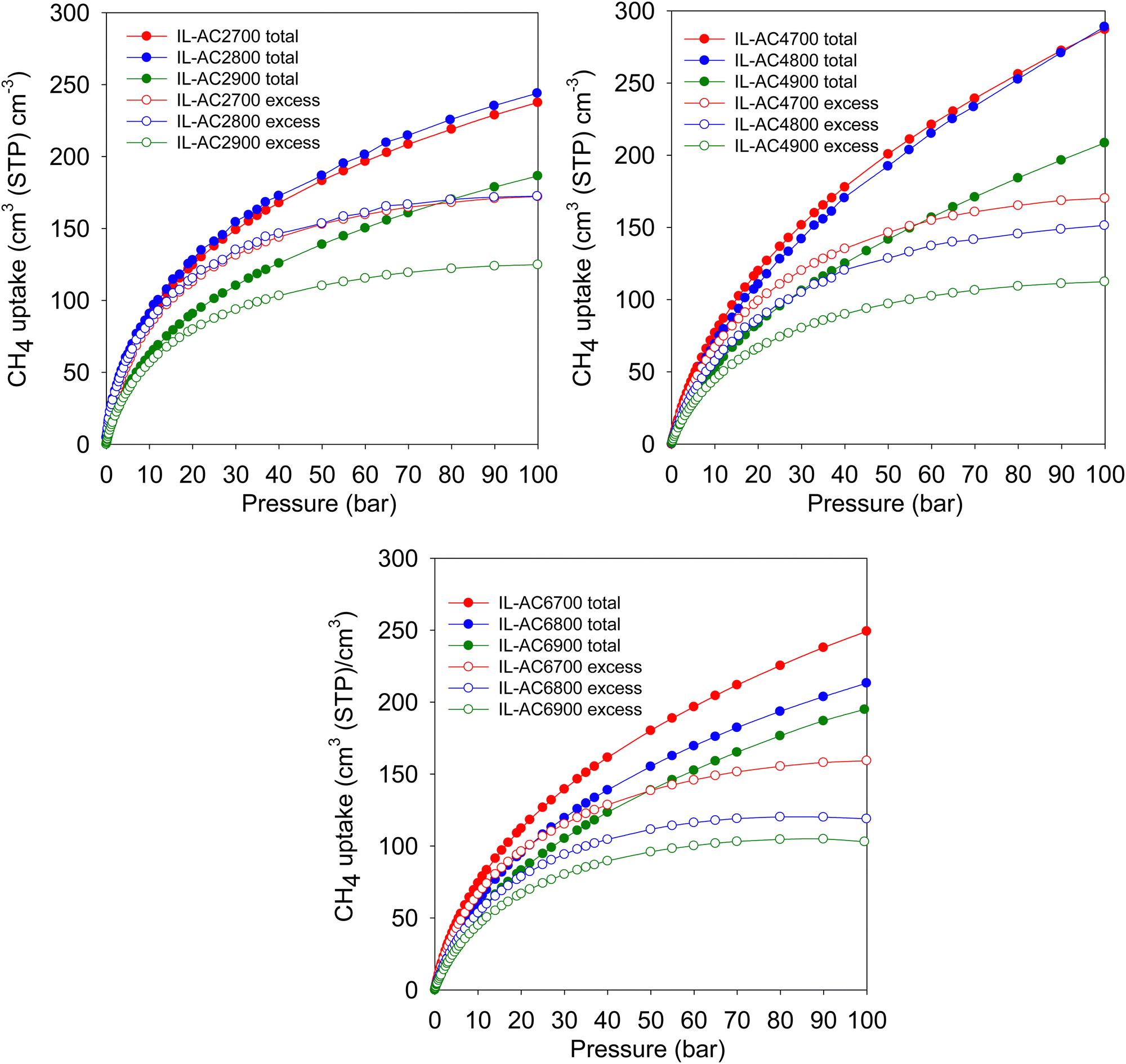 | ||
| Fig. 5 Volumetric methane uptake isotherms. Excess and total volumetric methane uptake at 25 °C for IL-ACxT activated carbons. | ||
Regarding the nature of adsorption of methane, the FTIR spectra of IL-ACxT carbons (Fig. S11, ESI†) indicate that there are no functional groups on the activated carbons except those associated with remnants of the imidazolium backbone and which give rise to stretching and bending vibrations of the aromatic structure. FTIR spectra of an activated carbon taken before and after methane uptake (Fig. S11, ESI†) indicates that the sorption process does not cause any variation in the functional groups on the surface, i.e., IR spectra remains unchanged. This is consistent with the uptake being based on physisorption rather than any strong interaction with surface functional groups.
From the foregoing discussion, it is clear that the methane uptake is chiefly dependent on the porosity of the carbons. Further insights on the performance of the carbons may be obtained from the isosteric heat of adsorption (Qst). Fig. 6 shows the Qst of the carbons as a function of methane loading. The Qst is in the range of 23 to 13 kJ mol−1 for methane loading of up to 7 mmol g−1. The Qst at low coverage, which is the best guide of the interaction between methane molecules and the carbons’ surface, is in the range of 23 to 15 kJ mol−1. This is within the range (10–25 kJ mol−1) previously reported for activated carbons and other porous materials, including MOFs.8,57 There seems to be a correlation between the Qst and pore size of the carbons, meaning that the Qst order is IL-AC2T > IL-AC4T > IL-AC6T, except for sample IL-AC6700, which shows anomalously high Qst. The observed correlation between Qst and pore size is consistent with the fact that smaller pores can enhance the interaction between methane and the carbon pore walls. However, whilst a high Qst can favour enhanced methane storage capacity, it can also act to limit the working capacity (deliverable methane) due to high uptake at low pressure, i.e., the deliverable pressure of 5 bar. Indeed, a closer look at the data in Table 4 indicates that the working capacity of IL-AC2T carbons is a lower proportion of the total uptake compared IL-AC4T and IL-AC6T carbons. For example, for a 35 to 5 bar swing the deliverable proportion is ca. 63% for IL-AC2T compared to at least 71% for IL-AC4T. For a 65 to 5 bar swing, the deliverable proportion is ca. 71% for IL-AC2T and 80% for IL-AC4T while for 100 to 5 bar it is 75% and 85% for IL-AC2T and IL-AC4T, respectively. This trend can be explained by the proportionately higher uptake at 5 bar for the IL-AC2T samples due to their higher Qst. In this regard, the porosity and Qst of the IL-AC4T carbons appears to offer a more optimised balance between low pressure uptake and total uptake, which translates to excellent working capacity.
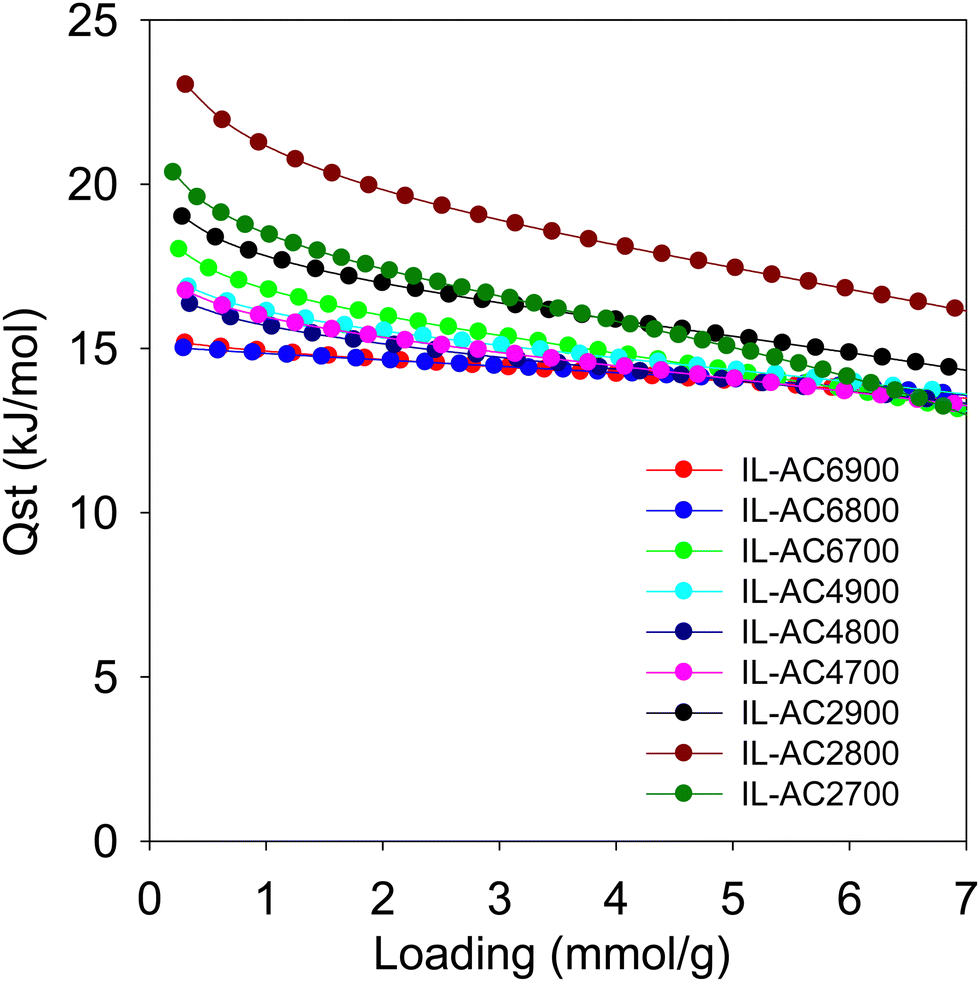 | ||
| Fig. 6 Isosteric heat of adsorption (Qst) of methane. Qst of methane on IL-ACxT carbons as a function of uptake, which is given as loading in mmol of methane per gram of carbon. | ||
To be viable for methane storage, porous materials need to be used over several cycles, which means that cyclability is an important consideration. We assessed the performance of one of the best performing carbons, IL-AC4800, over several cycles (Fig. S28, ESI†). The assessment also allowed us to clarify on any impact of compaction on the methane uptake. In this way, the first use (cycle 1) was measured for powdered IL-AC4800, i.e., before compaction, while the subsequent cycles (2, 3 and 4) were measured in pellet form after compaction of the activated carbon at 370 MPa for 10 min in a 1.3 cm (diameter) die. The uptake data over several cycles (Fig. S28, ESI†) show a high level of recyclability with no reduction in storage capacity. Furthermore, it is also clear that compaction had no impact on the methane uptake, which is consistent with the fact that the porosity (surface area, pore volume and pore size) remains largely unchanged. Our findings are in agreement with previous reports on the mechanical stability of high surface area activated carbons.47 The non-changing porosity means that compaction will also not affect the kinetics of adsorption and desorption of the methane. The pores of the IL-ACxT carbons are wide enough to facilitate easy ingress and egress of methane meaning that diffusion will not be a limiting factor in the performance of the carbons as storage materials. This is possible because the majority of pores of the IL-ACxT carbons are in the range of 10 to 50 Å, compared to 3.8 Å for the methane molecule. Indeed, previous studies have shown that the average methane mass transfer coefficient (MTC) for carbons, such as AX21, with porosity similar to that of the IL-ACxT samples is 12.8 min−1 at 27 °C, along with an effective diffusivity (Deff) of 1.79 × 10−13 m2 s−1.73 Most porous materials (activated carbons, MOFs and zeolites) exhibit such MTC and Deff values.74 This means that for the IL-ACxT carbons, the methane diffusion coefficients, under ambient temperature conditions, are at a level that is suitable for methane storage (uptake) at high pressure and release at lower pressures via pressure swing operations.73,74
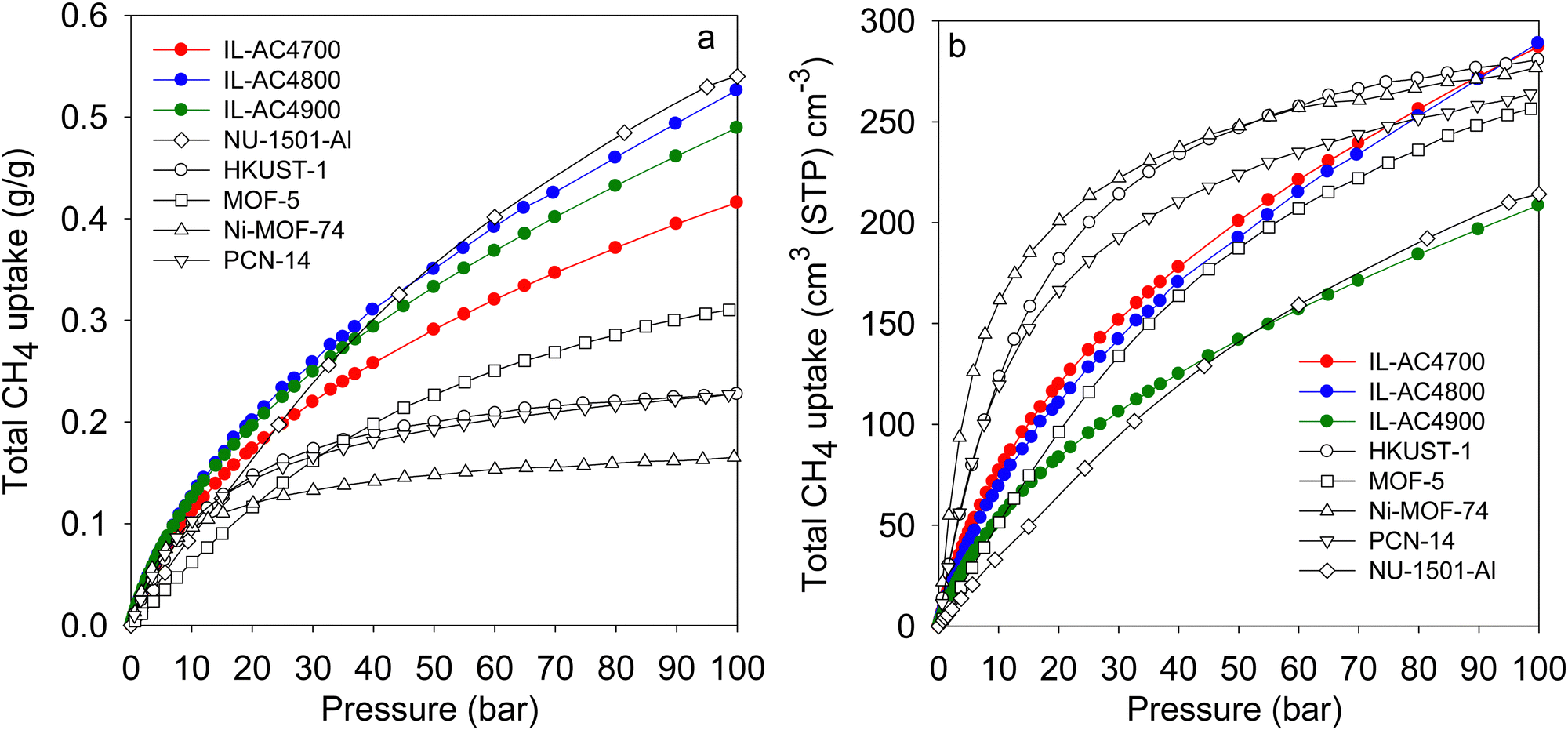 | ||
| Fig. 7 Comparative methane uptake of IL-AC4T activated carbons. (a) Total gravimetric methane uptake at 25 °C of IL-AC4T carbons compared to benchmark MOF materials. (b) Total volumetric methane storage capacity at 25 °C of IL-AC4T carbons compared to that of MOFs, for which the volumetric uptake was calculated using crystallographic density. Data for Nu-1501-A is from ref. 60, HKUST-1 and Ni-MOF-74 from ref. 57, MOF-5 from ref. 72 and PCN-14 from ref. 75 and 76. | ||
Most studies on methane storage in porous materials have been performed at ambient temperature (typically 25 °C), which is considered the most relevant for vehicular applications. However, other storage conditions have been explored. A notable study is that of Rozyyev and co-workers who reported on a flexible aromatic network porous polymer, COP-150, with attractive methane storage at 0 °C.68 The nature of COP-150 is such that it has very low volumetric and gravimetric uptake at 25 °C, which in combination with a relatively low packing density (0.34 g cm−3) suggest that it is not competitive. However, due to flexible porosity, COP-150 has attractive gravimetric and volumetric methane storage capacity of 0.64 g g−1 and 301 cm3 (STP) cm−3, respectively, at 0 °C and 100 bar. To have a fuller picture of the performance of the IL-ACxT carbons we measured the methane uptake for IL-AC4T samples at 0 °C (Fig. S31, ESI†). As expected the methane uptake at 0 °C is higher than that at 25 °C. At 100 bar, the total gravimetric methane uptake is 29.7 mmol g−1 (0.48 g g−1) for IL-AC4700, 37.3 mmol g−1 (0.60 g g−1) for IL-AC800, and 34.8 mmol g−1 (0.56 g g−1) for IL-AC4900. The volumetric uptake at 100 bar is, therefore, 329 cm3 (STP) cm−3 for IL-AC4700, 326 cm3 (STP) cm−3 for IL-AC4800 and 245 cm3 (STP) cm−3 for IL-AC4900. Thus, the storage capacity of IL-AC4800 matches that of COP-150 both in terms of gravimetric and volumetric uptake. The working capacity for IL-AC4800 for a pressure swing of 100 to 5 bar is 0.5 g g−1 and 273 cm3 (STP) cm−3, which is comparable to that of COP-150.
Next, we consider how the mix and level of porosity of the three samples (ACDS4800, PPY4800 and IL-AC4800) affects their gravimetric methane uptake. This is best achieved by plotting the total gravimetric methane uptake (Table S10, ESI†) at selected pressures (35, 65 and 100 bar) as a function of the surface area or pore volume of the carbons (Fig. S33, ESI†). The total gravimetric methane storage capacity at any uptake pressure increases linearly with both the surface area and pore volume. This finding indicates that the high gravimetric methane uptake of the IL-AC4800 carbon is due to its ultra-high porosity. Furthermore, the high gravimetric uptake when combined with the relatively undiminished packing density means that the IL-AC4800 carbon also exhibits very attractive volumetric methane storage capacity. On the other hand, ACDS4800 has high volumetric uptake due to its higher packing density but its overall storage performance is let down by low gravimetric uptake because of lower surface area and pore volume. For PPY4800, the moderate gravimetric uptake translates to low or moderate volumetric uptake due to the diminished packing density. The unique nature of IL-AC4800, in terms of porosity and packing density, means that it achieves both high gravimetric and volumetric methane storage.
The effect of adding N to the activation mix on the porosity of activated carbons is already well known.21–24 The presence of N in the framework of a carbonaceous precursor plays a role in enhancing the surface area and pore volume of activated carbons as demonstrated in this study and prior reports.21–24 This is because N acts as an in situ porogen, which favours generation of carbons that tend towards higher mesoporosity and high surface area. This arises from the fact that N is very readily lost during the activation process as it is more easily oxidised and eliminated than C. Moreover, the effect of N as an in situ porogen is also evidenced by the fact that although the IL-C precursor has low O/C ratio, and therefore should yield low to moderate surface area carbons, it shows anomalous behaviour by generating carbons with high surface area. This anomalous behaviour (Fig. S26 and S27, ESI†) is due to the presence of N in the IL-C precursor. The overall effect of having a precursor with low O/C ratio and high N content is to generate activated carbons that have just the right combination of porosity (i.e., high surface area and high pore volume arising from a mix of mesoporosity and microporosity) and high packing density required for enhanced methane storage for both gravimetric and volumetric uptake.
4. Conclusions
Activated carbons with a mix of micro/mesoporosity have been prepared via carbonisation of an ionic liquid, IL, (1-butyl-3-methylimidazolium tricyanomethanide, [BMIm][C(CN)3]) followed by chemical activation of the resulting carbonaceous matter with KOH. On carbonisation, the IL yields carbonaceous matter with the unusual combination of both a high N content and low O/C atomic ratio. On activation, the IL-derived carbonaceous matter (IL-C) generates activated carbons with a mix of micro and mesoporosity that have ultra-high surface area of up to ∼4000 m2 g−1 and pore volume of up to 3.3 cm3 g−1, along with comparatively high packing density. The porosity and packing density of the activated carbons is a consequence of the elemental composition of the IL-C precursor. In particular, the low O/C ratio and high N content of the IL-C precursor play a role in determining the porosity. In this way, the IL-C precursor shows anomalous activation behaviour when compared to other traditional carbonaceous matter. The IL-derived activated carbons are ideal for the storage of methane because of their well-balanced combination of microporosity and mesoporosity, high packing density, and large surface area. The best carbons achieve methane uptake of up to 0.53 g g−1 and 289 cm3 (STP) cm−3 at 25 °C and 100 bar. Such uptake exceeds the storage targets for both gravimetric and volumetric uptake set by the US Department of Energy of 0.5 g g−1 and 263 cm3 (STP) cm−3, which would allow widespread use for vehicular transport and other purposes. In addition, the IL-derived carbons achieve attractive deliverable methane capacity (working capacity) of up to 0.46 g g−1 and 248 cm3 (STP) cm−3 for a pressure swing of 100 to 5 bar at 25 °C. The best performing IL-derived activated carbons meet both gravimetric and volumetric methane storage targets.Author contributions
N. A: methodology, formal analysis, investigation, writing. L. S. B.: methodology, formal analysis, validation. R. M.: conceptualisation, writing – review & editing, supervision, funding acquisition, resources.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Nanoscale and Microscale Research Centre (nmRC) at the University of Nottingham for assistance with XPS and microscopy analysis. We thank Taibah University, Yanbu Al Bahr, 46423, Saudi Arabia, for funding a PhD studentship for Nawaf Albeladi. RM thanks the Royal Society for a Research Grant, and for a Royal Society Wolfson Research Merit Award.References
- International Energy Agency (IEA), Global CO2 emissions rebounded to their highest level in history in 2021, https://www.iea.org/reports/global-energy-review-co2-emissions-in-2021, (accessed March 8, 2022).
- C. F. Schleussner, J. Rogelj, M. Schaeffer, T. Lissner, R. Licker, E. M. Fischer, R. Knutti, A. Levermann, K. Frieler and W. Hare, Nat. Clim. Change, 2016, 6, 827–835 CrossRef.
- M. S. Dresselhaus and I. L. Thomas, Nature, 2001, 414, 332–337 CrossRef CAS PubMed.
- S. Choi, J. H. Drese and C. W. Jones, ChemSusChem, 2009, 2, 796–854 CrossRef CAS PubMed.
- U. Eberle, B. Muller and R. von Helmolt, Energy Environ. Sci., 2012, 5, 8780–8798 RSC.
- K. V. Kumar, K. Preuss, M. M. Titirici and F. Rodriguez-Reinoso, Chem. Rev., 2017, 117, 1796–1825 CrossRef CAS PubMed.
- I. Angelidaki, L. Treu, P. Tsapekos, G. Luo, S. Campanaro, H. Wenzel and P. G. Kougias, Biotechnol. Adv., 2018, 36, 452–466 CrossRef CAS PubMed.
- T. A. Makal, J.-R. Li, W. Lu and H.-C. Zhou, Chem. Soc. Rev., 2012, 41, 7761–7779 RSC.
- D. Lozano-Castello, J. Alcaniz-Monge, M. A. de la Casa-Lillo, D. Cazorla-Amoros and A. Linares-Solano, Fuel, 2002, 81, 1777–1803 CrossRef CAS.
- J. Romanos, S. Sweany, T. Rash, L. Firlej, B. Kuchta, J. C. Idrobo and P. Pfeifer, Adsorpt. Sci. Technol., 2014, 32, 681–691 CrossRef CAS.
- D. Z. Li, L. Chen, G. Liu, Z. Y. Yuan, B. F. Li, X. Zhang and J. Q. Wei, New Carbon Mater., 2021, 36, 468–492 CrossRef CAS.
- A. Alonso, J. Moral-Vico, A. A. Markeb, M. Busquets-Fité, D. Komilis, V. Puntes, A. Sánchez and X. Font, Sci. Total Environ., 2017, 595, 51–62 CrossRef CAS PubMed.
- A. Altwala and R. Mokaya, J. Mater. Chem. A, 2022, 10, 13744–13757 RSC.
- A. Altwala and R. Mokaya, Energy Environ. Sci., 2020, 13, 2967–2978 RSC.
- M. Sevilla and R. Mokaya, Energy Environ. Sci., 2014, 7, 1250–1280 RSC.
- L. S. Blankenship and R. Mokaya, Mater. Adv., 2022, 3, 1905–1930 RSC.
- Z. H. Hu, M. P. Srinivasan and Y. M. Ni, Carbon, 2001, 39, 877–886 CrossRef CAS.
- M. Sevilla, R. Mokaya and A. B. Fuertes, Energy Environ. Sci., 2011, 4, 2930–2936 RSC.
- N. Balahmar and R. Mokaya, J. Mater. Chem. A, 2019, 7, 17466–17479 RSC.
- A. M. Aljumialy and R. Mokaya, Mater. Adv., 2020, 1, 3267–3280 RSC.
- J. V. Guerrera, J. N. Burrow, J. E. Eichler, M. Z. Rahman, M. V. Namireddy, K. A. Friedman, S. S. Coffman, D. C. Calabro and C. B. Mullins, Energy Fuels, 2020, 34, 6101–6112 CrossRef CAS.
- M. Sevilla, A. S. M. Al-Jumialy, A. B. Fuertes and R. Mokaya, ACS Appl. Mater. Interfaces, 2018, 10, 1623–1633 CrossRef CAS PubMed.
- N. Tsubouchi, M. Nishio and Y. Mochizuki, Appl. Surf. Sci., 2016, 371, 301–306 CrossRef CAS.
- M. Sevilla, W. Sangchoom, N. Balahmar, A. B. Fuertes and R. Mokaya, ACS Sustainable Chem. Eng., 2016, 4, 4710–4716 CrossRef CAS.
- S. G. Zhang, K. Dokko and M. Watanabe, Mater. Horiz., 2015, 2, 168–197 RSC.
- P. F. Fulvio, J. S. Lee, R. T. Mayes, X. Wang, S. M. Mahurin and S. Dai, Phys. Chem. Chem. Phys., 2011, 13, 13486–13491 RSC.
- J. P. Paraknowitsch, J. Zhang, D. Su, A. Thomas and M. Antonietti, Adv. Mater., 2010, 22, 87–92 CrossRef CAS PubMed.
- J. S. Lee, X. Wang, H. Luo and S. Dai, Adv. Mater., 2010, 22, 1004–1007 CrossRef CAS PubMed.
- J. S. Lee, X. Wang, H. Luo, G. A. Baker and S. Dai, J. Am. Chem. Soc., 2009, 131, 4596–4597 CrossRef CAS PubMed.
- J. Rouquerol, P. Llewellyn and F. Rouquerol, Stud. Surf. Sci. Catal., 2006, 160, 49–56 CrossRef.
- A. Galarneau, D. Mehlhorn, F. Guenneau, B. Coasne, F. Villemot, D. Minoux, C. Aquino and J. P. Dath, Langmuir, 2018, 34, 14134–14142 CrossRef CAS PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- P. Iacomi and P. L. Llewellyn, Adsorption, 2019, 25, 1533–1542 CrossRef CAS.
- P. B. Whittaker, X. Wang, K. Regenauer-Lieb and H. T. Chua, Phys. Chem. Chem. Phys., 2013, 15, 473–482 RSC.
- M. Sevilla, P. Valle-Vigon and A. B. Fuertes, Adv. Funct. Mater., 2011, 21, 2781–2787 CrossRef CAS.
- J. Kim, J. H. Jeong, H. J. Ahn, J. S. Lee and K. C. Roh, ChemElectroChem, 2020, 7, 2410–2417 CrossRef CAS.
- J. P. Paraknowitsch, A. Thomas and M. Antonietti, J. Mater. Chem., 2010, 20, 6746–6758 RSC.
- X. Zhang, D. Lin and W. Chen, RSC Adv., 2015, 5, 45136–45143 RSC.
- B. Adeniran and R. Mokaya, Nano Energy, 2015, 16, 173–185 CrossRef CAS.
- M. Cox and R. Mokaya, Sustain. Energy Fuels, 2017, 1, 1414–1424 RSC.
- A. S. Jalilov, Y. L. Li, J. Tian and J. M. Tour, Ad. Energy Mater., 2017, 7, 1600693 CrossRef.
- J. W. M. Osterrieth, J. Rampersad, D. Madden, N. Rampal, L. Skoric, B. Connolly, M. D. Allendorf, V. Stavila, J. L. Snider, R. Ameloot, J. Marreiros, C. Ania, D. Azevedo, E. Vilarrasa-Garcia, B. F. Santos, X.-H. Bu, Z. Chang, H. Bunzen, N. R. Champness, S. L. Griffin, B. Chen, R.-B. Lin, B. Coasne, S. Cohen, J. C. Moreton, Y. J. Colón, L. Chen, R. Clowes, F.-X. Coudert, Y. Cui, B. Hou, D. M. D'Alessandro, P. W. Doheny, M. Dincă, C. Sun, C. Doonan, M. T. Huxley, J. D. Evans, P. Falcaro, R. Ricco, O. Farha, K. B. Idrees, T. Islamoglu, P. Feng, H. Yang, R. S. Forgan, D. Bara, S. Furukawa, E. Sanchez, J. Gascon, S. Telalović, S. K. Ghosh, S. Mukherjee, M. R. Hill, M. M. Sadiq, P. Horcajada, P. Salcedo-Abraira, K. Kaneko, R. Kukobat, J. Kenvin, S. Keskin, S. Kitagawa, K. Otake, R. P. Lively, S. J. A. DeWitt, P. Llewellyn, B. V. Lotsch, S. T. Emmerling, A. M. Pütz, C. Martí-Gastaldo, N. M. Padial, J. García-Martínez, N. Linares, D. Maspoch, J. A. Suárez del Pino, P. Moghadam, R. Oktavian, R. E. Morris, P. S. Wheatley, J. Navarro, C. Petit, D. Danaci, M. J. Rosseinsky, A. P. Katsoulidis, M. Schröder, X. Han, S. Yang, C. Serre, G. Mouchaham, D. S. Sholl, R. Thyagarajan, D. Siderius, R. Q. Snurr, R. B. Goncalves, S. Telfer, S. J. Lee, V. P. Ting, J. L. Rowlandson, T. Uemura, T. Iiyuka, M. A. van der Veen, D. Rega, V. V. Speybroeck, S. M. J. Rogge, A. Lamaire, K. S. Walton, L. W. Bingel, S. Wuttke, J. Andreo, O. Yaghi, B. Zhang, C. T. Yavuz, T. S. Nguyen, F. Zamora, C. Montoro, H. Zhou, A. Kirchon and D. Fairen-Jimenez, Adv. Mater., 2022, 35, 2201502 CrossRef PubMed.
- T. S. Blankenship and R. Mokaya, Energy Environ. Sci., 2017, 10, 2552–2562 RSC.
- W. Sangchoom, D. A. Walsh and R. Mokaya, J. Mater. Chem. A, 2018, 6, 18701–18711 RSC.
- J. J. He, J. W. F. To, P. C. Psarras, H. P. Yan, T. Atkinson, R. T. Holmes, D. Nordlund, Z. N. Bao and J. Wilcox, Adv. Energy Mater., 2016, 6, 1502491 CrossRef.
- E. Haffner-Staton, N. Balahmar and R. Mokaya, J. Mater. Chem. A, 2016, 4, 13324–13335 RSC.
- I. Alali and R. Mokaya, Energy Environ. Sci., 2022, 15, 4710–4724 RSC.
- E. A. Hirst, A. Taylor and R. Mokaya, J. Mater. Chem. A, 2018, 6, 12393–12403 RSC.
- M. Sevilla, A. B. Fuertes and R. Mokaya, Energy Environ. Sci., 2011, 4, 1400–1410 RSC.
- L. S. Blankenship, N. Balahmar and R. Mokaya, Nat. Commun., 2017, 8, 1545 CrossRef PubMed.
- W. Sangchoom and R. Mokaya, ACS Sustainable Chem. Eng., 2015, 3, 1658–1667 CrossRef CAS.
- H. M. Coromina, D. A. Walsh and R. Mokaya, J. Mater. Chem. A, 2016, 4, 280–289 RSC.
- N. Balahmar, A. S. Al-Jumialy and R. Mokaya, J. Mater. Chem. A, 2017, 5, 12330–12339 RSC.
- B. Adeniran and R. Mokaya, J. Mater. Chem. A, 2015, 3, 5148–5161 RSC.
- T. Tian, Z. Zeng, D. Vulpe, M. E. Casco, G. Divitini, P. A. Midgley, J. Silvestre-Albero, J. C. Tan, P. Z. Moghadam and D. Fairen-Jimenez, Nat. Mater., 2018, 17, 174–179 CrossRef CAS PubMed.
- E. Masika and R. Mokaya, Energy Environ. Sci., 2014, 7, 427–434 RSC.
- J. A. Mason, M. Veenstra and J. R. Long, Chem. Sci., 2014, 5, 32–51 RSC.
- B. Li, H. M. Wen, W. Zhou, J. Q. Xu and B. L. Chen, Chem, 2016, 1, 557–580 CAS.
- Y. He, W. Zhou, G. Qian and B. Chen, Chem. Soc. Rev., 2014, 43, 5657–5678 RSC.
- Z. Chen, P. Li, R. Anderson, X. Wang, X. Zhang, L. Robison, L. R. Redfern, S. Moribe, T. Islamoglu, D. A. Gomez-Gualdron, T. Yildirim, J. F. Stoddart and O. K. Farha, Science, 2020, 368, 297–303 CrossRef CAS PubMed.
- P. Pfeifer, L. Aston, M. Banks, S. Barker, J. Burress, S. Carter, J. Coleman, S. Crockett, C. Faulhaber, J. Flavin, M. Gordon, L. Hardcastle, Z. Kallenborn, M. Kemiki, C. Lapilli, J. Pobst, R. Schott, P. Shah, S. Spellerberg, G. Suppes, D. Taylor, A. Tekeei, C. Wexler, M. Wood, P. Buckley, T. Breier, J. Downing, S. Eastman, P. Freeze, S. Graham, S. Grinter, A. Howard, J. Martinez, D. Radke, T. Vassalli and J. Ilavsky, CHAOS, 2007, 17, 041108 CrossRef CAS PubMed.
- D. A. Gómez-Gualdrón, C. E. Wilmer, O. K. Farha, J. T. Hupp and R. Q. Snurr, J. Phys. Chem. C, 2014, 118, 6941–6951 CrossRef.
- M. E. Casco, F. Rey, J. L. Jorda, S. Rudic, F. Fauth, M. Martinez-Escandell, F. Rodriguez-Reinoso, E. V. Ramos-Fernandez and J. Silvestre-Albero, Chem. Sci., 2016, 7, 3658–3666 RSC.
- M. E. Casco, M. Martinez-Escandell, K. Kaneko, J. Silvestre-Albero and F. Rodriguez-Reinoso, Carbon, 2015, 93, 11–21 CrossRef CAS.
- C. M. Simon, J. Kim, D. A. Gomez-Gualdron, J. S. Camp, Y. G. Chung, R. L. Martin, R. Mercado, M. W. Deem, D. Gunter, M. Haranczyk, D. S. Sholl, R. Q. Snurr and B. Smit, Energy Environ. Sci., 2015, 8, 1190–1199 RSC.
- B. M. Connolly, M. Aragones-Anglada, J. Gandara-Loe, N. A. Danaf, D. C. Lamb, J. P. Mehta, D. Vulpe, S. Wuttke, J. Silvestre-Albero, P. Z. Moghadam, A. E. H. Wheatley and D. Fairen-Jimenez, Nat. Commun., 2019, 10, 2345 CrossRef CAS PubMed.
- B. M. Connolly, D. G. Madden, A. E. H. Wheatley and D. Fairen-Jimenez, J. Am. Chem. Soc., 2020, 142, 8541–8549 CrossRef CAS PubMed.
- V. Rozyyev, D. Thirion, R. Ullah, J. Lee, M. Jung, H. Oh, M. Atilhan and C. T. Yavuz, Nat. Energy, 2019, 4, 604–611 CrossRef CAS.
- S. Bracco, D. Piga, I. Bassanetti, J. Perego, A. Comotti and P. Sozzani, J. Mater. Chem. A, 2017, 5, 10328–10337 RSC.
- Y. Peng, V. Krungleviciute, I. Eryazici, J. T. Hupp, O. K. Farha and T. Yildirim, J. Am. Chem. Soc., 2013, 135, 11887–11894 CrossRef CAS PubMed.
- M. E. Casco, M. Martínez-Escandell, E. Gadea-Ramos, K. Kaneko, J. Silvestre-Albero and F. Rodríguez-Reinoso, Chem. Mater., 2015, 27, 959–964 CrossRef CAS.
- H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. O. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424–428 CrossRef CAS PubMed.
- N. Bimbo, J. P. Smith, H. Aggarwal, A. J. Physick, A. Pugsley, L. J. Barbour, V. P. Ting and T. J. Mays, Chem. Eng. Res. Des., 2021, 169, 153–164 CrossRef CAS.
- D. Cao, X. Zhang, J. Chen and J. Yun, Carbon, 2003, 41, 2653–2689 CrossRef.
- S. Ma, D. Sun, J. M. Simmons, C. D. Collier, D. Yuan and H.-C. Zhou, J. Am. Chem. Soc., 2008, 130, 1012–1016 CrossRef CAS PubMed.
- S. Dutta, A. Bhaumik and K. C. W. Wu, Energy Environ. Sci., 2014, 7, 3574–3592 RSC.
- I. Alali and R. Mokaya, J. Mater. Chem. A, 2023, 11, 6952–6965 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary Information accompanying this paper is available. Data that support the findings of this study are available within the paper (and its Supplementary Information Files) and from the corresponding author on reasonable request. See DOI: https://doi.org/10.1039/d3ee03957a |
| This journal is © The Royal Society of Chemistry 2024 |